

Abstract
Increasingly, repeat expansions are being identified as part of the complex genetic architecture of amyotrophic lateral sclerosis. To date, several repeat expansions have been genetically associated with the disease: intronic repeat expansions in C9orf72, polyglutamine expansions in ATXN2 and polyalanine expansions in NIPA1. Together with previously published data, the identification of an amyotrophic lateral sclerosis patient with a family history of spinocerebellar ataxia type 1, caused by polyglutamine expansions in ATXN1, suggested a similar disease association for the repeat expansion in ATXN1. We, therefore, performed a large-scale international study in 11 700 individuals, in which we showed a significant association between intermediate ATXN1 repeat expansions and amyotrophic lateral sclerosis (P = 3.33 × 10−7). Subsequent functional experiments have shown that ATXN1 reduces the nucleocytoplasmic ratio of TDP-43 and enhances amyotrophic lateral sclerosis phenotypes in Drosophila, further emphasizing the role of polyglutamine repeat expansions in the pathophysiology of amyotrophic lateral sclerosis.
Keywords: amyotrophic lateral sclerosis, trinucleotide repeat expansions, DNA repeat expansion, genetic association study
Repeat expansions are being identified as part of the complex genetic architecture of amyotrophic lateral sclerosis. This study shows a significant association between intermediate ATXN1 repeat expansions and amyotrophic lateral sclerosis, possibly via mislocalization of TDP-43, further emphasizing the role of polyglutamine expansions in the pathophysiology of amyotrophic lateral sclerosis.
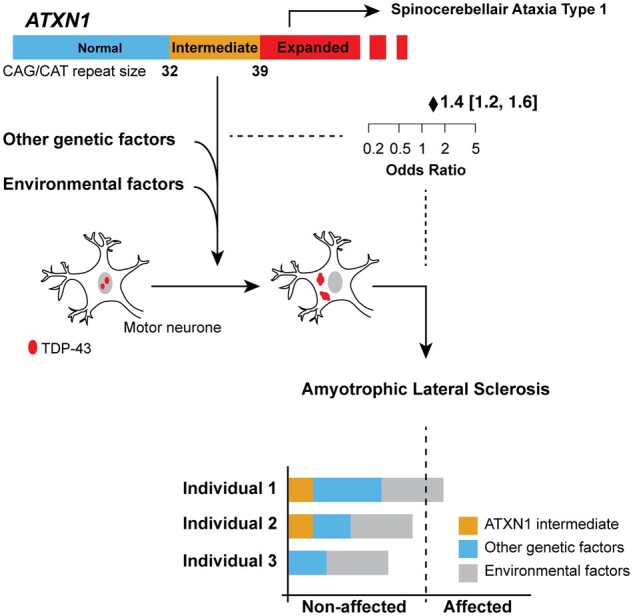
Graphical Abstract
Graphical Abstract.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder characterized by the loss of motor neurones leading to progressive weakness and spasticity (Brown and Al-Chalabi, 2017; van Es et al., 2017). Genetically, ALS is a highly heterogeneous disease with many underpinning factors (Al-Chalabi et al., 2017). In 5–15% of patients, there is a positive family history and it is assumed that there is a single causal mutation (Andersen and Al-Chalabi, 2011; Byrne et al., 2011). However, familial ALS mutations have also been identified in patients without a clear family history and multiple studies show that the genetic contribution to the risk of developing sporadic ALS is considerable (±40–60%) (Al-Chalabi et al., 2010; Wingo et al., 2011; Ryan et al., 2019). To date, over 40 different genes have been linked to ALS, mostly containing (rare) point mutations that significantly increase the risk of disease (Al-Chalabi et al., 2017). However, over the last few years, repeat expansions in several genes have also been implicated in ALS, including C9orf72, NIPA1 and ATXN2 (Blauw et al., 2010; Elden et al., 2010; DeJesus-Hernandez et al., 2011; Ajroud-Driss et al., 2015). ATXN2, for instance, contains trinucleotide repeat motif of CAG repeats, coding for a stretch polyglutamine (polyQ), and was first implicated as a risk factor in ALS after the discovery of it being a potent modifier of TDP-43 toxicity, an important step in ALS pathogenesis (Elden et al., 2010). A large expansion (>34) of the number of CAG repeats in ATXN2 (normally 22 or 23) is known to cause spinocerebellar ataxia type 2, whereas intermediate-length (29–33) repeats are associated with ALS (Elden et al., 2010; Rub et al., 2013).
In our outpatient clinic, we came across an ALS patient who had a positive family history for spinocerebellar ataxia type 1 (SCA1) (Fig. 1), a neurodegenerative disease caused by a polyQ repeat expansion in the ATXN1 gene (Banfi et al., 1994; Rub et al., 2012). There are some interesting similarities between ATXN2 and ATXN1, most importantly the presence of the coding CAG repeat motif. SCA1 patients may also have upper motor neurone signs, and autopsy studies show prominent loss of Betz cells, suggesting phenotypic overlap with ALS (Seidel et al., 2012; Rub et al., 2013; Saberi et al., 2015; Genc et al., 2017). Interestingly, a similar Italian ALS-SCA1 pedigree was reported a few years ago (Spataro and La Bella, 2014). This phenotypic overlap, as well as the co-occurrence of ALS and SCA1 in two unlinked pedigrees, makes ATXN1 a plausible candidate gene for ALS.
Figure 1.

Pedigree with co-occurrence of SCA1 and ALS. The index patient (arrow) was diagnosed with ALS and reported a positive family history for spinocerebellar ataxia type 1 (SCA1) in four other family members. No DNA samples from family members diagnosed with SCA1 were available for analysis.
Three previous studies have already explored this possible association between ATXN1 expansions and ALS (Lee et al., 2011; Conforti et al., 2012; Lattante et al., 2018). However, these studies have produced conflicting results, which are difficult to compare, due to the use of different repeat size cut-offs for expanded alleles; their conclusions mostly rely on nominal significance. Therefore, we set out to perform a large-scale genetic association study using data from 11 700 individuals and explore the possible role of ATXN1 in ALS.
Materials and methods
Subjects
All participants gave written informed consent, and approval was obtained from the local, relevant ethical committees for medical research. Genotyping experiments were performed on a total of 5088 DNA samples from four populations. All patients were diagnosed according to the revised El Escorial criteria. Control subjects were from ongoing population-based studies on risk factors in ALS (Huisman et al., 2011). All related individuals were excluded from further analysis.
PCR, sequencing and genotyping
Samples were analysed using polymerase chain reaction (PCR) according to protocols described previously, and results were analysed in a blinded and automated fashion. To confirm PCR fragment length, 850 samples were additionally analysed with Sanger sequencing. Primers: 5′-CAGTCTGAGCCAGACGCCGGGACACAAG-3′ (forward) and 5′-CGGTGTTCTGCGGAGAACTGGAAATGTGG-3′ (reverse).
To further increase sample size, we analysed ATXN1 repeat size in whole-genome sequencing (WGS) data, available to us through Project MinE using ExpansionHunter (Dolzhenko et al., 2017; Van Rheenen et al., 2017). There was a 1129 sample overlap in genotypes obtained from ExpansionHunter and PCR/Sanger sequencing, showing a 97.7% concordance in allele genotypes (2207/2258). In 30 of the 51 discordant alleles, there was only a single repeat unit difference between PCR and WGS, and of the remaining 21, at least 16 could simply be explained by mix-up of 8 samples. Considering this high percentage of concordance between ExpansionHunter and Sanger/PCR results, we did not perform additional validation experiments on the WGS samples and proceeded with the ExpansionHunter calls. C9orf72 status had been determined previously for 4530 ALS samples.
To identify the number and position of CAT interruptions in the CAG trinucleotide repeat of ATXN1, we analysed the Dutch WGS data of 353 control and 547 ALS cases sequenced using the HiseqX Sequencing System (resulting in 150 bp reads, able to span the entire repeat). All 150 bp reads mapped to the genomic region of ATXN1 (chr6: 16,327,000–16,329,000; hg19) were isolated and spanning reads were genotyped after the recognition of both the start- and end-motif within a single read. Two samples (one ALS, one control) did not contain any spanning reads and were, therefore, excluded. We only included repeat genotypes with two or more supporting reads and found repeat size predictions in 95% of alleles (1345/1418) comparable to genotypes determined with PCR and/or ExpansionHunter.
Cell culture and immunohistochemistry
mCherry-Ataxin-1 constructs were synthesized by Genscript (Piscataway, USA). HeLa cells expressing mNeongreen fusion to the endogeneous KPNA2 were created by CRISPR-mediated non-homologous endjoining of an mNeongreen-P2A-puromycin PCR product at the last codon of the KPNA2 CDS in its genomic locus. HeLa cells (ATCC) were cultured in high glucose DMEM (Invitrogen) supplemented with 10% foetal bovine serum (Greiner), 4 mM Glutamax (Invitrogen), penicillin (100 U/ml), streptomycin (100 μg/ml) and non-essential amino acids (1%). Cells were grown at 37°C in a humidified atmosphere with 5% CO2. Cells were transiently transfected using Lipofectamine F3000 (Invitrogen) according to manufacturer’s instructions. Cells were fixed 24 h after transfection in 4% formaldehyde in Phosphate Buffered Saline (PBS) and stained according to standard protocols (including methanol fixation and permeabilization by Phosphate buffered saline with Tween-20 (PBS-T) 0.04%). Rabbit anti-TDP-43 (12892-1-AP; Proteintech) was used to stain for TDP-43. AlexaFluor 488 secondary antibodies (Life Technologies) were used. Nuclei were visualized using NucBlue counterstaining (Thermo Scientific). Slides were mounted using ProLong Gold antifade reagent (Life Technologies).
Confocal images were obtained using a Zeiss LSM 510 Meta NLO confocal microscope. Images were analysed, formatted and quantified with FIJI software.
In brief, transfected cells from three independent transfections were analysed for their nuclear cytoplasmic ratio of TDP-43 or KPNA2 and scored for the presence of cytoplasmic inclusion bodies (only observed in ATXN1, but not mCherry transfected cells). All data were aggregated, and statistical analyses were carried out using Prism software.
Fly strains
Drosophila was maintained on a 12:12 light/dark cycle on a standard sugar-yeast medium (15 g/l agar, 50 g/l sugar, 100 g/l autolyzed yeast, 30 ml/l nipag and 3 ml/l propionic acid) at 25 °C. The following transgenic Drosophila strains were used in this study: GMR-TARDBP (# 51370) and UAS-GR36 (# 58692). All fly strains used were obtained from the Bloomington Drosophila Stock Center at Indiana University (BDSC) or the Vienna Drosophila RNAi Center (VDRC). The UAS-GR36 strain was crossed with balancer CyO and driver GMR, to obtain a balanced fly stock expressing the DPR construct in the eye.
Drosophila eye phenotype analysis
To assess the effect of ataxin-1 (ATXN1) repeat length, we crossed the GMR-TARDBP and the GMR-GR36 stocks with fly lines carrying UAS constructs expressing various sizes of the ATXN1 polyQ repeat.
Following strains were used: UAS-ATX1.2Q (# 39738), UAS-ATX1.30Q (# 39739), UAS-ATX1.82Q (# 37940) and UAS-eGFP (# 5428). For each cross, the collected offspring were divided by sex and the genotypes counted according to the balancers. We used a slightly modified eye phenotype analysis protocol as described in Boeynaems et al. (2016). Briefly, each fly was individually scored in a blinded fashion for the presence of necrotic spots using the following scoring scale (not affected = 0, mild = 1, medium = 2, heavy = 3, extreme = 4). We crossed each line at least three times independently to validate the specific phenotype. Eye phenotypes were imaged by light microscopy (Zeiss imager. M1), and the made Z-stacks were processed with ImageJ with the extended depth of field algorithm.
Statistical analysis
All statistical procedures were carried out in R 3.3.0 (http://www.r-project.org). Mantel–Haenszel method meta-analysis of odds ratios (ORs) was performed on subgroup and pooled data using ‘metafor’ 2.0 package. For the joint analysis on individual data, a generalized linear model was used with fixed-effects covariates: method of genotyping and country of origin. We additionally applied generalized linear mixed model to account for possible random effects, which gave similar results as the generalized linear model.
The effect on disease survival after onset and age at onset of the disease were tested using multivariate Cox regression with sex at birth, site of onset, age at onset (for survival only) and C9orf72 status as covariates. To calculate the expected frequency of co-occurring variants, we used the frequency of one variant in the unaffected population and multiplied this with the number of carriers of the other variant in the affected population. A binomial test was performed to compare the observed frequency of co-occurring variants in ALS patients with the calculated expected frequency.
The orthogonal data of the Drosphila eye images were analysed with the lbl_test of the coin package in R. This linear by linear association test takes into account the gradual scoring scale, where a score of 4 impacts the P-value more than a score of 1.
Data and materials availability
Genetic data generated from these cohorts has been placed for public access on the Project MinE Data Browser (http://databrowser.projectmine.com). DNA tissue samples can be obtained by contacting the corresponding author (M.A.v.E) or Project MinE ALS Sequencing Consortium. Fly strains can be obtained by contacting P.V.D.
Results
Genetic association of increased ATXN1 repeat size (≥33) with ALS
In our analysis, we included data from three different sources. First, we genotyped the repeat expansion in ATXN1 using PCR in 2672 ALS patients and 2416 geographically matched control samples from four different cohorts (Belgium, France, Ireland and The Netherlands; Supplementary Table 1). In this sample series, we found the most frequent alleles contained 29 or 30 trinucleotide repeats in both cases and controls (69.8% and 71.3%, respectively). In SCA1, ATXN1 repeat sizes ≥39 CAG/CAT are considered ‘expanded’ (Rub et al., 2013). We hypothesized that, similar to previous findings in ATXN2, ‘intermediate’ repeat sizes (between normal and expanded) could be associated with an increased risk of motor neurone disease. We determined the cut-off for these intermediate size expansions to be 33 or more CAG/CAT repeats using receiver operating characteristics and allele distribution analysis (with 94.7% of control alleles being within the ‘normal’ range) (Fig. 2). In this sample series, 12.2% of ALS patients (328/2,672) and 10.1% of controls (244/2,416) carried at least one ATXN1 allele with an expanded repeat size (i.e. above the ≥33 CAG/CAT cut-off). The fixed-effect meta-analysis of these four cohorts indicated an association between the presence of at least one expanded allele in ATXN1 and ALS status with an OR = 1.37 (95% CI = 1.18–1.60, P = 1.21 × 10−5; Fig. 3).
Figure 2.

Distribution of ATXN1 CAG/CAT repeat length. Proportion of total alleles grouped per ATXN1 repeat length determined via PCR analysis in a cohort of 2672 individuals affected with ALS (gray) and 2416 geographically matched controls (black) from four different cohorts (Belgium, France, Ireland and The Netherlands).
Figure 3.

ATXN1 polyglutamine repeat expansion meta-analysis. Forest plot for the fixed-effect Mantel–Haenszel meta-analysis of the effect of expanded (≥33) ATXN1 CAG/CAT repeats on ALS risk in three different datasets grouped per country of origin: previous reports, PCR-genotyped cohort and WGS-genotyped cohort. In addition, individual-level data of all three datasets were combined in a single logistic regression analysis (Joint analysis), which was corrected for the country of origin and method of genotyping. Weights depending on number of participants. CI, confidence interval. *Conforti et al. used a different cut-off for expanded/non-expanded status (≥32 CAG/CAT repeats). However, since the most frequent alleles in their data [28/29] seem to also have shifted one repeat unit compared to the Italian population in Lattante et al. and our data [29/30], we did not alter the expansion status.
Second, we investigated the association with ATXN1 repeat expansions in an independent cohort of 2048 ALS cases and 891 controls using WGS (Van Rheenen et al., 2017). ATXN1 repeat sizes were estimated from WGS data using ExpansionHunter (Dolzhenko et al., 2017). We confirmed a subset (n = 1129) of the ExpansionHunter genotypes using PCR and found 98% concordance between the two methods. Using the same cut-off for (intermediate) expanded alleles as in the PCR cohort (≥33 CAG/CAT), we found the direction of effect and allele frequency to be similar in all cohorts; expanded alleles were observed in 12.0% of cases (248/2048) compared to 8.8% in controls (78/891), resulting in an OR = 1.38 (95% CI = 1.02–1.88, P = 0.037; Fig. 3).
Lastly, we performed a fixed-effects meta-analysis on all available data, in which we also included the data from all three studies that previously reported on ATXN1 in ALS (totalling 2346 cases and 1327 controls). Using a Mantel–Haenszel meta-analysis, we found improved evidence of an association with ATXN1 expansions and ALS status with P = 3.55 × 10−6; and OR = 1.38, 95% CI = 1.20–1.57 (Fig. 3). We additionally applied a generalized linear model with correction for country of origin and method of genotyping on the pooled data of 7066 ALS patients and 4634 controls and found our results to be essentially unchanged (OR = 1.41, 95% CI = 1.24–1.61, P = 3.33 × 10−7).
No differences in CAT interruptions
In SCA1, the presence or absence of CAT interruptions in the CAG repeat can influence disease risk and/or phenotype (Menon et al., 2013). We explored the possibility that differences between cases and controls could be attributed to differences in CAT interruptions by analysing the WGS sequencing data in a subset of 352 control and 546 ALS cases. Almost all repeats contained one or more CAT interruptions, with only one affected and one non-affected individual carrying an uninterrupted repeat (13 and 30 CAG repeats, respectively). The majority of the ATXN1 repeats in both cases and controls contained two CAT interruptions (Fig. 4A), with 99.9% (1267/1268) having a (CAG)n1(CAT)(CAG)(CAT)(CAG)n2 interruption pattern. Because of this minimal variation in the interruption number and position, we found a similar correlation and distribution of uninterrupted CAG repeat size compared to that of the full-length repeat (Fig. 4B).
Figure 4.

Presence and number of CAT interruptions in ATXN1 CAG repeat expansion. (A and B) Plots show the results after genotyping 1418 repeat alleles (849 ALS; 569 control) from 150 bp WGS reads that span the full repeat. (A) Number of CAT interruptions per repeat allele. (B) Correlation between the total repeat size, including both CAG and CAT, and the longest stretch of uninterrupted CAG per allele for both ALS affected (blue) and unaffected (orange). CAT interruptions usually and exclusively appear after the first 12–17 CAG repeats, resulting in a significant correlation between the total and uninterrupted CAG repeat size (Kendall’s tau cor., P < 2.2e−16 for both ALS and controls) and therefore a similar distribution (margin panels; prop.tot = proportion of total alleles). There were two exceptions (red border): one ALS-affected allele had no interruptions, probably because of its short length (13), and one unaffected sample seemed to carry an uninterrupted stretch of 30 CAG.
No effect on age at onset or survival
Several ALS-associated risk factors also affect the clinical phenotype. We investigated the effect of ATXN1 repeat expansions on survival and age at onset in a subset of 1890 ALS patients for whom clinical data were available but found no significant effects (Fig. 5A and B).
Figure 5.

Effect of ATXN1 repeat expansion on survival and age at onset in ALS. (A and B) Plots of time-dependent probabilities in 1890 ALS patients with either ATXN1 normal (<33, orange) or expanded (≥33, blue) CAG/CAT repeat expansion. (A) Survival after the onset of disease in months, corrected for: sex, age at onset, bulbar site of onset and presence of C9ORF72 expansion. (B) Age at onset of the disease in years corrected for: sex, site of onset and the presence of a C9orf72 repeat expansion. No significant effects were found.
Ataxin-1 overexpression perturbs nucleocytoplasmic transport of TDP-43
The pathological hallmark of ALS is the aggregation and cytoplasmic mislocalization of the RNA binding protein, TDP-43(Neumann et al., 2006). It is thought that the mislocalization of TDP-43 leads to both a nuclear loss-of-function as well as a cytoplasmic toxic gain-of-function. However, the exact mechanisms underpinning TDP-43-mediated neurodegeneration have not yet been fully elucidated. Considering the large number of genes that have been implicated in ALS to date, it seems likely that TDP-43 pathology may arise through multiple pathways. Recent evidence shows that ataxin-2 drives localization of TDP-43 to cytoplasmic stress granules; this process, because of the subsequent incapacity to disassemble these stress granules, has been proposed as the first stepping stone towards the formation of pathological aggregates (Ramaswami et al., 2013; Becker et al., 2017). Given that ataxin-1 has similarities with ataxin-2, we initially considered that similar mechanisms would be involved.
Ataxin-2 is, however, a cytoplasmic stress granule protein known to interact with TDP-43 (Elden et al., 2010), whereas this is not the case for ataxin-1. We, therefore, explored other disease mechanisms for ataxin-1 in ALS and started by performing simple overexpression studies of wild type/normal-length ataxin-1 in HeLa cells. Overexpression of ataxin-1 did not alter endogenous TDP-43 expression (Fig. 6A and B) and resulted in the formation of nuclear and cytoplasmic ataxin-1 inclusion bodies, negative for TDP-43 (Fig. 6C, top panel). We also observed that some cells overexpressing ataxin-1 showed cytoplasmic mislocalization of TDP-43 (Fig. 6C, bottom panel). Interestingly, this TDP-43 mislocalization significantly correlated with the presence of the cytoplasmic ataxin-1 inclusion bodies (Fig. 6D).
Figure 6.

HeLA cells were transfected with mCherry-tagged ataxin-1 containing 27 polyglutamine repeats (mCherry-Atx227Q) or control vector (mCherry). (A) TDP-43 protein levels are not altered in ataxin-1 expressing cells (uncropped blot image in Supplementary Fig. 1). (B) Quantification of TDP-43 levels normalized to loading control GAPDH (glyceraldehyde 3-phosphate dehydrogenase). Unpaired t-test, two-sided, P-value: 0.1312. (C) The presence of cytoplasmic inclusion bodies (IB) correlates with TDP-43 mislocalization in ataxin-1-expressing cells. TDP-43 does not accumulate in nuclear or cytoplasmic ataxin-1 IB but does mislocalize to the cytoplasm in cells with IB. (D) Quantification of TDP-43 mislocalization in controls cells (mCherry) and cells without (−IBcyto) or with cytoplasmic ataxin-1 IB (+IBcyto). (E) Cytoplasmic IB also correlate with GFP-tagged KNPA2 mislocalization to the cytoplasm. (F) Quantification of KPNA2 mislocalization. (D and F) One-way ANOVA, ****P < 0.0001.
A possible mechanism for mislocalization of TDP-43, recently implicated in ALS pathogenesis, is that of misregulation of nucleocytoplasmic transport, making TDP-43 unable to (re)enter the nucleus and as a result become trapped in the cytoplasm (Woerner et al., 2016). We hypothesized that ataxin-1 cytoplasmic accumulation could perturb the nuclear import system and subsequently investigated importin-α2 (KPNA2; karyopherin subunit alpha 2), which is involved in importing TDP-43 into the nucleus (Nishimura et al., 2010). Similar to TDP-43, we indeed found significant mislocalization of endogenous KPNA2 in HeLa cells containing ataxin-1 inclusion bodies (Fig. 6E and F).
Co-expression of human TDP-43 with ataxin-1 aggravates the phenotype in Drosophila
Considering the modest effect of intermediate ATXN1 expansions in our genetic analysis, we do not presume that they have a directly pathogenic effect, but rather that they are a contributing factor in the multi-step process towards developing the disease. Based on this hypothesis, we postulate that expanded ATXN1 CAG repeats would aggravate the phenotype in an in vivo model of TDP-43 pathology. We, therefore, turned to Drosophila, a suitable model organism for genetic experiments, the fly eye being widely used to evaluate neurodegeneration (Fig. 7A;Freibaum et al., 2015; Zhang et al., 2015; Boeynaems et al., 2016). Expression of the human TDP-43 gene in the Drosophila eye using GMR-GAL4 results in a ‘rough eye’ phenotype (Choksi et al., 2014). This rough eye phenotype is mainly characterized by a progressive, age-dependent degeneration of the structure, which ultimately results in depigmentation by retinal degeneration. To increase the chance of an observable effect, we tested with ATXN1 containing either an exaggerated normal (2Q) or expanded (82Q) polyQ repeat length. Co-expression of human TDP-43 with ataxin-1 polyQ constructs with a repeat length of 82 aggravated the phenotype, with the formation of necrotic spots (Fig. 7B), whereas expressing ataxin-1 2Q or 82Q alone did not result in an eye phenotype. Scoring the severity of the eye abnormalities via a graduated scoring table showed a significant increase in the score in TDP-43-expressing flies that jointly expressed the 82Q repeat, indicating a synergistic effect of ataxin-1 on TDP-43 toxicity in Drosophila (P = 2.65x10−4; Fig. 7C).
Figure 7.

Ataxin-1 polyQ modifies eye phenotypes in Drosophila. (A) Scheme indicating assessment of genetic modifiers. (B) Effect of eye phenotype after co-expression of eGFP, 2Q ataxin-1 and 82Q ataxin-1 in wild type (top) and TDP-43- (middle) and GR36 (bottom)-expressing flies. (C) Fraction of flies per necrotic eye score rank (darker shading equals higher score). Right panel: flies overexpressing ATXN1.82Q only show a clear degenerative phenotype characterized by a moderate rough eye phenotype, but only very small necrotic spots. Middle panel: flies co-expressing TDP-43 and ATXN1 polyQ with a repeat length of 82 with a severe eye phenotype are significantly enriched compared to flies expressing TDP-43 and ATXN1 with a polyQ repeat length of 2 (P = 2.65 × 10−4); there was no significant difference with eGFP and 2 polyQ. Left panel: flies co-expressing GR(36) and ATXN1 polyQ with a repeat length of 82 with a severe eye phenotype are significantly enriched compared to flies expressing GR(36) and ATXN1 with a polyQ repeat length of 2 (P < 2.0 × 10−16); there was no significant difference with eGFP and 2 polyQ. Statistical analysis using linear by linear association test, n > 50 per genotype.
ATXN1 polyQ also aggravates the phenotype in a Drosophila model for C9orf72
The co-occurrence of variants in multiple ALS genes within a single case is observed frequently (van Blitterswijk et al., 2012; Bury et al., 2016). In particular, this co-occurrence of multiple variants has been reported for patients carrying repeat expansions in C9orf72 (van Blitterswijk et al., 2014a, b; Dekker et al., 2016), which to some degree might also explain the phenotypic heterogeneity associated with this gene (including ALS, frontotemporal dementia, parkinsonism and psychosis; Cooper-Knock et al., 2015). A previous study on ATXN1 in ALS reported the co-occurrence of ATXN1 and C9orf72 expansions (Lattante et al., 2018). In our cohort, we identified a total of 23 patients carrying both expansions (6.4% of all C9orf72-positive patients also had an ATXN1 expansion ≥33 CAG/CAT) and also came across a familial ALS pedigree in which two ALS-affected first degree relatives carried both repeat expansions (Supplementary Fig. 2). We, therefore, explored whether co-expression of ATXN1 polyQ constructs in a Drosophila model for C9orf72 [expressing toxic glycine-arginine (GR36) dipeptide repeats] would aggravate the rough eye phenotype (Mizielinska et al., 2014). Indeed, these flies show a strong eye phenotype, characterized by eye depigmentation and necrotic spots (Fig. 7B). When ATXN1 82Q, but not 2Q, was co-expressed in the eye, we observed a significant enrichment of the severely affected eyes (P < 2.0 × 10−16; Fig. 7C). Almost 50% of the scored flies showed a harsh degenerated eye with numerous necrotic spots, indicating that ataxin-1 polyglutamine expansions also aggravate the GR-mediated neurodegeneration. These findings suggest an interaction of expanded ataxin-1 polyQ with pathological events in the disease.
Discussion
In this study, we demonstrate an association between intermediate polyQ expansions in ATXN1 and risk of ALS. We observed similar allele frequencies and direction of effect across international cohorts and the increase in sample size resulted in stronger statistical evidence compared to previous reports, indicating a robust association.
Using a generalized linear model with correction for country of origin and method of genotyping on the pooled data of 7066 ALS patients and 4634 controls, we found a P-value of 3.33 × 10−7. Empirical significance thresholds have been set for studies analysing common genetic polymorphisms across the genome, such as genome-wide significance (P = 5.0 × 10−8) for genome-wide studies and exome-wide significance (P = 5.0 × 10−7) for studies that only focus on coding single-nucleotide variants. However, no such thresholds have been set for genetic studies looking at repeat expansions on a genome/exome-wide level. We, therefore, considered three different cut-off values for significance: (i) Bonferroni correction for the number of previously reported polymorphic polyQ stretches of 6 and longer in the genome (P = 0.05/85 = 5.9 × 10−4) (Kozlowski et al., 2010), (ii) correcting for the total number of genes in the genome containing a homo-amino acid stretch (P = 0.05/878 = P = 5.6 × 10−5) (Kozlowski et al., 2010) or (iii) simply applying the level for exome-wide significance, as polyQ repeats are a coding form of genetic variation. A valid argument can be made for all three thresholds and as more association studies on structural variation on a genome-wide level become available, it seems likely that empirical significance thresholds will be determined. For now, our findings are significant regardless of which threshold is applied.
It is still unclear as to how ATXN1 polyQ expansions could have a contributing effect on ALS development. We sought to provide the first steps by performing functional experiments investigating the effect of ATXN1 polyQ on the cellular processing of the nuclear RNA binding protein TDP-43, the pathological hallmark of ALS. Ataxin-2 plays an important role in stress granule formation and in ALS; these stress granules fail to disassemble, hereby forming the precursors of TDP-43 aggregates(Elden et al., 2010; Hart et al., 2012). As ATXN1 is largely homologous to ATXN2 and both contain an expanded polyQ stretch, this was our initial hypothesis. There is, however, no literature implicating ATXN1 in stress granule formation and in our in vitro model, we did not observe co-localization with TDP-43. We did, however, observe a cytoplasmic mislocalization that seemed to be dependent on the disruption of ataxin-1. Since mislocalization was observed in both expanded as well as wild-type (Q27) ATXN1 HeLa cell models, disruption is possibly due to overexpression itself (Supplementary Fig. 3); this is similar to observations in ATXN2, where the effects of wild-type overexpression on TDP-43 was an important first step for further investigation (Elden et al., 2010). Although a HeLa cell overexpression model is far from representative for ALS, the current consensus that both a nuclear loss- and cytoplasmic gain-of-function of TDP-43 play a key role in ALS pathogenesis led us to shift our focus to nucleocytoplasmic transport, another mechanism that has recently been implicated in ALS (Neumann et al., 2006; Ling et al., 2013). In vitro studies have shown that disruption of the classical nuclear import pathway (which includes KPNA2) in neurones leads to the cytoplasmic accumulation of TDP-43, and also in post-mortem studies of ALS and frontotemporal dementia cases; KPNA2 levels were found to be decreased in both brain and spinal cord (Nishimura et al., 2010). Similarly, our in vitro results show that overexpression of normal-length ataxin-1 can cause mislocalization of TDP-43 and KPNA2. This suggests that pathological ataxin-1 effects could be mediated via perturbed nucleocytoplasmic transport.
Given the multifactorial aetiology of ALS, the modest genetic effect and a possible pathological effect through TDP-43, we lastly explored whether ATXN1 polyQ would aggravate the phenotype in an in vivo model of TDP-43 pathology. For this, we used existing Drosophila models that indeed show an aggravated phenotype when expanded ATXN1 is co-expressed with human TDP-43. Since there is only a relatively small difference in the size of the polyQ tract between normal and intermediate expansions, we deliberately chose two extreme values (Q2 and Q82) to maximize the possible phenotypic effect of ATXN1 polyQ on TDP-43 pathology. Despite this exaggeration, the absence of a necrotic eye phenotype in ATXN1 Q82 alone suggests a neurotoxic effect via TDP-43 and since co-expression of ATXN1.82Q, but not ATXN1.2Q, dramatically enhanced the degenerative eye phenotype; this suggest that TDP-43 or GR36 overexpression-induced toxicity by ATXN1 occurs in a repeat-length dependent manner.
As the co-occurrence of C9orf72 and ATXN1 expansions was observed in multiple ALS patients, we performed a similar Drosophila experiment in which we co-expressed ATXN1 polyQ with GR36 (toxic dipeptide repeat associated with C9orf72) and again found synergistic toxic effect in these flies. There is high phenotypic variability among individuals carrying repeat expansions in C9orf72, which includes ALS, frontotemporal dementia, parkinsonism and psychosis (Cooper-Knock et al., 2015). It has been proposed that additional genetic factors influence the C9orf72 phenotype. For instance, there is evidence suggesting that SNPs in TMEM106b protect against dementia (Nicholson and Rademakers, 2016), whereas other variants in other genes may give rise to ALS (van Blitterswijk et al., 2014a, b; Dekker et al., 2016). Our data suggest that expanded ATXN1 polyQ alleles influence the phenotype associated with C9orf72.
In conclusion, we demonstrate a robust genetic association between ATXN1 repeat expansions with the risk of ALS and provide evidence suggesting that this contributes to ALS pathophysiology through perturbed nucleocytoplasmic transport. In line with the multistep and oligogenic hypothesis for ALS, we show that ATXN1 polyQ aggravates the phenotype in multiple transgenic fly models (hTDP-43 and GR36). As the ATXN1 polyQ expansion is likely to result in a gain-of-function, silencing the expanded allele and perhaps thereby (partially) restoring nucleocytoplasmic transport could prove to be an interesting therapeutic approach.
Supplementary Material
Acknowledgements
The authors thank the patients and unaffected individuals for participation in the study and thank the Project MinE ALS Sequencing Consortium for providing access to the WGS database.
Funding
This study was supported by the ALS Foundation Netherlands, the Belgian ALS Liga and National Lottery, Agency for Innovation by Science and Technology (IWT), and the MND Association (UK) (Project MinE, www.projectmine.com). Research leading to these results has received funding from the European Community’s Health Seventh Framework Programme (FP7/2007–2013). This study was supported by ZonMW under the frame of E-Rare-2, the ERA Net for Research on Rare Diseases (PYRAMID). This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no 772376—EScORIAL). The collaboration project is co-funded by the PPP Allowance made available by Health∼Holland, Top Sector Life Sciences & Health, to stimulate public-private partnerships. This is an EU Joint Programme—Neurodegenerative Disease Research (JPND) project (STRENGTH, BRAIN-MEND, SOPHIA, ALS-CarE). The project is supported through the following funding organizations under the aegis of JPND: UK, Medical Research Council (MR/L501529/1; MR/R024804/1) and Economic and Social Research Council (ES/L008238/1); Ireland, Health Research Board; Netherlands, ZonMw; Belgium, FWO-Vlaanderen. Samples used in this research were in part obtained from the UK National DNA Bank for MND Research, funded by the MND Association and the Wellcome Trust. This project was supported by the MND Association of England, Wales and Northern Ireland and the Netherlands Organisation for Health Research and Development (Vici scheme to L.H.v.d.B. and Veni scheme to M.A.v.E.). NDAL cordially thanks Suna and Inan Kirac Foundation for their generous support. Funding was provided by US National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NINDS) (R01NS073873, J.E.L.) and the American ALS Association (J.E.L.). S.B. holds an EMBO long-term fellowship. M.A.v.E. is supported by the Thierry Latran Foundation, the Dutch ALS Foundation and the Rudolf Magnus Brain Center Talent Fellowship. A.A.-C. receives salary support from the National Institute for Health Research (NIHR) Dementia Biomedical Research Unit and Biomedical Research Centre in Mental Health at South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. O.H. is funded by the Health Research Board Clinician Scientist Programme and Science Foundation Ireland. R.L.M. is supported by the Thierry Latran Foundation (ALSIBD) and the ALS Association (2284). P.V.D. holds a senior clinical investigatorship from FWO-Vlaanderen and is supported by the ALS Liga België, Een hart voor ALS and the Laevers fund for ALS research. S.M. is supported by the Association française contre les myopathies (AFM) and the Association pour la Recherche sur la Sclérose latérale amyotrophique et autres maladies du motoneurone (ARSla).
Competing interests
The authors declare no competing financial interests.
Glossary
- ALS =
amyotrophic lateral sclerosis
- OR =
odds ratio
- polyQ =
polyglutamine
- SCA1 =
spinocerebellar ataxia type 1
- WGS =
whole-genome sequencing
Appendix I: Project MinE ALS Sequencing Consortium
Fulya Akçimen, Ahmad Al Khleifat, Ammar Al-Chalabi, Peter Andersen, A. Nazli Basak, Denis C. Bauer, Ian Blair, William J. Brands, Ross P. Byrne, Andrea Calvo, Yolanda Campos Gonzalez, Adriano Chio, Jonothan Cooper-Knock, Philippe Corcia, Philippe Couratier, Mamede de Carvalho, Annelot M. Dekker, Vivian E. Drory, Chen Eitan, Alberto Garcia Redondo, Cinzia Gellera, Jonathan D. Glass, Marc Gotkine, Orla Hardiman, Eran Hornstein, Alfredo Iacoangeli, Kevin P. Kenna, Brandon Kenna, Matthew C. Kiernan, Cemile Kocoglu, Maarten Kooyman, John E. Landers, Victoria López Alonso, Russell L. McLaughlin, Bas Middelkoop, Jonathan Mill, Miguel Mitne-Neto, Matthieu Moisse, Jesus S. Mora Pardina, Karen E. Morrison, Susana Pinto, Marta Gromicho, Monica Povedano Panadés, Sara L. Pulit, Antonia Ratti, Wim Robberecht, Raymond D. Schellevis, Aleksey Shatunov, Christopher E. Shaw, Pamela J. Shaw, Vincenzo Silani, William Sproviero, Christine Staiger, Gijs H. P. Tazelaar, Nicola Ticozzi, Ceren Tunca, Nathalie A. Twine, Philip van Damme, Leonard H. van den Berg, Rick A. van der Spek, Perry T. C. van Doormaal, Kristel R. van Eijk, Michael A. van Es, Wouter van Rheenen, Joke J. F. A. van Vugt, Jan H. Veldink, Peter M. Visscher, Patrick Vourc’h, Markus Weber, Kelly L. Williams, Naomi Wray, Jian Yang, Mayana Zatz and Katharine Zhang.
Members are listed in alphabetical order, a full list of members with affiliations is found in Supplementary List 1.
Contributor Information
Project MinE ALS Sequencing Consortium:
Fulya Akçimen, Ahmad Al Khleifat, Ammar Al-Chalabi, Peter Andersen, A Nazli Basak, Denis C Bauer, Ian Blair, William J Brands, Ross P Byrne, Andrea Calvo, Yolanda Campos Gonzalez, Adriano Chio, Jonothan Cooper-Knock, Philippe Corcia, Philippe Couratier, Mamede de Carvalho, Annelot M Dekker, Vivian E Drory, Chen Eitan, Alberto Garcia Redondo, Cinzia Gellera, Jonathan D Glass, Marc Gotkine, Orla Hardiman, Eran Hornstein, Alfredo Iacoangeli, Kevin P Kenna, Brandon Kenna, Matthew C Kiernan, Cemile Kocoglu, Maarten Kooyman, John E Landers, Victoria López Alonso, Russell L McLaughlin, Bas Middelkoop, Jonathan Mill, Miguel Mitne-Neto, Matthieu Moisse, Jesus S Mora Pardina, Karen E Morrison, Susana Pinto, Marta Gromicho, Monica Povedano Panadés, Sara L Pulit, Antonia Ratti, Wim Robberecht, Raymond D Schellevis, Aleksey Shatunov, Christopher E Shaw, Pamela J Shaw, Vincenzo Silani, William Sproviero, Christine Staiger, Gijs H P Tazelaar, Nicola Ticozzi, Ceren Tunca, Nathalie A Twine, Philip van Damme, Leonard H van den Berg, Rick A van der Spek, Perry T C van Doormaal, Kristel R van Eijk, Michael A van Es, Wouter van Rheenen, Joke J F A van Vugt, Jan H Veldink, Peter M Visscher, Patrick Vourc’h, Markus Weber, Kelly L Williams, Naomi Wray, Jian Yang, Mayana Zatz, and Katharine Zhang
References
- Ajroud-Driss S, Fecto F, Ajroud K, Lalani I, Calvo SE, Mootha VK, et al. Mutation in the novel nuclear-encoded mitochondrial protein CHCHD10 in a family with autosomal dominant mitochondrial myopathy. Neurogenetics 2015; 16: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Chalabi A, Fang F, Hanby MF, Leigh PN, Shaw CE, Ye W, et al. An estimate of amyotrophic lateral sclerosis heritability using twin data. J Neurol Neurosurg Psychiatry 2010; 81: 1324–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Chalabi A, van den Berg LH, Veldink J.. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol 2017; 13: 96–104. [DOI] [PubMed] [Google Scholar]
- Andersen PM, Al-Chalabi A.. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol 2011; 7: 603–15. [DOI] [PubMed] [Google Scholar]
- Banfi S, Servadio A, Chung MY, Kwiatkowski TJ Jr., McCall AE, Duvick LA, et al. Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat Genet 1994; 7: 513–20. [DOI] [PubMed] [Google Scholar]
- Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017; 544: 367–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blauw HM, Al-Chalabi A, Andersen PM, van Vught PW, Diekstra FP, van Es MA, et al. A large genome scan for rare CNVs in amyotrophic lateral sclerosis. Hum Mol Genet 2010; 19: 4091–9. [DOI] [PubMed] [Google Scholar]
- Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovicic A, et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep 2016; 6: 20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RH, Al-Chalabi A.. Amyotrophic lateral sclerosis. N Engl J Med 2017; 377: 162–72. [DOI] [PubMed] [Google Scholar]
- Bury JJ, Highley JR, Cooper-Knock J, Goodall EF, Higginbottom A, McDermott CJ, et al. Oligogenic inheritance of optineurin (OPTN) and C9ORF72 mutations in ALS highlights localisation of OPTN in the TDP-43-negative inclusions of C9ORF72-ALS. Neuropathology 2016; 36: 125–34. [DOI] [PubMed] [Google Scholar]
- Byrne S, Walsh C, Lynch C, Bede P, Elamin M, Kenna K, et al. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2011; 82: 623–7. [DOI] [PubMed] [Google Scholar]
- Choksi DK, Roy B, Chatterjee S, Yusuff T, Bakhoum MF, Sengupta U, et al. TDP-43 Phosphorylation by casein kinase Iepsilon promotes oligomerization and enhances toxicity in vivo. Hum Mol Genet 2014; 23: 1025–35. [DOI] [PubMed] [Google Scholar]
- Conforti FL, Spataro R, Sproviero W, Mazzei R, Cavalcanti F, Condino F, et al. Ataxin-1 and ataxin-2 intermediate-length PolyQ expansions in amyotrophic lateral sclerosis. Neurology 2012; 79: 2315–20. [DOI] [PubMed] [Google Scholar]
- Cooper-Knock J, Kirby J, Highley R, Shaw PJ.. The spectrum of C9orf72-mediated neurodegeneration and amyotrophic lateral sclerosis. Neurotherapeutics 2015; 12: 326–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011; 72: 245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker AM, Seelen M, van Doormaal PT, van Rheenen W, Bothof RJ, van Riessen T, et al. Large-scale screening in sporadic amyotrophic lateral sclerosis identifies genetic modifiers in C9orf72 repeat carriers. Neurobiol Aging 2016; 39: 220 e9–15. [DOI] [PubMed] [Google Scholar]
- Dolzhenko E, van Vugt J, Shaw RJ, Bekritsky MA, van Blitterswijk M, Narzisi G, et al. ; The US–Venezuela Collaborative Research Group. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res 2017; 27: 1895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010; 466: 1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015; 525: 129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genc B, Jara JH, Lagrimas AK, Pytel P, Roos RP, Mesulam MM, et al. Apical dendrite degeneration, a novel cellular pathology for Betz cells in ALS. Sci Rep 2017; 7: 41765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart MP, Brettschneider J, Lee VM, Trojanowski JQ, Gitler AD.. Distinct TDP-43 pathology in ALS patients with ataxin 2 intermediate-length polyQ expansions. Acta Neuropathol 2012; 124: 221–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisman MH, de Jong SW, van Doormaal PT, Weinreich SS, Schelhaas HJ, van der Kooi AJ, et al. Population based epidemiology of amyotrophic lateral sclerosis using capture-recapture methodology. J Neurol Neurosurg Psychiatry 2011; 82: 1165–70. [DOI] [PubMed] [Google Scholar]
- Kozlowski P, de Mezer M, Krzyzosiak WJ.. Trinucleotide repeats in human genome and exome. Nucleic Acids Res 2010; 38: 4027–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattante S, Pomponi MG, Conte A, Marangi G, Bisogni G, Patanella AK, et al. ATXN1 intermediate-length polyglutamine expansions are associated with amyotrophic lateral sclerosis. Neurobiol Aging 2018; 64: 157.e1–157.e5. [DOI] [PubMed]
- Lee T, Li YR, Chesi A, Hart MP, Ramos D, Jethava N, et al. Evaluating the prevalence of polyglutamine repeat expansions in amyotrophic lateral sclerosis. Neurology 2011; 76: 2062–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW.. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 2013; 79: 416–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon RP, Nethisinghe S, Faggiano S, Vannocci T, Rezaei H, Pemble S, et al. The role of interruptions in polyQ in the pathology of SCA1. PLoS Genet 2013; 9: e1003648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014; 345: 1192–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314: 130–3. [DOI] [PubMed] [Google Scholar]
- Nicholson AM, Rademakers R.. What we know about TMEM106B in neurodegeneration. Acta Neuropathol 2016; 132: 639–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura AL, Zupunski V, Troakes C, Kathe C, Fratta P, Howell M, et al. Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain 2010; 133: 1763–71. [DOI] [PubMed] [Google Scholar]
- Ramaswami M, Taylor JP, Parker R.. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 2013; 154: 727–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rub U, Burk K, Timmann D, den Dunnen W, Seidel K, Farrag K, et al. Spinocerebellar ataxia type 1 (SCA1): new pathoanatomical and clinico-pathological insights. Neuropathol Appl Neurobiol 2012; 38: 665–80. [DOI] [PubMed] [Google Scholar]
- Rub U, Schols L, Paulson H, Auburger G, Kermer P, Jen JC, et al. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog Neurobiol 2013; 104: 38–66. [DOI] [PubMed] [Google Scholar]
- Ryan M, Heverin M, McLaughlin RL, Hardiman O.. Lifetime risk and heritability of amyotrophic lateral sclerosis. JAMA Neurol 2019; 76: 1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saberi S, Stauffer JE, Schulte DJ, Ravits J.. Neuropathology of amyotrophic lateral sclerosis and its variants. Neurol Clin 2015; 33: 855–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel K, Siswanto S, Brunt ERP, den Dunnen W, Korf H-W, Rüb U.. Brain pathology of spinocerebellar ataxias. Acta Neuropathol 2012; 124: 1–21. [DOI] [PubMed] [Google Scholar]
- Spataro R, La Bella V.. A case of amyotrophic lateral sclerosis with intermediate ATXN-1 CAG repeat expansion in a large family with spinocerebellar ataxia type 1. J Neurol 2014; 261: 1442–3. [DOI] [PubMed] [Google Scholar]
- van Blitterswijk M, Mullen B, Heckman MG, Baker MC, DeJesus-Hernandez M, Brown PH.. Ataxin-2 as potential disease modifier in C9ORF72 expansion carriers. Neurobiol Aging 2014. a; 35: 2421 e13–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Blitterswijk M, Mullen B, Wojtas A, Heckman MG, Diehl NN, Baker MC, et al. Genetic modifiers in carriers of repeat expansions in the C9ORF72 gene. Mol Neurodegener 2014. b; 9: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Blitterswijk M, van Es MA, Hennekam EA, Dooijes D, van Rheenen W, Medic J, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet 2012; 21: 3776–84. [DOI] [PubMed] [Google Scholar]
- van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, et al. Amyotrophic lateral sclerosis. Lancet 2017; 390: 2084–98. [DOI] [PubMed] [Google Scholar]
- Van Rheenen W, Pulit SL, Dekker AM, Al Khleifat A, Brands WJ, Iacoangeli A, et al. Project MinE: study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis. Eur J Hum Genet 2018; 26: 1537–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingo TS, Cutler DJ, Yarab N, Kelly CM, Glass JD.. The heritability of amyotrophic lateral sclerosis in a clinically ascertained United States research registry. PLoS One 2011; 6: e27985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 2016; 351: 173–6. [DOI] [PubMed] [Google Scholar]
- Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015; 525: 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.