

SUMMARY
Caspases regulate cell death, immune responses, and homeostasis. Caspase-6 is categorized as an executioner caspase but shows key differences from the other executioners. Overall, little is known about the functions of caspase-6 in biological processes apart from apoptosis. Here, we show that caspase-6 mediates innate immunity and inflammasome activation. Furthermore, we demonstrate that caspase-6 promotes the activation of programmed cell death pathways including pyroptosis, apoptosis, and necroptosis (PANoptosis) and plays an essential role in host defense against influenza A virus (IAV) infection. In addition, caspase-6 promoted the differentiation of alternatively activated macrophages (AAMs). Caspase-6 facilitated the RIP homotypic interaction motif (RHIM)-dependent binding of RIPK3 to ZBP1 via its interaction with RIPK3. Altogether, our findings reveal a vital role for caspase-6 in facilitating ZBP1-mediated inflammasome activation, cell death, and host defense during IAV infection, opening additional avenues for treatment of infectious/autoinflammatory diseases and cancer.
In Brief
Caspase-6 modulates innate immunity, inflammasome activation, and multiple cell death pathways
Graphical Abstract
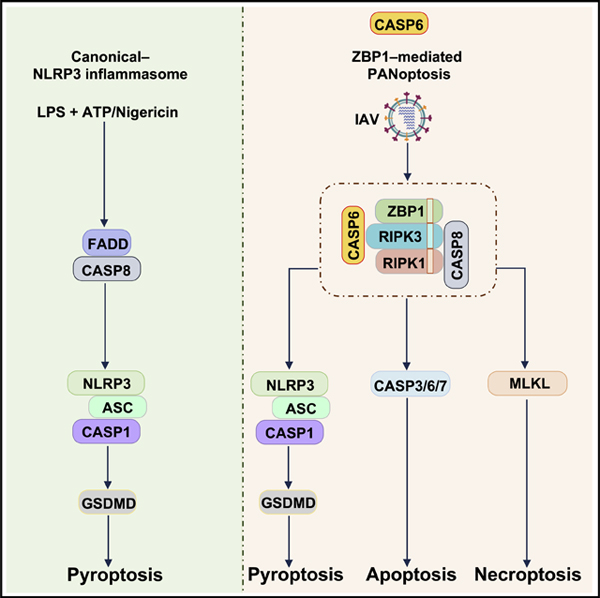
INTRODUCTION
Members of the caspase family of cysteine proteases play crucial roles in controlling host cell death, innate immune responses, and homeostasis (Galluzzi et al., 2016; Man and Kanneganti, 2016). Caspases are expressed in cells as inactive zymogens composed of an amino-terminal prodomain and a carboxy-terminal protease domain containing large and small catalytic subunits (Nicholson and Thornberry, 1997). To date, 14 caspases have been identified in mammals. Based on their functions, caspases can be grouped as inflammatory caspases (caspase-1, −4, −5, and −11) and apoptotic caspases (caspase-3, −6, −7, −8, −9, and −10) (Man and Kanneganti, 2016). Apoptotic caspases are further functionally classified as initiator caspases (caspase-8, −9, and −10) and effector (or executioner) caspases (caspase-3, −6, and −7).
Caspases are involved in multiple cell death pathways in response to damage-associated or pathogen-associated molecular patterns (DAMPs or PAMPs). One of these cell death pathways, pyroptosis, is executed by processed gasdermin D (GSDMD) in a caspase-1-dependent manner following inflammasome assembly. Pyroptosis allows the efficient release of inflammasome-dependent cytokines, interleukin (IL)-1β and IL-18 (Karki and Kanneganti, 2019). While this cell death pathway can lead to inflammation, apoptotic cell death activated by the apoptotic caspases is a non-inflammatory process (Nagata, 2018). In response to apoptotic cell death signals, initiator caspases undergo proximity-induced self-activation, allowing them to subsequently process the effector caspases to execute apoptotic function.
Emerging evidence suggests that the crosstalk between apoptotic caspases and inflammatory caspases regulates inflammasome activation and both the apoptotic and pyroptotic forms of cell death (Gurung et al., 2014). In the absence of caspase-1, the nucleotide-binding domain, leucine-rich repeat-containing (NLR) family pyrin domain-containing protein 1b (NLRP1b), the NLR family CARD domain-containing protein 4 (NLRC4), and the absent in melanoma 2 (AIM2) inflammasome have been shown to recruit caspase-8 to induce apoptosis (Mascarenhas et al., 2017; Pierini et al., 2012; Van Opdenbosch et al., 2017), suggesting a compensatory role for apoptotic caspases during inflammasome activation. The inflammatory caspase caspase-1 can also initiate apoptosis in cells lacking GSDMD via the Bid-caspase-9-caspase-3 axis (Tsuchiya et al., 2019). In addition to the role of the inflammasome and caspase-1 in apoptosis and pyroptosis, apoptotic caspases have also been shown to regulate pyroptosis. Caspase-8 can cleave GSDMD, eliciting pyroptotic cell death during Yersinia infection (Orning et al., 2018; Sarhan et al., 2018). The activation of effector caspase-3 by apoptotic triggers or chemotherapy drugs is able to process GSDME, another member of the gasdermin family, to induce pyroptosis (Rogers et al., 2017; Wang et al., 2017). Additionally, both caspase-3 and −7 can inactivate GSDMD by cleaving the amino-terminal domain, suggesting a counteracting effect of apoptosis on pyroptosis (Taabazuing et al., 2017). However, the role of the effector caspase-6 in regulating programmed cell death and inflammasome activation has remained enigmatic.
To understand the biological role of caspase-6 during an immune insult, we have examined the effect of caspase-6 deficiency on inflammasome activation after stimulation with various DAMPs and PAMPs or in response to different infections. Here we report that caspase-6 is a vital component for innate immunity, Z-DNA binding protein 1 (ZBP1)-mediated inflammasome activation, cell death, and host defense during influenza A virus (IAV) infection.
RESULTS
Caspase-6 Promotes ZBP1-Mediated NLRP3 Inflammasome Activation
The NLR family pyrin domain-containing protein 3 (NLRP3) inflammasome can be activated either in a canonical manner via infectious and endogenous ligands such as pore-forming toxins, ATP, and uric acid crystals (Kanneganti et al., 2006a, 2006b; Mariathasan et al., 2006; Martinon et al., 2006) or in a non-canonical manner through lipopolysaccharide (LPS) sensing by caspase-11 (or its human counterparts caspase-4/5) (Hagar et al., 2013; Kayagaki et al., 2011, 2013; Shi et al., 2014). The initiator caspase-8 has been shown to play critical roles in both canonical and non-canonical NLRP3 inflammasome activation (Chauhan et al., 2018; Gurung et al., 2014; Lawlor et al., 2015; Vince et al., 2012, 2018). Furthermore, the executioners caspase-3/7 have also been reported to act upstream of NLRP3 inflammasome activation (Chen et al., 2019; Vince et al., 2018). To investigate whether caspase-6 is also involved in NLRP3 inflammasome activation, we treated LPS-primed wild-type (WT) and caspase-6-deficient (Casp6−/−) bone-marrow-derived macrophages (BMDMs) with ATP or nigericin. BMDMs from both genotypes showed similar cleavage of caspase-1, production of the inflammasome effector cytokines IL-1β and IL-18, and pyroptosis, suggesting that caspase-6 is dispensable for canonical NLRP3 inflammasome activation (Figures 1A–1D and S1A). Priming of BMDMs with other Toll-like receptor (TLR) agonists Pam3CSK4 and poly(I:C) followed by ATP treatment did not affect NLRP3 inflammasome activation in Casp6−/− BMDMs compared with WT BMDMs (Figure S1B), further supporting that caspase-6 is not required for canonical NLRP3 inflammasome activation. Moreover, caspase-6 was also dispensable for non-canonical NLRP3 inflammasome activation triggered by LPS transfection under primed condition (Figures 1E–1H). We previously have shown that the NLRP3 inflammasome can also be activated upon ZBP1 sensing of IAV (Kesavardhana et al., 2017; Kuriakose et al., 2016). To investigate whether caspase-6 contributes to ZBP1-mediated NLRP3 inflammasome activation, we infected WT and Casp6−/− BMDMs with IAV. In contrast to our findings with the NLRP3 inflammasome triggers above, NLRP3 inflammasome activation as measured by caspase-1 cleavage and IL-1β and IL-18 production was significantly reduced in Casp6−/− BMDMs compared with WT BMDMs, and cell death was reduced in Casp6−/− BMDMs in response to IAV infection (Figures 1I–1L), suggesting that caspase-6 promotes ZBP1-mediated NLRP3 inflammasome activation. Next, we infected WT and Casp6−/− BMDMs with Pseudomonas aeruginosa and Salmonella enterica subspecies enterica serovar Typhimurium (S. typhimurium) to investigate the role of caspase-6 in NLRC4 inflammasome activation (Karki et al., 2018; Mariathasan et al., 2004). The similar levels of caspase-1 activation, IL-1β and IL-18 production, and cell death observed in both WT and Casp6−/− BMDMs (Figures 1M–1P and S1C) suggested there is no role for caspase-6 in NLRC4 inflammasome activation. This was further supported by the observation of similar cleavage of caspase-1 in WT and Casp6−/− BMDMs transfected with flagellin (Figure S1D). To study the role of caspase-6 in AIM2 inflammasome activation, WT and Casp6−/− BMDMs were transfected with poly(dA:dT) or infected with mouse cytomegalovirus (MCMV). Caspase-1 cleavage in poly(dA:dT)-transfected and MCMV-infected BMDMs from WT and Casp6−/− mice was comparable (Figures S1E and S1F). Our group has previously demonstrated that activation of the AIM2 inflammasome by Francisella tularensis subspecies novicida (F. novicida), but not by poly(dA:dT) or MCMV, requires interferon (IFN) signaling (Man et al., 2015, 2016). When infecting BMDMs from WT and Casp6−/− mice with F. novicida, we observed comparable caspase-1 cleavage, IL-1β and IL-18 production, and cell death (Figures 1Q–1T). These results suggested that caspase-6 was dispensable for AIM2 inflammasome activation. Toxin from Clostridium difficile (C. difficile) AB+ strain has been shown to induce PYRIN inflammasome activation (Karki et al., 2018; Xu et al., 2014). In response to this trigger, the caspase-1 cleavage in Casp6−/− BMDMs was similar to that in WT BMDMs (Figure S1G), suggesting that caspase-6 does not play a role in PYRIN inflammasome activation. Collectively, these results indicate that caspase-6 promotes ZBP1-mediated NLRP3 inflammasome activation but is dispensable for NLRP3, NLRC4, AIM2, and PYRIN inflammasome activation.
Figure 1. Caspase-6 Promotes IAV-Induced ZBP1-Mediated NLRP3 Inflammasome Activation.

(A) Immunoblots analysis of pro- (p45, black arrow) and cleaved caspase-1 (p20, red arrow; CASP1) in LPS plus ATP-treated bone-marrow-derived macrophages (BMDMs).
(B) Assessment of IL-18 release by ELISA following LPS plus ATP treatment.
(C) Assessment of IL-1β release by ELISA following LPS plus ATP treatment.
(D) Images of BMDMs after LPS plus ATP treatment.
(E–H) Immunoblot analysis of CASP1 (E), IL-18 release (F), IL-1β release (G), and cell images (H) from BMDMs primed with LPS and then transfected with LPS.
(I–L) Immunoblot analysis of CASP1 (I), IL-18 release (J), IL-1β release (K), and cell images (L) from BMDMs after influenza A virus (IAV) infection for 16 h.
(M–P) Immunoblot analysis of CASP1 (M), IL-18 release (N), IL-β release (O), and cell images (P) from BMDMs after P. aeruginosa (MOI, 2) infection for 2–3 h.
(Q–T) Immunoblot analysis of CASP1 (Q), IL-18 release (R), IL-1β release (S), and cell images (T) of BMDMs after F. novicida (MOI, 100) infection for 20 h.
The red arrows indicate pyroptotic cells (D, H, L, P, T). NS, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 (one-way ANOVA). Data are representative of three independent experiments. Data are shown as mean ± SEM (B, C, F, G, J, K, N, O, R, and S). The original magnification of all images is 320. See also Figure S1.
The reduced NLRP3 inflammasome activation seen in Casp6−/− BMDMs compared with WT BMDMs might be due to caspase-6 playing a role in the upregulation of the inflammasome components during IAV infection. However, we observed similar expression of NLRP3 and pro-IL-1β in IAV-infected BMDMs from WT and Casp6−/− mice (Figure 2A), suggesting that caspase-6 does not regulate the expression of inflammasome components. This was consistent with the observation of similarly activated inflammatory signaling pathways in WT and Casp6−/− BMDMs during IAV infection (Figure 2A). Furthermore, prior priming with Pam3CSK4 did not rescue caspase-1 cleavage in IAV-infected Casp6−/− BMDMs (Figure 2B), suggesting a priming-independent role for caspase-6 in facilitating ZBP1-mediated NLRP3 inflammasome activation. Additionally, the activation of STAT1 and expression of ZBP1 were not impaired in Casp6−/− BMDMs compared with WT BMDMs during IAV infection (Figure 2C), suggesting that caspase-6 regulates the NLRP3 inflammasome independently of IFN signaling and ZBP1 expression. The reduced NLRP3 inflammasome activation in Casp6−/− BMDMs might be due to less viral replication in Casp6−/− BMDMs. However, both WT and Casp6−/− BMDMs displayed comparable protein levels of viral non-structural protein NS1 and structural protein M1 (Figure 2C). Consistent with this, both transcription and replication as indicated by the M1 mRNA and vRNA levels were not attenuated in Casp6−/− BMDMs (Figure 2D). Moreover, the single-cycle replication assay directly demonstrated that IAV replication was not affected in Casp6−/− BMDMs (Figure 2E). Altogether, these findings demonstrate that caspase-6 regulates ZBP1-mediated NLRP3 inflammasome activation independently of NLRP3 priming and viral replication in BMDMs.
Figure 2. Deficiency of Caspase-6 Does Not Influence NLRP3 Priming and Viral Replication in Bone-Marrow-Derived Macrophages.

(A) Immunoblot analysis of phosphorylated IκBα (pIκBα), total IκBα (tIκBα), phosphorylated ERK (pERK), total ERK (tERK), NLRP3, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), pro–IL-1β, and pro- caspase-6 (pro-CASP6) in bone-marrow-derived macrophages (BMDMs) after influenza A virus (IAV) infection at the indicated time points. Actin is used as the internal control.
(B) Immunoblot analysis of pro- and cleaved caspase-1 (CASP1) in BMDMs primed with or without Pam3CSK4 (PAM3) for 5 h and then infected with IAV for 16 h.
(C) Immunoblot analysis of phosphorylated STAT1 (pSTAT1), total STAT1 (tSTAT1), Z-DNA binding protein 1 (ZBP1), influenza non-structural protein 1 (NS1), and influenza matrix protein 1 (M1) in BMDMs after IAV infection at the indicated time points. Actin is used as the internal control.
(D) Real-time PCR analysis of IAV M1 mRNA or vRNA in BMDMs after infection at the indicated time points, presented relative to levels of the host gene actin.
(E) Endpoint replication of IAV (MOI, 10) in BMDMs for 8 h. NS, not significant; *p < 0.05; ***p < 0.001 (two-way ANOVA).
Data are shown as mean ± SEM (D and E). Data are representative of three independent experiments.
Caspase-6 Is Required for IAV-Induced Pyroptosis, Apoptosis, and Necroptosis
Aside from regulating NLRP3 inflammasome activation, ZBP1 also regulates various cell death pathways following IAV infection (Kesavardhana et al., 2017; Kuriakose and Kanneganti, 2018; Kuriakose et al., 2016; Thapa et al., 2016). Therefore, we next interrogated how caspase-6 affected cell death during IAV infection. Building on our observation that there was reduced cell death in IAV-infected Casp6−/− BMDMs by microscopy (Figure 1L), we found that cell death induced by IAV infection was quantitatively reduced in Casp6−/− BMDMs compared with WT BMDMs (Figures 3A and 3B), suggesting that caspase-6 deficiency not only influences ZBP1-mediated NLRP3 inflammasome activation but also affects ZBP1-mediated programmed cell death pathways. To confirm these findings, we used CRISPR gene editing to knockout caspase-6 in mouse embryonic fibroblasts (MEFs) (denoted as Casp6KO; Figure 3C) and observed reduced cell death following IAV infection compared with WT MEFs (Figures 3D and 3E). Given that ZBP1 controls the activation of multiple cell death pathways including pyroptosis, apoptosis, and necroptosis (PANoptosis) (Kuriakose et al., 2016; Malireddi et al., 2019; Thapa et al., 2016), we then investigated the contribution of caspase-6 in each of those cell death pathways during IAV infection. We observed decreased pyroptosis as evidenced by decreased caspase-1 cleavage and IL-1β and IL-18 release in Casp6−/− BMDMs following IAV infection (Figures 1I–1K), which was further confirmed by decreased gasdermin D cleavage in Casp6−/− BMDMs following IAV infection (Figure 3F). We then explored the impact of caspase-6 deficiency on apoptosis and found that cleavage of initiator caspase-8 and executioner caspase-3 and −7 was attenuated following IAV infection in Casp6−/− BMDMs and Casp6KO MEFs compared with WT BMDMs and MEFs, respectively (Figures 3F–3I). To understand whether caspase-6 globally regulates apoptosis, we treated WT and Casp6−/− BMDMs with the classic apoptosis inducers etoposide and ultraviolet (UV) radiation. We observed similar cleavage of caspase-8, −3, and −7 in WT and Casp6−/− BMDMs treated with these classic apoptosis inducers (Figures S2A–S2D), suggesting that caspase-6 uniquely regulates IAV-induced ZBP1-mediated apoptosis. However, we consistently observed reduced amounts of the p25 form of caspase-3 after etoposide and UV stimulation in Casp6−/− BMDMs, suggesting that caspase-6 may contribute to caspase-3 cleavage at certain sites. Unlike the apoptotic cell death pathway that is elicited by initiator caspases, necroptosis is induced by activation of receptor-interacting protein kinase 3 (RIPK3) and the pursuant RIPK3-mediated phosphorylation of the pseudokinase mixed lineage kinase domain-like (MLKL) (Kang et al., 2013; Sun et al., 2012; Wang et al., 2012; Wu et al., 2013; Zhao et al., 2012). We observed reduced MLKL phosphorylation in IAV-infected Casp6−/− BMDMs compared with WT BMDMs (Figure 3J). In contrast, the dynamics of cell death induced by the classic necroptotic triggers TNF plus zVAD or LPS plus zVAD were similar between WT and Casp6−/− BMDMs (Figures S2E and S2F), implying that caspase-6 promotes IAV-induced ZBP1-mediated necroptosis specifically. Altogether, these results imply that caspase-6 plays a critical role in IAV-induced ZBP1-mediated cell death pathways, including pyroptosis, apoptosis, and necroptosis.
Figure 3. Caspase-6 Promotes IAV-Induced Pyroptosis, Apoptosis, and Necroptosis.

(A) Real-time analysis of cell death in bone-marrow-derived macrophages (BMDMs) using the IncuCyte imaging system and SYTOX Green nucleic acid staining after infection with influenza A virus (IAV) for 12 h. The original magnification is ×20. The red denotes the cells counted as dead in the analysis.
(B) Quantification of the cell death observed in (A).
(C) Immunoblot analysis of caspase-6 (CASP6) in mouse embryonic fibroblasts (MEFs) following CRISPR-directed deletion.
(D) Microscopic analysis of cell death in MEFs infected with IAV for 24 h. The original magnification is ×10.
(E) Quantification of the cell death observed in (D).
(F) Immunoblot analysis of the pro-and cleaved forms of caspase-3 (CASP3), caspase-7 (CASP7), and gasdermin D (GSDMD) in BMDMs after IAV infection for 9 h. Actin is used as the internal control.
(G) Immunoblot analysis of the pro- and cleaved forms of CASP3 and CASP7 in MEFs after IAV infection for 24 h. Actin is used as the internal control.
(H) Immunoblot analysis of pro- and cleaved caspase-8 (CASP8) in BMDMs after IAV infection for 9 h. Actin is used as the internal control.
(I) Immunoblot analysis of pro- and cleaved CASP8 in MEFs after IAV infection for 24 h. Actin is used as the internal control.
(J) Immunoblot analysis of phosphorylated mixed lineage kinase domain-line (pMLKL) and total MLKL (tMLKL) in BMDMs after IAV infection at the indicated time points. Actin is used as the internal control.
****p < 0.0001. Analysis was performed using the Student’s t test (B) or one-way ANOVA (E). Data are shown as mean ± SEM (B and E). See also Figure S2.
Caspase-6 Deficiency Increases Mouse Susceptibility to IAV Infection
Because ZBP1 is known to play a role in host defense against IAV (Kuriakose et al., 2016) and we observed that caspase-6 is critically involved in IAV-induced ZBP1-mediated processes, we examined the role of caspase-6 in host defense against IAV infection. We infected mice intranasally with IAV (A/Puerto Rico/8/34, H1N1 [PR8]) and monitored body-weight change and survival. Weight loss was similar in both WT and Casp6−/− mice over the first week, but Casp6−/− mice had slow weight recovery in the second week compared with WT mice (Figure 4A). Additionally, significantly more Casp6−/− mice succumbed to IAV infection than did WT mice (Figure 4B), suggesting an increased susceptibility to IAV infection in the absence of caspase-6. To further confirm this effect, we infected both Casp6+/− and Casp6−/− littermate mice with IAV and found that more Casp6−/− mice succumbed to IAV infection than did Casp6+/− mice (Figure 4C). We found greater viral spread in the Casp6−/− mice, as indicated by a remarkable increase in viral NP staining in the lungs of Casp6−/− mice at day 5 post-infection compared with WT mice (Figure 4D), suggesting that caspase-6 plays a role in controlling IAV spread in the lungs. To further evaluate viral replication in Casp6−/− mice, we collected the lungs of infected mice and measured virus titers. There was no notable difference in the viral load at day 3 post-infection between WT and Casp6−/− mice (Figure 4E). However, there was a significant increase in viral titer at day 7 post-infection in the lungs of Casp6−/− mice compared with WT mice (Figure 4F).
Figure 4. Caspase-6 Contributes to Host Protection against IAV Infection In Vivo.

(A) Body weight of 6 to 8-week-old wild-type (WT) and Casp6−/− mice infected intranasally with 50 plaque forming units (PFUs) of influenza A virus (IAV).
(B) Survival of 6- to 8-week-old WT and Casp6−/− mice infected intranasally with 50 PFUs of IAV.
(C) Survival of 6- to 8-week-old littermate Casp6+/− and Casp6−/− mice infected intranasally with 125 PFUs of IAV.
(D) Immunohistochemistry staining of viral nucleoprotein (NP) in the lungs collected from WT and Casp6−/− mice at day 5 post-infection.
(E and F) Lung viral titers in WT and Casp6−/− mice infected with IAV for 3 days (E) or 7 days (F).
(G and H) IL-1β levels in the bronchoalveolar lavage fluid (BALF) from WT and Casp6−/− mice infected with IAV for 3 days (G) or 7 days (H).
(I) Quantification of the percentage of the lung lesioned in WT and Casp6−/− mice infected with IAV for 5 days.
(J) Microscopic analysis of Arg1+ cells in the lung tissue of WT and Casp6−/− mice infected with IAV for 7 days.
The original magnification is ×10 and ×40 as indicated. NS, not significant; *p < 0.05, **p < 0.01, and ***p < 0.001. Analysis was performed using the Student’s t test (E–I), two-way ANOVA (A), and log-rank test (B and C). Data are shown as mean ± SEM (A and E–I). Data are pooled from 4 independent experiments (A and B).
We also assessed levels of IL-1b in the bronchoalveolar lavage fluid (BALF) at day 3 and 7 post-infection. While no difference was observed between WT and Casp6−/− mice at day 3 (Figure 4G), Casp6−/− mice exhibited a decrease in IL-1β levels in the BALF at day 7 (Figure 4H), suggesting there may be defective inflammasome activation in the lungs of Casp6−/− mice in response to IAV infection. Additionally, Casp6−/− mice showed significantly more lung damage at day 5 post-infection (Figure 4I). It has been shown that caspase-6 promotes the activation of alternatively activated macrophages (AAMs) (Yao et al., 2016). Consistent with this, fewer Arg1+ AAMs were detected in the lungs of Casp6−/− mice after IAV infection compared with WT mice (Figure 4J). In sum, these results indicate that caspase-6 is involved in host defense against IAV infection and loss of caspase-6 impairs viral clearance.
Caspase-6 Binds to RIPK3 to Enhance the Interaction between RIPK3 and ZBP1
Based on our findings both in vitro and in vivo that caspase-6 is critically involved in the ZBP1-mediated response to IAV infection, we sought to understand the interaction of caspase-6 with ZBP1 and its associated proteins. Consistent with our previous report (Kuriakose et al., 2016), we observed impaired cleavage of initiator caspase-8 and executioner caspase-3 and −7 in IAV-infected Zbp1−/− BMDMs and Zbp1KO MEFs compared with WT BMDMs and MEFs, respectively (Figures 5A, S3A, and S3B), along with decreased cell death in Zbp1KO MEFs (Figures S3C and S3D), demonstrating that ZBP1 is required to initiate apoptosis. We also observed impaired GSDMD cleavage and MLKL phosphorylation in IAV-infected Zbp1−/− BMDMs compared with WT BMDMs, confirming that ZBP1 is also required to drive pyroptosis and necroptosis, respectively (Figures 5A and 5B). Next, we asked whether ZBP1 contributed to caspase-6 activation during IAV infection. Consistent with the effects observed on the other executioner caspases, the cleavage of caspase-6 was reduced in IAV-infected Zbp1−/− BMDMs compared with WT BMDMs (Figures 5A and S4A), suggesting that ZBP1 regulates caspase-6 activation during IAV infection. ZBP1 recruits RIPK3 and RIPK1 via the receptor-interacting protein homotypic interaction motif (RHIM) to form a cell death complex in which FADD and caspase-8 are subsequently recruited (Figure 5C) (Kuriakose and Kanneganti, 2018; Thapa et al., 2016). Accordingly, RIPK3 has been shown to activate parallel pathways of necroptosis and apoptosis upon IAV infection (Nogusa et al., 2016; Thapa et al., 2016). In line with previous findings, we observed impaired activation of apoptotic caspases and necroptotic MLKL in IAV-infected Ripk3−/− BMDMs compared with WT BMDMs (Figures 5D and 5E). Moreover, the impaired cleavage of GSDMD in IAV-infected Ripk3−/− and Ripk3−/−Casp8−/− BMDMs compared with WT BMDMs suggests an additional role for RIPK3 in ZBP1-mediated pyroptosis. We next interrogated the role of RIPK3 and caspase-8 in caspase-6 activation during IAV infection. The cleavage of caspase-6 was reduced in IAV-infected Ripk3−/− and Ripk3−/−- Casp8−/− BMDMs compared with WT BMDMs (Figure 5D), indicating that RIPK3 alone promotes caspase-6 activation. Because the inflammasome has been shown to activate caspase-6 in the neuronal context (Kaushal et al., 2015), we next assessed whether the NLRP3 inflammasome is able to activate caspase-6 in the context of IAV infection. We found that cleavage of caspase-6 was not affected in the absence of GSDMD or NLRP3 after IAV infection (Figure S4B). Additionally, cleavage of lamin A/C, a caspase-6-specific substrate (Ruchaud et al., 2002), was impaired in Casp6−/− BMDMs but was unaffected in Gsdmd−/− and Nlrp3−/− BMDMs (Figures S4A and S4B). These results suggest that the NLRP3 inflammasome does not activate caspase-6.
Figure 5. Caspase-6 Is Critical for Enhancing the Interaction between RIPK3 and ZBP1.

(A) Immunoblot analysis of pro- and cleaved forms of caspase-6 (CASP6), −8 (CASP8), −7 (CASP7), −3 (CASP3), and gasdermin D (GSDMD) in bone-marrow-derived macrophages (BMDMs) after influenza A virus (IAV) infection for 12 h. Actin is used as the internal control.
(B) Immunoblot analysis of phosphorylated mixed lineage kinase domain-like (pMLKL) and total MLKL (tMLKL) in BMDMs after IAV infection at the indicated time points. Actin is used as the internal control.
(C) Schematic depiction of the relationship between receptor-interacting protein homotypic interaction motif (RHIM) domain-containing proteins in the Z-DNA binding protein 1 (ZBP1)-initiated cell death complex and caspase-6 after IAV infection. The red boxes indicate the RHIM domains.
(D) Immunoblot analysis of pro- and cleaved forms of CASP6, CASP8, CASP7, CASP3, and GSDMD in BMDMs after IAV infection for 9 h. Actin is used as the internal control.
(E) Immunoblot analysis of pMLKL and tMLKL in BMDMs after IAV infection at the indicated time points. Actin is used as the internal control.
(F) Immunoprecipitates and total lysates from 293T cells after co-transfection of FLAG-CASP6 with receptor-interacting protein kinase (RIPK) 1, RIPK3-GFP, ZBP1, or CASP8 for 30 h.
(G) Immunoprecipitates and total lysates from 293T cells after co-transfection with RIPK1, RIPK3-GFP, and ZBP1 in the absence or presence of FLAG-CASP6 for 30 h.
(H) Immunoprecipitates and total lysates from BMDMs with IAV infection for 16 h. Data are representative of three independent experiments. See also Figures S3, S4, and S5.
Since caspase-6 regulated all three cell death pathways that are driven by ZBP1 and RIPK3, we hypothesized that caspase-6 may act at the level of the ZBP1-RIPK3 cell death complex (Figure 5C). To determine whether this was the case, we overexpressed caspase-6 along with different components of the ZBP1-RIPK3 cell death complex in 293T cells, as endogenous caspase-6 in BMDMs cannot be pulled down with the currently available reagents, to evaluate the interaction of caspase-6 with the components of the cell death complex. Since overexpression of WT caspase-6-induced significant cell death in 293T cells, we used a catalytically dead mutant of caspase-6 (caspase-6-C146A; abbreviated caspase-6-CA). We did not detect any binding of caspase-6 to ZBP1, RIPK1, or caspase-8 (Figure 5F). However, RIPK3 was co-immunoprecipitated by caspase-6 and vice versa (Figures 5F and 5G, red arrow), suggesting that caspase-6 may be recruited to the cell death complex via RIPK3. Using the uncleavable mutant of caspase-6 (caspase-6-D5A/D162A/D175A; abbreviated caspase-6-DA) to co-immunoprecipitate RIPK3, no interaction was detected between them (Figure S5A). However, this was not true of the ability of RIPK3 to co-immunoprecipitate caspase-6-DA (Figure S5B). One possible explanation is that the antibody used for caspase-6-DA immunoprecipitation blocks the binding site of RIPK3 on caspase-6. To confirm that the interaction between caspase-6 and RIPK3 occurs under endogenous conditions, we transduced Casp6−/− immortalized BMDMs (iBMDMs) with a doxycycline-inducible FLAG-caspase-6 construct and performed co-immunoprecipitation. In line with what we observed in the overexpression system (Figure 5F), caspase-6 was able to immunoprecipitate RIPK3 during IAV infection (Figure S5C). Based on these findings, we hypothesized that caspase-6 recruitment to RIPK3 may modulate the binding of RIPK3 to ZBP1 or RIPK1. We found that the expression of caspase-6 did not affect the amount of ZBP1 and RIPK1 that is co-immunoprecipitated by RIPK3 in this system (Figures S5D and S5E). Previous work showed that the binding of RIPK3 to ZBP1 is greatly enhanced in cells lacking a functional RIPK1 RHIM domain (Lin et al., 2016; Newton et al., 2016), suggesting that RIPK1 and RIPK3 may compete for binding to ZBP1. We hypothesized that caspase-6 may influence this competition. To test this, we co-expressed RIPK1, RIPK3, and ZBP1 in the same co-immunoprecipitation assay with increasing amounts of caspase-6. While the amount of RIPK1 co-immunoprecipitated by RIPK3 was not altered by the amount of caspase-6 expressed, the amount of ZBP1 co-immunoprecipitated by RIPK3 was notably increased as the amount of caspase-6 increased in the overexpression system (Figure 5G), suggesting that caspase-6 is able to enhance the interaction between ZBP1 and RIPK3. Additionally, because this enhancement was achieved with the catalytically dead mutant of caspase-6, these results suggest a protease activity-independent scaffolding role for caspase-6 in this process. To verify that the ability of caspase-6 to enhance the interaction between ZBP1 and RIPK3 can occur outside the overexpression system, we performed endogenous co-immunoprecipitation using BMDMs from WT and Casp6−/− mice. We again observed that the absence of caspase-6 reduced the amount of ZBP1 co-immunoprecipitated by RIPK3 (Figures 5H, red arrow, and S5F), further supporting that caspase-6 enhances the interaction between ZBP1 and RIPK3. Consistent with a previous report (Thapa et al., 2016), no interaction between RIPK3 and ZBP1 was detected in the uninfected cells (Figure S5F).
To determine whether other executioner caspases could also enhance the RIPK3-ZBP1 interaction in the presence of RIPK1, we used our overexpression system and performed co-immunoprecipitations with caspase-6, caspase-3, or caspase-7 with ZBP1, RIPK3, and RIPK1. We observed that only caspase-6 increased the amount of ZBP1 in the complex (Figure S5G, red arrow), demonstrating that the interaction between RIPK3 and caspase-6 was selective. Additionally, both caspase-3 and −7 could not be immunoprecipitated by RIPK3 (Figure S5G).
Both the Large and Small Subunits of Caspase-6 Are Critical for the Binding of RIPK3 to ZBP1
We next sought to understand the nature of the interaction between caspase-6 and RIPK3. Under physiological conditions, caspase-6 can be cleaved into large and small subunits (Figure 6A). On the other hand, RIPK3 is composed of an amino terminal kinase domain and a RHIM-containing carboxy-terminal domain (Figure 6A). To further characterize the interaction between caspase-6 and RIPK3, we generated truncated mutants of caspase-6 and RIPK3 in 293T cells to map the domains required for their interaction. Unexpectedly, both the amino-terminal domain of caspase-6 (CASP6-N) and the carboxy-terminal domain of caspase-6 (CASP6-C) were able to interact with both the amino-terminal and carboxy-terminal domains of RIPK3 (Figure 6B, red arrow). Also, both CASP6-N and CASP6-C showed a strong interaction with full-length RIPK3 (Figure 6B, red arrow). In the absence of these two caspase-6 constructs, neither of the RIPK3 domains was pulled down by the antibody used, suggesting that the interaction observed between the domains of caspase-6 and domains of RIPK3 was specific. However, it is unclear how caspase-6 could interact with RIPK3 despite the lack of common homotypic interaction domains in these two proteins. By analyzing the protein sequences of caspase-6 and RIPK3 with the Protein DisOrder prediction System (PrDOS) (Ishida and Kinoshita, 2007), we noted that both proteins contain intrinsically disordered regions (IDRs) (Figures S6A and S6B). It has been suggested that the disordered regions of RIPK3 are critical for its RHIM domain-mediated interactions (Li et al., 2012; Wright and Dyson, 2015). Because the IDRs are distributed in both the amino-and carboxy-terminal domains of the two proteins, these IDRs could likely mediate the interactions observed between the domains of caspase-6 and RIPK3. Given the ability of both the amino- and carboxy-terminal domains of caspase-6 and RIPK3 to facilitate binding between the two proteins, we also investigated whether these interactions promoted the binding of RIPK3 to ZBP1. Consistent with our findings above (Figure 5G), full-length caspase-6 was able to facilitate the interaction between RIPK3 and ZBP1, as were both the N-terminal and C-terminal caspase-6 domains in the overexpression system (Figure 6C, red arrow). Collectively, these results imply that caspase-6 interacts with RIPK3 via the putative IDRs to facilitate the interaction between ZBP1 and RIPK3.
Figure 6. Both N- and C-Terminal Domains of Caspase-6 Are Critical for the Binding of RIPK3 to ZBP1.

(A) Schematic depiction of the domains in mouse CASP6 and RIPK3 in the full-length construct (F), N-terminal construct (N), and C-terminal construct (C). The aspartic acid sites shown denote the mouse CASP6 cleavage sites, and the cysteine denotes the catalytic site. L, large subunit; RHIM, receptor-interacting protein homotypic interaction motif; S, small subunit.
(B) Immunoprecipitates and total lysates from 293T cells after co-transfection with the indicated forms of CASP6 and RIPK3 for 30 h.
(C) Immunoprecipitates and total lysates from 293T cells after co-transfection with RIPK1, RIPK3-GFP, and Z-DNA binding protein 1 (ZBP1) in the absence or presence of full-length or truncated CASP6 for 30 h. Data are representative of three independent experiments. CA, CASP6-C146A.
See also Figure S6.
The Caspase Activity of Caspase-6 Is Dispensable for ZBP1-Mediated Inflammasome Activation
Based on the observation that the ability of caspase-6 to enhance the interaction between ZBP1 and RIPK3 was independent of its caspase activity (Figures 5G and 6), we sought to address whether the observed functions of caspase-6 in ZBP1-mediated inflammasome activation were caspase activity-dependent or independent. We reconstituted the expression and function of caspase-6, as evidenced by cleavage of lamin A/C, in Casp6−/− iBMDMs using a doxycycline-inducible system to express WT caspase-6 (Figure S6C) or caspase-6-CA (Figure S6D). We found that both the WT and catalytically dead forms of caspase-6 increased IAV-induced caspase-1 activation (Figures S6E and S6F), indicating that the caspase activity of caspase-6 was not required for IAV-induced inflammasome activation. This further supports that the caspase activity of caspase-6 is dispensable for it to carry out its functions and implicates a potential scaffolding role instead.
DISCUSSION
Caspase-6 has remained as a mystery caspase for decades. While it has been classified as an apoptotic executioner caspase and linked to several neurological disorders including Alzheimer’s disease and Huntington disease, its function and role in other biological processes remains enigmatic. In this study, we identified a role for caspase-6 in regulating ZBP1-mediated NLRP3 inflammasome activation and programmed cell death. However, caspase-6 was dispensable for canonical NLRP3, AIM2, NLRC4, and PYRIN inflammasome activation. Additionally, caspase-6 was not required for caspase-11-mediated non-canonical NLRP3 inflammasome activation induced under TLR2 priming followed by LPS transfection. Also, caspase-6 was not required for apoptosis and necroptosis induced by classic triggers. Mechanistically, caspase-6 interacted with RIPK3 to enhance the interaction between RIPK3 and ZBP1 during IAV infection. The ZBP1-RIPK3 complex induced during IAV infection was dramatically reduced in the absence of caspase-6. The residual RIPK3 and ZBP1 complex observed in Casp6−/− BMDMs possibly explains the reduced, but not abolished, inflammasome activation and cell death in those cells during IAV infection. The fact that caspase-6 was required for the efficient interaction between ZBP1 and RIPK3 provides a clue into the mysterious relationship among RIPK1, RIPK3, and ZBP1, three RHIM domain-containing proteins. It has been proposed that RIPK1 could counteract the interaction between ZBP1 and RIPK3 to inhibit necroptosis in the perinatal period (Lin et al., 2016; Newton et al., 2016). In cells without a functional RIPK1 RHIM domain, the binding of RIPK3 to ZBP1 was found to be greatly enhanced, suggesting that RIPK1 indeed has an inhibitory effect on the binding of RIPK3 to ZBP1. However, under physiological conditions, no interaction has been detected between RIPK1 and ZBP1 (Lin et al., 2016; Newton et al., 2016). We also noted that without RIPK1, the interaction between RIPK3 and ZBP1 was much stronger than it was with RIPK1 co-expression (data not shown), further confirming the competitive relationship between RIPK1 and RIPK3. Although there is no direct evidence that the interaction between RIPK3 and caspase-6 is mediated via their IDRs, the disordered regions of RIPK3 have been shown to be critical for its RHIM domain-mediated interactions (Li et al., 2012; Wright and Dyson, 2015). It has been suggested that the RHIM domain of RIPK3 is buried in the IDRs, but the process of exposing the RHIM domain during signaling activation is not well understood (Li et al., 2012). It is possible that the binding specificity of the RHIM domain may change depending on specific IDR-containing proteins involved in this process.
Similar to the phenotype of Ripk3−/− mice (Nogusa et al., 2016), loss of caspase-6 in mice increased susceptibility to IAV infection. While both proteins are critical for IAV-induced cell death in vitro, the mechanisms underlying the in vivo phenotype need more investigation. It has been shown that RIPK3 can interact with mitochondrial antiviral signaling protein (MAVS) to increase the expression of type I IFNs during IAV infection (Downey et al., 2017). In contrast, the executioners caspase-3 and −7 negatively regulate type I IFN expression by cleaving cyclic GMP-AMP synthase (cGAS), MAVS, and IFN regulatory factor 3 (IRF3) (Ning et al., 2019). Similar to the function of these two executioner caspases, we observed that caspase-6 also negatively regulates type I IFN expression, as evidenced by consistently higher IFN-b expression (data not shown) and elevated pSTAT1 levels in Casp6−/− BMDMs compared with WT BMDMs. Additionally, we observed that caspase-6 is required for the activation of lung AAMs, which are critical for inhibiting IAV-induced lethal inflammation (Wang et al., 2013), suggesting that the role of caspase-6 in AAM activation also contributes to host defense against IAV infection.
Despite being categorized as an executioner caspase, the substrate specificity of caspase-6 is similar to that of the initiator caspases caspase-8 and caspase-9 and different from that of the other two executioners, caspase-3 and caspase-7 (Thornberry et al., 1997). In addition, inhibitor of apoptosis (IAP) family proteins can inhibit the activity of caspase-3 and caspase-7 but have little effect on the activity of caspase-6 (Deveraux et al., 1997; Roy et al., 1997). While caspase-3 and caspase-7 play redundant roles in the development of C57BL/6 mice (Lakhani et al., 2006) and intrinsic and extrinsic apoptosis in human leukemic cell lines (McComb et al., 2019), caspase-6 is dispensable for all these processes. Also, the non-apoptotic function of caspase-6 in the pathogenesis of several neurological disorders has already been demonstrated. Our study extends these findings to highlight the unique biological capabilities of this caspase. The fact that the catalytically dead mutant of caspase-6 can enhance the interaction between RIPK3 and ZBP1 and can facilitate caspase-1 cleavage in our doxycycline-inducible system suggests a scaffolding role for caspase-6. The finding that the uncleavable mutant of caspase-6 is able to potentiate the interaction between RIPK3 and ZBP1 in the overexpression system reinforces the concept that caspase-6 can function as a scaffold during IAV infection. But this function is not unique to caspase-6. Among all the caspase proteins, caspase-8 has also been reported to possess the scaffold function (Henry and Martin, 2017; Kang et al., 2015). It has been suggested that caspase-8 can scaffold to promote cell death and inflammatory responses, whereas the mechanism of how caspase-8 protease activity is regulated during the scaffolding process is largely unknown. Given its enigmatic role in apoptosis, it is highly possible that caspase-6 could serve as a non-apoptotic caspase under different biological processes.
The inflammatory caspases are well studied for their critical roles in inflammasome activation and cleavage of GSDMD to cause pyroptosis. Although not shown to be directly involved in pyroptosis, caspase-7 and poly(ADP-ribose) polymerase 1 (PARP1) were also shown to be cleaved by caspase-1 downstream of the NLRP3 inflammasome (Lamkanfi et al., 2008; Malireddi et al., 2010). The executioner caspases, caspase-3 and caspase-7, have recently been reported to be able to process GSDMD and GSDME. We observed that GSDMD processing was reduced in Casp6−/− BMDMs compared with WT BMDMs. Despite the fact that the other apoptotic executioners can directly cleave GSDMD, caspase-6 was unable to cleave GSDMD directly in our overexpression studies (data not shown), suggesting that caspase-6 regulates GSDMD processing via ZBP1-mediated inflammasome activation during IAV infection.
To date, how the NLRP3 inflammasome is activated in response to different kinds of triggers has not been fully understood, including specifically IAV-induced NLRP3 inflammasome activation. We have previously shown that ZBP1 is the ribonucleoprotein (RNP) complex sensor that facilitates IAV-induced cell death and inflammasome activation (Kesavardhana et al., 2017; Kuriakose et al., 2016). Additionally, viral M2 protein has been evidenced to be critical for IAV-induced NLRP3 inflammasome activation (Ichinohe et al., 2010). It is known that the IAV M2 protein is a proton channel and that it is essential for IAV particles to release the RNP complex from the M1 coat during infection. M2 deficiency will lead to defective release of the RNP complex, which could decrease the PAMPs available for sensing by ZBP1. Our study shows that caspase-6 also plays a crucial role in the IAV-induced NLRP3 inflammasome activation, adding to our understanding of this process. However, future studies will be needed to further understand the specific, central mechanism by which the NLRP3 inflammasome is activated.
Overall, we identified a previously unknown role for caspase-6 in regulating ZBP1-mediated NLRP3 inflammasome activation and programmed cell death in a protease activity-independent manner. Caspase-6 was dispensable for canonical NLRP3, AIM2, NLRC4, and PYRIN inflammasome activation and for apoptosis and necroptosis induced by classic triggers. Importantly, caspase-6 was required for host defense in a murine model of IAV infection. On a molecular level, caspase-6 interacted with RIPK3 to enhance the interaction between RIPK3 and ZBP1. This improved mechanistic understanding of the role of caspase-6 in PANoptosis, which paves the way for developing therapies to treat diseases where cell death pathways go awry.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for reagents may be directed to, and will be fulfilled by the lead contact Thirumala-Devi Kanneganti (thirumala-devi.kanneganti@stjude.org). Primer sequences generated for this study have been provided in this manuscript, and this study did not generate any other unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Casp6—/— (Jackson Laboratory, 006236), Nlrp3—/— (Kanneganti et al., 2006a), Casp11—/— (Kayagaki et al., 2011), Gsdmd−/− (Karki et al., 2018), Nlrc4—/— (Mariathasan et al., 2004), Aim2—/— (Jones et al., 2010), Rip3−/− (Newton et al., 2004), Rip3−/−Casp-8−/− (Oberst et al., 2011), Pyrin−/− (Van Gorp et al., 2016), Zbp1—/— (Ishii et al., 2008) mice have been described previously. All mice were bred and maintained in a speciic pathogen-free facility at the Animal Resources Center at St. Jude Children’s Research Hospital and were backcrossed to the C57BL/6 background. Single nucleotide polymorphisms (SNPs) were sequenced to confirm the genomic background. Both male and female mice were used in this study; age- and sex-matched 6- to 8-week old mice were used for in vivo and 6- to 12-week old mice were used for in vitro studies. Mice were maintained with a 12 h light/dark cycle and were fed standard chow. Cohoused animals were used for in vivo analyses. Littermates were used where indicated. Animal studies were conducted under protocols approved by the St. Jude Children’s Research Hospital committee on the Use and Care of Animals.
Bone marrow-derived macrophages (BMDMs)
Both male and female mice were used to collect BMDMs. Primary BMDMs were cultivated for 6 days in DMEM (Thermo Fisher Scientific, 11995–073) supplemented with 10% FBS (Biowest, S1620), 30% L929-conditioned medium, 1% non-essential amino acids (Thermo Fisher Scientific, 11140–050), and 1% penicillin and streptomycin (Thermo Fisher Scientific, 15070–063). BMDMs were then seeded into 12-well plates with antibiotic-free medium at a density of 1 million cells per well and incubated overnight before use.
Immortalized BMDMs (iBMDMs)
iBMDMs were established as previously described (Zhu et al., 2018). Briefly, J2 Cre retrovirus was added into the BMDMs at day 0 and day 3. At day 7, BMDMs were split, and the percentage of L929-conditioned media was reduced by half with each passage. To reconstitute the expression of the indicated caspase-6 clone in Casp6—/— iBMDMs, pCW57.1 was used to construct the clones. Then, together with pMD2.G and psPAX2, lentiviral particles containing those caspase-6 clones were rescued in 293T cells. Casp6−/− iBMDMs were transduced with lentivirus and then selected with puromycin at a concentration of 5 mg/mL. To induce the expression of the indicated caspase-6 clones, doxycycline was used at a concentration of 100 ng/mL.
Mouse embryonic fibroblasts (MEFs)
Both male and female mice were used to collect MEFs. MEFs were immortalized by serial passage and cultured in DMEM supplemented with 10% FBS and 1% penicillin and streptomycin. MEFs were then seeded into 12-well plates at a density of 2 × 105 cells per well and incubated overnight before use.
Influenza A virus culture
The influenza A virus (A/Puerto Rico/8/34, H1N1 [PR8]) was generated by reverse genetics as previously described (Hoffmann et al., 2000). Virus stocks were propagated by inoculation of seed virus into allantoic cavity of 9- to 11-day old embryonated chicken eggs. Virus titer was measured by plaque assay in MDCK cells.
Bacterial culture
P. aeruginosa strain PAO1 (a gift from Dr. Joseph Mougous) and S. Typhimurium strain SL1344 were grown in Luria-Bertani (LB) broth (MP Biomedicals, 3002–031) under aerobic conditions at 37○C overnight. Bacteria were then subcultured separately at a ratio of 1:10 at 37○C for 3 h in fresh LB broth to reach the log phase. Single colony isolates of F. novicida strain U112 were cultured under aerobic conditions at 37○C for 16 h in BBL Trypticase Soy Broth (BD Biosciences, 211768) supplemented with 0.2% L-cysteine (Thermo Fisher Scientific, BP376–100). Then the bacteria were subcultured at a ratio of 1:10 at 37○C for another 4 h in fresh medium before being resuspended in PBS (Thermo Fisher Scientific, 14190–250) for infection. C. difficile strain r20291 AB‒ and AB+ strains (provided by Dr. N. Minton) were incubated on brain heart infusion agar (BD Biosciences, 21165) under anaerobic conditions at 37○C for 16 h. Then a single colony was cultured overnight in tryptone-yeast extract medium at 37○C under anaerobic conditions. The supernatant with or without the toxins was collected via centrifugation followed by passing through a 0.2 μm filter.
METHOD DETAILS
CRISPR deletion of proteins in MEFs
Lentiviral particles expressing two different gRNAs targeting caspase-6 or ZBP1 were prepared by transfecting HEK293T cells with gRNA-expressing construct, psPAX2 and pMD2.G plasmids. Lentiviruses were harvested 48 h after transfection, supplemented with 10% FBS and stored at ‒80○C. MEFs expressing the Cas9 protein were transduced with gRNA-lentiparticles in combination with 6–8 mg/mL polybrene. Then, 24 h after transduction, lentivirus-containing media was replaced with fresh DMEM with FBS and incubated for another 24 h. Cells were subjected to puromycin selection (5 μg/mL; InvivoGen, ant-pr-1) to isolate caspase-6 or ZBP1 knockout MEFs.
Cell stimulation/infection
For canonical NLRP3 inflammasome activation, BMDMs were primed for 4 h with 500 ng/mL ultrapure LPS from E. coli (0111:B4) (Invivogen, tlrl-3pelps), 1 μg/mL Pam3CSK4 (Invivogen, tlrl-pms) or 2 μg/mL poly(I:C) (Invivogen, tlrl-picw) and then stimulated for 45 min with 5 mM ATP (Roche, 10127531001) or for 2 h with 20 μM nigericin (Sigma, N7143). For non-canonical NLRP3 inflammasome activation, BMDMs were primed for 4 h with 500 ng/mL ultrapure LPS from E. coli (0111:B4) and then transfected with 2 μg of LPS per well using Xfect polymer (Clontech Laboratories, Inc, 631318) according to the manufacturer’s instructions. For IAV infection, BMDMs and MEFs were infected at an MOI of 20 in DMEM plain media (Sigma, D6171). After absorption for 2 h, cells were supplemented with 10% FBS and then incubated for the indicated time. For poly(dA:dT) transfection, BMDMs were transfected with 2 mg of poly(dA:dT) per well using Xfect polymer according the manufacturer’s instructions. For flagellin transfection, DOTAP (Roche, 11202375001) was used, and 1 μg of flagellin per well was transfected according to the manufacturer’s instructions. For bacterial infection, the BMDMs were infected separately with the following bacteria: F. novicida at an MOI of 100 for 20 h, P. aeruginosa at an MOI of 2 for 2‒3h (Hood et al., 2010) and S. Typhimurium at an MOI of 2 for 2‒3 h. During F. novicida infection, 100 μg/mL gentamicin (Thermo Fisher Scientific, 15750–060) was added at 4 h post-infection and incubated for 1 h to kill the extracellular bacteria. Then spent media was removed and fresh media supplemented with 10 μg/mL gentamicin was added to replace the spent media. For MCMV infection, BMDMs were infected at an MOI of 10 for 16 h.
Immunoblot analysis
For caspase-1 analysis, BMDMs were lysed along with the supernatant using 50 mL caspase lysis buffer (1 × protease inhibitors, 1 × phosphatase inhibitors, 10% NP-40 and 25 mM DTT) followed by the addition of 150 μL4 × SDS loading buffer. For signaling analysis, the supernatants were removed at the indicated time points, and cells were washed once with PBS, after which cells were lysed with RIPA buffer. Electrophoresis was used to separate proteins in 8%‒15% polyacrylamide gels. After transfer of the proteins onto PVDF membranes, the blots were blocked with 5% skim milk. Primary antibodies were incubated overnight at 4○C, and secondary HRP antibodies were incubated for 1 h at room temperature. Images were acquired using a GE Amersham Imager 600.
The antibodies used were as follows: anti-caspase-1 (AdipoGen, AG-20B-0042, 1:2000), anti-caspase-6 (Cell Signaling Technology [CST], #9762, 1:1000), anti-cleaved caspase-6 (CST, #9761, 1:1000), anti-caspase-3 (CST, #9662, 1:1000), anti-cleaved cas-pase-3 (CST, #9661, 1:1000), anti-caspase-7 (CST, #9492, 1:1000), anti-cleaved caspase-7 (CST, #9491, 1:1000), anti-caspase-8 (CST, #4927, 1:1000), anti-cleaved caspase-8 (CST, #8592, 1:1000), anti-ZBP1 (AdipoGen, AG-20B-0010-C100, 1:2000), anti-pERK (CST, #9101, 1:1000), anti-pIκB (CST, #2859, 1:1000), anti-ERK (CST, #9102, 1:1000), anti-IκB (CST, #9242, 1:1000), anti-pSTAT1 (CST, #7649, 1:1000), anti-STAT1 (CST, #14994, 1:1000), anti-NLRP3 (AdipoGen, AG-20B-0014, 1:2000), anti-pro–IL-1β (CST, #12507, 1:1000), anti-RIPK1 (CST, #3493, 1:1000), anti-RIPK3 (CST, #95702, 1:1000 or ProSci, #2283, 1:1000), anti-pMLKL (CST, #37333, 1:1000), anti-MLKL (Abgent, AP14272b, 1:1000), anti-GFP (Santa Cruz Biotechnology, sc-8334, 1:1000), anti-Flag (Sigma, F1804, 1:5000), anti-GSDMD (Abcam, ab209845, 1:1000), anti-IAV M1 (Abcam, #ab20910, 1:2000), anti-IAV NS1 (Santa Cruz Biotechnology, NS1–23-1; sc-130568, 1:1000), anti-lamin A/C (CST; #2032, 1:1000), anti-NP (Bio-Rad, MCA400; 1:1000), anti-β-actin (Proteintech, 66009–1-IG, 1:5000), and HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, anti-rabbit [111–035-047], 1:5000; anti-mouse [315–035-047], 1:5000).
Real-time (RT-PCR) analysis
RNA was extracted with TRIzol (Thermo Fisher Scientific, 15596026). cDNA was synthesized by using the First Strand cDNA Synthesis Kit (Applied Biosystems, 4368814) according to the manufacturer’s instructions. Real-time qPCR was performed with 2 × SYBR Green (Applied Biosystems, 4368706) on an ABI 7500 RT-PCR instrument. Primers used were as follows: Actin: 5’-CAG CTT CTT TGC AGC TCC TT-3’, 5’-GAG CTA TGT CTT GGC CTT CC-3’; Influenza M1: 5’-TGA GTC TTC TAA CCG AGG TC-3’, 5’-GGT CTT GTC TTT AGC CAT TCC-3’.
Real-time cell death analysis
Real-time cell death analysis was performed as previously described (Malireddi et al., 2018). In brief, BMDMs were seeded in 12-well plates (106 cells/well) were infected with IAV and after absorption 100 nM SYTOX Green (Thermo Fisher Scientific, S7020) was added together with FBS. Images were analyzed using IncuCyte S3 software.
Lactate dehydrogenase (LDH) assay
Cell supernatants were collected from MEFs at the indicated time points after IAV infection at an MOI of 20. Lactate dehydrogenase activity was measured using the Promega cytotoxicity kit (G180A) according to the manufacturer’s instructions.
Cytokine analysis
Cytokines were detected by using multiplex ELISA (Millipore, MCYTOMAG-70K) or IL-18 ELISA (Invitrogen, BMS618–3) according to the manufacturer’s instructions.
Co-immunoprecipitation assay in 293T overexpression system
When wild-type caspase-expressing plasmids were transfected, 25 μM zVAD (Millipore) was added. After transfection for 30 h, 293T cells were lysed in NP-40 lysis buffer (0.1% NP-40, 150 mM NaCl, 50 mM HEPES), and 20 min later lysates were centrifuged at 13000 rpm for 10 min. Supernatant was collected and incubated with 1–2 μg of the indicated primary antibody on a rocking platform at 4○C. After overnight incubation, protein A/G PLUS-Agarose (Santa Cruz Biotechnology) beads were added, and 1 h later the beads were collected by centrifugation. Finally, samples were harvested by boiling in 1 × SDS loading buffer at 100○C for 5 min. All the plasmids used for caspase-6 expression are the catalytically dead mutant unless specifically indicated. The sex of the HEK293T cell line is female.
Endogenous co-immunoprecipitation assay
Primary BMDMs were seeded into 10-cm dishes and infected with IAV at an MOI of 10 for 16 h. After washing with 1 × PBS, cells were lysed in NP-40 lysis buffer as described above. Supernatant was collected and incubated with 2 μg of RIPK3 antibody on a rocking platform at 4○C. After 24 h, protein A/G PLUS-Agarose beads were added and incubated for 2–4 h. Then samples were harvested by boiling in 1 × SDS loading buffer at 100○C for 5 min.
Animal infection
Age- and sex-matched, 6- to 8-week old wild-type and Casp6−/− mice were anesthetized with 250 mg/kg Avertin and then infected intranasally with PR8 in 50 μL PBS containing around 50 PFU. For littermate control experiments, Casp6+/− and Casp6-/− littermates were used and infected with 125 PFU of IAV. Infected mice were monitored over a period of 14 days for survival study. Lungs harvested at the indicated time points were fixed with formalin for histological analysis or homogenized in 1 mL PBS for viral titers to be enumerated by plaque assays. Lung sections were processed by the Veterinary Pathology Core at St. Jude Children’s Research Hospital and stained for Arg1 and NP, and histological analysis was carried out by one pathologist blinded to the experimental groups.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism 7.0 software was used for data analysis. Data are presented as mean ± SEM. The Student’s t tests (two-tailed) for two groups or ANOVA (with Dunnett’s multiple comparisons test, Dunn’s multiple comparisons test, or Tukey’s multiple comparisons test) for three or more groups were used to determine the statistical significance. The log-rank test was used to compare the survival curves. P values less than 0.05 were considered statistically significant where *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
DATA AND CODE AVAILABILITY
The published article includes all datasets generated or analyzed during this study.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | DENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-caspase-1 | AdipoGen | Cat# AG-2GB-GG42 |
| anti-caspase-6 | Cell Signaling Technology | Cat# 9762 |
| anti-cleaved caspase-6 | Cell Signaling Technology | Cat# 9761 |
| anti-caspase-3 | Cell Signaling Technology | Cat# 9662 |
| anti-cleaved caspase-3 | Cell Signaling Technology | Cat# 9661 |
| anti-caspase-7 | Cell Signaling Technology | Cat# 9492 |
| anti-cleaved caspase-7 | Cell Signaling Technology | Cat# 9491 |
| anti-caspase-8 | Cell Signaling Technology | Cat# 4927 |
| anti-cleaved caspase-8 | Cell Signaling Technology | Cat# S592 |
| anti-ZBP1 | AdipoGen | Cat# AG-2GB-GG1G-C1GG |
| anti-pERK | Cell Signaling Technology | Cat# 91G1 |
| anti-plkB | Cell Signaling Technology | Cat# 2S59 |
| anti-ERK | Cell Signaling Technology | Cat# 91G2 |
| anti-IkB | Cell Signaling Technology | Cat# 9242 |
| anti-pSTAT1 | Cell Signaling Technology | Cat# 7649 |
| anti-STAT1 | Cell Signaling Technology | Cat# 14994 |
| anti-NLRP3 | AdipoGen | Cat# AG-2GB-GG14 |
| anti-pro-IL-1b | Cell Signaling Technology | Cat# 125G7 |
| anti-RIPK1 | Cell Signaling Technology | Cat# 3493 |
| anti-RIPK3 | Cell Signaling Technology | Cat# 957G2 |
| anti-RIPK3 | proSci | Cat# 22S3 |
| anti-pMLKL | Cell Signaling Technology | Cat# 37333 |
| anti-MLKL | Abgent | Cat# AP14272b |
| anti-GFP | Santa Cruz Biotechnology | Cat# sc-8334 |
| anti-Flag | Sigma | Cat# F1SG4 |
| anti-GSDMD | Abcam | Cat# ab2G9S45 |
| anti-IAV M1 | Abcam | Cat# ab2G91G |
| anti-IAV NS1 | Santa Cruz Biotechnology | Cat# sc-13G56S |
| anti-lamin A/C | Cell Signaling Technology | Cat# 2G32 |
| anti-NP | 3io-Rad | Cat# MCA4GG |
| anti-b-actin | proteintech | Cat# 66GG9–1-IG |
| HRP-conjugated secondary anti-rabbit | Jackson ImmunoResearch Laboratories | Cat# 111-G35-G47 |
| HRP-conjugated secondary anti-mouse | Jackson ImmunoResearch Laboratories | Cat# 315-G35-G47 |
| Bacterial and Virus Strains | ||
| influenza A virus A/Puerto Rico/8/34, H1N1 [PR8] | Hoffmann et al., 2000 | N/A |
| Pseudomonas aeruginosa strain PAO1 | Dr. Joseph Mougous (Hood et al., 2010) | N/A |
| Salmonella Typhimurium SL1344 | Man et al., 2015 | N/A |
| Francisella novicida strain U112 | Man et al., 2015 | N/A |
| C. difficile strain r20291 | Dr. N. Minton | N/A |
| MCMV | Dr. Edward Mocarski | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM | Thermo Fisher Scientific | Cat# 11995-G73 |
| Fetal bovine serum | Biowest | Cat# S162G |
| non-essential amino acids | Thermo Fisher Scientific | Cat# 11140–050 |
| penicillin and streptomycin | Thermo Fisher Scientific | Cat# 15070–063 |
| puromycin | Invivogen | Cat# ant-pr-1 |
| doxycline | Cayman Chemical Co. | Cat# 14422 |
| Luria-Bertani broth | MP Biomedicals | Cat# 3002–031 |
| BBL Trypticase Soy Broth | BD Biosciences | Cat# 211768 |
| L-cysteine | Thermo Fisher Scientific | Cat# BP376–100 |
| PBS | Thermo Fisher Scientific | Cat# 14190–250 |
| brain heart infusion agar | BD Biosciences | Cat# 21165 |
| ultrapure LPS from E. coli (0111:B4) | Invivogen | Cat# tlrl-3pelps |
| Pam3CSK4 | Invivogen | Cat# tlrl-pms |
| poly(I:C) | Invivogen | Cat# tlrl-picw |
| ATP | Roche | Cat# 10127531001 |
| nigericin | Sigma | Cat# N7143 |
| DMEM plain media | Sigma | Cat# D6171 |
| Poly(dA:dT) | Invivogen | Cat# tlrl-patn |
| Flagellin | Invivogen | Cat# tlrl-epstfla-5 |
| DOTAP Liposomal Transfection Reagent | Roche | Cat# 11202375001 |
| gentamicin | Thermo Fisher Scientific | Cat# 15750–060 |
| Protease inhibitor | Roche | Cat# 11697498001 |
| Phosphatase inhibitor | Roche | Cat# 04906837001 |
| TRIzol | Thermo Fisher Scientific | Cat# 15596026 |
| SYBR Green | Applied Biosystems | Cat# 4368706 |
| SYTOX Green | Thermo Fisher Scientific | Cat# S7020 |
| zVAD | Millipore | Cat# 187389–52-2 |
| Critical Commercial Assays | ||
| Xfect transfection kit | Clontech Laboratories, Inc | Cat# 631318 |
| First Strand cDNA Synthesis Kit | Applied Biosystems | Cat# 4368814 |
| Cytotoxicity kit | Promega | Cat# G180A |
| Multiplex ELISA | Millipore | Cat# MCYTOMAG-70K |
| IL-18 ELISA | Invitrogen | Cat# BMS618–3 |
| protein A/G PLUS-Agarose beads | Santa Cruz Biotechnology | Cat# sc-2003 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat# CRL-3216; RRID:CVCL_0063 |
| MDCK | ATCC | Cat# CCL-34 |
| Experimental Models: Organisms/Strains | ||
| Casp6−/− mice | Jackson Laboratory | Cat# 006236 |
| Nlrp3−/− mice | Kanneganti et al., 2006a | N/A |
| Casp11−/− mice | Kayagaki et al., 2011 | N/A |
| Gsdmd−/− mice | Karki et al., 2018 | N/A |
| Nlrc4−/− mice | Mariathasan et al., 2004 | N/A |
| Aim2−/− mice | Jones et al., 2010 | N/A |
| Rip3−/− mice | Newton et al., 2004 | N/A |
| Rip3−/− Casp-8−/− mice | Oberst et al., 2011 | N/A |
| Pyrin−/− mice | Van Gorp et al., 2016 | N/A |
| Zbp1−/− mice | Ishii et al., 2008 | N/A |
| Oligonucleotides | ||
| RT-PCR Actin Forward 5'-CAG CTT CTT TGC AGC TCC TT-3' | This paper | N/A |
| RT-PCR Actin Reverse 5'-GAG CTA TGT CTT GGC CTT CC-3' | This paper | N/A |
| RT-PCR influenza M1 Forward 5'-TGA GTC TTC TAA CCG AGG TC-3' | This paper | N/A |
| RT-PCR influenza M1 Reverse 5'-GGT CTT GTC III AGC CAT TCC-3' | This paper | N/A |
| Casp6 gRNA 1 5'-CAG GTT GTC TCT GTC TGC GTN GG-3' | This paper | N/A |
| Casp6 gRNA 2 5'-CGT TGG TGC CCC GCC TCT CTN GG-3' | This paper | N/A |
| Zbp1 gRNA 1 5'-TGC AGG TGT TGA GCG ATG ACG G-3' | This paper | N/A |
| Zbp1 gRNA 2 5'-GTC CTT TAC CGC CTG AAG AAG G-3' | This paper | N/A |
| Recombinant DNA | ||
| pCW57.1 vector | David Root Lab, https://www.addgene.org/browse/article/6041 | Addgene Cat# 41393 |
| pMD2.G vector | Didier Trono Lab, https://www.addgene.org/ browse/article/836/ | Addgene Cat# 12259 |
| psPAX2 vector | Didier Trono Lab, https://www.addgene.org/ browse/article/836/ | Addgene Cat# 12260 |
| RIPK3-GFP vector | Cho et al., 2009 | Addgene Cat# 41382 |
| Software and Algorithms | ||
| GraphPad Prism 7.0 | GraphPad Software, Inc. | https://www.graphpad.com/ |
| Other | ||
| Embryonated chicken eggs | Charles River | Cat# 10100326 |
Highlights.
Caspase-6 is required for innate immunity and ZBP1-NLRP3 inflammasome activation
Caspase-6 promotes ZBP1-mediated pyroptosis, apoptosis, and necroptosis
Caspase-6 binds RIPK3 and enhances the interaction between RIPK3 and ZBP1
Mice deficient in caspase-6 were more susceptible to IAV infection
ACKNOWLEDGMENTS
We thank all the members of the Kanneganti laboratory for their comments and suggestions. We thank A. Burton (St. Jude Children’s Research Hospital) for her technical support, R. Webby (St. Jude Children’s Research Hospital) for plasmids rescuing IAV, E.S. Mocarski for MCMV, and D. Root for the pCW57.1 plasmid. psPAX2 (Addgene plasmid #12260) and pMD2.G (Addgene plasmid #1225) were gifts from D. Trono. RIPK3-GFP (Addgene plasmid #41382) was a gift from Francis Chan. We also thank R. Tweedell, PhD, for scientific editing and writing support. Work from our laboratory is supported by the National Institutes of Health (AI101935, AI124346, AR056296, and CA163507 to T.-D.K.) and the American Lebanese Syrian Associated Charities (to T.-D.K.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Chauhan D, Bartok E, Gaidt MM, Bock FJ, Herrmann J, Seeger JM, Broz P, Beckmann R, Kashkar H, Tait SWG, et al. (2018). BAX/BAK-Induced Apoptosis Results in Caspase-8-Dependent IL-1beta Maturation in Macrophages. Cell Rep. 25, 2354–2368. [DOI] [PubMed] [Google Scholar]
- Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, Pelczar P, and Broz P. (2019). Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 38 Published online May 15, 2019. 10.15252/embj.2019101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, and Chan FK (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveraux QL, Takahashi R, Salvesen GS, and Reed JC (1997). X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388, 300–304. [DOI] [PubMed] [Google Scholar]
- Downey J, Pernet E, Coulombe F, Allard B, Meunier I, Jaworska J, Qureshi S, Vinh DC, Martin JG, Joubert P, and Divangahi M. (2017). RIPK3 interacts with MAVS to regulate type I IFN-mediated immunity to Influenza A virus infection. PLoS Pathog. 13, e1006326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Ló pez-Soto A, Kumar S, and Kroemer G. (2016). Caspases Connect Cell-Death Signaling to Organismal Homeostasis. Immunity 44, 221–231. [DOI] [PubMed] [Google Scholar]
- Gurung P, Anand PK, Malireddi RK, Vande Walle L, Van Opdenbosch N, Dillon CP, Weinlich R, Green DR, Lamkanfi M, and Kanneganti TD (2014). FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol 192, 1835–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar JA, Powell DA, Aachoui Y, Ernst RK, and Miao EA (2013). Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CM, and Martin SJ (2017). Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol. Cell 65, 715–729. [DOI] [PubMed] [Google Scholar]
- Hoffmann E, Neumann G, Kawaoka Y, Hobom G, and Webster RG (2000). A DNA transfection system for generation of influenza A virus from eight plasmids. Proc. Natl. Acad. Sci. USA 97, 6108–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood RD, Singh P, Hsu F, vener T, Carl MA, Trinidad RRS, Silver-man JM, Ohlson BB, Hicks KG, Plemel RL, et al. (2010). A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe 7, 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinohe T, Pang IK, and Iwasaki A. (2010). Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol 11, 404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T, and Kinoshita K. (2007). PrDOS: prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 35, W460–W464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, Uematsu S, Takeuchi O, Takeshita F, Coban C, and Akira S. (2008). TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 451, 725–729. [DOI] [PubMed] [Google Scholar]
- Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, et al. (2010). Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. USA 107, 9771–9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T-B, Yang S-H, Toth B, Kovalenko A, and Wallach D. (2013). Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38, 27–40. [DOI] [PubMed] [Google Scholar]
- Kang S, Fernandes-Alnemri T, Rogers C, Mayes L, Wang Y, Dillon C, Roback L, Kaiser W, Oberst A, Sagara J, et al. (2015). Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun 6, 7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanneganti T-D, Ozö ren N, Body-Malapel M, Amer A, Park J-H, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, et al. (2006a). Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236. [DOI] [PubMed] [Google Scholar]
- Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, Taraporewala ZF, Miller D, Patton JT, Inohara N, and Nú ñ ez G. (2006b). Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem 281, 36560–36568. [DOI] [PubMed] [Google Scholar]
- Karki R, and Kanneganti TD (2019). Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 19, 197–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki R, Lee E, Place D, Samir P, Mavuluri J, Sharma BR, Balakrishnan A, Malireddi RKS, Geiger R, Zhu Q, et al. (2018). IRF8 Regulates Transcription of Naips for NLRC4 Inflammasome Activation. Cell 173, 920–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal V, Dye R, Pakavathkumar P, Foveau B, Flores J, Hyman B, Ghetti B, Koller BH, and LeBlanc AC (2015). Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 22, 1676–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszyński, A., et al. (2013). Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249. [DOI] [PubMed] [Google Scholar]
- Kesavardhana S, Kuriakose T, Guy CS, Samir P, Malireddi RKS, Mishra A, and Kanneganti TD (2017). ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J. Exp. Med 214, 2217–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriakose T, and Kanneganti TD (2018). ZBP1: Innate Sensor Regulating Cell Death and Inflammation. Trends Immunol. 39, 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, Neale G, Vogel P, and Kanneganti TD (2016). ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol 1, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhani SA, Masud A, Kuida K, Porter GA Jr., Booth CJ, Mehal WZ, Inayat I, and Flavell RA (2006). Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311, 847–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Kanneganti TD, Van Damme P, Vanden Berghe T, Vanoverberghe I, Vandekerckhove J, Vandenabeele P, Gevaert K, and Nú ñ ez G. (2008). Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol. Cell. Proteomics 7, 2350–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL, et al. (2015). RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun 6, 6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao Y-S, Damko E, Moquin D, Walz T, McDermott A, et al. (2012). The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, Polykratis A, and Pasparakis M. (2016). RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 540, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malireddi RKS, Ippagunta S, Lamkanfi M, and Kanneganti T-D (2010). Cutting edge: proteolytic inactivation of poly(ADP-ribose) polymerase 1 by the Nlrp3 and Nlrc4 inflammasomes. J. Immunol 185, 3127–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malireddi RKS, Gurung P, Mavuluri J, Dasari TK, Klco JM, Chi H, and Kanneganti T-D (2018). TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J. Exp. Med 215, 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malireddi RKS, Kesavardhana S, and Kanneganti T-D (2019). ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol 9, 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, and Kanneganti TD (2016). Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol 16, 7–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Karki R, Malireddi RKS, Neale G, Vogel P, Yamamoto M, Lamkanfi M, and Kanneganti T-D (2015). The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat. Immunol 16, 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J, Frase S, Zhu Q, Malireddi RKS, Kuriakose T, et al. (2016). IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell 167, 382–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, and Dixit VM (2004). Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, and Dixit VM (2006). Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232. [DOI] [PubMed] [Google Scholar]
- Martinon F, Pé trilli V, Mayor A, Tardivel A, and Tschopp J. (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241. [DOI] [PubMed] [Google Scholar]
- Mascarenhas DPA, Cerqueira DM, Pereira MSF, Castanheira FVS, Fernandes TD, Manin GZ, Cunha LD, and Zamboni DS (2017). Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5/ NLRC4/ASC inflammasome. PLoS Pathog. 13, e1006502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McComb S, Chan PK, Guinot A, Hartmannsdottir H, Jenni S, Dobay MP, Bourquin J-P, and Bornhauser BC (2019). Efficient apoptosis requires feedback amplification of upstream apoptotic signals by effector caspase-3 or −7. Sci. Adv 5, u9433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata S. (2018). Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol 36, 489–517. [DOI] [PubMed] [Google Scholar]
- Newton K, Sun X, and Dixit VM (2004). Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol. Cell. Biol 24, 1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, Lill JR, Roose-Girma M, Warming S, Solon M, et al. (2016). RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 540, 129–133. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, and Thornberry NA (1997). Caspases: killer proteases. Trends Biochem. Sci 22, 299–306. [DOI] [PubMed] [Google Scholar]
- Ning X, Wang Y, Jing M, Sha M, Lv M, Gao P, Zhang R, Huang X, Feng JM, and Jiang Z. (2019). Apoptotic Caspases Suppress Type I Interferon Production via the Cleavage of cGAS, MAVS, and IRF3. Mol. Cell 74, 19–31. [DOI] [PubMed] [Google Scholar]
- Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH 3rd, Ingram JP, Rodriguez DA, Kosoff R, Sharma S, Sturm O, et al. (2016). RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe 20, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, and Green DR (2011). Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, et al. (2018). Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierini R, Juruj C, Perret M, Jones CL, Mangeot P, Weiss DS, and Henry T. (2012). AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 19, 1709–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, and Alnemri ES (2017). Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun 8, 14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy N, Deveraux QL, Takahashi R, Salvesen GS, and Reed JC (1997). The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. 16, 6914–6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruchaud S, Korfali N, Villa P, Kottke TJ, Dingwall C, Kaufmann SH, and Earnshaw WC (2002). Caspase-6 gene disruption reveals a requirement for lamin A cleavage in apoptotic chromatin condensation. EMBO J. 21, 1967–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR, and Poltorak A. (2018). Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 115, E10888–E10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, and Shao F. (2014). Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192. [DOI] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, and Wang X. (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. [DOI] [PubMed] [Google Scholar]
- Taabazuing CY, Okondo MC, and Bachovchin DA (2017). Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem. Biol 24, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, Sridharan H, Kosoff R, Shubina M, Landsteiner VJ, et al. (2016). DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 20, 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, et al. (1997). A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem 272, 17907–17911. [DOI] [PubMed] [Google Scholar]
- Tsuchiya K, Nakajima S, Hosojima S, Thi Nguyen D, Hattori T, Manh Le T, Hori O, Mahib MR, Yamaguchi Y, Miura M, et al. (2019). Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun 10, 2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gorp H, Saavedra PH, de Vasconcelos NM, Van Opdenbosch N, Vande Walle L, Matusiak M, Prencipe G, Insalaco A, Van Hauwermeiren F, Demon D, et al. (2016). Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation. Proc. Natl. Acad. Sci. USA 113, 14384–14389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Opdenbosch N, Van Gorp H, Verdonckt M, Saavedra PHV, de Vasconcelos NM, Gonc A, Vande Walle, L., Demon D, Matusiak M, Van Hauwermeiren F, et al. (2017). Caspase-1 Engagement and TLR-Induced c-FLIP Expression Suppress ASC/Caspase-8-Dependent Apoptosis by Inflammasome Sensors NLRP1b and NLRC4. Cell Rep. 21, 3427–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O’Reilly L, Mason K, Gross O, Ma S, Guarda G, et al. (2012). Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 36, 215–227. [DOI] [PubMed] [Google Scholar]
- Vince JE, De Nardo D, Gao W, Vince AJ, Hall C, McArthur K, Simpson D, Vijayaraj S, Lindqvist LM, Bouillet P, et al. (2018). The Mitochondrial Apoptotic Effectors BAX/BAK Activate Caspase-3 and −7 to Trigger NLRP3 Inflammasome and Caspase-8 Driven IL-1beta Activation. Cell Rep. 25, 2339–2353. [DOI] [PubMed] [Google Scholar]
- Wang Z, Jiang H, Chen S, Du F, and Wang X. (2012). The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243. [DOI] [PubMed] [Google Scholar]
- Wang J, Li F, Sun R, Gao X, Wei H, Li L-J, and Tian Z. (2013). Bacterial colonization dampens influenza-mediated acute lung injury via induction of M2 alveolar macrophages. Nat. Commun 4, 2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, and Shao F. (2017). Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103. [DOI] [PubMed] [Google Scholar]
- Wright PE, and Dyson HJ (2015). Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol 16, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X, et al. (2013). Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 23, 994–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Yang J, Gao W, Li L, Li P, Zhang L, Gong Y-N, Peng X, Xi JJ, Chen S, et al. (2014). Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513, 237–241. [DOI] [PubMed] [Google Scholar]
- Yao Y, Shi Q, Chen B, Wang Q, Li X, Li L, Huang Y, Ji J, and Shen P. (2016). Identification of Caspase-6 as a New Regulator of Alternatively Activated Macrophages. J. Biol. Chem 291, 17450–17466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, and Liu Z-G (2012). Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 109, 5322–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Zheng M, Balakrishnan A, Karki R, and Kanneganti TD (2018). Gasdermin D Promotes AIM2 Inflammasome Activation and Is Required for Host Protection against Francisella novicida. J. Immunol 201, 3662–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study.