

Abstract
Previous studies demonstrate that activation of pancreatic ER kinase (PERK) protects oligodendrocytes against inflammation in the experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis (MS). Interestingly, data indicate that the cytoprotective effects of PERK activation on oligodendrocytes during EAE are not mediated by activating transcription factor 4 (ATF4) but are accompanied by activation of nuclear factor κB (NF-κB). NF-κB plays a critical role in MS and EAE; however, the effects of NF-κB activation on oligodendrocytes in these diseases remain elusive. Herein, we generated a mouse model that allow for activation of NF-κB specifically in oligodendrocytes and found that enhanced NF-κB activation in oligodendrocytes had a minimal effect on their viability and function under normal conditions (both male and female mice). Interestingly, we found that enhanced NF-κB activation in oligodendrocytes attenuated EAE disease severity and ameliorated EAE-induced oligodendrocyte loss, demyelination, and axon degeneration, without affecting inflammation (female mice). Moreover, we showed that the detrimental effects of PERK inactivation in oligodendrocytes in EAE were accompanied by impaired NF-κB activation in oligodendrocytes, and were completely rescued by enhanced NF-κB activation in oligodendrocytes (female mice). These findings suggest that NF-κB activation accounts for the cytoprotective effects of PERK activation on oligodendrocytes in MS and EAE.
SIGNIFICANCE STATEMENT Nuclear factor κB (NF-κB) is activated in oligodendrocytes in multiple sclerosis (MS) and its animal model experimental autoimmune encephalomyelitis (EAE); however, the role of NF-κB activation in oligodendrocytes in MS and EAE remains elusive. Herein, we generated a mouse model that allows for activation of NF-κB selectively in oligodendrocytes and demonstrated that NF-κB activation prevented oligodendrocyte death and myelin damage in the EAE model. We further demonstrated that NF-κB activation contributed to the protective effects of pancreatic ER kinase (PERK) activation on oligodendrocytes in the EAE model. As such, this work will facilitate the development of new treatments that enhance oligodendrocyte survival in MS patients by targeting the PERK-NF-κB pathway.
Keywords: A20, demyelination, EAE, NF-κB, oligodendrocyte, PERK
Introduction
Transcription factor Nuclear factor κB (NF-κB) is a hetero or homodimer of Rel family proteins, including p65, c-Rel, RelB, p50, and p52. In the quiescent state, NF-κB remains inactive and is sequestered in the cytoplasm by binding to NF-κB inhibitors (IκBs). Upon stimulation, NF-κB becomes activated though dissociation from IκBs and translocates into the nucleus where NF-κB binds to the κB consensus DNA sequence and enhances the expression of genes that control inflammation and cell viability. A variety of stimuli, including inflammatory mediators, growth factors, and cellular stresses, can activate NF-κB through various mechanisms, including the IκB kinase 2 (IKK2)-dependent canonical pathway, the non-canonical pathway, and atypical pathways (Hayden and Ghosh, 2012; Mincheva-Tasheva and Soler, 2013; Yue et al., 2018).
Multiple sclerosis (MS) is an autoimmune inflammatory demyelinating disease of the CNS. NF-κB is activated in T cells, microglia/macrophages, astrocytes, oligodendrocytes, and neurons in MS and experimental autoimmune encephalomyelitis (EAE; Gveric et al., 1998; Bonetti et al., 1999; Stone et al., 2019). Data indicate that NF-κB activation in inflammatory cells, including T cells and microglia/macrophages, enhances inflammation and facilitates MS and EAE development (Mc Guire et al., 2013; Yue et al., 2018). Conversely, several studies suggests that NF-κB activation protects oligodendrocytes against inflammatory mediators (Nicholas et al., 2001; Hamanoue et al., 2004; Lin et al., 2012; Stone et al., 2017). Our previous study showed that mice expressing IκBαΔN (a supersuppressor of NF-κB) specifically in oligodendrocytes develop extremely severe EAE with very high mortality. Because of early, sudden death of these EAE mice, we could not collect CNS tissues to determine the effects of IκBαΔN expression on oligodendrocyte during EAE (Stone et al., 2017). Nevertheless, another study showed that oligodendrocyte-specific deletion of RelB attenuates EAE-induced oligodendrocyte death and demyelination (Gupta et al., 2019). Moreover, a report mentioned that IKK2 deletion in oligodendrocytes, using IKK2loxP/loxP;MOG/Cre mice, does not affect EAE development but fell short to show the efficiency and specificity of Cre-mediated recombination of floxed IKK2 alleles in these mice (Raasch et al., 2011). Thus, the role of NF-κB activation in oligodendrocytes in MS and EAE remains unclear.
Upon endoplasmic reticulum (ER) stress, activation of pancreatic ER kinase (PERK) phosphorylates eukaryotic translation initiation factor 2α (eIF2α), which inhibits global protein translation but stimulates numerous cytoprotective gene expression through induction of the transcription factor activating transcription factor 4 (ATF4; Walter and Ron, 2011; Wang and Kaufman, 2016). The PERK-eIF2α pathway regulates oligodendrocyte viability in various myelin disorders (Lin and Popko, 2009; Lin and Stone, 2020). Several studies showed that enhanced PERK activation in oligodendrocytes attenuates oligodendrocyte death and demyelination during EAE and that decreased PERK activation in oligodendrocytes exacerbates oligodendrocyte death and demyelination during EAE (Hussien et al., 2014; Lin et al., 2007, 2013, 2014a). Surprisingly, a recent study showed that ATF4 inactivation specifically in oligodendrocytes does not alter oligodendrocyte death or demyelination in EAE mice, suggesting the minimal involvement of ATF4 in the cytoprotective effects of PERK activation on oligodendrocytes during EAE (Yue et al., 2019). Therefore, it remains elusive how PERK activation protects oligodendrocytes against inflammation in MS and EAE.
The PERK-eIF2α pathway also activates NF-κB by suppressing translation of its inhibitor IκBα (Deng et al., 2004; Ron and Walter, 2007). Interestingly, our previous in vitro studies showed that PERK activation triggers NF-κB activation in oligodendrocytes by suppressing translation of IκBα through phosphorylation of eIF2α (Lin et al., 2012, 2013). We also found that PERK activation induces NF-κB activation in oligodendrocytes during EAE (Lin et al., 2013). This study sought to test the possibility that the cytoprotective effects of PERK activation on oligodendrocytes in MS and EAE are mediated by NF-κB. We first demonstrated the cytoprotective effects of NF-κB activation on oligodendrocytes during EAE. We further showed that enhanced NF-κB activation specifically in oligodendrocytes completely rescued the detrimental effects of PERK inactivation in oligodendrocytes in EAE. These findings indicate that NF-κB activation contributes significantly to the cytoprotective effects of PERK activation on oligodendrocytes during EAE.
Materials and Methods
Mice
CNP/Cre mice (Lappe-Siefke et al., 2003), R26StopFLikk2ca mice (Sasaki et al., 2006; The Jackson Laboratory stock #008242), and PERKloxP mice (Zhang et al., 2002; The Jackson Laboratory stock #023066) were on the C57BL/6J background. CNP/Cre mice were crossed with R26StopFLikk2ca mice to obtain R26StopFLikk2ca; CNP/Cre mice (IKK2ca cKI mice) and control mice (WT mice), including R26StopFLikk2ca mice, CNP/Cre mice, and wild type mice. CNP/Cre mice were crossed with PERKloxP mice to obtain PERKloxP; CNP/Cre mice, and R26StopFLikk2ca mice were crossed with PERKloxP mice to obtain PERKloxP; R26StopFLikk2ca mice. PERKloxP; CNP/Cre mice were further crossed with PERKloxP; R26StopFLikk2ca mice to obtain R26StopFLikk2ca; PERKloxP/loxP; CNP/Cre mice (Double mice), R26StopFLikk2ca; CNP/Cre mice (IKK2ca cKI mice), PERKloxP/loxP; CNP/Cre mice (PERK cKO (knock out) mice), and control mice (WT mice), including PERKloxP/loxP mice, R26StopFLikk2ca mice, CNP/Cre mice, and wild type mice. Genotypes of mice were determined by PCR from DNA extracted from tail tips as described previously (Zhang et al., 2002; Lappe-Siefke et al., 2003; Sasaki et al., 2006). To assess Cre-mediated recombination in the tissues of these mice, genomic DNA was isolated from the indicated tissues, and PCR was performed as described previously (Zhang et al., 2002; Sasaki et al., 2006). All animal procedures were conducted in complete compliance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Minnesota.
EAE immunization
Eight-week-old female mice were injected subcutaneously in the flank/tail base with 200 μg of MOG35-55 peptide emulsified in complete Freund's adjuvant (BD Biosciences) supplemented with 600 μg of mycobacterium tuberculosis (strain H37Ra; BD Biosciences). Two intraperitoneal injections of 400-ng pertussis toxin (List Biological Laboratories) were given 0 and 48 h later. Clinical scores (0 = healthy, 1 = flaccid tail, 2 = ataxia and/or paresis of hindlimbs, 3 = paralysis of hindlimbs and/or paresis of forelimbs, 4 = tetraparalysis, 5 = moribund or death) were recorded daily as described previously (Lin et al., 2007, 2013; Stone et al., 2017, 2018). Aggregate EAE clinical score was the sum of daily clinical scores of each individual mouse during the period of observation.
Immunohistochemistry (IHC)
Anesthetized mice were perfused through the left cardiac ventricle with 4% paraformaldehyde in PBS. Half sagittal brain and the cephalic half of the lumbar spinal cord (SP; L1-L3) were postfixed in 4% paraformaldehyde for 2 h, cryoprotected in 30% sucrose for 48 h, embedded in optimum cutting temperature compound, and frozen on dry ice. Frozen sections were cut using a cryostat at a thickness of 10 µm. The other half sagittal brain and the caudal half of the lumbar SP (L3-L5) were postfixed in 4% paraformaldehyde for 72 h, dehydrated through graded alcohols, and embedded in paraffin wax. Paraffin sections were cut using a microtome at a thickness of 5 µm. Fluorescein (1:100, Vector Laboratories, anti-rabbit, RRID:AB_2336197; anti-mouse, RRID:AB_2336176), Cy3 (1:500, Millipore, anti-mouse, RRID:AB_11213281; anti-rat, RRID:AB_90854; anti-rabbit, RRID:AB_92489), or enzyme-labeled secondary antibodies (1:200, Vector Laboratories, anti-rat, RRID:AB_2336202; anti-mouse, RRID:AB_2313581) were used for detection. Immunohistochemical detection of CC1 (APC7, 1:50; EMD Biosciences, RRID:AB_2057371), myelin basic protein (MBP, 1:1000; Sternberger Monoclonals, RRID:AB_10120129), CD3 (1:50; Santa Cruz Biotechnology, RRID:AB_627010), CD11b (1:50; Millipore, RRID:AB_92930), the active form of p65 (1:50; Millipore, RRID:AB_2178887), aspartoacylase (ASPA; 1:500; Millipore, catalog ABN1698), Flag (1:250; Sigma, RRID: AB_1957945), SMI32 (1:1000; Covance, RRID: AB_509997), glial fibrillary acidic protein (GFAP; 1:1000; Covance, RRID:AB_2314542), and A20/TNFAIP3 (1:100; Santa Cruz Biotechnology, catalog sc-166692) were performed. Fluorescent stained sections were mounted with Vectashield mounting medium with DAPI (Vector Laboratories) and visualized with an Olympus FV1000 confocal microscope. To quantify cells and axons in the lumbar SP, we counted immunopositive cells or axons in an area of 0.1 mm2 within the anterior funiculus medially next to the anterior median fissure in the lumbar SP as described previously (Lin et al., 2007, 2013; Stone et al., 2018). For quantitative MBP IHC analysis, we calculated the percentage of the demyelinated area in the lumbar SP by normalizing the demyelinated area against the total white matter area. The total white matter area and the demyelinated area in the lumbar SP were measured with the NIH ImageJ software (http://rsb.info.nih.gov/ij/; RRID:SCR_003070) as described previously (Lin et al., 2007, 2013; Stone et al., 2018).
Western blot analysis
Brain was harvested from mice and rinsed in ice-cold PBS. After homogenization using a motorized homogenizer, samples were incubated on ice for 15 min and then centrifuged at 14,000 rpm for 30 min twice as described previously (Lin et al., 2013; Stone et al., 2017, 2018). The protein concentration was measured using the DC Protein Assay (Bio-Rad Laboratories). The extracts (50 µg) were separated by SDS-PAGE gels and transferred to nitrocellulose membranes. The membranes were incubated with a primary antibody against Flag (1:1000; Sigma, RRID: AB_1957945), A20/TNFAIP3 (1:1000; Santa Cruz Biotechnology, catalog sc-166692), or β-actin (1:5000, Sigma-Aldrich, RRID:AB_476694) overnight, followed by an horseradish peroxidase-conjugated secondary antibody (1:1000, Vector Laboratories, anti-mouse, catalog #PI-2000; anti-rabbit, #PI-1000). The chemiluminescent signal was detected using the ECL Detection Reagents (GE Healthcare Biosciences). The intensity of the chemiluminescence signals of Flag, A20/TNFAIP3, and β-actin were quantified using the NIH ImageJ software.
Real-time PCR
The SP was harvested from mice and rinsed in ice-cold PBS. RNA was extracted from the SP using TRIzol reagent (Invitrogen) and treated with DNaseI (Invitrogen) to eliminate genomic DNA. Reverse transcription was performed using the iScript cDNA Synthesis kit (Bio-Rad Laboratories). Real-time PCR was performed using LightCycler 480 Probes Master (Roche Diagnostics Corporation) on the LightCycler 480 System (Roche) as described previously (Lin et al., 2013; Stone et al., 2017, 2018).
T cell proliferation and viability assay and cytokine assays
Spleen was isolated from EAE mice at postimmunization day (PID)10 and homogenized into a single-cell suspension. The single-spleen cell suspensions were plated in 96-well microtiter plates in triplicate, and incubated with MOG35-55 peptide (0, 1, 10, or 100 µg/ml) at 37°C and 5% CO2. For T cell proliferation viability assay, 20 µl of bromodeoxyuridine (BrdU)-labeling solution (Millipore) was added to the culture media after 48 h and incubated for a further 24 h. Cell proliferation was determined using the Colorimetric BrdU Cell Proliferation kit (Millipore) according to manufacturer's instructions. For T cell viability assay, the cells were incubated at 37°C and 5% CO2 for 72 h. Cell viability was determined by the MTT assay kit (Promega) according to the manufacturer's instructions. For cytokine assay, cytokines in the culture supernatants were quantified using the ELISA kits (IFN-γ, IL-4, IL-17A, BioLegend) according to the manufacturer's instructions.
Toluidine blue staining
Mice were deeply anaesthetized and perfused with PBS containing 4% paraformaldehyde and 2.5% glutaraldehyde. The lumbar SP was removed, processed, and embedded in resin; 1-μm sections were cut and stained with toluidine blue as described previously (Lin et al., 2007, 2013; Stone et al., 2018). The demyelinated area and total white matter area in the lumbar SP were measured using the NIH ImageJ software, and the percentage of the demyelinated area was calculated by normalizing the demyelinated area against the total white matter area.
RNA-sequencing analysis
The optic nerves were harvested from mice and rinsed in ice-cold PBS. RNA was extracted from the optic nerves using RNeasy Plus Mini kit (QIAGEN). SMARTer Stranded Total RNA-Seq kit (Takara Bio) was used to construct poly(A) selected paired-end sequencing library. Sequencing was conducted with the Illumina NextSeq 550 Sequencer at The University of Minnesota Genomics Center. Raw paired-end RNA-seq reads in fastq format was assessed using gopher-pipeline software (Rnaseq-Pipeline version 1.4). Read mapping was performed via Hisat2 using the mouse genome (ensembl GRCm38) as reference. Cuffdiff 2.2.1 was used to quantify the expression level of each known gene in units of fragments per kilobase of exon model per million reads mapped (FPKM). Differentially expressed genes were identified using the edgeR (negative binomial) feature in CLC GWB (QIAGEN) using raw read counts. Genes that show a minimum 2× absolute fold change and raw p < 0.05 were considered differentially expressed.
Experimental design and statistical analysis
Sample size for each individual experiment can be found in the corresponding figure legends. For EAE experiments, only female mice were used because of the well-known sex differences in EAE. For other experiments, both male and female mice were used. EAE clinical score data are presented as mean ± SEM. All other data are expressed as mean ± SD. Comparison between two groups was statistically evaluated by t test using GraphPad Prism 6 (GraphPad Software, RRID:SCR_002798). Multiple comparisons were statistically evaluated by the one-way ANOVA with a Tukey's post hoc test using GraphPad Prism 6; p < 0.05 was considered significant.
Results
Enhanced NF-κB activation does not affect oligodendrocyte viability or function under normal conditions
Previous studies showed that attenuated NF-κB activation in oligodendrocytes does not alter myelin formation or maintenance in the CNS under normal conditions (Hilliard et al., 1999, 2002; van Loo et al., 2006; Stone et al., 2017; Blank and Prinz, 2014; Kretz et al., 2014). The canonical IKK2-dependent NF-κB pathway is activated by inflammatory mediators, which bind to specific receptors and induce activation of the IKK complex (a trimeric complex of IKK1, IKK2, and NEMO). Then the activated IKK complex phosphorylates IκBα, leads to ubiquitination and degradation of IκBα, and subsequently results in release and activation of the NF-κB p65/p50 heterodimer (hereafter referred to as p65 NF-κB; Hayden and Ghosh, 2012; Mincheva-Tasheva and Soler, 2013; Yue et al., 2018). To determine the effects of enhanced NF-κB activation on oligodendrocytes, we generated a mouse model that expresses a constitutively active form of IKK2 (IKK2ca; Mercurio et al., 1997) specifically in oligodendrocytes in the CNS. R26StopFLikk2ca mice (Sasaki et al., 2006) that carry a floxed conditional allele containing a floxed STOP cassette, Flag-tagged IKK2ca, and IRES-EGFP were crossed with CNP/Cre knock-in mice (Lappe-Siefke et al., 2003) that express Cre selectively in oligodendrocytes in the CNS to obtain R26StopFLikk2ca; CNP/Cre mice (IKK2ca cKI mice) and control mice (WT mice), including R26StopFLikk2ca, CNP/Cre mice, and wild type mice. IKK2ca cKI mice looked healthy and were indistinguishable from littermate WT mice. PCR analysis showed that the floxed STOP cassette that prevents translation of Flag-tagged IKK2ca and EGFP was removed by Cre recombination in the CNS of IKK2ca cKI mice but not in other organs of IKK2ca cKI mice or in any organs of WT mice (Fig. 1A). Western blot analysis showed that Flag-tagged IKK2ca was undetectable in the CNS of WT mice but became detectable in the CNS of IKK2ca cKI mice (Fig. 1B). CC1 (a marker for oligodendrocytes) and Flag double immunostaining showed that the immunoreactivity of Flag was undetectable in the CNS of WT mice (Fig. 1F–I). Importantly, the majority of oligodendrocytes stained positive for Flag in the CNS of IKK2ca cKI mice, and all Flag-positive cells were CC1 positive oligodendrocytes in these mice (p < 0.0001; Fig. 1C–E,I). These data demonstrate that IKK2ca is expressed specifically in oligodendrocytes in the CNS of IKK2ca cKI mice. Nevertheless, we found that the fluorescent signal of EGFP was barely detectable in the CNS of IKK2ca cKI mice (data not shown), which suggests very low expression of EGFP and prohibits us from using EGFP as a marker to identify IKK2ca expressing cells. Similarly, RNA-sequencing analysis showed that the level of IKK2 mRNA (the sum of the endogenous IKK2 and the transgenic IKK2ca) was increased <2-fold in the optic nerve of IKK2ca cKI mice compared with WT (Table 1), suggesting modest expression of the transgenic IKK2ca in oligodendrocytes of IKK2ca cKI mice. Thus, these results suggest that the expression of the transgenic IKK2ca and EGFP in oligodendrocytes of IKK2ca cKI mice is modest.
Figure 1.

Expression of IKK2ca led to p65 NF-κB activation specifically in oligodendrocytes. A, PCR analysis using genomic DNA showed that the floxed IKK2ca allele was present in all tissues in IKK2ca cKO mice and WT mice, but the IKK2ca knock-in (KI) allele was only present in the CNS of IKK2ca cKI mice. 1, brain; 2, SP; 3, heart; 4, liver; 5, spleen; 6, lung. B, Western blot analysis showed that Flag-IKK2ca was detectable in the brain of eight-week-old IKK2ca cKI mice but not in WT mice. C–I, CC1 and Flag double immunostaining showed that the immunoreactivity of Flag-IKK2ca became detectable selectively in oligodendrocytes in the brain of eight-week-old IKK2ca cKI mice, as compared with WT mice. J–P, ASPA and the active form of p65 double immunostaining showed that the number of oligodendrocytes that stained positive for the active form of p65 was dramatically increased in the brain of eight-week-old IKK2ca cKI mice compared with WT mice. N = 3 animals. Scale bars: 20 μm. Statistical analyses were done with a t test. Error bars represented SD; *p < 0.05.
Table 1.
Differentially expressed genes in the optic nerve of IKK2ca cKI mice compared with WT mice
| Gene ID | Gene expression, fold change | p value |
|---|---|---|
| Ikbkb (IKK2) | 1.949744 | 7.98E-13 |
| Nfkbia (IκBα) | 1.559935 | 2.57E-05 |
| Dffb | 2.127083 | 0.006999 |
| Exo1 | 2.779978 | 0.014126 |
| Fgf16 | 10.19853 | 0.004981 |
| Hist1 h2bj | 2.004363 | 0.04836 |
| Hist1 h3c | 3.514332 | 0.03688 |
| Nox3 | 13.52964 | 0.090603 |
| Poln | 2.257956 | 0.036469 |
| Pyhin1 | 2.365904 | 0.004892 |
| Samd3 | 3.385923 | 0.020414 |
| Sis | 2.694653 | 0.008233 |
| Tnfaip3 (A20) | 2.127618 | 5.69E-06 |
| Ube2c | 2.344501 | 0.040445 |
N = 3 animals. Boldface indicates the upregulation of A20.
ASPA (another marker for oligodendrocytes) and the active form of p65 double immunostaining showed that the immunoreactivity of the active form of p65 was undetectable in oligodendrocytes in the CNS of WT mice but became detectable in a number of oligodendrocytes in IKK2ca cKI mice (p < 0.0001; Fig. 1J–P). These data demonstrate that enforced expression of IKK2ca induces p65 NF-κB activation specifically in oligodendrocytes. Nevertheless, CC1 IHC showed that the number of oligodendrocytes in the CNS of three-week-old IKK2ca cKI mice was comparable with WT mice [SP p = 0.6283, corpus collosum (CC) p = 0.8891, cerebellum (CE) p = 0.7969; Fig. 2A,B,Q]. MBP IHC showed a comparable degree of myelination in the CNS of three-week-old IKK2ca cKI mice and WT mice (Fig. 2E,F). Robust myelination occurs in the rodent CNS between postnatal d 7 and 21 (Baumann and Pham-Dinh, 2001). These data suggest that enhanced p65 NF-κB activation has a minimal effect on actively myelinating oligodendrocytes during developmental myelination. Moreover, CC1 IHC and MBP IHC showed that enhanced p65 NF-κB activation in oligodendrocytes did not change oligodendrocyte numbers (SP p = 0.6138, CC p = 0.7643, CE p = 0.9713) or the degree of myelination in the CNS of adult mice (Fig. 2I,J,M,N,R). Taken together, these results suggest that enhanced p65 NF-κB activation does not affect oligodendrocyte viability or function under normal conditions.
Figure 2.

Enhanced NF-κB activation had a minimal effect on oligodendrocytes under normal conditions. A–D, Q, CC1 IHC showed a comparable number of oligodendrocytes in the CNS of three-week-old IKK2ca cKI mice, WT mice, PERK cKO mice, and Double mice. E–H, MBP IHC showed a comparable degree of myelination in the SP of three-week-old IKK2ca cKI mice, WT mice, PERK cKO mice, and Double mice. I–L, R, CC1 IHC showed a comparable number of oligodendrocytes in the CNS of eight-week-old IKK2ca cKI mice, WT mice, PERK cKO mice, and Double mice. M–P, MBP IHC showed a comparable degree of myelination in the SP of eight-week-old IKK2ca cKI mice, WT mice, PERK cKO mice, and Double mice. N = 4 animals. Scale bars: 40 μm (A–D, I–L) and 140 μm (E–H, M–P). Statistical analyses were done with a one-way ANOVA with a Tukey's post hoc test. Error bars represented SD; n.s., not significant.
Enhanced NF-κB activation in oligodendrocytes protects mice against EAE
To determine the effects of NF-κB activation on oligodendrocytes during EAE, eight-week-old female IKK2ca cKI mice and WT mice were immunized with MOG35-55 peptide to induce EAE. As expected, WT mice developed a typical EAE disease course (Fig. 3A). Although disease onset in IKK2ca cKI mice were comparable to WT mice, these IKK2ca cKI mice displayed significantly milder EAE clinical symptoms compared with WT mice [p = 0.0109–0.0450 (Fig. 3A); p = 0.0290 (Fig. 3B)]. CNS tissues were prepared from these EAE mice at the peak of disease, at PID19. ASPA and the active form of p65 showed that a few ASPA positive oligodendrocytes stained positive for the active form of p65 in the lumbar SP of WT mice at PID19, and that the number of oligodendrocytes that stained positive for the active form of p65 was significantly increased in the lumbar SP of IKK2ca cKI mice compared with WT mice (p < 0.0001; Fig. 3C,D,G). Thus, these results suggest that enhanced p65 NF-κB activation specifically in oligodendrocytes attenuates EAE disease severity.
Figure 3.

Enhanced NF-κB activation specifically in oligodendrocytes rendered mice resistant to EAE and abrogated the increased susceptibility of PERK cKO mice to EAE. A, Mean EAE clinical score. Error bars represent SEM. B, The mean aggregate EAE clinical score was significantly decreased in IKK2ca cKI mice (N = 16 animals) but significantly increased in PERK cKO mice (N = 16 animals), as compared with WT mice (N = 20 animals). The mean aggregate EAE clinical score of Double mice (N = 16 animals) was significantly decreased as compared with PERK cKO mice and WT mice. Error bars represent SD. C–G, ASPA and the active form of p65 double immunostaining showed that the number of oligodendrocytes (arrows) that stained positive for the active form of p65 was significantly increased in the lumbar SP of IKK2ca cKI mice and Double mice compared with WT mice at PID19 and was significantly decreased in the lumbar SP of PERK cKO mice compared with WT mice and Double mice at PID19. N = 4 animals. Error bars represent SD. Scale bars: 20 μm (C–F). Statistical analyses were done with a one-way ANOVA with a Tukey's post hoc test; *p < 0.05.
We further assessed demyelination, oligodendrocyte loss, axon degeneration, and inflammation in the CNS of these EAE mice. MBP IHC revealed substantial myelin damage in the lumbar SP of WT mice at PID19 (Fig. 4B). Interestingly, myelin damage was significantly attenuated in the lumbar SP of IKK2ca cKI mice compared with WT mice (p = 0.0006; Fig. 4A,B,M). Similarly, toluidine blue staining showed that the percentage of demyelinated area in the white matter of the lumbar SP in IKK2ca cKI mice was significantly reduced compared with WT mice at PID19 (p = 0.0002; Fig. 4E,F,N). Non-phosphorylated neurofilament-H (SMI-32) immunostaining showed that there was substantial axon degeneration in the lumbar SP of WT mice at PID19 (Fig. 4J) and that the number of degenerating axons was significantly reduced in the lumbar SP of IKK2ca cKI mice compared with WT mice (p = 0.0006; Fig. 4I,J,O). Importantly, CD3 and ASPA double immunostaining showed that there were a few remaining oligodendrocytes in inflammatory lesions in the lumbar SP of WT mice at PID19, and that the number of remaining oligodendrocytes were significantly increased in inflammatory lesions in IKK2ca cKI mice compared with WT mice (p = 0.0008; Fig. 5A,B,M). Nevertheless, the number of CD3-positive T cells was comparable in the lumbar SP of IKK2ca cKI mice and WT mice at PID19 (p = 0.8899; Fig. 5A,B,N). CD11b immunostaining also showed that the number of CD11b-positive microglia/macrophages was comparable in the lumbar SP of IKK2ca cKI mice and WT mice at PID19 (p = 0.9297; Fig. 5E,F,O). Moreover, GFAP immunostaining showed that the number of GFAP-positive astrocytes was comparable in the lumbar SP of IKK2ca cKI mice and WT mice at PID19 (p = 0.9583; Fig. 5I,J,P). Additionally, real-time PCR analysis showed that enhanced p65 NF-κB activation specifically in oligodendrocytes did not significantly alter the expression of immune cytokines, including IFN-γ (p = 0.8766), TNF-α (p = 0.8970), iNOS (p = 0.9956), IL-2 (p = 0.9735), IL-5 (p = 0.9387), and IL-10 (p = 0.8454), in the SPs of EAE mice at PID19 (Fig. 5Q). Collectively, these data suggest that enhanced p65 NF-κB activation specifically in oligodendrocytes attenuates oligodendrocyte death, demyelination, and axon degeneration but does not alter inflammation in the CNS of EAE mice.
Figure 4.
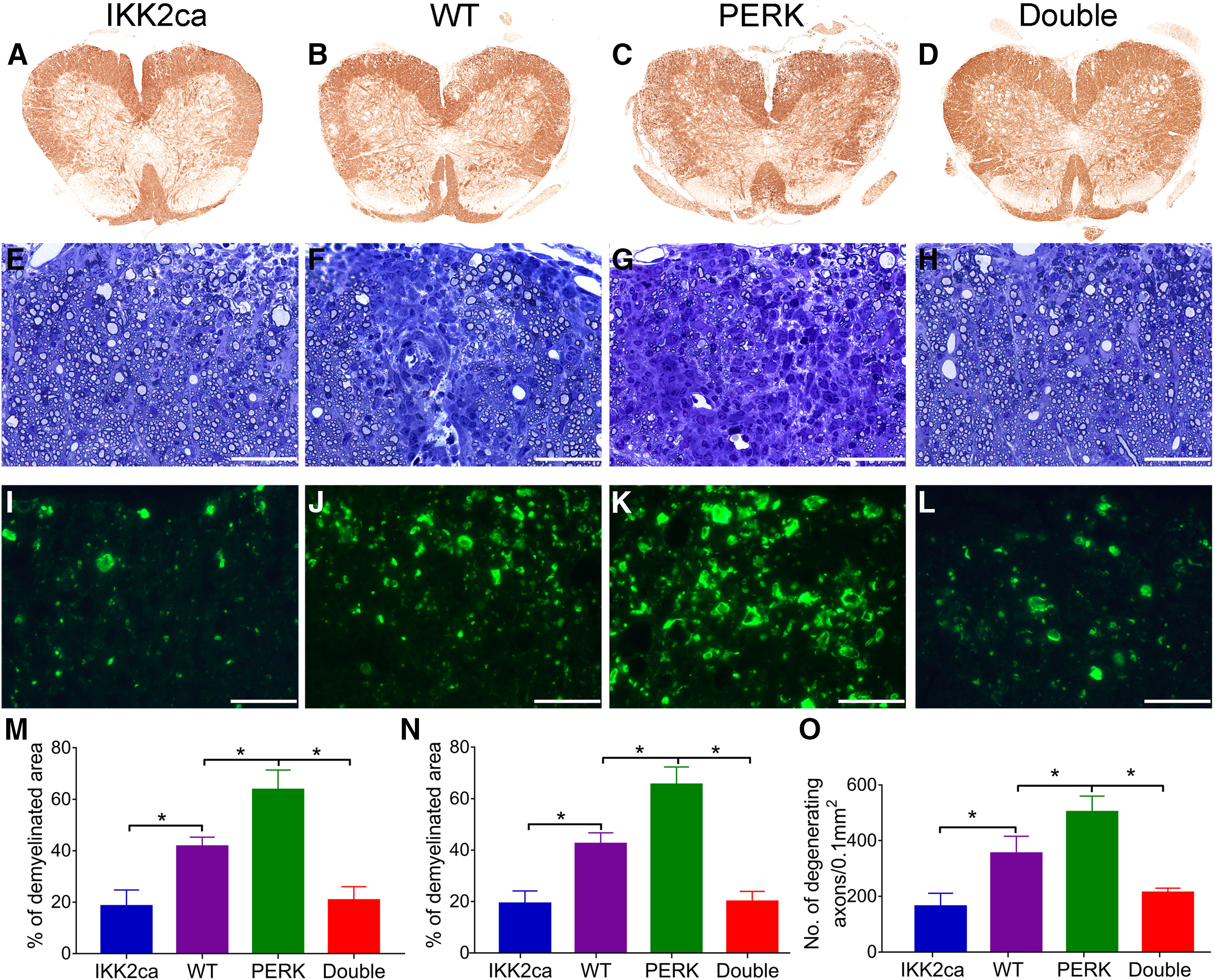
Enhanced NF-κB activation specifically in oligodendrocytes attenuated EAE-induced demyelination and axon degeneration, and reversed the exacerbation of demyelination and axon degeneration in PERK cKO mice undergoing EAE. A–D, M, MBP IHC showed that the percentage of demyelinated area in the lumbar SP was significantly decreased in IKK2ca cKI mice but was significantly increased in PERK cKO mice at PID19, as compared with WT mice. The percentage of demyelinated area in the lumbar SP of Double mice was significantly decreased as compared with PERK cKO mice and WT mice at PID19. E–H, N, Toluidine blue staining showed that the percentage of demyelinated area in the lumbar SP was significantly decreased in IKK2ca cKI mice but was significantly increased in PERK cKO mice at PID19, as compared with WT mice. The percentage of demyelinated area in the lumbar SP of Double mice was significantly decreased as compared with PERK cKO mice and WT mice at PID19. I–L, O, SMI-32 immunostaining showed that the number of degenerating axons was significantly decreased in the lumbar SP of IKK2ca cKI mice but was significantly increased in PERK cKO mice at PID19, as compared with WT mice. The number of degenerating axons was significantly decreased in the lumbar SP of Double mice as compared with PERK cKO mice and WT mice at PID19. N = 4 animals. Scale bars: 40 μm (E–L). Statistical analyses were done with a one-way ANOVA with a Tukey's post hoc test. Error bars represent SD; *p < 0.05.
Figure 5.
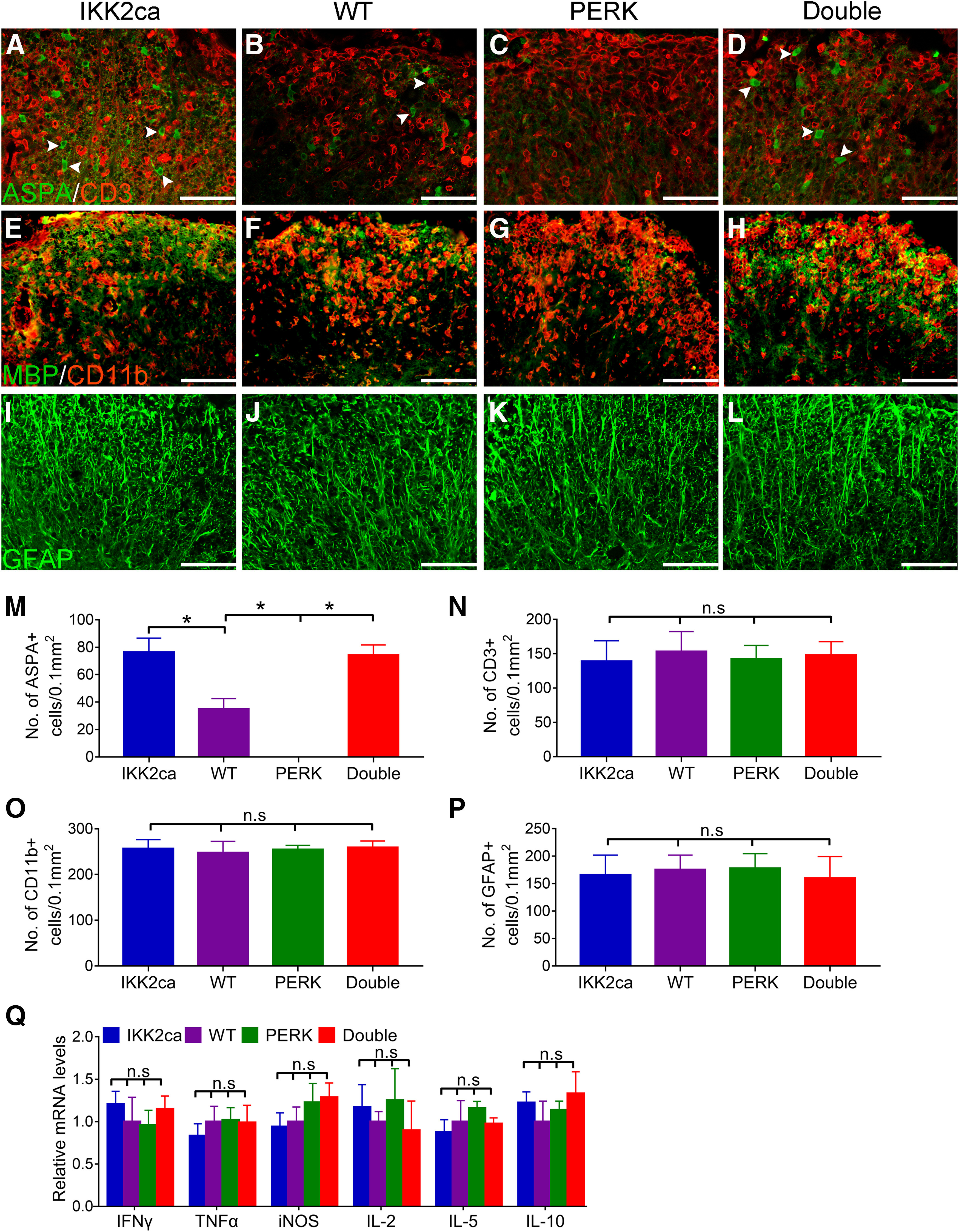
Enhanced NF-κB activation specifically in oligodendrocytes attenuated EAE-induced oligodendrocyte loss, and reversed the exacerbation of oligodendrocyte loss in PERK cKO mice undergoing EAE. A–D, M, ASPA and CD3 double immunostaining showed that the number of oligodendrocytes (arrowheads) was significantly increased in inflammatory lesions in the lumbar SP of IKK2ca cKI mice but was significantly decreased in PERK cKO mice at PID19, as compared with WT mice. The number of oligodendrocytes was significantly increased in inflammatory lesions in the lumbar SP of Double mice as compared with PERK cKO mice and WT mice at PID19. A–D, N, ASPA and CD3 double immunostaining showed a comparable number of CD3-positive T cells in the lumbar SP of IKK2ca cKI mice, WT mice, PERK cKO mice, and Double mice at PID19. E–H, O, CD11b and MBP double immunostaining showed a comparable number of CD11b-positive microglia/macrophages in the lumbar SP of IKK2ca cKI mice, WT mice, PERK cKO mice, and Double mice at PID19. I–L, P, GFAP immunostaining showed a comparable number of GFAP-positive astrocytes in the lumbar SP of IKK2ca cKI mice, WT mice, PERK cKO mice, and Double mice at PID19. Q, Real-time PCR analysis showed that the levels of IFN-γ, TNF-α, iNOS, IL-2, IL-5, and IL-10 were comparable in the lumbar SP of IKK2ca cKI mice, WT mice, PERK cKO mice, and Double mice at PID19. N = 4 animals. Scale bars: 40 μm (A–D), 60 μm (E–H), and 60 μm (I–L). Statistical analyses were done with a one-way ANOVA with a Tukey's post hoc test. Error bars represent SD; *p < 0.05; n.s., not significant.
PCR analysis showed that the floxed STOP cassette that prevents translation of Flag-tagged IKK2ca was not removed in leukocytes in the spleen of IKK2ca cKI mice (Fig. 1A), suggesting that IKK2ca is not expressed in leukocytes of IKK2ca cKI mice. We sought to rule out the possibility that T cell priming is altered in the peripheral immune system of IKK2ca cKI mice during EAE. We cultured leukocytes from the spleen of IKK2ca cKI mice and WT mice before onset of clinical symptom, at PID10 and then analyzed their in vitro response to a secondary exposure to MOG35-55 peptide. BrdU cell proliferation assay and MTT cell viability assay showed that the proliferation and viability of T cells generated from IKK2ca cKI mice and WT mice were comparable in response to MOG35-55 peptide, respectively [BrdU p = 0.7536–0.8963 (Fig. 6A); MTT p = 0.7134–0.8281 (Fig. 6B)]. ELISA assay showed that T cells generated from IKK2ca cKI mice and WT mice produced comparable levels of IFN-γ (p = 0.5324), IL-4 (p = 0.9889), and IL-17A (p = 0.9963) in response to MOG35-55 peptide (Fig. 6C–E). These results suggest that T cell priming in the peripheral immune system is not altered in IKK2ca cKI mice during EAE. Collectively, these data provide direct evidence that enhanced p65 NF-κB activation in oligodendrocytes protects mice against EAE without affecting inflammation, suggesting the cytoprotective effects of NF-κB activation on oligodendrocytes in MS and EAE.
Figure 6.

Neither oligodendrocyte-specific expression of IKK2ca nor oligodendrocyte-specific PERK inactivation influenced T cell priming in the peripheral immune system during EAE. A, BrdU cell proliferation assay showed comparable T cell proliferation in IKK2ca cKI mice, WT mice, PERK cKO mice, and Double mice in response to MOG35-55 peptide. B, MTT cell viability assay showed comparable T cell viability in IKK2ca cKI mice, WT mice, PERK cKO mice, and Double mice in response to MOG35-55 peptide. C–E, ELISA analysis showed comparable ability of T cells to produce the cytokines IFN-γ, IL-4, or IL-17A in IKK2ca cKI mice, WT mice, PERK cKO mice, and Double mice in response to MOG35-55 peptide. N = 4 animals. Statistical analyses were done with a one-way ANOVA with a Tukey's post hoc test. Error bars represent SD; n.s., not significant.
Enhanced NF-κB activation in oligodendrocytes completely rescues the detrimental effects of PERK inactivation in oligodendrocytes in EAE
Previous studies demonstrated the cytoprotective effects of PERK activation on oligodendrocytes during EAE (Lin et al., 2007, 2013, 2014a; Hussien et al., 2014). However, a recent study suggested that ATF4, the master transcription factor of the PERK-eIF2α pathway, is not involved in the cytoprotective effects of PERK activation on oligodendrocytes during EAE (Yue et al., 2019). Besides ATF4, NF-κB is another major transcription factor activated by the PERK-eIF2α pathway (Deng et al., 2004; Ron and Walter, 2007). Interestingly, our previous study demonstrated that PERK activation induces p65 NF-κB activation in oligodendrocytes during EAE (Lin et al., 2013). We sought to determine the involvement of p65 NF-κB in the cytoprotective effects of PERK activation on oligodendrocytes during EAE. Using PERKloxP/loxP; CNP/Cre mice (PERK cKO mice), a previous study demonstrated that PERK inactivation specifically in oligodendrocytes increases EAE disease severity and exacerbates EAE-induced oligodendrocyte death, demyelination, and axon degeneration, without affecting inflammation (Hussien et al., 2014). PERK cKO mice were crossed with IKK2ca cKI mice to generate R26StopFLikk2ca; PERKloxP/loxP; CNP/Cre mice (Double mice). As expected, PERK cKO mice looked healthy and did not display oligodendrocyte or myelin abnormalities in the CNS at the age of three weeks (SP p = 0.7450, CC p = 0.9723, CE p = 0.8671; Fig. 2B,C,F,G,Q) and eight weeks (SP p = 0.6138, CC p = 0.7643, CE p = 0.9713; Fig. 2J,K,N,O,R). Moreover, Double KO mice appeared healthy and were indistinguishable from PERK cKO mice, IKK2ca cKI mice, and WT mice. CC1 IHC showed that the number of oligodendrocytes was comparable in the CNS of Double KO mice, PERK cKO mice, IKK2ca cKI mice, and WT mice at the age of three weeks (SP p = 0.7738, CC p = 0.7069, CE p = 0.7253; Fig. 2A–D,Q) and eight weeks (SP p = 0.5836, CC p = 0.2417, CE p = 0.7915; Fig. 2I–L,R). MBP IHC showed that the degree of myelination was comparable in the CNS of Double KO mice, PERK cKO mice, IKK2ca cKI mice, and WT mice at the age of three and eight weeks (Fig. 2E–H,M–P). These data suggest that the combination of PERK inactivation and p65 NF-κB activation in oligodendrocytes does not affect their viability or function under normal conditions.
Eight-week-old female Double mice, IKK2ca cKI mice, PERK cKO mice and WT mice were immunized with MOG35-55 peptide to induce EAE. In accordance with the previous study (Hussien et al., 2014), PERK cKO mice developed a more severe EAE disease course compared with WT mice, including significantly earlier onset of disease and more severe clinical symptoms [p = 0.0002–0.0435 (Fig. 3A); p = 0.0445 (Fig. 3B)]. Importantly, the EAE clinical symptoms displayed by Double mice was significantly milder than PERK cKO mice and WT mice [Double mice vs PERK cKO mice p = 0.0002 to p < 0.0001, Double mice vs WT mice p = 0.0002–0.048 (Fig. 3A); Double mice vs PERK cKO mice, p < 0.0001, Double mice vs WT mice p = 0.0033 (Fig. 3B)]. ASPA and the active form of p65 double immunostaining showed that the number of oligodendrocytes that stained positive for the active form of p65 was significantly decreased in the lumbar SP of PERK cKO mice compared with WT mice (p < 0.0001; Fig. 3D,E,G), and was significantly increased in the lumbar SP of Double mice compared with PERK cKO mice and WT mice (Double mice vs PERK cKO mice p < 0.0001, Double mice vs WT mice p < 0.0001; Fig. 3D–G). These results suggest that enhanced p65 NF-κB activation specifically in oligodendrocytes abrogates the increased susceptibility of PERK cKO mice to EAE.
Consistent with the previous study (Hussien et al., 2014), we found that PERK inactivation specifically in oligodendrocytes exacerbated demyelination, oligodendrocyte loss, and axon degeneration, but did not alter inflammation during EAE (Figs. 4–6). Interestingly, MBP IHC showed that myelin damage was significantly attenuated in the lumbar SP of Double mice compared with PERK cKO mice and WT mice at PID19 (Double mice vs PERK cKO mice p < 0.0001, Double mice vs WT mice p = 0.0013; Fig. 4B–D,M). Similarly, toluidine blue staining showed that the percentage of demyelinated area in the white matter of the lumbar SP in Double mice was significantly decreased compared with PERK cKO mice and WT mice at PID19 (Double mice vs PERK cKO mice p < 0.0001, Double mice vs WT mice p = 0.0002; Fig. 4F–H,N). SMI-32 immunostaining showed that the number of degenerating axons in the lumbar SP of Double mice was significantly reduced compared with PERK cKO mice and WT mice at PID19 (Double mice vs PERK cKO mice p < 0.0001, Double mice vs WT mice p = 0.0018; Fig. 4J–L,O). Importantly, CD3 and ASPA double immunostaining showed that the number of remaining oligodendrocytes was significantly increased in inflammatory lesions in the lumbar SP of Double mice compared with PERK cKO mice and WT mice at PID19 (Double mice vs PERK cKO mice p < 0.0001, Double mice vs WT mice p = 0.0008; Fig. 5B–D,M). Conversely, the number of CD3-positive T cells was comparable in the lumbar SP of Double mice, IKK2ca cKI mice, PERK cKO mice, and WT mice at PID19 (p = 0.8978; Fig. 5A–D,N). CD11b immunostaining showed that the number of CD11b-positive microglia/macrophages was comparable in the lumbar SP of Double mice, IKK2ca cKI mice, PERK cKO mice, and WT mice at PID19 (p = 0.8730; Fig. 5E–H,O). GFAP immunostaining also showed that the number of GFAP-positive astrocytes was comparable in the lumbar SP of Double mice, IKK2ca cKI mice, PERK cKO mice, and WT mice at PID19 (p = 0.8974; Fig. 5I–L,P). Moreover, real-time PCR analysis showed comparable levels of immune cytokines, including IFN-γ (p = 0.7859), TNF-α (p = 0.8556), iNOS (p = 0.5074), IL-2 (p = 0.8210), IL-5 (p = 0.6424), and IL-10 (p = 0.6741), in the SP of Double mice, IKK2ca cKI mice, PERK cKO mice, and WT mice at PID19 (Fig. 5Q). Additionally, in vitro recall assays showed that the abilities of T cells to proliferate, to survive, and to produce immune cytokines in response to MOG35-55 peptide were comparable in Double mice, IKK2ca cKI mice, PERK cKO mice, and WT mice (BrdU p = 0.4884–0.8926, MTT p = 0.5793–0.8130, IFN-γ p = 0.5462, IL-4 p = 0.9388, IL-17A p = 0.9621; Fig. 6A–E). These results suggest that enhanced p65 NF-κB activation specifically in oligodendrocytes completely reverses the exacerbation of demyelination, oligodendrocyte death, and axon degeneration in the CNS of PERK cKO mice undergoing EAE, without altering inflammation. Collectively, these data demonstrate that the detrimental effects of PERK inactivation in oligodendrocytes in EAE are accompanied by impaired p65 NF-κB activation in oligodendrocytes, and are completely rescued by enhanced p65 NF-κB activation in oligodendrocytes.
The cytoprotective effects of NF-κB activation on oligodendrocytes during EAE are associated with upregulation of A20/TNFAIP3
There is evidence that NF-κB activation exerts its cytoprotective effects by inducing anti-apoptotic genes (Pahl, 1999; Karin and Lin, 2002; Oeckinghaus et al., 2011; Mincheva-Tasheva and Soler, 2013). It is known that ∼60% of cells in the optic nerve are oligodendrocytes (Vaughn, 1969; Lin et al., 2014b). To determine the mechanisms responsible for the cytoprotective effects of p65 NF-κB activation on oligodendrocytes, we employed an unbiased, whole-genome RNA-sequencing approach using the RNA isolated from the optic nerve of eight-week-old IKK2ca cKI mice and WT mice. Unexpectedly, differential gene expression analysis showed that there were only 12 significantly upregulated genes (increase >2-fold and p < 0.05) in the optic nerve of IKK2ca cKI mice compared with WT mice (Table 1). As described above, we found that both the transgenic IKK2ca and EGFP were expressed modestly in oligodendrocytes of IKK2ca cKI mice. Moreover, RNA-sequencing analysis showed that the level of IκBα mRNA, a major p65 NF-κB responsive gene (Pahl, 1999), was modestly increased (1.559935-fold) in the optic nerve of IKK2ca cKI mice compared with WT mice (Table 1). Taken together, these data suggest that modest p65 NF-κB activation induced by modest IKK2ca expression does not significantly increase (>2-fold) the expression of the majority of NF-κB-responsive genes in oligodendrocytes of IKK2ca cKI mice.
Among 12 significantly upregulated genes, only A20/TNFAIP3 (tumor necrosis factor α-induced protein 3) is a well-established NF-κB-responsive, anti-apoptotic gene (Pahl, 1999; Beyaert et al., 2000; Catrysse et al., 2014). Interestingly, western blot analysis showed that the level of A20/TNFAIP3 was significantly increased in the CNS of eight-week-old IKK2ca cKI mice compared with WT mice (p = 0.0451; Fig. 7A,B). Moreover, ASPA and A20/TNFAIP3 double immunostaining showed that the immunoreactivity of A20/TNFAIP3 was markedly increased in oligodendrocytes in the CNS of eight-week-old IKK2ca cKI mice compared with WT mice (p < 0.0001; Fig. 7C–I). Importantly, ASPA and A20/TNFAIP3 double immunostaining showed that there were a few A20/TNFAIP3-positive oligodendrocytes in the lumbar SP of WT mice with EAE and that the number of A20/TNFAIP3-positive oligodendrocytes was significantly increased in the lumbar SP of IKK2ca cKI mice with EAE compared with WT mice with EAE (p < 0.001; Fig. 7J,K,N). Additionally, the number of A20/TNFAIP3-positive oligodendrocytes was significantly decreased in the lumbar SP of PERK cKO mice with EAE compared with WT mice with EAE (p < 0.0001; Fig. 7K,L,N) and was significantly increased in the lumbar SP of Double mice with EAE compared with PERK cKO mice with EAE and WT mice with EAE (Double mice vs PERK cKO mice p < 0.0001, Double mice vs WT mice p < 0.0001; Fig. 7K–N). These results demonstrate that enhanced p65 NF-κB activation stimulates the expression of the anti-apoptotic gene A20/TNFAIP3 in oligodendrocytes. Collectively, these data suggest the possibility that induction of A20/TNFAIP3 contributes to the cytoprotective effects of p65 NF-κB activation on oligodendrocytes during EAE.
Figure 7.
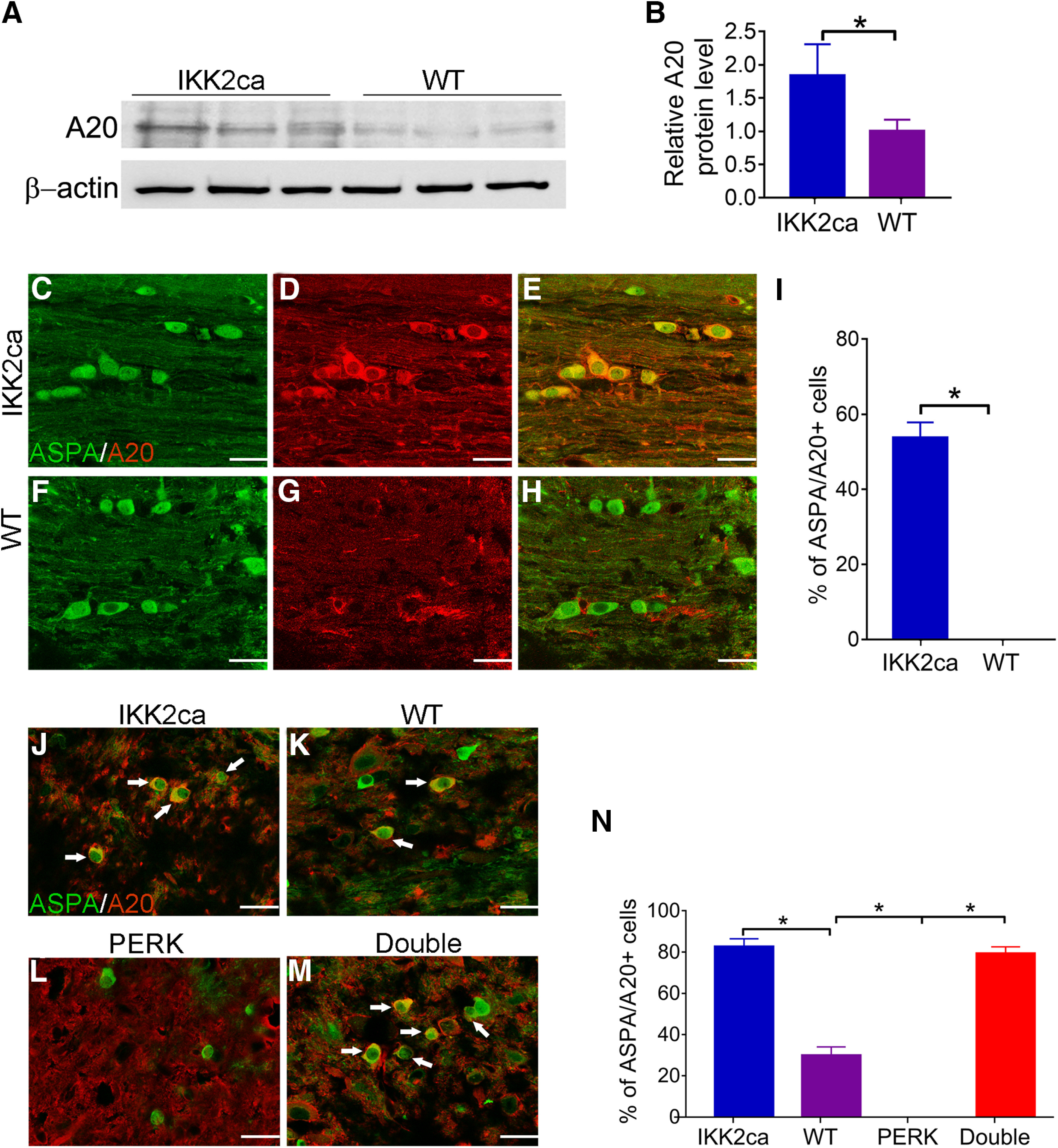
Enhanced NF-κB activation stimulated the expression of A20/TNFAIP3 in oligodendrocytes. A, B, Western blot analysis showed that the level of A20/TNFAIP3 was significantly increased in the brain of eight-week-old IKK2ca cKI mice compared with WT mice. N = 3 animals. C–I, ASPA and A20/TNFAIP3 double immunostaining showed that the number of oligodendrocytes that stained positive for A20/TNFAIP3 was significantly increased in the brain of eight-week-old IKK2ca cKI mice compared with WT mice. N = 4 animals. J–N, ASPA and A20/TNFAIP3 double immunostaining showed that the number of oligodendrocytes (arrows) that stained positive for A20/TNFAIP3 was significantly increased in the lumbar SP of IKK2ca cKI mice and Double mice compared with WT mice at PID19 and was significantly decreased in the lumbar SP of PERK cKO mice compared with WT mice and Double mice at PID19. N = 4 animals. Scale bars: 20 μm. Statistical analyses were done with a t test or a one-way ANOVA with a Tukey's post hoc test. Error bars represent SD; *p < 0.05.
Discussion
NF-κB is the major transcription factor that regulates inflammation and cell viability in inflammatory diseases, including MS and EAE (Oeckinghaus et al., 2011; Mc Guire et al., 2013; Herrington et al., 2016; Yue et al., 2018). It is known that NF-κB is activated in oligodendrocytes in MS and EAE (Bonetti et al., 1999; Lin et al., 2013). Using a mouse model that expresses IκBαΔN (a supersuppressor of NF-κB) specifically in oligodendrocytes, our previous study showed that IκBαΔN expression blocks p65 NF-κB activation and render (re)myelinating oligodendrocytes sensitive to the cytotoxicity of IFN-γ in the young, developing mice and in the cuprizone model (Stone et al., 2017). We also found that oligodendrocyte-specific expression of IκBαΔN dramatically increases EAE diseases severity; however, we were not able to collect tissues to determine the effects of IκBαΔN expression on oligodendrocytes during EAE because of early, sudden death of these EAE mice (Stone et al., 2017). Moreover, a report showed that oligodendrocyte-specific deletion of RelB attenuates EAE-induced oligodendrocyte death and demyelination and that the beneficial effects of RelB deletion in oligodendrocytes during EAE are associated with p65 NF-κB activation (Gupta et al., 2019). In this study, we generated a mouse model that expresses IKK2ca specifically in oligodendrocytes. We found that IKK2ca expression induced p65 NF-κB activation selectively in oligodendrocytes, but did not affect oligodendrocyte viability or function under normal conditions. Importantly, we found that enhanced p65 NF-κB activation specifically in oligodendrocytes ameliorated EAE disease severity and attenuated EAE-induced oligodendrocyte loss, demyelination, and axon degeneration, without affecting inflammation. There is evidence that oligodendrocytes support axon integrity under normal and diseases conditions through myelin-dependent and independent mechanisms (Bankston et al., 2013; Saab et al., 2013). Several studies using oligodendrocyte conditional transgenic mice showed that enhancement of oligodendrocyte survival results in attenuation of axon degeneration during EAE (Ren et al., 2011; Lin et al., 2013). Thus, these data suggest that p65 NF-κB activation in oligodendrocytes protects oligodendrocytes and myelin against inflammation, and subsequently results in attenuation of axon degeneration during EAE.
The PERK-eIF2α pathway is a major player in regulating oligodendrocyte viability and function under normal and disease conditions (Lin and Popko, 2009; Volpi et al., 2016; Lin and Stone, 2020; Stone et al., 2020). Previous studies demonstrated the cytoprotective effects of PERK activation on oligodendrocytes during EAE (Lin et al., 2007, 2013, 2014a; Hussien et al., 2014). ATF4 is regarded as the master transcription factor of the PERK-eIF2α pathway (Walter and Ron, 2011; Wang and Kaufman, 2016); however, a recent study suggested that ATF4 is not involved in the cytoprotective effects of PERK activation on oligodendrocytes during EAE (Yue et al., 2019). Data indicate that the PERK-eIF2α pathway also activates NF-κB in ER-stressed cells (Deng et al., 2004; Ron and Walter, 2007). Interestingly, our previous study showed that the cytoprotective effects of PERK activation on oligodendrocytes during EAE are accompanied with NF-κB activation (Lin et al., 2013). Thus, these data raise a possibility that the cytoprotective effects of PERK activation on oligodendrocytes during EAE are mediated by NF-κB. To test this possibility, one strategy is to determine whether NF-κB inactivation in oligodendrocytes diminishes the cytoprotective effects of PERK activation on oligodendrocytes during EAE using a mouse model that allows for activation of PERK and inactivation of NF-κB (via expression of IκBαΔN) specifically in oligodendrocytes. Using mice that allow for temporally-controlled activation of PERK specifically in oligodendrocytes, we have demonstrated the cytoprotective effects of PERK activation on oligodendrocytes during EAE (Lin et al., 2013). Nevertheless, mice expressing IκBαΔN specifically in oligodendrocytes develop extremely severe EAE with very high mortality (Stone et al., 2017). We have no choice but to give up this strategy. On the other hand, using a mouse model that allows for inactivation of PERK specifically in oligodendrocytes, a previous study showed that PERK inactivation in oligodendrocytes exacerbates EAE-induced oligodendrocyte death, demyelination, and axon degeneration (Hussien et al., 2014). Importantly, we showed here that the detrimental effects of PERK inactivation in oligodendrocytes in EAE were accompanied by impaired NF-κB activation in oligodendrocytes. Therefore, the other strategy is to determine whether enhanced NF-κB activation in oligodendrocytes rescues the detrimental effects of PERK inactivation on the cells during EAE. We generated a mouse model that allows for PERK inactivation and NF-κB activation specifically in oligodendrocytes, and showed that the exacerbation of oligodendrocyte death, demyelination, and axon degeneration induced by PERK inactivation in oligodendrocytes during EAE were completed rescued by enhanced NF-κB activation in oligodendrocytes. Collectively, these results demonstrate the significant contribution of NF-κB activation to the cytoprotective effects of PERK activation on oligodendrocytes during EAE.
Evidence suggests that NF-κB activation exerts cytoprotection through induction of anti-apoptotic genes, including cIAPs, cFLIP, Bcl-2, Bcl-xL, and A20/TNFAIP3 (Pahl, 1999; Karin and Lin, 2002; Oeckinghaus et al., 2011; Mincheva-Tasheva and Soler, 2013). Herein, we showed that enhanced NF-κB activation stimulated the expression of A20/TNFAIP3 in oligodendrocytes in naive mice and mice undergoing EAE. Since A20/TNFAIP3 is a well-established NF-κB-responsive, anti-apoptotic gene (Pahl, 1999; Beyaert et al., 2000; Catrysse et al., 2014), it is possible that NF-κB activation protects oligodendrocytes against inflammation during EAE by upregulating A20/TNFAIP3. Interestingly, genome-wide association studies show that polymorphism of the Tnfaip3 gene is associated with an increased risk for MS (De Jager et al., 2009; Gilli et al., 2011; Hoffjan et al., 2015). Recent studies suggest that A20/TNFAIP3 is a potent inhibitor of inflammation in autoimmune inflammatory diseases, including MS and EAE, by suppressing inflammatory cell activation (Vereecke et al., 2009; Voet et al., 2018; Malynn and Ma, 2019). However, the role of A20/TNFAIP3 in oligodendrocytes in MS and EAE remains unexplored. An investigation into the role of A20/TNFAIP3 in oligodendrocytes and its contribution to the cytoprotective effects of NF-κB activation on oligodendrocytes during EAE is warranted.
Data indicate that oligodendrocytes actively influences inflammation in the CNS in MS and EAE by producing cytokines and chemokines (Balabanov et al., 2007; Ren et al., 2011; Jäkel et al., 2019). Evidence suggests that NF-κB activation stimulates the expression of cytokines and chemokines (Newton and Dixit, 2012; Mc Guire et al., 2013). However, we found that enhanced NF-κB activation in oligodendrocytes attenuated oligodendrocyte death, demyelination, and axon degeneration, but did not alter inflammation in the CNS of EAE mice. Accordingly, RNA- sequencing analysis showed that enhanced NF-κB activation induced only 12 significantly upregulated genes in oligodendrocytes, which did not include any cytokines or chemokines. Moreover, we found that the expression of transgenic IKK2ca and EGFP in oligodendrocytes of IKK2ca cKI mice was modest. Thus, it is possible that modest NF-κB activation induced by modest IKK2ca expression does not significantly increase the expression of cytokines and chemokines in oligodendrocytes, and results in minimal alteration of CNS inflammation in IKK2ca cKI mice with EAE. On the other hand, the dogma of MS and EAE pathogenesis is that these diseases are initiated by autoimmune inflammation against oligodendrocytes and myelin, and then CNS tissue damage enhances inflammation, forming a vicious circle (Frohman et al., 2006; Bradl and Lassmann, 2010). The alternative, but not mutually exclusive, possibility is that enhanced NF-κB activation in oligodendrocytes is a double-edged sword in regulating inflammation in the CNS of EAE mice: (1) promotes inflammation through induction of cytokines and chemokines; (2) suppresses inflammation by attenuating CNS tissue damage. Neutralization of these two opposite effects results in the minimal effects of enhanced NF-κB activation in oligodendrocytes on CNS inflammation during EAE.
In summary, using a mouse model that expresses IKK2ca specifically in oligodendrocytes in the CNS, we demonstrated the cytoprotective effects of NF-κB activation on oligodendrocytes during EAE. The results presented in this study also suggest that NF-κB activation accounts for the cytoprotective effects of PERK activation on oligodendrocytes during EAE. Because NF-κB activation in inflammatory cells enhances inflammation and facilitates the development of MS and EAE, NF-κB is not an ideal therapeutic target for MS. In contrast, several lines of evidence have suggested that the PERK-eIF2α pathway is a potential therapeutic target for MS (Way and Popko, 2016; Lin and Stone, 2020). Moreover, recent studies showed that treatment with small chemical compounds that enhance activation of the PERK-eIF2α pathway protects oligodendrocytes against inflammation during EAE (Way et al., 2015; Chen et al., 2019). Thus, this study will facilitate the development of treatments targeting the PERK-eIF2α pathway for MS patients by defining the mechanism responsible for the cytoprotective effects of PERK activation on oligodendrocytes in MS and EAE.
Footnotes
The authors declare no competing financial interests.
This study was supported by National Institutes of Health Grants NS094151 and NS105689 (to W.L.). We thank Dr. Klaus-Armin Nave (Department of Neurogenetics, Max Planck Institute of Experimental Medicine, Göttingen, Germany) for providing the CNP/Cre mice. We also thank Dr. Juan E. Abrahante Lloréns (University of Minnesota Informatics Institute) for conducting bioinformatics analysis of RNA-sequencing data.
References
- Balabanov R, Strand K, Goswami R, McMahon E, Begolka W, Miller SD, Popko B (2007) Interferon-gamma-oligodendrocyte interactions in the regulation of experimental autoimmune encephalomyelitis. J Neurosci 27:2013–2024. 10.1523/JNEUROSCI.4689-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankston AN, Mandler MD, Feng Y (2013) Oligodendroglia and neurotrophic factors in neurodegeneration. Neurosci Bull 29:216–228. 10.1007/s12264-013-1321-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann N, Pham-Dinh D (2001) Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev 81:871–927. 10.1152/physrev.2001.81.2.871 [DOI] [PubMed] [Google Scholar]
- Beyaert R, Heyninck K, Van Huffel S (2000) A20 and A20-binding proteins as cellular inhibitors of nuclear factor-kappa B-dependent gene expression and apoptosis. Biochem Pharmacol 60:1143–1151. 10.1016/s0006-2952(00)00404-4 [DOI] [PubMed] [Google Scholar]
- Blank T, Prinz M (2014) NF-κB signaling regulates myelination in the CNS. Front Mol Neurosci 7:47. 10.3389/fnmol.2014.00047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetti B, Stegagno C, Cannella B, Rizzuto N, Moretto G, Raine CS (1999) Activation of NF-kappaB and c-jun transcription factors in multiple sclerosis lesions. Implications for oligodendrocyte pathology. Am J Pathol 155:1433–1438. 10.1016/s0002-9440(10)65456-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradl M, Lassmann H (2010) Oligodendrocytes: biology and pathology. Acta Neuropathol 119:37–53. 10.1007/s00401-009-0601-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catrysse L, Vereecke L, Beyaert R, van Loo G (2014) A20 in inflammation and autoimmunity. Trends Immunol 35:22–31. 10.1016/j.it.2013.10.005 [DOI] [PubMed] [Google Scholar]
- Chen Y, Podojil JR, Kunjamma RB, Jones J, Weiner M, Lin W, Miller SD, Popko B (2019) Sephin1, which prolongs the integrated stress response, is a promising therapeutic for multiple sclerosis. Brain 142:344–361. 10.1093/brain/awy322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jager PL, Jia X, Wang J, de Bakker PIW, Ottoboni L, Aggarwal NT, Piccio L, Raychaudhuri S, Tran D, Aubin C, Briskin R, Romano S, Baranzini SE, McCauley JL, Pericak-Vance MA, Haines JL, Gibson RA, Naeglin Y, Uitdehaag B, Matthews PM, et al. (2009) Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet 41:776–782. 10.1038/ng.401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, Ron D (2004) Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol 24:10161–10168. 10.1128/MCB.24.23.10161-10168.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman EM, Racke MK, Raine CS (2006) Multiple sclerosis–the plaque and its pathogenesis. N Engl J Med 354:942–955. 10.1056/NEJMra052130 [DOI] [PubMed] [Google Scholar]
- Gilli F, Navone ND, Perga S, Marnetto F, Caldano M, Capobianco M, Pulizzi A, Malucchi S, Bertolotto A (2011) Loss of braking signals during inflammation: a factor affecting the development and disease course of multiple sclerosis. Arch Neurol 68:879–888. 10.1001/archneurol.2011.32 [DOI] [PubMed] [Google Scholar]
- Gupta AS, Biswas DD, Brown LSN, Mockenhaupt K, Marone M, Hoskins A, Siebenlist U, Kordula T (2019) A detrimental role of RelB in mature oligodendrocytes during experimental acute encephalomyelitis. J Neuroinflammation 16:161. 10.1186/s12974-019-1548-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gveric D, Kaltschmidt C, Cuzner ML, Newcombe J (1998) Transcription factor NF-kappaB and inhibitor I kappaBalpha are localized in macrophages in active multiple sclerosis lesions. J Neuropathol Exp Neurol 57:168–178. 10.1097/00005072-199802000-00008 [DOI] [PubMed] [Google Scholar]
- Hamanoue M, Yoshioka A, Ohashi T, Eto Y, Takamatsu K (2004) NF-kappaB prevents TNF-alpha-induced apoptosis in an oligodendrocyte cell line. Neurochem Res 29:1571–1576. 10.1023/B:NERE.0000029571.39497.56 [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S (2012) NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 26:203–234. 10.1101/gad.183434.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington FD, Carmody RJ, Goodyear CS (2016) Modulation of NF-κB signaling as a therapeutic target in autoimmunity. J Biomol Screen 21:223–242. 10.1177/1087057115617456 [DOI] [PubMed] [Google Scholar]
- Hilliard B, Samoilova EB, Liu TS, Rostami A, Chen Y (1999) Experimental autoimmune encephalomyelitis in NF-kappa B-deficient mice:roles of NF-kappa B in the activation and differentiation of autoreactive T cells. J Immunol 163:2937–2943. [PubMed] [Google Scholar]
- Hilliard BA, Mason N, Xu L, Sun J, Lamhamedi-Cherradi S-E, Liou HC, Hunter C, Chen YH (2002) Critical roles of c-Rel in autoimmune inflammation and helper T cell differentiation. J Clin Invest 110:843–850. 10.1172/JCI15254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffjan S, Okur A, Epplen JT, Wieczorek S, Chan A, Akkad DA (2015) Association of TNFAIP3 and TNFRSF1A variation with multiple sclerosis in a German case-control cohort. Int J Immunogenet 42:106–110. 10.1111/iji.12183 [DOI] [PubMed] [Google Scholar]
- Hussien Y, Cavener DR, Popko B (2014) Genetic inactivation of PERK signaling in mouse oligodendrocytes: normal developmental myelination with increased susceptibility to inflammatory demyelination. Glia 62:680–691. 10.1002/glia.22634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäkel S, Agirre E, Mendanha Falcão A, van Bruggen D, Lee KW, Knuesel I, Malhotra D, Ffrench-Constant C, Williams A, Castelo-Branco G (2019) Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 566:543–547. 10.1038/s41586-019-0903-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Lin A (2002) NF-kappaB at the crossroads of life and death. Nat Immunol 3:221–227. 10.1038/ni0302-221 [DOI] [PubMed] [Google Scholar]
- Kretz A, Herrmann K-H, Fischer S, Engelmann C, Witte OW, Reichenbach JR, Weih F, Haenold R (2014) Dysfunctional NF-κB and brain myelin formation. Eur J Hum Genet 22:724–725. 10.1038/ejhg.2013.240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave K-A (2003) Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33:366–374. 10.1038/ng1095 [DOI] [PubMed] [Google Scholar]
- Lin W, Popko B (2009) Endoplasmic reticulum stress in disorders of myelinating cells. Nat Neurosci 12:379–385. 10.1038/nn.2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Stone S (2020) Unfolded protein response in myelin disorders. Neural Regen Res 15:636–645. 10.4103/1673-5374.266903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Bailey SL, Ho H, Harding HP, Ron D, Miller SD, Popko B (2007) The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J Clin Invest 117:448–456. 10.1172/JCI29571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Lin Y, Li J, Fenstermaker AG, Way SW, Clayton B, Jamison S, Harding HP, Ron D, Popko B (2013) Oligodendrocyte-specific activation of PERK signaling protects mice against experimental autoimmune encephalomyelitis. J Neurosci 33:5980–5991. 10.1523/JNEUROSCI.1636-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Jamison S, Lin W (2012) Interferon-γ activates nuclear factor-κ B in oligodendrocytes through a process mediated by the unfolded protein response. PLoS One 7:e36408. 10.1371/journal.pone.0036408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Huang G, Jamison S, Li J, Harding HP, Ron D, Lin W (2014a) PERK activation preserves the viability and function of remyelinating oligodendrocytes in immune-mediated demyelinating diseases. Am J Pathol 184:507–519. 10.1016/j.ajpath.2013.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Pang X, Huang G, Jamison S, Fang J, Harding HP, Ron D, Lin W (2014b) Impaired eukaryotic translation initiation factor 2B activity specifically in oligodendrocytes reproduces the pathology of vanishing white matter disease in mice. J Neurosci 34:12182–12191. 10.1523/JNEUROSCI.1373-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malynn BA, Ma A (2019) A20: a multifunctional tool for regulating immunity and preventing disease. Cell Immunol 340:103914. 10.1016/j.cellimm.2019.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mc Guire C, Prinz M, Beyaert R, van Loo G (2013) Nuclear factor kappa B (NF-κB) in multiple sclerosis pathology. Trends Mol Med 19:604–613. 10.1016/j.molmed.2013.08.001 [DOI] [PubMed] [Google Scholar]
- Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A (1997) IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 278:860–866. 10.1126/science.278.5339.860 [DOI] [PubMed] [Google Scholar]
- Mincheva-Tasheva S, Soler RM (2013) NF-κB signaling pathways: role in nervous system physiology and pathology. Neuroscientist 19:175–194. 10.1177/1073858412444007 [DOI] [PubMed] [Google Scholar]
- Newton K, Dixit VM (2012) Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol 4:a006049 10.1101/cshperspect.a006049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas RS, Wing MG, Compston A (2001) Nonactivated microglia promote oligodendrocyte precursor survival and maturation through the transcription factor NF-kappa B. Eur J Neurosci 13:959–967. 10.1046/j.0953-816x.2001.01470.x [DOI] [PubMed] [Google Scholar]
- Oeckinghaus A, Hayden MS, Ghosh S (2011) Crosstalk in NF-κB signaling pathways. Nat Immunol 12:695–708. 10.1038/ni.2065 [DOI] [PubMed] [Google Scholar]
- Pahl HL. (1999) Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18:6853–6866. 10.1038/sj.onc.1203239 [DOI] [PubMed] [Google Scholar]
- Raasch J, Zeller N, van Loo G, Merkler D, Mildner A, Erny D, Knobeloch KP, Bethea JR, Waisman A, Knust M, Del Turco D, Deller T, Blank T, Priller J, Brück W, Pasparakis M, Prinz M (2011) IkappaB kinase 2 determines oligodendrocyte loss by non-cell-autonomous activation of NF-kappaB in the central nervous system. Brain 134:1184–1198. 10.1093/brain/awq359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Wang Y, Tao D, Liebenson D, Liggett T, Goswami R, Clarke R, Stefoski D, Balabanov R (2011) Overexpression of the dominant-negative form of interferon regulatory factor 1 in oligodendrocytes protects against experimental autoimmune encephalomyelitis. J Neurosci 31:8329–8341. 10.1523/JNEUROSCI.1028-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8:519–529. 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- Saab AS, Tzvetanova ID, Nave K-A (2013) The role of myelin and oligodendrocytes in axonal energy metabolism. Curr Opin Neurobiol 23:1065–1072. 10.1016/j.conb.2013.09.008 [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Derudder E, Hobeika E, Pelanda R, Reth M, Rajewsky K, Schmidt-Supprian M (2006) Canonical NF-kappaB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity 24:729–739. 10.1016/j.immuni.2006.04.005 [DOI] [PubMed] [Google Scholar]
- Stone S, Jamison S, Yue Y, Durose W, Schmidt-Ullrich R, Lin W (2017) NF-κB activation protects oligodendrocytes against inflammation. J Neurosci 37:9332–9344. 10.1523/JNEUROSCI.1608-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone S, Wu S, Jamison S, Durose W, Pallais JP, Lin W (2018) Activating transcription factor 6α deficiency exacerbates oligodendrocyte death and myelin damage in immune-mediated demyelinating diseases. Glia 66:1331–1345. 10.1002/glia.23307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone S, Yue Y, Stanojlovic M, Wu S, Karsenty G, Lin W (2019) Neuron-specific PERK inactivation exacerbates neurodegeneration during experimental autoimmune encephalomyelitis. JCI Insight 4:e124232 10.1172/jci.insight.124232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone S, Wu S, Nave KA, Lin W (2020) The UPR preserves mature oligodendrocyte viability and function in adults by regulating autophagy of PLP. JCI Insight 5:e132364 10.1172/jci.insight.132364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loo G, De Lorenzi R, Schmidt H, Huth M, Mildner A, Schmidt-Supprian M, Lassmann H, Prinz MR, Pasparakis M (2006) Inhibition of transcription factor NF-kappaB in the central nervous system ameliorates autoimmune encephalomyelitis in mice. Nat Immunol 7:954–961. 10.1038/ni1372 [DOI] [PubMed] [Google Scholar]
- Vaughn JE. (1969) An electron microscopic analysis of gliogenesis in rat optic nerves. Z Zellforsch Mikrosk Anat 94:293–324. 10.1007/BF00319179 [DOI] [PubMed] [Google Scholar]
- Vereecke L, Beyaert R, van Loo G (2009) The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol 30:383–391. 10.1016/j.it.2009.05.007 [DOI] [PubMed] [Google Scholar]
- Voet S, Mc Guire C, Hagemeyer N, Martens A, Schroeder A, Wieghofer P, Daems C, Staszewski O, Vande Walle L, Jordao MJC, Sze M, Vikkula HK, Demeestere D, Van Imschoot G, Scott CL, Hoste E, Gonçalves A, Guilliams M, Lippens S, Libert C, et al. (2018) A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat Commun 9:2036. 10.1038/s41467-018-04376-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpi VG, Touvier T, D'Antonio M (2016) Endoplasmic reticulum protein quality control failure in myelin disorders. Front Mol Neurosci 9:162. 10.3389/fnmol.2016.00162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334:1081–1086. 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- Wang M, Kaufman RJ (2016) Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529:326–335. 10.1038/nature17041 [DOI] [PubMed] [Google Scholar]
- Way SW, Popko B (2016) Harnessing the integrated stress response for the treatment of multiple sclerosis. Lancet Neurol 15:434–443. 10.1016/S1474-4422(15)00381-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way SW, Podojil JR, Clayton BL, Zaremba A, Collins TL, Kunjamma RB, Robinson AP, Brugarolas P, Miller RH, Miller SD, Popko B (2015) Pharmaceutical integrated stress response enhancement protects oligodendrocytes and provides a potential multiple sclerosis therapeutic. Nat Commun 6:6532. 10.1038/ncomms7532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Stone S, Lin W (2018) Role of nuclear factor κB in multiple sclerosis and experimental autoimmune encephalomyelitis. Neural Regen Res 13:1507–1515. 10.4103/1673-5374.237109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Stanojlovic M, Lin Y, Karsenty G, Lin W (2019) Oligodendrocyte-specific ATF4 inactivation does not influence the development of EAE. J Neuroinflammation 16:23. 10.1186/s12974-019-1415-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR (2002) The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol 22:3864–3874. 10.1128/mcb.22.11.3864-3874.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]