

Abstract
Microencapsulated phase change materials (PCMs) are attracting increasing attention as functional fillers in polymer matrices, to produce smart thermoregulating composites for applications in thermal energy storage (TES) and thermal management. In a polymer composite, the filler–matrix interfacial adhesion plays a fundamental role in the thermomechanical properties. Hence, this work aims to modify the surface of commercial PCM microcapsules through the formation of a layer of polydopamine (PDA), a bioinspired polymer that is emerging as a powerful tool to functionalize chemically inert surfaces due to its versatility and great adhesive potential in many different materials. Scanning electron microscopy (SEM) and atomic force microscopy (AFM) evidenced that after PDA coating, the surface roughness increased from 9 to 86 nm, which is beneficial, as it allows a further increase in the interfacial interaction by mechanical interlocking. Spectroscopic techniques allowed investigating the surface chemistry and identifying reactive functional groups of the PDA layer and highlighted that, unlike the uncoated microcapsules, the PDA layer is able to react with oxirane groups, thereby forming a covalent bond with the epoxy matrix. Hot-stage optical microscopy and differential scanning calorimetry (DSC) highlighted that the PDA modification does not hinder the melting/crystallization process of the paraffinic core. Finally, SEM micrographs of the cryofracture surface of epoxy composites containing neat or PDA-modified microcapsules clearly evidenced improved adhesion between the capsule shell and the epoxy matrix. These results showed that PDA is a suitable coating material with considerable potential for increasing the interfacial adhesion between an epoxy matrix and polymer microcapsules with low surface reactivity. This is remarkably important not only for this specific application but also for other classes of composite materials. Future studies will investigate how the deposition parameters affect the morphology, roughness, and thickness of the PDA layer and how the layer properties influence the capsule–matrix adhesion.
1. Introduction
Thermal energy storage (TES) has recently become an important tool for energy management and an effective way to address environmental concerns related to fossil fuel depletion, air pollution, and greenhouse gas emission. TES systems store excess thermal energy for later use, thereby providing an effective solution toward more efficient use of energy. They reduce the mismatch between energy demand and availability, enhance the reliability of energy distribution, and improve the management of renewable energy sources, which often exhibit an intermittent nature (e.g., solar, wind, geothermal).1−3
The most important class of materials for TES in the temperature range between 0 and 100 °C is represented by the organic solid–liquid phase change materials (PCMs), such as paraffin waxes, fatty acids, and poly(ethylene glycol)s. Thanks to their high enthalpy of fusion/crystallization, these PCMs can store and release a considerably high amount of energy as latent heat during melting and freezing, in a relatively small mass and volume.4,5 Since the temperature of the storage medium is maintained nearly constant throughout the whole phase change, these PCMs are also suitable for applications of temperature regulation, e.g., in indoor environments, and as passive cooling media for electronic devices.1,6−8 Additional advantages include their chemical inertness, thermal stability, small volume change at phase transition, large availability, and low cost.5,9
However, organic PCMs exhibit low thermal conductivity and need to be confined to avoid leakage above the melting temperature.4,10,11 There are many strategies to address these drawbacks: the PCM can be encapsulated in macro-, micro-, or nanoshells, or “shape-stabilized” using polymer matrices, porous or layered materials, or nanofiller networks.2,12−14 However, one of the most effective strategies to address both the critical shortcomings of PCMs is microencapsulation,4,15−18 intended as the inclusion of the PCM in a stable polymeric or inorganic shell of micrometric dimensions. The shell not only prevents PCM leakage but also inhibits undesired reactions with the surrounding environment and contributes toward accommodating volume changes during phase transition. Microencapsulation also increases the specific surface area of the storage medium, thereby enhancing the thermal exchange surface and increasing the charge/discharge efficiency, which partially addresses the issue of the low thermal conductivity.19 A PCM can be microencapsulated through several physical, physical–chemical, and chemical techniques. Among these, the most diffused, researched, and developed on an industrial scale is in situ polymerization, which results in the inclusion of the PCM droplets in thin and lightweight polymer shells and allows great versatility in terms of capsule size (0.05–1000 μm), shell thickness, and chemical composition.20−22
Microencapsulation eases PCM handling and dispersion in solid and liquid media. PCM microcapsules have been traditionally added to construction materials to enhance the thermal management of buildings,23,24 but innovative approaches involve their embedment in polymer matrices to produce smart thermoregulating polymer composites for multiple applications, including fibers,25 packaging materials,26 solar energy collectors,19 anti-icing coatings,27 and materials for electronics.28
In a polymer composite, the interface between the discontinuous phase(s) and the matrix plays a fundamental role in determining the final mechanical properties. Defects at the filler/matrix interface can originate from poor chemical compatibility and/or a mismatch in the thermal expansion coefficients of the involved phases, as the production process of polymer composites generally requires steps at high temperatures.29 In a composite material containing PCM microcapsules, an additional issue is represented by the repeated thermal cycles, which cause repeated volume variations of the PCM. This could further damage the interphase zone, thereby causing microcracks and debonding phenomena that deteriorate the mechanical properties and thermal conductivity and shorten the service life of the component. The damage at the interface could also undermine the mechanical integrity of the capsule shell, thereby favoring PCM leakage and reducing the energy storage capability of the composite.30−33
Despite being such a critical aspect, the adhesion between PCM microcapsules and a polymer matrix has not been the subject of intensive investigations so far. One of the few research studies is that of Su et al.,31,32 who prepared paraffin microcapsules with a melamine–formaldehyde shell and studied the effect of methanol addition during the capsule preparation. They concluded that the methanol modification enhanced the adhesion with an epoxy resin due to the increasing molecular interaction at the interface, which led to better mechanical performance also after several thermal cycles. A more detailed insight on the study of the interfacial adhesion between polymer matrices and polymer microcapsules can be found in the field of self-healing composites. In these materials, a good capsule/matrix adhesion is fundamental, as it determines crack propagation across the capsules and not at the interface, thereby allowing the release of healing agents in the crack zone.34 The scientific literature reports numerous studies on the modification of polymer microcapsules via the addition of silane compounds during (in situ)35,36 or after (a posteriori)37−40 the microcapsule synthesis. However, these works focus almost exclusively on the modification of urea–formaldehyde resin, while there is considerable lack of investigation on melamine–urea–formaldehyde (MUF) or melamine–formaldehyde (MF) microcapsules, which are by far the most common and commercially available shells containing a PCM, due to the low cost, thermal stability, and the easily controlled production process.31
Previous research41−47 highlighted nonoptimal adhesion between MF microcapsules and polymers such as epoxy, acrylic resins, and polyamides, which resulted in a considerable decrease in the mechanical properties of the produced composite. Due to the lack of reactive functional groups of a fully cured MF surface, the a posteriori modification of the microcapsule surface presents considerable challenges, especially when addressed with traditional surface modification techniques like silane chemistry.
Among the possible alternative treatments, a method that has attracted increasing attention in the last decade is the deposition of polydopamine (PDA).48 PDA is a bioinspired synthetic polymer that has emerged as one of the most powerful tools for broad applications, as it allows the production of continuous layers with relative simplicity and noteworthy versatility and exhibits great adhesion onto a great variety of metallic, polymeric, and ceramic surfaces.49−53 It is particularly appealing to functionalize chemically inert surfaces; it has been employed to improve the fiber/matrix interfacial adhesion of highly inert reinforcing fibers, such as ultra-high-molecular-weight polyethylene (UHMWPE) and poly(p-phenylene-2,6-benzobisoxazole) (PBO) fibers.54,55 Produced by the self-polymerization of dopamine, PDA contains catechol and amine groups and presents a chemical composition analogous to that of the mussel adhesive proteins.56,57 Due to its unique chemistry and the interesting assortment of reactive functional groups, this bioinspired coating can act as a compatibilizer in multiple filler/matrix systems and can be subsequently functionalized.54,58 PDA has been proven to be effective in improving the filler–matrix adhesion with several types of fillers (e.g., nanoclays,59 carbon nanotubes60), but a very limited number of articles can be found on the application of this coating procedure to formaldehyde-based resins and, to the best of the authors’ knowledge, no articles have been published on the coating of PCM microcapsules with PDA.
The aim of the present work is to modify the surface of commercial PCM microcapsules, which have a shell of formaldehyde-based resin, through the deposition of dopamine. The PDA layer, which was instantly formed by the oxidative polymerization of the applied dopamine, improved the interfacial adhesion with an epoxy matrix. Therefore, the novelty of this work is not the synthesis of the PCM microcapsules but the successful coating of such microcapsules with a layer of PDA, and the proof that this coating remarkably improves the adhesion with an epoxy matrix. In fact, these commercial microcapsules have been observed to have great potential as PCMs for thermal management applications, but they have poor adhesion with an epoxy matrix. This work specifically focuses on employing microscopy techniques to study how the interphase between the microcapsules and the epoxy matrix varies between uncoated and coated capsules, and on investigating how the PDA layer influences the heat storage properties and the mechanical stiffness of the microcapsules. This work expands the potentialities of PDA coatings to the field of polymers containing PCM microcapsules, as the coating procedure was proven to be mild enough to preserve the integrity of the thin microcapsule shell and effective in increasing the interaction with an epoxy resin.
2. Materials and Methods
2.1. Materials
The microencapsulated PCM Microtek MPCM43D was procured from Microtek Laboratories (Dayton, OH). The PCM phase consists of paraffin wax with a melting temperature of 43 °C, encapsulated inside a formaldehyde-based resin shell constituting 10–15% of the mass. These microcapsules are characterized by an average diameter of 17–20 μm, and the declared melting enthalpy is 190–200 J/g. This microencapsulated PCM will henceforth be labeled MC. The bicomponent epoxy system Elan-tech EC157/W342 was kindly provided by Elantas Europe S.r.l. (Collecchio, Italy). Dopamine hydrochloride and amino-2-(hydroxymethyl)propane-1,3-diol (TRIS buffer, Trizma) were purchased from Sigma-Aldrich (Steinheim, Germany). Acetone was supplied by Fisher Scientific (Nidderau, Germany). Deionized water was prepared employing a PURELAB Chorus 2+ water purification system (Elga LabWater, High Wycombe, U.K.). Bisphenol A diglycidyl ether (2-[[4-[2-[4-(oxiran-2-ylmethoxy)phenyl]propan-2-yl]phenoxy]methyl]oxirane, DGEBA) was supplied by Sigma-Aldrich (Steinheim, Germany).
2.2. Preparation of PDA-Coated Microcapsules
Dopamine hydrochloride (2.0 g, as supplied) was added to 600 mL of TRIS solution (0.1 mol/L) in an uncovered beaker and dissolved in an ultrasonic bath (VWR International, Leuven, Belgium) at room temperature. Then, 15 g microcapsules were slowly added to the dopamine solution under vigorous magnetic stirring (1000 rpm). In the uncovered beaker, the suspension was stirred for 24 h at room temperature. The formation of PDA became visible by the umber staining of both the suspension and the microcapsule surfaces. The PDA-coated microcapsules were filtered off under vacuum by a Büchner funnel lined with a Sartorius 389 filter (Sartorius AG, Göttingen, Germany). The coated microcapsules were rinsed twice with 50 mL of deionized water before each cycle of drying under vacuum at room temperature for 4 h. Washing and drying cycles were repeated to a constant weight of the surface-modified microcapsules. After their modification, the formerly white microcapsules were dark brown, which indicated the presence of insoluble and irreversibly bonded PDA layers on their surfaces, as evidenced in Figure 1.
Figure 1.

Photograph of neat and PDA-coated microcapsules.
The PDA reference material was synthesized by adding 20 g of dopamine hydrochloride (as supplied) to 600 mL of TRIS solution (0.1 mol/L) in an uncovered beaker. Dopamine polymerization was performed at room temperature for 24 h. The black-brownish polymer was filtered, washed twice with deionized water (100 mL), and dried under vacuum to a constant weight.
2.3. Reaction of Neat and PDA-Coated Microcapsules with an Oxirane-Carrying Molecule
The surface reactivity of the PDA-coated microcapsules was assessed by investigating the cross-linking reaction with an oxirane-carrying molecule, namely, the DGEBA. Coated microcapsules (1 g) were introduced into a 20 mL glass vial. Then, 5 mL of DGEBA was added. The suspension was stirred for 4 h utilizing a magnetic stirrer. The microcapsules were separated by vacuum filtration with a Sartorius 389 filter. To remove unbound DGEBA, the microcapsules were rinsed twice with 20 mL of acetone. The DGEBA-cross-linked PDA-coated microcapsules were dried under vacuum at room temperature for about 1 h. The surface investigation was performed through X-ray photoelectron spectroscopy (XPS), as described in Section 2.5.3.
2.4. Preparation of the MC-Filled Epoxy Composites
MC-filled epoxy composites were produced by mixing the epoxy base EC157 and the hardener W342 in a weight ratio of 100:30, as suggested by the manufacturer.61 Neat or PDA-coated microcapsules were added to the epoxy preparation in a weight fraction of 10 wt % and stirred with a SpeedMixer DAC 400 FVZ (Landrum, SC) at 1000 rpm for 1 min, to ensure a homogeneous dispersion. The mixtures were degassed for 5 min, cast in stainless-steel molds, and cured at 100 °C for 10 h.61 The curing was followed by slow cooling to room temperature.
2.5. Characterization of the Neat and PDA-Coated Microcapsules
2.5.1. Scanning Electron Microscopy (SEM)
The morphology and roughness of the neat and PDA-coated microcapsules were studied by a Zeiss Gemini Neon 40ESB scanning electron microscope (Oberkochen, Germany). The powder samples were prepared on SEM stubs equipped with conductive tape. Further application of conductive coating was avoided, so as not to alter the surface morphology of the samples. The microcapsules were analyzed under high-vacuum conditions, and a low acceleration voltage (1 kV) and a small beam aperture (20 μm) were employed to achieve the best image quality and avoid electrostatic charging. The images were acquired with the secondary electron (SE) sensor.
2.5.2. Atomic Force Microscopy (AFM)
AFM tests were carried out to study the morphology and surface roughness before and after PDA coating. Neat and PDA-coated microcapsules were glued with double-sided tape on glass slides. The measurements were done in the peak force tapping mode by a Dimension FastScan atomic force microscope (Bruker-Nano, Santa Barbara) equipped with silicon nitride sensors ScanAsyst Fluid+ (Bruker) with a nominal spring constant of 0.7 N/m and a tip radius of 2 nm. The setpoint was 0.04 V. The mean roughness Sa was calculated through the program NanoScope Analysis 1.9 (Bruker-Nano, Santa Barbara) after removing the curvature of the spheres by a sixth-order flatten command.
2.5.3. X-ray Photoelectron Spectroscopy (XPS)
XPS was carried out to study the surface chemistry and functional groups of the neat and PDA-coated microcapsules, and to study the different surface reactivities, molecular interactions, and chemical bond formations when the microcapsules were reacted with DGEBA (see Section 2.3). Four samples were analyzed with this technique, namely, (i) the neat microcapsules, (ii) the PDA-coated microcapsules, (iii) the PDA-coated microcapsules reacted with DGEBA, and (iv) the PDA-coated microcapsules reacted with DGEBA. All of the XPS studies were carried out by means of an Axis Ultra photoelectron spectrometer (Kratos Analytical, Manchester, U.K.). The spectrometer was equipped with a monochromatic Al Kα (hν = 1486.6 eV) X-ray source of 300 W at 15 kV. The kinetic energy of photoelectrons was determined with a hemispheric analyzer set to a pass energy of 160 eV for wide-scan spectra and 20 eV for high-resolution spectra. Employing a double-sided adhesive tape (3M Company, Maplewood, MN), the powder samples were prepared as thick films on a sample holder that allowed their introduction in the recipient of the XPS spectrometer. During all of the measurements, electrostatic charging of the sample was avoided by means of a low-energy electron source working in combination with a magnetic immersion lens. All of the recorded peaks were shifted by the same value that was necessary to set the C 1s peak to 285.00 eV. Quantitative elemental compositions were determined from peak areas using experimentally determined sensitivity factors and the transmission function of the spectrometer. Spectrum background was subtracted according to Shirley.62 The high-resolution spectra were deconvoluted by means of Kratos spectra deconvolution software (Kratos Analytical, Ltd., Manchester, U.K.). Free parameters of component peaks were their binding energy (BE), height, full width at half-maximum (FWHM), and the Gaussian–Lorentzian ratio. The interpretation of the origins of the component peaks was performed according to the determined binding energy values.63
2.5.4. Fourier Transform Infrared Spectroscopy (FTIR)
The FTIR spectra of the neat microcapsules, PDA-coated microcapsules, and PDA as the reference coating material were recorded in the attenuated-total reflection (ATR) mode (Golden Gate accessory with diamond crystal, Specac Limited, Orpington, England) using an FTIR spectrometer Tensor 27 (Bruker Optics GmbH, Ettlingen, Germany). All samples were placed on the horizontal ATR diamond crystal. A reference spectrum was recorded from the pure ATR crystal. A total of 100 interferograms were co-added at a spectral resolution of 2 cm–1. The interferograms were Fourier transformed applying Blackman–Harris-3 apodization and zero filling factor of 2.
Calculated absorbance spectra were subjected to atmospheric compensation, limited to the fingerprint region (1900 cm–1 to about 600 cm–1), the baseline function of OPUS software was used (2 pt. baseline—two iterations concave rubber band method), and normalized to the height of the band appearing at 716 cm–1.
2.5.5. Colloidal Probe AFM Tests (CP-AFM)
Mechanical characterization of microcapsules was performed on MFP-3D (Asylum Research, Oxford Instruments, Santa Barbara, CA) equipped with an optical microscope Axio Observer Z1 (Zeiss, Oberkochen, Germany). For colloidal probe preparation, silica beads (5 μm, SiO2 probe particles, microParticles GmbH, Berlin, Germany) were glued with a bicomponent epoxy adhesive (UHU Plus Endfest, Buhl, Germany) on cantilevers with spring constants of 25.6 and 24.4 nN/nm (NCS35/NoAl/tipples, Mikromasch Europe, Wetzlar, Germany) and thermally treated overnight at 80 °C. Spring constant calibration was performed by the thermal noise method according to Hutter and Bechhoefer.64 Calibration of the cantilever sensitivity was performed on a hard surface before and after the measurements. Single-microcapsule force–deformation experiments were done at a velocity of 1 μm/s at the apex of the microcapsules. Microcapsules were fixed to the surface by a thin layer of poly(ethyleneimine) and measured in water at 50 °C after an equilibration time of 1 h to ensure the complete melting of the paraffinic core. The microcapsule size was measured by the inverted microscope before the deformation experiments. The stiffness of the microcapsule shells was calculated from the linear slope at a small deformation regime.65
2.5.6. Differential Scanning Calorimetry (DSC)
DSC was performed to measure the most important thermal properties of a PCM, such as the phase transition enthalpies and temperatures, and to evaluate their changes after the coating process. Powder samples of approx. 5 mg were placed in standard aluminum DSC pans and analyzed with a TA Instruments Q2000 calorimeter (New Castle, DE). Each sample was subjected to a heating/cooling/heating cycle at 10 °C/min from −20 to 80 °C, under a nitrogen atmosphere. The test allowed the measurement of the peak temperature and enthalpy of each phase transition and an evaluation of the variation in the core-to-shell mass ratio after the coating.
2.5.7. Thermogravimetric Analysis (TGA)
TGA was performed to assess the evolution of the thermal resistance of the coated capsules. A TA Instruments Q500 thermo-balance (New Castle, DE) was employed to record thermograms from approx. 5 mg of the powder samples. The samples were heated up to 700 °C at a heating rate of 10 °C/min, under a nitrogen atmosphere. The tests allowed the determination of the temperatures corresponding to mass losses of 1, 3, and 5 wt % (T1%, T3%, T5%), the temperature at the maximum degradation rate, corresponding to the peak of the mass loss derivative (Td), and the residual mass after the test (mr).
2.5.8. Temperature-Controlled (Hot-Stage) Optical Microscopy
The melting of the core of the microcapsules depending on the temperature has been investigated by an optical microscope equipped with a hot stage. MC was placed on a light microscopy glass slide on a temperature-controlled stage LTS-420 (Linkam, Surrey, U.K.) and heated from rt to 50 °C and recooled to rt. Visual images have been recorded with an Axio Imager (Carl Zeiss Microscopy GmbH, Jena, Germany). Temperature-controlled microscopy was performed on the neat microcapsules and the PDA-coated microcapsules.
2.6. Characterization of the MC-Filled Epoxy Composites
SEM micrographs of the fracture surface of the prepared composites were recorded to visually observe the modification of the adhesion and the interfacial region after PDA coating, to study the fracture morphology and propagation, and to measure the thickness of the coating as a function of the deposition parameters. The MC-containing epoxy composites were prepared as described in Section 2.4. Then, the composites were cryofractured in liquid nitrogen and the fracture surface was investigated through SEM. The specimens were fixed on SEM stubs with conductive tape and investigated with a Zeiss Supra 60 field emission scanning electron microscope (FESEM; Oberkochen, Germany), after Pt–Pd sputtering.
3. Results and Discussion
3.1. Surface Morphology of Neat and PDA-Coated Microcapsules
Figure 2a–f shows the SEM micrographs of the neat and PDA-coated samples. The neat microcapsules appear spherical and with a smooth surface, while the surface roughness increases considerably for the PDA-coated microcapsules. The PDA layer presents a rough morphology with a globular appearance, which could contribute to the filler–matrix interfacial strength by mechanical interlocking. The PDA is deposited nearly uniformly on the whole MC surface, as is observable from Figure 2d. The coating led to a change in the microcapsule color, from white to brown, but this is not an issue for the proposed application.
Figure 2.

SEM micrographs of neat microcapsules (a, c, e) and PDA-coated microcapsules (b, d, f) at different magnifications.
The surface morphology and roughness of the PDA coating were studied quantitatively with AFM. From Figure 3a,b, it can be seen that the neat microcapsule has a relatively smooth surface, while the PDA-coated microcapsule shows a nodular surface structure with varying nodule sizes. The mean surface roughness Sa increased from 9 nm for the neat MC, to 86 nm for the PDA-coated MC.
Figure 3.

Three-dimensional (3D) AFM images of (a) neat microcapsules and (b) PDA-coated microcapsules. Lateral image dimension 10 μm × 10 μm, color-coded height range 4 μm.
3.2. FTIR Spectroscopy
Although FTIR is a widely used nondestructive method for many compounds, complex samples, and coatings, an accurate investigation of the structure of PDA through FTIR is still a challenge. The PDA polymer consists of a number of different monomer units with various oxidation states and chemical and physical linking, organized in different hierarchical aggregates.66 Thus, absorption spectra of PDA do not contain well-resolved bands, as observed for single functional groups in defined compounds. On the contrary, wide and featureless spectra are often detected, which show the overlapping contributions of different functional units with absorption energies close to each other.
This is clearly observable from the ATR-FTIR spectrum acquired on PDA, shown in Figure 4 by a dashed line. This spectrum is reported with a constant vertical translation to facilitate the interpretation of the results. A complex PDA spectrum with broad bands was obtained as a result of different binding and oxidation states, a variety of cyclic hetero- or aromatic structures, and different bridging bonds and positions. Figure 4 also reports the ATR-FTIR spectra of the neat and PDA-modified microcapsules with a dotted and solid line, respectively. Both spectra show narrow hydrocarbon bands of the paraffin core, which is crystalline, and broader bands of the melamine–formaldehyde shell, as elucidated in Table 1. The PDA modification of the microcapsules can be evaluated by the increase in intensity between 1650 and 1050 cm–1. The subtraction of the spectrum of neat microcapsules from that of the PDA-modified microcapsules results in good approximation of the PDA spectrum. Most characteristic band positions and intensities are recovered for both the PDA spectrum and the subtracted one, shown as a red line. The spectral band positions of the PDA phase, reported in Table 2, are consistent with other reports,66−70 and assignments are based on literature data and on the comparison with pyrrole, indole, and catechol structures.71 These results prove that the ATR-FTIR technique is a fast and cost-effective characterization method to detect and investigate polymer shells and thin PDA coatings, even though they represent a small fraction of the total sample mass and their spectroscopic signals are not identified by sharp peaks.
Figure 4.
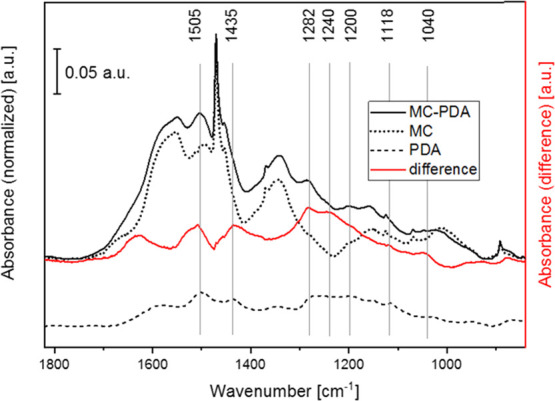
ATR-FTIR spectra of neat microcapsules (MC, dotted line), PDA-coated microcapsules (MC-PDA, solid line), and a polydopamine layer (PDA, dashed line). The difference spectrum (red line) was calculated by the subtraction of MC from MC-PDA.
Table 1. Band Assignments for the Neat Microcapsulesa.
| wavenumber (cm–1) | intensity | band assignment | molecular structure | material | refs |
|---|---|---|---|---|---|
| 1653 | vw, shoulder | δ(N–H) | N–H | melamine–formaldehyde | (72) |
| 1548 | s, broad | ν | triazine ring | melamine–formaldehyde | (73) |
| 1500 | s, broad | δ(C–H) | C–H, in CH2, CH2–OH, CH2–O– | melamine–formaldehyde | (71, 74) |
| 1470 | vs | asymmetric, δ(C–H) | CH2 | paraffin | (72) |
| 1455 | s, broad | δ(C–H) | C–H, in CH2, CH2–OH, CH2–O– | melamine–formaldehyde | (71, 73, 74) |
| 1367 | w | symmetric, δ(C–H), | C–H, in CH2, CH2–OH, CH2–O–CH2, bridge | melamine–formaldehyde | (72) |
| 1341 | s, broad | ν(C–N) | aromatic C–N | melamine–formaldehyde | (73) |
| 1156 | s, broad | ν(C–N) | C–N/–CH2–O– | melamine–formaldehyde | (73) |
| 1070 | vw | symmetric, ν(C–O) | –CH2–O–CH2–, ether | melamine–formaldehyde | (73) |
| 891 | m | twisting | C–H in CH2 | paraffin | (72) |
| 813 | s | δ | triazine ring | melamine–formaldehyde | (73) |
| 755 | w | δ | N–H, in amines, triazine ring | melamine–formaldehyde | (73) |
| 716 | vs | δ, in-plane rocking | C–H in CH2 | paraffin | (72) |
Abbreviations for band intensities. vw: very weak; w: weak; m: medium; s: strong; and vs: very strong.
Table 2. Band Assignments for the PDA Phaseb.
| wavenumber (cm–1) | intensity | band assignment | molecular structure | structure unit | refs |
|---|---|---|---|---|---|
| 3612–3140 | w-m, broad | ν(OH) | O–H | phenolic 36 | (66) |
| ν(NH) | N–H | ||||
| 3033 | m, broad | ν(CH) | aromatic C–H | aromatic ring, pyrrole, indole | (66) |
| 2963 | w | νas(CH) | CH3 | (66) | |
| 2954 | w | νas(CH) | CH2 | (66) | |
| 2916 | m | νs(CH) | CH3 | (66) | |
| 2850 | m | νs(CH) | CH2 | (66) | |
| 1725 | w-m | ν(C=O) | carbonyl group | (66, 68) | |
| 1574 | s | ν(C–C), δ(C–H) | ring | aromatic ring, pyrrole, indole, catechol | (66, 70) |
| 1502 | vs | ν(C–C), ν(C=N) | aromatic C–C, C=N | aromatic ring, pyrrole, indole | (66, 68) |
| 1472 | w | ν(C=C) | C–H | pyrrole, indole ring | (68, 75) |
| 1436 | s | νring(C–C) + ν(C–N) + δ(O–H), δ(C–H) + δ(N–H) + ν(C–N) | C–H, C–N, O–H | aliphatic CHx indole ring | (66, 67, 69) |
| 1347 | m | νring(C–C) + ν(C–N) + δ(O–H) | C–N, O–H, aromatic ring, C–N–C | aliphatic CHx, indole ring | (66, 67, 75) |
| 1266 | w | ν(C–O) + δ(N–H) + δ(C–H), ν(C–O) + δ(C–H) + δring | C–O | catechol | (66, 68−70) |
| 1198 | s | δ(OH) + δ(CH), ν(C–O) | C–O–C, C–O | (66) | |
| 1153 | w | δ (OH) + δ(CH), ν(C–O) | C–O | (66) | |
| 1115 | w | δ(OH) + δ(CH) +ν(C–O) | C–O | (66) | |
| 1035 | w | δ(CH) + δ(NH) + ν(C–O) | aromatic ring | indole ring | (66) |
| 954 | m | δring + δ(C–H) | indole ring | (66) | |
| 864 | m | γ(C–H) | indole ring | (66, 67) | |
| 807 | s | γ(C–H) + γring | indole ring | (66, 67) | |
| 632 | w | ||||
| 589 | w | ||||
| 455 | m |
Abbreviations for band intensities. vw: very weak; w: weak; m: medium; s: strong; vs: very strong.
3.3. XPS Characterization
The results of the XPS investigation are summarized in Figure 5. The C 1s spectrum of a cured melamine–formaldehyde sample should present peaks relative to the carbon atoms of the 1,3,5-triazine ring networks (expected at about 287.15 eV), the carbon atoms carrying amino groups (expected at about 286 eV), and—as usually found on all surfaces—saturated hydrocarbons (at 285.00 eV).63 However, the recorded C 1s spectrum of the neat microcapsules (Figure 5a) did not show the presence of amino groups; neither primary amino groups (C–NH2) nor secondary amino groups (C–NH–C) were detected, as better explained below. Amino groups are able to open the oxirane rings of the epoxy molecules and initiate the formation of epoxy networks. Their absence on the surfaces of the microcapsules explains the poor adhesion in epoxy polymer matrices, as it hinders the formation of covalent bonds at the microcapsule–matrix interface.76
Figure 5.

Wide-scan (left column), C 1s (middle column), and N 1s (right column) high-resolution XPS spectra recorded from (a) neat microcapsules, (b) neat microcapsules reacted with DGEBA, (c) PDA-coated microcapsules, and (d) PDA-coated microcapsules reacted with DGEBA.
According to the shape of the recorded C 1s spectrum, it can be assumed that the melamine–formaldehyde was cured in the presence of urea. During the hardening reaction, the urea molecules were condensed on the −OH groups of the intermediately formed hexamethylolmelamine. The chemical structure of the reaction product shown in Figure 6 confirms the C 1s spectrum recorded from the neat microcapsules depicted in Figure 5a.
Figure 6.

Presumed chemical structure (cut out from the polymeric network) of the surface of the neat microcapsules derived from the C 1s high-resolution XPS spectrum (Figure 5a). The italic letters indicate the assignment of the carbon or nitrogen atoms to the component peaks of the C 1s (B, C, and F) and N 1s (K and L) spectra.
Photoelectrons escaped from the carbon atoms of the 1,3,5-triazine rings contributed to component peak B at 287.16 eV. The binding energy value found for that component peak was slightly lower than the value of the tertiary amino groups ([CC−]3N, component peak C at 287.91 eV), which were not involved in the conjugated electron-rich π–electron systems of the triazine rings. According to the stoichiometry of structure 1 (Figure 6), the intensity of component peak C was equal to that of component peak F at 289.03 eV that represents the carbonyl carbon atoms of the condensed urea molecules ([NH−]2FC=O). Moreover, a further component peak A was observed at 285.00 eV. This component peak was caused by photoelectrons from carbon atoms bonded as saturated hydrocarbons (ACxHy). As mentioned above, saturated hydrocarbons were always observed when the samples were prepared under ambient conditions or came in contact with the atmosphere.63 However, this peak component could also be due to some paraffin diffused out of the capsules and partly covering the outer shell surfaces. The chemical inertness of the paraffin film also contributes to limit the chemical interaction between the epoxy matrix and the microcapsule surfaces. The corresponding N 1s spectrum (Figure 5a, right column) was clearly composed of the two component peaks K and L. The binding energy value of the nitrogen atoms involved in the conjugated π–electron systems of the triazine rings (component peak K) was found at 398.63 eV, at a slightly lower energy than the value for the tertiary amines ([C−]3LN) and the urea units ([LNH−]2C=O), which were observed as component peak L at 399.84 eV.
To investigate in more detail the poor chemical reactivity of the neat microcapsule surface, an attempt was made to directly graft DGEBA on the untreated MC shell, via the sample preparation route described in Section 2.3. Figure 5b shows the C 1s and N 1s spectra of these samples. As can be seen, neither the C 1s nor the N 1s spectrum showed significant differences in the spectra recorded from the neat microcapsules. In the case of successful grafting of DGEBA, the corresponding C 1s spectrum should be characterized by an intensive component peak at approx. 286.4 eV, resulting from alcohol (C–OH) and ether (C–O–C) groups formed after the ring-opening reaction of the oxirane groups. From the absence of this characteristic component peak, it can be concluded that DGEBA was not bonded to the surface of the microcapsule sample, which implies the impossibility of direct grafting of oxirane group-carrying substances on the microcapsule surface.76
After the deposition of dopamine and its polymerization to PDA, the C 1s spectrum of the PDA-coated microcapsules (Figure 5c) showed the characteristic shape found for PDA.77 Electron transitions between π- and π*-orbitals cause not only the dark color observed after the polymerization of dopamine but also intense shake-up peaks (Sh) in the high-resolution C 1s spectrum (Figure 5c, middle column). The C 1s area remaining after subtraction of the shake-up peaks was deconvoluted into five component peaks, which confirmed the number of different structural units in the PDA molecules. At low energy, the component peak Ph (284.53 eV) evidences the presence of sp2-hybridized carbon atoms of the PDA phenyl rings, whereas the B1C–N bonds in the amino groups led to component peak B1 at 285.83 eV. Since its intensity was two-thirds of component peak Ph, it can be concluded that preferably secondary amino (B1C–NH–B1C) groups and cyclic imide (B1C=N–B1C) were formed during the polymerization of dopamine (Figure 7). Component peak C1 at 286.49 eV was assigned to the carbon atoms carrying the phenolic OH groups of the catechol units (C1C–OH). Its intensity was smaller than that of component peak C1, even though, according to chemical structures 2 and 3 suggested in Figure 7, the intensities should be equal, [C1] = [B1]. This may be due to the fact that some of the catechol groups were present in their oxidized form. During the polymerization of dopamine, oxidation reactions are in equilibrium with reduction reactions. Photoelectrons escaping from the carbon atoms of remaining quinonelike structures 4 (D1C=O) were collected as component peak D1 at 287.29 eV. The sum of the intensities of component peaks D1 and C1 exactly equaled the intensity of component peak B ([D1] + [C1] = [B1]), which corresponds to the stoichiometric ratio in the binding states of the carbon atoms suggested in Figure 7. The findings also supported the assumption that cyclic amine and imine structures were preferably formed during the oxidative polymerization of dopamine.
Figure 7.

Characteristic subunits from the chemical structure of PDA. Italic letters denote the assignment of the carbon and nitrogen atoms to the component peaks in the C 1s and N 1s high-resolution spectra.
Also, the N 1s spectrum of the PDA-coated microcapsules is different from that recorded on the neat microcapsules. Besides component peaks K and L, it was necessary to introduce a third component peak M (401.65 eV), showing protonated amino groups (C–MN+H2–C). Unlike the reacted urea groups in structure 1 (Figure 6), the secondary amines of PDA (2) contain a basic nitrogen atom that can be easily protonated in an aqueous environment. The nonprotonated nitrogen atoms of the amino groups were identified as component peak L at 399.93 eV.
The ratio of the two component peaks [M]:[N] illustrates the protonation/deprotonation equilibrium of the amino groups. For the subsequent reaction with epoxides, it is essential that the intensity of component peak M is small compared to that of component peak L, because the protonated species can neither initiate the opening of the oxirane rings nor be involved in the epoxy cross-linking reactions. Due to the high electron density at the nitrogen atom of imide group (3) (Figure 7), which is involved in the delocalized π–electron system, component peak K was shifted to a lower binding energy value (398.74 eV). From the high-resolution element spectra (Figure 5c), it can be concluded that the surfaces of the microcapsules were fully coated by thick layers of PDA. These findings are supported by the complete absence of potassium in the wide-scan spectrum of the PDA-modified sample and the brownish color of the microcapsules after the deposition of dopamine and its polymerization.
The model reaction of the PDA-modified microcapsules with DGEBA (Figure 5d) strongly reduced the relative amount of nitrogen ([N]:[C]|PDA = 0.094 → [N]:[C]|PDA+DGEBA = 0.044), while the [O]:[C] ratio was not significantly affected ([O]:[C]|PDA = 0.226 → [O]:[C]|PDA+DGEBA = 0.238). Obviously, the epoxy resin, which is formed on the surfaces of the microcapsules, inelastically scatters the photoelectrons emitted from the nitrogen atoms bonded to the microcapsule surface. The inelastically scattered photoelectrons did not contribute to the spectral information but were counted in the spectrum background. The high-resolution C 1s and N 1s spectra provided more information about the success of the grafting of the DGEBA molecules on the PDA-modified microcapsule surfaces. The shape of the C 1s spectrum was changed by the epoxy resin formed during the ring-opening reactions of the oxirane units and their subsequent self-cross-linking. The recorded C 1s spectrum is very characteristic of weakly cured epoxy resins. Beside the main component peak A (285.00 eV), which showed saturated hydrocarbons, an intense component peak C2 arose at 286.67 eV. This component peak resulted from C2C–O bonds of alcohol (C2C–OH) and ether (C2C–O–C2C) groups, the typical reaction products of the opened oxirane rings.76 Between component peaks A and C2, component peak B1 (285.84 eV) shows the C–N bonds of amine groups. The intensity of this component peak ([B1] = 0.123) is significantly larger than twice the [N]:[C]|PDA+DGEBA ratio ([N]:[C]|PDA+DGEBA = 0.044), determined from the wide-scan spectrum. This indicates that the secondary amino groups of the PDA molecules were involved as tertiary amines in the depth of the epoxy resin network. In principle, the −OH groups of the catechol units are also able to open oxirane rings and form covalent bonds with the epoxy resin. Nonreacted oxirane groups were detected from component peak D2 at 287.79 eV. Such groups, frequently observed in weakly cured epoxy resins, can be reacted by a thermal curing process.78 Even though its origin is unclear, the presence of component peak F2 at 289.16 eV shows the presence of carboxylate ester groups in the epoxy resin network. While component peak F resulted from photoelectrons that escaped from the carbonyl carbon atoms (O = F2C–O–C2C), the photoelectrons of the corresponding alcohol-sided carbon atoms (O = F2C–O–C2C) contributed to component peak C2. The peak area above 290 eV can be assigned to shake-up peaks (Sh), indicating the small contribution of the PDA interphase to the C 1s spectrum. The shape of the N 1s spectrum recorded from the epoxy-reacted, PDA-coated microcapsules was not very different from that acquired from the PDA-coated microcapsules. The origin of the three component peaks K, L, and M is the same as that discussed for the spectrum in Figure 5c. However, it is interesting to observe that the relative intensities of component peaks K were equal in the two N 1s spectra ([K] = 0.166), which indicates that the imide-bonded nitrogen atoms (C = KN–C) were not reacted with the oxirane species and, therefore, not bonded to the resin.
This detailed investigation further establishes the XPS technique as a powerful tool to study the chemical structure, morphology, and reactivity of surfaces and contributed toward elucidating how the potentialities of this technique can be applied to study PDA coatings.
3.4. CP-AFM Analysis
The PCM microcapsules investigated in this work are intended to be included in a polymer matrix, and the resulting mechanical properties of any composite materials strongly depend not only on the interfacial adhesion but also on the intrinsic mechanical properties of each phase. Hence, it is important to investigate the considered microcapsules also from the point of view of the mechanical performance, especially to assess if the PDA coating impairs their mechanical stiffness and strength.
The mechanical response of the microcapsules was investigated through colloidal probe AFM.79 Measurements were performed in water at 50 °C to avoid the impact of solid wax on the deformation behavior. Microcapsules with a mean radius of 10 μm were chosen for the characterization to keep the size-dependent shell thickness variations stable. The corresponding force–deformation curves are presented in Figure 8a. Both capsule types have a broad natural distribution of mechanical behavior under compression including very stiff microcapsules with a steep slope and some softer ones with high deformation at low forces. Thus, there is no visible impact of surface modification on the mechanical stability. The stiffness was calculated as the ratio between the applied force and the measured deformation in the small deformation range, in the initial linear portion of the load–deformation curve, for deformation values up to 15–20% of the shell thickness. Under these conditions, the resulting stiffness depends on the mechanical properties of the shell as well as the surface structure and modification.65,80Figure 8b demonstrates the calculated stiffness from the linear part of the force–deformation curves, with mean values of 12.6 ± 6.0 N/m for the modified microcapsules and 13.3 ± 4.9 N/m for the nonmodified microcapsules. In the range of standard deviation, the stiffness of both microcapsule types is comparable, so it can be concluded that the PDA coating does not modify significantly the mechanical properties of the MC shell.
Figure 8.

Mechanical characterization of microcapsules. (a) Force–deformation curves of modified and unmodified microcapsules. (b) Stiffness of microcapsules from the linear slope at small deformations; mean values with the standard deviation are presented in black.
3.5. Hot-Stage Optical Microscopy
The results of the melting/crystallization test performed on an optical microscope with hot-stage equipment are reported in Figure 9, which shows selected micrographs of the neat and PDA-coated microcapsules during heating and cooling ramps. This test was performed to follow the melting/crystallization process of both samples and to visually detect any differences in the behavior due to the PDA coating. At the beginning of the test, both the samples are solid and crystalline, as observable from the bright cores of the capsules in Figure 9a,d. As the temperature rises, the core starts melting, and above the melting point, all of the capsule cores are melted, and so almost transparent under the optical microscope, as is observable from Figure 9b,e acquired at 46 °C. During cooling, the recrystallization starts when the temperature falls below the crystallization temperature of the paraffinic core, as is observable from Figure 9c,f, in which some of the capsules have already crystallized. This crystallization temperature is different from that found via differential scanning calorimetry (DSC) (see Section 3.6.1), due to the different heating/cooling rate. The analysis did not show any appreciable differences in the behavior of the neat and PDA-coated microcapsules, which suggests that the PDA coating does not hinder the melting/crystallization process of the core.
Figure 9.

Thermal-controlled microscopy of microcapsules at different temperatures: (a–c) unmodified microcapsules and (d–f) PDA-coated microcapsules.
3.6. Thermal Analysis of Neat and PDA-Coated Microcapsules
3.6.1. DSC Characterization
Figure 10 shows the DSC thermograms of the first heating scan and cooling scan on the neat and PDA-coated microcapsules, while the most important DSC results are reported in Table 3. The neat microcapsules show an endothermic peak at approx. 45 °C, indicating the melting of the PCM contained in the microcapsules, while the cooling scan shows the corresponding crystallization peak at approx. 29 °C. Both the phase change peaks are composed of at least two component peaks, which are more appreciable in the cooling scan and would be more resolvable by decreasing the heating/cooling rate of the test. As already observed in previous studies investigating a similar PCM,44,45,47,81 the presence of multiple peaks stems from a sequence of transitions on heating and cooling. The first smaller peak encountered on heating is probably associated with the solid–solid transition from the solid crystalline phase to the so-called “rotator phase,” while the second peak could be ascribed to the solid–liquid transition; analogous transitions are observable on cooling. This description is in good agreement with the thorough investigation on the thermal transitions of n-docosane (C22H46, Tm = 44.4 °C) performed by Wang et al.82 In any case, since all of the mentioned phase transitions contribute to the total phase change enthalpy and occur in a relatively narrow temperature range, the precise assignment of the DSC peak is beyond the scope of this work. In previous work, these paraffin microcapsules have been proven to be resistant to repeated (up to 50) heating/cooling DSC tests without losing their heat storage ability.46 The DSC thermogram of the PDA-coated microcapsules shows the same features of the neat sample, since the shape of the thermograms and the transition temperatures are not significantly different, as is also appreciable from Table 3. Since the PDA coating does not contribute to the phase change enthalpy, as it does not show any phase transitions in the investigated temperature range, it is reasonable to expect that the crystallization enthalpy measured on the PDA-coated microcapsules (192.1 J/g) is lower than that of the uncoated sample (224.1 J/g). Since the coating process was performed under mild conditions of temperature, pressure, and stirring speed, it can be assumed that the capsules were not damaged during the process and the paraffin did not leak out of the capsule shell, as also confirmed by the SEM micrographs and the XPS results. Therefore, the whole enthalpy decrease can be imputed to the mass gain due to PDA coating, since the specific enthalpy is calculated by normalization of the absorbed or released heat by the total sample mass. If this is the case, the total PDA weight fraction can be calculated through eq 1 as
| 1 |
where ΔHcneat and ΔHc are the crystallization enthalpies of the neat and PDA-coated microcapsules, respectively. The PDA mass fraction calculated in this way was 14.3 wt %.
Figure 10.

DSC thermograms of the neat and PDA-coated microcapsules (first heating scan and cooling scan).
Table 3. Main Results of the DSC Tests on the Neat and PDA-Coated Microcapsulesa.
| sample | Tm (°C) | ΔHm (J/g) | Tc (°C) | ΔHc (J/g) | MC (wt %) | PDA (wt %) | Tm,2 (°C) | ΔHm,2 (J/g) |
|---|---|---|---|---|---|---|---|---|
| MC | 44.7 | 227.2 | 29.0 | 224.1 | 100 | 0 | 44.4 | 224.0 |
| MC-PDA | 44.3 | 189.2 | 29.7 | 192.1 | 85.7 | 14.3 | 43.8 | 190.9 |
Tm, ΔHm = melting temperature and enthalpy (first heating scan); MC (wt %), PDA (wt %) = weight fractions of MC and PDA, calculated from the measured melting enthalpy; Tc, ΔHc = crystallization temperature and enthalpy (cooling scan); and Tm,2, ΔHm,2 = melting temperature and enthalpy (second heating scan).
3.6.2. TGA Characterization
Figure 11 shows the TGA thermograms on the neat and PDA-coated microcapsules, while the most important TGA results are reported in Table 4. As already observed in previous studies,81 the thermal degradation of the neat microcapsules starts with several small steps between 100 and 250 °C. They can be related to the thermal degradation of some free (nonencapsulated) paraffin and low-molecular-weight compounds, not completely removed after the capsule synthesis. On the other hand, the degradation steps at high temperature are related to the degradation of the capsule core and shell. The considerably narrow peak of the mass loss derivative signal suggests that this degradation is fast and sudden, and it can be related to the degradation of the shell and the abrupt release of the core, as extensively investigated in previous studies on the same paraffin microcapsules.43−46 This spike, also observed in repeated measurements, is lower and broader in the thermograms of the PDA-coated microcapsules. Moreover, the peak temperature was 22 °C higher than that of the neat sample, which indicates an improvement in the thermal resistance. However, the temperatures T1%, T3%, and T5% are lower than those of the neat MC sample, which suggests that the thermal degradation begins at lower temperatures, as reported in Table 4 and also observable in Figure 11. The decrease is probably related to the thermal degradation of the PDA coating, but further studies are needed to fully explain this decrease. Nevertheless, the temperature at the beginning of thermal degradation is well above the processing temperature of the epoxy/MC composites (100 °C) and far above the intended service temperatures, which are around the PCM phase change interval (40–50 °C).
Figure 11.

TGA thermograms of the neat and PDA-coated microcapsules. Residual mass (solid lines) and mass loss derivative (dashed lines) as a function of temperature.
Table 4. Main Results of the TGA Tests on the Neat and PDA-Coated Microcapsulesa.
| sample | T1% (°C) | T3% (°C) | T5% (°C) | Td (°C) | mr (%) |
|---|---|---|---|---|---|
| MC | 118.4 | 164.1 | 241.2 | 421.2 | 0.34 |
| MC-PDA | 112.5 | 158.2 | 172.5 | 443.2 | 9.24 |
T1%, T3%, T5% = temperature corresponding to a mass loss of 1, 3, or 5 wt %, Td = temperature at the maximum of the mass loss derivative, and mr = residual mass.
3.7. Interfacial Adhesion and Characterization of the MC/Epoxy Composites
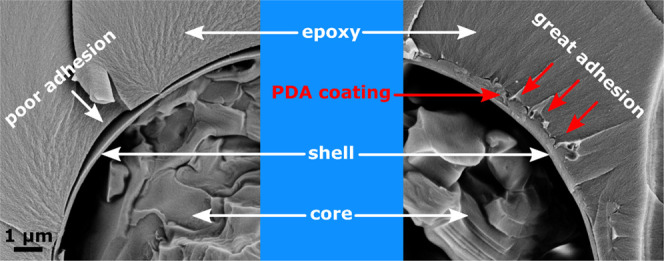
Figures 12a–f and 13 show SEM micrographs of the cryofracture surfaces of epoxy composites containing neat and PDA-modified microcapsules. Most of the capsules are broken due to the cryofracturing process, and this clearly confirms their core–shell structure. From the low-magnification micrographs (Figure 12a,b), it is evident that both the neat and the PDA-coated microcapsules are uniformly dispersed in the epoxy matrix, which is essential for producing a component with homogeneous thermomechanical properties. This suggests that the PDA coating does not induce agglomeration of the microcapsules. What is remarkably different between the two samples is the interfacial adhesion between the capsule shell and the surrounding epoxy matrix, as is appreciable from the medium- and high-magnification micrographs (Figure 12c–f). The PDA coating reduces the gap caused by filler–matrix debonding by promoting the chemical and mechanical interactions between the capsule and the surrounding matrix. The morphology of the coating can be better appreciated in Figure 13; the PDA coating has an irregular surface morphology and the thickness varies significantly from point to point. The thickness was measured on several zones of different microcapsules with software ImageJ and was 265 ± 85 nm, comparable with the thickness of the shell. If it is assumed that the PDA has a density of 1.52 g/cm383 and that the microcapsules have an average diameter of 20 μm and a density of 0.9 g/cm3 (data from the manufacturer’s datasheet), the mass fraction of PDA in the PDA-coated capsules can be calculated as 12.1 wt %, in good agreement with the results from DSC. The details about the calculation of this result are reported in Figure 14.
Figure 12.

SEM micrographs of the cryofracture surface of epoxy matrices containing neat microcapsules (a, c, e) and PDA-modified microcapsules (b, d, f) at different magnifications.
Figure 13.

SEM micrograph of the cryofracture surface of an epoxy matrix containing PDA-modified microcapsules showing details at the high-magnification level.
Figure 14.
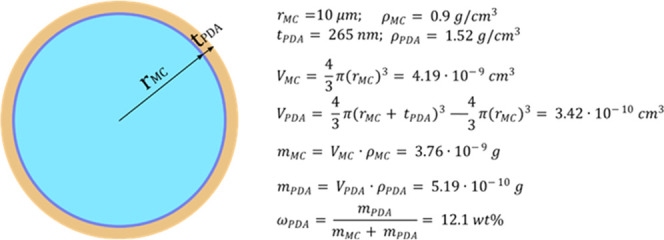
Approximate calculation of the PDA mass fraction on a single microcapsule. rMC = total microcapsule radius (core + shell), tPDA = PDA layer thickness, ρPDA = PDA density, ρMC = density of the microcapsules, VMC = volume of a microcapsule, VPDA = volume of the PDA phase deposited onto a microcapsule, mMC = mass of a microcapsule, mPDA = mass of the PDA phase deposited onto a microcapsule, and ωPDA = mass fraction of PDA on a microcapsule.
Although the PDA layer does not directly contribute toward increasing the total energy that can be stored and released in a TES process involving these PCMs, this remarkable increase in adhesion can benefit the thermal properties of the host composite. As evidenced by Su et al.84 and recently confirmed in a study by our group dealing with the same microcapsules and the same epoxy matrix as those used in this work,46 adhesion enhancement could significantly increase the thermal conductivity of the whole composite, as suggested by many studies from the literature, but further tests are needed to prove this hypothesis for the samples of the present study.
The XPS analysis suggests that the main mechanism of adhesion enhancement is the creation of covalent bonds with the epoxy matrix during curing. Therefore, the same improvement of adhesion could probably be achieved with a thinner PDA layer; this would also reduce the total deposited PDA mass, which would decrease the PDA weight fraction, thereby increasing the total phase change enthalpy. The optimization of the PDA deposition parameters to tune the PDA layer thickness will be the subject of upcoming studies.
4. Conclusions
In this work, a PDA layer was successfully deposited onto the outer shell surface of paraffin microcapsules, and the resulting coating considerably enhanced the adhesion with an epoxy matrix. The morphology and roughness of the PDA layer were studied through microscopy techniques such as SEM and AFM, which evidenced that after PDA coating the surface roughness increased from 9 to 86 nm, which is beneficial, as it allows a further increase in the interfacial interaction by mechanical interlocking. FTIR and XPS techniques have been employed to comprehensively study the surface chemistry and assortment of reactive functional groups of the PDA layer to elucidate its complex chemical structure and configuration. XPS also highlighted that, unlike the uncoated microcapsule shells, the PDA layer is able to react with oxirane groups, thereby evidencing the possibility of forming a covalent bond with the epoxy matrix during the curing step. Hot-stage optical microscopy and DSC tests pointed out that the PDA modification does not hinder the melting/crystallization process of the paraffinic core. DSC results were also useful to estimate the deposited PDA mass fraction, which was 14.3 wt % of the total PDA-coated microcapsule sample. Finally, the SEM micrographs of the cryofracture surface of epoxy composites containing neat or PDA-modified microcapsules clearly evidenced improved adhesion between the capsule shell and the epoxy matrix and allowed an estimation of the PDA layer thickness as 265 ± 85 nm. These results establish the PDA as a coating material with considerable potential for increasing the interfacial adhesion in epoxy–matrix composites containing polymer microcapsules, which is remarkably important not only for this specific application but also in the field of self-healing composites or toughened polymers. Future work will focus on investigating how the parameters of PDA deposition influence the morphology, roughness, and thickness of the PDA layer, and how these features in turn affect the enhancement of the interfacial adhesion. Moreover, attention will be focused on evaluating how the PDA layer and the interface resist multiple thermal cycles, and how this affects the mechanical and thermal properties of the host epoxy matrix.
Acknowledgments
The authors wish to thank Hannes Kettner (Center for Multi-Scale Characterization of the Leibniz Institute of Polymer Research Dresden) and Steffi Preßler (Department of Processing of the Leibniz Institute of Polymer Research Dresden) for performing the SEM measurements, Maria Auf der Landwehr (Department of Processing of the Leibniz Institute of Polymer Research Dresden) for temperature-controlled microscopy investigations, and André Krauss (Department of Reactive Processing of the Leibniz Institute of Polymer Research Dresden) for carrying out the PDA modification.
The authors declare no competing financial interest.
References
- Pielichowska K.; Pielichowski K. Phase change materials for thermal energy storage. Prog. Mater. Sci. 2014, 65, 67–123. 10.1016/j.pmatsci.2014.03.005. [DOI] [Google Scholar]
- Fleischer A. S.Thermal Energy Storage using Phase Change Materials - Fundamentals and Applications; Springer Briefs in Applied Science and Technology; Thermal Engineering and Applied Science: Minneapolis, MN, 2015. [Google Scholar]
- Khadiran T.; Hussein M. Z.; Zainal Z.; Rusli R. Encapsulation techniques for organic phase change materials as thermal energy storage medium: A review. Sol. Energy Mater. Sol. Cells 2015, 143, 78–98. 10.1016/j.solmat.2015.06.039. [DOI] [Google Scholar]
- Zhao C. Y.; Zhang G. H. Review on microencapsulated phase change materials (MEPCMs): Fabrication, characterization and applications. Renewable Sustainable Energy Rev. 2011, 15, 3813–3832. 10.1016/j.rser.2011.07.019. [DOI] [Google Scholar]
- Kahwaji S.; Johnson M. B.; Kheirabadi A. C.; Groulx D.; White M. A. A comprehensive study of properties of paraffin phase change materials for solar thermal energy storage and thermal management applications. Energy 2018, 162, 1169–1182. 10.1016/j.energy.2018.08.068. [DOI] [Google Scholar]
- Pereira da Cunha J.; Eames P. Thermal energy storage for low and medium temperature applications using phase change materials – A review. Appl. Energy 2016, 177, 227–238. 10.1016/j.apenergy.2016.05.097. [DOI] [Google Scholar]
- Ostry M.; Charvat P. In Materials for Advanced Heat Storage in Buildings, 11th International Conference on Modern Building Materials, Structures and Techniques (MBMST 2013), Vilnius, Lithuania; Procedia Engineering: Vilnius, Lithuania, 2013; pp 837–843.
- Fumani N. M. R.; Roghabadi F. A.; Alidaei M.; Sadrameli S. M.; Ahmadi V.; Najafi F. Prolonged Lifetime of Perovskite Solar Cells Using a Moisture-Blocked and Temperature-Controlled Encapsulation System Comprising a Phase Change Material as a Cooling Agent. ACS Omega 2020, 5, 7106–7114. 10.1021/acsomega.9b03407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anghel E. M.; Georgiev A.; Petrescu S.; Popov R.; Constantinescu M. Thermo-physical characterization of some paraffins used as phase change materials for thermal energy storage. J. Therm. Anal. Calorim. 2014, 117, 557–566. 10.1007/s10973-014-3775-6. [DOI] [Google Scholar]
- Peng H.; Zhang D.; Ling X.; Li Y.; Wang Y.; Yu Q.; She X.; Li Y.; Ding Y. n-Alkanes phase change materials and their microencapsulation for thermal energy storage: a critical review. Energy Fuels 2018, 32, 7262–7293. 10.1021/acs.energyfuels.8b01347. [DOI] [Google Scholar]
- Mishra A. K.; Lahiri B. B.; Philip J. Effect of Surface Functionalization and Physical Properties of Nanoinclusions on Thermal Conductivity Enhancement in an Organic Phase Change Material. ACS Omega 2018, 3, 9487–9504. 10.1021/acsomega.8b01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Gu X. B.; Bian L.; Cheng X. F.; Peng L. H.; He H. C. Thermal Properties and Enhanced Thermal Conductivity of Capric Acid/Diatomite/Carbon Nanotube Composites as Form-Stable Phase Change Materials for Thermal Energy Storage. ACS Omega 2019, 4, 2964–2972. 10.1021/acsomega.8b03130. [DOI] [Google Scholar]
- Liu P.; Gu X. B.; Zhang Z. K.; Rao J.; Shi J. P.; Wang B.; Bian L. Capric Acid Hybridizing Fly Ash and Carbon Nanotubes as a Novel Shape-Stabilized Phase Change Material for Thermal Energy Storage. ACS Omega 2019, 4, 14962–14969. 10.1021/acsomega.9b01746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X.; Guan Y.; Li Y.; Yu H.-Y.; Marek J.; Wang D.; Militky J.; Zou Z.-Y.; Yao J. Shape-Stabilized Cellulose Nanocrystal-Based Phase-Change Materials for Energy Storage. ACS Appl. Nano Mater. 2020, 3, 1741–1748. 10.1021/acsanm.9b02441. [DOI] [Google Scholar]
- Zhao J.; Yang Y.; Li Y.; Zhao L.; Wang H.; Song G.; Tang G. Microencapsulated phase change materials with TiO2-doped PMMA shell for thermal energy storage and UV-shielding. Sol. Energy Mater. Sol. Cells 2017, 168, 62–68. 10.1016/j.solmat.2017.04.014. [DOI] [Google Scholar]
- Zhang H.; Wang X.; Wu D. Silica encapsulation of n-octadecane via sol-gel process: a novel microencapsulated phase-change material with enhanced thermal conductivity and performance. J. Colloid Interface Sci. 2010, 343, 246–255. 10.1016/j.jcis.2009.11.036. [DOI] [PubMed] [Google Scholar]
- Yu S.; Wang X.; Wu D. Microencapsulation of n-octadecane phase change material with calcium carbonate shell for enhancement of thermal conductivity and serving durability: Synthesis, microstructure, and performance evaluation. Appl. Energy 2014, 114, 632–643. 10.1016/j.apenergy.2013.10.029. [DOI] [Google Scholar]
- Fredi G.; Dirè S.; Callone E.; Ceccato R.; Mondadori F.; Pegoretti A. Docosane-organosilica microcapsules for structural composites with thermal energy storage/release capability. Materials 2019, 12, 1286 10.3390/ma12081286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.; Zhu C.; Fang G. Synthesis and properties of microencapsulated stearic acid/silica composites with graphene oxide for improving thermal conductivity as novel solar thermal storage materials. Sol. Energy Mater. Sol. Cells 2019, 189, 197–205. 10.1016/j.solmat.2018.10.005. [DOI] [Google Scholar]
- Tyagi V. V.; Kaushik S. C.; Tyagi S. K.; Akiyama T. Development of phase change materials based microencapsulated technology for buildings: A review. Renewable Sustainable Energy Rev. 2011, 15, 1373–1391. 10.1016/j.rser.2010.10.006. [DOI] [Google Scholar]
- Jamekhorshid A.; Sadrameli S. M.; Farid M. A review of microencapsulation methods of phase change materials (PCMs) as a thermal energy storage (TES) medium. Renewable Sustainable Energy Rev. 2014, 31, 531–542. 10.1016/j.rser.2013.12.033. [DOI] [Google Scholar]
- Konuklu Y.; Ostry M.; Paksoy H. O.; Charvat P. Review on using microencapsulated phase change materials (PCM) in building applications. Energy Build. 2015, 106, 134–155. 10.1016/j.enbuild.2015.07.019. [DOI] [Google Scholar]
- Chen Z.; Cao L.; Shan F.; Fang G. Preparation and characteristics of microencapsulated stearic acid as composite thermal energy storage material in buildings. Energy Build. 2013, 62, 469–474. 10.1016/j.enbuild.2013.03.025. [DOI] [Google Scholar]
- Cao V. D.; Pilehvar S.; Salas-Bringas C.; Szczotok A. M.; Valentini L.; Carmona M.; Rodriguez J. F.; Kjøniksen A.-L. Influence of microcapsule size and shell polarity on thermal and mechanical properties of thermoregulating geopolymer concrete for passive building applications. Energy Convers. Manage. 2018, 164, 198–209. 10.1016/j.enconman.2018.02.076. [DOI] [Google Scholar]
- Gao X.-y.; Han N.; Zhang X.-x.; Yu W.-y. Melt-processable acrylonitrile–methyl acrylate copolymers and melt-spun fibers containing MicroPCMs. J. Mater. Sci. 2009, 44, 5877–5884. 10.1007/s10853-009-3830-z. [DOI] [Google Scholar]
- Singh S.; Gaikwad K. K.; Lee Y. S. Phase change materials for advanced cooling packaging. Environ. Chem. Lett. 2018, 16, 845–859. 10.1007/s10311-018-0726-7. [DOI] [Google Scholar]
- Zhu K.; Li X.; Su J.; Li H.; Zhao Y.; Yuan X. Improvement of anti-icing properties of low surface energy coatings by introducing phase-change microcapsules. Polym. Eng. Sci. 2018, 58, 973–979. 10.1002/pen.24654. [DOI] [Google Scholar]
- Tomizawa Y.; Sasaki K.; Kuroda A.; Takeda R.; Kaito Y. Experimental and numerical study on phase change material (PCM) for thermal management of mobile devices. Appl. Therm. Eng. 2016, 98, 320–329. 10.1016/j.applthermaleng.2015.12.056. [DOI] [Google Scholar]
- Awaja F.; Zhang S.; Tripathi M.; Nikiforov A.; Pugno N. Cracks, microcracks and fracture in polymer structures: Formation, detection, autonomic repair. Prog. Mater. Sci. 2016, 83, 536–573. 10.1016/j.pmatsci.2016.07.007. [DOI] [Google Scholar]
- Keblinski P.; Phillpot S. R.; Choi S. U. S.; Eastman J. A. Mechanisms of heat flow in suspensions of nano-sized particles (nanofluids). Int. J. Heat Mass Transfer 2002, 45, 855–863. 10.1016/S0017-9310(01)00175-2. [DOI] [Google Scholar]
- Su J.-F.; Wang X.-Y.; Wang S.-B.; Zhao Y.-H.; Zhu K.-Y.; Yuan X.-Y. Interface stability behaviors of methanol-melamine-formaldehyde shell microPCMs/epoxy matrix composites. Polym. Compos. 2011, 32, 810–820. 10.1002/pc.21102. [DOI] [Google Scholar]
- Su J.-F.; Zhao Y.-H.; Wang X.-Y.; Dong H.; Wang S. B. Effect of interface debonding on the thermal conductivity of microencapsulated-paraffin filled epoxy matrix composites. Composites, Part A 2012, 43, 325–332. 10.1016/j.compositesa.2011.12.003. [DOI] [Google Scholar]
- Burger N.; Laachachi A.; Ferriol M.; Lutz M.; Toniazzo V.; Ruch D. Review of thermal conductivity in composites: Mechanisms, parameters and theory. Prog. Polym. Sci. 2016, 61, 1–28. 10.1016/j.progpolymsci.2016.05.001. [DOI] [Google Scholar]
- Mauldin T. C.; Kessler M. R. Self-healing polymers and composites. Int. Mater. Rev. 2010, 55, 317–346. 10.1179/095066010X12646898728408. [DOI] [Google Scholar]
- Wang R.; Li H.; Hu H.; He X.; Liu W. Preparation and characterization of self-healing microcapsules with poly(urea-formaldehyde) grafted epoxy functional group shell. J. Appl. Polym. Sci. 2009, 113, 1501–1506. 10.1002/app.30001. [DOI] [Google Scholar]
- Wang R.; Li H.; Liu W.; He X. Surface modification of poly(urea-formaldehyde) microcapsules and the effect on the epoxy composites performance. J. Macromol. Sci., Part A: Pure Appl.Chem. 2010, 47, 991–995. 10.1080/10601325.2010.507982. [DOI] [Google Scholar]
- Tong X.-M.; Zhang M.; Wang M.-S.; Fu Y. Effects of surface modification of self-healing poly(melamine-urea-formaldehyde) microcapsules on the properties of unsaturated polyester composites. J. Appl. Polym. Sci. 2013, 127, 3954–3961. 10.1002/app.37711. [DOI] [Google Scholar]
- Cai X.; Fu D.; Qu A. Effects of surface modification on the properties of microcapsules for self-healing. J. Wuhan Univ. Technol., Mater. Sci. Ed.. 2015, 30, 1234–1239. 10.1007/s11595-015-1301-8. [DOI] [Google Scholar]
- Cai X. L.; Fu D. T.; Qu A. L. Effects of surface modification on properties of nanocapsules for self-healing materials. Plast., Rubber Compos. 2014, 43, 161–165. 10.1179/1743289814Y.0000000081. [DOI] [Google Scholar]
- Mirabedini S. M.; Esfandeh M.; Farnood R. R.; Rajabi P. Amino-silane surface modification of urea-formaldehyde microcapsules containing linseed oil for improved epoxy matrix compatibility. Part I: Optimizing silane treatment conditions. Prog. Org. Coat. 2019, 136, 105242 10.1016/j.porgcoat.2019.105242. [DOI] [Google Scholar]
- Fredi G.; Dorigato A.; Pegoretti A. Multifunctional glass fiber/polyamide composites with thermal energy storage/release capability. eXPRESS Polym. Lett. 2018, 12, 349–364. 10.3144/expresspolymlett.2018.30. [DOI] [Google Scholar]
- Dorigato A.; Fredi G.; Pegoretti A. In Novel Phase Change Materials using Thermoplastic Composites, AIP Conference Proceedings of the 9th International Conference “Times of Polymers and Composites” (TOP), 2018; pp 020044/1–020044/4.
- Fredi G.; Dorigato A.; Unterberger S.; Artuso N.; Pegoretti A. Discontinuous carbon fiber/polyamide composites with microencapsulated paraffin for thermal energy storage. J. Appl. Polym. Sci. 2019, 136, 47408 10.1002/app.47408. [DOI] [Google Scholar]
- Fredi G.; Dorigato A.; Pegoretti A. Novel reactive thermoplastic resin as a matrix for laminates containing phase change microcapsules. Polym. Compos. 2019, 40, 3711–3724. 10.1002/pc.25233. [DOI] [Google Scholar]
- Dorigato A.; Fredi G.; Pegoretti A. Application of the thermal energy storage concept to novel epoxy/short carbon fiber composites. J. Appl. Polym. Sci. 2019, 136, 47434 10.1002/app.47434. [DOI] [Google Scholar]
- Fredi G.; Dorigato A.; Fambri L.; Pegoretti A. Detailed experimental and theoretical investigation of the thermo-mechanical properties of epoxy composites containing paraffin microcapsules for thermal management. Polym. Eng. Sci. 2020, 60, 1202–1220. 10.1002/pen.25374. [DOI] [Google Scholar]
- Fredi G.; Dorigato A.; Pegoretti A. Dynamic-mechanical response of carbon fiber laminates with a reactive thermoplastic resin containing phase change microcapsules. Mech. Time-Depend. Mater. 2019, 30, 313 10.1007/s11043-019-09427-y. [DOI] [Google Scholar]
- Ryu J. H.; Messersmith P. B.; Lee H. Polydopamine Surface Chemistry: A Decade of Discovery. ACS Appl. Mater. Interfaces 2018, 10, 7523–7540. 10.1021/acsami.7b19865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynge M. E.; Van Der Westen R.; Postmab A.; Stadler B. Polydopamine—a nature-inspired polymer coating for biomedical science. Nanoscale 2011, 3, 4916–4928. 10.1039/c1nr10969c. [DOI] [PubMed] [Google Scholar]
- Jeong Y. K.; Park S. H.; Choi J. W. Mussel-inspired coating and adhesion for rechargeable batteries: a review. ACS Appl. Mater. Interfaces 2018, 10, 7562–7573. 10.1021/acsami.7b08495. [DOI] [PubMed] [Google Scholar]
- Posati T.; Nocchetti M.; Kovtun A.; Donnadio A.; Zambianchi M.; Aluigi A.; Capobianco M. L.; Corticelli F.; Palermo V.; Ruani G.; Zamboni R.; Navacchia M. L.; Melucci M. Polydopamine Nanoparticle-Coated Polysulfone Porous Granules as Adsorbents for Water Remediation. ACS Omega 2019, 4, 4839–4847. 10.1021/acsomega.8b02900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue H. Y.; Du T.; Wang O. X.; Shi Z. P.; Dong H. Y.; Cao Z. X.; Qiao Y.; Yin Y. H.; Xing R. M.; Yang S. T. Biomimetic Synthesis of Polydopamine Coated ZnFe2O4 Composites as Anode Materials for Lithium-Ion Batteries. ACS Omega 2018, 3, 2699–2705. 10.1021/acsomega.7b01752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.; Zhang M. Y.; Wang J. T.; Liu G. H.; Liu H. R.; Jiang Y. J. Bioinspired Modification of Layer-Stacked Molybdenum Disulfide (MoS2) Membranes for Enhanced Nanofiltration Performance. ACS Omega 2019, 4, 4012–4022. 10.1021/acsomega.9b00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y.; Dong W.; Tang L.; Zhang Y.; Kong J.; Gu J. Fabrication and investigations on the polydopamine/KH-560 functionalized PBO fibers/cyanate ester wave-transparent composites. Compos. Commun. 2018, 8, 36–41. 10.1016/j.coco.2018.03.006. [DOI] [Google Scholar]
- Shanmugam L.; Feng X.; Yang J. Enhanced interphase between thermoplastic matrix and UHMWPE fiber sized with CNT-modified polydopamine coating. Compos. Sci. Technol. 2019, 174, 212–220. 10.1016/j.compscitech.2019.03.001. [DOI] [Google Scholar]
- Kwon I. S.; Bettinger C. J. Polydopamine Nanostructures as Biomaterials for Medical Applications. J. Mater. Chem. B 2018, 6, 6895–6903. 10.1039/C8TB02310G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.; Zeng G.; Wang K.; Wan Q.; Tao L.; Zhang X.; Wei Y. Recent developments in polydopamine: an emerging soft matter for surface modification and biomedical applications. Nanoscale 2016, 8, 16819–16840. 10.1039/C5NR09078D. [DOI] [PubMed] [Google Scholar]
- Zheng Z. F.; Cui Z. X.; Si J. H.; Yu S. R.; Wang Q. T.; Chen W. Z.; Turng L. S. Modification of 3-D Porous Hydroxyapatite/Thermoplastic Polyurethane Composite Scaffolds for Reinforcing Interfacial Adhesion by Polydopamine Surface Coating. ACS Omega 2019, 4, 6382–6391. 10.1021/acsomega.9b00404. [DOI] [Google Scholar]
- Yang L.; Phua S. L.; Teo J. K.; Toh C. L.; Lau S. K.; Ma J.; Lu X. A biomimetic approach to enhancing interfacial interactions: polydopamine-coated clay as reinforcement for epoxy resin. ACS Appl. Mater. Interfaces 2011, 3, 3026–3032. 10.1021/am200532j. [DOI] [PubMed] [Google Scholar]
- Ling Y.; Li W.; Wang B.; Gan W.; Zhu C.; Brady M. A.; Wang C. Epoxy resin reinforced with nanothin polydopamine-coated carbon nanotubes: a study of the interfacial polymer layer thickness. RSC Adv. 2016, 6, 31037–31045. 10.1039/C5RA26539H. [DOI] [Google Scholar]
- Epoxy Resins and Structural Adhesives for Composite Materials; Product Catalog. ELANTAS Europe s.r.l. (5/2017). Collecchio, Italy.
- Shirley D. A. High-resolution X-ray photoemission spectrum of the valence bands of gold. Phys. Rev. B 1972, 5, 4709–4714. 10.1103/PhysRevB.5.4709. [DOI] [Google Scholar]
- Beamson G.; Briggs D.. High Resolution XPS of Organic Polymers - The Scienta ESCA300 Database; John Wiley & Sons: Chichester, U.K., 1992. [Google Scholar]
- Hutter J. L.; Bechhoefer J. Calibration of atomic-force microscope tips. Rev. Sci. Instrum. 1993, 64, 1868–1873. 10.1063/1.1143970. [DOI] [Google Scholar]
- Kolesnikova T. A.; Gorin D. A.; Fernandes P.; Kessel S.; Khomutov G. B.; Fery A.; Shchukin D. G.; Mohwald H. Nanocomposite microcontainers with high ultrasound sensitivity. Adv. Funct. Mater. 2010, 20, 1189–1195. 10.1002/adfm.200902233. [DOI] [Google Scholar]
- Roldán M. L.; Centeno S. A.; Rizzo A. An improved methodology for the characterization and identification of sepia in works of art by normal Raman and SERS, complemented by FTIR, Py-GC/MS, and XRF. J. Raman Spectrosc. 2014, 45, 1160–1171. 10.1002/jrs.4620. [DOI] [Google Scholar]
- Sun G.; Zu F.; Koch N.; Rappich J.; Hinrichs K. In situ infrared spectroscopic monitoring and characterization of the growth of polydopamine (PDA) films. Phys. Status Solidi B 2019, 256, 1800308 10.1002/pssb.201800308. [DOI] [Google Scholar]
- Perna G.; Lasalvia M.; Capozzi V. Vibrational spectroscopy of synthetic and natural eumelanin. Polym. Int. 2016, 65, 1323–1330. 10.1002/pi.5182. [DOI] [Google Scholar]
- Furtado L. M.; Ando R. A.; Petri D. F. S. Polydopamine-coated cellulose acetate butyrate microbeads for caffeine removal. J. Mater. Sci. 2020, 55, 3243–3258. 10.1007/s10853-019-04169-1. [DOI] [Google Scholar]
- Al Aani S.; Haroutounian A.; Wright C. J.; Hilal N. Thin Film Nanocomposite (TFN) membranes modified with polydopamine coated metals/carbon-nanostructures for desalination applications. Desalination 2018, 427, 60–74. 10.1016/j.desal.2017.10.011. [DOI] [Google Scholar]
- Socrates G.Infrared and Raman Characteristic Group Frequencies, 3rd ed.; Wiley-VCH Verlag: Weinheim, Germany, 2004. [Google Scholar]
- Salaün F.; Devaux E.; Bourbigot S.; Rumeau P. Influence of process parameters on microcapsules loaded with n-hexadecane prepared by in situ polymerization. Chem. Eng. J. 2009, 155, 457–465. 10.1016/j.cej.2009.07.018. [DOI] [Google Scholar]
- Weiss S.; Urdl K.; Mayer H. A.; Zikulnig-Rusch E. M.; Kandelbauer A. IR spectroscopy: Suitable method for determination of curing degree and crosslinking type in melamine–formaldehyde resin. J. Appl. Polym. Sci. 2019, 136, 47691 10.1002/app.47691. [DOI] [Google Scholar]
- Günzler H.; Gremlich H.-U.. IR-Spektroskopie: Eine Einführung, 4th ed.; Wiley-VCH Verlag: Weinheim, Germany, 2003. [Google Scholar]
- Wang L.; Yan L.; Gao X. Effect of electron beam irradiation on polydopamine and its application in polymer solar cells. Int. J. Energy Res. 2018, 42, 3496–3505. 10.1002/er.4087. [DOI] [Google Scholar]
- Parker R. E.; Isaacs N. S. Mechanisms of epoxide reactions. Chem. Rev. 1959, 59, 737–799. 10.1021/cr50028a006. [DOI] [Google Scholar]
- Silva C.; Simon F.; Friedel P.; Pötschke P.; Zimmerer C. Elucidating the chemistry behind the reduction of graphene oxide using a green approach with polydopamine. Nanomaterials 2019, 9, 902 10.3390/nano9060902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenzel R.; Schiefer T.; Jansen I.; Simon F.; Calvimontes A.; Grundke K.; Häußler L.; Beyer E. Polyelectrolytes to promote adhesive bonds of laser-structured aluminium. Int. J. Adhes. Adhes. 2015, 61, 35–45. 10.1016/j.ijadhadh.2015.05.001. [DOI] [Google Scholar]
- Butt H. J.; Cappella B.; Kappl M. Force measurements with the atomic force microscope: Technique, interpretation and applications. Surf. Sci. Rep. 2005, 59, 1–152. 10.1016/j.surfrep.2005.08.003. [DOI] [Google Scholar]
- Neubauer M. P.; Poehlmann M.; Fery A. Microcapsule mechanics: from stability to function. Adv. Colloid Interface Sci. 2014, 207, 65–80. 10.1016/j.cis.2013.11.016. [DOI] [PubMed] [Google Scholar]
- Fredi G.; Brünig H.; Vogel R.; Scheffler C. Melt-spun polypropylene filaments containing paraffin microcapsules for multifunctional hybrid yarns and smart thermoregulating thermoplastic composites. eXPRESS Polym. Lett. 2019, 13, 1071–1087. 10.3144/expresspolymlett.2019.93. [DOI] [Google Scholar]
- Wang S.; Tozaki K.; Hayashi H.; Hosaka S.; Inaba H. Observation of multiple phase transitions in n-C22H46 using a high resolution and super-sensitive DSC. Thermochim. Acta 2003, 408, 31–38. 10.1016/S0040-6031(03)00312-5. [DOI] [Google Scholar]
- Nishizawa N.; Kawamura A.; Kohri M.; Nakamura Y.; Fujii S. Polydopamine particle as a particulate emulsifier. Polymers 2016, 8, 62 10.3390/polym8030062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J.-F.; Wang X.-Y.; Huang Z.; Zhao Y.-H.; Yuan X.-Y. Thermal conductivity of microPCMs-filled epoxy matrix composites. Colloid Polym. Sci. 2011, 289, 1535–1542. 10.1007/s00396-011-2478-9. [DOI] [Google Scholar]