

Abstract
Alzheimer’s disease (AD) is the most common form of dementia and the most prevalent neurodegenerative disease. Genome-wide association studies have linked PICALM to AD risk. PICALM has been implicated in Aβ42 production and turnover, but whether it plays a direct role in modulating Aβ42 toxicity remains unclear. We found that increased expression of the Drosophila PICALM orthologue lap could rescue Aβ42 toxicity in an adult-onset model of AD, without affecting Aβ42 level. Imbalances in the glutamatergic system, leading to excessive, toxic stimulation, have been associated with AD. We found that Aβ42 caused the accumulation of presynaptic vesicular glutamate transporter (VGlut) and increased spontaneous glutamate release. Increased lap expression reversed these phenotypes back to control levels, suggesting that lap may modulate glutamatergic transmission. We also found that lap modulated the localization of amphiphysin (Amph), the homologue of another AD risk factor BIN1, and that Amph itself modulated postsynaptic glutamate receptor (GluRII) localization. We propose a model where PICALM modulates glutamatergic transmission, together with BIN1, to ameliorate synaptic dysfunction and disease progression.
Graphical Abstract
Graphical Abstract.
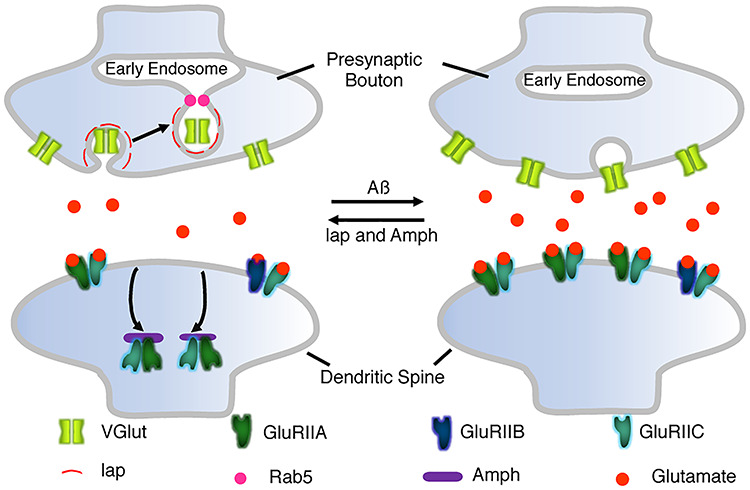
lap and Amph in glutamate metabolism. Aβ affects the localization of both VGlut and GluRIIA, leading to increased glutamate signalling. Lap and Amph restore glutamate signalling, potentially by promoting presynaptic endocytosis of VGlut via lap and/or Rab5/EndoA, and postsynaptic retrival of GluRIIA via Amph.
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disease. Its symptoms include progressive memory loss, cognitive impairment, difficulties in abstract reasoning and decision-making and complete social dependence. AD is characterized by two main neuropathological hallmarks, extracellular amyloid plaques, composed of amyloid-β peptides (Aβ), and intracellular neurofibrillary tangles of hyperphosphorylated tau protein (1).
Most cases of AD are sporadic and strongly age-related. Several genome-wide association studies (GWAS) have linked specific genetic variants to sporadic AD. One of these has been linked to phosphatidylinositol-binding clathrin assembly protein (PICALM) and another to bridging integrator 1 (BIN1), both of which are involved in clathrin-dependent endocytosis (2,3). PICALM interacts with AP2 and clathrin to recruit cargos to membranes (4). It also physically binds to the soluble NSF attachment protein receptors (SNAREs), such as VAMP2 and VAMP8, and interacts with Rab5 and Rab11 to guide synaptic vesicles from the plasma membrane to endosomes (5–8). PICALM expression is decreased in the brains of AD patients (9,10). However, the role of PICALM in AD remains controversial. On the one hand, PICALM knockdown can reduce Aβ42 generation, by limiting APP or γ-secretase internalization, and promotes tau degradation, by triggering autophagy (6,11–13). On the other hand, PICALM overexpression has been reported to modulate APP degradation via elevated autophagy or Aβ42 clearance through transcytosis (4,8). Although these studies have implicated PICALM in Aβ42 production and turnover (11), little is known of its contribution to modulating Aβ toxicity.
BIN1 plays a crucial role in intracellular endosome trafficking, through the interaction with the GTPase dynamin (14). Mice lacking BIN1 present learning deficits (15). It is not clear whether BIN1 is increased or decreased in AD (16,17), and its role in AD pathology is still unclear. It has been implicated mainly in Tau pathology (18), although its exact role remains to be elucidated. Some studies suggest that the upregulation blocks Tau spread (19), others that downregulation ameliorates Tau toxicity (17). BIN1 also plays a role in Aβ production, with reduced expression linked to increased BACE1 and Aβ production (20,21). However, whether BIN1 plays a role in Aβ toxicity has also not been investigated. Recently, BIN1 was shown to be involved in neurotransmitter release in mouse hippocampal neurons (22).
Glutamate excitotoxicity has long been thought to play an important role in AD aetiology (23). Glutamatergic neurons are severely affected in AD, and it has been speculated that the disease might be caused, at least in part, by over-activation of glutamatergic neurons (24). Aβ oligomers enhance glutamate release (25–27) and impair glutamate reuptake by astrocytes (28,29), leading to increased extracellular glutamate and activation of extra-synaptic NMDAR receptors and synaptic damage (25,30). Aβ also affects the composition of glutamate receptors (GluRII), with a reduction of GluA1 and GluA2 subunits of the AMPA receptor, and both GluN1 and GluN2A of the NMDA receptor (31–34). Up-regulation of these has been linked to suppression of Aβ toxicity (35–38). On the other hand, downregulation of GluA3 or GluN2B can ameliorate Aβ toxicity (25,30,39). These studies suggest that amyloid beta peptides (Aß) can exert neurotoxic effects both through increased glutamatergic excitotoxicity and through altered composition of postsynaptic glutamate receptors.
Here we demonstrate that overexpression of the Drosophila PICALM orthologue, like AP180 (lap), ameliorates Aβ42-induced shortened lifespan and locomotor defects in a fly AD model, importantly without affecting Aβ42 levels. Because these findings implicated a gene involved in endocytosis/exocytosis in Aβ42 toxicity, we next performed a small-scale, targeted, genetic screen of endocytic/exocytic genes (Table 1) and identified Rab5 and EndoA as partial suppressors of Aβ42-induced toxicity. Given that Rab5 and EndoA are involved in cargo translocation from the plasma membrane to the early endosome, our findings suggested that early steps in endocytosis, including those mediated by lap, are crucial to AD progression. Aβ expression also led to the accumulation of vesicular glutamate transporters (VGlut) at the presynaptic region, and increased lap expression restored their wild-type localization. Concordantly, lap reduced the increased glutamate release during spontaneous activity associated with Aβ expression. lap also restored the localization of the BIN1 orthologue, amphiphysin (Amph), which was disrupted upon Aβ expression. Moreover, Amph modulated the localization of glutamate receptors (GluRII), which was also disrupted by Aβ expression. We therefore propose a novel model where PICALM and BIN1 can cooperate to restore the distribution of pre- and postsynaptic proteins involved in glutamatergic neurotransmission and thus ameliorate aberrant glutamatergic transmission and neurotoxicity in the presence of Aβ42.
Table 1.
Screen for endocytic–exocytic genes on Drosophila lifespan
| Drosophila homologue | Human gene | Function | Effect on lifespan |
|---|---|---|---|
| Cindr | CD2AP | Actin remodelling | Decreased |
| Clc | CLC | Vesicle formation | Decreased |
| Shi | DYNAMIN | Vesicle scission | None |
| EndoA | ENDOA | Vesicle formation | Increased |
| Eps15 | EPS15 | Vesicle formation | Decreased |
| Iqf | EPSIN | Vesicle formation | Decreased |
| Rab4 | RAB4 | Endocytic recycling | Decreased |
| Rab5 | RAB5 | Plasma membrane to early endosome | Increased |
| Rab7 | RAB7 | Early-to-late endosome | Decreased |
| Rab8 | RAB8 | Endocytic recycling | Decreased |
| Rab10 | RAB10 | Endocytic recycling | Decreased |
| Rab11 | RAB11 | Endocytic recycling | Decreased |
| Rab14 | RAB14 | Endocytosis | None |
| Snap25 | SNAP25 | Vesicle fusion | None |
| nSyb | VAMP2 | Vesicle fusion | None |
Summary table of the effect of overexpression of a number of genes on the lifespan of Aß-expressing flies.
Results
lap reduces Aβ42 pathology
To explore the role of PICALM in AD aetiology in vivo, we examined the role of the Drosophila homologue lap (40) in Aβ pathology. We generated an adult-onset model of Aβ toxicity that expressed two copies of wild-type Aβ42 (41) under the control of an inducible, pan-neuronal driver (elavGS) (42). ElavGS is induced by the drug RU486 (42), which was added to the fly food only after eclosion, thus restricting the expression of Aβ to adult neurons. The Aβ42-expressing flies had a shortened lifespan and displayed locomotor deficits (Fig. 1A and B), suggesting that wild-type Aβ42 is toxic to adult neurons, as previously reported (43). In humans, two SNPs near the gene PICALM, rs3851179 and rs541458, which are associated with decreased levels of AD occurrence in a number of patient cohorts (44,45) are associated with higher levels of expression of PICALM (using the LIBD eQTL browser (46)), potentially suggesting that PICALM has a protective role in AD. To test this, we generated flies overexpressing lap under the control of the UAS promoter and confirmed the overexpression under induction of ElavGS by qPCR (Fig. 1D). Co-overexpression of lap in Aβ42-expressing flies attenuated Aβ42 toxicity, ameliorating both the reduction in lifespan and the impaired locomotion as assessed by negative geotaxis (climbing) assays (Fig. 1A and B). Importantly, increased lap expression did not alter Aβ42 protein levels (Fig. 1C), suggesting that lap acts downstream of Aβ42 generation or degradation. Conversely, inhibition of lap by RNA interference enhanced Aβ42 toxicity, leading to further shortening of lifespan (Fig. 1F).
Figure 1.
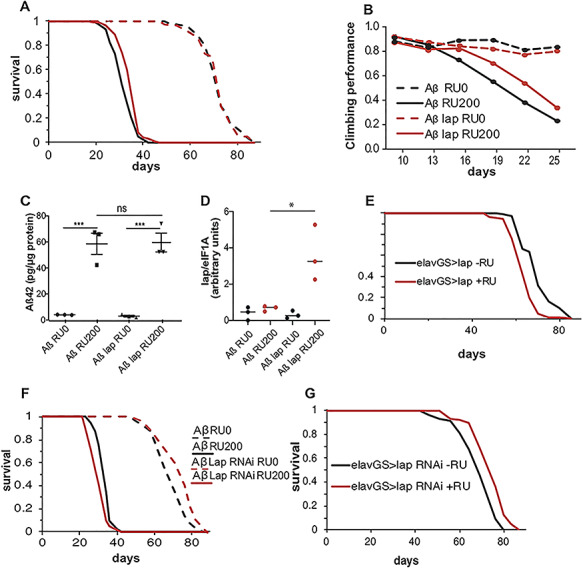
lap alleviates Aβ42 toxicity. (A) Survival curves of flies expressing Aβ, with (red lines, UAS-Aβ/UAS-lap; elavGS/+) and without (black lines, UAS-Aβ/+; elavGS/+) lap co-overexpression, in adult neurons (RU200, solid lines) and uninduced controls (RU0, dashed lines). Expression of Aβ in neurons shortened lifespan, and lap co-overexpression significantly improved this shortened lifespan. n > 120 per condition. Aβ RU0 versus Aβ RU200, P = 1.1E−72; Aβ RU0 versus Aβ lap RU0, ns, not significant; Aβ lap RU0 versus Aβ lap RU200, P = 2.44E−47; Aβ RU200 versus Aβ lap RU200, P = 1.8E−5, determined by log–rank test. There was a significant interaction of genotype and RU by Cox proportional hazard analysis, P = 0.016, indicating that expression of lap significantly extended the lifespan of Aß-expressing flies. (B) Locomotor performance index of flies of the same genotypes as in (A). Aβ caused a climbing defect, which was significantly improved by the co-overexpression of lap, n = ~50 flies per condition. There was a statistically significant interaction between RU and genotype by ordinal logistics, P = 0.00040826, indicating that the expression of lap significantly improved the climbing of Aß-expressing flies. (C) Aβ42 protein levels, measured by ELISA, in the heads of 21-day-old flies expressing Aβ with or without co-overexpression of lap in neurons (RU200) and uninduced controls (RU0). lap co-overexpression had no effect on Aβ42 levels. Means ± SEM, n = 3 biological replicates of 10 heads per replicate per condition. F(3,8) = 34.53, P < 0.0001 by one-way ANOVA;***P < 0.01, ns, not significant, comparison by Tukey’s post hoc test. (D) qPCR of lap mRNA levels in fly heads expressing Aß, Aß and lap (RU200) and their uninduced controls (RU0), showing overexpression of lap in the Aß, lap expressing flies (F(3,8) = 1.987) by one-way ANOVA *P = 0.0088 for comparison between the two induced conditions. (E) Adult survival curves of lap overexpression in adult neurons (RU200, red line) and uninduced controls (RU0, black line), n > 120 per condition, P = 4.5E−16, by log–rank test. (F) Adult survival of flies harbouring Aβ with (red lines, UAS-Aβ/+; elavGS/UAS-lap-RNAi) or without (black lines, UAS-Aβ/+; elavGS/+) lap RNAi in adult neurons (RU200, solid lines) and uninduced controls (RU0, dashed lines). Inhibition of lap reduces longevity of Aβ-expressing flies. n > 120 per condition. Aβ RU0 versus Aβ RU200, P = 7.6254E−72; Aβ RU0 versus Aβ lap RU0, P = 1.48871E−06; Aβ lap RU0 versus Aβ lap RU200, P = 1.65991E−69; Aβ RU200 versus Aβ lap RU200, P = 3.2225E−10, by log–rank test. There was a significant interaction of genotype and RU by Cox proportional hazard analysis, P = 9.5E−06, indicating that downregulation of lap significantly shortened the lifespan of Aß-expressing flies. (G) Adult survival curves of lap RNAi flies in adult neurons (RU200, red line) and uninduced controls (RU0, black line), n > 120 per condition. P = 1.08638E−09, by log–rank test.
The protective allele for SNP rs3851179 is also enriched in Italian centenarians, suggesting a role for PICALM/lap in healthy ageing as well as AD (47). We therefore examined the effect of lap on healthy ageing, by both overexpressing and downregulating its expression in neurons in the absence of Aβ42. In contrast to our findings with Aβ42 expression, lap overexpression on its own shortened lifespan, while RNAi against lap extended lifespan (Fig. 1E and G), indicating that the neuroprotective effect of lap overexpression was specific to Aβ toxicity and not due to a broader effect on ageing.
lap regulates glutamate release
AD is associated with glutamate excitotoxicity (23,48), and Aβ oligomers lead to increased glutamate release (25). To check if this was also the case in our fly model, we used the fluorescent extracellular glutamate reporter intensity-based glutamate sensing fluorescent reporter (iGluSnFR) to detect extracellular glutamate levels (49,50). We expressed UAS-iGluSnFR in larval motor neurons and observed transient, local bursts of fluorescence, presumably associated with spontaneous local release of glutamate (Fig. 2A and E). We occasionally also observed waves of fluorescence running along the anterior–posterior axis, similar to those previously reported (50) (Supplementary Material, Movie S1). Expression of Aβ led to a dramatic increase in the intensity of local bursts (Supplementary Material, Movie S2, Fig. 2B and F), suggesting that Aβ42 increased the release of glutamate, consistent with a previous study observing increased glutamate neurotransmission in APP/PS1 transgenic mice (51). However, we did not observe any change in the number of glutamate release events per minute with Aβ expression (Fig. 2J). Strikingly, lap co-expression reduced the intensity of local fluorescence bursts back to control levels (Fig. 2D, H, I and Supplementary Material, Movie S4), whereas expression of lap alone has no effect (Fig 2C, G, I and Supplementary Material, Movie S3). Taken together, these findings suggest that Aβ compromises components of glutamatergic signalling, leading to increased glutamate release, and that lap overexpression acts to reinstate healthy levels of glutamatergic signalling.
Figure 2.
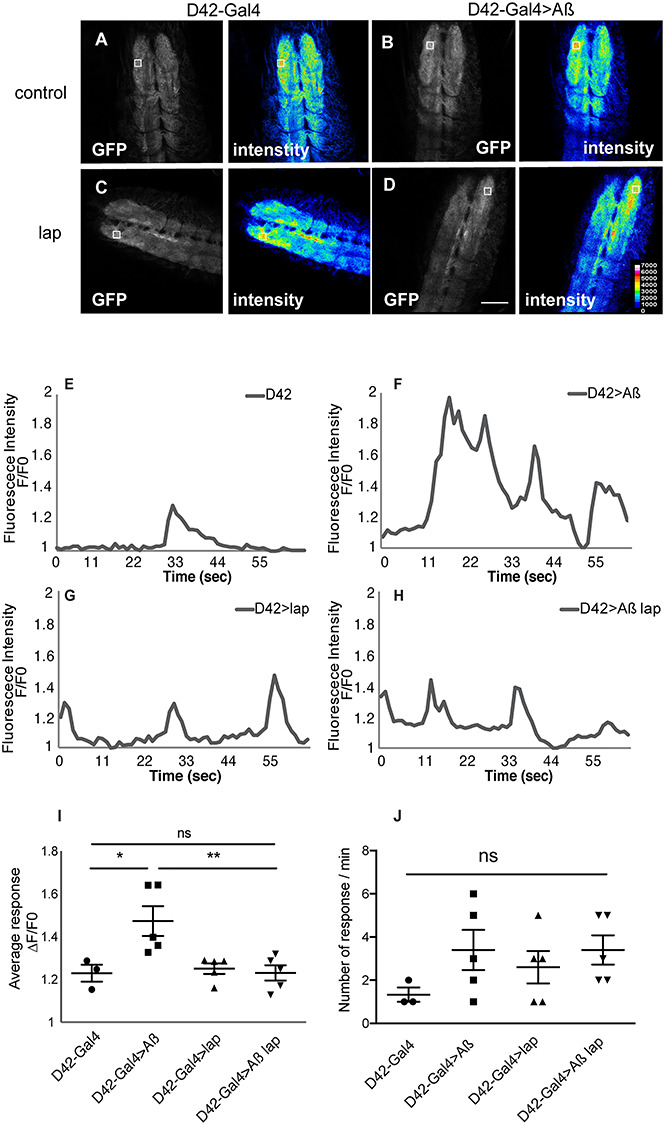
Lap reduces excess glutamate release upon Aβ expression. (A–D) Single confocal images from movies of intact L2 larvae expressing iGluSnFR in neurons, showing local and transient increases in fluorescence reflecting the release of glutamate. The image is a representative burst of fluorescence from a movie (in grey) and an intensity map of the same frame (in colour). Genotypes: (A) D42-Gal4/UAS-iGluSnFR, (B) UAS-Aβ/+; D42-Gal4/UAS-iGluSnFR, (C) UAS-lap/+; D42-Gal4/UAS-iGluSnFR, (D) UAS-Aβ/UAS-lap; D42-Gal4/UAS-iGluSnFR. Scale bar, 20 μm. (E–H) Traces of changes in iGluSnFR fluorescence display the spontaneous activity in the neuropil. Fluorescence signals are normalized to minimum fluorescence in each trace and expressed relative to baseline. Genotypes are as above. The arrow indicates the time point shown in (A–D) (I) Quantification of the amplitude of the glutamate burst (described as changes in fluorescence relative to baseline) in the neuropil of L2 larvae shown in A–D, expressing driver alone with the reporter or together with Aβ, lap or both driven, plotted as means per animal ± SEM, n > 3 animals. Genotypes are as above. F(3,14) = 6.503, P = 0.0056, determined by one-way ANOVA; **P < 0.001, ns, not significant, comparison by Tukey’s post hoc test. (J) Quantification of the number of glutamate release events of the same larvae in (I), plotted as means ± SEM, n > 3 per condition. Genotypes: D42-Gal4/UAS-iGluSnFR, UAS-Aβ; D42-Gal4/UAS-iGluSnFR, UAS-lap; D42-Gal4/UAS-iGluSnFR, UAS-Aβ/UAS-lap; D42-Gal4/UAS-iGluSnFR. F(3,14) = 1.248, P = 0.3299, determined by one-way ANOVA; ns, not significant, comparison by Tukey’s post hoc test.
Next we investigated the molecular mechanisms by which lap reduced Aβ42 toxicity. PICALM plays a major role in endocytosis (52,53), which is important for accurate neurotransmitter signalling. To determine whether endocytosis could play a wider role in the rescue of Aβ toxicity, we performed a targeted genetic screen of well-characterized components regulating endocytosis (Table 1). Of the genes tested, only the overexpression of Rab5 and EndoA ameliorated the shortened lifespan (Fig. 3A and B). However, Rab5 did not ameliorate climbing (Fig. 3C), while EndoA worsened the overall climbing ability of Aß-expressing flies but slowed down the rate of decline, suggesting it was slowing the development of Aß toxicity but possibly had some direct detrimental effect on climbing. In contrast, the overexpression of Rab4, Rab7, Rab8, Rab10 and Rab11 exacerbated Aβ42 toxicity (Table 1). These findings suggest that the upregulation of early steps of clathrin-mediated endocytosis up to the early endosome could play some part in the amelioration of Aβ toxicity, consistent with a study in yeast (54). However, given the contrasting effects we observed for different Rab genes in our small screen, we hypothesized that lap’s rescue could be mediated by additional key factors.
Figure 3.
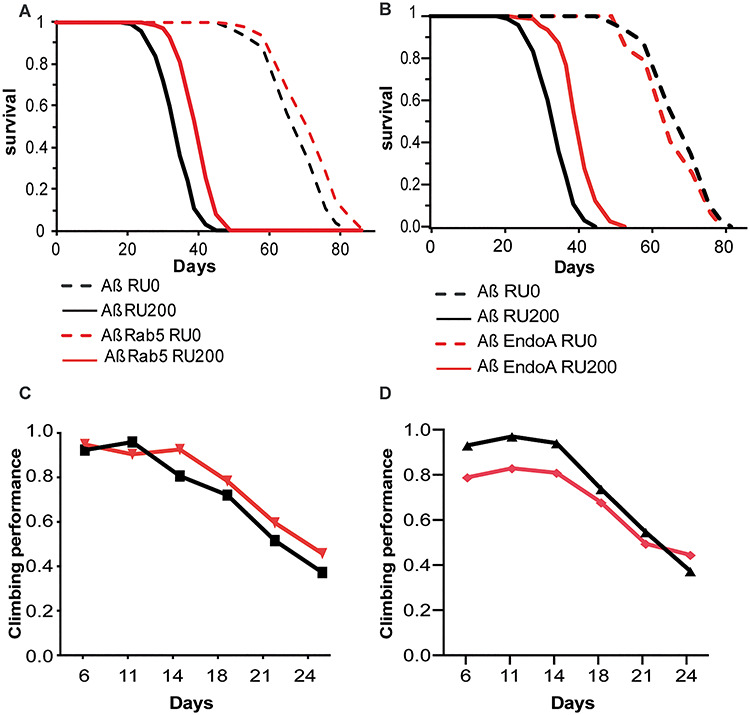
Rab5 and Endo A ameliorates Aβ42 toxicity. (A) Survival curves of flies expressing Aβ, with (red lines, UAS-Aβ/+; elavGS/UAS-Rab5) and without (black lines, UAS-Aβ/+; elavGS/+) Rab5 co-overexpression, in adult neurons (RU200, solid lines) and uninduced controls (RU0, dashed lines). Co-overexpression of Rab5 extended lifespan of Aβ-expressing flies. n > 120 per condition. Aβ RU0 versus Aβ RU200, P = 7.46192E−76; Aβ RU0 versus Aβ Rab5 RU0, P = 6.07338E−05; Aβ Rab5 RU0 versus Aβ Rab5 RU200, P = 1.54498E−70; Aβ RU200 versus Aβ Rab5 RU200 P = 6.06826E−22, determined by log–rank test. There was a significant interaction of genotype and RU by Cox proportional hazard analysis, P = 0.000338, indicating that expression of Rab5 significantly extended the lifespan of Aß-expressing flies. (B) Survival curves of flies expressing Aβ, with (red lines, UAS-Aβ/UAS-EndoA; elavGS/+) and without (black lines, UAS-Aβ/+; elavGS/+) EndoA co-overexpression, in adult neurons (RU200, solid lines) and uninduced controls (RU0, dashed lines). Co-overexpression of EndoA extended lifespan of Aβ-expressing flies. n > 120 per condition. Aβ RU0 versus Aβ RU200, P = 7.46192E−76; Aβ RU0 versus Aβ EndoA RU0, P = 0.007893292; Aβ EndoA RU0 versus Aβ EndoA RU200, P = 5.2333E−70; Aβ RU200 versus Aβ EndoA RU200 P = 1.42894E−26, determined by log–rank test. There was a significant interaction of genotype and RU by Cox proportional hazard analysis, P < 2E−16, indicating that expression of EndoA significantly extended the lifespan of Aß-expressing flies. (C) Locomotor performance index of induced flies of the same genotypes as in (A). Co-overexpression of Rab5 did not effect climbing ability by ordinal logistics regression (P = 0.62), n = ~50 flies per condition. (D) Locomotor performance index of induced flies of the same genotypes as in (B). Co-overexpression of EndoA worsened the climbing ability of Aß flies but slowed down the decline relative to Aß-expressing flies alone, P = 3.3867E−06 for effect of genotype (z value = −4.6), P = 0.00011043 for interaction between genotype and time (z value = 3.9), n = ~50 flies per condition.
lap mediates VGlut localization
Defects in endocytosis caused by Aβ have been reported to disrupt the trafficking of transmembrane proteins to their proper destination (54), and alterations in endocytic trafficking can disrupt the delivery or recycling of synaptic proteins (55). lap collaborates with clathrin to recycle synaptic vesicles, regulating the efficiency of synaptic vesicle endocytosis and vesicle size (56). lap is also required for recruitment of synaptic vesicle proteins (40,57) and is known to bind to vesicular glutamate transporters (VGlut) (57). We confirmed this by co-immunoprecipitating VGlut with lap in heads of wild type adult flies (Fig. 4D). VGlut is involved in loading glutamate into synaptic vesicles and regulates glutamate release events. We therefore determined whether the protective role of lap was mediated by VGlut. VGlut expression was not altered by Aβ42 or lap expression (Fig. 4C). Next we assessed VGlut distribution. Adult Drosophila fly brains have a high density of neurons, making it difficult to monitor individual synapses. We therefore turned to the larval neuromuscular junctions (NMJ), which provide an excellent model system for monitoring individual synapses, and are extensively used to analyse cellular and molecular mechanisms of synaptic development and neurotransmission (58). We observed that VGlut was abnormally accumulated at the presynaptic terminal of the NMJ upon Aβ42 expression (Fig. 4A and B), potentially impairing glutamatergic synaptic transmission. This accumulation was reduced by lap co-overexpression (Fig. 4A and B), suggesting that lap overexpression acts to reinstate wild-type glutamatergic signalling by directly binding (Fig. 4D) and regulating the localization of vesicular transporters (Fig. 4) and therefore affecting the release of glutamate as previously observed (Fig. 2).
Figure 4.
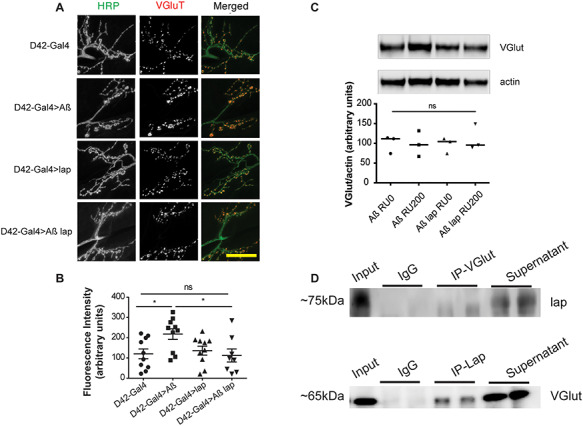
Lap reduces the Aβ-induced accumulation of VGlut at the larval NMJ. (A) Confocal images of the NMJs of wandering third-instar larvae expressing the D42-Gal4 driver alone (+/+; D42-Gal4/+), and Aβ (UAS-Aβ/+; D42-Gal4/+), lap (UAS-lap/+; D42-Gal4/+), or Aβ + lap (UAS-Aβ/UAS-lap; D42-Gal4/+) driven by D42-Gal4. Scale bar, 20 μm. (B) Fluorescence intensity scores are plotted as means ± SEM, n > 7 per condition. F(3,34) = 3.576, P = 0.0238 determined by one-way ANOVA; *P < 0.05, ns, not significant, comparison by Tukey’s post hoc test. (C) Western blot and quantification of VGlut relative to actin in adult Drosophila heads expressing Aß or Aß and lap (RU200) and their uninduced controls (RU 0). Genotypes: UAS-Aβ; elavGS/+, UAS-Aβ/UAS-lap; elavGS/+. (D) Western blots of fractions of a co-IP for VGlut (upper) and lap (lower) from the heads of wild-type adult flies, probed for lap (upper) and VGlut (lower). Samples are head extracts before the IP was started (input), bead-only pull-down (IgG) showing no non-specific binding; pull-down with the indicated antibody (IP-VGlut, IP-lap), showing successful pull-down of the binding partner; supernatant after the pull-down (supernatant), showing only partial depletion; please see Materials and Methods for details. These IP show that VGlut can pull-down lap and lap can pull-down VGlut. All co-IPs were run in duplicate.
Postsynaptic loss of Amph is rescued by lap
BIN1, another AD modifier identified by GWAS in humans (59), also plays a role in endocytosis (60) and has recently been shown to regulate neurotransmitter release in mouse glutamatergic neurons (22). The overexpression of Amph, the fly homologue of BIN1, led to a slight rescue in lifespan (Fig. 5A) and climbing (Fig. 5B), without affecting Aß levels (Fig. 5C).
Figure 5.
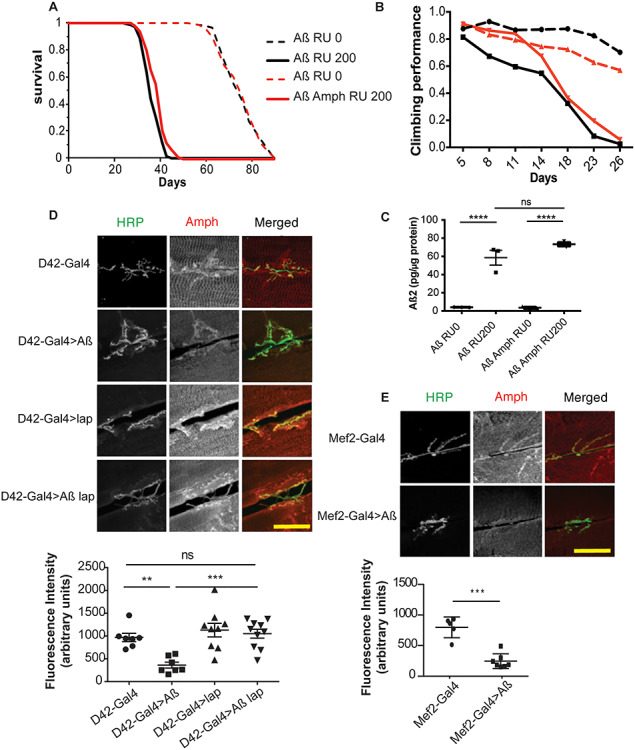
Lap regulates Amph localization. (A) Survival curves of flies expressing Aβ, with (red lines, UAS-Aβ/UAS-Amph; elavGS/+) and without (black lines, UAS-Aβ/+; elavGS/+) lap co-overexpression, in adult neurons (RU200, solid lines) and uninduced controls (RU0, dashed lines). Expression of Aβ in neurons shortened lifespan, and lap co-overexpression significantly rescued this shortened lifespan. n > 145 per condition. Aβ RU0 versus Aβ RU200, P = 3.29E−73; Aβ RU0 versus Aβ Amph RU0, ns, not significant; Aβ Amph RU0 versus Aβ Amph RU200, P = 1.25E−82; Aβ RU200 versus Aβ lap RU200, P = 8.8E−7, determined by log–rank test. (B) Locomotor performance index of flies of the same genotypes as in (A). Aβ caused a climbing defect, which was significantly rescued by co-overexpression of Amph, n = ~50 flies per condition. There was a statistically significant interaction between RU and genotype by ordinal logistics, P = 1.0908E−12, indicating that expression of Amph significantly improved the climbing specifically of of Aß-expressing flies. (C) Aβ42 protein levels, measured by ELISA, in the heads of 21-day-old flies expressing Aβ with or without co-overexpression of Amph in neurons (RU200) and uninduced controls (RU0). Amph co-overexpression had no effect on Aβ42 levels. Means ± SEM, n = 3 biological replicates of 10 heads per replicate per condition. F(3,8) = 0.9406, P < 0.0001 by one-way ANOVA; ****P < 0.001, ns, not significant, comparison by Tukey’s post hoc test. (D) Confocal images and quantification of Amph fluorescence at the NMJ of wandering third-instar larvae expressing the D42-Gal4 driver alone (+/+; D42-Gal4/+), and Aβ (UAS-Aβ/+; D42-Gal4/+), lap (UAS-lap/+; D42-Gal4/+), or Aβ + lap (UAS-Aβ/UAS-lap; D42-Gal4/+) driven by D42-Gal4, plotted as means ± SEM, n > 7 per condition. Scale bar, 20 μm. F(3,29) = 8.954, P = 0.0002, determined by one-way ANOVA; **P < 0.01, ***P < 0.001, ns, not significant, comparison by Tukey’s post hoc test. (E) Confocal images and quantification of Amph fluorescence at the NMJ of wandering third-instar larvae expressing the Mef2-Gal4 driver alone (Mef2-Gal4/+) or Aβ driven by Mef2-Gal4 (UAS-Aβ/+; Mef2-Gal4/+), plotted as means ± SEM, n > 5 per condition. Scale bar, 20 μm. ***P < 0.001, comparison by Student’s test.
In flies, lap is expressed presynaptically (40,56), whereas Amph is expressed postsynaptically (61,62), implying no direct interaction between lap and Amph. To investigate the interplay between them, we assessed Amph localization at the larval NMJ. We expressed Aβ either presynaptically, with D42-Gal4, or postsynaptically, with Mef2-Gal4, and saw that, in both cases, Amph abundance was significantly decreased upon Aβ42 expression (Fig. 5D and E). Overexpression of Aß at the NMJ does not affect the bouton size or dendritic branching (63). We therefore hypothesized that the changes in signalling were responsible for changed in Amph localization via a yet unknown mechanism. Overexpression of lap presynaptically rescued Amph localization at the NMJ (Fig. 5D), suggesting that presynaptic lap can affect the postsynaptic localization of Amph, potentially through the effects on VGlut and glutamate release observed above.
Amph and lap modulate GluR accumulation
Amph modulates the localization of Dlg, the Drosophila homologue of PSD-95, which is known to stabilize glutamate receptors (61,62). The fly NMJ ionotropic glutamate receptor GluRII is composed of regulatory subunits GluRIIA, GluRIIB and constitutive GluRIIC, with GluRIIA increasing postsynaptic sensitivity and GluRIIB decreasing it (64–67). We determined whether Aβ42 regulated GluRII localization in larvae by expressing Aβ42 postsynaptically, and we found that GluRIIA substantially accumulated on the plasma membrane while GluRIIB decreased (Fig. 6A–D), in line with previous studies showing that the upregulation of GluRIIA leads to a decrease in GluRIIB (68). GluRIIC localization remained unchanged (Fig. 6E). These findings suggest that Aβ expression can alter the composition of GluRII receptors postsynaptically.
Figure 6.
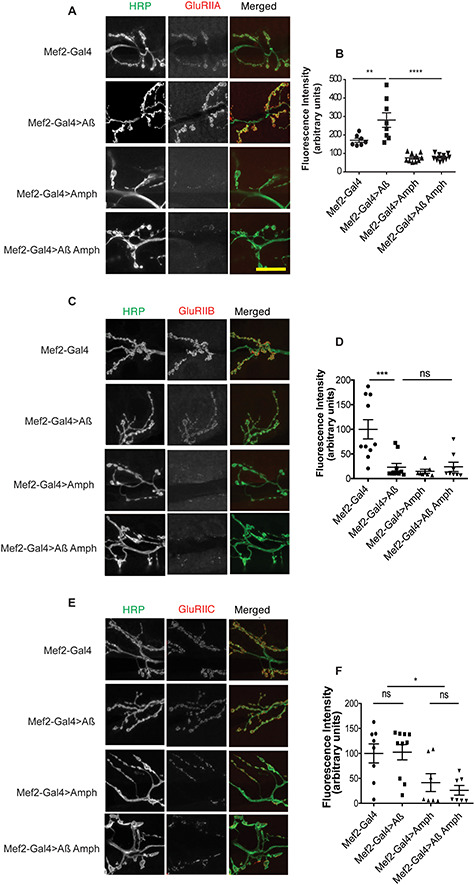
Amph effects GluRII localization. (A) Confocal images of the NMJ of wandering third-instar larvae expressing the Mef2-Gal4 driver alone (+/+; Mef2-Gal4/+), and Aβ (UAS-Aβ/+; Mef2-Gal4/+), Amph (Mef2-Gal4/UAS-Amph), or Aβ + lap (UAS-Aβ/+; Mef2-Gal4/UAS-Amph) driven by Mef2-Gal4, stained for GluRIIA. (B) Fluorescence intensity scores from (A) are plotted as means ± SEM, n > 7 per condition. F(3,31) = 25.04, P < 0.0001, determined by one-way ANOVA; **P < 0.01, ***P < 0.001, ****P < 0.0001, comparison by Tukey’s post hoc test. (C) GluRIIB fluorescence at the NMJ of wandering third-instar larvae expressing driver alone and Aβ, Amph or both driven by Mef2-Gal4 (genotypes as in A). (D) Fluorescence intensity scores from (C) are plotted as means ± SEM, n > 7 per condition. Scale bar, 20 μm. F(3,32) = 10.66, P < 0.001, by a one-way ANOVA; ***P < 0.001, ns, not significant, by Tukey’s post hoc test. (E) GluRIIC fluorescence at the NMJ of wandering third-instar larvae expressing driver alone, and Aβ, Amph or both driven by Mef2-Gal4 (genotypes as in A). (F) Fluorescence intensity scores from (E) are plotted as means ± SEM, n > 7 per condition. Scale bar, 20 μm. F(3,28) = 5.847, P = 0.0031 by one-way ANOVA; *P < 0.05, ns, not significant, by Tukey’s post hoc test.
We next assessed whether increased Amph expression could rescue these deficits in the composition of GluRII receptors. However, we found that overexpression of Amph led to a dramatic decrease in the localization of all the GluRII subunits at the NMJ (Fig. 6), indicating that Amph likely modulates glutamate receptor localization rather than composition.
Lap and Amph therefore both regulate the localization of key components of glutamatergic signalling, which could contribute to their rescue of Aβ toxicity.
Given Amph expression postsynaptically affects the localization of GluRIIA while Aβ42 and lap expression presynaptically affects Amph localization postsynaptically, we checked whether the expression of Aß and lap presynaptically affected GluRIIA. Indeed the expression of Aβ42 presynaptically increased GluRIIA levels postsynaptically, and co-overepxression of lap reduced GluRIIA levels (Fig. 7), suggesting that lap, via Amph, can affect GluRIIA localization. How this is mediated will require further investigation.
Figure 7.
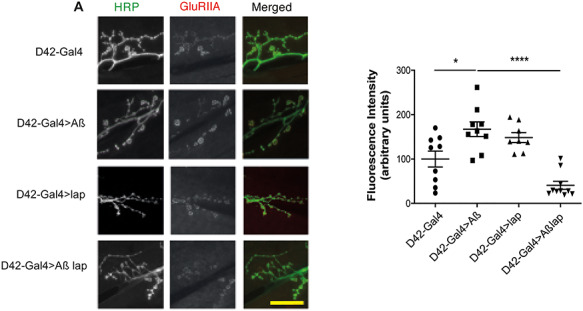
Lap affects GluRII localization. (A) Confocal images and (B) quantification of GluRIIA fluorescence at the NMJ of wandering third-instar larvae expressing the D42-Gal4 driver alone (+/+; D42-Gal4/+), and Aβ (UAS-Aβ/+; D42-Gal4/+), lap (UAS-lap/+; D42-Gal4/+), or Aβ + lap (UAS-Aβ/UAS-lap; D42-Gal4/+) driven by D42-Gal4, plotted as means ± SEM, n > 8 per condition. Scale bar, 20 μm. F(3,32) = 1.558, P ≤ 0.0001, determined by one-way ANOVA; *P < 0.05, ****P < 0.0001, ns, not significant, comparison by Tukey’s post hoc test.
Discussion
Several publications have described the role of PICALM in Aβ42 production and clearance. However, its role in Aβ42 toxicity remains less explored. Our findings provide a novel link between the Drosophila homologues of PICALM and BIN1, lap and Amph, respectively, and glutamatergic transmission in an AD model.
We showed that Aβ expression leads to an increase in spontaneous local burst of glutamate, possibly because of an increase in presynaptic VGlut, which could lead to excessive glutamate release (69). Based on the known effects of VGlut overexpression in increasing vesicle size rather than altering glutamate concentration within the vesicle (69), and the ability of lap to reduce vesicle size (56), it is likely that increased presynaptic vesicle size is one mechanism by which Aβ increases glutamate release. In addition, we find that Aβ expression increases postsynaptic GluRIIA, which can lead to an alteration in postsynaptic sensitivity of the GluRII receptor (67). Both of these changes could lead to aberrant glutamatergic signalling and neurotoxicity. Defects in glutamatergic signalling are a key feature of AD (23), with expression of glutamate receptors and transporters altered in sporadic AD patients (28) and mouse AD models (70). Furthermore, treatment of mouse hippocampal slices and cultured neurons with Aß oligomers leads to excess extracellular glutamate (71,72). We have shown that lap and Amph interact from both sides of the synapse to restore wild-type levels and localization of effectors of glutamatergic synaptic transmission. lap decreases VGlut levels presynaptically, whereas Amph lowers GluRIIA levels postsynaptically. Moreover, lap can restore postsynaptic Amph localization, which is disrupted by Aβ expression, suggesting that lap, via Amph, could possibly modulate GluRIIA localization too.
lap has been shown to bind VGlut and mediate its endocytosis from the plasma membrane (57). We also showed that Rab5 and EndoA, which play a role in mediating the formation of clathrin coated pits at the plasma membrane (73), as well as in fusion of endocytic vesicles with early endosomes (74,75), could also rescue Aβ42 shortened lifespan. This finding suggests that lap’s endocytic function may contribute to its ability to rescue Aβ42 toxicity, by removing excess VGlut from the synaptic terminal. In contrast, PICALM endocytic function has not been directly linked to synaptic vesicle proteins or to glutamatergic signalling, and it will be interesting to determine whether PICALM plays a similar role in mammalian AD models and can directly modulate Aβ42 toxicity. It is interesting to note the opposing effect of lap in normal ageing as opposed to a disease context. It is possible that, since glutamate receptors have been shown to decrease during ageing (76,77), a decrease in lap and possibly also endocytosis helps maintain glutamate signalling in the context of ageing, but in a pathological context characterized by excess glutamate signalling, an increase in lap is beneficial. One caveat in our experiments is we overexpressed a single lap isoform, whereas in flies there are nine isoforms; it will be interesting to determine the effect of overexpressing the other isoforms.
Amph also plays an important role in endocytosis, but its role in Aβ toxicity remains unexplored. Our study highlights a role for Amph in regulating the localization of glutamate receptor GluRII at the synapse. GRIK4, the human homologue of GluRIIA, is increased in AD patients (78), and it would therefore be interesting to verify whether BIN1 can modulate its localization.
In summary, we identified a novel role of two prominent AD-associated GWAS hits, PICALM and BIN1, as modulators of glutamatergic signalling, which could contribute to their role in AD aetiology. It would be interesting to investigate whether this role is conserved in mammalian models of AD, thus potentially opening the possibility of targeting PICALM and BIN1 as modulators of Aβ toxicity in sporadic AD.
Materials and Methods
Fly husbandry and stocks
All flies were reared at 25°C on a 12-h:12-h light–dark (LD) cycle at constant humidity and on standard sugar-yeast-agar (SYA) medium (agar, 15 g/l; sugar, 50 g/l; autolyzed yeast, 100 g/l; nipagin, 30 ml/l (of 10% solution in ethanol) and propionic acid, 2 ml/l). ElavGS, derived from the original elavGS 301.2 line (42), was a gift from Dr H. Tricoire (CNRS), the UAS-Aβ42 stock was a gift from Dr P. Fernandez-Funez (University of Minnesota, Twin Cities, USA) (41), and the UAS-Cindr line was a gift from Dr H. Stenmark (University of Oslo, Oslo, Norway) (79). All UAS lines for the genetic screen (including UAS-Rab5-GFP and UAS-Amph), lap RNAi (TRiP.HMS01939), D42-Gal4, Mef2-Gal4 and iGluSnFR were obtained from the Bloomington Drosophila Stock Center. UAS-Rab8-HA and UAS-Rab11-HA were obtained from the FlyORF. The Amph RNAi (v9264) line was obtained from the Vienna Drosophila RNAi Center.
To generate the UAS-lap transgenic line, a 1.4-kb DNA fragment containing the lap A isoform was amplified by PCR using primers CACCATGACCATGGCAGGG and TTACTGTGCGGCGCCG from cDNA clone RH47395 from Drosophila Genomics Resource Center (NIH Grant 2P40OD010949), gateway cloned into an entry vector and transferred into a gateway compatible pUAST vector according to standard protocols (80). The UAS-lap construct was randomly inserted into the w1118 background, and two clones on chromosome 2 were used for the analysis. To ensure a homogeneous genetic background between all lines, all transgenes used, unless otherwise stated, were backcrossed to the wDah stock for six generations. All experiments were carried out on mated females, unless otherwise stated.
Use of mifepristone (RU486) to induce transgene expression by elavGS
For all experiments involving RU486 addition to fly food, the compound was dissolved in a stock solution of ethanol and added to the fly food while it was still liquid but had cooled to 50°C. The stock solution was added to the food, mixed well, dispensed into individual fly vials and allowed to cool to room temperature overnight before storage at 4°C. On the day of experiments, food vials were warmed to room temperature before being used. RU486 (Sigma, stock solution 100 mm in ethanol) was added to the food at a final concentration of 200 μm, with 2 ml/l ethanol used as the vehicle control condition. To induce gene expression with RU486, 24–48 h after eclosion, female flies carrying a heterozygous copy of elavGS and at least one UAS construct were transferred to vials containing either vehicle control food or food supplemented with 200-μm mifepristone (RU486). Flies were maintained on either control food or RU486-containing food throughout their lifespan.
Lifespan analysis
Flies were raised at standard density in 200-ml bottles. After eclosion, flies were allowed to mate for 24–48 h. For each experiment at least 130–150 females per condition were split into groups of 15 and housed in vials containing SYA medium with or without RU486. Deaths were scored, and flies tipped onto fresh food three times a week. Data are presented as cumulative probability of survival, and survival rates were compared using log–rank tests and Cox proportional hazards. All lifespans were measured at 25°C, unless otherwise stated.
Negative geotaxis assay
The climbing assay was performed as previously described (81). Briefly, 50 flies were housed in a glass-walled chamber 25-cm tall, and flies were tapped to the bottom and allowed to climb for 20 s before scoring. The numbers of flies in the top 5 cm, centre 15 cm, and bottom 5 cm were scored. A performance index was calculated for each time point and plotted using the following formula:
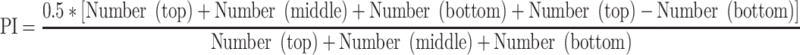 |
Statistical analysis was performed in R using ordinal logistics package.
RT-qPCR
Total RNA was extracted from 20 to 25 fly heads per sample using TRIzol® (GIBCO) according to the manufacturer’s instructions. The concentration of total RNA purified for each sample was measured using an Eppendorf BioPhotometer. One microgram of total RNA was then subjected to DNA digestion using DNase I (Ambion), immediately followed by reverse transcription using the SuperScript® II system (Invitrogen) with oligo(dT) primers. Quantitative PCR was performed using the PRISM 7000 sequence–detection system (Applied Biosystems), SYBR® Green (Molecular Probes), ROX Reference Dye (Invitrogen) and HotStarTaq (Qiagen) by following the manufacturer’s instructions. Each sample was analysed in duplicate, and the values are the mean of three independent biological repeats ± SEM. The primers used were as follows:
eIF1A ATCAGCTCCGAGGATGACGC + GCCGAGACAGACGTTCCAGA.
lap GCACTTGGACTATTTGGTGCAC + GCATAAATCGCTCATTGCCATATG.
Western blotting
Fifteen flies per sample were flash frozen in liquid nitrogen. Heads were isolated by vortexing and separation through a small sieve. Protein samples were prepared by homogenizing heads in 2× SDS Laemmli sample buffer [4% SDS, 20% glycerol, 120 mm Tris-HCl (pH 6.8), 200 mm DTT with bromophenol blue] and boiled at 95°C for 10 min. Samples were separated on pre-cast 4–12% Invitrogen Bis-Tris gels (NP0322) or 10% Bis-Tris gels and blotted onto PVDF or nitrocellulose membrane (for VGlut) in Tris-glycine buffer supplemented with 20% methanol. Membranes were blocked in 5% milk in TBST (Tris-buffered saline with 0.1% Tween-20) for 1 h at room temperature (RT) and then incubated with primary antibodies overnight at 4°C. Primary antibody dilutions used were as follows: anti-actin, 1:10 000 (Abcam, ab1801); anti-VGlut, 1:10 000 (a gift from Dr A. DiAntonio, Washington University, St. Louis); and anti-PICALM, 1:1000 (Abcam ab127551). Secondary antibodies used were anti-rabbit and anti-mouse HRP (Abcam, ab6789 and ab6721) at 1:10 000 dilutions for 1 h at RT. Bands were visualized with Luminata Forte (Millipore) and imaged with ImageQuant LAS4000 (GE Healthcare Life Sciences). Quantification was carried out with ImageQuant software or ImageJ.
Aβ42 ELISA
Five fly heads were homogenized in 50 μl GnHCl extraction buffer (5 M guanidinium HCl, 50 mm HEPES (pH 7.3), 1:10 dilution of protease inhibitor cocktail (Roche, P8340) and 5 mm EDTA) and centrifuged at 21 000 × g for 5 min at 4°C, and cleared supernatant retained as the total fly Aβ42 sample. Aβ42 levels were measured with an ELISA kit (Thermal Fisher, KHB3441), according to the manufacturer’s instructions, and total protein levels were measured with a Bradford assay (Bio-Rad protein assay reagent). The amount of Aβ42 in each sample was expressed relative to the total protein content (picograms per microgram of total protein). Data are expressed as the mean ± SEM obtained from three biological repeats for each genotype.
Cross-linking of antibody to beads
50 μl of resuspended Dynabeads® Protein G (Life Technologies, 10004D) were precipitated on a magnet for 1 min, the supernatant removed, and the beads washed 4 times in 1 ml of 0.2 M triethanolamine, pH 8.2. The beads were then resuspended in 1 ml of fresh 20 mm DMP (dimethyl pimelimidate dihydrochloride, Pierce #21666) in 0.2 M triethanolamine, pH 8.2 (5.4 mg DMP/ml buffer), together with the antibody, DVGlut (11 000) and PICALM (1:500), and incubated overnight at 4°C. The beads were then precipitated, and the cross-linking reaction stopped by addition of 1 ml of 50 mm Tris, pH 7.5 for 15 min. The beads were then washed with 1 ml PBS pH 7.4 and resuspended in PBS (pH 7.4) until required.
Co-immunoprecipitation
Forty adult fly heads were isolated from WDah flies and homogenized in 600-μl of lysis buffer [50 mm Tris (pH 7.5), 150 mm NaCl, 0.5% NP-40 (v/v), 0.1 mm MgCl2, 0.1 mm Na3VO4, 5 mm NEM, 1:10 dilution of protease inhibitor cocktail (Roche, P8340), 1:100 dilution of phosphatase inhibitor cocktail 2 (Sigma P5726) and 1 mm phenylmethylsulfonyl fluoride (PMSF)] and spun at 13 000 rpm (17 949 × g) for 15 min at 4°C. 50 μl of the supernatant was taken for ‘input fraction’, while 200 uL of the remaining supernatant was subjected to IP. The lysates were precleared with 10 uL of protein G dynabeads (Life Technologies, 10004D) for 10 min at 4°C and then incubated with 50 ul of the same beads cross-linked with anti-PICALM or anti-dVGlut antibody overnight at 4°C. A bead-only control was also run and treated like the IP samples. Beads were collected by a magnetic stand, and 200 μl of supernatant was removed (supernatant fraction). The beads were washed for five times with 1-ml PBS with 0.5% BSA (PBSB). The beads were then heated to 70°C for 5 min with 20 ul 2× SDS sample buffer and 200 mm DTT, and the supernatant was collected as the ‘IP fraction’ or ‘IgG fraction’ in the case of the bead-only control. Input and supernatant were diluted in 2× SDS sample buffer to a 1× final concentration with 200 mm DTT.
Immunohistochemistry
Wandering third-instar larvae were dissected in HL3 solution and fixed in Bouin’s fixative (for VGlut and GluRII) or 4% paraformaldehyde (for Amph) for 20 min. Larvae were then rinsed in PBS with 0.2% Triton-100 (PBST) and blocked in 5% BSA in PBST for 1 h at RT and then incubated with primary antibodies overnight at 4°C and washed in PBST three times. Primary antibody dilutions used were as follows: anti-GluRIIA, 1:100 (8B4D2 obtained from the Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA); anti-VGlut, 1:1000; anti-GluRIIB, 1:2500; anti-GluRIIC, 1:1000 (a gift from Dr A. DiAntonio, Washington University, St. Louis); and anti-Amph, 1:2500 (a gift from Dr A. Zelhof, Indiana University, Bloomington). Secondary antibodies were anti-HRP, 1:200 (Jackson ImmunoResearch, West Grove, PA); anti-rabbit Alexa Fluor 568, 1:1000 (Thermal Fisher, A11036); and anti-mouse Alexa Fluor 568, 1:1000 (Thermal Fisher, A11019), all incubated for 1 h at RT. After washing, larvae were mounted in Vectashield (Vector Laboratories, Burlingame, CA).
Image acquisition and analysis
All images were acquired as stacks on a Zeiss LSM700 inverted confocal microscope using a 63× objective and are shown as maximum intensity projections of the complete Z-stack. The 10-μm stack was taken from muscles 7 and 6 of segments A2-A4. All images for one experiment were taken at the same microscope settings to reduce variability, and all larvae dissected were imaged. All images from one experiment were processed in the same way, setting the threshold so that all the background intensities would be the same across all samples. Mean fluorescence intensity of each slice of the NMJ was measured with ImageJ. Values shown are the averages for 5–10 NMJ ± SEM. Samples were compared by one-way ANOVA followed by Tukey’s post hoc test.
Live imaging of glutamate release
Heterozygous D42-Gal4 > UAS-iGluSnFR L2 larvae were prepared for live imaging as described previously (50) and imaged on a Zeiss LSM880 airyscan confocal microscope. We immobilized larvae by gently squeezing them under a cover glass in halocarbon oil. Images were collected at a rate of 0.92 frame/s. A single plane was taken from the ventral nerve cord (VNC). All images for one experiment were taken at the same microscopy settings and motion-corrected using the Fiji plug-in, MoCo (82). The mean fluorescence intensity of a specific ROI per VNC was measured with Fiji for each time point.
Glutamate signals for each region of interest (ROI) were defined as deviations from the average fluorescence intensity inside each ROI in each frame, Ft, measured as a function of time (ΔF/F = (Ft − F0)/F0), where F0 is minimal fluorescence intensity recorded. Glutamate ‘events’ were defined as ΔF/F > 0.1. Glutamate event onsets were set as the first frame in the rising phase of the signal. Glutamate event could be either single events or bursts. Glutamate ‘bursting’ was defined by a continuous glutamate transient with ≥2 glutamate events, with the offset of each burst set as the first local minimum in the trace when the falling F/F0 was within 0.2 of the rising onset. Glutamate bursting onsets for all glutamate events were set as the first frame in the rising phase of the signal. For glutamate bursts only the highest peak was scored.
Samples were compared by one-way ANOVA followed by Tukey’s post hoc test.
Statistical analysis
Statistical analysis is described in each section above. The interaction between genotype and RU were analysed by Cox proportional hazards (for lifespans) and ordinal logistics analysis (for climbings) in R. One-way ANOVAs were carried using GraphPad Prism v8.0 software. Parameters are reported in the figure legends.
Supplementary Material
Acknowledgements
We thank Daniel Ciantar for advice on imaging analysis and Carina Kern for discussion. We thank Weiyu Yu for analysis of locomotor performance, Dr T. Stork and Dr M.R. Freeman for advice on live imaging.
Conflict of Interest statement. The authors declare no conflict of interest.
Funding
China Scholar Council (to Y.Y.); Wellcome Trust Strategic Award (WT098565/Z/12/Z to L.P.); Alzheimer’s Society junior fellowship (AS-JF-17b-011 to N.S.W.). ARUK Senior Fellowship (ARUK-SRF2018A-003 to T.N.).
References
- 1. Spires T.L. and Hyman B.T. (2005) Transgenic models of Alzheimer's disease: learning from animals. NeuroRx, 2, 423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harold D., Abraham R., Hollingworth P., Sims R., Gerrish A., Hamshere M.L., Pahwa J.S., Moskvina V., Dowzell K., Williams A. et al. (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat. Genet., 41, 1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lambert J.C., Ibrahim-Verbaas C.A., Harold D., Naj A.C., Sims R., Bellenguez C., DeStafano A.L., Bis J.C., Beecham G.W., Grenier-Boley B. et al. (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat. Genet., 45, 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tian Y., Chang J.C., Fan E.Y., Flajolet M. and Greengard P. (2013) Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer's APP-CTF for terminal degradation via autophagy. Proc. Natl. Acad. Sci. U. S. A., 110, 17071–17076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Koo S.J., Markovic S., Puchkov D., Mahrenholz C.C., Beceren-Braun F., Maritzen T., Dernedde J., Volkmer R., Oschkinat H. and Haucke V. (2011) SNARE motif-mediated sorting of synaptobrevin by the endocytic adaptors clathrin assembly lymphoid myeloid leukemia (CALM) and AP180 at synapses. Proc. Natl. Acad. Sci. U. S. A., 108, 13540–13545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moreau K., Fleming A., Imarisio S., Lopez Ramirez A., Mercer J.L., Jimenez-Sanchez M., Bento C.F., Puri C., Zavodszky E., Siddiqi F. et al. (2014) PICALM modulates autophagy activity and tau accumulation. Nat. Commun., 5, 4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koo S.J., Kochlamazashvili G., Rost B., Puchkov D., Gimber N., Lehmann M., Tadeus G., Schmoranzer J., Rosenmund C., Haucke V. et al. (2015) Vesicular Synaptobrevin/VAMP2 levels guarded by AP180 control efficient neurotransmission. Neuron, 88, 330–344. [DOI] [PubMed] [Google Scholar]
- 8. Zhao Z., Sagare A.P., Ma Q., Halliday M.R., Kong P., Kisler K., Winkler E.A., Ramanathan A., Kanekiyo T., Bu G. et al. (2015) Central role for PICALM in amyloid-beta blood-brain barrier transcytosis and clearance. Nat. Neurosci., 18, 978–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ando K., Brion J.P., Stygelbout V., Suain V., Authelet M., Dedecker R., Chanut A., Lacor P., Lavaur J., Sazdovitch V. et al. (2013) Clathrin adaptor CALM/PICALM is associated with neurofibrillary tangles and is cleaved in Alzheimer's brains. Acta Neuropathol., 125, 861–878. [DOI] [PubMed] [Google Scholar]
- 10. Parikh I., Fardo D.W. and Estus S. (2014) Genetics of PICALM expression and Alzheimer's disease. PLoS One, 9, e91242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xiao Q., Gil S.C., Yan P., Wang Y., Han S., Gonzales E., Perez R., Cirrito J.R. and Lee J.M. (2012) Role of phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia (PICALM) in intracellular amyloid precursor protein (APP) processing and amyloid plaque pathogenesis. J. Biol. Chem., 287, 21279–21289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu C., Nwabuisi-Heath E., Laxton K. and Ladu M.J. (2010) Endocytic pathways mediating oligomeric Abeta42 neurotoxicity. Mol. Neurodegener., 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kanatsu K., Morohashi Y., Suzuki M., Kuroda H., Watanabe T., Tomita T. and Iwatsubo T. (2014) Decreased CALM expression reduces Abeta42 to total Abeta ratio through clathrin-mediated endocytosis of gamma-secretase. Nat. Commun., 5, 3386. [DOI] [PubMed] [Google Scholar]
- 14. Takei K., Slepnev V.I., Haucke V. and De Camilli P. (1999) Functional partnership between amphiphysin and dynamin in clathrin-mediated endocytosis. Nat. Cell Biol., 1, 33–39. [DOI] [PubMed] [Google Scholar]
- 15. Di Paolo G., Sankaranarayanan S., Wenk M.R., Daniell L., Perucco E., Caldarone B.J., Flavell R., Picciotto M.R., Ryan T.A., Cremona O. et al. (2002) Decreased synaptic vesicle recycling efficiency and cognitive deficits in amphiphysin 1 knockout mice. Neuron, 33, 789–804. [DOI] [PubMed] [Google Scholar]
- 16. Glennon E.B., Whitehouse I.J., Miners J.S., Kehoe P.G., Love S., Kellett K.A. and Hooper N.M. (2013) BIN1 is decreased in sporadic but not familial Alzheimer's disease or in aging. PLoS One, 8, e78806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chapuis J., Hansmannel F., Gistelinck M., Mounier A., Van Cauwenberghe C., Kolen K.V., Geller F., Sottejeau Y., Harold D., Dourlen P. et al. (2013) Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry, 18, 1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tan M.S., Yu J.T. and Tan L. (2013) Bridging integrator 1 (BIN1): form, function, and Alzheimer's disease. Trends Mol. Med., 19, 594–603. [DOI] [PubMed] [Google Scholar]
- 19. Calafate S., Flavin W., Verstreken P. and Moechars D. (2016) Loss of Bin1 promotes the propagation of tau pathology. Cell Rep., 17, 931–940. [DOI] [PubMed] [Google Scholar]
- 20. Ubelmann F., Burrinha T., Salavessa L., Gomes R., Ferreira C., Moreno N. and Guimas Almeida C. (2017) Bin1 and CD2AP polarise the endocytic generation of beta-amyloid. EMBO Rep., 18, 102–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miyagawa T., Ebinuma I., Morohashi Y., Hori Y., Young Chang M., Hattori H., Maehara T., Yokoshima S., Fukuyama T., Tsuji S. et al. (2016) BIN1 regulates BACE1 intracellular trafficking and amyloid-beta production. Hum. Mol. Genet., 25, 2948–2958. [DOI] [PubMed] [Google Scholar]
- 22. De Rossi P., Nomura T., Andrew R.J., Masse N.Y., Sampathkumar V., Musial T.F., Sudwarts A., Recupero A.J., Le Metayer T., Hansen M.T. et al. (2020) Neuronal BIN1 regulates presynaptic neurotransmitter release and memory consolidation. Cell Rep., 30, 3520–3535 e3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hynd M.R., Scott H.L. and Dodd P.R. (2004) Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer's disease. Neurochem. Int., 45, 583–595. [DOI] [PubMed] [Google Scholar]
- 24. Campos-Peña V. and Meraz-Ríos M. (2014) In Heinbockel T. (ed), Neurochemistry, Ch11.
- 25. Talantova M., Sanz-Blasco S., Zhang X., Xia P., Akhtar M.W., Okamoto S., Dziewczapolski G., Nakamura T., Cao G., Pratt A.E. et al. (2013) Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. U. S. A., 110, E2518–E2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abramov E., Dolev I., Fogel H., Ciccotosto G.D., Ruff E. and Slutsky I. (2009) Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat. Neurosci., 12, 1567–1576. [DOI] [PubMed] [Google Scholar]
- 27. Cummings D.M., Liu W., Portelius E., Bayram S., Yasvoina M., Ho S.H., Smits H., Ali S.S., Steinberg R., Pegasiou C.M. et al. (2015) First effects of rising amyloid-beta in transgenic mouse brain: synaptic transmission and gene expression. Brain, 138, 1992–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jacob C.P., Koutsilieri E., Bartl J., Neuen-Jacob E., Arzberger T., Zander N., Ravid R., Roggendorf W., Riederer P. and Grunblatt E. (2007) Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer's disease. J. Alzheimers Dis., 11, 97–116. [DOI] [PubMed] [Google Scholar]
- 29. Pirttimaki T.M., Codadu N.K., Awni A., Pratik P., Nagel D.A., Hill E.J., Dineley K.T. and Parri H.R. (2013) Nicotinic receptor-mediated astrocytic gliotransmitter release: Aβ effects in a preclinical Alzheimer's mouse model. PLoS One, 8, alpha7, e81828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li S., Jin M., Koeglsperger T., Shepardson N.E., Shankar G.M. and Selkoe D.J. (2011) Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci., 31, 6627–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hsieh H., Boehm J., Sato C., Iwatsubo T., Tomita T., Sisodia S. and Malinow R. (2006) AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron, 52, 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shankar G.M., Bloodgood B.L., Townsend M., Walsh D.M., Selkoe D.J. and Sabatini B.L. (2007) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci., 27, 2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Snyder E.M., Nong Y., Almeida C.G., Paul S., Moran T., Choi E.Y., Nairn A.C., Salter M.W., Lombroso P.J., Gouras G.K. et al. (2005) Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci., 8, 1051–1058. [DOI] [PubMed] [Google Scholar]
- 34. Wei W., Nguyen L.N., Kessels H.W., Hagiwara H., Sisodia S. and Malinow R. (2010) Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci., 13, 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cisse M., Halabisky B., Harris J., Devidze N., Dubal D.B., Sun B., Orr A., Lotz G., Kim D.H., Hamto P. et al. (2011) Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature, 469, 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim T., Vidal G.S., Djurisic M., William C.M., Birnbaum M.E., Garcia K.C., Hyman B.T. and Shatz C.J. (2013) Human LilrB2 is a beta-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer's model. Science, 341, 1399–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Um J.W., Kaufman A.C., Kostylev M., Heiss J.K., Stagi M., Takahashi H., Kerrisk M.E., Vortmeyer A., Wisniewski T., Koleske A.J. et al. (2013) Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron, 79, 887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Um J.W., Nygaard H.B., Heiss J.K., Kostylev M.A., Stagi M., Vortmeyer A., Wisniewski T., Gunther E.C. and Strittmatter S.M. (2012) Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci., 15, 1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Reinders N.R., Pao Y., Renner M.C., Silva-Matos C.M., Lodder T.R., Malinow R. and Kessels H.W. (2016) Amyloid-beta effects on synapses and memory require AMPA receptor subunit GluA3. Proc. Natl. Acad. Sci. U. S. A., 113, E6526–E6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bao H., Daniels R.W., MacLeod G.T., Charlton M.P., Atwood H.L. and Zhang B. (2005) AP180 maintains the distribution of synaptic and vesicle proteins in the nerve terminal and indirectly regulates the efficacy of Ca2+-triggered exocytosis. J. Neurophysiol., 94, 1888–1903. [DOI] [PubMed] [Google Scholar]
- 41. Casas-Tinto S., Zhang Y., Sanchez-Garcia J., Gomez-Velazquez M., Rincon-Limas D.E. and Fernandez-Funez P. (2011) The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum. Mol. Genet., 20, 2144–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Osterwalder T., Yoon K.S., White B.H. and Keshishian H. (2001) A conditional tissue-specific transgene expression system using inducible GAL4. Proc. Natl. Acad. Sci. U. S. A., 98, 12596–12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Burnouf S., Gorsky M.K., Dols J., Gronke S. and Partridge L. (2015) Abeta43 is neurotoxic and primes aggregation of Abeta40 in vivo. Acta Neuropathol., 130, 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Masri I., Salami A., El Shamieh S. and Bissar-Tadmouri N. (2019) rs3851179G>A in PICALM is protective against Alzheimer's disease in five different countries surrounding the Mediterranean. Curr. Aging. Sci., 12: 1. [DOI] [PubMed] [Google Scholar]
- 45. Zeng F.F., Liu J., He H., Gao X.P., Liao M.Q., Yu X.X., Liu Y.H., Zhu S. and Jing C.X. (2019) Association of PICALM gene polymorphisms with Alzheimer's disease: evidence from an updated meta-analysis. Curr. Alzheimer Res., 16, 1196–1205. [DOI] [PubMed] [Google Scholar]
- 46. BrainSeq A.H.B.G.C.E.a.d.l.o. and BrainSeq A.H.B.G.C. (2015) BrainSeq: neurogenomics to drive novel target discovery for neuropsychiatric disorders. Neuron, 88, 1078–1083. [DOI] [PubMed] [Google Scholar]
- 47. Piaceri I., Bagnoli S., Lucenteforte E., Mancuso M., Tedde A., Siciliano G., Piacentini S., Bracco L., Sorbi S. and Nacmias B. (2011) Implication of a genetic variant at PICALM in Alzheimer's disease patients and centenarians. J. Alzheimers Dis., 24, 409–413. [DOI] [PubMed] [Google Scholar]
- 48. Ping Y., Hahm E.T., Waro G., Song Q., Vo-Ba D.A., Licursi A., Bao H., Ganoe L., Finch K. and Tsunoda S. (2015) Linking abeta42-induced hyperexcitability to neurodegeneration, learning and motor deficits, and a shorter lifespan in an Alzheimer's model. PLoS Genet., 11, e1005025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marvin J.S., Borghuis B.G., Tian L., Cichon J., Harnett M.T., Akerboom J., Gordus A., Renninger S.L., Chen T.W., Bargmann C.I. et al. (2013) An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat. Methods, 10, 162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stork T., Sheehan A., Tasdemir-Yilmaz O.E. and Freeman M.R. (2014) Neuron-glia interactions through the heartless FGF receptor signaling pathway mediate morphogenesis of Drosophila astrocytes. Neuron, 83, 388–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hefendehl J.K., LeDue J., Ko R.W., Mahler J., Murphy T.H. and MacVicar B.A. (2016) Mapping synaptic glutamate transporter dysfunction in vivo to regions surrounding Abeta plaques by iGluSnFR two-photon imaging. Nat. Commun., 7, 13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tebar F., Bohlander S.K. and Sorkin A. (1999) Clathrin assembly lymphoid myeloid leukemia (CALM) protein: localization in endocytic-coated pits, interactions with clathrin, and the impact of overexpression on clathrin-mediated traffic. Mol. Biol. Cell, 10, 2687–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Maritzen T., Koo S.J. and Haucke V. (2012) Turning CALM into excitement: AP180 and CALM in endocytosis and disease. Biol. Cell., 104, 588–602. [DOI] [PubMed] [Google Scholar]
- 54. Treusch S., Hamamichi S., Goodman J.L., Matlack K.E., Chung C.Y., Baru V., Shulman J.M., Parrado A., Bevis B.J., Valastyan J.S. et al. (2011) Functional links between Abeta toxicity, endocytic trafficking, and Alzheimer's disease risk factors in yeast. Science, 334, 1241–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Voglmaier S.M., Kam K., Yang H., Fortin D.L., Hua Z., Nicoll R.A. and Edwards R.H. (2006) Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron, 51, 71–84. [DOI] [PubMed] [Google Scholar]
- 56. Zhang B., Koh Y.H., Beckstead R.B., Budnik V., Ganetzky B. and Bellen H.J. (1998) Synaptic vesicle size and number are regulated by a clathrin adaptor protein required for endocytosis. Neuron, 21, 1465–1475. [DOI] [PubMed] [Google Scholar]
- 57. Vanlandingham P.A., Barmchi M.P., Royer S., Green R., Bao H., Reist N. and Zhang B. (2014) AP180 couples protein retrieval to clathrin-mediated endocytosis of synaptic vesicles. Traffic, 15, 433–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Keshishian H., Broadie K., Chiba A. and Bate M. (1996) The Drosophila neuromuscular junction: a model system for studying synaptic development and function. Annu. Rev. Neurosci., 19, 545–575. [DOI] [PubMed] [Google Scholar]
- 59. Seshadri S., Fitzpatrick A.L., Ikram M.A., DeStefano A.L., Gudnason V., Boada M., Bis J.C., Smith A.V., Carassquillo M.M., Lambert J.C. et al. (2010) Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA, 303, 1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Leprince C., Romero F., Cussac D., Vayssiere B., Berger R., Tavitian A. and Camonis J.H. (1997) A new member of the amphiphysin family connecting endocytosis and signal transduction pathways. J. Biol. Chem., 272, 15101–15105. [DOI] [PubMed] [Google Scholar]
- 61. Razzaq A., Robinson I.M., McMahon H.T., Skepper J.N., Su Y., Zelhof A.C., Jackson A.P., Gay N.J. and O'Kane C.J. (2001) Amphiphysin is necessary for organization of the excitation-contraction coupling machinery of muscles, but not for synaptic vesicle endocytosis in Drosophila. Genes Dev., 15, 2967–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Won S., Incontro S., Nicoll R.A. and Roche K.W. (2016) PSD-95 stabilizes NMDA receptors by inducing the degradation of STEP61. Proc. Natl. Acad. Sci. U. S. A., 113, E4736–E4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lopez-Arias B., Monedero I., Turiegano E. and Torroja L. (2017) The Drosophila adult neuromuscular junction as a model for unravelling amyloid peptide influence on synapse dynamics. Neural Regen. Res., 12, 1987–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schuster C.M., Ultsch A., Schloss P., Cox J.A., Schmitt B. and Betz H. (1991) Molecular cloning of an invertebrate glutamate receptor subunit expressed in Drosophila muscle. Science, 254, 112–114. [DOI] [PubMed] [Google Scholar]
- 65. Petersen S.A., Fetter R.D., Noordermeer J.N., Goodman C.S. and DiAntonio A. (1997) Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron, 19, 1237–1248. [DOI] [PubMed] [Google Scholar]
- 66. Davis G.W., DiAntonio A., Petersen S.A. and Goodman C.S. (1998) Postsynaptic PKA controls quantal size and reveals a retrograde signal that regulates presynaptic transmitter release in Drosophila. Neuron, 20, 305–315. [DOI] [PubMed] [Google Scholar]
- 67. DiAntonio A., Petersen S.A., Heckmann M. and Goodman C.S. (1999) Glutamate receptor expression regulates quantal size and quantal content at the Drosophila neuromuscular junction. J. Neurosci., 19, 3023–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sigrist S.J., Thiel P.R., Reiff D.F. and Schuster C.M. (2002) The postsynaptic glutamate receptor subunit DGluR-IIA mediates long-term plasticity in Drosophila. J. Neurosci., 22, 7362–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Daniels R.W., Collins C.A., Gelfand M.V., Dant J., Brooks E.S., Krantz D.E. and DiAntonio A. (2004) Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J. Neurosci., 24, 10466–10474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schallier A., Smolders I., Van Dam D., Loyens E., De Deyn P.P., Michotte A., Michotte Y. and Massie A. (2011) Region- and age-specific changes in glutamate transport in the AbetaPP23 mouse model for Alzheimer's disease. J. Alzheimers Dis., 24, 287–300. [DOI] [PubMed] [Google Scholar]
- 71. Li S., Hong S., Shepardson N.E., Walsh D.M., Shankar G.M. and Selkoe D. (2009) Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron, 62, 788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang R. and Reddy P.H. (2017) Role of glutamate and NMDA receptors in Alzheimer's disease. J. Alzheimers Dis., 57, 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. McLauchlan H., Newell J., Morrice N., Osborne A., West M. and Smythe E. (1998) A novel role for Rab5-GDI in ligand sequestration into clathrin-coated pits. Curr. Biol., 8, 34–45. [DOI] [PubMed] [Google Scholar]
- 74. Bucci C., Parton R.G., Mather I.H., Stunnenberg H., Simons K., Hoflack B. and Zerial M. (1992) The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell, 70, 715–728. [DOI] [PubMed] [Google Scholar]
- 75. Horiuchi H., Lippe R., McBride H.M., Rubino M., Woodman P., Stenmark H., Rybin V., Wilm M., Ashman K., Mann M. et al. (1997) A novel Rab5 GDP/GTP exchange factor complexed to Rabaptin-5 links nucleotide exchange to effector recruitment and function. Cell, 90, 1149–1159. [DOI] [PubMed] [Google Scholar]
- 76. Magnusson K.R., Brim B.L. and Das S.R. (2010) Selective vulnerabilities of N-methyl-D-aspartate (NMDA) receptors during brain aging. Front. Aging Neurosci., 2, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Menard C., Quirion R., Vigneault E., Bouchard S., Ferland G., El Mestikawy S. and Gaudreau P. (2015) Glutamate presynaptic vesicular transporter and postsynaptic receptor levels correlate with spatial memory status in aging rat models. Neurobiol. Aging, 36, 1471–1482. [DOI] [PubMed] [Google Scholar]
- 78. Hondius D.C., Nierop P., Li K.W., Hoozemans J.J., Schors R.C., Haastert E.S., Vies S.M., Rozemuller A.J. and Smit A.B. (2016) Profiling the human hippocampal proteome at all pathologic stages of Alzheimer's disease. Alzheimers Dement., 12, 654–668. [DOI] [PubMed] [Google Scholar]
- 79. Haglund K., Nezis I.P., Lemus D., Grabbe C., Wesche J., Liestol K., Dikic I., Palmer R. and Stenmark H. (2010) Cindr interacts with anillin to control cytokinesis in Drosophila melanogaster. Curr. Biol., 20, 944–950. [DOI] [PubMed] [Google Scholar]
- 80. Brand A.H., Manoukian A.S. and Perrimon N. (1994) Ectopic expression in Drosophila. Methods Cell Biol., 44, 635–654. [DOI] [PubMed] [Google Scholar]
- 81. Niccoli T., Cabecinha M., Tillmann A., Kerr F., Wong C.T., Cardenes D., Vincent A.J., Bettedi L., Li L., Gronke S. et al. (2016) Increased glucose transport into neurons rescues Abeta toxicity in Drosophila. Curr. Biol., 26, 2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dubbs A., Guevara J. and Yuste R. (2016) Moco: fast motion correction for calcium imaging. Front. Neuroinform., 10, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.