

Abstract
Synaptojanin1 (synj1) is a phosphoinositide phosphatase with dual SAC1 and 5′-phosphatase enzymatic activities in regulating phospholipid signaling. The brain-enriched isoform has been shown to participate in synaptic vesicle (SV) recycling. More recently, recessive human mutations were identified in the two phosphatase domains of SYNJ1, including R258Q, R459P and R839C, which are linked to rare forms of early-onset Parkinsonism. We now demonstrate that Synj1 heterozygous deletion (Synj1+/−), which is associated with an impaired 5′-phosphatase activity, also leads to Parkinson’s disease (PD)-like pathologies in mice. We report that male Synj1+/− mice display age-dependent motor function abnormalities as well as alpha-synuclein accumulation, impaired autophagy and dopaminergic terminal degeneration. Synj1+/− mice contain elevated 5′-phosphatase substrate, PI(4,5)P2, particularly in the midbrain neurons. Moreover, pharmacological elevation of membrane PI(4,5)P2 in cultured neurons impairs SV endocytosis, specifically in midbrain neurons, and further exacerbates SV trafficking defects in Synj1+/− midbrain neurons. We demonstrate down-regulation of SYNJ1 transcript in a subset of sporadic PD brains, implicating a potential role of Synj1 deficiency in the decline of dopaminergic function during aging.
Introduction
Synaptojanin1 (synj1, encoded by Synj1) is an important phosphoinositide phosphatase enriched in nerve terminals, where it regulates synaptic vesicle (SV) recycling and synaptic protein targeting (1–4). Synj1 hydrolyzes membrane phosphoinositide (e.g. PI(4,5)P2, PI(3,4)P2, PI3P and PI4P) via its two phosphatase domains (the SAC1-like enzyme and the 5′-phosphatase enzyme) and binds to endophilin via the proline-rich domain (Fig. 1A). Recently, three mutations—R258Q (5–7) and R459P (8) in the SAC1 domain, and R839C (9) in the 5′-phosphatase domain of SYNJ1—have been identified in families with juvenile atypical Parkinsonism with epilepsy. These patients typically develop progressive tremor, dystonia and rigidity with varying degrees of seizure and cognitive decline. Further neuroimaging studies revealed severe nigrostriatal dopaminergic (DAergic) defects in some of these patients (6, 7). Because of this association with Parkinsonism, SYNJ1 is a member of the PARK gene family and is also known as PARK20. Knock-in (KI) mice carrying the homozygous R258Q disease allele, which abolishes the SAC1 activity required for hydrolyzing PI3P and PI4P (5), recapitulated parkinsonian symptoms and exhibited defective clathrin uncoating of SVs and dystrophic changes in the nigrostriatal terminals (10). A recent study of the R258Q KI flies suggests the impaired PI3P metabolism as a potential pathogenic mechanism for its essential role in autophagosome formation (11). These studies highlighted the roles of the SAC1 enzyme activity in synj1-mediated motor abnormality. However, the identification of a new mutation (R839C) in the 5′-phosphatase domain (9), which leads to similar clinical parkinsonian symptoms in affected siblings, suggests that SYNJ1-mediated Parkinsonism may be more complex than previously proposed (10, 11).
Figure 1.
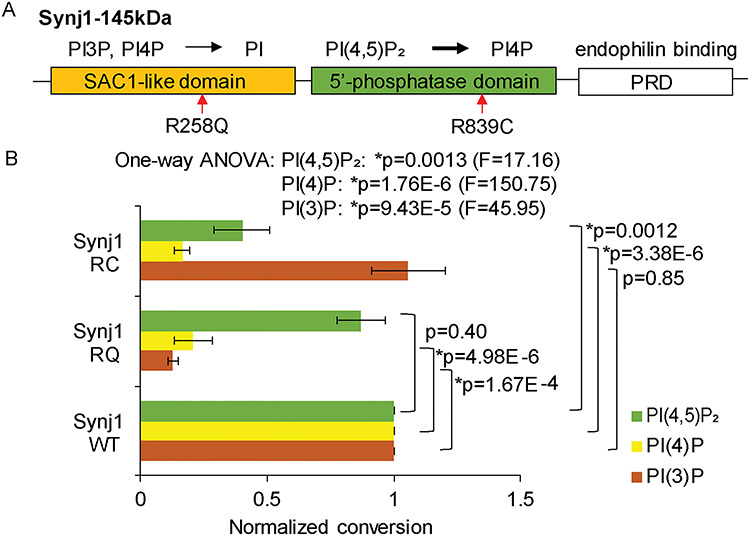
Synj1 R839C mutation impairs dual phosphatase activities. (A) Domain structure of the brain-enriched isoform of synj1 with their specified functions (on top). Red arrows point to two known Parkinsonism mutations. (B) Normalized lipid conversion, representing enzymatic activities of synj1 R258Q and R839C in comparison with WT synj1. Data obtained using malachite green-based phosphatase assay, from three repetitions. One-way ANOVA was performed for each lipid substrate to compare wildtype (WT) and mutant (RC and RQ) synj1 proteins. P-values are from Turkey’s post-hoc analyses.
We previously showed that heterozygous deletion of Synj1 (Synj1+/−) disrupts SV endocytosis selectively in MB neurons (12). Here, we report that haploinsufficiency of Synj1 in mice leads to motor function abnormality, impairment of striatal dopamine (DA) metabolism, loss of DAergic terminals and increase in alpha-synuclein levels. We also find an increase in PI(4,5)P2, the primary substrate of synj1 5′-phosphatase domain, in the MB of Synj1+/− mice, and vulnerability of the Synj1+/− MB neurons to the accumulation of PI(4,5)P2. These findings expand our previous understanding of SYNJ1-mediated Parkinsonism and link the reduced Synj1 expression to the vulnerability of the nigrostriatal DAergic pathway.
Results
SYNJ1 R839C mutation impairs the dual phosphatase and reduced SYNJ1 expression in a subset of PD brains
We have previously shown that the SYNJ1 R258Q mutation abolishes the SAC1 activity in hydrolyzing PI3P and PI4P (5). To determine the biochemical consequences of the newly identified Parkinsonism mutation, R839C ((9), Fig. 1A), we performed in vitro phosphatase assays as described earlier (5). We found that the R839C mutation not only impaired the hydrolysis of PI4P (Tukey’s post hoc between synj1 WT and synj1 RC: P = 3.38E-6) but also reduced 50% of the 5′-phosphatase activity (Tukey’s post hoc between synj1 WT and synj1 RC: P = 0.0012, Fig. 1B), which represents the major enzymatic activity of synj1 (1, 13). This new finding suggests that a loss in either synj1 enzymatic activity (SAC1 or 5′-phosphatase) can cause dysregulation of the basal ganglia function and the development of parkinsonian symptoms. In our previous study, we found that the Parkinson’s disease (PD)-linked LRRK2 G2019S mutation signals at least in part through reducing synj1 activity and that heterozygous deletion of Synj1 (Synj1+/−) disrupts SV endocytosis selectively in the mouse midbrain (MB) neurons (12). These findings, taken together, raise the possibility that reduced expression of Synj1 per se could lead to MB dysfunction and DAergic vulnerability.
We then reviewed genome-wide expression datasets for post-mortem sporadic PD brains in the public domain (PMID: 20926834) (Material and Methods/human data analysis). Three of eight datasets (corresponding to 43 out of a total 154 PD cases) showed significant downregulation of SYNJ1 using multiple comparison error-corrected Student’s t test (Supplementary Material, Fig. S1, Supplementary Material, Table S1). A reduction of SYNJ1 was revealed in various brain regions including the prefrontal cortex, the striatum and the substantia nigra (SN) while the synaptic marker, SYP (encoding synaptophysin), was largely unaltered in the same subjects except for the lateral SN (Supplementary Material, Fig. S1).
Synj1+/− mice display motor function abnormalities, impaired striatal dopamine metabolism, and progressive reduction of DAergic innervation
To investigate the impact of reduced expression of Synj1, we examined Synj1+/− mice, which have partial loss of both phosphatase activities and was originally thought to be functionally tolerated (14). Mice with complete deletion of Synj1 are not viable and die shortly after birth. Synj1+/− mice, however, appear normal with regard to body size and life span. Unlike the homozygous R258Q KI mice (RQ KI), some of which display severe movement impairment and tonic–clonic seizures (10), Synj1+/− mice do not display seizures or apparent gait difficulties up to 18 months. We evaluated the general locomotor functions of a cohort of male mice in the open-field assay. The 7-month-old Synj1+/− mice exhibited hyperactivity (Fig. 2A) compared with their littermate control (Synj1+/+) (Tukey’s post hoc: P = 0.037). Both Synj1+/+ and Synj1+/− mice displayed an age-related deterioration in their activity levels at 12 months (two-way ANOVA for age: P = 1.19E-13) and Synj1+/− mice appeared no different than the wildtype mice at this age (Tukey’s post hoc: P = 0.91, Fig. 2A). However, when Synj1+/− mice were challenged on the accelerated Rota-rod, they exhibited impaired motor coordination at 12 months (Student’s t test: P = 0.024, Fig. 2B). Such decline in motor functions following hyperactivity is reminiscent of the findings in several other PD models (15–18), although the underlying cause is not fully understood. High-performance liquid chromatography (HPLC) was used to measure striatal DA content and its two major metabolites—DOPAC and HVA from flash frozen striatal tissues of Synj1+/− and Synj1+/+ mice at 7 and 12 months. Not surprisingly, DA and the two metabolites were substantially decreased by age (Fig. 2C). Interestingly, a significant reduction of the striatal DOPAC level was observed in the Synj1+/− mice at 7 months (Tukey’s post hoc: P = 0.021 following two-way ANOVA, Fig. 2C, middle); and DA, DOPAC and HVA levels were all trending to be less abundant in the Synj1+/− mice at 12 months (Fig. 2C).
Figure 2.
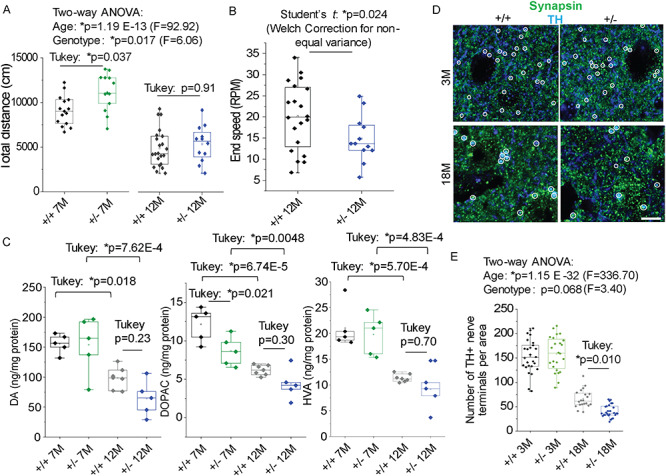
Synj1+/− mice display motor function abnormalities and progressive reduction of DAergic innervation. (A) Total distance measured in the open-field assay for male Synj1+/+ and Synj1+/− littermate mice at 7 months (7 M) and 12 months (12 M), respectively. 7 M cohort: Synj1+/+ (N = 15) and Synj1+/− (N = 13); 12 M cohort: Synj1+/+ (N = 22) and Synj1+/− (N = 13). Two-way ANOVA was performed, followed by Tukey’s post-hoc analyses. (B) Accelerated Rota-rod assay summarizing the end speed before mice fell off the rotating bar. P-values are from two-sample Student’s t tests with Welch’s correction. (C) Dopamine (DA) content and its metabolites levels in the striatum of 7-month-old (Synj1+/+ N = 5; Synj1+/− N = 5) and 12-month-old male mice (Synj1+/+ N = 7; Synj1+/− N = 5) measured by HPLC. Two-way ANOVA was performed, followed by Turkey’s post-hoc analyses. (D) Representative 3-month-old (3 M) and 18-month-old (18 M) striatal slices from a Synj1+/+ mouse and a Synj1+/− mouse immunolabeled with anti-TH and anti-synapsin1/2. Only puncta with dual immunofluorescence for TH (blue) and synapsin1/2 (green) were considered DAergic terminals (while circles) and were included in the double-blinded analysis. Scale bar, 5 μm. (E) Summary for the number of DAergic terminals analyzed from 3 male mice. DAergic terminals were sampled from 10–20 images from 2–3 brain slices for each individual mouse. Two-way ANOVA was performed, followed by Turkey’s post-hoc analyses.
We further investigated the integrity of DAergic terminals by immunohistochemical analysis of the striatal brain slices from young (3 months) and aged (18 months) mice. Co-immunolabeling of the synaptic marker, synapsin1/2, and the DA neuron marker, tyrosine hydroxylase (TH), was used to define DAergic terminals (white circles indicating colocalized structures, Fig. 2D). We found an age-related progressive reduction (from 3 to 18 months) of DAergic terminals in these mice (two-way ANOVA for age: P = 1.15E-32), while a more profound reduction of DAergic terminals (~75%) was noted in Synj1+/− mice when compared with Synj1+/+ mice at 18 months (Tukey’s post hoc: P = 0.010, Fig. 2E, right). However, stereological analysis of DAergic cell bodies in the ventral MB revealed no difference in aged (18 months) Synj1+/− mice (Supplementary Material, Fig. S2). These results are consistent with the abnormal motor behavior and DA metabolism we found in Synj1+/− mice, suggesting a significant impact of Synj1 heterozygous deletion on disrupting the nigrostriatal pathway.
Aged Synj1+/− mice exhibit accumulation of alpha-synuclein
Increased levels of alpha-synuclein (alpha-syn) and deposition of alpha-syn in Lewy bodies are known as the pathological hallmark of PD. Although it is not fully understood, alpha-syn increase or pathology is absent in many recessive PD mouse models, including the Synj1 RQ KI mice (10). It has been shown that alpha-syn recruited into pathological inclusions undergoes extensive phosphorylation at Ser129 (19, 20). In 18-month-old Synj1+/− brains, we found a significant increase in pS129 alpha-syn immunofluorescence (IF) in the striatum (Student’s t test: P = 0.022, Fig. 3A and B). To verify the IF analysis and explore the pathology in other brain regions, we performed DAB enhanced immunohistochemistry for pS129 alpha-syn. Consistently, the cortex and substantia nigra (SN) areas of the Synj1+/− brain exhibited increased DAB staining, and it appeared to be localized to the cytosol (Fig. 3C). In homogenized brain samples from the 18-month-old brains, we observed a larger fraction of Triton-insoluble material, including synj1 itself (Fig. 3D), in the Synj1+/− brains compared with the control, suggesting an increased amount of protein aggregates due to Synj1 deficiency. The pSer129 alpha-syn was observed in the soluble fraction and was increased most significantly in the striatum of the Synj1+/− brain (Tukey’s post hoc: P = 3.92E-4, Fig. 3D and E). Furthermore, we detected an increase in total alpha-syn in the cortex (Tukey’s post hoc: P = 0.0018), MB (Tukey’s post hoc: P = 0.017), and striatum (Tukey’s post hoc: P = 2.56E-8) of the Synj1+/− brain compared with control (Fig. 3D and E), suggesting alpha-syn-associated pathology in multiple brain regions of the Synj1+/− mice.
Figure 3.
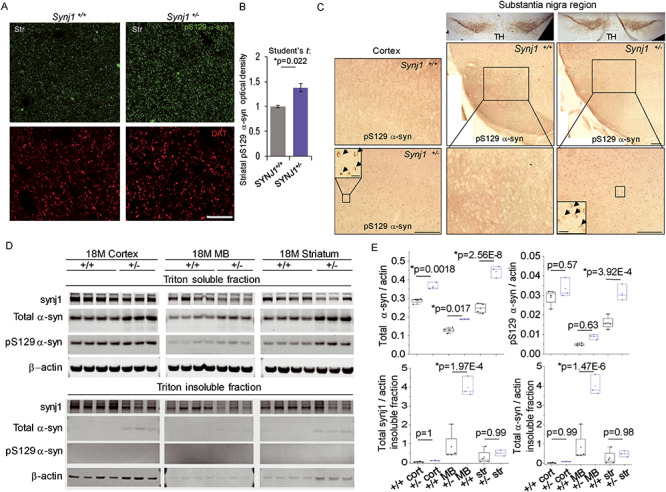
Increased total and phosphorylated alpha-syn levels in aged Synj1+/− mice. (A) Representative images from the striatum of Synj1+/+ and Synj1+/− mice immunolabeled with pS129 alpha-syn (green) and DAT (red). Scale bar: 200 μm. (B) Summary for the relative immunofluorescence (IF) intensity in the striatal slices from 18-month-old Synj1+/+ and Synj1+/− mice. Three images were taken at randomly selected areas of the striatum for each animal and a total of 3 male mice were analyzed. P-value is from two-sample Student’s t tests. (C) Representative immunohistochemical images of pS129 alpha-syn (DAB enhanced) from the cortex (left) as well as TH and pS129 alpha-syn (DAB enhanced) from the substantia nigra (right) of 18-months-old Synj1+/+ and Synj1+/− mice. Insets, high magnification images of the boxed areas in the corresponding Synj1+/− brain slices. Scale bars: 100 μm and 10 μm (inset). (D) Western blot for 18-month-old Synj1+/+ (N = 4) and Synj1+/− (N = 3) brains regions. Triton-soluble and -insoluble fractions were prepared from the same amount of brain homogenate for each brain region. Substantially larger pellets, enriched with alpha-syn and beta-actin, were obtained for all Synj1+/− samples compared with Synj1+/+ samples from the same brain region. (E) Quantitative analysis for total alpha-syn and pS129 alpha-syn in Triton-soluble as well as Triton-insoluble fractions (see Methods/alpha-synuclein analysis). All blots are repeated two to three times and the average was taken to represent the sample value. Two-way ANOVA was performed for genotype and brain region for all proteins analyzed. Significant difference was observed between Synj1+/+ and Synj1+/− mice for total alpha-syn (P = 2.56E-9), pS129 alpha-syn (P = 1.06E-4), insoluble synj1 (P = 0.0013) and insoluble alpha-syn (P = 3.02E-5). P-values shown in the box plots are from Tukey’s post-hoc analyses.
Impaired autophagy function in Synj1+/− neurons
A recent study reported a role of synj1 in autophagy regulation (11). We, therefore, examined autophagy markers in aged Synj1+/− brains. Using brain lysates of 18-month-old Synj1+/− mice, we found that in the soluble fractions of the brain lysate, neither LC3B lipidation (measured by LC3B-II/LC3B-I) nor p62, an autophagy substrate, were changed compared with control mice (Fig. 4A and B). Strikingly, total LC3B and p62 proteins were both accumulated in the Triton-insoluble fractions from the cortex, MB and striatum of the Synj1+/− mice (two-way ANOVA for brain region and genotype, genotype P = 9.99E-4 for LC3B; P = 2.23E-7 for p62, Fig. 4A and B). We also performed IF analysis for the fixed and perfused brain slices. We found enhanced p62 levels in the 18-month-old Synj1+/− mice in all brain regions examined, including the cortex, striatum as well as the DAergic cell bodies in the MB (Tukey’s post hoc: P = 0.00 for all regions, Fig. 4C and D). Consistently, in cultured MB neurons (days in vitro 14), we observed a higher level of p62 in the soma of the Synj1+/− DA neurons (Supplementary Material, Fig. S3), which can be reversed by inhibition of mTOR using rapamycin (Supplementary Material, Fig. S3). Our data suggest an impairment of autophagy in the Synj1+/− neurons, consistent with the previous study (11). However, the underlying mechanism of autophagy impairment in the Synj1+/− model awaits further investigation.
Figure 4.
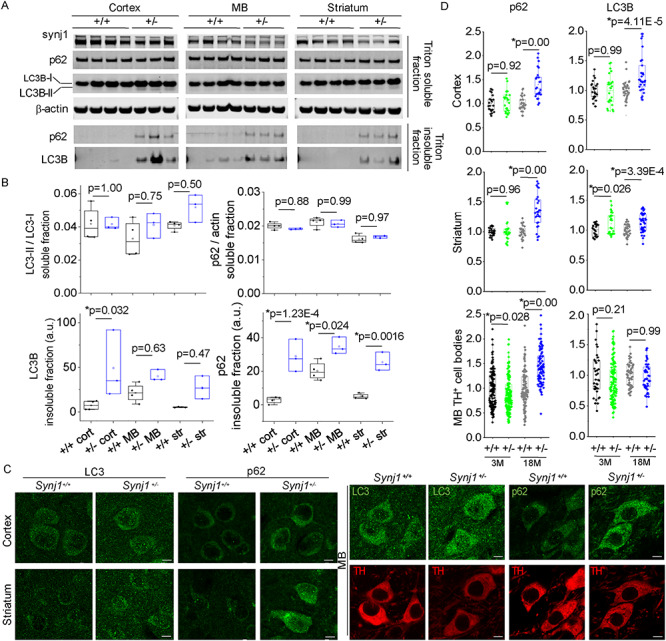
Impaired autophagy in aged Synj1+/− brains. (A) Western blots for 18-month-old Synj1+/+ (N = 4) and Synj1+/− (N = 3) brains regions. Blots from the same protein gel as the ones shown in Figure 3D. (B) Quantitative analysis for LC3B, p62, mTOR and pS2481 mTOR in Triton-soluble as well as Triton-insoluble fractions. Two-way ANOVA was performed for genotype and brain region for all proteins analyzed. P-values are from Tukey’s post-hoc analyses. (C) Representative confocal images of the 18-month cortex and striatum immunolabeled with LC3B or p62 (left) and images of the MB immunolabeled with either LC3 and TH or p62 and TH (right). Scale bar, 5 μm. (D) Summary for p62 and LC3B in the cortex and the striatum (measured by optical density) as well as those in the MB (measured by immunofluorescence at the TH+ cell bodies). Data from 10–20 images for each individual mouse, and three male mice were examined in each group. P-values are from Tukey’s post-hoc analyses following two-way ANOVA for age and genotype.
MB displays specific vulnerability to PI(4,5)P2 metabolism
We next measured various substrates of synj1—PIP2, PIP and PI in the cortex, striatum and MB of 1-year-old male Synj1+/− and Synj1+/+ littermate mice using HPLC. In a large cohort of wildtype mice (N = 17), we found that these phosphoinositide species were differentially enriched among different brain regions. The PI and PIP were most abundant in the cortex, while the PIP2 level was significantly higher in the MB compared with the striatum and cortex (Fig. 5A). In Synj1+/− mice, the PIP2 level in the MB was further elevated by 20% (Tukey’s post hoc: P = 0.038) compared with Synj1+/+ littermates (Fig. 5B). In contrast, a less notable change was observed in the cortex (Tukey’s post hoc: P = 0.12, Fig. 5B). These results suggest that the MB is more sensitive than the cortex to reduced synj1 phosphatase activity in PIP2 metabolism (Tukey’s post hoc for MB and cortex: P = 0.00; for MB and striatum: P = 0.00 following two-way ANOVA for brain region: P = 2.58E-15). The amount of PIP2 increase in the Synj1+/− MB is commensurate to a previous study, which found approximately 12% elevation in the whole brain samples of Synj1+/− mice (14).
Figure 5.
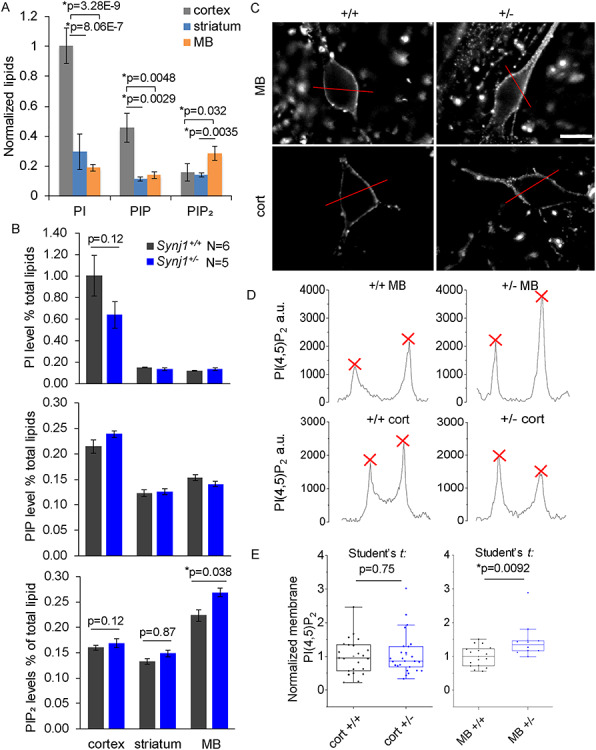
MB neuron-specific elevation of membrane PI(4,5)P2 in the Synj1+/− mice. (A) Comparison of phosphoinositide contents measured by HPLC in different brain regions (cortex, MB and striatum) of male wildtype mice at 12-months old (N = 17). One-way ANOVA was performed for each lipid across different brain regions. P-values are from Tukey’s post-hoc tests. (B) Summary of PI, PIP and PIP2 levels from 12-months-old Synj1+/− mice and Synj1+/+ mice. P-values are from Tukey’s post-hoc analyses following two-way ANOVA for each lipid. (C) Representative images of cultured MB and cortical neurons from Synj1+/+ mice and Synj1+/− mice immunolabeled with PI(4,5)P2. Red lines are representative selections used for profile plot for each cell. Scale bar, 20 μm. (D) Line profile plots for the four representative neurons in C. Red crosses are peak signals used for averaging and calculating membrane PI(4,5)P2 for the cell. (E) Summary of the relative membrane PI(4,5)P2 for different neurons normalized to the average of the Synj1+/+ neurons. Cort +/+ N = 23; Cort +/− N = 25; MB +/+ N = 16; MB +/− N = 10. P-values are calculated by two-sample Student’s t tests.
To determine that the increase in PIP2 in the Synj1+/− MB represents PI(4,5)P2, we examined cultured neurons using a monoclonal antibody against PI(4,5)P2. Despite controversies on using immunochemistry for analyzing membrane lipids, the monoclonal anti-PI(4,5)P2 antibody (clone 2C11 IgM) from Echelon Biosciences has been validated in multiple studies (21–23). We further verified the specificity of the antibody using cultured Synj1+/+ and Synj1−/− cortical neurons, as well as Synj1+/+ neurons treated with a PI3K inhibitor, LY294002 or A66 (Supplementary Material, Fig. S4). Both inhibitors significantly increased membrane PI(4,5)P2 in wildtype neurons by 50% (Tukey’s post hoc following one-way ANOVA for control and LY294002: P = 1.23E-6; for control and A66: P = 1.92E-3) and a similar increase in membrane PI(4,5)P2 was observed in Synj1−/− neurons (Tukey’s post hoc for control and Synj1−/− neurons: P = 1.23E-6, Supplementary Material, Fig. S4). In agreement with the HPLC measurement for the whole brains samples, cultured Synj1+/− MB neurons exhibited a significant elevation in membrane PI(4,5)P2 (Student’s t test: P = 0.0092, Fig. 5C–E), while the Synj1+/− cortical neurons did not show any changes compared with control (Student’s t test: P = 0.75, Fig. 5C–E).
Synaptic vesicle recycling in Synj1+/− MB neurons is vulnerable to upregulation of the membrane PI(4,5)P2
To understand the impact of membrane PI(4,5)P2 elevation in membrane trafficking, we sought to examine SV recycling, as recent evidence indicated potential crosstalk between SV recycling and the autophagy pathway (24–27). The optical reporter, pHluorin, when tagged to SV proteins, provides the ability to quantitatively measure the kinetics of exocytosis and endocytosis (28–30). Fusion protein vMAT2-pHluorin or vGLUT1-pHluorin was expressed in cultured MB or cortical neurons, respectively, and 10-hertz (10 Hz), 10-s (10 s) field stimulations were applied to trigger SV recycling. We previously showed that cultured MB, but not cortical neurons, from the Synj+/− mice displayed slowed endocytosis (12), suggesting that heterozygous deletion of Synj1 affects SV endocytosis preferentially in the MB neurons. Further analysis indicated that the altered SV recycling in Synj1+/− MB neurons was irrelevant to TH immunoreactivity (Tukey’s post hoc following two-way ANOVA for genotype and TH: TH+ vs. TH− neurons in the Synj1+/− group: P = 0.79 for endocytosis and P = 0.93 for exocytosis, Supplementary Material, Fig. S5). To verify MB neuron-selective vulnerability to altered phosphoinositide levels, we treated cultured WT neurons with the PI3K inhibitor LY249002 (LY), which blocks the conversion of PI(4,5)P2 to PI(3,4,5)P3 on the plasma membrane (31), resulting in accumulation of PI(4,5)P2 (Fig. 6A, Supplementary Material, Fig. S4). We found that SV endocytosis in MB neurons was substantially slower after a 10–15-min incubation of LY and remained slow during the 30-min drug treatment (paired Student’s t test: P = 0.0013, Fig. 6B and C). In contrast, cortical neurons showed minimal responsiveness to a 30-min LY incubation (paired Student’s t test: P = 0.15, Fig. 6B and C). Exocytosis, however, was not different after the drug treatment in either type of neurons. Our data suggest that SV trafficking in MB neurons is more susceptible to PI(4,5)P2 accumulation than cortical neurons. We next applied LY in cultured Synj1+/− MB neurons, whose endocytosis is impaired compared with littermate wildtype neurons ((12), Supplementary Material, Fig. S5). The additional increase in PI(4,5)P2 resulting from the blocking of PI3K in these neurons led to progressive exacerbation of SV recycling defects (Fig. 6D and E), which was reflected by a further slowing of SV endocytosis after a 10-min incubation with LY (paired Student’s t test: P = 0.035) and a significant reduction in exocytosis after 20 min of incubation with LY (paired Student’s t test: P = 0.036). In three batches of cultures examined, we again did not observe any difference between TH+ and TH− neurons in their responses to LY (Fig. 6C and E, TH+ neurons are indicated in green symbols). Thus, our data suggest that reduced Synj1 expression, which results in an aberrant accumulation of PI(4,5)P2 in specific brain regions or neuron populations, may contribute to impaired synaptic function and vulnerability.
Figure 6.
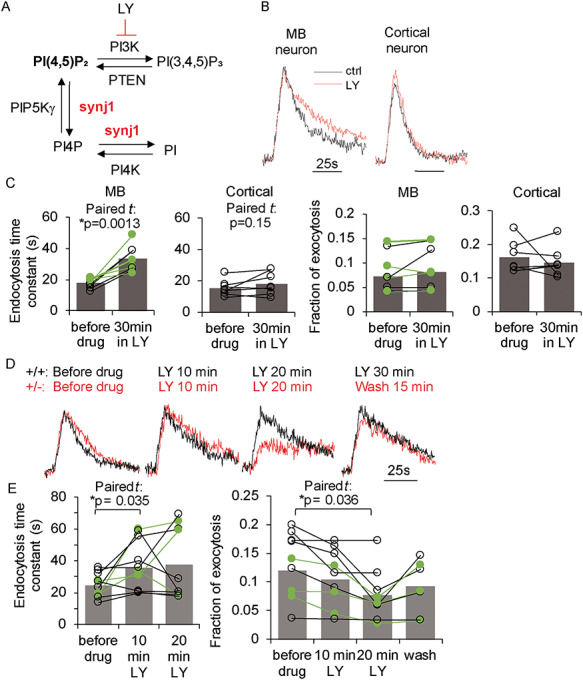
Nerve terminals of MB neurons are more vulnerable to PI(4,5)P2 accumulation. (A) Illustration for the role of synj1 in regulating phosphoinositide metabolism and the role of LY249002 (LY) in inhibiting the conversion of PI(4,5)P2 to PI(3,4,5)P3. (B) Representative pHluorin traces for cultured wildtype MB and cortical neurons at 10 Hz, 10 s stimulation before (black) and after LY (50 μM) incubation (red). (C) Summary of endocytosis kinetics and exocytosis fraction (the amplitude of the pHluorin trace at 10 Hz, 10 s stimulation normalized to the pHluorin response of that neuron to pH 7.4 NH4Cl solution perfusion) before and after LY treatment in wildtype neurons. Green symbols and lines represent DAergic neurons by post-hoc analysis. (D) PHluorin traces from a Synj1+/+ MB neuron (black) and a Synj1+/− MB neuron (red) before drug treatment and at various times after LY incubation. (E) Summary of endocytosis and exocytosis kinetics before and during LY treatment in Synj1+/− MB neuron neurons. Green symbols and lines represent DAergic neurons by post-hoc analysis. P-values in C and E are calculated by paired Student’s t tests.
Taken together, our study suggests that down-regulation of Synj1 expression may lead to PD-like pathological changes and impairment in DAergic signaling in mice.
Discussion
Our previous study and others have demonstrated a causal link of rare SYNJ1 (Synj1) mutations to the dysfunctional and dystrophic nigrostriatal DAergic pathway (5–11). We now show that haploinsufficiency of the Synj1 dual phosphatase activities leads to parkinsonian-like pathologies including progressive loss of DAergic terminals, increased alpha-synuclein and altered striatal dopamine metabolism associated with motor function abnormalities in mice. Thus, our study demonstrates that intact Synj1 expression is required for maintaining the functional DAergic pathway and/or for preventing DAergic degeneration in the aged animal. We show accumulation of the synj1 5′-phosphatase substrate, PI(4,5)P2 in the Synj1+/− MB neurons, which could contribute to dysfunctional SV recycling. We also report an impairment in autophagy in Synj1+/− neurons, which is consistent with the findings in the disease mutation (R258Q) KI model (11). These data suggest that lipid deregulation, likely resulting from Synj1 haploinsufficiency, may impair normal function of the MB and promote the development of PD-like pathologies.
The molecular mechanisms underlying striatal DAergic degeneration and alpha-synuclein accumulation in Synj1+/− mice have yet to be fully elucidated. Our current study suggests that the elevation of PI(4,5)P2 in the MB of Synj1+/− mice may trigger dysfunctional SV recycling at the nerve terminal, which could in turn subject these nerve terminals to selective elimination (32). Considering the lack of difference in the synaptic phenotype between TH+ and TH− neurons ((12), Fig. 6), it is likely that additional factors, such as different calcium burdens, further loss of the SAC1 activity, or neuroinflammatory responses (10, 30, 33, 34), may contribute to the vulnerable nigral DAergic axons. It was recently reported that loss of synj1 SAC1 activity leads to impaired autophagosome maturation in flies due to abnormal accumulation of PI3P (11). In the Synj1+/− mice, the total PIP concentration (including PI3P), however, was not altered in the cortex, the MB or the striatum (Fig. 5B). Our data indicate that the autophagy impairment in the Synj1+/− brain is likely due to other mechanisms than previously reported. For example, it is possible that increases in PI(4,5)P2 due to Synj1-deficiency further activates the Akt/mTOR pathway (35, 36) and contributes to the autophagy inhibition. It remains to be clarified in greater detail whether the PIP2 in the cortex and the striatum is converted to PIP3, and whether PI3K/mTOR activation contributes to DAergic neurodegeneration or alpha-syn accumulation.
Despite the lack of human data directly linking SYNJ1 haploinsufficiency to Parkinsonism or sporadic PD, compelling evidence suggests a common disease mechanism involving a network of genes with roles in autophagy and synaptic trafficking (26, 27, 37, 38). Indeed, a number of the PD linked proteins may be co-regulated in the presynaptic terminal, as De Camilli and colleagues reported elevated levels of both PARK2/parkin and DNAJC6/PARK19/auxilin levels in the endophilin mutant mouse brains (39) and the close binding partner of synj1, SH3GL2/endophilinA1, is now a known risk gene for sporadic PD (40). Whether Synj1 deficiency interferes with the functions of other PD variants has yet to be determined. We previously reported a potential genetic interaction between LRRK2 disease mutation G2019S and Synj1 haploinsufficiency in mice and found LRRK2 mediates phosphorylation of synj1, which exacerbates SV trafficking defects in MB neurons as well as motor functions in aged mice (12). Recent studies have also shown that LRRK2 phosphorylates endophilin A and synj1 (12, 41–43), suggesting the potential involvement of the LRRK2-endophilinA-synj1 complex in regulating membrane trafficking in neurotransmission and autophagy, two essential processes which may go awry in the pathogenesis of PD.
Materials and methods
Animals
Mice were housed in the pathogen-free Center for Comparative Medicine at The Icahn School of Medicine at Mount Sinai and The Rutgers Robert Wood Johnson Medical School. Handling procedures were in accordance with the National Institutes of Health guidelines and approved by the Mount Sinai Institutional Animal Care and Use Committee (IACUC).
Behavioral assays
Male Synj1+/− mice and their littermate controls were tested for general locomotor activity in an open field chamber in a dark room for 60 min. Motor coordination was assessed by accelerated Rota-rod assay. All mice were subjected to a 1-h habituation in the test room with food and water supply prior to testing. Open-field test—each mouse was placed in the center of a 16 × 16-inch chamber equipped with a Versamax monitor system (Accuscan) in a quiet dark room. The horizontal and vertical movements of the mouse were monitored and recorded for 60 min by a grid of 32 infrared beams at ground level and 16 elevated (3 inch) beams. Quinpirole test—mice were randomly divided into two groups for each genotype which were then subjected to peritoneal injection of a D2R agonist quinpirole (0.05 mg/kg) or 1x PBS before being placed into the open-field chamber. Movement was recorded for the following 60 min in the dark room as described above. Accelerated Rota-rod test—the mouse was placed on a rotating rod with increasing acceleration from 4 to 40 RPM over 5 min. Each mouse was trained for two trials before the test. The duration of time a mouse spent on the accelerated Rota-rod was calculated by averaging consecutive three trials spaced by 15 min.
Cryostat, immunochemistry and antibodies
Mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) and perfused transcardially with 4% fresh paraformaldehyde, and post-fixed with 4% paraformaldehyde. Dissected brains were cryoprotected in 30% sucrose prior to flash-freezing in the OCT-compound media (SAKURA). Coronal sections were sliced at 40 μm thickness on a Leica CM 3050 S cryostat. IHC was carried out following a standard protocol as previously described (44). Briefly, tissue slices were blocked in 5% goat serum, primary antibodies diluted in 5% goat serum were applied and incubated overnight at 4°C, followed by Alexa Fluor® secondary antibodies (Invitrogen™). The following primary antibodies were used: anti-TH antibody (Sigma-Aldrich, T2928, EMD Millipore, AB152), rat anti-DAT (EMD Millipore, MAB369), rabbit anti-synj1 (Novus Biologicals, NBP1-87842), guinea pig anti-synapsin 1/2 (Synaptic System, 106 004), rabbit anti-pS129 alpha-synuclein (clone EP1536Y, Abcam ab51253), mouse anti-alpha-synuclein (BD bioscience, 610 787), guinea pig anti-p62 (Progene, GPP62-C; MBL, PM045), rabbit anti-LC3B (Abcam, ab48394), mouse anti-beta-actin (Cell signaling, 3700), rabbit anti-mTOR (Cell signaling, 2983) and rabbit anti-pS2481 mTOR (Cell signaling, 2974). For post-hoc analysis of cultured MB neurons, a standard protocol was used for immunocytochemistry (12). Chicken anti-GFP (Invitrogen, A10262) and mouse anti-TH were used to label the pHluorin-expressing neurons. Immunolabeling for PI(4,5)P2 was following the company’s suggested and optimized protocol (45). Briefly, fixation was performed using 4% paraformaldehyde and 0.2% glutaraldehyde. Cell membrane was permeabilized using Saponin. Mouse anti-PI(4,5)P2 (Echelon Biosciences, ZP045) was used along with rabbit anti-beta III tubulin (Tuj, Abcam, ab18207) or rabbit anti-TH (Novus Biologicals, NB300-109). Blocking, permeabilization and staining were performed on ice. 50 mM NH4Cl solution was used for washing and 2% paraformaldehyde was used for post fixation before mounting.
Stereology microscopy
For stereological counting, one in every five slices was selected and a total of eight brain slices were used from each mouse for IHC labeling. Zeiss Axioplan2 was used for tissue slice imaging with a 20X objective, and Stereo Investigator was used for data analysis using the following parameters: frame sizes: 150 × 150 μm; grid sizes: 250 × 250 μm; top guard zone height: 2 μm and optical dissector height: 8 μm.
alpha-Synuclein analysis
Brains regions from age-matched Synj1+/+ and Synj1+/− mice were collected using mouse brain matrices (ASI, RBM-2000C) and homogenized in a sucrose-buffered solution containing 0.32 M sucrose, 20 mM HEPES, pH 7.4, 1 mM NaHCO3, 2.5 mM CaCl2, 1 mM MgCl2 and protease and phosphatase inhibitor cocktail (Pierce, A32961). The same amount of brain homogenate (~1 mg) from both Synj1+/+ and Synj1+/− brains were lysed on ice for 30 min using Triton-based lysis buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton as well as protease and phosphatase inhibitors. After centrifugation at 16 000 rcf, 4°C for 30 min, supernatant was collected as the ‘Triton-soluble fraction.’ The pellet (the Triton-insoluble fraction) was washed three times in a PBST solution containing 1% Triton and solubilized in 1% SDS solution at 60°C for 60 min. For better alpha-synuclein detection, the PVDF membrane was incubated with fresh-made 4% PFA for 30 min at room temperature immediately after transfer.
HPLC lipid analysis and monoamine analysis
Different brain regions were dissected using rodent brain matrices (ASI, RBM-2000C). Flash frozen mouse brain samples were used for lipid extraction, followed by anion-exchange high-pressure liquid chromatography quantification as described previously (46–49). Striatal samples were collected from freshly dissected brains using 2 mm reusable biopsy punch (World Precision Instrument, 504529) and flash frozen for further analyzed by the Vanderbilt University Neurochemistry Core.
Cell culture and transfection
MB cultures (30, 45) and cortical cultures (3) were prepared as described previously. Ventral MBs (containing both VTA and SN) or cortices were dissected from P0-1 mouse pups and digested using papain (Worthington, LK003178), or trypsin (Sigma, T1005) supplemented with DNase (Sigma, D5025), respectively. MB neurons were then prepared according to our previously published protocol plated at a cell density of 199 000 cells/cm2 and grown in the Neurobasal-A based medium supplemented with GDNF (10 ng/ml, EMD Millipore, GF030). Cortical neurons were plated at 142 000 cells/cm2 and grown in the MEM-based medium supplemented with insulin (24 μg/ml, Sigma, I6634) and transferrin (0.1 mg/ml, Calbiochem, 616 420). Typically, four P0-P1 mouse brains are required for a MB culture. Calcium phosphate was used for transfection to achieve sparse expression and to ensure analysis of single neurons during the imaging experiments. Transfection was carried out at DIV 3–5 for MB neurons and at DIV 5–6 for cortical neurons, after which the medium was supplemented with an antimitotic agent, ARA-C (Sigma-Aldrich, C6645).
Optical imaging
For live cell imaging, cells were mounted on a custom-made laminar-flow stimulation chamber with constant perfusion (at a rate of ~0.2–0.3 ml/min) of a Tyrode’s salt solution containing 119 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 25 mM HEPES, 30 mM Glucose, 10 μM 6-cyano-7- nitroquinoxaline-2,3-dione (CNQX) and 50 μM D, L-2-amino-5-phosphonovaleric acid (AP5) and buffered to pH 7.40. NH4Cl solution containing 50 mM NH4Cl, 70 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 25 mM HEPES, 30 mM Glucose, 10 μM CNQX and 50 μM AP5, buffered to pH 7.40 was used to reveal total pHluorin expression for normalizing exocytosis. All chemicals were purchased from Sigma-Aldrich. Temperature was clamped at 30.0°C at the objective throughout the experiment. Field stimulations were delivered at 10 V/cm by A310 Accupulser and A385 stimulus isolator (World Precision Instruments). A 1-ms pulse was used to evoke single action potentials. Images were acquired using a highly sensitive, back-illuminated EM-CCD camera (iXon+ Model # DU-897E-BV, Andor Corp., CT, USA). Olympus IX73 microscope was modified for laser illumination. A solid-state 488 nm OPSL smart laser at 50 mW (used at 10% and output at ~2 mW at the back aperture) was built into a laser combiner system for millisecond on/off switching and camera blanking control (Andor Corp). PHluorin fluorescence excitation and collection were through an Olympus PLAPON 60XO 1.42 NA objective using 525/50 m emission filter and 495LP dichroic filters (Chroma, 49 002). Images were sampled at 2 Hz with an Andor Imaging Workstation driven by Andor iQ-CORE-FST (ver 2.x) iQ3.0 software.
Confocal microscopy
An LSM780 upright confocal microscope driven by the Zeiss Zen Black software was used to examine immunofluorescence in brain slices. Images were acquired at 1024X1024 pixel resolution using single scans by a 10X (for striatal and cortical synj1 expression analysis) or a 63X lens (for nerve terminal analysis). Immunofluorescence of these images was analyzed using ImageJ.
Human data analysis
Sporadic PD brain transcriptome data were downloaded from PMID: 20926834. Among the 17 genome-wide expression datasets (GEO accession numbers GSE6613, GSE7621, GSE8397 (two data sets), GSE20141, GSE20146, GSE20153, GSE20159, GSE20163, GSE20164, GSE20168, GSE20291, GSE20292, GSE20314, GSE20333 and GSE24378; and GSE28894), we only examined datasets with a sample size greater than 15 in each group to ensure the statistical power. Only eight of these datasets met the selection criterion and three of the eight datasets, GSE28894, GSE20168, GSE8397, were found to exhibit statistical difference (P < 0.05, two-sample Student’s t test) in SYNJ1 levels in multiple brain regions. Clinical information of the post-mortem brain tissue samples can be found in the following articles: PMID 15 965 975 and 16 344 956.
Experimental design and statistical analysis
All statistical tests were performed in OriginPro 8.2, except the Kolmogorov–Smirnov test, which uses a built-in function at http://www.physics.csbsju.edu/stats/KS-test.n.plot_form.html. Descriptive statistical tests were carried out to determine the distribution of the data sets. All data sets conforming to the normal distribution were subjected to two-sample Student’s t test or multiple-sample ANOVA test followed by Tukey’s post-hoc tests. P-values less than 0.05 were considered statistically significant. For quantification of DAergic terminals in striatal slices (Fig. 2E), circular regions of interests (ROIs) were manually placed on all colocalized puncta on the 135 × 135 μm image. To reduce the error of this relatively arbitrary measurement, the following strategies were used: (1) Analysis was performed when the experimenter was blinded to the identity of the sample. (2) The immunostaining procedure, the confocal imaging settings and the gain/contrast of the images during analysis were kept the same for a matching number of Synj1+/+ and Synj1+/− samples. (3) A matching number of images from Synj1+/+ and Synj1+/− mice were assigned for analysis at a single time. (4) Counts were compared between two analysts. For cell body p62 fluorescence qualification (Supplementary Material, Fig. S3), we circled the cell bodies of cultured midbrain neurons in images of max intensity projections from Z-stack capturing and performed background subtraction. For p62 punctate fluorescence qualification, ROIs were placed on p62 puncta, defined by structures <5 × 5 pixels. For PI(4,5)P2 analysis (Fig. 5), two adjacent images with the most optimum focus for the cell body were selected from a Z-stack to obtain a Max projection image. All images were background subtracted and a line profile plot was generated, providing the measurement for two randomly selected opposite points on the plasma membrane, which is then averaged and taken as PI(4,5)P2 membrane immunofluorescence for the cell. For pHluorin imaging study (Fig. 6), data were collected from at least three batches of cultures. Each data point represents an average of 2–3 stable trials spaced by 3 min on a single cell before and after drug treatment (connected by lines). Typically, 15–50 nerve terminals with consistent responses were selected for analysis for each cell.
Supplementary Material
Acknowledgements
The work was funded by a Cote Early Investigator Award (PD12-00011) and a NINDS R01 grant (R01NS112390) to P.-Y.P., an International Research grant (PDF-IRG-1447) from the Parkinson’s Disease Foundation (PDF), R21NS109895-01A1 and P50NS094733-01 to Z.Y., and a NINDS R21 grant (1R21NS095155-01) to P.-Y.P. and Z.Y.
Conflict of Interest statement: The authors declare no conflict of interests.
References
- 1. McPherson P.S., Garcia E.P., Slepnev V.I., David C., Zhang X., Grabs D., Sossin W.S., Bauerfeind R., Nemoto Y. and De Camilli P. (1996) A presynaptic inositol-5-phosphatase. Nature, 379, 353–357. [DOI] [PubMed] [Google Scholar]
- 2. Micheva K.D., Kay B.K. and McPherson P.S. (1997) Synaptojanin forms two separate complexes in the nerve terminal. Interactions with endophilin and amphiphysin. J. Biol. Chem., 272, 27239–27245. [DOI] [PubMed] [Google Scholar]
- 3. Mani M., Lee S.Y., Lucast L., Cremona O., Di Paolo G., De Camilli P. and Ryan T.A. (2007) The dual phosphatase activity of synaptojanin1 is required for both efficient synaptic vesicle endocytosis and reavailability at nerve terminals. Neuron, 56, 1004–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dong Y., Gou Y., Li Y., Liu Y. and Bai J. (2015) Synaptojanin cooperates in vivo with endophilin through an unexpected mechanism. elife, 4, e05660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krebs C.E., Karkheiran S., Powell J.C., Cao M., Makarov V., Darvish H., Di Paolo G., Walker R.H., Shahidi G.A., Buxbaum J.D. et al. (2013) The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive parkinsonism with generalized seizures. Hum. Mutat., 34, 1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Quadri M., Fang M., Picillo M., Olgiati S., Breedveld G.J., Graafland J., Wu B., Xu F., Erro R., Amboni M. et al. (2013) Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset parkinsonism. Hum. Mutat., 34, 1208–1215. [DOI] [PubMed] [Google Scholar]
- 7. Olgiati S., De Rosa A., Quadri M., Criscuolo C., Breedveld G.J., Picillo M., Pappatà S., Quarantelli M., Barone P., De Michele G. et al. (2014) PARK20 caused by SYNJ1 homozygous Arg258Gln mutation in a new Italian family. Neurogenetics, 15, 183–188. [DOI] [PubMed] [Google Scholar]
- 8. Kirola L., Behari M., Shishir C. and Thelma B.K. (2016) Identification of a novel homozygous mutation Arg459Pro in SYNJ1 gene of an Indian family with autosomal recessive juvenile parkinsonism. Parkinsonism Relat. Disord., 31, 124–128. [DOI] [PubMed] [Google Scholar]
- 9. Taghavi S., Chaouni R., Tafakhori A., Azcona L.J., Firouzabadi S.G., Omrani M.D., Jamshidi J., Emamalizadeh B., Shahidi G.A., Ahmadi M. et al. (2017) A clinical and molecular genetic study of 50 families with autosomal recessive parkinsonism revealed known and novel gene mutations. Mol. Neurobiol., 55, 3477–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cao M., Wu Y., Ashrafi G., McCartney A.J., Wheeler H., Bushong E.A., Boassa D., Ellisman M.H., Ryan T.A. and De Camilli P. (2017) Parkinson sac domain mutation in Synaptojanin 1 impairs Clathrin uncoating at synapses and triggers dystrophic changes in dopaminergic axons. Neuron, 93, 882–896.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vanhauwaert R., Kuenen S., Masius R., Bademosi A., Manetsberger J., Schoovaerts N., Bounti L., Gontcharenko S., Swerts J., Vilain S. et al. (2017) The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J., 36, 1392–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pan P.Y., Li X., Wang J., Powell J., Wang Q., Zhang Y., Chen Z., Wicinski B., Hof P., Ryan T.A. et al. (2017) Parkinson's disease associated LRRK2 hyperactive kinase mutant disrupts synaptic vesicle trafficking in ventral midbrain neurons. J. Neurosci., 37, 11366–11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Woscholski R., Finan P.M., Radley E., Totty N.F., Sterling A.E., Hsuan J.J., Waterfield M.D. and Parker P.J. (1997) Synaptojanin is the major constitutively active phosphatidylinositol-3,4,5-trisphosphate 5-phosphatase in rodent brain. J. Biol. Chem., 272, 9625–9628. [DOI] [PubMed] [Google Scholar]
- 14. Voronov S.V., Frere S.G., Giovedi S., Pollina E.A., Borel C., Zhang H., Schmidt C., Akeson E.C., Wenk M.R., Cimasoni L. et al. (2008) Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down's syndrome. Proc. Natl. Acad. Sci. U. S. A., 105, 9415–9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Unger E.L., Eve D.J., Perez X.A., Reichenbach D.K., Xu Y., Lee M.K. and Andrews A.M. (2006) Locomotor hyperactivity and alterations in dopamine neurotransmission are associated with overexpression of A53T mutant human alpha-synuclein in mice. Neurobiol. Dis., 21, 431–443. [DOI] [PubMed] [Google Scholar]
- 16. Lam H.A., Wu N., Cely I., Kelly R.L., Hean S., Richter F., Magen I., Cepeda C., Ackerson L.C., Walwyn W. et al. (2011) Elevated tonic extracellular dopamine concentration and altered dopamine modulation of synaptic activity precede dopamine loss in the striatum of mice overexpressing human α-synuclein. J. Neurosci. Res., 89, 1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chesselet M.F. and Richter F. (2011) Modelling of Parkinson's disease in mice. Lancet Neurol., 10, 1108–1118. [DOI] [PubMed] [Google Scholar]
- 18. Chesselet M.F., Richter F., Zhu C., Magen I., Watson M.B. and Subramaniam S.R. (2012) A progressive mouse model of Parkinson's disease: the Thy1-aSyn ("line 61") mice. Neurotherapeutics, 9, 297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fujiwara H., Hasegawa M., Dohmae N., Kawashima A., Masliah E., Goldberg M.S., Shen J., Takio K. and Iwatsubo T. (2002) Alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol., 4, 160–164. [DOI] [PubMed] [Google Scholar]
- 20. Samuel F., Flavin W.P., Iqbal S., Pacelli C., Sri Renganathan S.D., Trudeau L.E., Campbell E.M., Fraser P.E. and Tandon A. (2016) Effects of serine 129 phosphorylation on α-Synuclein aggregation, membrane association, and internalization. J. Biol. Chem., 291, 4374–4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hammond G.R., Schiavo G. and Irvine R.F. (2009) Immunocytochemical techniques reveal multiple, distinct cellular pools of PtdIns4P and PtdIns(4,5)P(2). Biochem. J., 422, 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Esteban-Pretel G., Marín M.P., Romero A.M., Timoneda J., Ponsoda X., Ballestín R. and Renau-Piqueras J. (2013) Polyphosphoinositide metabolism and Golgi complex morphology in hippocampal neurons in primary culture is altered by chronic ethanol exposure. Alcohol Alcohol., 48, 15–27. [DOI] [PubMed] [Google Scholar]
- 23. Choi S., Chen M., Cryns V.L. and Anderson R.A. (2019) A nuclear phosphoinositide kinase complex regulates p53. Nat. Cell Biol., 21, 462–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vijayan V. and Verstreken P. (2017) Autophagy in the presynaptic compartment in health and disease. J. Cell Biol., 216, 1895–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Azarnia Tehran D., Kuijpers M. and Haucke V. (2018) Presynaptic endocytic factors in autophagy and neurodegeneration. Curr. Opin. Neurobiol., 48, 153–159. [DOI] [PubMed] [Google Scholar]
- 26. Pan P.Y., Zhu Y., Shen Y. and Yue Z. (2019) Crosstalk between presynaptic trafficking and autophagy in Parkinson's disease. Neurobiol. Dis., 122, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sheehan P. and Yue Z. (2019) Deregulation of autophagy and vesicle trafficking in Parkinson's disease. Neurosci. Lett., 697, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sankaranarayanan S., De Angelis D., Rothman J.E. and Ryan T.A. (2000) The use of pHluorins for optical measurements of presynaptic activity. Biophys. J., 79, 2199–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ariel P. and Ryan T.A. (2010) Optical mapping of release properties in synapses. Front. Neural Circuits, 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pan P.Y. and Ryan T.A. (2012) Calbindin controls release probability in ventral tegmental area dopamine neurons. Nat. Neurosci., 15, 813–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Milosevic I., Sørensen J.B., Lang T., Krauss M., Nagy G., Haucke V., Jahn R. and Neher E. (2005) Plasmalemmal phosphatidylinositol-4,5-bisphosphate level regulates the releasable vesicle pool size in chromaffin cells. J. Neurosci., 25, 2557–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stevens B., Allen N.J., Vazquez L.E., Howell G.R., Christopherson K.S., Nouri N., Micheva K.D., Mehalow A.K., Huberman A.D., Stafford B. et al. (2007) The classical complement cascade mediates CNS synapse elimination. Cell, 131, 1164–1178. [DOI] [PubMed] [Google Scholar]
- 33. Sulzer D., Alcalay R.N., Garretti F., Cote L., Kanter E., Agin-Liebes J., Liong C., McMurtrey C., Hildebrand W.H., Mao X. et al. (2017) T cells from patients with Parkinson's disease recognize α-synuclein peptides. Nature, 546, 656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Surmeier D.J., Obeso J.A. and Halliday G.M. (2017) Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci., 18, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dall’Armi C., Devereaux K.A. and Di Paolo G. (2013) The role of lipids in the control of autophagy. Curr. Biol., 23, R33–R45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. George A.A., Hayden S., Stanton G.R. and Brockerhoff S.E. (2016) Arf6 and the 5'phosphatase of Synaptojanin 1 regulate autophagy in cone photoreceptors. Inside Cell, 1, 117–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Trinh J. and Farrer M. (2013) Advances in the genetics of Parkinson disease. Nat. Rev. Neurol., 9, 445–454. [DOI] [PubMed] [Google Scholar]
- 38. Schirinzi T., Madeo G., Martella G., Maltese M., Picconi B., Calabresi P. and Pisani A. (2016) Early synaptic dysfunction in Parkinson's disease: insights from animal models. Mov. Disord., 31, 802–813. [DOI] [PubMed] [Google Scholar]
- 39. Cao M., Milosevic I., Giovedi S. and De Camilli P. (2014) Upregulation of parkin in endophilin mutant mice. J. Neurosci., 34, 16544–16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chang D., Nalls M.A., Hallgrímsdóttir I.B., Hunkapiller J., Brug M., Cai F., International Parkinson's Disease Genomics Consortium; 23andMe Research Team, Kerchner G.A., Ayalon G. et al. (2017) A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat. Genet., 49, 1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Matta S., Van Kolen K., Cunha R., Bogaart G., Mandemakers W., Miskiewicz K., De Bock P.J., Morais V.A., Vilain S., Haddad D. et al. (2012) LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron, 75, 1008–1021. [DOI] [PubMed] [Google Scholar]
- 42. Arranz A.M., Delbroek L., Van Kolen K., Guimarães M.R., Mandemakers W., Daneels G., Matta S., Calafate S., Shaban H., Baatsen P. et al. (2015) LRRK2 functions in synaptic vesicle endocytosis through a kinase-dependent mechanism. J. Cell Sci., 128, 541–552. [DOI] [PubMed] [Google Scholar]
- 43. Islam M.S., Nolte H., Jacob W., Ziegler A.B., Pütz S., Grosjean Y., Szczepanowska K., Trifunovic A., Braun T., Heumann H. et al. (2016) Human R1441C LRRK2 regulates the synaptic vesicle proteome and phosphoproteome in a drosophila model of Parkinson's disease. Hum. Mol. Genet., 25, 5365–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lu J., He L., Behrends C., Araki M., Araki K., Wang Q.J., Catanzaro J.M., Friedman S.L., Zong W.X., Fiel M.I. et al. (2014) NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L-linked phosphatidylinositol-3 kinase III activity. Nat. Commun., 5, 3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mani M. and Ryan T.A. (2009) Live imaging of synaptic vesicle release and retrieval in dopaminergic neurons. Front. Neural Circuits, 3, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Berman D.E., Dall'Armi C., Voronov S.V., McIntire L.B., Zhang H., Moore A.Z., Staniszewski A., Arancio O., Kim T.W. and Di Paolo G. (2008) Oligomeric amyloid-beta peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat. Neurosci., 11, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Landman N., Jeong S.Y., Shin S.Y., Voronov S.V., Serban G., Kang M.S., Park M.K., Di Paolo G., Chung S. and Kim T.W. (2006) Presenilin mutations linked to familial Alzheimer's disease cause an imbalance in phosphatidylinositol 4,5-bisphosphate metabolism. Proc. Natl. Acad. Sci. U. S. A., 103, 19524–19529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhu L., Zhong M., Elder G.A., Sano M., Holtzman D.M., Gandy S., Cardozo C., Haroutunian V., Robakis N.K. and Cai D. (2015) Phospholipid dysregulation contributes to ApoE4-associated cognitive deficits in Alzheimer's disease pathogenesis. Proc. Natl. Acad. Sci. U. S. A., 112, 11965–11970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cao J., Gaamouch F.E., Meabon J.S., Meeker K.D., Zhu L., Zhong M.B., Bendik J., Elder G., Jing P., Xia J. et al. (2017) ApoE4-associated phospholipid dysregulation contributes to development of tau hyper-phosphorylation after traumatic brain injury. Sci. Rep., 7, 11372. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.