

Abstract
Protein synthesis is quickly and tightly regulated in cells to adapt to the ever-changing extracellular and intracellular environment. Accurate quantitation of rapid protein synthesis changes can provide insights into protein functions and cellular activities, but it is very challenging to achieve because of the lack of effective analysis methods. Here, we developed an effective mass spectrometry-based method named quantitative O-propargyl-puromycin tagging (QOT) by integrating O-propargyl-puromycin (OPP) labeling, bioorthogonal chemistry, and multiplexed proteomics for global and quantitative analysis of rapid protein synthesis. The current method enables us to accurately quantitate rapid changes of newly synthesized proteins because, unlike amino acids and their analogs, OPP can be utilized by the ribosome immediately without being activated and conjugated to tRNA, and thus cell starvation or pretreatment is not required. This method was applied to quantitate rapid changes of protein synthesis in THP-1 macrophages treated with lipopolysaccharide (LPS). For 15-min labeling, >3000 proteins were quantitated, and the synthesis of 238 proteins was significantly altered, including transcription factors and cytokines. The results demonstrated that protein synthesis was modulated to facilitate protein secretion in macrophages in response to LPS. Considering the importance of protein synthesis, this method can be extensively applied to investigate rapid changes of protein synthesis in the biological and biomedical research fields.
Graphical Abstract
Prompt and tight regulation of protein synthesis is essential to maintain proteostasis and cellular activities, and its dysregulation is correlated with aging and diseases.1 Protein expression is quickly altered in response to extracellular nutrient changes and various stimuli, such as pathogens and heat shock, during which multiple signaling pathways are involved, including the mammalian target of rapamycin (mTOR) and the mitogen-activated protein kinase (MAPK) pathways.2,3 The rate of protein synthesis is precisely controlled at the transcription and translation levels. Despite the complexity of this regulation, the rate of protein synthesis can be modulated in a fast and efficient manner.4,5 Recent studies showed that the synthesis rates of over 30 proteins in mouse macrophages were dramatically affected under a 1-h lipopolysaccharide (LPS) treatment, and 24 proteins in HeLa cells had increased synthesis rates after a 15-min epidermal growth factor (EGF) treatment.6,7
Two indirect methods, i.e., RNA sequencing and ribosome profiling, have been widely employed to obtain information about protein expression,8,9 but neither present accurate changes on protein synthesis. It has been shown that the correlation between the mRNA and protein abundances is low.10,11 In addition, the comparison of proteomic results with those from the ribosome profiling shows a low correlation between the ribosome footprint density and the abundance of newly synthesized proteins in cells under bortezomib-induced stress.12 With the advances in mass spectrometry (MS)-based proteomics, several methods have been developed to directly study newly synthesized proteins and protein synthesis, especially those based on stable isotope labeling by amino acids in cell culture (SILAC). For example, pulsed SILAC, where newly synthesized proteins are labeled with heavy isotopic amino acids, was applied to study protein synthesis in different biological systems.10,12 Noncanonical amino acids, such as azidohomoalanine (AHA), were reported to label proteins, and together with bioorthogonal chemistry, newly synthesized proteins were enriched and studied.7,13–16 Quantitative noncanonical amino acid tagging (QuaNCAT) was developed for quantifying stimuli-induced proteome dynamics in primary cells by combining bioorthogonal noncanonical amino acid tagging (BONCAT) and SILAC, and >600 newly synthesized proteins were quantitated in primary resting T cells subjected to activation stimuli.17
However, there are several limitations that hamper the quantitation accuracy and sensitivity of SILAC-based methods and prevent the investigation of rapid protein synthesis. First, metabolic labeling with stable isotope and/or noncanonical amino acids normally requires the depletion of the corresponding amino acids through cell starvation for at least 30 min to increase the labeling efficiency, which can induce cell stress responses and thus alter protein synthesis. Second, stored and recycled canonical amino acids can also be incorporated into newly synthesized proteins, which impacts the quantitation accuracy. Third, stable isotope and noncanonical amino acids need to be conjugated to a specific tRNA in cells to form the aminoacyl-tRNA before they can be utilized by the ribosome. This lowers the labeling efficiency, especially in the early stage of metabolic labeling. It is often compensated by metabolically labeling cells for at least 30 min to achieve more efficient labeling, which prevents the quantification of rapid changes of protein synthesis. Fourth, protein degradation exists during the studies, and newly synthesized proteins collected at different time points are affected differently by the degradation pathways, especially for a longer period of labeling time.
To overcome these issues, we developed an effective method termed quantitative O-propargyl-puromycin tagging (QOT) for accurate and sensitive investigation of rapid changes of protein synthesis in human cells by combining OPP labeling, bioorthogonal chemistry, and multiplexed proteomics. OPP is an alkyne analog of puromycin that can be rapidly incorporated into nascent polypeptide chains and has been extensively employed to visualize and measure protein synthesis in the cells and in vivo.18–22 Unlike metabolic labeling with AHA or SILAC, OPP labeling does not require any pretreatment or amino acid depletion before the labeling, which makes it well-suited to accurately analyze rapid changes of protein synthesis. In this work, by combining OPP labeling with multiplexed proteomics, we quantitated the rapid protein synthesis rates at six different time points over 6 h from THP-1 macrophages treated with LPS. Newly synthesized proteins were labeled with OPP for 15 min at each of the six time points, which is short and minimizes the influence of protein degradation. Nascent polypeptides were subsequently enriched through the click reaction. To further eliminate the effect of nonspecific binding, a background sample, which serves as a negative control, was analyzed without the catalytic reagents in the parallel experiments. With 15-min labeling, more than 3000 newly synthesized proteins were quantitated in each of the triplicate experiments, and a total of over 3700 proteins were quantitated. The synthesis rates of 238 proteins were significantly altered in the cells treated with LPS, including many transcription factors, cytokines, cluster of differentiation (CD) proteins, and other proteins related to the inflammatory process. The synthesis of some proteins was dramatically affected at the first and second time points, i.e., 0 and 0.5 h, during the LPS treatment, indicating that the method can quantify immediate changes of newly synthesized proteins. Furthermore, the synthesis of multiple proteins related to the classical secretory pathway, including five enzymes for protein N-glycosylation, was markedly changed in the treated macrophages. Considering the importance of protein expression and the unique feature of the current method for analysis of rapid protein synthesis, this method can be widely applied to investigate rapid changes of protein synthesis in the biological and biomedical research fields.
METHODS
Cell Culture, THP-1 Cell Differentiation, Lipopolysaccharide (LPS) Treatment, and Time-Course O-Propargyl-Puromycin (OPP) Labeling.
THP-1 human monocytes (ATCC) were cultured in RPMI-1640 medium (Sigma-Aldrich) containing 10% fetal bovine serum (FBS, Corning) and 1.0% penicillin-streptomycin in an incubator with 5% CO2 at 37 °C. Cells were equally split into eight flasks once the density reached ∼7 × 105 cells/mL. Phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich) was added to the medium to the final concentration of 100 ng/mL to differentiate the monocytes into M0 macrophages for 48 h as previously reported.23 After that, the medium was removed, and the adherent macrophages were rested in the normal RPMI medium without PMA for 24 h.23 For the time-course OPP labeling samples, LPS (Sigma-Aldrich) was added to six flasks to the final concentration of 1 μg/mL. After 0, 0.5, 1, 2, 3, and 6 h, respectively, the macrophages were treated with 30 μM OPP (Click Chemistry Tools) for 15 min. The medium was removed after the 15-min OPP treatment, and the cells were washed twice with ice-cold PBS before harvesting. The cells were centrifuged at 300g for 5 min, and the supernatant was removed. For the control and background samples, 30 μM OPP was added to the medium without the treatment of LPS, and the other steps for cell harvest are the same.
Cell Lysis, Enrichment of Newly Synthesized Proteins, On-Bead Digestion, and Peptide Purification.
The cells were lysed and the OPP-labeled nascent proteins were enriched through the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction. For the background sample, the catalytic reagents (CuSO4 and THPTA) were not added to the solution. The reaction lasted for 2 h in the dark and was quenched by adding 10 mM dithiothreitol (DTT; Sigma-Aldrich). The enriched proteins were further reduced at 56 °C for 25 min and alkylated with 14 mM iodoacetamide at room temperature in the dark for 30 min. The beads from all samples were stringently washed to remove the nonspecific binding proteins. The enriched newly synthesized proteins were digested on beads with trypsin (Promega) at 37 °C overnight. The digestion was quenched with trifluoroacetic acid (Millipore), and the pH was adjusted to ~2 before desalting. The supernatant containing eluted peptides was collected, and the peptides were purified using a tC18 Sep-Pak Vac Cartridge (Waters). The purified peptides were dried in a vacuum concentrator. The detailed information is included in the Supporting Information.
TMT Labeling of Peptides and the Fractionation Using High Performance Liquid Chromatography (HPLC).
The dried peptides from 8 samples were resuspended in 100 μL of 100 mM HEPES, pH = 8.5, and 30 μL ACN. The TMT10plex reagents were dissolved in 41 μL of anhydrous ACN, and 5 μL was added to each sample (127N-0 h; 127C-0.5 h; 128N-1 h; 128C-2 h; 129N-3 h; 129C-6 h; 130N-control; 130C-background), respectively. The reaction was performed for 1 h at room temperature with shaking and subsequently quenched with 10 μL of 5% hydroxylamine hydrochloride. The labeled peptides were combined, purified, and dried before fractionation. High-pH reversed-phase HPLC was employed to fractionate the combined sample using an XBridge C18 3.5 μm, 4.6 × 250 mm column (Waters) with an 80 min gradient of 5–60% ACN containing 10 mM ammonium formate (pH = 10). The peptides were consolidated into 20 samples and each sample was purified using the StageTip method described previously.24
LC-MS/MS Analysis.
The dried peptides were resuspended, separated by reversed-phase HPLC. Data were acquired with a hybrid dual-cell quadrupole linear ion trap-Orbitrap mass spectrometer (LTQ Orbitrap Elite, Thermo Scientific, with Xcalibur 3.0.63 software) using a data-dependent Top15 method. The detailed information is included in the Supporting Information.
Database Searching, Data Filtering, and Quantitation of Protein Relative Synthesis Rates.
The raw files were converted into the mzXML format. The mass spectra were searched using the SEQUEST algorithm (version 28)25 against the human proteome (Homo sapiens) database encompassing sequences of all proteins downloaded from UniProt (https://www.uniprot.org). The following parameters were used for the search: 20 ppm precursor mass tolerance; 0.025 Da product ion mass tolerance; fully digested with trypsin; up to 3 missed cleavages; variable modifications: oxidation of methionine (+15.9949); fixed modifications: carbamidomethylation of cysteine (+57.0214) and the TMT labeling of lysine and the N-terminus (+229.1630). The target-decoy method was employed to evaluate and control the false discovery rates (FDRs) of peptide and protein identifications.26 The detailed information is included in the Supporting Information.
The ion intensities for the eight TMT channels were recorded and corrected using the isotopic information provided by Thermo. The abundance of each newly synthesized protein in every sample was calculated from the median TMT intensity of all peptides from this protein. The experiment was performed in technical triplicates. In each replicate, the abundances of proteins in the background sample were subtracted from the abundances of proteins in the other seven samples to eliminate the possible influence from nonspecific binding. For every protein, the relative synthesis rate at each point in each replicate was calculated between the LPS-treated and the control samples. Eventually, the final relative synthesis rate and the standard deviation were calculated from the average of the three replicates.
Statistical Analysis.
Gene Ontology (GO)-based enrichment analysis was performed based on cellular component, molecular function, and biological process using the Protein Analysis Through Evolutionary Relationships (PANTHER) classification system.27 P-values were calculated using Fisher’s exact test. Proteins that changed the relative synthesis rate by at least 1.5-fold were considered to be significantly affected by the treatment.
RESULTS AND DISCUSSION
Principle for Quantitation of Rapid Protein Synthesis Changes Using Quantitative O-Propargyl-Puromycin Tagging (QOT).
In this work, we employed OPP, an alkyne-bearing puromycin derivative, to effectively and rapidly label newly synthesized proteins. It has been shown that OPP can be incorporated into newly synthesized proteins in a short incubation time.18,19 The incorporation of OPP into newly synthesized proteins generates a chemical handle for their enrichment using bioorthogonal chemistry (Figure 1A).28–31 The enriched newly synthesized proteins are covalently bound to the agarose resins, which enables stringent washes to minimize nonspecific binding.
Figure 1.

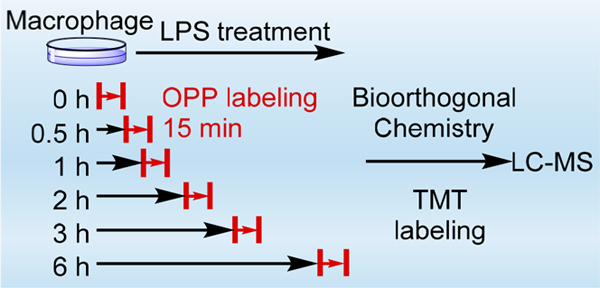
Experimental procedure for enrichment and quantitation of rapid changes of newly synthesized proteins in cells. (A) Enrichment of newly synthesized proteins via the click reaction between OPP-conjugated newly synthesized proteins and azide agarose resins. (B) Experimental procedure for the quantitation of the relative synthesis rates in macrophages treated with LPS.
Here, THP-1 cells were equally passaged to eight flasks and differentiated into M0 macrophages with phorbol 12-myristate 13-acetate (PMA) as reported previously.23 Then, the macrophages were treated with LPS, except those designated as the control and background samples. The cells were treated with OPP at different time points, i.e., 0, 0.5, 1, 2, 3, and 6 h after the LPS challenge, for 15 min without starvation or any pretreatment before the labeling to ensure that the cells are under no stress. The short labeling time also minimizes the effects of protein degradation on the quantitation of the synthesis rate. Similarly, the cells in the control and background samples were also labeled with OPP for 15 min. The cells were then harvested and lysed, and the labeled newly synthesized proteins from all samples were enriched. For the background sample, the catalytic reagents were not added during the click reaction and thus proteins quantified are those only resulted from nonspecific binding.
After stringent washes, the enriched proteins were digested on beads, and the resulted peptides were further labeled with different tandem mass tag (TMT) reagents, respectively, i.e., 127N for 0 h, 127C for 0.5 h, 128N for 1 h, 128C for 2 h, 129N for 3 h, 129C for 6 h, 130N for the control sample, and 130C for the background sample. Multiplexed proteomics allows for simultaneous quantitation of all these samples, therefore increasing the throughput and quantitation accuracy. After fractionation and purification, the peptides were analyzed by an online LC-MS system (Figure 1B). The intensities of nonspecific binding proteins were subtracted from those of the corresponding proteins in all other samples (analysis of nonspecific binding proteins on the azide agarose resins including Figures S1 and S2 is in the Supporting Information), and the relative synthesis rates were calculated from the ratios between the protein intensities from each time point and those in the control sample.
The incorporation of OPP prevents the elongation of newly synthesized proteins and causes the premature release of truncated proteins. The labeled proteins are then enriched and analyzed using bottom-up proteomics. The measurement of protein synthesis is affected by the degradation of the labeled proteins if the labeling time is too long in the experiments. However, previous work showed that the abundances of OPP-containing proteins were not significantly reduced after 30 min while their degradation needed several hours.22,32 In this work, newly synthesized proteins are labeled for 15 min and the labeling time is the same for all time points. This minimizes the influence of protein degradation and increases the quantitation accuracy.
Because OPP labeling causes premature termination of protein synthesis, the identified peptides might be biased toward the N-terminus of proteins. To evaluate whether OPP labeling is biased, we performed more data analysis for all identified peptides in one experiment, and the distribution of the starting positions of peptides in their proteins is shown in Figure 2. The number of identified peptides is almost equally distributed across the whole length of proteins, except fewer at both N- and C-termini. The same results were found in other experiments. The possible reason for fewer N- and C-terminal peptides is that the peptides at the N- and C-termini of proteins are too short after the trypsin digestion, and peptides with the length of less than seven amino acid residues were discarded for the quality control of peptide identification. Additionally, the N-termini of proteins may be modified, and in this work we did not add any possible modification on the N-terminus. Therefore, modified N-terminal peptides were not identified. The results demonstrate that the current method does not bias toward the N-termini of nascent proteins.
Figure 2.
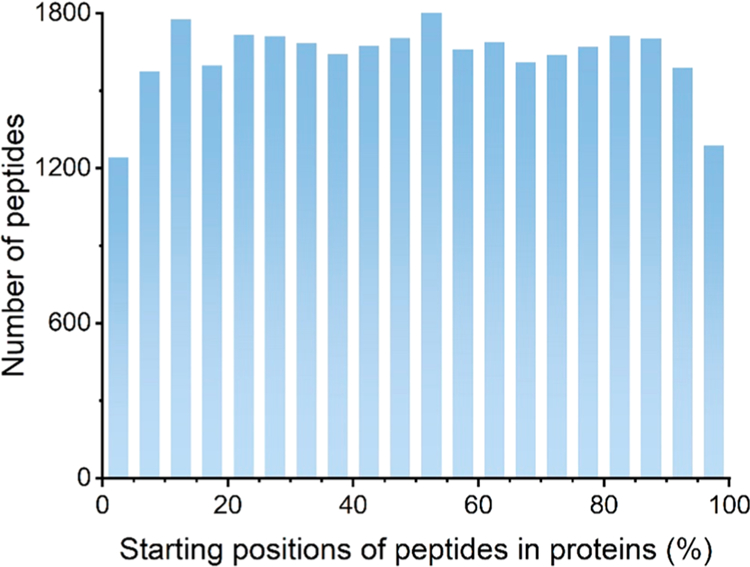
Distribution of the starting positions of all identified peptides in their proteins. The starting position was converted into a percentage where 0% means the peptide starting at the N-terminus and 100% refers to the peptide starting at the C-terminus. Each bin represents the number of peptides within the range of 5%. For example, the first column shows the number of peptides starting between 0% and 5% in proteins.
Identification of Newly Synthesized Proteins in LPS-Treated M0 THP-1 Macrophages.
During the activation induced by LPS, M0 THP-1 macrophages produce and secrete numerous proteins including inflammatory cytokines, enzymes, and growth factors.33 The LPS receptor complex consists of toll-like receptor 4 (TLR4), myeloid differentiation protein-2 (MD-2), and CD14. Its interaction with LPS results in the activation of IκB kinase (IKK), mitogen-activated protein kinase (MAPK), extracellular signal-regulated kinase (ERK) 1 and ERK2, c-Jun N-terminal kinases (JNK), and other signaling pathways.34–37 As a consequence, a number of transcription factors, such as nuclear factor κB (NF-κB) and activator protein-1 (AP-1), are activated, which promote the synthesis of many proteins related to the innate immune response.38–40 The response of macrophages is fast, as shown in the previous study that the phosphoproteome was greatly altered within the first 15 min after the LPS treatment.41 Therefore, conventional methods are not suitable to accurately and sensitively quantify the quick changes of protein synthesis in macrophages in response to LPS.
Using the proposed method, we identified 3758 newly synthesized proteins within the labeling time of 15 min after the LPS treatment in three replicates (Figure 3A). Among those, 2589 proteins quantitated commonly at all time points in all triplicates were chosen for further analysis (Table S1). Gene Ontology (GO) clustering of all newly synthesized proteins was performed using the Protein Analysis Through Evolutionary Relationships (PANTHER) classification system based on cellular component, molecular function, and biological process.27 P-values were calculated using Fisher’s exact test. Nearly 600 newly synthesized proteins located in the extracellular exosome were highly enriched with a very low P-value of 6.01 × 10−71. These extracellular exosomes released from macrophages play vital roles in intercellular communication and inflammatory response, and it is well-known that the LPS treatment promotes the secretion of extracellular exosomes from macrophages.42,43
Figure 3.

Newly synthesized proteins identified in macrophages treated with LPS and some results from protein clustering. (A) Newly synthesized proteins identified in the triplicate experiments. (B) Clustering of newly synthesized proteins related to protein secretion. (C) Clustering of newly synthesized proteins related to the immune response.
In addition, newly synthesized proteins involved in protein transport (P = 3.59 × 10−58), exocytosis (P = 1.89 × 10−31), and protein targeting to membrane (P = 2.02 × 10−28) were overrepresented (Table S2 and Figure 3B). These results demonstrated that protein synthesis related to the secretion of extracellular exosomes was highly promoted in cells by LPS. After the activation of macrophages, multiple pro-inflammatory cytokines were rapidly synthesized and released, such as tumor necrosis factor (TNF). The classical secretion pathway is mainly responsible for the secretion of TNF, and the Golgi transport complex is an important component of this pathway.33,44 In the current data set, 8 out of 13 proteins involved in the Golgi transport complex were quantitated, indicating that protein synthesis related to this pathway is greatly activated. Another way to release TNF is through the recycling endosome,44,45 and the newly synthesized proteins related to the recycling endosome were overrepresented as well.
Furthermore, newly synthesized proteins involved in the activation of the innate immune response were enriched, indicating a successful activation of THP-1 macrophages by LPS (Figure 3C). The activation of macrophages resulted in the enrichment of newly synthesized proteins related to NF-κB signaling, Wnt signaling, and Rab protein signal transduction, which are critical for the regulation of immune response by macrophages.46,47 Subsequently, the pro-inflammatory cytokines were synthesized and released from the LPS-induced macrophages, and these cytokines can further activate the cells. For example, TNF acts on TNF receptor 1 (TNFR1) and TNF receptor 2 (TNFR2), which activates the macrophages through the TNF-mediated signaling pathway.48 Here, thirty-nine newly synthesized proteins quantitated are related to the TNF-mediated signaling pathway. Similarly, proteins related to the interleukin-1 (IL-1)-mediated signaling pathway were also overrepresented. The results correspond very well with the previous studies that macrophages can be activated by TNF and IL-1.48,49
Quantitation of the Protein Synthesis Rates in Macrophages Treated with LPS.
After the protein abundance correction using the background sample, the relative synthesis rate for each protein at each time point was calculated based on the ratios between the protein abundances from the cells treated with LPS and the control sample, which is under the normal synthesis rates for the resting macrophages (Figure S3A). To compensate for the technical variabilities, the raw relative synthesis rates were normalized based on three commonly used proteins, i.e., tubulin beta chain (TUBB), β-actin (ACTB), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which were not affected during the LPS-induced macrophage activation. In each replicate, the median relative synthesis rate of these three proteins was obtained at each time point and used for the normalization of all other relative synthesis rates at that time point (Figure S3B). The distribution of relative synthesis rates for all proteins after the normalization in one replicate is shown in Figure S3C. Eventually, the final relative synthesis rate for each protein at every time point is the average value of the relative synthesis rates from three replicates (Table S1 and Figure S4A). The distribution of relative standard deviation shows that the triplicate experiments were in reasonably good agreement (Figure S4B). The results show that protein synthesis is relatively more promoted at 0.5 and 6 h during the LPS treatment.
Overall, the relative synthesis rates of 238 proteins were dramatically affected by the LPS treatment (the relative synthesis rate changed by at least 1.5-fold). Among those, the synthesis of 229 proteins was promoted, while 9 proteins had reduced synthesis rates. GO analysis demonstrates that proteins related to protein transport, Golgi vesicle transport, innate immune response, and immune system process were highly enriched. In addition, many proteins related to the two major pathways in the immune response, i.e., the NF-κB signaling pathway and the TNF signaling pathway, were drastically affected by the LPS treatment (Table S2 and Figure S5).
The synthesis of those proteins markedly affected by the LPS treatment was classified into four groups based on their response kinetics (Table S4 and Figure 4). Group I (“immediate increase”) consists of 17 proteins, including TNF and transcription factor p65 (RELA). These proteins had notably increased relative synthesis rates at the beginning (0 and 0.5 h) of the LPS treatment. Group II (“intermediate increase”) includes 13 proteins, such as IL-1 beta (IL1B) and TNF receptor-associated factor 1 (TRAF1). These proteins had increased relative synthesis rates between 1 and 3 h during the LPS treatment. One hundred and ninety-nine proteins were clustered in Group III (“delayed increase”), which increased their relative synthesis rates at 6 h of the LPS treatment. On the contrary, several proteins had decreased relative synthesis rates, which are clustered in Group IV (“decrease”).
Figure 4.
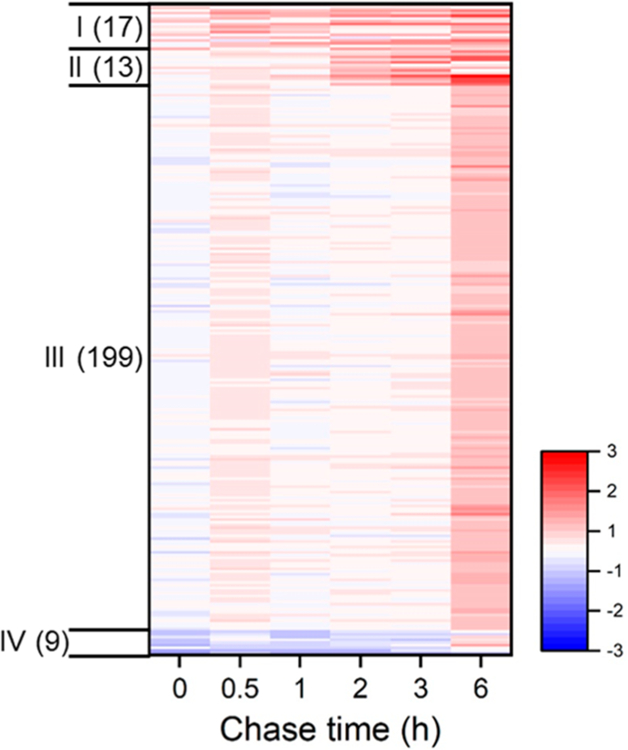
A heat map shows the relative synthesis rates of 238 proteins that were significantly affected by the LPS treatment. Proteins are clustered into four groups based on the response kinetics.
Translation of Cytokines and Transcription Factors in Macrophages in Response to LPS.
The synthesis rates of several cytokines, transcription factors, and other proteins related to the inflammatory process were greatly affected by the LPS treatment. Their relative synthesis rates with the error bars referring to the standard deviation at each time point are shown in Figures 5 and S6. The translation of these proteins in macrophages is known to be promoted by the LPS treatment; thus, they are the direct proof that the M0 THP-1 macrophages were activated by LPS. The rapid changes of the protein synthesis within 15 min at each time point during the treatment were successfully determined by the current method.
Figure 5.

Relative synthesis rates of transcription factors, cytokines, and other proteins in macrophages treated with LPS. (A) Heat map showing the relative synthesis rates of several example proteins at each time point during the LPS treatment. (B–G) The relative synthesis rates with the error bars referring to the standard deviation at each time point for (B) c-REL, (C) CCL5, (D) IL1B, (E) SOD2, (F) IGFBP3, and (G) ICAM1 (CD54).
NF-κB as a transcription factor is critical for the regulation of immune response. NF-κB is activated by the LPS treatment, which subsequently promotes the expression of many cytokines, transcription factors, and other proteins related to the inflammatory process. In this work, two subunits of NF-κB, i.e., RELA and c-REL, were quantitated and classified in Group I. The previous studies found that these two subunits could bind to about 190 genes and were essential for the LPS-induced immune response in macrophages.36,38,40,50 The relative synthesis rates of the two subunits were upregulated during the whole process of the LPS treatment (Figures 5B and S6A). Notably, the relative synthesis rate of RELA was 1.53 ± 0.19 at 0 h during the LPS treatment, while that of c-REL was 2.00 ± 0.18 at 0.5 h. Here, newly synthesized proteins were labeled with OPP for 0.25 h (15 min) at each time point. Therefore, the relative synthesis rate at 0 h reflects the protein synthesis from 0 to 0.25 h.
The expression of many cytokines, including TNF, IL1B, C–C motif chemokine 5 (CCL5), and C–C motif chemokine 20 (CCL20), was largely promoted by the LPS treatment through the activation of NF-κB and other transcription factors. CCL5 (or regulated upon activation, normal T cell expressed and secreted (RANTES)) plays critical roles in inflammatory responses by activating and recruiting T cells and macrophages. The expression of CCL5 is induced through the activation of IFN regulatory factor-3 (IRF3) after the LPS treatment.51 In this work, the relative synthesis rate significantly increased at 1 h and reached nearly 7-fold at 6 h during the LPS treatment compared with the normal synthesis rate (Figure 5C). Similarly, the expression of CCL20, also known as macrophage inflammatory protein-3α, significantly increased at 0.5 h, and the relative synthesis rate was 4.54 ± 0.91 at 6 h (Figure S6B). The increased expression of CCL20 was reported in a previous study using an MS-independent method.52 TNF and IL1B are two key regulators of the inflammatory response.48,49 It is well-known that the synthesis and secretion of TNF and IL1B were promoted through the activation of NF-κB in the LPS-induced macrophage activation.34,39,53 The relative synthesis rate of TNF was >1.5 at 0.5 h, and that of IL1B was 4.42 ± 0.88 at 6 h (Figures 5D and S6C). The results correspond very well with previous studies and further validate the effectiveness of the current method.
Synthesis Alteration of Other Important Proteins in Macrophages Treated with LPS.
Besides transcription factors and cytokines, proteins with various functions were also found to be increasingly synthesized during the LPS treatment. For example, cell stress-related superoxide dismutase [Mn] (SOD2), a protein responsible for destroying superoxide anion radicals and one of the target genes of NF-κB,54 increased its synthesis rate at 2 h after the LPS treatment (Figure 5E). Similarly, the synthesis rate of prostaglandin G/H synthase 2 (PTGS2), also known as COX-2, elevated over 8-fold during the LPS treatment (Figure S6D), which is known to be strongly induced by LPS.55 The synthesis of some proteins with anti-inflammatory functions altered in macrophages treated with LPS. For example, insulin-like growth factor-binding protein 3 (IGFBP3) is known to induce cell apoptosis and inhibit the NF-κB signaling pathway.56 The synthesis of IGFBP3 was slightly decreased at the beginning of the LPS treatment (0.80 ± 0.09), while the relative synthesis rate increased to 2.64 ± 0.47 at 6 h (Figure 5F). The result demonstrates that although the anti-inflammatory process is suppressed at the beginning, a prolonged LPS treatment may promote the synthesis of anti-inflammatory proteins to counteract the inflammatory response induced by LPS.
Intercellular adhesion molecule-1 (ICAM1, CD54) is vital in the cellular response to LPS, TNF, and IL-1. The relative synthesis rate of CD54 is 1.60 ± 0.16 at 6 h during the LPS treatment (Figure 5G). Besides CD54, two other CD proteins, i.e., urokinase plasminogen activator surface receptor (PLAUR, CD87) and disintegrin and metalloproteinase domain-containing protein 17 (ADAM17, CD156b), were also significantly affected by LPS. A previous study showed that the mRNA abundance of CD87 increased after 4 h of the LPS treatment,57 and here an increased relative synthesis rate was found at 6 h during the LPS treatment (Figure S6E). ADAM17 is a surface enzyme, which cleaves and releases the TNF precursor.58 This protein is involved in LPS-induced inflammation, and its synthesis was greatly promoted (Figure S6F). In addition, the synthesis of 9 proteins was reduced during the LPS treatment (Figures S6G and S6H). Their functions are not directly related to the inflammatory process, and further study is needed to uncover the mechanism of the translation suppression of these proteins.
Protein Synthesis Promoted in the Classical Secretory Pathway.
After the LPS treatment, more cytokines and other proteins related to the inflammatory response, such as TNF, IL1B, CCL5, CCL20, ICAM1 (CD54), and PTGS2 (COX-2), are secreted through the classical secretory pathway. They may be regulated at the transcription, translation, post-translational modification, and protein translocation levels.44,45 We found that the synthesis of many proteins related to this pathway was significantly induced during the LPS treatment, and some examples are displayed in Figure 6.
Figure 6.

Proteins related to the classical secretory pathway with altered relative synthesis rates were clustered and displayed based on biological process or cellular compartment. Proteins in Group I (“immediate increase”), Group II (“intermediate increase”), and Group III (“delayed increase”) are colored in green, red, and yellow, respectively.
Apart from RELA and c-REL, several transcription factors related to the inflammatory process were markedly affected by LPS, such as A-kinase anchor protein 8 (AKAP8), Y-box-binding protein 1 (YBX1), and zinc finger protein 384 (ZNF384), while some of them may not be associated with the inflammatory process, including putative oxidoreductase GLYR1 and high mobility group protein HMG-I/HMG-Y (HMGA1). AKAP8 plays a role in the anti-inflammatory process and suppresses the expression of TNF in macrophages.59 YBX1 is important for transcription regulation, and its expression was promoted by LPS in a previous study using mice as a model.60 ZNF384, also known as CIZ, binds with the DNA sequence (G/C)AAAAA and activates the expression of matrix metalloproteinases (MMPs) which are upregulated in the LPS-induced inflammatory process.61 The relative synthesis rate of HMGA1 drastically increased at 0.5 h during the LPS treatment, and the expression of HMGA1 was previously reported to be promoted in activated macrophages by transcriptomic analysis.62 However, its role in the inflammatory process remains to be explored.
RNA splicing is a critical step to generate the mature mRNA, which is accomplished by the spliceosome. The synthesis of many proteins related to the spliceosome, such as splicing factors (RBM22, SRSF10, SRSF7, and SF3B2), ribonucleoproteins (HNRNPC and HNRNPU), and RNA helicase (DDX5), was largely induced during the LPS treatment. In addition, the synthesis of several nuclear pore complex proteins (NUP98, NUP107, NUP153, NUP160, and RAN) was also promoted to enhance the translocation of mature mRNA to the ER-bounded ribosome.
The ER is an essential component for the synthesis and trafficking of secreted proteins.63 Here, many proteins related to protein secretion in the ER and the ribosomes, and N-glycosylation were quantitated and their synthesis rates were greatly altered by the LPS treatment. Ribosome-binding protein 1 (RRBP1) mediates the interaction between the ribosomes and the ER membrane, which is critical for the classical secretory pathway. The relative synthesis rate was 1.71 ± 0.14 at 6 h during the LPS treatment. The increased synthesis of RRBP1 suggests the activation of secreted protein synthesis. In addition, the synthesis of proteins related to protein folding (DNAJA1, DNAJA2, and DNAJB11), quality control (SEL1L), and translocation (SEC24B and SEC63) was also promoted by LPS.
Most secreted proteins are N-glycosylated, and N-glycosylation often determines protein folding, trafficking, and quality control.64,65 The oligosaccharyl transferase (OST) complex is responsible for the en bloc transfer of Glc3Man9GlcNAc2 to the asparagine (Asn) residue in the sequence of N-X-S/T (X is any amino acid residues but not proline) of newly synthesized secreted proteins. Dolichyl-diphosphooligosaccharide-protein glycosyltransferase subunit STT3B is a catalytic subunit of the OST complex, and the synthesis of STT3B was increased during the LPS treatment. Similarly, an accessory component of the OST complex, tumor suppressor candidate 3 (TUSC3), was induced in the activated macrophages. In addition, the synthesis of proteins involved in the formation of precursor oligosaccharide (ALG1) and the modification of N-glycans (B4GALT1 and PRKCSH) was also upregulated in the LPS-induced macrophages. The results suggest that protein N-glycosylation is activated by the LPS treatment.
CONCLUSIONS
In this work, an effective method termed quantitative O-propargyl-puromycin tagging (QOT) was developed to investigate rapid protein synthesis changes in cells. OPP labeling has been proved to be a powerful method to measure rapid protein synthesis. By integrating OPP labeling with multiplexed proteomics, the current method enables labeling and selective enrichment of rapid translation products for quantitative analysis by MS. Compared with other commonly used approaches, the current method has several advantages. First, OPP can be efficiently incorporated into nascent polypeptides without prior cell starvation or pretreatment. This greatly minimizes the influence on cellular metabolism caused by the stress and reflects the real changes of protein synthesis in response to the stimulus. Second, OPP can be utilized by the ribosome immediately after entering cells, while heavy isotopic and/or noncanonical amino acids need to be activated and conjugated to tRNA before being incorporated into newly synthesized proteins. Therefore, the labeling efficiency of OPP is much higher than those amino acids or their analogs, especially in the early labeling stage, enabling us to label newly synthesized proteins in a short period of time (such as 15 min used here). Third, a short labeling time eliminates (at least minimizes) the effect on quantitation of protein synthesis by the protein degradation. Fourth, one OPP molecule is incorporated into one newly synthesized protein, which allows us to fairly enrich newly synthesized proteins with different sequences.
The current method was applied to study the rapid changes of protein synthesis in macrophage treated by LPS. More than 3000 newly synthesized proteins were identified with 15-min labeling in M0 THP-1 macrophages after the LPS treatment. The synthesis rates of 238 proteins were dramatically altered, including transcription factors, cytokines, CD proteins, and other proteins related to the inflammatory process. This method enabled us to quantitate protein synthesis immediately after the LPS treatment and to identify proteins with altered synthesis rates in a short period of time (15 min) after the treatment. The results also demonstrate that the protein synthesis was modulated to facilitate protein secretion during the macrophage activation, supported by the increased synthesis of multiple proteins related to the classical secretory pathway, including five enzymes for protein N-glycosylation. This method can be widely applied to investigate rapid changes of protein synthesis in cells.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM118803.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c01823.
Complete description of methods, analysis of nonspecific binding proteins, and supporting figures (PDF) Quantitation of the relative synthesis rates of 2589 proteins in LPS-induced M0 THP-1 macrophages (Table S1) (XLSX)
Clustering of newly synthesized proteins in LPS-induced THP-1 M0 macrophages (Table S2) (XLSX) Quantitation of the ratios of the abundances between nonspecific binding proteins and enriched ones (Table S3) (XLSX)
Quantitation of the relative synthesis rates of 238 significantly affected proteins (Table S4) (XLSX)
The authors declare no competing financial interest.
REFERENCES
- (1).Labbadia J; Morimoto RI Annu. Rev. Biochem 2015, 84, 435–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Roux PP; Topisirovic I Signaling pathways involved in the regulation of mRNA translation, Mol. Cell. Biol 2018, 38 (12), 10.1128/MCB.00070-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wang X; Proud CG Physiology 2006, 21, 362–369. [DOI] [PubMed] [Google Scholar]
- (4).Clemens MJ; Bushell M; Jeffrey IW; Pain VM; Morley SJ Cell Death Differ 2000, 7 (7), 603–615. [DOI] [PubMed] [Google Scholar]
- (5).Dever TE; Kinzy TG; Pavitt GD Genetics 2016, 203 (1), 65–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Eichelbaum K; Krijgsveld J Mol. Cell. Proteomics 2014, 13 (3), 792–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Rothenberg DA; Taliaferro JM; Huber SM; Begley TJ; Dedon PC; White FM iScience 2018, 9, 367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ingolia NT Nat. Rev. Genet 2014, 15 (3), 205–213. [DOI] [PubMed] [Google Scholar]
- (9).Wang Z; Gerstein M; Snyder M Nat. Rev. Genet 2009, 10 (1), 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Schwanhausser B; Busse D; Li N; Dittmar G; Schuchhardt J; Wolf J; Chen W; Selbach M Nature 2011, 473 (7347), 337–342. [DOI] [PubMed] [Google Scholar]
- (11).Liu YS; Beyer A; Aebersold R Cell 2016, 165 (3), 535–550. [DOI] [PubMed] [Google Scholar]
- (12).Liu TY; Huang HH; Wheeler D; Xu Y; Wells JA; Song YS; Wiita AP Cell Syst 2017, 4 (6), 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dieterich DC; Link AJ; Graumann J; Tirrell DA; Schuman EM Proc. Natl. Acad. Sci. U. S. A 2006, 103 (25), 9482–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ma YH; McClatchy DB; Barkallah S; Wood WW; Yates JR Nat. Protoc 2018, 13 (8), 1744–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chen W; Smeekens JM; Wu R Chem. Sci 2016, 7 (2), 1393–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tong M; Xiao H; Smeekens J; Wu R Chem. Sci 2020, 11, 3557–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Howden AJM; Geoghegan V; Katsch K; Efstathiou G; Bhushan B; Boutureira O; Thomas B; Trudgian DC; Kessler BM; Dieterich DC; Davis BG; Acuto O Nat. Methods 2013, 10 (4), 343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nagelreiter F; Coats MT; Klanert G; Gludovacz E; Borth N; Grillari J; Schosserer M Biotechnol. J 2018, 13 (4), e1700492. [DOI] [PubMed] [Google Scholar]
- (19).Forester CM; Zhao Q; Phillips NJ; Urisman A; Chalkley RJ; Oses-Prieto JA; Zhang L; Ruggero D; Burlingame AL Proc. Natl. Acad. Sci. U. S. A 2018, 115 (10), 2353–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Liu J; Xu Y; Stoleru D; Salic A Proc. Natl. Acad. Sci. U. S. A 2012, 109 (2), 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Barrett RM; Liu HW; Jin H; Goodman RH; Cohen MS ACS Chem. Biol 2016, 11 (6), 1532–1536. [DOI] [PubMed] [Google Scholar]
- (22).Hidalgo San Jose L; Signer RA J. Nat. Protoc 2019, 14 (2), 441–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chanput W; Mes J; Vreeburg RA; Savelkoul HF; Wichers HJ Food Funct 2010, 1 (3), 254–261. [DOI] [PubMed] [Google Scholar]
- (24).Rappsilber J; Mann M; Ishihama Y Nat. Protoc 2007, 2 (8), 1896–1906. [DOI] [PubMed] [Google Scholar]
- (25).Eng JK; McCormack AL; Yates JR J. Am. Soc. Mass Spectrom 1994, 5 (11), 976–989. [DOI] [PubMed] [Google Scholar]
- (26).Elias JE; Gygi SP Nat. Methods 2007, 4 (3), 207–214. [DOI] [PubMed] [Google Scholar]
- (27).Mi H; Muruganujan A; Huang X; Ebert D; Mills C; Guo X; Thomas PD Nat. Protoc 2019, 14 (3), 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wang Q; Chan TR; Hilgraf R; Fokin VV; Sharpless KB; Finn MG J. Am. Chem. Soc 2003, 125 (11), 3192–3193. [DOI] [PubMed] [Google Scholar]
- (29).Chen WX; Smeekens JM; Wu RH Chem. Sci 2015, 6 (8), 4681–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Xiao HP; Suttapitugsakul S; Sun FX; Wu RH Acc. Chem. Res 2018, 51 (8), 1796–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Suttapitugsakul S; Ulmer LD; Jiang CD; Sun FX; Wu RH Anal. Chem 2019, 91 (10), 6934–6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Signer RAJ; Magee JA; Salic A; Morrison SJ Nature 2014, 509 (7498), 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Stow JL; Low PC; Offenhauser C; Sangermani D Immunobiology 2009, 214 (7), 601–612. [DOI] [PubMed] [Google Scholar]
- (34).Xiong Y; Hales DB Endocrinology 1993, 133 (6), 2568–2573. [DOI] [PubMed] [Google Scholar]
- (35).Fujihara M; Muroi M; Tanamoto K; Suzuki T; Azuma H; Ikeda H Pharmacol. Ther 2003, 100 (2), 171–194. [DOI] [PubMed] [Google Scholar]
- (36).Schreiber J; Jenner RG; Murray HL; Gerber GK; Gifford DK; Young RA Proc. Natl. Acad. Sci. U. S. A 2006, 103 (15), 5899–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hambleton J; Weinstein SL; Lem L; DeFranco AL Proc. Natl. Acad. Sci. U. S. A 1996, 93 (7), 2774–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ouaaz F; Li M; Beg AA J. Exp. Med 1999, 189 (6), 999–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Pahl HL Oncogene 1999, 18 (49), 6853–6866. [DOI] [PubMed] [Google Scholar]
- (40).Sharif O; Bolshakov VN; Raines S; Newham P; Perkins ND BMC Immunol 2007, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Weintz G; Olsen JV; Fruhauf K; Niedzielska M; Amit I; Jantsch J; Mages J; Frech C; Dolken L; Mann M; Lang R Mol. Syst. Biol 2010, 6, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Bhatnagar S; Shinagawa K; Castellino FJ; Schorey JS Blood 2007, 110 (9), 3234–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Singh PP; Smith VL; Karakousis PC; Schorey JS J. Immunol 2012, 189 (2), 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Murray RZ; Stow JL Front. Immunol 2014, 5, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Murray RZ; Kay JG; Sangermani DG; Stow JL Science 2005, 310 (5753), 1492–1495. [DOI] [PubMed] [Google Scholar]
- (46).Lee H; Bae S; Choi BW; Yoon Y Immunopharmacol. Immunotoxicol 2012, 34 (1), 56–65. [DOI] [PubMed] [Google Scholar]
- (47).Pei G; Bronietzki M; Gutierrez MG J. Leukocyte Biol 2012, 92 (1), 41–50. [DOI] [PubMed] [Google Scholar]
- (48).Bradley JR J. Pathol 2008, 214 (2), 149–160. [DOI] [PubMed] [Google Scholar]
- (49).Dinarello CA Blood 2011, 117 (14), 3720–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wang Y; Rickman BH; Poutahidis T; Schlieper K; Jackson EA; Erdman SE; Fox JG; Horwitz BH J. Immunol 2008, 180 (12), 8118–8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Qian X; Zhang J; Liu J J. Biol. Chem 2011, 286 (3), 2111–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Schutyser E; Struyf S; Menten P; Lenaerts JP; Conings R; Put W; Wuyts A; Proost P; Van Damme J J. Immunol 2000, 165 (8), 4470–4477. [DOI] [PubMed] [Google Scholar]
- (53).Netea MG; Nold-Petry CA; Nold MF; Joosten LA; Opitz B; van der Meer JH; van de Veerdonk FL; Ferwerda G; Heinhuis B; Devesa I; Funk CJ; Mason RJ; Kullberg BJ; Rubartelli A; van der Meer JW; Dinarello CA Blood 2009, 113 (10), 2324–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Morgan MJ; Liu ZG Cell Res 2011, 21 (1), 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Eliopoulos AG; Dumitru CD; Wang CC; Cho JH; Tsichlis PN EMBO J 2002, 21 (18), 4831–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Williams AC; Smartt H; AM H. Z.; Macfarlane M; Paraskeva C; Collard TJ Cell Death Differ 2007, 14 (1), 137–145. [DOI] [PubMed] [Google Scholar]
- (57).Scott MG; Rosenberger CM; Gold MR; Finlay BB; Hancock RE J. Immunol 2000, 165 (6), 3358–3365. [DOI] [PubMed] [Google Scholar]
- (58).Gooz M Crit. Rev. Biochem. Mol. Biol 2010, 45 (2), 146–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Wall EA; Zavzavadjian JR; Chang MS; Randhawa B; Zhu XC; Hsueh RC; Liu J; Driver A; Bao XR; Sternweis PC; Simon MI; Fraser IDC. Suppression of LPS-induced TNF-alpha production in macrophages by camp is mediated by PKA-AKAP95-p105. Sci. Signaling 2009, 2 (75), ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Hanssen L; Alidousty C; Djudjaj S; Frye BC; Rauen T; Boor P; Mertens PR; van Roeyen CR; Tacke F; Heymann F; Tittel AP; Koch A; Floege J; Ostendorf T; Raffetseder U J. Immunol 2013, 191 (5), 2604–2613. [DOI] [PubMed] [Google Scholar]
- (61).Nakamoto T; Yamagata T; Sakai R; Ogawa S; Honda H; Ueno H; Hirano N; Yazaki Y; Hirai H Mol. Cell. Biol 2000, 20 (5), 1649–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Xue J; Schmidt SV; Sander J; Draffehn A; Krebs W; Quester I; De Nardo D; Gohel TD; Emde M; Schmidleithner L; Ganesan H; Nino-Castro A; Mallmann MR; Labzin L; Theis H; Kraut M; Beyer M; Latz E; Freeman TC; Ulas T; Schultze JL Immunity 2014, 40 (2), 274–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Benham AM Cold Spring Harbor Perspect. Biol 2012, 4 (8), a012872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Aebi M Biochim. Biophys. Acta, Mol. Cell Res 2013, 1833 (11), 2430–2437. [DOI] [PubMed] [Google Scholar]
- (65).Roth J Chem. Rev 2002, 102 (2), 285–303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.