

Abstract
Avoidance memory reactivation at recall triggers theta-gamma hippocampal phase amplitude coupling (hPAC) only when it elicits hippocampus-dependent reconsolidation. However, it is not known whether there is a causal relationship between these phenomena. We found that in adult male Wistar rats, silencing the medial septum during recall did not affect avoidance memory expression or maintenance but abolished hPAC and the amnesia caused by the intrahippocampal administration of reconsolidation blockers, both of which were restored by concomitant theta burst stimulation of the fimbria–fornix pathway. Remarkably, artificial hPAC generated by fimbria–fornix stimulation during recall of a learned avoidance response naturally resistant to hippocampus-dependent reconsolidation made it susceptible to reactivation-dependent amnesia. Our results indicate that hPAC mediates the destabilization required for avoidance memory reconsolidation and suggest that the generation of artificial hPAC at recall overcomes the boundary conditions of this process.
SIGNIFICANCE STATEMENT Theta-gamma hippocampal phase-amplitude coupling (hPAC) increases during the induction of hippocampus-dependent avoidance memory reconsolidation. However, whether hPAC plays a causal role in this process remains unknown. Using behavioral, electrophysiological, optogenetic, and biochemical tools in adult male Wistar rats, we demonstrate that reactivation-induced hPAC is necessary for avoidance memory destabilization, and that artificial induction of this patterned activity during recall of reconsolidation-resistant aversive memories renders them liable to the amnesic effect of reconsolidation inhibitors.
Keywords: C/EBPβ, deep brain stimulation, fear, forgetting, PTSD, retrieval
Introduction
Memories are initially susceptible to disruption, but, as time passes, they consolidate and become resistant to interference (McGaugh, 1966; Dudai, 2004). Nevertheless, reactivation during recall can destabilize consolidated memories, which must then be restabilized through a gene expression and protein synthesis-dependent reconsolidation process to persist (Spear, 1973; Przybyslawski and Sara, 1997; Nader et al., 2000; Haubrich and Nader, 2018).
Post-traumatic stress disorder (PTSD) is a highly prevalent mental illness triggered by witnessing or suffering distressing events (Bryant, 2019). PTSD patients show exacerbated avoidance when they face situations reminding them of the trauma and are usually treated with anxiolytic or antidepressant medication in combination with psychotherapy (Pratchett et al., 2011; Polak et al., 2012; Friedman and Bernardy, 2017), although the relapse and recurrence rates are high (Batelaan et al., 2017). Because reconsolidation may mediate the incorporation of new information into unstable reactivated memories (Nader and Hardt, 2009; Nader and Einarsson, 2010; Haubrich and Nader, 2018), clinical interventions that are able to destabilize avoidance memories and enable their reconsolidation could help PTSD patients to resignify aversive recollections and recontextualize traumatic experiences (Schwabe et al., 2014; Dunbar and Taylor, 2017; Elsey and Kindt, 2017; Waits and Hoge, 2018). However, reactivation at recall does not necessarily destabilize memory (Cammarota et al., 2004; Sevenster et al., 2012; Bos et al., 2014; Merlo et al., 2014; Thome et al., 2016), but there are conditions that constrain this process and limit the occurrence of reconsolidation (Nader and Hardt, 2009).
Oscillatory activity is a fundamental property of brain function (Buzsáki, 2006). In particular, the hippocampus shows prominent oscillations in the theta and gamma frequency bands (Buzsáki, 2002; Colgin et al., 2009). Theta and gamma oscillations interact with each other (Lisman and Jensen, 2013). In fact, gamma amplitude is modulated by the phase of theta (Jensen and Colgin, 2007), and it is theorized that theta-gamma hippocampal phase-amplitude coupling (hPAC) coordinates neuronal activity at the timescale required for memory processing in the hippocampus (Skaggs et al., 1996; Lisman et al., 2005), although the actual role of this interaction remains unclear (Lisman and Buzsáki, 2008). In this respect, hPAC strength predicts correct choice probability over the course of associative learning (Tort et al., 2009) and increases when animals evaluate choice-relevant information (Amemiya and Redish, 2018), which seems to be essential for the destabilization of some memory types at the moment of recall, including avoidance memory (Radiske et al., 2017; Yang et al., 2019). Interestingly, we recently showed that hPAC strength is associated with hippocampus-dependent avoidance memory reconsolidation in rats (Radiske et al., 2017). Here, we determined that there is a causal relationship between hPAC induction and avoidance memory destabilization, and demonstrated that the generation of artificial hPAC during retrieval can make reconsolidation-resistant avoidance memories susceptible to reactivation-targeted amnesic manipulations.
Materials and Methods
Subjects.
We used a total of 785 3-month-old naive male Wistar rats weighing 300–350 g, including those used for pilot experiments. They were maintained in a vivarium on a 12 h light/dark cycle (lights on at 6:00 A.M.) at 23°C and housed in groups of five per cage with free access to food and water. Experiments were performed during the light phase of the cycle and conducted by researchers blinded to the behavioral condition and treatment of animals. Procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the local institutional ethics committee (Comissão de Ética no Uso de Animais-CEUA).
Surgical procedures.
Anesthetized rats (ketamine 80 mg/kg; xylazine 10 mg/kg) were implanted with 22 gauge stainless steel guide cannulas aimed at the CA1 region of the dorsal hippocampus [anteroposterior (AP), −4.2 mm; laterolateral (LL), ±3.0 mm; dorsoventral (DV), −3.0 mm] and/or the medial septum (MS; AP, −0.2 mm; LL, −1.1 mm; DV, −6.0 mm; 10° angle insertion). For experiments involving electrical stimulation of the fimbria-fornix (FFx) or the corpus callosum, bipolar electrodes were lowered to AP, −1.3 mm; LL, +1.4 mm; DV, −4.0 mm; and AP, −1.3 mm; LL, +1.4 mm; and DV, −3.0 mm coordinates, respectively. For optogenetically silencing the MS, we delivered the adeno-associated viral vector AAV-CAG-ArchT-GFP (UNC Vector Core; 2 × 1011 particles/ml) using a Hamilton syringe coupled to an infusion pump (3 × 0.5 μl at a rate of 0.2 μl/min) aimed at three consecutive depths in the MS (AP, −0.2 mm; LL, −1.1 mm; DV,−5.5/−6.0/−6.5 mm; 10° angle insertion). Two weeks later, optical fibers (200 μm in diameter) were implanted in the MS. To record hippocampal local field potential (LFP) signals, animals were implanted with 16-channel electrode arrays [50 μm tungsten wires coated with paraformaldehyde (PFA) arranged in a 2 × 8 250-μm-spaced configuration] aimed to dorsal CA1 (AP, −3.6 mm; LL, +2.4 mm; DV, −3.6 mm). Two screws were twisted into the parietal bone to establish ground connection. Stereotaxic coordinates were chosen based on previous reports (Shirvalkar et al., 2010; Radiske et al., 2017). After surgery, animals received meloxicam (0.2 mg/kg, s.c.). Rats implanted with electrode arrays were housed individually. Experiments began no fewer than 10 d after surgery.
Drugs and infusion procedures.
Drug doses were based on previous studies and pilot experiments. CCAAT/Enhancer Binding Protein β (C/EBPβ) antisense (ASO;59-CCAGCAGGCGGTGCATGAAC-3′; 2 nmol/µl) and C/EBPβ scrambled antisense oligonucleotides (sASO; 59-TCGGAGACTAAGCGCGGCAC-3′; 2 nmol/µl) were phosphorothioated on the three terminal bases to avoid nuclease degradation. Oligonucleotides, anisomycin (ANI; 160 µg/µl), and muscimol (MUS; 0.2 µg/µl) were dissolved on arrival, aliquoted, and stored at −20°C until use. Stock aliquots were thawed and diluted to working concentration in sterile saline, pH ∼7.2, on the day of the experiment. For intracerebral drug delivery, infusion cannulas were fitted into the guides, and infusions were conducted using a Hamilton syringe coupled to an infusion pump (1 μl/side at a rate of 0.5 μl/min and 0.5 μl at a rate of 0.25 μl/min for intradorsal CA1 and intra-MS infusions, respectively). Infusion cannulas were left in place for one additional minute to minimize backflow.
Habituation.
Before the experiments involving optogenetic/electrical stimulation and/or electrophysiological recordings, animals were habituated to mounting the optic fiber, the stimulator connector and/or the headstage without anesthesia, and to move freely in the recording cage with the cables and/or optical fibers attached to the corresponding implants.
Step-down inhibitory avoidance task.
The training apparatus consisted of a Plexiglas box (50 × 25 × 25 cm) with a wood platform (5 × 8 × 25 cm) on the left end of a series of bronze bars that made up the floor of the box. Before training, rats were handled for 5 min/d during 5 d (HAN animals) or put on the step-down inhibitory avoidance (SDIA) training box platform and allowed to freely explore the SDIA training box for 5 min/d during 5 d (PEX animals). One day after the end of these procedures, the animals were placed on the platform facing the left rear corner of the apparatus, and, when they stepped down and placed their four paws on the grid, received a 2-s-long 0.8 mA scrambled footshock [training (TR) session]. Immediately after training, the animals were returned to their home cages and 24 h later placed again on the training box platform for 40 s to reactivate SDIA memory [reactivation (RA) session]. During these 40 s, the animals explored the platform, avoiding stepping down from it. We did not observe any significant behavioral difference between HAN and PEX animals during the reactivation session. Avoidance memory retention was evaluated 1 d after training or reactivation by placing the animals on the training box platform and measuring their latency to step down (test session, TEST). The test session finished when the rats stepped down to the grid (footshock omitted) or after 500 s. Nontreated HAN and PEX animals behaved similarly during the test session. They stayed on the platform in a minimal movement state showing avoidance behavior. Animals were trained and tested only once.
Optogenetic stimulation.
For optogenetic experiments, rats were injected with AAV-CAG-ArchT-GFP and implanted with an optic fiber in the MS, as described above. Optic fibers were coupled to blue light-emitting (470 nm) or yellow light-emitting (565 nm) LEDs (ThorLabs). Light delivery was driven using a DC4104 LED driver (ThorLabs). The MS was continuously illuminated during the reactivation session.
Electrical stimulation.
FFx stimulation was performed using bipolar electrodes (stainless steel PFA-coated wire; 50 μm diameter) with tips separated vertically 0.5 mm. A voltage/current stimulator (STG4004, Multichannel Systems) was used to generate stimuli. For theta stimulation, single pulses were delivered at 7.7 Hz. For high-frequency stimulation (HFS), single pulses were delivered at 100 Hz. For theta-burst stimulation (TBS), four single-pulse trains at 500 Hz were delivered at 7.7 trains/s (7.7 Hz). Stimulation protocols used 10–50 µA biphasic pulses (pulse width, 500 µs). Stimulation intensity was determined in pilot experiments; 10–50 µA was the smallest current range able to induce hPAC with modulation index (MI) values similar to those observed in PEX animals during the reactivation session.
In vivo electrophysiology.
Electrophysiological signals were acquired using the Cerebus Neural Signal Processor system (Blackrock Microsystems). Signals were amplified, filtered at cutoff frequencies of 0.3 and 250 Hz, and sampled at 1 kHz. Data were analyzed offline in MATLAB (RRID:SCR_001622) using built-in and custom-written routines (Signal Processing Toolbox). Anatomical localization of the electrodes was identified from LFP features as well as from histologic confirmation of the electrode tracks, as previously described (Brankack et al., 1993; Bragin et al., 1995). Baseline field potentials were acquired in a familiar recording cage 1 h before memory reactivation. We used the first 40 s of stable signals in the recording cage during which the animal was awake and in a minimal movement state with mean velocity <1 cm/s, similar to the behavior observed during the memory reactivation session. Electrical stimulation using squared pulses can contaminate LFP signals with broadband noise. To minimize this interference, we wrote an algorithm of peak detection using the Signal Processing Toolbox. This algorithm detected artifacts by identifying the local maxima around stimulation times. Since the longest continuous period of stimulation was 8 ms (corresponding to four pulses at 500 Hz), we excluded from further analysis data in a window of ±10 ms around the artifacts detected. Power spectra were computed using the Welch periodogram method (4 s Hamming windows, 75% overlap). To construct spectrograms, the power spectrum was calculated using sliding windows of 4 s and 1 s steps. To avoid spurious comparisons of power values resulting from differences in the absolute raw signal magnitudes, we normalized each power spectrum by the power of the frequency band under 250 Hz computed from the spectrum obtained before memory reactivation. Theta, slow gamma (Sgamma) and fast gamma (Fgamma) band power were defined as the average power in the frequency range of 5–10, 35–55, and 65–100 Hz, respectively. To determine the effect of intra-MS MUS infusion on spontaneous hippocampal oscillatory activity, power variation was calculated as postinfusion band power/preinfusion band power * 100, considering 10-min-long intervals. For theta-gamma phase-amplitude cross-frequency coupling analysis, LFP signals were filtered at theta, Sgamma, and Fgamma frequency bands. Theta phase and gamma amplitude were extracted using the Hilbert transform. Theta phase was binned into 18 intervals of 20°. The mean amplitude of gamma bands was computed for each theta phase bin and normalized by the sum of amplitude values over all bins. The MI was computed using an adaptation of the Kullback–Leibler distance between the uniform distribution and the probability function derived from the mean amplitude per phase distribution, as described in the study by Tort et al. (2009). Comodulograms were obtained by expressing the MI of several frequency band pairs (4 Hz bandwidths, 1 Hz steps for phase frequencies; 10 Hz bandwidths, 2 Hz steps for amplitude frequencies) in a bidimensional pseudocolor plot (Tort et al., 2009). Mean MIs were obtained by averaging the corresponding MI values in the (5–10 Hz) × (35–55 Hz) or (5–10 Hz) × (65–100 Hz) regions of the comodulograms. To calculate MI, we used LFP recordings from the entire 40-s-long reactivation session. Events of Sgamma and Fgamma amplitude were defined as time intervals when gamma power surpassed by 2.5 SD their respective time-averaged power. These events were identified, and then the theta phase associated with each event was determined, as in the study by Colgin et al. (2009). To avoid the analysis of artifactual events, time intervals with power above 6 SDs in the computations were discarded. Events separated by <100 ms were merged and considered as a single event. Theta phase at the time points corresponding to the maximum of each gamma event was extracted, and the circular mean was computed, obtaining a single-phase value associated with the occurrence of high amplitude. The time course for the distribution of Sgamma and Fgamma events over theta phase was obtained by averaging values in 5-s-long windows. To equalize theta and gamma power between HAN and PEX animals, we computed theta, Sgamma, and Fgamma power in consecutive windows based on their power spectrums (Welch periodogram method, consecutive windows of 1 s in 0.5 s steps), excluded windows with power outside boundaries defined for each group (below the 75th percentile for PEX animals and above that percentile for HAN animals), and used the remaining windows to compute MI values. Digital video cameras fixed above the SDIA apparatus were used for tracking the position of the animals. Video data were acquired at 30 frames/s and analyzed using TopScan system (RRID:SCR_017141). During RA, all animals stayed on the platform in a minimal movement state, thus excluding the possibility that speed-dependent variations in hippocampal LFP activity account for our results.
Immunoblotting.
Tissue punches from dorsal CA1 were homogenized in ice-chilled homogenization buffer (10 mm Tris-HCl, pH 7.4, containing 0.32 m sucrose, 1 mm EDTA, 1 mm EGTA, 1 mm PMSF, 10 μg/ml aprotinin, 15 μg/ml leupeptin, 10 μg/ml bacitracin, 10 μg/ml pepstatin, 50 mm NaF, and 1 mm sodium orthovanadate). Protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Equal amounts of total protein (15 μg/lane) were resolved on SDS/PAGE (10% gels) under reducing conditions and electrotransferred onto PVDF membranes. Blots were blocked for 2 h in TTBS (100 mm Tris-HCl, 0.9% NaCl, 0.1% Tween 20, pH 7.6), incubated overnight at 4°C with anti-C/EBPβ (1:20,000; RRID:AB_626772) or anti-Zif268 (1:5000; RRID:AB_2231020) antibodies, washed with TTBS, and incubated with HRP-coupled anti-IgG secondary antibody (1:200,000; RRID:AB_641171 or RRID:AB_641180, respectively). ECL Prime Western Blotting Detection Reagent (GE Healthcare) was used to detect immunoreactivity. For loading control, blots were stripped, incubated overnight at 4°C with anti-GAPDH (1:20,000; RRID:AB_307275), and then incubated with HRP-coupled anti-IgG secondary antibody. Immunoreactivity was detected as described above. Blots were acquired and quantified using GE Healthcare Imager 600 and ImageQuant TL 8.1 analysis software.
Immunofluorescence.
Animals were deeply anesthetized with thiopental and transcardially perfused with 4% PFA, at pH ∼7.2. Brains were removed, placed in PFA at 4°C overnight, transferred to 30% sucrose, and allowed to settle for 48 h. Brains were cut in 50 µm coronal sections in a cryostat. Free-floating slices from the rostral-caudal extent of the dorsal hippocampus and the medial septum were rinsed with PBS, incubated in PBST (0.2% Triton X-100) for 1 h, and blocked for 2 h at room temperature. Slices were incubated with anti-GFP (1:1000; RRID:AB_300798) overnight at 4°C and then washed in PBST and incubated with Alexa Fluor 488 anti-chicken (1:1000; RRID:AB_2534096) for 2 h at room temperature. Slices were counterstained with DAPI (1:1000; RRID:AB_2629482), mounted with Fluoromount-G, and stored at −20°C until image acquisition. Images were acquired using a Leica DM5000 fluorescence microscope and a CoolSNAP HQ2 CCD Camera in 16 bit grayscale. For visualization, pseudocolor was applied (linear look-up table covered full images, Image-Pro Plus 7.0 software).
Histology.
To verify cannula placement, we infused 4% methylene blue into the MS (0.5 µl) or dorsal CA1 (1 µl) after the end of the behavioral tests. Rats were killed 30 min thereafter, and the dye spread was taken as an indication of the diffusion of the drug previously injected. Data from animals with incorrect cannula implants (4%) were excluded from statistical analysis. Rats with cannula and electrode implants were deeply anesthetized and transcardially perfused with 4% PFA. Brains were removed and left in 30% sucrose for 48 h, and coronal sections were cut with a cryostat (50 μm). Relevant sections were selected and stained with Cresyl violet to confirm implant location.
Quantification and statistical analysis.
Statistical analyses wereperformed using GraphPad Prism 8 software (RRID:SCR_002798). Significance was set at p < 0.05. The sample size for each experiment is indicated in the figures (dots). No statistical methods were used to determine sample sizes; the number of subjects per group was based on previous reports and the experience for each experiment to yield high power to detect specific effects. Subjects were randomly assigned to experimental groups. Training step-down latencies were expressed as mean ± SEM and analyzed using unpaired Student's t test (Table 1). Because of the 500 s ceiling imposed, test step-down latencies were expressed as the median ± interquartile range (IQR), analyzed by two-tailed Mann–Whitney U test, and Spearman's rank correlation used to determine the strength and direction of the putative monotonic association between the mnemonic effect of postreactivation manipulations and hippocampal oscillatory activity during recall. Immunoblots and electrophysiological data were expressed as the mean ± SEM and analyzed using a one-sample t test with a theoretical mean of 100, paired t test, unpaired Student'st test, one-way repeated-measures (RM) ANOVA, or two-way ANOVA followed by Bonferroni's multiple-comparisons test, Rayleigh test, or Watson-Williams multisample test, as appropriate.
Table 1.
Step-down latency during SDIA training
| Figure | Behavioral condition | Treatment | Latency (s) (per group) | n | Latency (s) (per condition) | p Value (HAN vs PEX) |
|---|---|---|---|---|---|---|
| 1B | HAN | VEH 5′ | 11.75 ± 2.11 | 8 | 13.34 ± 0.93 | <0.0001 |
| ANI 5′ | 15.00 ± 2.77 | 8 | ||||
| sASO 5′ | 16.88 ± 2.68 | 8 | ||||
| ASO 5′ | 12.88 ± 2.66 | 8 | ||||
| VEH 6 h | 12.25 ± 2.62 | 8 | ||||
| ANI 6 h | 14.00 ± 3.47 | 8 | ||||
| sASO 6 h | 11.88 ± 2.07 | 8 | ||||
| ASO 6 h | 12.13 ± 3.03 | 8 | ||||
| PEX | VEH 5′ | 4.88 ± 0.85 | 9 | 5.68 ± 0.50 | ||
| ANI 5′ | 5.00 ± 0.57 | 9 | ||||
| sASO 5′ | 7.44 ± 2.39 | 9 | ||||
| ASO 5′ | 5.88 ± 1.41 | 9 | ||||
| VEH 6 h | 5.55 ± 1.58 | 9 | ||||
| ANI 6 h | 4.66 ± 1.29 | 9 | ||||
| sASO 6 h | 7.00 ± 1.70 | 9 | ||||
| ASO 6 h | 5.00 ± 1.25 | 9 | ||||
| 1D–I | HAN | 12.67 ± 1.58 | 6 | 0.0132 | ||
| PEX | 5.33 ± 1.85 | 6 | ||||
| 1J | PEX | VEH | 6.22 ± 1.02 | 9 | ||
| ANI | 6.11 ± 1.30 | 9 | ||||
| 2A | HAN | VEH | 13.25 ± 1.27 | 8 | 12.81 ± 0.91 | <0.0001 |
| MUS | 12.38 ± 1.37 | 8 | ||||
| PEX | VEH | 5.12 ± 0.85 | 8 | 5.37 ± 0.52 | ||
| MUS | 5.62 ± 0.65 | 8 | ||||
| 2B | HAN | VEH + VEH | 11.00 ± 1.43 | 8 | 14.98 ± 0.89 | <0.0001 |
| VEH + ANI | 15.63 ± 2.71 | 8 | ||||
| MUS + VEH | 15.13 ± 2.72 | 8 | ||||
| MUS + ANI | 17.13 ± 2.81 | 8 | ||||
| VEH + sASO | 12.63 ± 2.41 | 8 | ||||
| VEH + ASO | 16.25 ± 2.44 | 8 | ||||
| MUS + sASO | 15.38 ± 2.51 | 8 | ||||
| MUS + ASO | 16.75 ± 3.29 | 8 | ||||
| PEX | VEH + VEH | 6.00 ± 1.64 | 8 | 6.08 ± 0.49 | ||
| VEH + ANI | 5.12 ± 0.63 | 8 | ||||
| MUS + VEH | 5.11 ± 0.73 | 9 | ||||
| MUS + ANI | 6.22 ± 1.09 | 9 | ||||
| VEH + sASO | 7.87 ± 2.67 | 9 | ||||
| VEH + ASO | 6.12 ± 1.58 | 8 | ||||
| MUS + sASO | 6.11 ± 1.36 | 9 | ||||
| MUS + ASO | 7.33 ± 1.40 | 9 | ||||
| 2D,E | HAN | VEH | 13.40 ± 2.65 | 5 | 14.40 ± 1.78 | <0.0001 |
| MUS | 15.40 ± 2.58 | 5 | ||||
| PEX | VEH | 3.80 ± 0.66 | 5 | 4.80 ± 0.71 | ||
| MUS | 5.80 ± 1.58 | 5 | ||||
| 3 | PEX | Light OFF/VEH | 6.77 ± 1.913 | 9 | ||
| Light OFF/ANI | 6.00 ± 0.897 | 9 | ||||
| Blue ON/VEH | 5.77 ± 1.362 | 9 | ||||
| Blue ON/ANI | 5.66 ± 0.866 | 9 | ||||
| Yellow ON/VEH | 5.88 ± 1.814 | 9 | ||||
| Yellow ON/ANI | 5.50 ± 1.327 | 10 | ||||
| 4C,D | PEX | No Stim./VEH | 6.80 ± 1.04 | 10 | ||
| No Stim./ANI | 6.90 ± 1.56 | 10 | ||||
| Theta Stim./VEH | 6.50 ± 1.15 | 8 | ||||
| Theta Stim./ANI | 7.25 ± 1.26 | 8 | ||||
| TBS RA/VEH | 6.70 ± 1.484 | 10 | ||||
| TBS RA/ANI | 4.90 ± 0.604 | 10 | ||||
| TBS RA(LFP)/VEH | 6.11 ± 1.160 | 9 | ||||
| TBS RA(LFP)/ANI | 6.00 ± 1.384 | 9 | ||||
| TBS after RA/VEH | 6.60 ± 1.50 | 10 | ||||
| TBS After RA/ANI | 6.30 ± 1.10 | 10 | ||||
| TBS No RA/VEH | 6.66 ± 0.95 | 9 | ||||
| TBS No RA/ANI | 5.88 ± 1.20 | 9 | ||||
| TBS 100 corpus callosum/VEH | 6.16 ± 1.10 | 6 | ||||
| TBS 100 corpus callosum/ANI | 5.66 ± 1.33 | 6 | ||||
| 4E | PEX | No Stim./sASO | 4.77 ± 0.70 | 9 | ||
| No Stim./ASO | 6.33 ± 0.95 | 9 | ||||
| TBS RA/sASO | 5.55 ± 1.40 | 9 | ||||
| TBS RA/ASO | 4.77 ± 0.87 | 9 | ||||
| 5A | HAN | No Stim./VEH | 10.88 ± 1.65 | 8 | ||
| No Stim./ANI | 12.38 ± 1.61 | 8 | ||||
| TBS RA/VEH | 15.67 ± 2.02 | 9 | ||||
| TBS RA/ANI | 13.89 ± 2.70 | 9 | ||||
| TBS RA(LFP)/VEH | 12.11 ± 1.95 | 9 | ||||
| TBS RA(LFP)/ANI | 15.67 ± 2.37 | 9 | ||||
| TBS after RA/VEH | 9.87 ± 1.99 | 8 | ||||
| TBS after RA/ANI | 14.50 ± 1.91 | 8 | ||||
| TBS no RA/VEH | 11.50 ± 2.00 | 8 | ||||
| TBS no RA/ANI | 12.25 ± 1.57 | 8 | ||||
| 5B | HAN | No Stim./sASO | 12.71 ± 1.61 | 7 | ||
| No Stim./ASO | 11.71 ± 2.56 | 7 | ||||
| TBS RA/sASO | 15.00 ± 1.79 | 7 | ||||
| TBS RA/ASO | 11.71 ± 2.48 | 7 |
Step-down latency during SDIA training. Repeated pre-exposure to the SDIA training box decreased step-down latency at training, indicating learning of SDIA-related nonaversive information during pre-expositions, as reported in Radiske et al. (2017). Latencies did not differ between groups for the same behavioral condition. Data are presented as the mean ± SEM and were analyzed using an unpaired t test or one-way ANOVA.
Results
Hippocampus-dependent avoidance memory reconsolidation requires prior learning of related nonaversive information and is accompanied by increased theta-gamma phase-amplitude coupling.
To analyze whether hPAC and avoidance memory reconsolidation are causally related, we trained adult male Wistar rats(3 months old; weight, 300–350 g) in a one-trial, SDIA learning paradigm, which generates a long-lasting fear-motivated avoidance memory (Bernabeu et al., 1995; Cammarota et al., 2000; Bekinschtein et al., 2007). SDIA memory reconsolidation engages the hippocampus only when animals acquired the avoidance response in an environment they previously considered safe (Radiske et al., 2017). Therefore, to differentiate reconsolidation-specific mechanisms from those related merely to memory recall, rats were handled (HAN group) or allowed to explore the SDIA training box freely during 5 min once daily for 5 d (PEX group). This last procedure induces learning of nonaversive SDIA-related information without affecting SDIA memory acquisition, strength, or persistence, and renders SDIA memory susceptible to hippocampus-dependent reconsolidation on nonreinforced reactivation (Radiske et al., 2017). Twenty-four hours later, the animals were trained in SDIA (0.8 mA/2 s footshock) and 1 d thereafter submitted to a 40-s-long nonreinforced memory RA session able to induce reconsolidation but not extinction of the learned avoidance response (Cammarota et al., 2004; Radiske et al., 2017). Protein synthesis dependency is a reconsolidation hallmark, and C/EBPβ is part of the molecular signature of this process (Milekic et al., 2007). In fact, it has been suggested that aversive memories resistant to postretrieval hippocampal protein synthesis inhibition are also immune to hippocampal C/EBPβ signaling disruption (Taubenfeld et al., 2001). In agreement with this suggestion, we found that intradorsal CA1 infusion of C/EBPβ-ASO at a dose that reduced basal C/EBPβ levels ∼30% (Fig. 1A; C/EBPβ: t(4) = 8.499, p = 0.0011 in one-sample t test; theoretical mean, 100) or of the protein synthesis inhibitor ANI, 5 min but not 6 h after RA, triggered amnesia in PEX but not in HAN animals [Fig. 1B; PEX 5′: U = 7.500, p = 0.0017 for vehicle (VEH) vs ANI; U = 12.50, p = 0.0093 for scrambled ASO (sASO) vs ASO; Mann–Whitney test]. Accordingly, memory reactivation increased dorsal CA1 C/EBPβ levels only in PEX animals (Fig. 1C; F(1,8) = 17.73, p = 0.0030 for interaction; F(1,8) = 3.795, p = 0.0872 for pre-exposition effect; F(1,8) = 21.46, p = 0.0017 for reactivation effect in two-way ANOVA; p < 0.001 for PEX RA vs PEX NR in Bonferroni's multiple-comparisons test). Confirming and extending previous findings, analysis of dorsal CA1 LFP during RA showed that Sgamma (35–55 Hz) power augmented in HAN and PEX animals, but theta and Fgamma (65–100 Hz) power increased only in PEX rats (Fig. 1D; HAN Sgamma power: t(5) = 2.838, p = 0.0363; PEX theta power: t(5) = 2.709, p = 0.0423; PEX Sgamma power: t(5) = 2.628, p = 0.0466; PEX Fgamma power: t(5) = 3.735, p = 0.0135 for before vs during RA in paired t test). hPAC was stronger in PEX than in HAN animals and independent of theta fluctuation (Fig. 1E,F; theta-Sgamma MI: t(10) = 4.340, p = 0.0015; theta-Fgamma MI: t(10) = 4.342, p = 0.0015; for HAN vs PEX in unpaired t test). The number of gamma events, defined as periods when the power of the gamma frequency sub-bands exceeded 2.5 SDs, did not differ between HAN and PEX animals during RA (Fig. 1G), but Sgamma and Fgamma events occurred preferentially around the peak of the theta cycle throughout the reactivation session only in PEX animals (Fig. 1H; Sgamma events: Z = 26.11, p < 0.0001; Fgamma events: Z = 27.62, p < 0.0001; Rayleigh test). The difference in coupling strength between PEX and HAN animals persisted even after equalizing theta, Sgamma, or Fgamma power (Fig. 1I; equalized theta power: theta-Sgamma MI: t(10) = 6.085, p = 0.0001; theta- Fgamma MI: t(10) = 3.231, p = 0.0090; equalized Sgamma power: theta-Sgamma MI: t(10) = 8.945, p < 0.0001; theta-Fgamma MI: t(10) = 2.969, p = 0.0141; equalized Fgamma power: theta-Sgamma MI: t(10) = 4.874, p = 0.0006; theta-Fgamma MI: t(10) = 2.631, p = 0.0251; HAN vs PEX in unpaired t test), indicating that it did not result from a power increase or improved phase detection, and suggesting that hPAC, but not theta or gamma activity per se, is associated with memory reconsolidation. In agreement with this assertion, theta-Sgamma MI and theta-Fgamma MI, but not theta, Sgamma, or Fgamma power, correlated negatively with memory retention in PEX animals that received ANI in dorsal CA1 5 min after RA (Fig. 1J).
Figure 1.
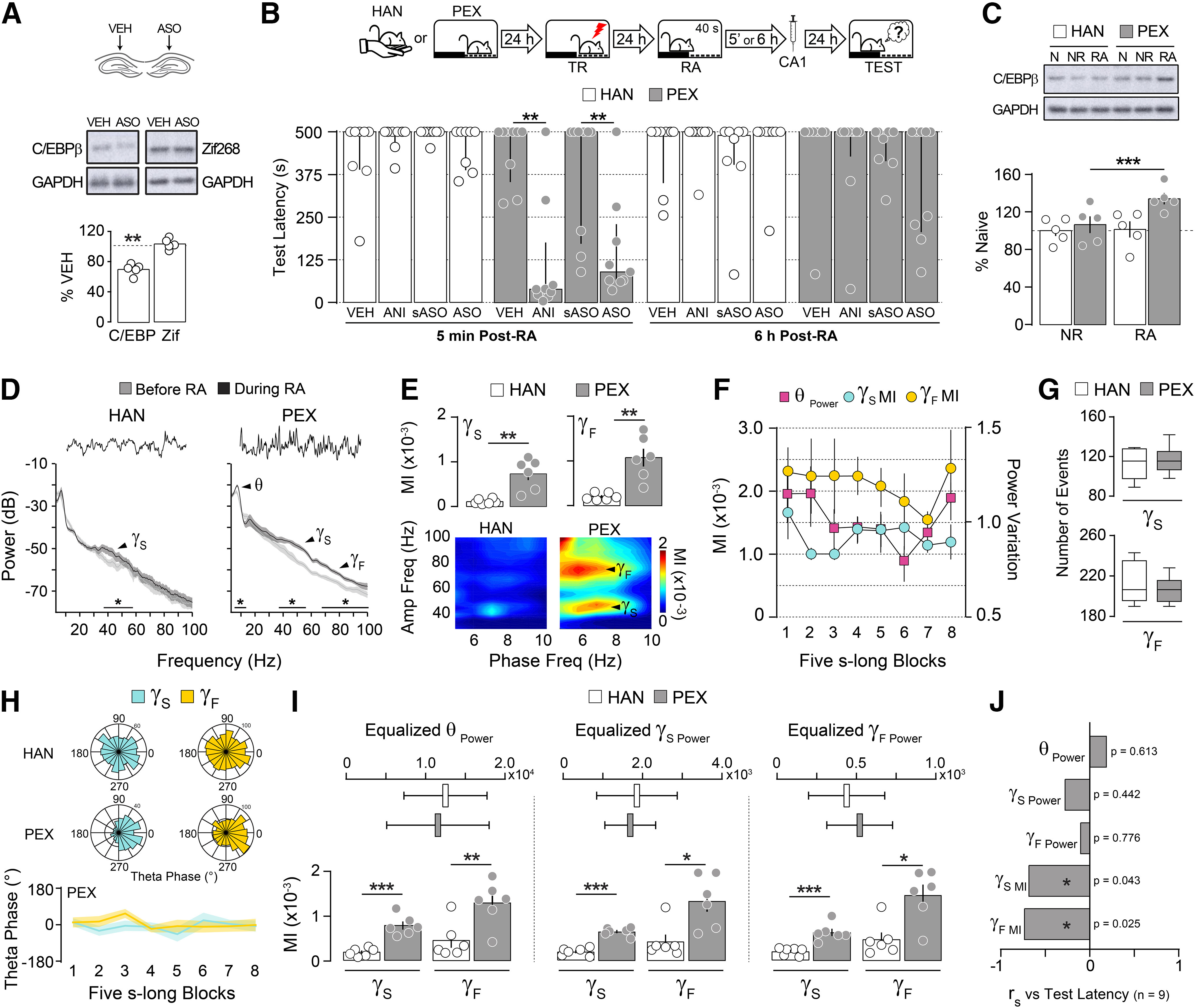
Prior nonaversive learning is a boundary condition for hippocampus-dependent avoidance memory reconsolidation. A, Naive rats received VEH (0.9% saline) in the dorsal CA1 region of one hemisphere and C/EBPβ-ASO (2 nmol/µl) in the other. Ninety minutes later, the animals were decapitated, and the dorsal CA1 region was dissected out and homogenized to determine C/EBPβ, Zif268, and GAPDH protein levels by immunoblotting (n = 5/group). B, Rats were handled (HAN animals) or allowed to freely explore the SDIA training box (PEX animals) during 5 min once daily for 5 consecutive days. Twenty-four hours after the last handling or pre-exposition session, the animals were trained in SDIA (0.8 mA/2 s), and 1 d later submitted to a 40-s-long nonreinforced memory RA session. Five minutes or 6 h after RA, the animals received bilateral intradorsal CA1 infusions of VEH, ANI (160 µg/side), C/EBPβ-ASO (2 nmol/µl), or sASO (2 nmol/µl). Retention was evaluated 1 d later (TEST; n = 8–9/group). C, HAN and PEX animals were trained as in B, but 24 h after training they were handled during 40 s (NR) or submitted to RA, killed by decapitation 90 min thereafter, and the dorsal CA1 region was dissected out and homogenized to determine C/EBPβ and GAPDH protein levels by immunoblotting (n = 5/group). D, Representative raw hippocampal LFP traces and power spectrum density plots for HAN and PEX animals for theta (θ; 5–10 Hz), Sgamma (γS; 35–55 Hz), and Fgamma (γF; 65–100 Hz) frequency bands showing reactivation-induced alterations in hippocampal oscillatory activity (n = 6/group). E, Mean theta-Sgamma and theta-Fgamma MI and representative phase-amplitude comodulograms for HAN and PEX animals during RA. F, MI and theta power computed in eight nonoverlapping blocks throughout RA for PEX animals. G, Mean number of Sgamma and Fgamma events for HAN and PEX animals during RA. H, Gamma events distribution over theta phase for HAN and PEX animals during RA. 0° = theta peak. I, MIs calculated using epochs with equalized theta, Sgamma, or Fgamma power (expressed as μV2/Hz) for HAN and PEX animals during RA. J, SDIA-trained PEX animals with cannulas and electrode arrays implanted in the CA1 region of the dorsal hippocampus were submitted to RA, during which LFP signals were recorded. Five minutes after RA, the animals received VEH or ANI in dorsal CA1. Retention was evaluated 1 d later (median = 500, IQR = 379–500 for VEH; median = 78, IQR = 25–235 for ANI; U = 4.00, p = 0.0004 for VEH vs ANI in Mann–Whitney test; n = 9/group). Bar plots represent Spearman's r correlation coefficient between test latency and normalized theta power (θ Power; mean = 0.373, SEM = 0.026), Sgamma power (γS Power; mean = 0.087, SEM = 0.007), Fgamma power (γF Power; mean = 0.029, SEM = 0.003), theta-Sgamma MI (γS MI; mean = 0.479 × 10−3, SEM = 0.008 × 10−3) or theta-Fgamma MI (γF MI; mean = 0.728 × 10−3, SEM = 0.156 × 10−3) during RA for animals that received ANI. Data are expressed as the median ± IQR or mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Medial septum inactivation impedes hippocampal theta-gamma phase-amplitude coupling and the amnesia caused by postretrieval hippocampal protein synthesis inhibition and C/EBPβ knockdown
MS inactivation reduces hippocampal theta rhythm, and it has been repeatedly used to analyze the involvement of hPAC in memory processing and behavior (Winson, 1978; Shirvalkar, et al., 2010; Brandon et al., 2014). In our preparation, intra-MS infusion of the GABAA agonist MUS (0.2 µg/side) diminished theta oscillations in dorsal CA1 for at least 3 h without affecting gamma activity (Fig. 2A, top; theta power variation: F(1.929,5.789) = 12.47, p = 0.0082 in RM one-way ANOVA; *p < 0.05, **p < 0.01 in Bonferroni's multiple-comparisons test) or SDIA memory retrieval (Fig. 2A, bottom). By itself, intra-MS MUS infusion before memory reactivation had no effect on SDIA memory retention, regardless of pretraining behavioral manipulations, but canceled the amnesia triggered by the postreactivation intra-CA1 administration of ANI and C/EBPβ-ASO (Fig. 2B; PEX: U = 8.000, p = 0.0079 for VEH+ VEH vs VEH+ANI; U = 12.00, p = 0.0275 for VEH+sASO vs VEH+ ASO in Mann–Whitney test) and impeded the reactivation-induced increase in C/EBPβ levels (Fig. 2C; t(8) = 3.589, p = 0.0071 for VEH vs MUS in unpaired t test). In PEX animals, MS inactivation decreased theta power, but not slow gamma and fast gamma power (Fig. 2D; theta power: HAN/MUS: t(4) = 3.9530, p = 0.0168; PEX/VEH: t(4) = 3.277, p = 0.0306; PEX/MUS: t(4) = 9.836, p = 0.0006; Sgamma power: HAN/VEH: t(4) = 3.721, p = 0.0205; HAN/MUS: t(4) = 6.153, p = 0.0035; PEX/VEH: t(4) = 3.586, p = 0.0230; PEX/MUS: t(4) = 3.317, p = 0.0295; Fgamma power: PEX/VEH: t(4) = 4.662, p = 0.0096; PEX/MUS: t(4) = 2.850, p = 0.0464 for before RA vs during RA in paired t test) and diminished the strong nested theta-gamma activity induced by SDIA memory reactivation (Fig. 2E; theta- Sgamma MI: F(1,16) = 12.09, p = 0.0031 for interaction; F(1,16) = 17.09, p = 0.0008 for pre-exposition effect; F(1,16) = 33.87, p < 0.0001 for treatment effect; theta- Fgamma MI: F(1,16) = 10.37, p = 0.0053 for interaction; F(1,16) = 19.56, p = 0.0004 for pre-exposition effect; F(1,16) = 16.22, p = 0.0010 for treatment effect in two-way ANOVA; p < 0.001 for HAN/VEH vs PEX/VEH, HAN/MUS vs PEX/VEH, or PEX/VEH vs PEX/MUS in Bonferroni's multiple-comparisons test).
Figure 2.
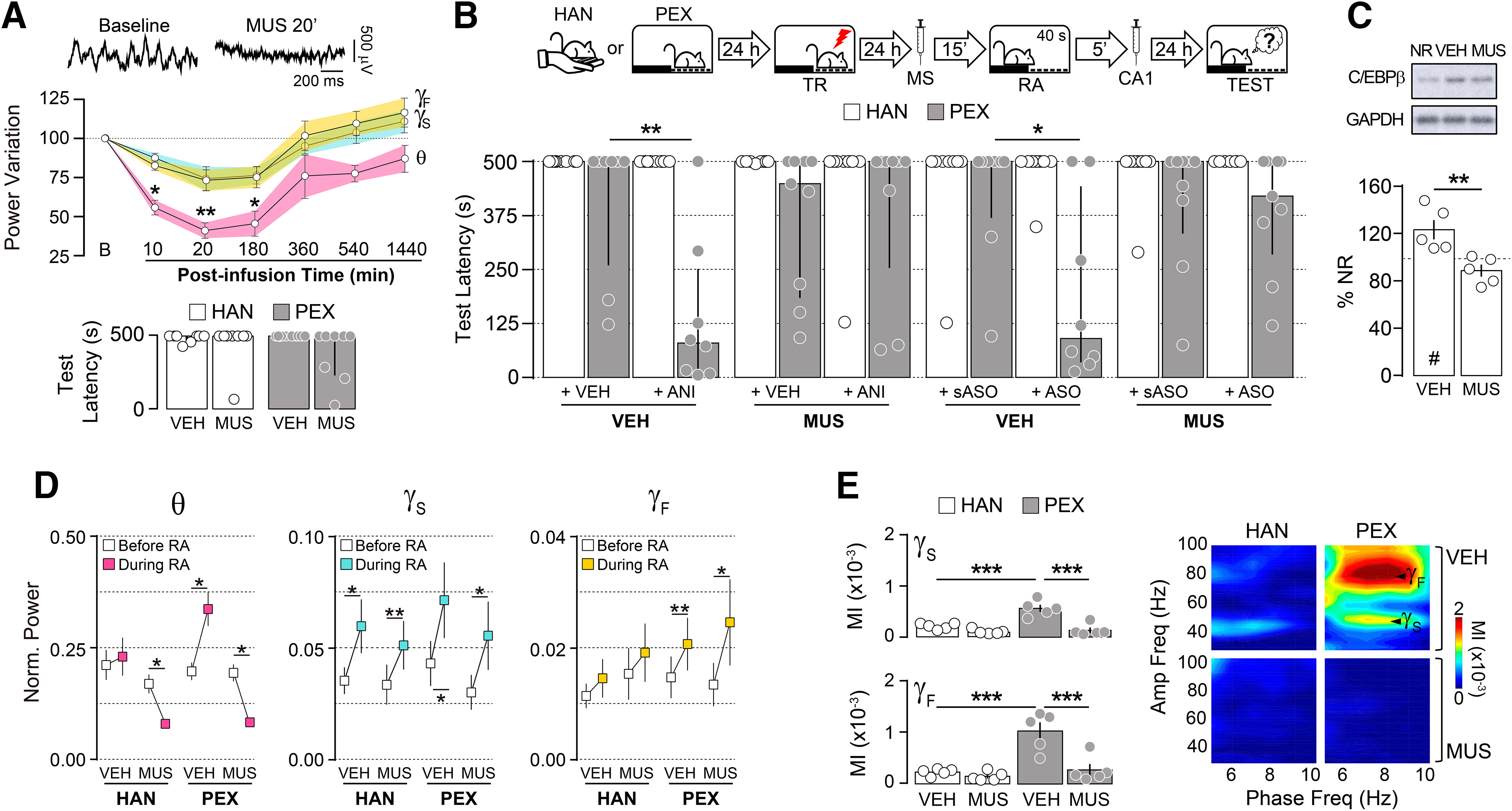
Medial septum inactivation during avoidance memory reactivation impedes hippocampal theta-gamma phase-amplitude coupling and memory destabilization. A, Top, Representative raw hippocampal LFP traces at baseline and 20 min postinfusion from animals given MUS (0.2 µg/µl) into the MS. Middle, Theta (θ; 5–10 Hz), Sgamma (γS; 35–55 Hz), and Fgamma (γF; 65–100 Hz) power variation from baseline (B) at different time points after intra-MS MUS infusion (n = 4). Bottom, Rats were handled (HAN animals) or allowed to freely explore the SDIA training box (PEX animals) during 5 min once daily for 5 consecutive days. Twenty-four hours after the last handling or pre-exposition session, animals were trained in SDIA(0.8 mA/2 s). One day later, they received injections of VEH (0.9% saline) or MUS into MS, and 15 min thereafter were submitted to a retention test session (n = 8/group). B, One day after training, HAN and PEX animals received VEH or MUS into MS, and 15 min thereafter were submitted to a 40-s-long nonreinforced memory RA session. Five minutes after RA, the animals received bilateral intradorsal CA1 infusions of VEH, ANI (160 µg/side), C/EBPβ-ASO (2 nmol/µl), or sASO (2 nmol/µl). Retention was evaluated 1 d later (TEST; n = 8–9/group). C, PEX animals were treated as in B, but 90 min after RA were killed by decapitation, and the dorsal CA1 region dissected out and homogenized to determine C/EBPβ and GAPDH protein levels by immunoblotting (n = 5/group). D, Normalized theta (θ), Sgamma (γS), and Fgamma (γF) power before and during RA for HAN and PEX animals that received intra-MS VEH or MUS 15 min before RA (n = 5/group). E, Mean theta-Sgamma and theta-Fgamma MI and representative phase-amplitude comodulograms for HAN and PEX animals during RA. Data are expressed as the median ± IQR or mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, #p < 0.05 in one-sample Student's t test with a theoretical mean = 100.
Optogenetic silencing of the medial septum during recall impedes avoidance memory destabilization
Our results corroborate and expand previous findings suggesting that increased hippocampal theta-gamma phase-amplitude coupling is functionally linked to memory reconsolidation and support the hypothesis that this interaction is necessary for SDIA memory destabilization during recall. However, in our preparation, the inhibitory effect of intra-MS MUS administration on hippocampal oscillatory activity extended well beyond the end of the reactivation session, making it difficult to link reactivation-induced hippocampal rhythms and memory destabilization irrefutably. To overcome this drawback and limit MS inactivation to the duration of the reactivation session, we virally expressed yellow light-sensing archaerhodopsin T (ArchT) in the MS of PEX animals to optogenetically silence this brain region. One day after the last pre-exposition session, PEX animals were trained in SDIA and 1 d later submitted to a memory reactivation session. During that session, one group of animals was used as nonstimulated control, while the other two groups were optically stimulated in the MS with blue light (470 nm) or yellow light (565 nm). Because ArchT responds to green-yellow light but not to blue light, stimulation at 470 nm was used as an additional control. Five minutes after memory reactivation, animals received bilateral intra-CA1 infusions of VEH or ANI, and retention was tested 1 d later. As expected, ANI caused SDIA amnesia in nonstimulated PEX animals as well as in PEX animals stimulated with blue light in the MS. Yellow light stimulation had no effect on SDIA memory retention, but hindered the amnesic effect of ANI and reduced hPAC (Fig. 3A; Light OFF: U = 10.00, p = 0.0046; Blue ON: U = 13.00, p = 0.0093 for VEH vs ANI in Mann–Whitney test), as well as theta power in the hippocampus (Fig. 3B; F(1.658,8.291) = 21.30, p = 0.0007 in RM one-way ANOVA; *p < 0.05, **p < 0.01 in Bonferroni's multiple-comparisons test). MS stimulation with yellow light did not alter hippocampal gamma oscillations (Fig. 3B).
Figure 3.

Medial septum optogenetic silencing impedes memory destabilization. A, Top, Rats expressing archaerhodopsin T in the MS were allowed to freely explore the SDIA training box during 5 min once daily for 5 consecutive days (PEX animals) and 24 h after the last pre-exposition session were trained in SDIA (TR; 0.8 mA/2 s). One day post-TR, animals were submitted to a 40-s-long nonreinforced memory RA session during which the MS was not stimulated (Light OFF) or optogenetically stimulated with blue light (470 nm; Blue ON) or yellow light (565 nm; Yellow ON). 5 min after RA, rats received bilateral injections of VEH (0.9% saline) or ANI (160 µg/side) in dorsal CA1. Retention was evaluated 1 d later (TEST; n = 9–10/group). Bottom, Representative phase-amplitude comodulograms during RA for each experimental group. B, Left top, Representative images showing archaerhodopsin T expression in MS and dorsal hippocampus reported by GFP. Right top, Representative raw hippocampal LFP trace and spectrogram plot showing the effect of MS yellow light stimulation on theta power. Bottom, Bars show mean hippocampal theta (θ; 5–10 Hz), Sgamma (γS; 35–55 Hz), and Fgamma (γF; 65–100 Hz) power before, during, and after light delivery to the MS (n = 6/group). Data are expressed as the median ± IQR or mean ± SEM. *p < 0.05, **p < 0.01.
Induction of artificial hippocampal theta-gamma phase-amplitude coupling during recall restores the amnesic effect of reconsolidation blockers in medial septum-inactivated animals
Our analyses demonstrate that the difference in hPAC strength between HAN and PEX animals does not result from increased oscillatory power or enhanced phase identification, but reflects an active memory process instead. However, the steep decline in theta activity produced by MS inactivation prevented us from decisively determining the existence of a causal connection between hPAC and memory destabilization. To get around this problem, we took advantage of the fact that the MS projects to the hippocampus through the FFx (Manseau et al., 2008; Müller and Remy, 2018), and it is known that electrical stimulation of this pathway can restore hippocampal theta rhythm and hPAC in MS-lesioned animals (Yoder and Pang, 2005; McNaughton et al., 2006) without affecting gamma activity (Shirvalkar et al., 2010). We found that FFx theta stimulation and FFx TBS, but not FFx high-frequency stimulation (100 Hz), bypassed MUS-induced MS inactivation and promoted theta activity in dorsal CA1 (Fig. 4A). However, only FFx TBS nested hippocampal gamma activity within theta envelopes (Fig. 4B; theta-Sgamma MI: t(4) = 4.443, p = 0.0113; theta-Fgamma MI: t(4) = 5.384, p = 0.0058 for before vs during FFx TBS in paired t test) and restored the amnesia provoked by the postreactivation intra-CA1 administration of ANI in PEX animals. This effect was seen only when FFx TBS was applied during RA, but not 5 min thereafter or in its absence, suggesting that it was contingent on memory reactivation. TBS delivered in the corpus callosum, 1 mm above the FFx, did not re-establish the capacity of ANI to induce amnesia [Fig. 4C,D; RA: U = 9.00, p = 0.0009; RA(LFP): U = 10.00, p = 0.0046 for VEH vs ANI in Mann–Whitney test; theta- Sgamma MI: t(8) = 3.670, p = 0.0063; theta-Fgamma MI: t(8) = 4.419, p = 0.0022 for before RA vs during RA in paired t test]. Notably, theta-Sgamma MI and theta-Fgamma MI, but not theta, Sgamma, or Fgamma power, correlated with the amnesic effect of ANI (Fig. 4D). We also found that FFx TBS renewed the amnesic effect of post-RA intra-CA1 C/EBPβ-ASO infusions in PEX animals that received intra-MS MUS 15 min before memory reactivation (Fig. 4E; TBS: U = 7.00 and p = 0.0012 for sASO vs ASO in Mann–Whitney test).
Figure 4.
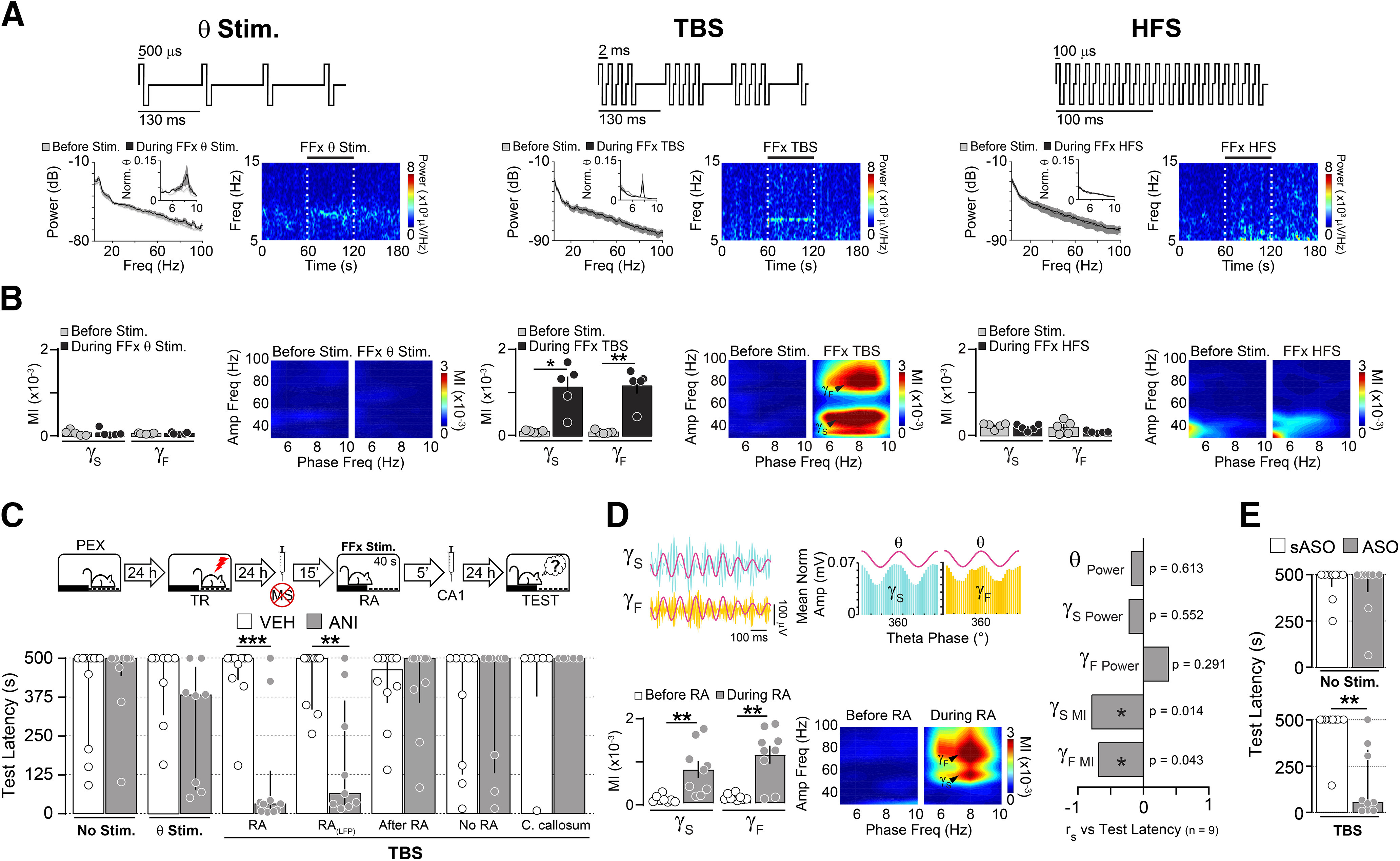
Theta-burst stimulation of the fimbria-fornix induces artificial hippocampal theta-gamma phase-amplitude coupling and restores the amnesic effect of reconsolidation blockers in MS-inactivated animals. A, Top panels, Schematic representation of FFx stimulation protocols: theta stimulation (θ Stim.), TBS, and HFS. Bottom panels, Animals received injections of muscimol (MUS; 0.2 µg/side) into the MS, and 15 min later the FFx pathway was stimulated using θ, TBS, or HFS protocols. Power spectrum density plots and spectrograms show the effect of FFx stimulation on dorsal CA1 LFPs before and during FFx stimulation. Dashed lines in spectrograms indicate stimulation intervals. B, Mean theta-Sgamma (γS) and theta-Fgamma (γF) MI and representative phase-amplitude comodulograms before and during FFx stimulation (n = 5/group). C, PEX rats were trained in the SDIA task (TR; 0.8 mA/2 s) 24 h after the last pre-exposition session and 1 d later received MUS in the MS. Fifteen minutes after MUS infusion, the animals were submitted to a 40-s-long nonreinforced memory RA session during which they received no stimulation (No Stim.), theta stimulation in the FFx (θ Stim.), or TBS in the FFx (RA). A group of PEX rats with electrode arrays implanted in dorsal CA1 [RA(LFP)] was treated and stimulated as animals in the RA group, except that LFPs were recorded throughout the reactivation session. Additional control groups received TBS either in the FFx during 40 s beginning immediately after memory reactivation (After RA), in the FFx during 40 s 24 h after training in the absence of RA (No RA), or in the corpus callosum during RA (C. callosum). Five minutes after these manipulations, rats received bilateral injections of VEH (0.9% saline) or ANI (160 µg/side) into dorsal CA1. Retention was evaluated 1 d later (TEST; n = 6–10/group). D, Left top, Filtered LFPs and normalized Sgamma and Fgamma amplitude distribution over theta. Left bottom, Mean theta-Sgamma and theta-Fgamma MI and representative phase-amplitude comodulograms before and during RA. Right, Spearman's r correlation coefficients between test latency (median = 65, IQR = 27–364) and normalized theta power (θ Power; mean = 0.147, SEM = 0.024), Sgamma power (γS Power; mean = 0.072, SEM = 0.016), Fgamma power (γF Power; mean = 0.019, SEM = 0.004), theta-Sgamma MI (γS MI; mean = 0.792 × 10−3, SEM = 0.194 × 10−3), or theta-Fgamma MI (γF MI; mean = 1.164 × 10−3, SEM = 0.216 × 10−3) during the reactivation session for the RA(LFP) animals that received ANI shown in C. E, PEX rats were trained and treated as in C, except that they received no stimulation (No Stim.) or FFx TBS during RA (TBS) and 5 min later were given C/EBPβ-ASO (2 nmol/µl) or sASO (2 nmol/µl) in dorsal CA1 (n = 9/group). Data are expressed as the median ± IQR or mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Induction of artificial theta-gamma phase-amplitude coupling during recall makes reconsolidation-resistant memory vulnerable to reactivation-dependent amnesia
We next investigated the effect of FFx TBS in rats resistant to hippocampus-dependent reconsolidation. We reasoned that if hPAC were indeed necessary for memory destabilization, then perhaps increasing hPAC artificially during recall could help to destabilize SDIA memory in HAN animals, making it vulnerable to postreactivation amnesic manipulations. To test this hypothesis, HAN animals were trained in SDIA and 1 d later submitted to a reactivation session during which they received TBS in FFx to induce artificial hPAC. Five minutes after memory reactivation, rats received bilateral intradorsal CA1 injections of VEH or ANI, and retention was evaluated 1 d later. As expected, unstimulated HAN animals given VEH or ANI retained the learned avoidance response. However, memory became susceptible to ANI in animals that received FFx TBS during reactivation. FFx TBS did not make memory vulnerable to ANI when delivered immediately after the reactivation session or in its absence [Fig. 5A; RA: U = 11.00, p = 0.0041; RA(LFP): U = 15.00, p = 0.0167 for VEH vs ANI in Mann–Whitney test]. The amnesic potency of ANI correlated with the strength of artificially induced hPAC, but not with theta, Sgamma, or Fgamma power at reactivation (Fig. 5A). FFx TBS also enabled RA-dependent SDIA amnesia in HAN animals that received intradorsal CA1 C/EBPβ-ASO, but not C/EBPβ-sASO, 5 min after RA (Fig. 5B; TBS, U = 4.000, p = 0.0047 for sASO vs ASO in Mann–Whitney test).
Figure 5.
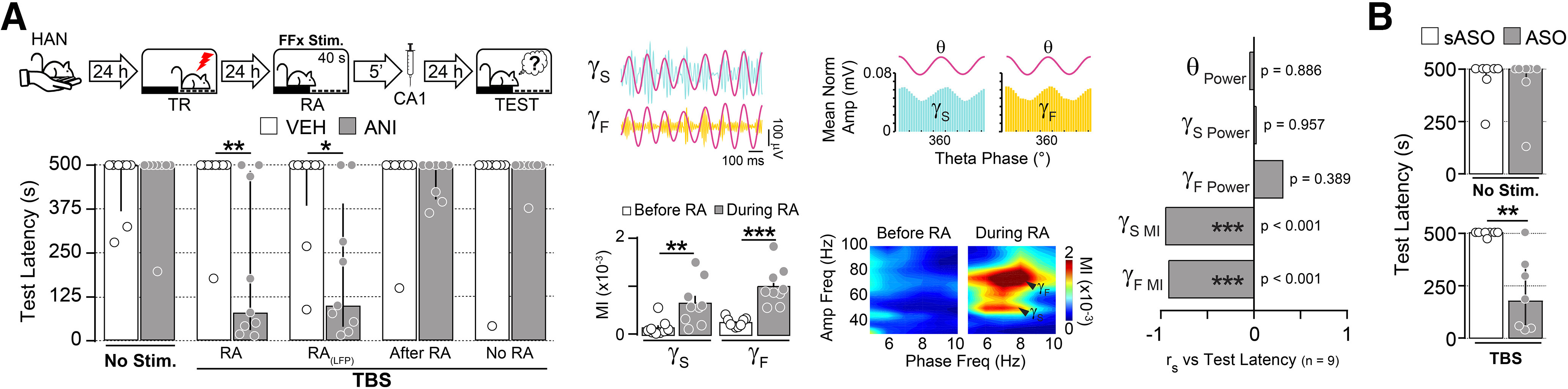
Induction of artificial hippocampal theta-gamma phase-amplitude coupling during RA overcomes the boundary conditions of avoidance reconsolidation and destabilizes reconsolidation-resistant memories. A, Left, HAN animals were trained in the SDIA task (TR; 0.8 mA/2 s) 24 h after the last handling session and 1 d later submitted to a 40-s-long nonreinforced memory reactivation session during which they received no stimulation (No Stim.) or FFx TBS (RA). A group of HAN rats with electrode arrays implanted in dorsal CA1 was treated and stimulated as animals in the RA group, except that LFPs were recorded throughout the reactivation session [RA(LFP)]. Additional control groups received TBS in the FFx during 40 s beginning immediately after memory reactivation (After RA) or TBS in the FFx during 40 s 24 h after TR in the absence of RA (No RA). Immediately after these manipulations, rats were given bilateral intra-CA1 injections of VEH (0.9% saline) or ANI (160 µg/side), and retention was evaluated 1 d later (TEST; n = 8–9/group). Middle top, Filtered LFPs and normalized Sgamma and Fgamma amplitude distribution over theta. Middle bottom, Mean theta-Sgamma and theta-Fgamma MI and representative phase-amplitude comodulograms before and during RA (theta-Sgamma MI: t(8) = 3.748, p = 0.0056; theta-Fgamma MI: t(8) = 5.548, p = 0.0005 for Before RA vs During RA in paired t test). Right, Spearman's r correlation coefficients test latency (median = 101, IQR = 40–391) and normalized theta power (θ Power; mean = 0.342, SEM = 0.056), Sgamma power (γS Power; mean = 0.060, SEM = 0.011), Fgamma power (γF Power; mean = 0.023, SEM = 0.005), theta-Sgamma MI (γS MI; mean = 0.670 × 10−3, SEM = 0.154 × 10−3), or theta-Fgamma MI (γF MI; mean = 1.019 × 10−3, SEM = 0.132 × 10−3) during the reactivation session for the RA(LFP) animals that received ANI, shown in the left panel. B, HAN rats were trained and treated as in A, except that they received no stimulation (No Stim.) or FFx TBS during RA (TBS) and 5 min later were given C/EBPβ-ASO (2 nmol/µl) or sASO (2 nmol/µl) in dorsal CA1 (n = 7/group). Data are expressed as the median ± IQR or mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Our data confirm that hPAC increases during avoidance memory reactivation only when this process induces hippocampus-dependent reconsolidation (Radiske et al., 2017) and show that this theta–gamma interaction, but not theta or gamma activity per se, is causally linked to destabilization of the avoidance response. Importantly, our results also demonstrate that artificial hPAC generation during recall can destabilize memories that are normally resistant to reconsolidation, making them liable to reactivation-targeted amnesic manipulations.
SDIA memory reactivation enables hippocampus-dependent reconsolidation only when it results in conflicting representations about the possible consequences of avoidance. Then, it is tempting to speculate that hPAC reflects the switch between aversive and nonaversive states that probably underlies the decision-making process that PEX animals undergo during the reactivation session. This hypothesis is in agreement with findings showing that hPAC increases for both error and correct trials during associative learning (Tort et al., 2009) and is correlated with state-dependent information processing during decision-making (Amemiya and Redish, 2018).
In view of the fact that network activity modulation is intertwined with long-term plasticity, and the onset of reconsolidation is linked to changes in synaptic weight driven by reversible adjustments in AMPAR function (Hong et al., 2013), the increased hPAC observed in PEX animals may reflect modifications in hippocampal synaptic efficacy. In fact, the pattern of oscillatory activity in the hippocampus of PEX rats during memory reactivation resembles that necessary for the induction of long-term potentiation (LTP) in dorsal CA1 (Thomas et al., 1998). Potentiated synapses may return to a protein synthesis-dependent state following LTP reactivation, and, when multiple inputs converge, the active synapses must compete for plasticity factors to stay potentiated (Fonseca et al., 2004, 2006). We propose that a similar mechanism is triggered when antagonistic representations clash over the control of behavior at recall, with the dominant trace becoming destabilized, as previously suggested (Eisenberg et al., 2003). Since FFx-TBS may induce LTP in CA1 (Li et al., 2005), such a mechanism could explain why artificial hPAC circumvented the inhibitory effect of MS inactivation on memory destabilization and made animals naturally resistant to reconsolidation become sensitive to postreactivation amnesia only when generated concomitantly with avoidance memory recall.
Targeting memory reconsolidation is a promising therapeutic approach to treat PTSD that offers the opportunity to erase intrusive recollections in a single session (Nader et al., 2013). However, recall does not always induce destabilization of the reactivated trace, which limits its translational value. Our findings have important implications for the implementation of reconsolidation-based therapies in clinical practice. They not only show that hPAC strength can be used as an estimator of memory destabilization, which could help optimize memory reactivation protocols and adjust pharmacological interventions to suit the needs of individual patients, but also indicate that it is feasible to overcome the constraints on reconsolidation through brain stimulation protocols similar to those used to alleviate motor and cognitive dysfunctions (Laxton et al., 2010; Lyons, 2011; Hao et al., 2015). This could be useful to treat patients refractory to conventional therapies or when the source of the trauma is unknown. However, it is important to note that the nature of conflicting signals at recall can differ among avoidance memory types. Therefore, our results should not be generalized to conclude that hPAC is involved in the destabilization of all avoidance memories. For example, while hippocampus-dependent SDIA reconsolidation is linked to reactivation of contradictory representations regarding the possible consequences of avoidance (Radiske et al., 2017), the induction of step-through avoidance (STA) memory reconsolidation involves competition between innate and learned responses, and requires protein synthesis and C/EBPβ expression in the amygdala instead of the hippocampus (Taubenfeld et al., 2001; Milekic et al., 2007). Given that similar molecular mechanisms underlie hippocampus-dependent and amygdala-dependent avoidance reconsolidation, and switches between fear and safety states engage distinct theta–gamma coupling patterns in the basolateral amygdala (Stujenske et al., 2014), it would be interesting to evaluate whether reactivation-induced theta–gamma interactions in the amygdala are associated with STA memory destabilization.
Footnotes
This study was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil)-Finance Code 001. We thank Jorge H. Medina for helpful discussion.
The authors declare no competing financial interests.
References
- Amemiya S, Redish AD (2018) Hippocampal theta-gamma coupling reflects state-dependent information processing in decision making. Cell Rep 25:3894–3897. 10.1016/j.celrep.2018.12.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batelaan NM, Bosman RC, Muntingh A, Scholten WD, Huijbregts KM, van Balkom A (2017) Risk of relapse after antidepressant discontinuation in anxiety disorders, obsessive-compulsive disorder, and post-traumatic stress disorder: systematic review and meta-analysis of relapse prevention trials. BMJ 358:j3927. 10.1136/bmj.j3927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernabeu R, Izquierdo I, Cammarota M, Jerusalinsky D, Medina JH (1995) Learning-specific, time-dependent increase in [3 H] phorbol dibutyrate binding to protein kinase C in selected regions of the rat brain. Brain Res 685:163–168. 10.1016/0006-8993(95)00310-m [DOI] [PubMed] [Google Scholar]
- Bekinschtein P, Cammarota M, Igaz LM, Bevilaqua LR, Izquierdo I, Medina JH (2007) Persistence of long-term memory storage requires a late protein synthesis- and BDNF- dependent phase in the hippocampus. Neuron 53:261–277. 10.1016/j.neuron.2006.11.025 [DOI] [PubMed] [Google Scholar]
- Bos MG, Beckers T, Kindt M (2014) Noradrenergic blockade of memory reconsolidation: a failure to reduce conditioned fear responding. Front Behav Neurosci 8:412. 10.3389/fnbeh.2014.00412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragin A, Jandó G, Nádasdy Z, Hetke J, Wise K, Buzsáki G (1995) Gamma (40–100 Hz) oscillation in the hippocampus of the behaving rat. J Neurosci 15:47–60. 10.1523/JNEUROSCI.15-01-00047.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon MP, Koenig J, Leutgeb JK, Leutgeb S (2014) New and distinct hippocampal place codes are generated in a new environment during septal inactivation. Neuron 82:789–796. 10.1016/j.neuron.2014.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brankack J, Stewart M, Fox SE (1993) Current source density analysis of the hippocampal theta rhythm: associated sustained potentials and candidate synaptic generators. Brain Res 615:310–327. 10.1016/0006-8993(93)90043-m [DOI] [PubMed] [Google Scholar]
- Bryant RA. (2019) Post-traumatic stress disorder: a state-of-the-art review of evidence and challenges. World Psychiatry 18:259–269. 10.1002/wps.20656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G. (2002) Theta oscillations in the hippocampus. Neuron 33:325–340. 10.1016/s0896-6273(02)00586-x [DOI] [PubMed] [Google Scholar]
- Buzsáki G. (2006) Rhythms of the brain. Oxford, UK: Oxford UP. [Google Scholar]
- Cammarota M, Bevilaqua LR, Ardenghi P, Paratcha G, Levi de Stein M, Izquierdo I, Medina JH (2000) Learning-associated activation of nuclear MAPK, CREB and Elk-1, along with Fos production, in the rat hippocampus after a one-trial avoidance learning: abolition by NMDA receptor blockade. Brain Res Mol Brain Res 76:36–46. 10.1016/s0169-328x(99)00329-0 [DOI] [PubMed] [Google Scholar]
- Cammarota M, Bevilaqua LR, Medina JH, Izquierdo I (2004) Retrieval does not induce reconsolidation of inhibitory avoidance memory. Learn Mem 11:572–578. 10.1101/lm.76804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgin LL, Denninger T, Fyhn M, Hafting T, Bonnevie T, Jensen O, Moser MB, Moser EI (2009) Frequency of gamma oscillations routes flow of information in the hippocampus. Nature 462:353–357. 10.1038/nature08573 [DOI] [PubMed] [Google Scholar]
- Dudai Y. (2004) The neurobiology of consolidations, or, how stable is the engram? Annu Rev Psychol 55:51–86. 10.1146/annurev.psych.55.090902.142050 [DOI] [PubMed] [Google Scholar]
- Dunbar AB, Taylor JR (2017) Reconsolidation and psychopathology: moving towards reconsolidation-based treatments. Neurobiol Learn Mem 142:162–171. 10.1016/j.nlm.2016.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg M, Kobilo T, Berman DE, Dudai Y (2003) Stability of retrieved memory: inverse correlation with trace dominance. Science 301:1102–1104. 10.1126/science.1086881 [DOI] [PubMed] [Google Scholar]
- Elsey JWB, Kindt M (2017) Tackling maladaptive memories through reconsolidation: from neural to clinical science. Neurobiol Learn Mem 142:108–117. 10.1016/j.nlm.2017.03.007 [DOI] [PubMed] [Google Scholar]
- Fonseca R, Nägerl UV, Morris RG, Bonhoeffer T (2004) Competing for memory: hippocampal LTP under regimes of reduced protein synthesis. Neuron 44:1011–1020. 10.1016/S0896-6273(04)00713-5 [DOI] [PubMed] [Google Scholar]
- Fonseca R, Nägerl UV, Bonhoeffer T (2006) Neuronal activity determines the protein synthesis dependence of long-term potentiation. Nat Neurosci 9:478–480. 10.1038/nn1667 [DOI] [PubMed] [Google Scholar]
- Friedman MJ, Bernardy NC (2017) Considering future pharmacotherapy for PTSD. Neurosci Lett 649:181–185. 10.1016/j.neulet.2016.11.048 [DOI] [PubMed] [Google Scholar]
- Hao S, Tang B, Wu Z, Ure K, Sun Y, Tao H, Gao Y, Patel AJ, Curry DJ, Samaco RC, Zoghbi HY, Tang J (2015) Forniceal deep brain stimulation rescues hippocampal memory in Rett syndrome mice. Nature 526:430–434. 10.1038/nature15694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubrich J, Nader K (2018) Memory reconsolidation. Curr Top Behav Neurosci 37:151–176. 10.1007/7854_2016_463 [DOI] [PubMed] [Google Scholar]
- Hong I, Kim J, Kim J, Lee S, Ko HG, Nader K, Kaang BK, Tsien RW, Choi S (2013) AMPA receptor exchange underlies transient memory destabilization on retrieval. Proc Natl Acad Sci U S A 110:8218–8223. 10.1073/pnas.1305235110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen O, Colgin LL (2007) Cross-frequency coupling between neuronal oscillations. Trends Cogn Sci 11:267–269. 10.1016/j.tics.2007.05.003 [DOI] [PubMed] [Google Scholar]
- Laxton AW, Tang-Wai DF, McAndrews MP, Zumsteg D, Wennberg R, Keren R, Wherrett J, Naglie G, Hamani C, Smith GS, Lozano AM (2010) A phase I trial of deep brain stimulation of memory circuits in Alzheimer's disease. Ann Neurol 68:521–534. 10.1002/ana.22089 [DOI] [PubMed] [Google Scholar]
- Li C, Maier DL, Cross B, Doherty JJ, Christian EP (2005) Fimbria-fornix lesions compromise the induction of long-term potentiation at the Schaffer collateral-CA1 synapse in the rat in vivo. J Neurophysiol 93:3001–3006. 10.1152/jn.00546.2004 [DOI] [PubMed] [Google Scholar]
- Lisman JE, Buzsáki G (2008) A neural coding scheme formed by the combined function of gamma and theta oscillations. Schizophr Bull 34:974–980. 10.1093/schbul/sbn060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Jensen O (2013) The θ-γ neural code. Neuron 77:1002–1016. 10.1016/j.neuron.2013.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Talamini LM, Raffone A (2005) Recall of memory sequences by interaction of the dentate and CA3: a revised model of the phase precession. Neural Netw 18:1191–1201. 10.1016/j.neunet.2005.08.008 [DOI] [PubMed] [Google Scholar]
- Lyons MK. (2011) Deep brain stimulation: current and future clinical applications. Mayo Clin Proc 86:662–672. 10.4065/mcp.2011.0045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manseau F, Goutagny R, Danik M, Williams S (2008) The hippocamposeptal pathway generates rhythmic firing of GABAergic neurons in the medial septum and diagonal bands: an investigation using a complete septohippocampal preparation in vitro. J Neurosci 28:4096–4107. 10.1523/JNEUROSCI.0247-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaugh JL. (1966) Time-dependent processes in memory storage. Science 153:1351–1358. 10.1126/science.153.3742.1351 [DOI] [PubMed] [Google Scholar]
- McNaughton N, Ruan M, Woodnorth MA (2006) Restoring theta-like rhythmicity in rats restores initial learning in the Morris water maze. Hippocampus 16:1102–1110. 10.1002/hipo.20235 [DOI] [PubMed] [Google Scholar]
- Merlo E, Milton AL, Goozée ZY, Theobald DE, Everitt BJ (2014) Reconsolidation and extinction are dissociable and mutually exclusive processes: behavioral and molecular evidence. J Neurosci 34:2422–2431. 10.1523/JNEUROSCI.4001-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milekic MH, Pollonini G, Alberini CM (2007) Temporal requirement of C/EBPbeta in the amygdala following reactivation but not acquisition of inhibitory avoidance. Learn Mem 14:504–511. 10.1101/lm.598307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller C, Remy S (2018) Septo-hippocampal interaction. Cell Tissue Res 373:565–575. 10.1007/s00441-017-2745-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader K, Einarsson EO (2010) Memory reconsolidation: an update. Ann N Y Acad Sci 1191:27–41. 10.1111/j.1749-6632.2010.05443.x [DOI] [PubMed] [Google Scholar]
- Nader K, Hardt O (2009) A single standard for memory: the case for reconsolidation. Nat Rev Neurosci 10:224–234. 10.1038/nrn2590 [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE (2000) Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406:722–726. 10.1038/35021052 [DOI] [PubMed] [Google Scholar]
- Nader K, Hardt O, Lanius R (2013) Memory as a new therapeutic target. Dialogues Clin Neurosci 15:475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak AR, Witteveen AB, Visser RS, Opmeer BC, Vulink N, Figee M, Denys D, Olff M (2012) Comparison of the effectiveness of trauma-focused cognitive behavioral therapy and paroxetine treatment in PTSD patients: design of a randomized controlled trial. BMC Psychiatry 12:166. 10.1186/1471-244X-12-166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratchett LC, Daly K, Bierer LM, Yehuda R (2011) New approaches to combining pharmacotherapy and psychotherapy for posttraumatic stress disorder. Expert Opin Pharmacother 12:2339–2354. 10.1517/14656566.2011.604030 [DOI] [PubMed] [Google Scholar]
- Przybyslawski J, Sara SJ (1997) Reconsolidation of memory after its reactivation. Behav Brain Res 84:241–246. 10.1016/s0166-4328(96)00153-2 [DOI] [PubMed] [Google Scholar]
- Radiske A, Gonzalez MC, Conde-Ocazionez SA, Feitosa A, Köhler CA, Bevilaqua LR, Cammarota M (2017) Prior learning of relevant nonaversive information is a boundary condition for avoidance memory reconsolidation in the rat hippocampus. J Neurosci 37:9675–9685. 10.1523/JNEUROSCI.1372-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe L, Nader K, Pruessner JC (2014) Reconsolidation of human memory: brain mechanisms and clinical relevance. Biol Psychiatry 76:274–280. 10.1016/j.biopsych.2014.03.008 [DOI] [PubMed] [Google Scholar]
- Sevenster D, Beckers T, Kindt M (2012) Retrieval per se is not sufficient to trigger reconsolidation of human fear memory. Neurobiol Learn Mem 97:338–345. 10.1016/j.nlm.2012.01.009 [DOI] [PubMed] [Google Scholar]
- Shirvalkar PR, Rapp PR, Shapiro ML (2010) Bidirectional changes to hippocampal theta-gamma comodulation predict memory for recent spatial episodes. Proc Natl Acad Sci U S A 107:7054–7059. 10.1073/pnas.0911184107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaggs WE, McNaughton BL, Wilson MA, Barnes CA (1996) Theta phase precession in hippocampal neuronal populations and the compression of temporal sequences. Hippocampus 6:149–173. 10.1002/(SICI)1098-1063(1996)6:2<149::AID-HIPO6>3.0.CO;2-K [DOI] [PubMed] [Google Scholar]
- Spear N. (1973) Retrieval of memory in animals. Psychol Rev 80:163–194. 10.1037/h0034326 [DOI] [Google Scholar]
- Stujenske JM, Likhtik E, Topiwala MA, Gordon JA (2014) Fear and safety engage competing patterns of theta–gamma coupling in the basolateral amygdala. Neuron 83:919–933. 10.1016/j.neuron.2014.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenfeld SM, Milekic MH, Monti B, Alberini CM (2001) The consolidation of new but not reactivated memory requires hippocampal C/EBPbeta. Nat Neurosci 4:813–818. 10.1038/90520 [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Watabe AM, Moody TD, Makhinson M, O'Dell TJ (1998) Postsynaptic complex spike bursting enables the induction of LTP by theta frequency synaptic stimulation. J Neurosci 18:7118–7126. 10.1523/JNEUROSCI.18-18-07118.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome J, Koppe G, Hauschild S, Liebke L, Schmahl C, Lis S, Bohus M (2016) Modification of fear memory by pharmacological and behavioural interventions during reconsolidation. PLoS One 11:e0161044. 10.1371/journal.pone.0161044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tort AB, Komorowski RW, Manns JR, Kopell NJ, Eichenbaum H (2009) Theta-gamma coupling increases during the learning of item-context associations. Proc Natl Acad Sci U S A 106:20942–20947. 10.1073/pnas.0911331106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waits WM, Hoge CW (2018) Reconsolidation of traumatic memories using psychotherapy. Am J Psychiatry 175:1145. 10.1176/appi.ajp.2018.18060646 [DOI] [PubMed] [Google Scholar]
- Winson J. (1978) Loss of hippocampal theta rhythm results in spatial memory deficit in the rat. Science 201:160–163. 10.1126/science.663646 [DOI] [PubMed] [Google Scholar]
- Yang Y, Jie J, Li J, Chen W, Zheng X (2019) A novel method to trigger the reconsolidation of fear memory. Behav Res Ther 122:103461. 10.1016/j.brat.2019.103461 [DOI] [PubMed] [Google Scholar]
- Yoder RM, Pang KC (2005) Involvement of GABAergic and cholinergic medial septal neurons in hippocampal theta rhythm. Hippocampus 15:381–392. 10.1002/hipo.20062 [DOI] [PubMed] [Google Scholar]