

Abstract
In murine experimental glioma models, TLR3 or TLR9 activation of microglial/macrophages has been shown to impair glioma growth, which could, however, not been verified in recent clinical trials. We therefore tested whether combined TLR3 and TLR9 activation of microglia/macrophages would have a synergistic effect. Indeed, combined TLR3/TLR9 activation augmented the suppression of glioma growth in organotypic brain slices from male mice in a microglia-dependent fashion, and this synergistic suppression depended on interferon β release and phagocytic tumor clearance. Combined TLR3/TLR9 stimulation also augmented several functional features of microglia, such as the release of proinflammatory factors, motility, and phagocytosis activity. TLR3/TLR9 stimulation combined with CD47 blockade further augmented glioma clearance. Finally, we confirmed that the coactivation of TLR3/TLR9 also augments the impairment of glioma growth in vivo. Our results show that combined activation of TLR3/TLR9 in microglia/macrophages results in a more efficient glioma suppression, which may provide a potential strategy for glioma treatment.
SIGNIFICANCE STATEMENT Glioma-associated microglia/macrophages (GAMs) are the predominant immune cells in glioma growth and are recently considered as antitumor targets. TLRs are involved in glioma growth, but the TLR3 or TLR9 ligands were not successful in clinical trials in treating glioma. We therefore combined TLR3 and TLR9 activation of GAMs, resulting in a strong synergistic effect of tumor clearance in vitro, ex vivo, and in vivo. Mechanisms of this GAM-glioma interaction involve IFNβ signaling and increased tumor clearance by GAMs. Interfering with CD47 signaling had an additional impact on tumor clearance. We propose that these signaling pathways could be exploited as anti-glioma targets.
Keywords: brain macrophage, costimulation, glioma, microglia, Toll-like receptor 3, Toll-like receptor 9
Introduction
Glioblastoma is the most common malignant tumor in the CNS (Gladson et al., 2010) with a median survival time of 12–15 months (Mellinghoff and Gilbertson, 2017). In the last years, it has become evident that the microenvironment of gliomahas an impact on tumor progression and treatment resistance (Hambardzumyan et al., 2016). Microglia and macrophages derived from the periphery infiltrate glioma tissue and make up to 30% of the tumor mass. There are various aspects in which interactions between glioma-associated microglia/macrophages (GAMs) and glioma cells play a role, including cytokine signaling and release and activation of extracellular matrix-degrading enzymes. As a consequence of these interactions, GAMs promote glioma growth (Hu et al., 2015). Moreover, glioma cells change GAM properties, resulting in a specific, protumorigenic phenotype that is distinct from the classical M1/M2 phenotype (Szulzewsky et al., 2015). Toll-like receptors (TLRs) and associated signaling pathways mediate glioma/GAM interaction. These pattern recognition receptors recognize pathogens (Trinchieri and Sher, 2007; Lehnardt, 2010) and in the CNS are prominently expressed on microglia. In human, the TLR family consists of 10 members (i.e., TLR1-TLR10). While TLR3 recognizes double-stranded RNA and subsequently initiates Type I interferon production (Barton and Medzhitov, 2003; Trinchieri and Sher, 2007), TLR9 preferentially detects single-stranded DNA and triggers signaling pathways, resulting in the release of proinflammatory cytokines (Barton and Medzhitov, 2003; Trinchieri and Sher, 2007). Poly(I:C) is structurally similar to double-stranded RNA and has been widely used as a TLR3 agonist, while oligodeoxynucleotides with unmethylated cytosine-phosphate-guanine (CpG) motifs as short single-stranded DNA represent an established TLR9 activator (Lehnardt, 2010).
TLR3 and TLR9 activation was previously described to suppress tumor progression, including glioma growth (Salaun et al., 2006; Krieg, 2007; Morikawa et al., 2007; Chiappinelli et al., 2015). Several clinical trials tested either Poly(I:C) or CpG treatment on patients with primary or recurrent glioma (Krieg, 2007; Butowski et al., 2009; Rosenfeld et al., 2010). The outcome so far was not promising. Since TLR3 and TLR9 activate distinct pathways, namely, signaling via IRF/TRIF adaptor protein and MyD88, respectively, we hypothesized that combined TLR3 and TLR9 activation leads to synergistic effects with regard to tumor growth suppression. We therefore evaluated the effects of combined treatment with Poly(I:C) and CpG on growth of different glioma cell lines, in a mouse glioma model, and on diverse microglial functions, including phagocytosis, cell motility, and cytokine release.
Material and Methods
Animals
For all experiments, we used C57BL6J mice or MacGreen mice on C57BL6J background. MacGreen mice were derived from a transgenic line expressing EGFP under the control of a promoter for macrophage colony-stimulating factor receptor (CSF-1R) (Sasmono and Williams, 2012). Mice of either sex were used for primary neonatal microglia culture, and male mice were used for organotypic brain slices and in vivo experiments. These animals were handled according to the regulations and rules of LaGeSo and Max-Delbrueck-Center. C57BL6J mice used for in vivo experiments were handled according to in vivo experiments, guidelines pertaining to animal experimentation approved by the Committee on Animal Research of Tongji Medical College of Huazhong University of Science and Technology, China.
Human material
All patients were operated at the Department of Neurosurgery, University Medical Center Schleswig-Holstein, Campus Kiel. The study was approved by the Ethics Committee of the University of Kiel (approval #D477/18) and was in accordance with the Helsinki Declaration of 1964 and its later amendments. Informed consent was obtained from all individual patients. Freshly resected tumor tissue was stored in DMEM at 4°C for < 24 h until further experimental workup.
Cell culture
Murine glioma cell line GL261 (American Type Culture Collection), human glioma cell lines U87 (ECACC), U251 (ECACC, #89181493), and LN229 (American Type Culture Collection, ATCC-CRL-2611) were cultured in DMEM with supplements (10% FCS), 50 units/ml penicillin, 50 µg/ml streptomycin, and 200 mm glutamine (all purchased from Invitrogen). THP1 cells (American Type Culture Collection) were cultured in RPMI-1640 medium with 10% FCS, 50 units/ml penicillin, 50 µg/ml streptomycin, 200 mm glutamine (Invitrogen), and 0.05 mm 2-mercaptoethanol (Invitrogen/Thermo Fisher Scientific). Before the experiment, THP1 cells were treated with PMA for 48 h. Primary microglia were prepared and harvested from neonatal C57BL6 (WT) mouse brain as previously described (Minelli et al., 2000). GL261mCherry, U87mCherry, U251mCherry, and LN229mCherry cells were generated as previously described (Vinnakota et al., 2013). For conditioned medium collection, primary microglia were seeded into a 6-well plate overnight until adherence, followed by adding 10 µg/ml Poly(I:C) (Invivogen) and 2 µM CpG (Invivogen) for 24 h. Supernatant was harvested and filtered using membranes with 0.2 µM pores (Corning).
Human GAMs isolation by magnetic activated cell sorting (MACS)
GAMs were freshly isolated by MACS as previously described (Vinnakota et al., 2013). Briefly, after washing with PBS, tumor tissue from human glioma samples was enzymatically digested into single-cell-suspension using Adult Brain Dissociation Kit (Miltenyi Biotec). Tissue was further dissociated, and debris was removed by applying a 40 µm cell strainer (Miltenyi Biotec). Next, cell suspension was incubated with CD11b microbeads in MACS buffer (PBS supplemented with 0.5% BSA and 2 mm EDTA) for 15 min. Cells were then loaded onto a MACS column (Miltenyi Biotec), after washing the column with MACS buffer. CD11b+ and CD11b– cells were eluted from the column. A fraction of the isolated cells was stained with CD11b antibody for FACS analysis to verify cell purity. Populations of CD11b+ and CD11b– cells were used for investigating gene expression changes by qRT-PCR.
Total RNA isolation and PCR
Total RNA was isolated using Promega RNA mini kit (Stratec) according to the manufacturer's instructions. Quality and yield were determined by NanoDrop 1000 (PeqLabBiotechnologie). cDNA was synthesized using 100 ng total RNA with SuperScript II reverse transcriptase kit (Invitrogen). RT-PCR gene amplification was performed in duplicate using SYBR Green PCR mix (Applied Biosystems) and 7500 Fast Real-Time PCR System (Applied Biosystems). Primer sequences generated by Biotex were listed as follows: interleukin 12 (il12: sense 5′-TGGTTTGCCATCGTTTTGCTG-3′, antisense 5′-ACAGGTGAGGTTCACTGTTTCT-3′), nitric oxide synthase 2 (nos2: sense 5′-TCACGCTTGGGTCTTGTTCA-3′, antisense 5′-TGAAGAGAAACTTCCAGGGGC-3′), interleukin 1β (il1β: sense 5′-GCAACTGTTCCTGAACTCAACT-3′, antisense 5′-ATCTTTTGGGGTCCGTCAACT-3′), Tumour Necrosis Factor alpha (tnfα: sense 5′-CCCTCACACTCAGATCATCTTCT-3′, antisense 5′-GCTACGACGTGGGCTACAG-3′), interferon β (ifnβ: sense 5′-CAGCTCCAAGAAAGGACGAAC-3′, antisense 5′- GGCAGTGTAACTCTTCTGCAT-3′), TATA-binding protein (tbp: sense 5′-AAGGGAGAATCATGGACCAG-3′, antisense 5′-CCGTAAGGCATCATTGGACT-3′), B-cell lymphoma 2 (bcl2: sense 5′-ATGCCTTTGTGGAACTATATGGC-3′, antisense 5′-GGTATGCACCCAGAGTGATGC-3′), myeloid cell leukemia 1 (mcl1: sense 5′-AAAGGCGGCTGCATAAGTC-3′, antisense 5′-TGGCGGTATAGGTCGTCCTC- 3'), Bcl-2-associated X protein (bax: sense 5′-TGAAGACAGGGGCCTTTTTG-3′, antisense 5′-AATTCGCCGGAGACACTCG-3′), Spp1 (sense 5-AGCAAGAAACTCTTCCAAGCAA-3′, antisense 5′-GTGAGATTCGTCAAGATTCATCCG-3′), Glycoprotein NMB (gpnmb: sense 5′-GCTGGTCTTCGGATGAAAATGA-3′, antisense 5′-CCACAAAGGTGATATTGGAACCC-3′), matrix metallopeptidase 9 (mmp9: sense 5′-CTGGACAGCCAGACACTAAAG-3′, antisense 5′- CTCGCGGCAAGTCTTCAGAG-3′), matrix metallopeptidase 14 (mmp14: sense 5′-CAGTATGGCTACCTACCTCCAG-3′, antisense 5′-GCCTTGCCTGTCACTTGTAAA-3′), cd86 (sense5′-TGTTTCCGTGGAGACGCAAG-3′, antisense 5′- TTGAGCCTTTGTAAATGGGCA-3′), cd206 (sense 5′-CTCTGTTCAGCTATTGGACGC-3′, antisense 5′-CGGAATTTCTGGGATTCAGCTTC-3′), bad (sense 5′-AAGTCCGATCCCGGAATCC-3′, antisense 5′-GCTCACTCGGCTCAAACTCT-3′), bim (sense5′-CCCGGAGATACGGATTGCAC-3′, antisense 5′-GCCTCGCGGTAATCATTTGC-3′), Toll like receptor 3 tlr3 (sense 5′-GTGAGATACAACGTAGCTGACTG-3′, antisense 5′-TCCTGCATCCAAGATAGCAAGT-3′), and tlr9 (sense 5′-ATGGTTCTCCGTCGAAGGACT-3′, antisense 5′-GAGGCTTCAGCTCACAGGG-3′). The results were analyzed by2-ΔΔCT ways normalized to tbp and were presented as fold change normalized to control group.
Protein extraction and Western blot
Whole-cell protein extracts were prepared from primary microglia treated with Poly(I:C) and/or CpG or GL261, U87, U251, and LN229 glioma cells using RIPA lysis buffer (Sigma Millipore) containing EDTA-free protease inhibitor cocktail tablets (Roche Diagnostics). Protein concentration was determined by a BCA protein assay kit (Thermo Fisher Scientific), and 20 µg of total protein of each sample was resolved on a 10% SDS-PAGE gel, followed by wet transfer of resolved proteins onto a PVDF membrane (GE Healthcare). Membranes were blocked and followed by overnight incubation at 4°C with rabbit anti-phosphorylated-Akt (Ser473) (CST), anti-GAPDH (CST), or anti-total-Akt antibodies (CST). For GL261, U87, U251, and LN229 glioma cell line, primary antibodies, including anti-CD47 (Thermo Fisher Scientific) and anti-GAPDH (CST), were used. Membranes were incubated with a secondary anti-rabbit HRP antibody (1:2000; Cell Signaling Technology), developed with SuperSignal West Pico Chemiluminescence substrate kit (Thermo Fisher Scientific). Signal was detected by Molecular Imager Gel Doc XR system (Bio-Rad).
Immunofluorescent staining and image processing
For murine tumor slices, mice brains were harvested and perfused with PBS followed by 4% PFA solution (Sigma Millipore); 40 µm free-floating tumor sections were prepared as previously described (Vinnakota et al., 2013). Human glioma specimens were prepared following the procedure previously described (Vinnakota et al., 2013). Slices were washed 3 times with PBS for 5 min and blocked with 5% of donkey serum and 0.1% Triton-X (Sigma Millipore). For glioma cell immunostaining, cells on coverslips treated with recombinant IFNβ were fixed with 4% PFA followed by blocking with 5% of donkey serum and 0.1% Triton-X. Primary antibodies were added overnight at 1:100 for cleaved caspase-3 (CST), 1:500 dilution for Iba-1, 1:200 for TLR3 (Abcam), or 1:100 for TLR9 (Abcam) at 4°C. TLR3 and TLR9 were detected using the secondary antibody Cy3-conjugated goat anti-rabbit IgG (1:200; Jackson ImmunoResearch Laboratories), and cleaved caspase-3 was detected using Alexa-488-conjugated donkey anti-mouse secondary antibody, while Iba-1 was detected using the secondary antibody anti-goat DyLight 488 (1:200; Jackson ImmunoResearch Laboratories). Nuclei were counterstained with DAPI (Sigma Millipore). Images were taken using a confocal microscope (LSM710, Carl Zeiss) with 20× or 40× oil objectives. For human glioma specimens, TLR3-, TLR9-, and Iba-1-labeled cells were counted using Imaris software (Bitplane).
Organotypic brain slice (OBS) model and tumor inoculation
OBSs were prepared as described previously (Markovic et al., 2009). Briefly, 14-d-old WT or MacGreen mice were decapitated, and brains were cut in coronal plane into 250 µm sections with a vibratome (Leica Microsystems, VT1000S). Brain slices were collected with a sterile plastic pipette (7 mm diameter) and transferred onto cell culture inserts with 0.4 µm pores (Becton Dickinson), which were fitted into wells of a 6-well plate; 1 ml culture medium containing DMEM supplemented with 10% heat-inactivated FCS, 0.2 mm glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin was added into each well; and after overnight incubation, medium was changed with cultivation medium containing 25% heat-inactivated FCS, 50 mm sodium bicarbonate, 2% glutamine, 25% Hanks balanced salt solution, 1 mg/ml insulin (Invitrogen), 2.46 mg/ml glucose (Braun Melsungen), 0.8 mg/ml vitamin C (Sigma Millipore), 100 U/ml penicillin, 100 mg/ml streptomycin, and 5 mm Tris in DMEM (all from Invitrogen). To deplete microglia in OBSs, liposome-encapsulated clodronate or liposome-encapsulated PBS diluted withculture medium (1:10) was added into the well. After 24 h incubation, medium containing clodronate liposomes was replaced by cultivation medium and incubated for another 48 h. Afterward, 5000 GL261mCherry cells were slowly injected into the caudate putamen region of the slice in 150 µm depth of both hemispheres. Careful control of the injection procedure ensured that no cells spilled onto the surface of the slice, which could migrate over the surface rather than invade through the tissue. After 5 d, slices were washed and fixed with 4% PFA. Tumor volumes were measured by confocal microscopy (LSM710, Carl Zeiss) with z-stack scanning and were reconstructed by Imaris into 3D model for exact volume evaluation. For determining microglial density in normal brain slices and tumor containing slices, we quantified the EGFP fluorescence intensity or numbers using Imaris software. To define the tumor area within the slices, we used the mCherry fluorescence. For treatment, Poly(I:C)/CpG, recombinant IFNβ (R&D Systems), normal IgG control (IgG, R&D Systems), IFNβ-neutralizing antibody (aIFN, R&D Systems), rat IgG2a isotype control (Isotype, Bioxcell), or CD47-neutralizing antibody (Bioxcell) were applied to the medium after tumor implantation, and every other day, fresh agonists, recombinant protein, antibody, or isotype were added during medium change.
For microglia isolation from organotypic brain slices 5 d after tumor inoculation, we harvested the tumor tissue with scalpel and transferred it into 1.5 ml Eppendorf tubes. For control microglia, cells were similarly collected from nontumor slices. Then the tissues were enzymatically digested to obtain a single-cell-suspension using Adult Brain Dissociation Kit (Miltenyi Biotec) followed by the microglia isolation procedure by MACS as described previously.
Glioma implantation and treatment administration in vivo
GL261 cells were inoculated into mouse brain as described previously (Hu et al., 2015; Szulzewsky et al., 2015). Briefly, anesthetized mice were immobilized and mounted onto a stereotactic head holder (David Kopf Instruments) in flat-skull position. After skin incision 1 mm anterior and 1.5 mm lateral to bregma, the skull was carefully drilled with a 20G needle tip. A 1 µl syringe with a blunt tip (Mikroliterspritze 7001N, Hamilton) was inserted to a depth of 4 mm and retracted to a depth of 3 mm from the dural surface into the right caudate putamen. Over 2 min, 0.5 µl (2 × 104 cells/µl) of glioma cell suspension was slowly injected into the brain. After surgery, mice were kept warm, and postoperative condition was monitored daily.
After 14 d of tumor inoculation, for Poly(I:C) administration, tumor-bearing mice were intraperitoneally injected with 200 µg Poly(I:C) every 3 d. For CpG administration, mice were first anesthetized, and 100 µg CpG in 2 µl volume was administered intratumorally. All animal experiments were performed at least in triplicate. For tumor volume quantification, mice were killed 28 d after tumor inoculation, and tumor volumes were determined by H&E staining every 12th slices according to the Cavalieri principle (Hu et al., 2015) as we used previously (Vinnakota et al., 2013).
CCK-8 kit assay
A total of 10,000 GL261 cells per well were seeded in a 96-well plate. After treatment with either conditioned medium from microglia or recombinant IFNβ for 6, 12, 24, or 48 h, CCK-8 reagent (Toyobo) was added (10 µl per well) in a 98-well plate and incubated for 2 h. Plates were measured with a multireader at 450 nm absorbance. Results were normalized to the absorbance of the control group at 6 h.
Assessment of phagocytosis
Primary microglia were seeded on coverslips in a 24-well plate for 6 h to adhere. Poly(I:C) and/or CpG was added to the medium for 24 h. Subsequently, YF fluorescent beads (Polysciences) were added for 30 min. Coverslips were then washed 3 times with PBS for 5 min and fixed with 4% PFA for 1 h at room temperature. Cells were stained with Iba-1 as primary antibody overnight in 4°C. After washing, cells on coverslips were incubated with secondary antibody Alexa-647 donkey anti-goat and DAPI at room temperature for 2 h. Coverslips were mounted with Aquapolymount (Polysciences), and images were taken by a confocal microscope (LSM710, Carl Zeiss). Phagocytosis index was calculated as total number of beads divided by 100 number of DAPI-positive cells.
To quantify phagocytosis of glioma cells by microglia, 5000 microglia were seeded on coverslips in a 24-well plate for 6 h and treated with Poly(I:C) and/or CpG for 24 h at 37°C. A total of 25,000 GL261mCherry cells were added to coverslips and incubated for 2 h. After washing, cells on coverslips were fixed and stained for Iba-1 as described above. Coverslips were scanned with a confocal microscope (LSM710, Carl Zeiss). The number of mCherry fluorescence within the iba1 volume was determined with Imaris software as a proxy for glioma phagocytosis.
Flow cytometry
Cultured primary microglia or THP1 macrophages were first treated with Poly(I:C), CpG, Poly(I:C) + CpG, or CD47 antibody (Bioxcell) for 24 h. GL261mCherry cells were added to primary microglia, whereas U87mCherry, U251mCherry, and LN229mCherry glioma cells were applied to THP1 macrophages. The ratio between microglia/macrophages to tumor cells was 1:1. After 2 h coincubation, cell suspension was washed and incubated with CD11b primary FACS antibody (BD Bioscience). Flow cytometry data were quantified for the population of cd11b and mCherry fluorescence-positive cells. Data were analyzed using FlowJo software (TreeStar).
Agarose spot assay and Boyden chamber assay
Agarose spot assay was performed as previously described (Ifuku et al., 2016). Briefly, 0.1 g of low-melting point agarose (Promega) was solved in 20 ml PBS. The solution was then heated until boiling and subsequently cooled down to 40°C. Afterward, 90 µl of agarose solution was mixed with 10 µl of PBS with or without Poly(I:C), CpG, and Poly(I:C) + CpG in a 0.5 ml Eppendorf tube; 10 µl of mixed solution was rapidly plated into 35 mm glass-bottom dishes (MAktek) and were cooled down for 10 min at 4°C. Four spots were pipetted in one dish: two containing PBS only and two with Poly(I:C) or CpG or Poly(I:C) + CpG. These substances were also added to the medium; 5 × 105 WT microglial cells were plated in the dish in 2 ml DMEM supplemented with 10% FCS and incubated at 37°C for 3 h. Subsequently, cells inside the spot were counted at the microscope. Six repetitions per individual experiment and three individual experiments were performed.
Boyden chamber assay were performed as previously described (Ifuku et al., 2016). Briefly, polycarbonate filter (8 µm pore size; Poretics) was used to separate upper and lower wells. Microglial cells in serum-free DMEM were added to the upper compartment, while the lower wells contained the TLR ligands in medium. Culture medium was used as a control. The chamber was incubated at 37°C and 5% CO2 for 6 h. Cells remaining on the upper surface of the membrane were removed by wiping, and cells in the lower compartment were fixed in methanol for 10 min and subjected to Diff-Quik stain (Medion Grifols Diagnostics). The rate of microglial migration was calculated by counting cells in four random fields of each well using a 20 × bright-field objective. The number of cells in each field wasnormalized to the average in control condition (100%). Five repetitions per individual experiment and three individual experiments were performed.
Experimental design and statistical analysis
We investigated the potential synergistic effect of microglial TLR3/TLR9 activation using neonatal primary cultures from P1–P3 C57BL6J mice. Each replication represents the fresh preparation of primary cultures. Using the TLR3 ligand Poly(I:C) and the TLR9 ligand CpG, we studied the combined application on microglial and macrophage cytokine release, motility, and phagocytosis activity. When comparisons were made between single or combined treatment to test synergistic effects, one-way ANOVA with Tukey post hoc test was used. Conditioned medium from TLR3/TLR9-stimulated microglia was tested on their effect on glioma proliferation and apoptosis. Comparisons were conducted between control conditioned medium and TLR3/TLR9-activated conditioned medium. When using neutralizing antibodies, comparisons were made to isotype IgG (Isotype or IgG) groups. Survival analysis was performed by Log-rank analysis. All data represent the average of at least three independent experiments. Error bars indicate SD. Datasets were analyzed statistically by Prism 7.0 software (GraphPad) and tested for normality by Shapiro-Wilks test. Parametric testing was done with Student's t test. Comparison between multiple groups was performed using one-way ANOVA with Tukey post hoc test.
Results
TLR3 and TLR9 costimulation impairs glioma growth in a microglia-dependent fashion
To test for the impact of TLR3 and TLR9 signaling on glioma growth, we applied the TLR3 and TLR9 ligands Poly(I:C) and CpG, respectively, to an organotypic brain slice glioma model. GL261mCherry glioma cells were injected into brain slices from mouse cortex, maintained for 5 d, and the volume occupied by mCherry-labeled glioma cells was subsequently determined by confocal microscopy. Application of Poly(I:C) or CpG alone during the entire tumor growth period resulted in a slight reduction of tumor volume (ctrl vs Poly(I:C), p = 0.0318, n = 18 per group; ctrl vs CpG, p = 0.018, n = 18 per group). When the two ligands were added in a combined manner, there was a stronger reduction in glioma expansion (ctrl vs Poly(I:C) + CpG, p = 0.0003, n = 18 per group; Fig. 1A,B).
Figure 1.

TLR3 and TLR9 costimulation impairs glioma growth mediated by microglia. A, OBS cultures were inoculated with GL261mCherry cells and treated with Poly(I:C), CpG, orPoly(I:C) + CpG compared with untreated control (ctrl). We compared slices with microglia (ctrl) with slices depleted of microglia by clodronate. Representative images of mCherry labeling are shown after 120 h of glioma growth. Scale bar, 500 μm. B, Tumor volumes in slices described in A were quantified by confocal microscopy with z-stack scanning and 3D reconstruction. C, Proliferation of GL261 glioma cells was determined by ki67 immunofluorescence staining after 48 h of treatment with medium conditioned by microglia (CM). Cultured microglia were stimulated with 10 μg/ml Poly(I:C), 2 µM CpG, or Poly(I:C) + CpG for 24 h compared with an unstimulated control (ctrl). After medium change, cells were maintained for another 24 h, and resulting conditioned medium was used to tumor cell incubation. Left, Images of samples. Right, Quantification of Ki67 staining. Scale bar, 50 μm. n = 5 per group. D, Proliferation rate of GL261 glioma cells was determined using CCK-8 cell counting kit. Glioma cells were treated as described in C for 24 and 48 h. Data are normalized to proliferation rate measured after a 6 h treatment with control medium from unstimulated microglia; n = 3 per group. E, Three different human glioma cell lines (i.e., U87, U251, and LN229) were treated for 24 h with medium harvested from THP1 cells treated with a similar paradigm as described in C. Proliferation rate of glioma cells was measured with CCK-8 cell counting kit assay and normalized to control stimulation; n = 6 per group. F, qPCR was used to evaluate both proapoptosis marker and antiapoptosis marker expression levels after 48 h treatment of GL261 glioma cells with supernatant harvested from microglia. Microglia were treated with the same paradigm as described in C. G, OBCs were inoculated with GL261mCherry cells. Yellow represents mCherry labeling. Immunofluorescence labeling with Ph3 (red), cleaved caspase-3 (green), and DAPI (blue). Right, The image in which all labeling is merged. Slices were either untreated (top row, Ctrl) or treated with Poly(I:C) + CpG (bottom row). Scale bar, 50 μm. H, Quantification of Ph3 (left) and cleaved caspase-3 (right) immunofluorescence after treatment with 10 µg/ml Poly(I:C), 2 µM CpG, or 10 µg/ml Poly(I:C) +2 µM CpG compared with untreated slices. C-Caspase 3 = cleaved-caspase 3. ns = no significance. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We verified previous observations that microglia accumulate in the glioma tissue and acquire a defined phenotype (Extended data Fig. 1-1). We found that infiltrating microglia expressed significant higher level of Secreted Phosphoprotein 1 (Spp1, p = 0.0001), Glycoprotein Nmb (Gpnmb, p = 0.0014), Matrix Metallopeptidase 14 (MMP14, p = 0.0243), and Matrix Metallopeptidase 9 (MMP9, p = 0.0204, n = 3, Extended data Fig. 1-1D), which is similar to our previous published RNA-sequencing datasets (Szulzewsky et al., 2015).
(A) Representative figures of mCherry GL261 (red) inoculated MacGreen (CSF1R-EGFP, green for microglia) organotypic brain slices. Scale bar, 200 µm. (B) Representative figures illustrated 3D reconstruction of microglial accumulating and infiltrating in tumor area. The quantification of microglial density between normal brain slices (n = 12) and tumor area within the slices is shown in the graph below (n = 18). (C) The working scheme illustrates the isolation procedure of tumor infiltrating microglia from organotypic brain slices. (D) Expression levels of genes as indicated of control microglia (Ex vivo ctrl) and glioma associated microglia (Ex vivo GAMs) isolated from organotypic brain slices. (*) p < 0.05, (**) p <0.01, (***) p < 0.001, (****) p < 0.0001. Download Figure 1-1, TIF file (15.9MB, tif) .
To test whether the observed tumor reduction was mediated by microglia, brain slices were treated with clodronate for 24 h, which resulted in a depletion of microglia as previously described (Markovic et al., 2009). Subsequently, glioma cells were inoculated and maintained for 5 d as described above. Confirming previous reports, tumors were smaller in microglia-depleted slices compared with control slices (ctrl vs clodronate, p = 0.0038, n = 18). However, combined treatment with Poly(I:C) + CpG did not significantly reduce tumor volume when microglia were not present (clodronate (n = 18) vs clodronate + Poly(I:C) + CpG (n = 19), p = 0.628; Fig. 1A,B).
Factors released from microglia/macrophages activated through TLR3/TLR9 impair glioma viability and induce apoptosis
To examine whether the impact of microglial TLR3/TLR9 signaling on glioma growth is because of factors released from microglia, cultured neonatal microglia were stimulated with Poly(I:C), CpG, or combination of Poly(I:C) + CpG for 24 h. Supernatant was harvested and added to GL261 glioma cells. Using Ki-67 immunofluorescence staining, we tested the impact of 48 h treatment with conditioned medium on proliferation of GL261 cells. Medium from Poly(I:C)-treated microglia significantly reduced glioma cell proliferation after 48 h compared with control medium (ctrl 83 ± 3% vs Poly(I:C) 65 ± 2%, p = 0.0032, n = 5), whereas medium from CpG-treated microglia did not induce such a change (ctrl 83 ± 3% vs CpG 80 ± 3%, n = 5). Medium of Poly(I:C) + CpG-stimulated microglia led to an increased reduction compared with Poly(I:C) alone (ctrl 83 ± 3% vs Poly(I:C) + CpG 42 ± 4%, n = 5, p < 0.0001, Poly(I:C) + CpG vs Poly(I:C), p = 0.0013, n = 5; Fig. 1C).
Next, we measured proliferation of GL261 cells via CCK-8 cell counting after treatment with conditioned medium for 6, 12, 24, and 48 h. After 6 and 12 h, there was no significant difference between the different media conditions (data not shown). After 24 h treatment, neither Poly(I:C) nor CpG conditioned media (CM) suppressed GL261 proliferation, whereas Poly(I:C) + CpG significantly inhibited GL261 cell proliferation (ctrl vs Poly(I:C) + CpG, p = 0.0281; n = 3, Fig. 1D). After 48 h treatment, Poly(I:C) + CpG CM was even more effective (ctrl vs Poly(I:C) + CpG, p = 0.0001, n = 3; Fig. 1D). To test for potential tumor-suppressing effects mediated by Poly(I:C)- and CpG-stimulated human monocytes, we stimulated the human macrophage cell line THP1 with Poly(I:C) and CpG alone and combined for 24 h and applied the conditioned medium to the human glioma cell lines U87, U251, and LN229 for 48 h. CM from Poly(I:C)-stimulated THP1 cells inhibited U87 (p = 0.0366, n = 6), U251 (p = 0.0182, n = 6), and LN229 (p = 0.0010, n = 6) human glioma cell growth (Fig. 1E). This inhibition was further enhanced when Poly(I:C) was applied in combination with CpG (U87: p = 0.0128, n = 6; U251: p = 0.0184, n = 6; LN229: p = 0.0291, n = 6; Fig. 1E). We also tested whether supernatant from Poly(I:C)-, CpG-, and Poly(I:C) + CpG-treated microglia triggers apoptosis in GL261 cells. To this end, we examined the expression of proapoptosis markers Bcl-2-like protein 11 (bim), Bcl-2-associated death promoter (bad), Bcl-2-associated X protein (bax), and antiapoptosis markers B-cell lymphoma 2 (Bcl2), myeloid cell leukemia 1(mcl1) by qPCR. As shown in Figure 1F, combined TLR3 and TLR9 stimulation led to maximum expression of all three proapoptosis markers (bad: p = 0.0001; bim: p < 0.0001; bax: p = 0.002, n = 3; Fig. 3F), with no significant changes of antiapoptosis genes (bcl2: p = 0.9984; mcl1: p = 0.9827, n = 3; Fig. 3F). Furthermore, we detected cleaved caspase-3, a further key molecule in apoptosis, on tumor slices by immunofluorescence (Fig. 1G), and quantified the number of cleaved caspase-3-positive cells. Combined application of Poly(I:C) and CpG induced much more cleaved caspase-3-positive cells within glioma tissue compared with single treatment (Fig. 1H). In addition, we tested for the proliferation marker Phospho-Histone H3 (Ph3) and found that Poly(I:C) and CpG synergistically suppress Ph3 expression in glioma tissue (Fig. 1G,H).
Figure 3.

Properties of IFNβ release from microglia after TLR3 and TLR9 costimulation and its impact on glioma growth. A, Left, Microglia were treated with 10 µg/ml Poly(I:C), 2 µm CpG, or 10 µg/ml Poly(I:C) + 2 µM CpG for 24 h, and IFNβ levels in the supernatant were measured by ELISA. A, Right, Microglia were pretreated with 10 µg/ml Poly(I:C) or 2 µM CpG for 3 or 6 h. After this pretreatment period and a medium change, cells were incubated with 10 µg/ml Poly(I:C) + 2 µM CpG for 24 h. IFNβ levels in the supernatant were measured by ELISA. B, Microglia were treated with different concentrations of Poly(I:C) from 0.1 to 20 µg/ml or 2 µM CpG for 6 h as indicated. At 10 µg/ml Poly(I:C), the ceiling level was reached. In an additional set of experiments shown in the same graph, microglia were treated with 10 µg/ml Poly(I:C) combined with 1, 2, or 4 µM CpG. IFNβ levels in the supernatant were determined by ELISA. C, GL261 tumor cells were treated with 1, 10, or 100 ng/ml of murine recombinant IFNβ or vehicle for different time periods as indicated, and proliferation was determined by CCK-8 kit. Results were normalized to control group treated for 6 h. D, OBS cultures were inoculated with GL261mCherry and treated with 1, 10, and 100 ng/ml murine recombinant IFNβ for 120 h. Subsequently, tumor volume was determined. E, Conditioned medium from microglia stimulated with Poly(I:C) + CpG along with or without IFNβ-neutralizing antibody (αIFN) and compared with an isotype control antibody (IgG) for 6, 24, or 48 h were added to GL261 cells. Proliferation of GL261 cells was determined by CCK-8 proliferation assay. F, Organotypic brain slices inoculated with GL261mCherry cells were treated with Poly(I:C) + CpG with or without αIFN and compared with IgG. Tumor volume was determined. G, Proliferation rate of human glioma cell lines U87, U251, and LN229 was determined with CCK-8 proliferation assay after treatment with 1, 10, and 100 ng/ml human recombinant IFNβ for 48 h. Data are normalized to proliferation rate of untreated cells. H, Gene expression of proapoptosis markers (Bad and Bim) and an antiapoptosis marker (bcl2) in GL261 glioma cells treated with 0.1, 1, 10, and 100 ng/ml mouse recombinant IFNβ for 48 h compared with the untreated control. I, Human glioma cell lines U87, U251, and LN229 were treated with human recombinant IFNβ for 48 h; subsequently, the percentage of cleaved-caspase 3 (C-C3, green) positive cells was determined by immunofluorescence staining normalizing to DAPI signals. Left, Representative figures. Right, quantification. Scale bar,50 μm. n = 5. ns = no significance. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
TLR3 and TLR9 costimulation induces microglial cytokine and NOS2 expression
Since we found that costimulation of microglia with Poly(I:C) and CpG suppressed tumor growth via soluble factor(s), we searched for potential candidates responsible for this effect. Primary microglia were treated with Poly(I:C), CpG, or Poly(I:C) + CpG for 6 h, and mRNA expression of TNFα, IFNβ, IL1β, NOS2, IL6, and IL12 was analyzed by qPCR (Fig. 2A). Poly(I:C) alone increased tnfα, ifnβ, il1β, nos2, and il6 expression, whereas CpG increased tnfα, il1β, and il6 expression. Combined application of Poly(I:C) + CpG augmented tnfα, ifnβ, nos2, and il12 expression compared with single stimulation with either Poly(I:C) or CpG (Fig. 2A). IL6 expression was augmented by CpG stimulation, but combined application of Poly(I:C) + CpG resulted in lower expression level compared with the condition where CpG alone was used. ELISA analysis revealed that Poly(I:C) and CpG costimulation augmented the release of IL12, nitric oxide (NO), and TNFα, whereas IL6 levels were lower compared with stimulation with CpG alone, thereby confirming the data on mRNA expression changes on protein level (Fig. 2B).
Figure 2.

TLR3 and TLR9 costimulation and cytokine release. A, Microglia were treated with 10 µg/ml Poly(I:C), 2 µM CpG, or 10 µg/ml Poly(I:C) + 2 µM CpG for 6 h and compared with untreated control. mRNA expression of il12, tnfα, ifnβ, il1β, nos2, and il6 were determined by qPCR (n = 3 per group). B, Microglia were treated as described in A. Subsequently, protein levels of TNFα, IL12, and IL6 were determined by ELISA (n = 3 per group). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Properties of microglial IFNβ release after TLR3 and TLR9 costimulation
We considered IFNβ as an interesting candidate to mediate the interaction between microglia and glioma via Poly(I:C)/CpG-induced TLR signaling, since TLR3 and TLR9 signaling both activate IRF/Type I IFN signaling. IFNβ as crucial member of Type I IFN has been identified as a potent antitumorogenic factor in multiple cancers (Borden, 2019). We therefore analyzed different time periods of stimulation and different agonist concentrations with respect to IFNβ release. According to the qPCR result shown in Figure 2A, mRNA of ifnβ was most prominently increased after costimulation with Poly(I:C) and CpG compared with control (58,548-fold upregulation). To confirm this effect on protein level, microglia were treated with Poly(I:C), CpG, or Poly(I:C) + CpG in combination for 3, 6, and 24 h, and IFNβ release was determined by ELISA. After both 3 and 6 h, Poly(I:C) increased IFNβ expression, whereas CpG had no effect compared with control (data not shown). After 24 h, however, the combined application of Poly(I:C) and CpG strongly enhanced IFNβ release compared with treatment with Poly(I:C) alone (Poly(I:C) 515.8 ± 115.8 vs Poly(I:C) + CpG 1135.0 ± 112.1, p = 0.0028, n = 3; Fig. 3A).
To test whether microglia priming with either Poly(I:C) or CpG plays a role in the observed IFNβ release, we first applied Poly(I:C) for 3 or 6 h alone and then added CpG to continue stimulation for 24 h. Conversely, we first applied CpG alone for 3 or 6 h and then added Poly(I:C) for the rest of the 24 h stimulation. To reach the synergistic effect of augmented IFNβ release, as described above, CpG had to be present in the beginning. When Poly(I:C) was applied for 3 or 6 h alone, subsequent addition of GpG did not result in such a synergistic effect (Fig. 3A).
To test whether the synergistic effect of IFNβ release described above is dose-dependent, we tested different Poly(I:C) concentrations in our experimental setup. While 0.1 µg/ml resulted in a small effect, IFNβ release was increased when higher dosages, such as 1 and 10 µg/ml, were used. Concentrations at 20 µg/ml did not further enhance the release. If 1 or 2 µM CpG was added to Poly(I:C) treatment, the release was augmented in a dose-dependent manner (10 µg/ml Poly(I:C) vs 10 µg/ml Poly(I:C) + 1 µM CpG, p = 0.0008, n = 4; 10 µg/ml Poly(I:C) +1 µM CpG vs 10 µg/ml Poly(I:C)+2 µM CpG, p < 0.0001, n = 4; Fig. 3B), while concentrations at 4 µm/ml induced similar effects as concentrations at 2 µM CpG (Fig. 3B).
IFNβ inhibits glioma growth and induces apoptosis of glioma cells
It was reported that IFNβ suppresses tumor growth (Doherty et al., 2017). Therefore, we tested the impact of IFNβ treatment on cultured GL261 cells. To this end, we added 1, 10, or 100 ng/ml IFNβ to the medium and determined the proliferation rate of GL261 cells using the CCK-8 assay. After 6 and 12 h, we did not observe any significant effect of INFβ on GL261 proliferation at any of the tested concentrations. However, after 48 h, 10 and 100 ng/ml IFNβ significantly suppressed the proliferation rate (vehicle vs 10 ng/ml IFNβ, p = 0.0017, n = 3; vehicle vs 100 ng/ml IFNβ, p = 0.0008, n = 3), whereas 1 ng/ml IFNβ had no significant effect (vehicle vs 1 ng/ml IFNβ, p = 0.0579, n = 3) (Fig. 3C). We observed similar findings analyzing GL261mCherry cells (data not shown).
Next, we tested the effect of IFNβ on glioma growth in a brain slice glioma model. GL261mCherry cells were inoculated into the slices, and IFNβ was added at 1, 10, or 100 ng/ml doses. Compared with control slices, tumor volumes significantly decreasedafter incubation with 10 ng/ml IFNβ (ctrl 20.1 ± 3.0 µm3 vs 10 ng/ml IFNβ, 11.8 ± 1.6 µm3, p = 0.0226, 50 ng/ml IFNβ 10.4 ± 1.6 µm3, p = 0.0086, 200 ng/ml IFNβ 5.5 ± 1.0 µm3, p = 0.0001; Fig. 3D).
To verify that IFNβ is the main factor mediating antiproliferating effects in the supernatant derived from Poly(I:C) + CpG-stimulated microglia, we added an IFNβ-neutralizing antibody (Yang et al., 2013) to the CM from microglia treated with Poly(I:C) + CpG. As shown in Figure 3E, proliferation of GL261 cells decreased when supernatant from Poly(I:C) + CpG-treated microglia was added to GL261 cells for 24 or 48 h. When the IFNβ-neutralizing antibody was added to the respective supernatant, proliferation rate was no longer reduced (24 h: p > 0.05, n = 5; 48 h: p > 0.05, n = 5; Fig. 3E), indicating that IFNβ mediates the main antiproliferative activity of Poly(I:C) + CpG-treated microglia (Fig. 3E).
We also determined the impact of IFNβ-neutralizing antibody on glioma growth in the glioma brain slice model. As a control, we used IgG antibody. We incubated the slices after inoculation with GL261mCherry cells with Poly(I:C) + CpG in the presence of IFNβ antibody and compared this treatment with control condition using IgG antibody instead. As described above, Poly(I:C) + CpG treatment decreased glioma growth, whereas antibody treatment significantly restored glioma growth (p = 0.0195, n = 12; Fig. 3F). Treatment with IFNβ antibody alone did not affect glioma growth. Next, we treated human glioma cells U87, U251, and LN229 with different concentrations of human recombinant IFNβ (hrIFNβ) for 48 h and analyzed their proliferation activity. hrIFNβ inhibited human glioma cell growth (Fig. 3G).
Finally, we tested whether glioma cell apoptosis can be induced by IFNβ. To this end, GL261 cells were treated with increasing concentrations (1, 10, 100 ng/ml) of murine IFNβ, for 48 h. Gene expression of proapoptosis markers (bad, bim) and an antiapoptosis marker (bcl2) was determined by qPCR. IFNβ treatment upregulated Bad and Bim expression in GL261 cells in a dose-dependent manner, while no significant change of bcl2 was observed at all concentrations (Fig. 3H). To further verify that glioma cell apoptosis is induced by IFNβ, glioma cell lines U87, U251, LN229, and GL261 were treated with 10 ng/ml IFNβ for 48 h, and protein level of cleaved caspase-3 was examined by immunofluorescence staining. IFNβ treatment significantly increased cleaved caspase-3 expression in all glioma cell lines (U87: p = 0.0006; U251: p < 0.0001; LN229: p = 0.0014; GL261: p = 0.0009, n = 5; Fig. 3I).
Synergistic TLR3 and TLR9 activation increases microglial phagocytosis activity
IFNβ blockade did not completely abolish Poly(I:C)- and CpG-attributed tumor suppression, indicating that TLR3 and TLR9 activation may affect additional microglial properties (Fig. 3F). Thus, we tested the impact of TLR3/TLR9 stimulation on microglial phagocytosis activity representing a key function of microglia. Cultured microglia were pretreated with single Poly(I:C) or CpG, or combined treatment for 24 h. Subsequently, we used a bead phagocytosis assay to quantify phagocytosis activity (Von Zahn et al., 1997). Application of Poly(I:C) or CpG alone increased the phagocytosis activity compared with control (ctrl 0.5 ± 0.1 vs Poly(I:C) 0.9 ± 0.2, p = 0.0475, CpG 1.1 ± 0.3, p = 0.0012, n = 9), and the combined treatment with both agonists led to increased phagocytosis (Poly(I:C) + CpG 1.8 ± 0.5, p < 0.0001, n = 9; Fig. 4A,B).
Figure 4.

Synergistic activation of TLR3 and TLR9 increases microglial phagocytosis activity. A, Cultured microglia were treated with 10 µg/ml Poly(I:C), 2 µM CpG, or 10 µg/ml Poly(I:C) + 2 µM CpG for 24 h. Subsequently, YFP-fluorescent beads were applied for 30 min, and microglial cells were stained with Iba-1. Yellow represents beads. Red represents Iba-1 labeling (n = 9). Scale bar, 50 μm. B, Quantification of phagocytosis from images as shown in A. C, Cultured microglia were pretreated with 10 µg/ml Poly(I:C) + 2 µM CpG for 24 h, and GL261mCherry cells were subsequently added for 2 h. Green represents Iba-1 labeling. Red represents GL261mCherry labeling. Blue represents DAPI labeling. DAPI labeled all nuclei. Scale bar, 50 μm. D, The number of Iba-1-labeled microglial cells containing GL261mCherry was determined by confocal microscopy with Imaris software analysis and given as phagocytosis index. E, In a similar experiment as described in C, cells were harvested from the culture. Subsequently, the cell suspension was labeled with CD11b and analyzed by flow cytometry. Cells were sorted according to mCherry and CD11b fluorescence to determine the microglia containing mCherry material. F, Quantification of phagocytosis activity from experiments as shown in E. The percentage of cells containing mCherry material is given. Cultured mouse microglia were exposed to GL261 cells (n = 6 per group), whereas THP1 macrophages were incubated with the human glioma lines U87, U251, and LN229 (n = 9 per group). G, Organotypic brain slices obtained from MacGreen mice, in which microglia are labeled by EGFP, were inoculated with GL261mCherry cells. The mCherry incorporation into microglia was analyzed by determining the overlap of red and green fluorescence. We compared control slices with slices treated with Poly(I:C) + CpG for 120 h. Using Imaris 3D rendering, we determined the incorporation of mCherry material (red) into microglia (green). Right, Two images represent magnifications as indicated and show microglial cells that had not incorporated mCherry material (top, Non-phago) and those with incorporation of mCherry material (bottom, Phago). Scale bars: Overview, 50 μm; Enlargement, 15 µm. n = 6. H, In experiments as described in G, phagocytosis activity was determined in untreated slices and slices treated with Poly(I:C), CpG, or Poly(I:C) + CpG. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We then analyzed the ability of microglia to phagocytose glioma cells. Microglia were seeded on coverslips and maintained for 6 h. To quantify this phagocytic activity, cells were subsequently fixed and analyzed with Imaris software. We determined the amount of mCherry-fluorescent material within the compartment of Iba1-labeled microglia. Next, we compared this microglial activity observed under control conditions with the microglial activity after treatment with Poly(I:C), CpG, or combined Poly(I:C) + CpG for 6 h before the exposure to GL261 cells. While Poly(I:C) alone did not change phagocytic activity compared with control, CpG alone increased phagocytic activity (p < 0.0001, n = 9; Fig. 4C,D). However, treatment with both Poly(I:C) + CpG induced a further increase in phagocytosis (p < 0.0001, n = 9; Fig. 4D). To validate the enhanced microglial phagocytosis of tumor cells induced by Poly(I:C) and CpG coadministration, we pretreated microglia with single agonists or combined Poly(I:C) and CpG treatment for 24 h, then added GL261mCherry cells to microglia for 2 h. Subsequently, we used FACS to quantify the number of microglial cells, which had phagocytosed glioma cells by determining the population of cells being double-positive for mCherry and CD11b. Similarly to the findings using immunofluorescence analysis described above, using flow cytometry, Poly(I:C) alone did not increase phagocytic activity, while a large population of mCherry-positive microglia was detected after CpG stimulation (ctrl 3.4 ± 0.3 vs CpG, 13.6 ± 1.0, p < 0.0001, n = 6). An even higher rate of mCherry-positive microglia was found in the combined treatment group (CpG13.6 ± 1.0 vs Poly(I:C) + CpG 24.0 ± 1.5, p < 0.0001, n = 6; Fig. 4E,F). We could confirm this finding for the human system by using the THP1 macrophage cell line as a proxy for microglia combined with three different mCherry-labeled human glioma lines (Fig. 4F). In addition, we used MacGreen mice (CSF1R-EGFP) (Sasmono and Williams, 2012) to label microglia in brain slices inoculated with GL261 glioma cells. Using z-stack confocal microscopy scanning and 3D rendering by Imaris, we quantified mCherry fluorescence within CSF1R-EGFP fluorescence as an indicator of glioma cell phagocytosis. Combined treatment with Poly(I:C) and CpG promoted phagocytosis of glioma cells (ctrl 0.271 ± 0.027 vs Poly(I:C) + CpG 0.551 ± 0.035, p < 0.0001, n = 18; Fig. 4G,H). As a previous study (Liu et al., 2019) reported on higher oxygen consumption as the metabolic reason of higher phagocytic activity induced by CpG, we also determined the microglial basal oxygen consumption and indeed found that treatment with Poly(I:C) + CpG significantly induced higher basic oxygen consumption (Extended data Fig. 4-1).
Microglia were pretreated with 10µg/ml Poly(I:C), 2µM CpG, or 10µg/ml Poly(I:C) + 2µM CpG for 24 h. Basal microglial oxygen consumption was measured within 35 min. Download Figure 4-1, TIF file (370.4KB, tif) .
Synergistic TLR3 and TLR9 activation enhances microglial motility via PI3K/Akt signaling
As microglia infiltrate glioma tissue (Gutmann and Kettenmann, 2019), we tested whether TLR3/TLR9 signaling would affect microglial infiltration. To this end, brain slices were generated from MacGreen mice whose microglia express the fluorescent marker EGFP. After inoculation of GL261mCherry glioma cells, slices were treated with Poly(I:C), CpG, or Poly(I:C) + CpG in combination. Subsequently, we determined the density of microglia within the glioma area by Imaris 3D rendering. The numbers of infiltrating microglia were significantly increased in slices incubated with Poly(I:C) + CpG combination (Fig. 5A,B).
Figure 5.

Synergistic activation of TLR3 and TLR9 enhances microglial motility via PI3K/Akt signaling. A, Organotypic brain slices were inoculated with GL261mCherry cells, and accumulation of microglia within the tumor areas without treatment and after 120 h treatment with Poly(I:C) +CpG is shown. Microglia were labeled with GFP as MacGreen mice were used (n = 6 per group). Scale bar, 100 μm. B, Quantification of total microglial number within the tumor area of organotypic slices of untreated and Poly(I:C)-, CpG-, and Poly(I:C) CpG-treated slices determined by confocal microscopy with Imaris 3D reconstruction. C, Using the Boyden chamber assay, we determined microglia motility activity by applying 10 µg/ml Poly(I:C), 2 µM CpG, or 10 µg/ml Poly(I:C) + 2 µM CpG into both compartments, and the number of cells migrating through the membrane was quantified. Left, Cells on the membrane were labeled with Diff-Quik kit. Right, Data are quantified as cells/mm2 (5 repetitions per n, n = 3 per group). D, Motility was evaluated using the agarose spot assay. Spots and fluid surrounding the spots contained either medium only or 10 µg/ml Poly(I:C), 2 µM CpG, or 10 µg/ml Poly(I:C) + 2 µM CpG. Left, Images from sample spots. Right, Quantification of migrating cells normalized to PBS control. Values are normalized to average migration in control medium (6 repetitions per n, n = 3). E, Protein lysate from cultured microglia treated with 10 µg/ml Poly(I:C), 2 µM CpG, or 10 µg/ml Poly(I:C) + 2 µM CpG for 24 h were analyzed for protein level of phosphor-Akt (p-Akt) and total Akt (t-Akt) by Western blot (n = 3). F, Quantification of p-Akt relative to t-Akt Western blot data as shown in E. G, Quantification of motility using the Boyden chamber of cells treated with 10 µg/ml Poly(I:C) + 2 μm CpG and with and without the PI3K inhibitors LY294002 and wortmannin (5 repetitions per n, n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Next, we tested the impact of TLR3/TLR9 signaling on motility using two assays, namely, Boyden chamber and agarose spot assay (Ifuku et al., 2016). Using the Boyden chamber, microglia exhibited enhanced migration across the membrane in the presence of both Poly(I:C) + CpG compared with treatment with Poly(I:C) or CpG alone (Poly(I:C) 41 ± 4 vs Poly(I:C) + CpG, 86 ± 5, p < 0.0001; CpG 43 ± 4 vs Poly(I:C) + CpG 86 ± 5, p < 0.0001; Fig. 5C). Similar results were obtained with the agarose spot assay (Poly(I:C) 3.5 ± 0.3 vs Poly(I:C) + CpG 5.7 ± 0.3, p < 0.0001; CpG 2.9 ± 0.2 vs Poly(I:C) + CpG 5.7 ± 0.3, p < 0.0001; Fig. 5D).
Since TLR3 and TLR9 activation increases microglial migration via PI3K/Akt signaling (Ifuku et al., 2016), we tested whether microglial migration in response to TLR3 and TLR9 costimulation involves PI3K/Akt activation. First, we determined the expression level of phosphor-Akt(p-Akt) in microglia treated with Poly(I:C), CpG, or Poly(I:C) + CpG combination by Western blot, using GAPDH as loading control. Normalized to Total-Akt (t-Akt) signal intensity, both Poly(I:C) and CpG significantly increased expression of p-Akt (Poly(I:C): p = 0.0134; CpG: p = 0.0015, n = 3), whereas combined treatment with Poly(I:C) + CpG further upregulated the p-Akt expression (Poly(I:C) vs Poly(I:C) + CpG, p = 0.0011; CpG vs Poly(I:C) + CpG, p = 0.007, n = 3; Fig. 5E,F). Next, we pretreated microglia with the PI3K/Akt inhibitors LY294002 or wortmannin and subsequently stimulated them with Poly(I:C) + CpG to test for migration in the Boyden chamber. Both LY294002 and wortmannin attenuated Poly(I:C) + CpG-increased microglial motility (Fig. 5G), indicating that Poly(I:C) + CpG- triggered microglial migration is mediated by PI3K/Akt signaling.
To determine microglial density within murine glioma in vivo, GL261 cells were inoculated into C57BL/6 mice. According to a previously reported Poly(I:C) and CpG delivery method (Jordan and Waxman, 2016; Garzon-Muvdi et al., 2018), 14 days after tumor inoculation, mice were then given CpG intratumorally once a week and Poly(I:C) peritoneally every 3 d for another 2 weeks (Fig. 6A). We assessed microglia density within the tumors of each treatment group. Tumors with combined CpG and Poly(I:C) treatment displayed a higher density of Iba-1+ cells compared with tumors within slices treated with CpG or Poly(I:C) alone (Poly(I:C) + CpG vs CpG, 39.8 ± 0.8% vs 34.5 ± 1.3%, p = 0.0398; Fig. 6B).
Figure 6.

TLR3 and TLR9 synergistically suppress tumor growth in vivo. A, Scheme represents the experimental paradigms of the glioma-injected and treated mice. Top, The survival experiment. Bottom, The determination of glioma volume and tissue analysis. Mice were inoculated with GL261 cells and peritoneally injected with 200 µg Poly(I:C) every 3 d and intratumorally injected with 100 µg CpG every 7 d. B, Twenty-eight days after tumor inoculation, slices were prepared and Iba-1 (green) immunofluorescence staining was used to label microglia/macrophages, DAPI (blue) for labeling nuclei. Bottom row represents the merged labeling. Left, Representative figures. Right, Quantification of Iba-1+ cells (n = 4 per group). Scale bar, 50 μm. C, Left, Twenty-eight days after tumor inoculation, slices were prepared, and tumor volumes were quantified by H&E staining and based on unbiased stereology (n = 6 per group). Right, Kaplan–Meier curves represent the cumulative survival of tumor mice with the different treatment paradigms (n = 11 per group). D, Left, Immunohistochemical labeling of glioma containing slices from mice killed 28 d after tumor inoculation. Top two rows represent proliferation marker Ph3 (red) and apoptosis marker cleaved caspase-3 (C-caspase3, green). Third row represents nucleus marker DAPI. Fourth row represents a merge of the three rows. Scale bar, 50 μm. Right, Immunofluorescence labeling of Ph3 and cleaved caspase-3 was quantified by measuring the fluorescence signal subsequently normalized to DAPI. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Synergistic TLR3 and TLR9 treatment suppresses tumor growth in vivo
To study the impact of TLR3/TLR9 signaling on glioma growth, we generated an immunocompetent mouse model by inoculating GL261 cells into the brain of 7-week-old C57BL/6 mice. Fourteen days after tumor inoculation, tumor-bearing mice were administered Poly(I:C), CpG, or combined Poly(I:C) + CpG. Treatment with Poly(I:C) alone diminished tumor volumes (Poly(I:C) 14.3 ± 1.1 vs ctrl, 19.9 ± 1.9 mm3, p = 0.0286, n = 6; Fig. 6C), while treatment with combined Poly(I:C) + CpG even further decreased tumor volumes compared with treatment with single agonist (Poly(I:C) + CpG 8.25 ± 1.0 mm3 vs Poly(I:C) 14.3 ± 1.1 mm3, p = 0.0069; Fig. 6C). Survival time of tumor-inoculated mice was monitored in a different cohort of mice. Mice were killed if the following symptoms became apparent: hunchback, low reaction to stimulation, tarnished fur, or movement problem. Mice that had received treatment with Poly(I:C) + CpG in combination survived longer compared with control mice, when 60 d was assigned as observation endpoint (ctrl 29 d vs Poly(I:C) + CpG 52 d, p < 0.0001; Fig. 6C). Mice treated with Poly(I:C) alone exhibited longer median survival (ctrl 29 d vs Poly(I:C) 45 d, p = 0.004; Fig. 6C), while treatment with Poly(I:C) + CpG further improved median survival (Poly(I:C) 45 d vs Poly(I:C) + CpG 52 d,p = 0.0059; Fig. 6C). In another series of experiments, we determined proliferation and apoptosis of tumor cells by Ph3 labeling and cleaved caspase-3 labeling, respectively. The percentage of Ph3-positive cells within tumors was significantly reduced in the group treated with Poly(I:C) + CpG in combination compared with mice that received single agonist. Further, we observed a higher cleaved caspase-3 signal in mice treated with Poly(I:C) + CpG in combination compared with treatment with single agonists (Fig. 6D).
CD47 blockade and TLR3/TLR9 costimulation synergistically improve microglia/macrophage-induced tumor clearance
CD47 was recognized as suppressor of phagocytosis in macrophages and microglia in the context of glioma (Zhang et al., 2016; Hutter et al., 2019). The Cancer Genome Atlas (TCGA)-GBM/LGG database (using GlioVis data portal for visualization and analysis of brain tumor expression datasets) (Bowman et al., 2017) indicates that CD47 mRNA expression is significantly increased in glioblastoma multiforme (GBM) compared with low-grade glioma (LGG) (Extended data Fig. 7-1). GBM from patients and control healthy brain cortex tissue from biopsy samples was immunolabelled with CD47. We found that CD47 is highly expressed in GBM specimen, whereas in normal brain tissue CD47 expression levels are low (Fig. 7A). Further, all tested GL261mCherry, U87, U251, and LN229 cell lines expressed CD47 (Fig. 7B).
Figure 7.
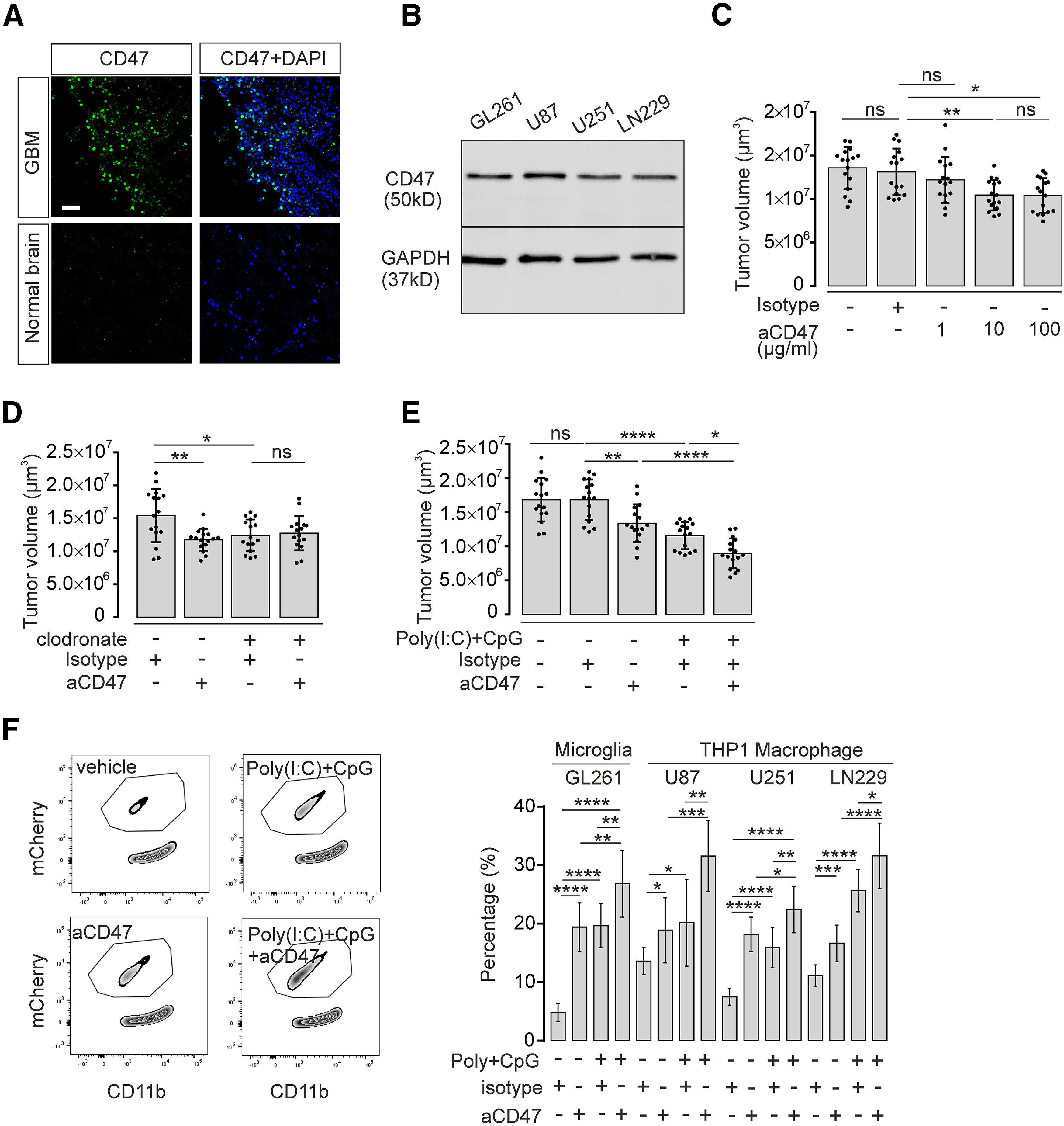
CD47 blockade and TLR3/TLR9 costimulation synergistically improve microglia/macrophage-induced tumor clearance. A, Immunofluorescence staining of CD47 expression (green) in human GBM tissue compared with normal human brain cortex tissue. Nuclei are counterstained with DAPI. Scale bar, 50 μm. B, CD47 protein level in murine GL261 and human U87, U251, and LN229 glioma cell lines was detected by Western blot. GAPDH expression served as reference. C, Organotypic brain slices inoculated with GL261mCherry cells were treated with 1, 10, or 100 µg/ml CD47 blocking antibody (aCD47) and compared with isotype control antibody. Tumor volumes were evaluated (n = 16 per group). D, Organotypic brain slices inoculated with GL261mCherry cells were pretreated with clodronate for 48 h to deplete intrinsic microglia. Subsequently, slices were treated with 10 µg/ml CD47 blocking antibody (aCD47) for 120 h and compared with isotype control antibody (n = 16 per group). E, Organotypic brain slices injected with GL261mCherry cells were treated with 10 µg/ml CD47 blocking antibody (aCD47) together with or without 10 µg/ml Poly(I:C) and 2 µM CpG, and tumor volumes were assessed (n = 16 per group). F, FACS assay was performed as described in Figure 4E to determine mCherry incorporation into cultured microglia treated with 10 µg/ml CD47 blocking antibody (aCD47), Poly(I:C) + CpG, or Poly(I:C) + CpG + CD47 blocking antibody (left). Right, Quantification of FACS data. Percentage of microglial cells that had incorporated mCherry material is given. In addition, using this paradigm described above, we tested the effect on the incorporation of the human glioma cell lines U87, U252, and LN229 into the macrophage cell line THP1. Isotype antibody served as a control (n = 9 per group). ns = no significance, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
mRNA expression level of CD47 in TCGA-LGG database (using GlioVis data portal for visualization and analysis of brain tumor expression datasets) shows that CD47 is expressed at higher levels in GBM compared to low grade glioma. Download Figure 7-1, TIF file (505.9KB, tif) .
To test for potential interaction between CD47 and TLR3/TLR9 signaling in glioma growth control mediated by microglia, we generated organotypic brain slices inoculated with GL261mCherry cells and directly applied increasing concentrations of CD47 blocking antibody (BioXCell) to slices, using 10 µg/ml isotype as control condition. Glioma volume was measured after 5 d. While the isotype did not affect tumor volume (ctrl. 13.5 ± 0.6 vs isotype 13.1 ± 0 0.7 × 106 µm3, p > 0.05; Fig. 7C), CD47 blocking antibody (10 µg/ml) reduced tumor growth (isotype 13.1 ± 0.7 × 106 µm3 vs 10 µg/ml aCD47 10.5 ± 0.4 × 106 µm3, p = 0.0090; Fig. 7C). An increase of CD47 blocking antibody dosage to 100 µg/ml did not further decrease tumor volume (10 µg/ml aCD47 10.5 ± 0.4 × 106 µm3 vs 100 µg/ml aCD47 10.4 ± 0.5 × 106 µm3, p > 0.05; Fig. 7C), while 1 µg/ml of CD47 antibody did not have a significant effect (Fig. 7C). To test whether the effect of CD47 interference is mediated by microglia, we depleted microglia in the slices described above by clodronate treatment. In microglia-depleted slices, CD47 blocking antibody did not affect glioma growth (clodronate 12.4 ± 0.7 × 106 vs clodronate + aCD47 13.1 ± 0.7 × 106 µm3, p > 0.05; Fig. 7D). Finally, we combined CD47 blockade with combined Poly(I:C) + CpG treatment. CD47 blockade and Poly(I:C) + CpG combination synergistically suppressed tumor volume compared with either mono-CD47 blockade or mono Poly(I:C) + CpG treatment (anti-CD47 + Poly(I:C) + CpG 9.0 ± 0.5 × 106 µm3 vs Poly(I:C) + CpG 11.6 ± 0.5 × 106 µm3; aCD47 + Poly(I:C) + CpG 9.0 ± 0.5 × 106 µm3 vs aCD47 13.4 ± 0.7 × 106 µm3; Fig. 7E).
To verify whether microglia/macrophage-mediated tumor phagocytosis is enhanced by combined treatment, including CD47 blocking antibody and Poly(I:C) + CpG, we stimulated cultured microglia with Poly(I:C) + CpG and CD47 antibody, and added GL261mCherry. We found that parallel application of CD47 antibody and Poly(I:C) + CpG promoted the engulfment of tumor cells by microglia. We determined the percentage of microglial cells that contained mCherry material (Poly(I:C) + CpG + aCD47 26.83 ± 1.90% vs Poly(I:C) + CpG. 19.64 ± 1.26%; Poly(I:C) + CpG+aCD47 26.83 ± 1.90% vs aCD47 19.41 ± 1.38%; Fig. 7F). Also, we tested the effect of combined Poly(I:C) and CpG treatment on the phagocytic activity of THP1 macrophages that were exposed to U87mCherry, U251mCherry, or LN229mCherry cells. Combined treatment with Poly(I:C) + CpG and CD47 antibody enhanced the engulfment of glioma cells. Thus, we propose that the combination of CD47 interference with TLR3/TLR9 coactivation leads to synergistic antitumor activity of microglia.
TLR3 and TLR9 are expressed on GAMs in human glioma tissues
Since our present study focused on TLR3/TLR9 activation, we determined the expression pattern of TLR3/TLR9 in microglia, GL261, and astrocytes by qPCR. TLR3 was expressed significantly higher in microglia compared with glioma cells (p = 0.0024, n = 4; Fig. 8A) and astrocytes (p = 0.0118, n = 4). TLR9 expression was at a similar level in microglia and glioma cells (p > 0.05, n = 4; Fig. 8D), but lower in astrocytes (p = 0.0128, n = 4). To compare the expression of TLR3 and TLR9 in human GAMs versus glioma cells, we separated CD11b+ cells and CD11b– cells by MACS from 12 GBM tissue samples and subsequently analyzed expression levels of TLR3 and TLR9 by qPCR. In almost all glioma samples, TLR3 expression was significantly higher in CD11b+ cells compared with CD11b– cells (p < 0.01; Fig. 8B,C). Using GlioVis data portal for visualization and analysis of brain tumor expression datasets (Bowman et al., 2017), we also found a significant correlation between the microglial/macrophage-specific marker AIF1 (Iba1) and TLR3 expression in the TCGA RNA-seq database (r = 0.56, p < 0.001; Extended data Fig. 8-1). In contrast, TLR9 expression level was similar in CD11b+ cells compared with CD11b– cells (Fig. 8E,F; p > 0.05), and correlation between Iba1 and TLR9 was not significant in TCGA (r = −0.14, p > 0.05; Extended data Fig. 8-1). In order to confirm the data on mRNA expression level, we used immunohistochemical analysis of TLR3 and TLR9 protein expression, combined with immunostaining using Iba-1 as a marker for GAMs. We identified TLR3 and TLR9 that colabeled with Iba-1 expression in human GBM tissue. TLR3 positivity was higher in Iba-1-positive cells compared with surrounding glioma cells (Iba-1-positive 76.81 ± 3.00% vs Iba-1-negative 5.69 ± 1.01%, p < 0.0001, n = 8; Fig. 8G), while TLR9 staining was found at comparable level in both Iba-1-positive and Iba-1-negative cells (Fig. 8G).
Figure 8.
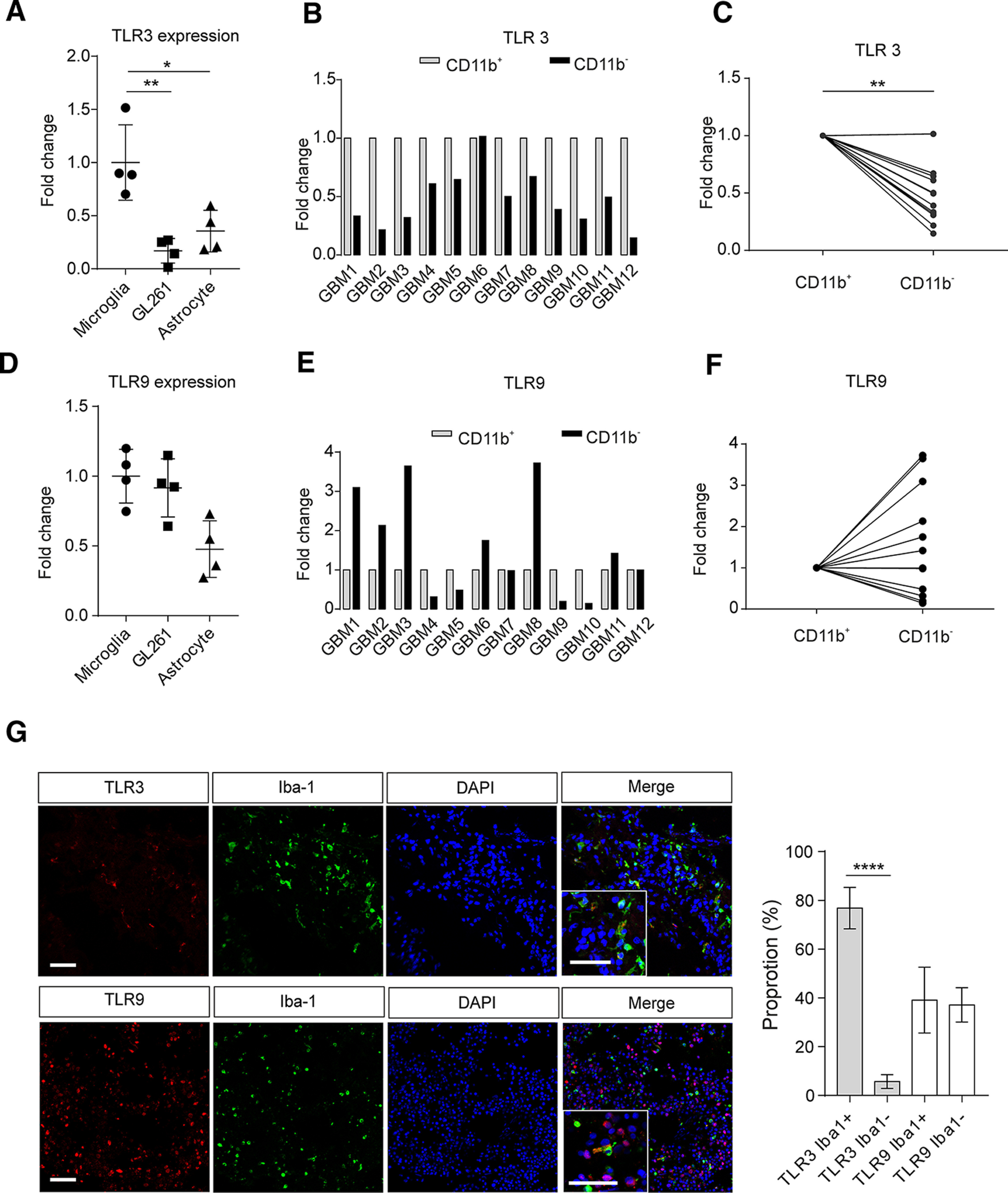
TLR3 and TLR9 expression in human GAMs and glioblastoma samples. TLR3 mRNA (A) and TLR9 mRNA (D) levels of microglia, GL261 glioma cell line, and murine primary astrocyte were determined by qPCR. GAMs from 12 human glioblastoma samples were isolated by MACS, and total RNA was extracted. TLR3 mRNA (B) and TLR9 mRNA (E) levels of CD11b+ and CD11b– cells were measured by qPCR. Results were normalized to the housekeeping gene GAPDH. Relative expression of TLR3 (C) and TLR9 (F) is given as fold-change of expression in CD11b– cells normalized to expression in CD11b+ cells. G, Left, Representative figures show that TLR3 (red) and TLR9 (red) protein expression in glioma samples was analyzed by immunofluorescence combined with the microglia/macrophage markers Iba-1 (green), and nuclei were stained with DAPI (blue) in slices of human GBM samples. Right, Proportion of TLR3 or TLR9 colocalized with Iba-1-positive or Iba-1-negative cells (n = 8). Scale bar, 50 µm. *p < 0.05, **p < 0.01, ****p < 0.0001.
The TCGA RNA-seq database (using GlioVis data portal for visualization and analysis of brain tumor expression datasets) was used to correlate the mRNA levels of TLR3 and TLR9 with Iba-1, a marker for GAMs. Download Figure 8-1, TIF file (839.5KB, tif) .
Discussion
In the last years, it has become apparent that the glioma microenvironment has a profound impact on tumor growth and infiltration. A number of mechanisms have been described explaining how glioma convert GAMs into a defined phenotype and how these cells influence glioma growth (Hambardzumyan et al., 2016; Poon et al., 2017; Gutmann and Kettenmann, 2019). One important signaling pathway for glioma with respect to GAM intercellular communication is mediated by TLRs. Glioma release endogenous factors, such as versican, which activate TLR2 resulting in an upregulation of the metalloproteases MMP9 and MMP14 (Vinnakota et al., 2013; Hu et al., 2014, 2015). Moreover, glioma stem cells upregulate IL-6 secretion in GAMs via TLR4 signaling (Dzaye et al., 2016). let-7 microRNAs suppress glioma growth by activating TLR7 (Buonfiglioli et al., 2019). Here, we demonstrate that TLR3 and TLR9 impair glioma growth in a synergistic fashion. Indeed, both TLRs have been previously considered as therapeutic targets tested in clinical trials. In preclinical studies administration of the TLR3 ligand Poly(I:C) strongly retarded the growth of human tumor xenograft in mice (Schaffert et al., 2011), and combined administration of Poly(I:C) with peptide-based vaccination improved the survival of tumor-bearing mice (Zhu et al., 2007). The involvement of microglia in TLR3-mediated antitumor effects has also been established (Kees et al., 2012). These preclinical observations have been translated into clinical studies. However, multiple trials using TLR3 ligand polyinosinic-polycytidylic acid-poly-l-lysine carboxymethylcellulose (Poly-ICLC) have not yielded benefit in survival (Butowski et al., 2009; Rosenfeld et al., 2010). Also, a TLR9 ligand, namely, synthetic CpG oligodeoxynucleotide, also has been used in glioma-related trials. CpG treatment has yielded mixed results in different types of lung tumor, non-Hodgkin lymphoma, and meningitis (Belani et al., 2013; Witzig et al., 2013; Ursu et al., 2015). In a Phase II trial, glioma patients treated with CpG reached very limited long-term survival benefit (Carpentier et al., 2010; Carpentier and Lambert, 2017). Thus, until now, single TLR3 or TLR9 activation in glioma patients did not result in predominant survival improvement. We now provide evidence that combined application of TLR3 and TLR9 agonists reveals strong synergistic action on inhibition of glioma growth and several microglial functional properties. That TLRs can have synergistic effects when applied in combination has already been demonstrated for different combined activation of TLR2, TLR4, TLR7, and TLR9, which result in different inflammatory responses compared with single activation of a given TLR (Rosenberger et al., 2014).
Combined activation of TLR3 and TLR9 leads to an augmented release of several cytokines, such as IL1β, IFNβ, IL12, or TNFα. Cytokines, such as IL12 and TNFα, are well-established factors that interfere with tumor growth (van Horssen et al., 2006; Lu, 2017). In the present study, we found that augmented IFNβ release from microglia by costimulation of TLR3 and TLR9 has a strong impact on glioma growth. We found that direct application of IFNβ mimicked the responses triggered by combined TLR3/TLR9 stimulation, and an antibody interfering with IFNβ signaling partially inhibited the TLR3/TLR9-mediated responses. We also found that, at the ceiling dosage of Poly(I:C) (i.e., at maximum of TLR3 stimulation), additional TLR9 stimulation by CpG led to a more than two fold IFNβ release. Indeed, Type I interferons (IFNα and IFNβ) mediate many direct (on cancer cells) and indirect (through immune effector cells and vasculature) effects on tumor, and a recent review highlights these interferon-directed therapeutic opportunities (Borden, 2019). Further, a number of studies indicate that IFNβ directly or indirectly induces tumor cell apoptosis, suppresses proliferation, decreases extravascular migration, and affects parameters relevant to tumor function (Sims et al., 2010; Doherty et al., 2017; Makowska et al., 2018).
We also found that combined TLR3/TLR9 activation augments microglial functional parameters that are relevant in the glioma context. We have previously reported on TLR9 activation as a chemotactic signal for microglia, and this chemotaxis activity depends on PI3K/Akt signaling (Ifuku et al., 2016). Here, we describe that combined stimulation of TLR3 and TLR9 leads to enhanced motility activity and that this effect depends on PI3K/Akt signaling. Interestingly, higher density of microglia was observed in Poly(I:C) + CpG-treated tumors, suggesting an enhanced invasion of microglia into the tumor tissue. A second glioma-relevant function that is affected by TLR3/TLR9 stimulation is phagocytosis activity. Our study shows that microglia have the capacity to phagocytose glioma cells and that this phagocytic activity is most efficiently stimulated by combined TLR3 and TLR9 activation. Obviously, under normal physiologic conditions, microglia do not phagocytose glioma cells, which is because of stop signals preventing phagocytes to attack cells in their environment. Such a stop signal is represented by the membrane protein CD47, which broadly inhibits phagocytosis and is abundantly expressed in all healthy cells (Alvey and Discher, 2017). We therefore used an approach to interfere with CD47 signaling and found that TLR3/TLR9-controlled phagocytic activity could be further enhanced. Indeed, tumor cells express CD47, which transduces inhibitory signals through SIRPα to myeloid cells (Weiskopf, 2017). CD47 antibodies have been used in clinical trials in non-Hodgkin's lymphoma and other cancers (Advani et al., 2018; Sikic et al., 2019). Interesting in this context is the observation that TLR9 stimulation triggers metabolic activity in macrophages that impart macrophages with antitumor potential capable of overcoming inhibitory CD47 on cancer cells (Liu et al., 2019).
In conclusion, combined microglial TLR3 and TLR9 activation triggers an antitumor phenotype of microglia, which affects glioma cells via release of cytokines, stimulates their phagocytic activity to directly attack glioma cells, and enhances migratory activity, which may explain increased accumulation of microglia in glioma tissue. Given the fact that single TLR3 and TLR9 stimulation has so far failed in clinical trials, treatment with combined stimulation of TLR3 and TLR9 in concert with CD47 inhibition might provide a novel approach for glioma therapy.
Footnotes
The authors declare no competing financial interests.
This work was supported by Einstein-Stiftung, the Helmholtz-Gemeinschaft, Zukunftsthema Immunology and Inflammation ZT-0027 to H.K., the Berlin Institute of Health, National Natural Science Foundation of China 81602202 to F.H., and China Scholarship Council and Medical Neuroscience graduate program of Charité, Berlin to Y.H. We thank Regina Piske, Maren Wendt, Nadine Scharek, and Michaela Seeger-Zografakis for technical assistance; and the microscope core facility of the Max-Delbrueck-Center (Advanced Light Microscopy) and FACS facility for technical assistance.
References
- Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, Kline J, Roschewski M, LaCasce A, Collins GP, Tran T, Lynn J, Chen JY, Volkmer JP, Agoram B, Huang J, Majeti R, Weissman IL, Takimoto CH, Chao MP, et al. (2018) CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin's lymphoma. N Engl J Med 379:1711–1721. 10.1056/NEJMoa1807315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvey C, Discher DE (2017) Engineering macrophages to eat cancer: from “marker of self” CD47 and phagocytosis to differentiation. J Leukoc Biol 102:31–40. 10.1189/jlb.4RI1216-516R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton GM, Medzhitov R (2003) Toll-like receptor signaling pathways. Science 300:1524–1525. 10.1126/science.1085536 [DOI] [PubMed] [Google Scholar]
- Belani CP, Nemunaitis JJ, Chachoua A, Eisenberg PD, Raez LE, Cuevas JD, Mather CB, Benner RJ, Meech SJ (2013) Phase 2 trial of erlotinib with or without PF-3512676 (CPG 7909, a Toll-like receptor 9 agonist) in patients with advanced recurrent EGFR-positive non-small cell lung cancer. Cancer Biol Ther 14:557–563. 10.4161/cbt.24598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden EC. (2019) Interferons α and β in cancer: therapeutic opportunities from new insights. Nat Rev Drug Discov 18:219–234. 10.1038/s41573-018-0011-2 [DOI] [PubMed] [Google Scholar]
- Bowman RL, Wang Q, Carro A, Verhaak RG, Squatrito M (2017) GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol 19:139–141. 10.1093/neuonc/now247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonfiglioli A, Efe IE, Guneykaya D, Ivanov A, Huang Y, Orlowski E, Krüger C, Deisz RA, Markovic D, Flüh C, Newman AG, Schneider UC, Beule D, Wolf SA, Dzaye O, Gutmann DH, Semtner M, Kettenmann H, Lehnardt S (2019) let-7 microRNAs regulate microglial function and suppress glioma growth through Toll-like receptor 7. Cell Rep 29:3460–3471.e7. 10.1016/j.celrep.2019.11.029 [DOI] [PubMed] [Google Scholar]
- Butowski N, Chang SM, Junck L, DeAngelis LM, Abrey L, Fink K, Cloughesy T, Lamborn KR, Salazar AM, Prados MD (2009) A phase II clinical trial of poly-ICLC with radiation for adult patients with newly diagnosed supratentorial glioblastoma: a North American Brain Tumor Consortium (NABTC01-05). J Neurooncol 91:175–182. 10.1007/s11060-008-9693-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier A, Lambert J (2017) Intracerebral injection of CpG oligonucleotide for patients with de novo glioblastoma: a phase II multicentric, randomised study. Eur J Cancer 73:30–37. 10.1016/j.ejca.2016.12.003 [DOI] [PubMed] [Google Scholar]
- Carpentier A, Metellus P, Ursu R, Zohar S, Lafitte F, Barrié M, Meng Y, Richard M, Parizot C, Laigle-Donadey F, Gorochov G, Psimaras D, Sanson M, Tibi A, Chinot O, Carpentier AF (2010) Intracerebral administration of CpG oligonucleotide for patients with recurrent glioblastoma: a phase II study. Neuro Oncol 12:401–408. 10.1093/neuonc/nop047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, Makarov V, Budhu S, Buhu S, Slamon DJ, Wolchok JD, Pardoll DM, Beckmann MW, Zahnow CA, Merghoub T, Mergoub T, et al. (2015) Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162:974–986. 10.1016/j.cell.2015.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty MR, Cheon HJ, Junk DJ, Vinayak S, Varadan V, Telli ML, Ford JM, Stark GR, Jackson MW (2017) Interferon-beta represses cancer stem cell properties in triple-negative breast cancer. Proc Natl Acad Sci USA 114:13792–13797. 10.1073/pnas.1713728114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzaye OD, Hu F, Derkow K, Haage V, Euskirchen P, Harms C, Lehnardt S, Synowitz M, Wolf SA, Kettenmann H (2016) Glioma stem cells but not bulk glioma cells upregulate IL-6 secretion in microglia/brain macrophages via toll-like receptor 4 signaling. J Neuropathol Exp Neurol 75:429–440. 10.1093/jnen/nlw016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon-Muvdi T, Theodros D, Luksik AS, Maxwell R, Kim E, Jackson CM, Belcaid Z, Ganguly S, Tyler B, Brem H, Pardoll DM, Lim M (2018) Dendritic cell activation enhances anti-PD-1 mediated immunotherapy against glioblastoma. Oncotarget 9:20681–20697. 10.18632/oncotarget.25061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladson CL, Prayson RA, Liu WM (2010) The pathobiology of glioma tumors. Annu Rev Pathol 5:33–50. 10.1146/annurev-pathol-121808-102109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutmann DH, Kettenmann H (2019) Microglia/brain macrophages as central drivers of brain tumor pathobiology. Neuron 104:442–449. 10.1016/j.neuron.2019.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Gutmann DH, Kettenmann H (2016) The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci 19:20–27. 10.1038/nn.4185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Dzaye OD, Hahn A, Yu Y, Scavetta RJ, Dittmar G, Kaczmarek AK, Dunning KR, Ricciardelli C, Rinnenthal JL, Heppner FL, Lehnardt S, Synowitz M, Wolf SA, Kettenmann H (2015) Glioma-derived versican promotes tumor expansion via glioma-associated microglial/macrophages Toll-like receptor 2 signaling. Neuro Oncol 17:200–210. 10.1093/neuonc/nou324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Ku MC, Markovic D, Dzaye OD, Lehnardt S, Synowitz M, Wolf SA, Kettenmann H (2014) Glioma-associated microglial MMP9 expression is upregulated by TLR2 signaling and sensitive to minocycline. Int J Cancer 135:2569–2578. 10.1002/ijc.28908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter G, Theruvath J, Graef CM, Zhang M, Schoen MK, Manz EM, Bennett ML, Olson A, Azad TD, Sinha R, Chan C, Kahn SA, Gholamin S, Wilson C, Grant G, He J, Weissman IL, Mitra SS, Cheshier SH (2019) Microglia are effector cells of CD47-SIRPα antiphagocytic axis disruption against glioblastoma. Proc Natl Acad Sci USA 116:997–1006. 10.1073/pnas.1721434116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ifuku M, Buonfiglioli A, Jordan P, Lehnardt S, Kettenmann H (2016) TLR2 controls random motility, while TLR7 regulates chemotaxis of microglial cells via distinct pathways. Brain Behav Immun 58:338–347. 10.1016/j.bbi.2016.08.003 [DOI] [PubMed] [Google Scholar]
- Jordan M, Waxman DJ (2016) CpG-1826 immunotherapy potentiates chemotherapeutic and anti-tumor immune responses to metronomic cyclophosphamide in a preclinical glioma model. Cancer Lett 373:88–96. 10.1016/j.canlet.2015.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kees T, Lohr J, Noack J, Mora R, Gdynia G, Tödt G, Ernst A, Radlwimmer B, Falk CS, Herold-Mende C, Régnier-Vigouroux A (2012) Microglia isolated from patients with glioma gain antitumor activities on poly (I:C) stimulation. Neuro Oncol 14:64–78. 10.1093/neuonc/nor182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg AM. (2007) Development of TLR9 agonists for cancer therapy. J Clin Invest 117:1184–1194. 10.1172/JCI31414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S. (2010) Innate immunity and neuroinflammation in the CNS: the role of microglia in toll-like receptor-mediated neuronal injury. Glia 58:253–263. 10.1002/glia.20928 [DOI] [PubMed] [Google Scholar]
- Liu M, O'Connor RS, Trefely S, Graham K, Snyder NW, Beatty GL (2019) Metabolic rewiring of macrophages by CpG potentiates clearance of cancer cells and overcomes tumor-expressed CD47−mediated 'don't-eat-me' signal. Nat Immunol 20:265–275. 10.1038/s41590-018-0292-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X. (2017) Impact of IL-12 in cancer. Curr Cancer Drug Targets 17:682–697. [DOI] [PubMed] [Google Scholar]
- Makowska A, Wahab L, Braunschweig T, Kapetanakis NI, Vokuhl C, Denecke B, Shen L, Busson P, Kontny U (2018) Interferon beta induces apoptosis in nasopharyngeal carcinoma cells via the TRAIL-signaling pathway. Oncotarget 9:14228–14250. 10.18632/oncotarget.24479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H, Stock K, Sliwa M, Lehmann S, Kälin R, Van Rooijen N, Holmbeck K, Heppner FL, Kiwit J, Matyash V, Lehnardt S, Kaminska B, Glass R, Kettenmann H (2009) Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci USA 106:12530–12535. 10.1073/pnas.0804273106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellinghoff IK, Gilbertson RJ (2017) Brain tumors: challenges and opportunities to cure. J Clin Oncol 35:2343–2345. 10.1200/JCO.2017.74.2965 [DOI] [PubMed] [Google Scholar]
- Minelli A, Lyons S, Nolte C, Verkhratsky A, Kettenmann H (2000) Ammonium triggers calcium elevation in cultured mouse microglial cells by initiating Ca2+ release from thapsigargin-sensitive intracellular stores. Pflugers Arch Eur J Physiol 439:370–377. 10.1007/s004240050952 [DOI] [PubMed] [Google Scholar]
- Morikawa T, Sugiyama A, Kume H, Ota S, Kashima T, Tomita K, Kitamura T, Kodama T, Fukayama M, Aburatani H (2007) Identification of toll-like receptor 3 as a potential therapeutic target in clear cell renal cell carcinoma. Clin Cancer Res 13:5703–5709. 10.1158/1078-0432.CCR-07-0603 [DOI] [PubMed] [Google Scholar]
- Poon CC, Sarkar S, Yong VW, Kelly JJ (2017) Glioblastoma-associated microglia and macrophages: targets for therapies to improve prognosis. Brain 140:1548–1560. 10.1093/brain/aww355 [DOI] [PubMed] [Google Scholar]
- Rosenberger K, Derkow K, Dembny P, Krüger C, Schott E, Lehnardt S (2014) The impact of single and pairwise Toll-like receptor activation on neuroinflammation and neurodegeneration. J Neuroinflammation 11:166. 10.1186/s12974-014-0166-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld MR, Chamberlain MC, Grossman SA, Peereboom DM, Lesser GJ, Batchelor TT, Desideri S, Salazar AM, Ye X (2010) A multi-institution phase II study of poly-ICLC and radiotherapy with concurrent and adjuvant temozolomide in adults with newly diagnosed glioblastoma. Neuro Oncol 12:1071–1077. 10.1093/neuonc/noq071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaun B, Coste I, Rissoan MC, Lebecque SJ, Renno T (2006) TLR3 can directly trigger apoptosis in human cancer cells. J Immunol 176:4894–4901. 10.4049/jimmunol.176.8.4894 [DOI] [PubMed] [Google Scholar]
- Sasmono RT, Williams E (2012) Generation and characterization of MacGreen mice, the Cfs1r-EGFP transgenic mice. Methods Mol Biol 844:157–176. 10.1007/978-1-61779-527-5_11 [DOI] [PubMed] [Google Scholar]
- Schaffert D, Kiss M, Rödl W, Shir A, Levitzki A, Ogris M, Wagner E (2011) Poly(I:C)-mediated tumor growth suppression in EGF-receptor overexpressing tumors using EGF-polyethylene glycol-linear polyethylenimine as carrier. Pharm Res 28:731–741. 10.1007/s11095-010-0225-4 [DOI] [PubMed] [Google Scholar]
- Sikic BI, Lakhani N, Patnaik A, Shah SA, Chandana SR, Rasco D, Colevas AD, O'Rourke T, Narayanan S, Papadopoulos K, Fisher GA, Villalobos V, Prohaska SS, Howard M, Beeram M, Chao MP, Agoram B, Chen JY, Huang J, Axt M, et al. (2019) First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J Clin Oncol 37:946–953. 10.1200/JCO.18.02018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims TL, McGee M, Williams RF, Myers AL, Tracey L, Hamner JB, Ng C, Wu J, Gaber MW, McCarville B, Nathwani AC, Davidoff AM (2010) IFN-β restricts tumor growth and sensitizes alveolar rhabdomyosarcoma to ionizing radiation. Mol Cancer Ther 9:761–771. 10.1158/1535-7163.MCT-09-0800 [DOI] [PubMed] [Google Scholar]
- Szulzewsky F, Pelz A, Feng X, Synowitz M, Markovic D, Langmann T, Holtman IR, Wang X, Eggen BJ, Boddeke H, Hambardzumyan D, Wolf SA, Kettenmann H (2015) Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS One 10:e0116644 10.1371/journal.pone.0116644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G, Sher A (2007) Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol 7:179–190. 10.1038/nri2038 [DOI] [PubMed] [Google Scholar]
- Ursu R, Taillibert S, Banissi C, Vicaut E, Bailon O, Le Rhun E, Guillamo JS, Psimaras D, Tibi A, Sacko A, Marantidou A, Belin C, Carpentier AF (2015) Immunotherapy with CpG-ODN in neoplastic meningitis: a phase I trial. Cancer Sci 106:1212–1218. 10.1111/cas.12724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Horssen R, ten Hagen TL, Eggermont AM (2006) TNF‐α in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist 11:397–408. 10.1634/theoncologist.11-4-397 [DOI] [PubMed] [Google Scholar]
- Vinnakota K, Hu F, Ku MC, Georgieva PB, Szulzewsky F, Pohlmann A, Waiczies S, Waiczies H, Niendorf T, Lehnardt S, Hanisch UK, Synowitz M, Markovic D, Wolf SA, Glass R, Kettenmann H (2013) Toll-like receptor 2 mediates microglia/brain macrophage MT1-MMP expression and glioma expansion. Neuro Oncol 15:1457–1468. 10.1093/neuonc/not115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Zahn J, Möller T, Kettenmann H, Nolte C (1997) Microglial phagocytosis is modulated by pro-and anti-inflammatory cytokines. Neuroreport 8:3851–3856. 10.1097/00001756-199712220-00003 [DOI] [PubMed] [Google Scholar]
- Weiskopf K. (2017) Cancer immunotherapy targeting the CD47/SIRPα axis. Eur J Cancer 76:100–109. 10.1016/j.ejca.2017.02.013 [DOI] [PubMed] [Google Scholar]
- Witzig TE, Wiseman GA, Maurer MJ, Habermann TM, Micallef IN, Nowakowski GS, Ansell SM, Colgan JP, Inwards DJ, Porrata LF, Link BK, Zent CS, Johnston PB, Shanafelt TD, Allmer C, Asmann YW, Gupta M, Ballas ZK, Smith BJ, Weiner GJ (2013) A phase I trial of immunostimulatory CpG 7909 oligodeoxynucleotide and 90yttrium ibritumomab tiuxetan radioimmunotherapy for relapsed B-cell non-Hodgkin lymphoma. Am J Hematol 88:589–593. 10.1002/ajh.23460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Lee SM, Gao B, Zhang J, Fang D (2013) Histone deacetylase sirtuin 1 deacetylates IRF1 protein and programs dendritic cells to control Th17 protein differentiation during autoimmune inflammation. J Biol Chem 288:37256–37266. 10.1074/jbc.M113.527531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Hutter G, Kahn SA, Azad TD, Gholamin S, Xu CY, Liu J, Achrol AS, Richard C, Sommerkamp P, Schoen MK, McCracken MN, Majeti R, Weissman I, Mitra SS, Cheshier SH (2016) Anti-CD47 treatment stimulates phagocytosis of glioblastoma by M1 and M2 polarized macrophages and promotes M1 polarized macrophages in vivo. PLoS One 11:e0153550 10.1371/journal.pone.0153550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Nishimura F, Sasaki K, Fujita M, Dusak JE, Eguchi J, Fellows-Mayle W, Storkus WJ, Walker PR, Salazar AM, Okada H (2007) Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models. J Transl Med 5:10 10.1186/1479-5876-5-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Representative figures of mCherry GL261 (red) inoculated MacGreen (CSF1R-EGFP, green for microglia) organotypic brain slices. Scale bar, 200 µm. (B) Representative figures illustrated 3D reconstruction of microglial accumulating and infiltrating in tumor area. The quantification of microglial density between normal brain slices (n = 12) and tumor area within the slices is shown in the graph below (n = 18). (C) The working scheme illustrates the isolation procedure of tumor infiltrating microglia from organotypic brain slices. (D) Expression levels of genes as indicated of control microglia (Ex vivo ctrl) and glioma associated microglia (Ex vivo GAMs) isolated from organotypic brain slices. (*) p < 0.05, (**) p <0.01, (***) p < 0.001, (****) p < 0.0001. Download Figure 1-1, TIF file (15.9MB, tif) .
Microglia were pretreated with 10µg/ml Poly(I:C), 2µM CpG, or 10µg/ml Poly(I:C) + 2µM CpG for 24 h. Basal microglial oxygen consumption was measured within 35 min. Download Figure 4-1, TIF file (370.4KB, tif) .
mRNA expression level of CD47 in TCGA-LGG database (using GlioVis data portal for visualization and analysis of brain tumor expression datasets) shows that CD47 is expressed at higher levels in GBM compared to low grade glioma. Download Figure 7-1, TIF file (505.9KB, tif) .
The TCGA RNA-seq database (using GlioVis data portal for visualization and analysis of brain tumor expression datasets) was used to correlate the mRNA levels of TLR3 and TLR9 with Iba-1, a marker for GAMs. Download Figure 8-1, TIF file (839.5KB, tif) .