

Abstract
As a follow‐up to the re‐evaluation of starch sodium octenyl succinate (SSOS; E 1450), the Panel on Food Additives and Flavourings (FAF) was requested to assess the safety of SSOS (E 1450) when used in food for infants below 16 weeks of age for food categories 13.1.5.1 and 13.1.1 and to address the data gaps identified during the re‐evaluation of the SSOS (E 1450). The process involved the publication of a call for data. The Panel considered it feasible to amend the specifications based on the analytical evidence submitted. In the call for data, clinical trials were submitted to support the safe use in this age group. In addition, the report of a postnatal piglet study was provided. Due to the low internal validity of the clinical studies, the Panel concluded that a reference point could not be derived from them. The Panel noted that the uncertainty surrounding the results of the piglet study precludes deriving a reference point from this study. On the other hand, both data sources did not clearly indicate an adverse effect due to SSOS (E 1450). Given the available data, the Panel concluded that at use levels of SSOS in food for infants below 16 weeks within the range reported in the clinical studies (up to 2,725 mg/kg body weight (bw) per day), there is no indication for safety concern and reiterated the conclusion of the Panel on Food Additives and Nutrient Sources added to Food (ANS) that there was no need for a numerical acceptable daily intake (ADI). When extrapolating this conclusion to the safety assessment of the food additive when used in food categories (FCs) 13.1.5.1 and 13.1.5.2 in food for infants above 16 weeks of age and young children, the Panel considered that there is no indication for safety concern also for these uses within the range reported in the clinical studies.
Keywords: Starch sodium octenyl succinate, E 1450, food additive, infants
Summary
In accordance with Regulation (EU) No 257/2010, the European Food Safety Authority (EFSA) is currently re‐evaluating the safety of food additives already permitted in the Union before 20 January 2009 and issuing scientific opinions on their safety when used in food as per Annexes II and III to Regulation (EC) No 1333/2008. The risk assessment approach followed in the re‐evaluation has not covered the use of food additives in food for infants below 12 weeks of age so far. Additionally, while re‐evaluating the safety of food additives referred to above, EFSA identified some concerns, namely (1) data gaps that have triggered recommendations in the (to be) published scientific opinions; and/or (2) data gaps that have increased uncertainties linked to the risk assessment and/or which prevented the panel from concluding on some aspects of it.
On 31 May 2017, EFSA published a guidance on the risk assessment of substances present in food intended for infants below 16 weeks of age, thus enabling EFSA to assess the safety of food additive used in food for infants below this age. The age up to 16 weeks was selected in the guidance because infants are exposed to formula feeding until this age as the only source of food since complementary feeding is not supposed to be introduced before.
As follow‐up of the above, this opinion addresses the data gaps previously identified during the re‐evaluation of starch sodium octenyl succinate (SSOS; E 1450) including the risk assessment of starch sodium octenyl succinate (E 1450) for the use as food additive in food according to FC 13.1.5.1 (dietary foods for infants for special medical purposes and special formulae for infants) and FC 13.1.5.2 (dietary foods for babies and young children for special medical purposes as defined in Directive 1999/21/EC) in infants above 16 weeks of age and young children up to 3 years and the safety in the special subpopulation of infants below 16 weeks of age.
The process which followed involved the publication of a dedicated call for data allowing all interested business operators to provide the requested information for completing the assessment as pointed out above and to confirm that the additive is used in FC 13.1.5.1 and is also present in infant formula (13.1.1) as a carryover resulting from the authorised use in Regulation (EC) No 1333/2008.
The data submitted in response to the call for data on SSOS (E 1450) comprise technical information on impurities of the additive, formulation examples for products on the market, use levels in relevant infant formulae, toxicity data such as a report on a 3‐week dietary study on piglets, clinical data and post‐marketing surveillance reports. No adequate data were submitted by the interested business operators which can serve as the basis to assess the safety of SSOS (E 1450) in the use according to FC 13.1.5.1 and 13.1.5.2 for uses in food for infants above 16 weeks and young children.
SSOS (E 1450) is a starch esterified with octenylsuccinic anhydride. Specifications for SSOS have been defined in Commission Regulation (EU) No 231/2012. Considering the data submitted in response to the call for data, the Panel considered it feasible to amend a number of specifications. This refers to lower existing limits for SO2, for toxic elements (heavy metals), as well as introducing new specifications for cadmium and microbiological criteria for the food additive.
Currently, SSOS (E 1450) is approved for use in dietary foods for infants for special medical purposes and special formulae for infants (FC 13.1.5.1) at a maximum level 20,000 mg/L (or mg/kg, as appropriate) only in infant formulae and follow‐on formulae according to Annex II to Regulation (EC) No 1333/2008. Additionally, SSOS (E 1450) is approved for use in dietary foods for babies and young children for special medical purposes as defined in Directive 1999/21/EC (FC 13.1.5.2) at a maximum level of 20,000 mg/L (or mg/kg, as appropriate) and 50,000 mg/L (or mg/kg, as appropriate; only processed cereal‐based foods and baby foods). According to Annex III, Part 5 of Regulation (EC) No 1333/2008, SSOS (E 1450) is also authorised as a food additive added in nutrients i.e. vitamin preparations and polyunsaturated fatty acid preparations, intended to be used in foods for infants and young children listed in point 13.1 of part E of Annex II, at carry‐over levels of 100 and 1,000 mg/kg, respectively.
An in vitro digestibility study and an in vivo study addressing the absorption, distribution, metabolism and excretion of SSOS were available from the former evaluation. Additionally, a study in juvenile rats was described by the World Health Organization (WHO) monograph and additional studies in humans were also available. Comparison among different species and data comparing young and old population were not available.
Regarding the microbiome, in infants, it is known that the microbiome depends on different factors including the mode of delivery, the feeding, the age, diet, host genetics, antibiotic usage and the birth environment of the infants, e.g. neonatal intensive care unit (NICU). According to the reviewed literature, more data are required for a better understanding of the interaction between the factors and what is necessary to maintain intestinal homoeostasis in terms of microbiome in the different population groups. The Panel noted that changes in the composition of the gut microbiota without measuring a specific health outcome are difficult to interpret.
In the animal studies evaluated by the Panel on Food Additives and Nutrient Sources added to Food (ANS), no indication of significant toxic effects of SSOS was observed. However, the FAF Panel considered that the 8‐week study in weanling rats and the 90‐day rat study were not appropriate for the evaluation of SSOS as a food additive in food for infants below 16 weeks of age. In the study in pups of Beagle dogs up to 10,000 mg SSOS/kg body weight (bw) per day for 6 weeks effects on body weight and food consumption were not described. The full study report was not available to the Panel, and therefore, reference point could not be derived from this study.
The results of the post‐natal study in piglets were considered by the FAF Panel as the most suitable animal data for the evaluation of SSOS as food additive in food for infants below 16 weeks of age. However, due to the absence of effects in female animals and a lack of a dose‐response in the effect on body weights of male piglets, the Panel could not identify a reference point for the hazard characterisation of SSOS based on the data from this study.
Further to the call for data, six clinical trials conducted in infants below 16 weeks of age were submitted by interested business operators. Two reviewers evaluated independently the six studies concerning the risk of bias applying an assessment tool modified from the OHAT RoB tool. Five of the studies were allocated to tier 3. Concerning the outcome of the assessment of RoB of the clinical studies, it is general agreement that studies allocated to tier 3 can only be used as supportive evidence. One study was allocated to tier 2 (moderate risk of bias).
Dietary exposure to SSOS (E 1450) from its use as a food additive was assessed based on (1) maximum permitted levels (MPLs) set out in the EU legislation (defined as the regulatory maximum level exposure assessment scenario) and (2) the reported use levels (defined as the refined exposure assessment scenario), (3) the levels in the formulas given as interventions in the clinical trials considered in this assessment. Both scenarios (1) and (2) are based on the recommended consumption levels from the Scientific Committee Guidance which recommends values of 200 and 260 mL formula/kg bw per day as conservative mean and high level consumption values for 14–27 days old infants.
For infants below 16 weeks of age consuming foods for special medical purposes (FSMP) (FC 13.1.5.1), mean exposure in the regulatory maximum level exposure assessment scenario was estimated at 4,000 mg/kg bw per day while at the high level was estimated at 5,200 mg/kg bw per day. As the maximum level provided by industry was equal to the MPL of 20,000 mg/kg, exposure estimates are the same for the refined scenario based on maximum levels of use provided by the interested business operators. For the scenario using the mean of the reported use levels from industry, exposure estimates were of 1,676 mg/kg bw per day at the mean and 2,179 mg/kg bw per day at the high level.
The lowest levels of exposure to SSOS (E 1450) estimated for the clinical trials were ■■■■■the highest level of exposure was about 2,700 mg/kg bw per day.
For infants below 16 weeks of age consuming infant formulae which could contain SSOS (E 1450) from carry‐over (FC 13.1.1), in the regulatory maximum level exposure assessment scenario, mean exposure was estimated at 220 mg/kg bw per day while at the high level was estimated at 286 mg/kg bw per day. For the scenario using the maximum level provided by industry, mean exposure was estimated at 77 mg/kg bw per day while at the high level was estimated at 100 mg/kg bw per day. The scenario using the mean of the reported use levels from industry, mean exposure was estimated 12 mg/kg bw per day at the mean and 16 mg/kg bw per day at the high level.
When considering the available information to set a reference point, studies in healthy infants, would be the preferred data source. However, most of them (five of the six studies provided) had low internal validity, reflected in the high risk of bias (tier 3). The study with a moderate risk of bias (tier 2) had very low content of SSOS in the formulae used in both study arms of ■■■■■. In addition, in all the studies, the composition of the control formula, used without SSOS (E 1450), differed from the composition of the experimental formula with SSOS (E 1450). The Panel explored the possibility to compare the studies concerning growth with non‐randomised comparisons (i.e. comparisons to historical controls, such as comparisons to growth reference charts the data of which are of observational nature), but considered that the available data on these growth reference charts were not sufficiently informative for the European population. Hence, the Panel concluded that a reference point could not be derived from the clinical studies. The Panel considered whether the results from the piglet study could be used for identifying a reference point but had to note that the uncertainty surrounding the results precludes deriving a reference point from this study. On the other hand, both data sources did not clearly indicate an adverse effect due to SSOS. Given the available data, the FAF Panel reiterated the conclusion of the ANS Panel that there was no need for a numerical acceptable daily intake (ADI) and considered that for exposure to SSOS of infants below 16 weeks, there is no indication for a concern when within the range reported in the clinical studies (up to 2,725 mg/kg bw per day). When extrapolating the conclusion above to the safety assessment of the food additive when used in FCs 13.1.5.1 and 13.1.5.2 in food for infants above 16 weeks of age and young children, the Panel considered that for exposure to SSOS for infants above 16 weeks and young children, there is no indication for a concern when within the range reported in the clinical studies (up to 2,725 mg/kg bw per day). The Panel noted that at the reported use levels, the estimates of exposure could exceed the higher end of the exposure in the clinical trials.
1. Introduction
The present opinion deals with:
the risk assessment of starch sodium octenyl succinate (E 1450) in food for infants below 16 weeks of age in the food category (FC) 13.1.5.1 (‘Dietary foods for special medical purposes and special formulae for infants’), and referring to carry over from indirect use in nutrients (Annex II and section B of part 5 of Annex III to the Regulation (EC) No 1333/20081 on food additives).
the follow‐up on issues that have been expressed in the conclusions and recommendations of the Scientific Opinion on the re‐evaluation of starch sodium octenyl succinate (E 1450) as a food additive (EFSA ANS Panel, 2017) including the risk assessment of starch sodium octenyl succinate (E 1450) for the use as food additive in food according to FC 13.1.5.1 and FC 13.1.5.2 (dietary foods for babies and young children for special medical purposes as defined in Directive 1999/21/EC) in infants above 16 weeks of age and young children up to 3 years.
1.1. Background and Terms of Reference as provided by the requestor
1.1.1. Background
The composition of food intended for infants and young children, as defined by Regulation (EU) No 609/20132, is regulated at EU level and such rules include requirements concerning the use of substances as food additives.
The use of food additives is regulated by Regulation (EC) No 1333/2008 on food additives. Only food additives that are included in the Union list, in particular in Annex II and III to that Regulation, may be placed on the market and used in food under the conditions of use specified therein.
In accordance with Regulation (EU) No 257/20103, EFSA is currently re‐evaluating the safety of food additives already permitted in the Union before 20 January 2009 and issuing scientific opinions on their safety when used in food as per Annexes II and III to Regulation (EC) No 1333/2008. However, the risk assessment approach followed until now has not covered the use of food additives in food for infants below 12 weeks of age. Consequently, EFSA published several scientific opinions on the re‐evaluation of the safety of food additives permitted in food category 13.1 but not addressing their use in food for infants below 12 weeks of age.
In addition, in these opinions EFSA identified some concerns, namely 1) Data gaps that have triggered recommendations in the (to be) published scientific opinions, and/or; 2) Data gaps that have increased uncertainties linked to the risk assessment and/or which prevented the Panel from concluding on some aspects of it.
On 31 May 2017, EFSA published a guidance document (EFSA Scientific Committee, 2017a,b) on the risk assessment of substances present in food intended for infants below 16 weeks of age, thus enabling EFSA to assess the safety of food additive used in food for infants below 12 weeks of age.4 Now EFSA is expected to launch dedicated calls for data to be able to perform such risk assessments.
The EC considers more effective that EFSA, in the context of these dedicated calls for data, also addresses all the issues and data gaps already identified in the relevant (to be) published scientific opinions on the re‐evaluation of the safety of food additives permitted in food category 13.1.
In accordance with the current EC approach for the follow‐up of EFSA's scientific opinions on the re‐evaluation of the safety of permitted food additives for which some concerns have been identified, a specific call for data would be published by the EC on DG SANTE's website5 on food additives and additional (missing) information would then be provided by interested business operators to the EC.
However, for those scientific opinions on the re‐evaluation of the safety of permitted food additives in food category 13.1 for which the risk assessment does not address their uses in food for infants below 12 weeks of age and for which some concerns have been identified by EFSA, the EC considers that for the sake of efficiency it would be appropriate to streamline the approach as described above.
Therefore, the EC requests EFSA to address all the issues and data gaps already identified in the relevant (to be) published scientific opinions of those food additives (or groups of additives that can be addressed simultaneously) as part of the upcoming work on the safety assessment of food additives uses in food for infants below 12 weeks of age.
This follow‐up aims at completing the re‐evaluation of the food additives in question for all food categories, and includes calls for data covering the actual use and usage levels of food additives in food for both infants below 12 or 16 weeks of age as well as for older infants, young children and other groups of the population for which EFSA has already finalised its assessment.
The future evaluations of EFSA should systematically address the safety of use of food additives for all age groups, including the infants below 12 or 16 weeks of age.
1.1.2. Terms of Reference
In accordance with Article 29(1)(a) of Regulation (EC) No 178/20026, and as part of EFSA's work in completing its risk assessments concerning the use of food additives in food for infants below 12 weeks of age, covered by the re‐evaluation programme and its terms of reference, the European Commission requests the European Food Safety Authority to address all the data gaps specified in the recommendations made in its scientific opinions on the re‐evaluation of the safety of food additives permitted in food category 13.1 (food for infants and young children) of annex II to Regulation (EC) No 1333/2008.
1.1.3. Interpretation of Terms of Reference
The Panel noted that the use of ingredients for infant formula and follow‐on formula is regulated by Commission Delegated Regulation (EU) 2016/127 of 25 September 2015 on infant formulae and follow‐on formulae,7 clearly stating that ‘The use of ingredients containing gluten shall be prohibited8’.
Therefore, the food additive starch sodium octenyl succinate (E 1450) in infant formulae and follow‐on formulae should not contain gluten.
Before the publication of the EFSA Scientific Committee Guidance on the risk assessment of substances present in food intended for infants below 16 weeks of age (EFSA Scientific Committee, 2017a,b), EFSA has taken 12 weeks as a cut off age for the applicability of the safety assessment. However, according to EFSA Scientific Committee (2017a,b), the assessment will include infants up to 16 weeks of age because they are exposed to formula feeding until this age as the only source of food since complementary feeding is not supposed to be introduced before this age (see EFSA Scientific Committee, 2017a,b).
1.2. Previous evaluations of starch sodium octenyl succinate (E 1450) for use in foods for infants
The safety of starch sodium octenyl succinate (E 1450), abbreviated as SSOS,9 for use as an emulsifier in infant formula and in formula for special medical purposes (FSMP) intended for use in infants was evaluated by the Joint FAO/WHO Expert Committee on Food Additives (JECFA) at its 79th meeting in 2014 (JECFA, 2015).
According to the JECFA evaluation (JECFA, 2015): ‘The fates of OSA‐modified starch 10 and the hydrolysed product OSA (octenyl succinic acid) are similar in rats, dogs and human infants with respect to enzyme hydrolysis in the digestive tract, followed by absorption, metabolism and elimination, with the only difference being the amount of OSA excreted unchanged in the urine. The clinical data indicate that infants are able to metabolise SSOS to a number of metabolites, including propane‐1,2-3‐tricarboxylic acid. While the degree of metabolism may differ among species, in general, the same metabolites are produced.’
Exposure estimates calculated on the basis of the energy intakes at the maximum proposed use level of 20 g/L formula (energy density of 67 kcal/100 mL) resulted in median and high levels of 3.7 g/kg body weight (bw) per day and 4.4 g/kg bw per day, respectively. The highest reported 95th percentile energy intakes were for infants aged 14–27 days, the median referred instead to infants aged 0–6 months (JECFA, 2015).
Two toxicity studies in neonatal animals were considered by JECFA in this assessment, one conducted in beagle pups and another in neonatal piglets. The latter was considered to be more relevant for the safety assessment because of the similarity between the digestive system of neonatal swine and human infants. From the study in neonatal piglets, a no‐observed‐adverse‐effect level (NOAEL) was identified at 10,000 mg/kg bw per day, corresponding to the highest dose tested (JECFA, 2015).
Data from a 90‐day oral toxicity study in rats were also reviewed, showing no treatment‐related adverse effects at doses up to 37,000 mg/kg bw per day. Data from post‐marketing monitoring and clinical studies in infants were also considered for the evaluation. Based on the above, in its evaluation, JECFA concluded that: ‘the consumption of OSA‐modified starch 8 in infant formula or formula for special medical purposes intended for infants is not of concern at use levels up to 20 g/L’ (JECFA, 2015).
1.3. Summary of the previous EFSA re‐evaluation of SSOS (E 1450) for uses in foods for all population groups except for infants below 12 weeks of age
Under the frame of Regulation (EC) No 257/2010, the EFSA Panel on Food Additives and Nutrient Sources added to Food (ANS) has re‐evaluated the safety of SSOS (E 1450) when used as a food additive (EFSA ANS Panel, 2017).
The scientific opinion on the safety of SSOS (E 1450) also encompassed the assessment of other starches used as food additives, namely: oxidised starch (E 1404), monostarch phosphate (E 1410), distarch phosphate (E 1412), phosphated distarch phosphate (E 1413), acetylated distarch phosphate (E 1414), acetylated starch (E 1420), acetylated distarch adipate (E 1422), hydroxypropyl starch (E 1440), hydroxypropyl distarch phosphate (E 1442), acetylated oxidised starch (E 1451) and starch aluminium octenyl succinate (E 1452). The assessment performed by the ANS Panel was, however, incomplete with respect to the safety of SSOS (E 1450) for use in food for infants below 16 weeks of age.
In its scientific opinion, the ANS Panel reviewed available technical, biological and toxicological data on the above‐mentioned modified starches used as food additives, including data on SSOS (E 1450). A combined dietary exposure estimate was calculated in the general population for the modified starches E 1404, E 1410, E 1412, E 1413, E 1414, E 1420, E 1422, E 1440, E 1442, E 1450 and E 1451 resulting in maximum intake levels of approximately 3 g/kg bw per day in toddlers at the 95th percentile in the brand‐loyal consumer scenario. The ANS Panel concluded that there was no safety concern for the use of modified starches as food additives at the reported uses and use levels and that there was no need for a numerical ADI for the general population (EFSA ANS Panel, 2017).
Based on the data reviewed, the ANS Panel had concluded that in humans modified starches, such as SSOS (E 1450), would not be absorbed intact but would be significantly hydrolysed by intestinal enzymes and then fermented by intestinal microbiota. In the specific case of SSOS (E 1450), the ANS Panel had concluded that the octenyl succinate moiety of the modified starch would either be metabolised to tricarboxylic acid or excreted unchanged (EFSA ANS Panel, 2017).
From the toxicological data set reviewed, which was considered to adequately cover all relevant toxicity endpoints, no treatment‐related effects of modified starches were observed even after long‐term treatment at high levels (up to 17,000 mg/kg bw per day) (EFSA ANS Panel, 2017).
Although no genotoxicity data on the modified starches were available for the re‐evaluation, the Panel had considered the lack of structural alerts based on the use of an in silico predictive tool as sufficient evidence to rule out possible genotoxic concerns (EFSA ANS Panel, 2017).
However, with respect to the uses of SSOS (E 1450) in the food categories ‘13.1.5.1 Dietary foods for special medical purposes and special formulae for infants’ and ‘13.1.5.2 Dietary foods for babies and young children for special medical purposes as defined by Commission Directive 1999/22/EC’ at the maximum use levels of 20,000 or 50,000 mg/kg, respectively, the ANS Panel had concluded that the available data did not allow for an adequate assessment of the safety.
In its scientific opinion, the ANS Panel, cited the JECFA evaluation (JECFA, 2015) reporting no effects on body weight and food intake in male and female neonatal pigs exposed to 10,000 mg/kg bw per day of SSOS (E 1450) in formula for 21 days. Furthermore, it was noted that in the JECFA (2015) evaluation SSOS (E 1450), up to a single dose of 25,000 mg/person was reported to be well tolerated by fasting healthy adults, but gastrointestinal symptoms were reported in infants with hypoallergenic formula containing 2% of SSOS (about 24,000 mg/person). The ANS Panel was also concerned with the possibility that infants and young children consuming foods belonging to the two food categories 13.1.5.1 and 13.1.5.2 may show a higher susceptibility to the gastrointestinal effects of modified starches than their healthy counterparts due to their underlying medical condition. The available information on the clinical studies in infants was considered too limited by the ANS Panel also because the results referred to the feeding of formula products containing SSOS in concentrations lower than the currently authorised maximum level. The level of exposure in populations consuming foods for special medical purposes and special formulae was estimated to be up to 5,286 mg/kg bw per day for infants at the 95th percentile (EFSA ANS Panel, 2017). The ANS Panel had therefore recommended the evaluation of additional data for completing the assessment for these uses.
From the former ANS opinion (EFSA ANS Panel, 2017) which dealt with the safety of SSOS (E 1450) in the general population, several issues required follow‐up and additional data that were requested in a dedicated call for data11 (see also Appendix B).
2. Data and methodologies
2.1. Data
EFSA launched a public call for data11 to collect relevant information from interested business operators.
The Panel based its assessment on the information submitted to EFSA following the public call for data, the information from previous evaluations and additional available literature up to 22 June 2020.
The Mintel's Global New Products Database (GNPD) is an online database which monitors new introductions of packaged goods in the market worldwide. It contains information of over 3 million food and beverage products of which more than 1,100,000 are or have been available on the European food market. Mintel started covering EU's food markets in 1996, currently having 24 out of its 27 member countries, and Norway and UK presented in the Mintel GNPD.12 The Mintel's GNPD database was consulted to verify the use of the food additive SSOS (E 1450) in food products.
2.2. Methodologies
This opinion was formulated following the principles described in the EFSA Guidance on transparency with regard to scientific aspects of risk assessment (EFSA Scientific Committee, 2009) and following the relevant existing guidance documents from the EFSA Scientific Committee including the EFSA Guidance of the Scientific Committee on the risk assessment of substances present in food intended for infants below 16 weeks of age (EFSA Scientific Committee, 2017a).
In order to conclude on the safety of SSOS (E 1450) for all population groups and to address the data gaps identified during the re‐evaluation, the FAF Panel assessed the information provided:
for the follow‐up on issues that have been raised in the conclusions and recommendations of the Scientific Opinion on the re‐evaluation of SSOS (E 1450) as a food additive including the risk assessment of starch sodium octenyl succinate (E 1450) for the use as food additive in food according to FC 13.1.5.1 and FC 13.1.5.2 (dietary foods for babies and young children for special medical purposes as defined in Directive 1999/21/EC) in infants above 16 weeks of age and young children up to 3 years (EFSA ANS Panel, 2017); and
for the risk assessment of SSOS (E 1450) in food for infants below 16 weeks of age in the FC 13.1.5.1 (Dietary foods for infants for special medical purposes and special formulae for infants), and referring to carry over from indirect use in nutrients (Annex II and section B of part 5 of Annex III to the Regulation (EC) No 1333/2008 on food additives).
When in animal studies, the test substance was administered in the feed or in drinking water, but doses were not explicitly reported by the authors as mg/kg bw per day based on actual feed or water consumption, the daily intake is calculated by the Panel using the relevant default values. In case of rodents, the values as indicated in the EFSA Scientific Committee Guidance document (EFSA Scientific Committee, 2012) are applied. In the case of other animal species, the default values by JECFA (2000) are used. In these cases, the dose was expressed as ‘equivalent to mg/kg bw per day’. When in human studies in adults (aged above 18 years) the dose of the test substance administered was reported in mg/person per day, the dose in mg/kg bw per day was calculated by the Panel using a body weight of 70 kg as default for the adult population as described in the EFSA Scientific Committee Guidance document (EFSA Scientific Committee, 2012).
The animal post‐natal study and the human clinical trials were assessed by two reviewers (members of the Working Group) applying an assessment tool modified from the OHAT RoB tool (NTP‐OHAT, 2015, 2019). The elements considered for the appraisal are described in Appendices C and D of this opinion, as well as the decision rule for assigning the studies to tiers of reliability.
Dietary exposure to SSOS (E 1450) from its use as a food additive in foods for infants below 16 weeks of age was estimated combining the mean and highest consumption figures reported for the period of 14–27 days of life which corresponds to values of 200 and 260 mL/kg bw per day, respectively, with the maximum levels according to Annex II and Annex III, Part 5 Section B to Regulation (EC) No 1333/2008 and/or reported use levels submitted to EFSA following a call for data. Different scenarios were used to calculate exposure (see Section 3.3.1). Uncertainties on the exposure assessment were identified and discussed.
As SSOS (E 1450) is also authorised in the food category 13.1.5.2, an exposure assessment considering FC 13.1.5.1 and FC 13.1.5.2 was performed to estimate the exposure of infants (above 16 weeks) and toddlers who may eat and drink these foods for special medical purposes (FSMP).
The consumption of these foods is not reported in the EFSA Comprehensive database. To consider potential exposure to SSOS (E 1450) via these foods, the Panel assumes that the amount of FSMP consumed by infants and toddlers resembles that of comparable foods in infants and toddlers from the general population. Thus, the consumption of FSMP categorised as FC 13.1.5 was assumed equal to that of formulae and food products categorised as FCs 13.1.1, 13.1.2, 13.1.3 and 13.1.4.
3. Assessment
3.1. Technical data
3.1.1. Identity of the substance
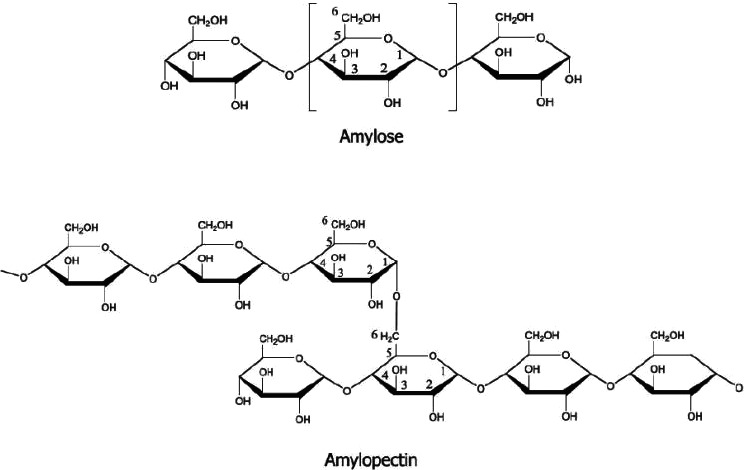
According to Commission Regulation (EU) No 231/201213, the food additive E 1450 is named as starch sodium octenyl succinate. A generic representation of the structure of starches is shown in Figure 1. To make the food additive with the desired functionality, starch is chemically modified by esterification of some of its hydroxyl groups with octenylsuccinic anhydride. This reaction is depicted in Figure 2, where in this example sodium hydroxide is used in the reaction to provide the sodium salt.
Figure 1.
Structural formula of starch as reproduced in EFSA ANS Panel (2017)
Figure 2.

Process for derivatisation of starch to generate SSOS (Image from RSC 14)
3.1.2. Specifications
The specifications for SSOS (E 1450) as defined in the Commission Regulation (EU) No 231/2012 and tentative specifications as proposed by JECFA (2018) are listed in Table 1. At its 79th meeting, JECFA recommended the separation of the combined specification for the modified starches into 16 separate specifications (JECFA, 2014). As a first step, 16 stand‐alone specification monographs were prepared at the 82nd JECFA meeting and published in FAO JECFA Monographs 19 (JECFA 2016a, 2017). For SSOS (E 1450), the resulting individual specification monograph was incomplete and therefore got the tentative status due to pending information (JECFA, 2016b). At the 86th JECFA meeting (JECFA, 2018), this concept was refined: all modified starches were included in a ‘modular monograph’ consisting of ‘general specifications’ that contains common specifications to all modified starches and eight ‘annexes’ with specifications applicable to each individual modified starch based on the chemical treatment(s) that native starches received. The status of these specifications is tentative. Furthermore, JECFA requested suitable microbiological acceptance criteria and supporting data for all modified starches (JECFA 2016a).
Table 1.
Specifications for SSOS (E 1450) according to Commission Regulation (EU) No 231/2012 and for modified starches, including INS 1450, tentatively proposed by JECFA (2018)
| Commission Regulation (EU) No 231/2012 | JECFA, tentative (2018) | |
|---|---|---|
| Definition | Starch sodium octenyl succinate is starch esterified with octenylsuccinic anhydride |
General for modified starches: Starch consists mainly of amylose and amylopectin. Amylose is a linear molecule of α‐d‐glucopyranosyl units linked by (1‐4)‐α‐linkages. Amylopectin is a highly branched polymer of α‐d‐glucopyranosyl units linked by (1‐4)‐α‐linkages and by (1‐6)‐α‐linkages that constitute the branch points. Each glucose unit possesses a maximum of three hydroxyls that can undergo chemical substitution Native starches can be physically (pre‐gelatinised starches) and/or chemically modified for improved functionality. The most common sources of native starch used in these modifications are various roots, tubers, cereals and legumes. Modified starches are used in applications requiring special properties not attainable by native starches Chemical modifications of native starches are often performed, in an aqueous suspension under controlled conditions of pH, time and temperature, unless otherwise indicated in the description of the respective annex. After sufficient reaction time, the modified starch is recovered by filtration or centrifugation, washed with water, dried and packaged. The relevant modification reactions can be, separately or in combination, fragmentations (hydrolysis, oxidation, enzymatic), bleaching, oxidation, esterification, etherification or phosphorylation of one or more of the hydroxyl groups of the α‐d‐glucopyranosyl units or crosslinking using polyfunctional agents |
| Treatment | – | Octenylsuccinic anhydride can be used for the esterification and either sodium hydroxide or sodium carbonate as a pH buffer for neutralisation |
| Synonym | SSOS | |
| CAS Numbers |
Starch sodium octenyl succinate 66829‐29‐6 (modified starch) 52906‐93‐1 (modified starch) 125109‐81‐1 (modified amylopectin) |
|
| Description | White or nearly white powder or granules or (if pregelatinised) flakes, amorphous powder or coarse particles | White or nearly white powder or granules or (if pregelatinised) flakes, or amorphous powder or coarse particles |
| Identification | ||
| Solubility | – | Insoluble in cold water (if not pregelatinised); forming typical colloidal solutions with viscous properties in hot water; insoluble in ethanol |
| Microscopic observation | Passes test (if not pregelatinised) | Passes test |
| Iodine staining | Passes test (dark blue to light red colour) | Passes test |
| Copper reduction | – | Passes test |
| Ester groups | – | Passes test |
| Purity | ||
| pH | 3.0‐9.0 | |
| Loss on drying |
Not more than 15.0% for cereal starch Not more than 21.0% for potato starch Not more than 18.0% for other starches |
Cereal starch: not more than 15.0% Potato starch: not more than 21.0% Other starches: not more than 18.0% Conditions: 120°, 4 h, vacuum not exceeding 100 mmHg |
| Octenylsuccinyl groups | Not more than 3% (on an anhydrous basis) | Not more than 3% on the dried basis |
| Octenylsuccinic acid residue | Not more than 0.3% (on an anhydrous basis) | Not more than 0.3% on the dried basis |
| Carboxyl groups | – | Not more than 0.1% on the dried basis |
| Sulfur dioxide |
Not more than 50 mg/kg for modified cereal starches (on an anhydrous basis) Not more than 10 mg/kg for other modified starches, unless otherwise specified (on an anhydrous basis) |
Not more than 50 mg/kg on the dried basis for modified cereal starches Not more than 10 mg/kg on the dried basis for other modified starches |
| Arsenic | Not more than 1 mg/kg | – |
| Lead | Not more than 2 mg/kg (on an anhydrous basis) |
Not more than 0.2 mg/kg on the dried basis Not more than 0.1 mg/kg on the dried basis for Starch sodium octenylsuccinate (INS 1450) for use in infant formula and formula for special medical purposes intended for infants |
| Mercury | Not more than 0.1 mg/kg | – |
| Manganese | – | Not more than 50 mg/kg on the dried basis |
| Microbiological criteria | ||
| Aerobic plate count | Not more than 1,000 CFU/g | |
| Yeasts and moulds | Not more than 1,000 CFU/g | |
| Total coliforms | Not more than 10 CFU/g | |
CFU: colony‐forming units.
3.1.2.1. Technical data from commercial samples of the food additive
Analytical data from 27 batches of commercial samples of SSOS (E 1450) have been provided by one of the interested parties in response to the call for data (Documentation provided to EFSA n. 1).
Based on the analytical data submitted along with the performance of the analytical methods used, the following purity criteria are proposed by Starch Europe:
Table 2.
Proposed purity criteria by Starch Europe (Documentation provided to EFSA n.1) for SSOS (E 1450) for uses in food for infants below 16 weeks of age
| Limit proposed by Starch Europe (only for uses in food for infants below 16 weeks of age): | Method(s) | |
|---|---|---|
| Arsenic | Not more than 0.05 mg/kg |
NMKL No.161, 1998, mod 13805:2014 Two further, in‐house, methods described, using open‐ or closed vessel acid digestion of samples followed by ICP‐MS determination of the elements |
| Cadmium | Not more than 0.01 mg/kg |
NMKL No.161, 1998, mod 13805:2014 Two further, in‐house, methods described, using open‐ or closed vessel acid digestion of samples followed by ICP‐MS determination of the elements |
| Lead | Not more than 0.03 mg/kg |
NMKL No.161, 1998, mod 13805:2014 Two further, in‐house, methods described, using open or closed vessel acid digestion of samples followed by ICP‐MS determination of the elements |
| Mercury | Not more than 0.05 mg/kg |
SS‐EN 16277:2012 Two further, in‐house, methods described, using open or closed vessel acid digestion of samples followed by ICP‐MS determination of the elements |
| Lowest technologically achievable levels proposed by Starch Europe | Method(s) | |
| SO2 | 10 mg/kg |
Three methods reported: SS‐EN‐1988‐2 NE EN 1185 990.28 AOAC (16th Edition, Chapter 47) |
| Octenylsuccinic acid residue | 0.3% |
Ranging from 0.06% to 0.3% Measured in 13 samples according to JECFA specifications for modified starches |
| Microbiological criteria | ||
| Salmonella spp. |
Negative in 25 g (3 samples; method: Rapid Salmonella Short prot) Negative in 375 g (5 samples: US FDA Bacteriological Analytical Manual (BAM) Chapter 5) Other 13 negative samples, method unclear |
|
| E. coli |
Negative in 10 g (3 samples; method: ISO 7251) Negative in 10 g (5 samples; method: Modified USP 62) Other 13 negative samples, method unclear |
|
| Aerobic plate count |
< 100 CFU/g (3 samples; method: NMKL 86) < 10 CFU/g (8 samples; ISO 4833) < 10 CFU/g (5 samples; Modified USP 61) Other samples tested, method unclear |
|
| Yeast |
< 100 CFU/g (3 samples; method: 3M Petrifilm Rapid) < 5 CFU/g (2 samples; method: NF v08‐059) < 10 CFU/g (3 samples; method: NF v08‐059) < 10 CFU/g (5 samples; method: Modified USP 61) Other samples tested, method unclear |
|
ICP‐MS: Inductively coupled plasma mass spectrometry; CFU: colony‐forming unit.
No analytical data and no proposal for specifications for use in food for other population groups than infants below the age of 16 weeks were provided as requested in part A.1 of the call for data11 (clarification letter by Starch Europe dated 29 October 2019).
Eight commercial samples of SSOS (E 1450) sold to baby food industry were tested for Cronobacter (Enterobacter) sakazakii using the method ISO/TS 22964. All tested samples were negative (Documentation provided to EFSA n.1).
The proposed revisions of the EU specifications are presented in Section 3.5.
3.1.2.2. Analytical data on toxic elements in final infant formulae
As part of the EFSA call for data,11 information on the concentrations of toxic elements for which legal limits are not in place in the final products – i.e. infant formulas made using SSOS (E 1450) was requested. Only limited information was provided, covering just a few samples and for an incomplete set of elements of potential concern (Documentation provided to EFSA by a Specialised Nutrition Europe (SNE) member). Nonetheless, considering the recommendation that specifications for the content of certain toxic elements should be tightened (see above) then further analytical data for the final infant formulae was not requested because if these specifications were applied, the concentrations of toxic elements in the final products would not raise concern.
3.1.3. Stability of the substance, and reaction and fate in food
SSOS (E 1450) is produced through the esterification of a food starch with octenylsuccinic anhydride. According to the European Commission specifications (Table 1), the content of octenylsuccinyl groups (i.e. esterified with the starch hydroxyl groups) should not exceed 3% w/w and the unreacted residue of octenylsuccinic acid (i.e. free, unbound) should not exceed 0.3% w/w.
Concerning the starch backbone, starch and OSA starches are stable powders with a long shelf‐life. There is no reason to believe that when mixed with other non‐active powder ingredients and used as intended, this would change.
Considering the possibility of hydrolysis, the additive is stable in infant formulae products. Under normal conditions of use for both powdered and liquid infant formulae products, it would not be expected that more than a minor fraction of the octenylsuccinyl group hydrolyse. This is supported by a study (Johns et al., 2014) in which the dissociation of octenylsuccinic acid from SSOS (E 1450) during the production of a hydrolysed protein‐based nutritional product was evaluated. Even under these severe manufacturing conditions (compared to normal use conditions for this additive), there was only a minor (~ 2%) conversion of the total amount of octenylsuccinic acid from the starch‐bound form to the free form (Documentation provided to EFSA n. 2). Since the additive may already contain about 10% free OSA (e.g. at the limit values, 0.3 free vs. 3% w/w bound, see above), such small further release is not considered to be significant.
Considering other potential reactions of the OSA group, it contains a C=C double bond at carbons C6 and C7 of the octyl chain (see Figure 2). This double bond is isolated insofar as it is not in conjugation with and cannot foreseeably become conjugated with (and thereby activated by) another site of unsaturation such as a carbonyl or other alkene bond. So, no reactivity at that site is foreseen when the additive is used in infant food products.
3.2. Authorised uses and use levels
Maximum levels of SSOS (E 1450) in foods for infants below 16 weeks of age have been defined in Annex II and III to Regulation (EC) No 1333/2008 on food additives, as amended. In this document, these levels are named maximum permitted levels (MPLs).
Currently, SSOS (E 1450) is approved for use in dietary foods for infants for special medical purposes and special formulae for infants (FC 13.1.5.1) at a maximum level 20,000 mg/L (or mg/kg, as appropriate) only in infant formulae and follow‐on formulae according to Annex II to Regulation (EC) No 1333/2008. Additionally, SSOS (E 1450) is approved for use in dietary foods for babies and young children for special medical purposes as defined in Directive 1999/21/EC at a maximum level of 20,000 mg/L (or mg/kg, as appropriate) and 50,000 mg/L (or mg/kg, as appropriate; only processed cereal‐based foods and baby foods), see Table 3.
Table 3.
MPLs of SSOS (E 1450) in foods for infants below 16 weeks of age according to the Annex II and Annex III to Regulation (EC) No 1333/2008
| Food category number | Food category name | E‐number | Restrictions/exception | MPL (mg/L or mg/kg as appropriate) |
|---|---|---|---|---|
| 13.1.5.1a | Dietary foods for infants for special medical purposes and special formulae for infants | E 1450 | Only in infant formulae and follow‐on formulae | 20,000 |
| 13.1.5.2b | Dietary foods for babies and young children for special medical purposes as defined in Directive 1999/21/EC | E 1450 | Only processed cereal‐based foods and baby foods | 50,000 |
| 13.1.5.2b | Dietary foods for babies and young children for special medical purposes as defined in Directive 1999/21/EC | E 1450 | Except processed cereal‐based foods and baby foods | 20,000 |
| Food category | E‐number | Nutrient to which the food additive may be added | ||
| Annex III, Part 5 Section B | In nutrients intended to be used in foodstuffs for infants and young children listed in point 13.1 of Part E of Annex II | E 1450 | Vitamin preparations | Carry‐over 100c |
| Polyunsaturated fatty acid preparations | Carry‐over 1,000c |
MPL: maximum permitted level.
This category covers dietary foods for infants for special medical purposes and special formulae such as premature infant formulae, hospital discharge formulae, low and very low birth weight formulae, and human breast milk fortifiers.
This category covers foods specially processed or formulated and intended for the dietary management of babies and young children, to be used under medical supervision. This includes, for example the dietary management of infants and young children with metabolic or gastrointestinal disorders, or single or multiple food allergies or intolerances (e.g. cow's milk protein allergy, protein mal‐absorption) and for general tube feeding. Baby foods are foodstuffs destined to children of at least 4 months (see Article 8 of Commission Directive 2006/125).
Two preparations can contain E 1450 as carry‐over. In the final formula, it is in theory possible that carry‐over from both preparations are present. Therefore, for the regulatory exposure scenario, a maximum carry‐over level of 1,100 mg/L or mg/kg as appropriate was used.
According to Annex III, Part 5 of Regulation (EC) No 1333/2008, SSOS (E 1450) is also authorised as a food additive added in nutrients i.e. vitamin preparations and polyunsaturated fatty acid preparations, intended to be used in foods for infants and young children listed in point 13.1 of part E of Annex II, at carry‐over levels of 100 and 1,000 mg/kg, respectively (see Table 3).
3.3. Exposure data
Some food additives are authorised in the EU for infant formulae as defined by Commission Delegated Regulation (EU) 2016/127/EC) (FC 13.1.1) and in ‘dietary foods for infants for special medical purposes and special formulae for infants’ (FC 13.1.5.1) and in ‘dietary foods for babies and young children for special medical purposes as defined in Directive 1999/21/EC’ (FC 13.1.5.2) at a specific MPL. However, a food additive may be used at a lower level than the MPL. Therefore, actual use levels are required for performing a more realistic exposure assessment.
In the framework of Regulation (EC) No 1333/2008 on food additives and of Commission Regulation (EU) No 257/2010 regarding the re‐evaluation of approved food additives, EFSA issued a public call15 for technical and toxicological data on SSOS (E 1450) for uses as a food additive in foods for all population groups including infants below 16 weeks of age. In response to this public call, updated information on the actual use levels of SSOS (E 1450) in foods was made available to EFSA by industry. No analytical data on the concentration of SSOS (E 1450) in foods were made available by the Member States.
3.3.1. Reported use levels in food category 13.1.1 as a carry‐over from the authorised use according to Annex III, Part 5, Section B
A theoretical maximum value for carry‐over of 1,100 mg/kg final formulae as fed was derived using the information in Table 6. This represents the carry‐over coming from two different preparations that could be used together in the final product. The first is the maximum authorised carry‐over of 100 mg/kg from vitamin preparations and the second is the maximum authorised carry‐over of 1,000 mg/kg from polyunsaturated fatty acids (PUFA) preparations.
Table 6.
Dietary exposure to starch sodium octenyl succinate (E 1450) for infants above 16 weeks of age and toddlers, according to the Annex II to Regulation (EC) No 1333/2008
| Infants (16 weeks to 11 months) | Toddlers (12–35 months) | |
|---|---|---|
| Refined scenario considering the whole diet (mg/kg bw per day) | ||
|
303–1,994 1,686–4,069 |
45–687 250–2,579 |
| Refined scenario considering only foods from FC 13.1.5.1 and 13.1.5.2 (mg/kg bw per day) | ||
|
303–1,990 1,686–4,068 |
42–672 245–2,562 |
EFSA has received information on the use levels (n = 12) of SSOS (E 1450) as a food additive in nutrients intended to be used in foodstuffs for infants and young children according to Annex III, Part 5 Section B of Regulation (EC) No 1333/2008, with respect to the carry over (FC 13.1.1) ■■■■■. The levels are for:
-
■■■■■
■■■■■
-
■■■■■
■■■■■
Other data on SSOS (E 1450) were received through the general call for data batch 4 in 2016. Use levels reported during that call are on average lower than those recently received and are not considered in the current opinion.
Appendix A provides a summary of the use levels of E 1450 in foods as reported by industry.
3.3.2. Reported use levels in food category 13.1.5.1
Use levels were also reported for SSOS (E 1450) as a food additive in the FC 13.1.5.1 (n = 11). These levels were provided by four companies (■■■■■).
■■■■■
Appendix A provides a summary of the use levels of E 1450 in foods as reported by industry.
3.3.3. Reported use levels in food category 13.1.5.2
Industry provided EFSA with three use levels of SSOS (E 1450) in FC 13.1.5.2. These levels of SSOS (E 1450) were provided by Specialised Nutrition Europe (SNE) during the call launched in 2015.16
FC 13.1.5.2 covers all foods for babies and young children (i.e. from 4 months up to 3 years). This includes:
-
–
formulae
-
–
processed cereal‐based foods and baby foods
-
–
other foods for young children (e.g. milk‐based products)
The use levels received for SSOS (E 1450) for FC 13.1.5.2 only refer to formulae.
3.3.4. Summarised data extracted from the Mintel's Global New Products Database
The Mintel's GNPD is an online database which monitors new introductions of packaged goods in the market worldwide. It contains information of over 3 million food and beverage products of which more than 1,100,000 are or have been available on the European food market. Mintel started covering EU's food markets in 1996, currently having 24 out of its 27 member countries, and Norway and UK presented in the Mintel's GNPD.12
For the purpose of this Scientific Opinion, the Mintel's GNPD17 was used for checking the labelling of food and beverage products and food supplements for SSOS (E 1450) within the EU's food market as the database contains the compulsory ingredient information on the label.
No products were found in the Mintel's GNPD as labelled with SSOS (E 1450). The additive is authorised for direct use (Annex II) in food for special medical purposes for infants below 16 weeks (FC 13.1.5.1) and for babies and young children above 16 weeks of age (FC 13.1.5.2) which products are most probably available from specialised outlets (e.g. pharmacy) not covered by the Mintel's GNPD. Labelling of infant formula (FC 13.1.1) with SSOS authorised according to Annex III to Regulation N°1333/2008 (carry‐over) is not mandatory.
3.4. Exposure estimates
3.4.1. Exposure estimates for infants below 16 weeks
Exposure to SSOS (E 1450) from its uses as a food additive in formulae for infants below 16 weeks was estimated. The scenarios are based on the recommended consumption levels from the EFSA Scientific Committee Guidance (EFSA Scientific Committee, 2017a). This guidance ‘recommends values of 200 and 260 mL formula 18 /kg bw per day as conservative mean and high level consumption values to be used for performing the risk assessments of substances which do not accumulate in the body present in food intended for infants below 16 weeks of age’. These recommended consumption levels correspond to 14‐ to 27‐day‐old infants’ consumption, at this age the consumption peaks when expressed on a body weight basis. For the regulatory maximum level exposure assessment scenario, the MPL for infant formulae (20,000 mg/kg for FC 13.1.5.1 and 1,100 mg/kg for FC 13.1.1) were used. For the refined scenario, reported use levels (mean and maximum) were considered. The density of infant formulae that is ready to feed is assumed to be 1 g/mL.
3.4.1.1. Dietary exposure to starch sodium octenyl succinate (E 1450) from FSMP formulae
Table 4 summarises the estimated exposure to SSOS (E 1450) from its use as a food additive in FC 13.1.5.1 for infants below 16 weeks of age.
Table 4.
Dietary exposure to starch sodium octenyl succinate (E 1450) in foods for infants below 16 weeks of age according to the Annex II to Regulation (EC) No 1333/2008 (i.e. considering FC 13.1.5.1) (in mg/kg bw per day)
| Infants (< 16 weeks of age) | |
|---|---|
| Regulatory maximum level exposure assessment scenario (20,000 mg/kg) | |
|
4,000 5,200 |
| Refined estimated exposure assessment scenario | |
| Scenario using maximum use level reported by industry (20,000 mg/kg) | |
|
4,000 5,200 |
| Scenario using mean of use levels reported by industry (8,379 mg/kg) | |
|
1,676 2,179 |
bw: body weight.
The maximum occurrence scenario was used in the assessment. The mean occurrence scenario is reported and indicates that there are products on the market giving lower exposure levels.
3.4.1.2. Dietary exposure to starch sodium octenyl succinate (E 1450) from carry‐over into infant formulae
Table 5 summarises the estimated exposure to SSOS (E 1450) from its use as a food additive in nutrient preparations, as carry‐over in FC 13.1.1 for infants below 16 weeks of age.
Table 5.
Dietary exposure to starch sodium octenyl succinate (E 1450) due to carry‐over into foods for infants below 16 weeks of age according to the Annex III to Regulation (EC) No 1333/2008 (i.e. considering FC 13.1.1) (in mg/kg bw per day)
| Infants (< 16 weeks of age) | |
|---|---|
| Regulatory maximum carry‐over scenario (1,100 mg/kg) | |
|
220 286 |
| Refined estimated exposure assessment scenario | |
| Scenario using maximum carry‐over level reported by industry (386 mg/kg) | |
|
77 100 |
| Scenario using mean of carry‐over levels reported by industry (61 mg/kg) | |
|
12 16 |
bw: body weight.
3.4.2. Exposure estimates for infants above 16 weeks of age and toddlers consuming FSMP
As SSOS (E 1450) is also authorised in the food categories 13.1.5.1 and 13.1.5.2, an additional exposure assessment scenario considering these two food categories was performed to estimate the exposure of infants (above 16 weeks) and toddlers (classified as young children in Commission Delegated Regulation (EU) 2016/127, age of 1–3 years) who may eat and drink these FSMP.
The consumption of these foods is not reported in the EFSA Comprehensive database. To consider potential exposure to SSOS (E 1450) via these foods, the Panel assumes that the amount of FSMP consumed by infants and toddlers resembles that of comparable foods in infants and toddlers from the general population. Thus, the consumption of FSMP categorised as FC 13.1.5 was assumed equal to that of formulae and food products categorised as FCs 13.1.1, 13.1.2, 13.1.3 and 13.1.4.
Use levels received for SSOS (E 1450) for FC 13.1.5.2 only refer to formulae. Therefore, use levels reported for the FC 13.1.3 and 13.1.4 were used for calculating dietary exposure to E 1450 for infants above 16 weeks of age and toddlers.
This scenario was estimated as follows:
-
–
Consumers only of FSMP were assumed to be exposed to SSOS (E 1450) present at the maximum reported use level on a daily basis via consumption of food categories 13.1.5.1 and 13.1.5.2.
-
–
For the remaining food categories, the mean of the typical reported use levels was used.
In the refined scenario considering the whole diet, dietary exposure to SSOS (E 1450) ranged from 45 mg/kg bw per day for toddlers to 1994 mg/kg bw per day for infants above 16 weeks of age. At the high level (95th percentile), dietary exposure to SSOS (E 1450) ranged from 250 mg/kg bw per day for toddlers to 4069 mg/kg bw per day for infants above 16 weeks of age.
For both infants and toddlers, the main contributing food categories were infant formulae as defined by Commission Delegated Regulation (EU) 2016/127 and processed cereal‐based foods and baby foods for infants and young children as defined by Directive 2006/125/EC.
3.4.3. Uncertainty analysis
In accordance with the guidance provided in the EFSA opinion related to uncertainties in dietary exposure assessment (EFSA, 2007), the following sources of uncertainties have been considered and summarised in Table 7.
Table 7.
Qualitative evaluation of influence of uncertainties on the dietary exposure estimate
| Sources of uncertainties | Directiona |
|---|---|
Consumption data:
|
+ +/– |
| Methodology used to estimate high percentiles (95th) long‐term (chronic) exposure based on data from food consumption surveys covering only a few days for subjects above 16 weeks of age | + |
| Correspondence of reported use levels to the food items in the EFSA Comprehensive Database: uncertainties to which types of food the levels refer | +/– |
| Uncertainty in possible national differences in use levels of food categories | +/– |
|
Regulatory maximum level exposure assessment scenario: – for infants below 16 weeks of age: exposure calculations based on the MPL according to Annex II to Regulation (EC) No 1333/2008 for FC 13.1.5.1 or according to Annex III to Regulation (EC) No 1333/2008 for FC 13.1 |
+ |
|
Refined exposure assessment scenarios: – exposure calculations based on the maximum or mean levels (reported use from industries) |
+/– |
MPL: maximum permitted level.
+, uncertainty with potential to cause overestimation of exposure; –, uncertainty with potential to cause underestimation of exposure.
SSOS (E 1450) is authorised in FC 13.1.5.1 and FC 13.1.5.2 according to Annex II to Reg N°1333/2008 and in foods for infants (FC 13.1) according to Annex III.
Based on the assumption that carers of children with allergies or any other medical disorder would be brand‐loyal to an infant formula for special medical purposes (FC 13.1.5.1) that suits his medical disorder, the refined scenario using maximum use level reported by industry (Table 4) would in general result in an average realistic estimation of exposure for infants below 16 weeks of age.
Based on the assumption that carers would anyway be brand‐loyal to an infant formula (FC 13.1.1), the refined scenario using maximum reported use level (Table 5) would also in general result in an average realistic estimation of exposure for infants below 16 weeks of age.
The Panel noted that information from the Mintel GNPD indicated that no FSMP products for infant and young children were labelled with SSOS (E 1450). Considering that the maximum reported levels were used for foods under FC 13.1.5.1 and 13.1.5.2 while mean reported use levels were used for the rest of the diet, the Panel considered that the dietary exposure to SSOS (E 1450) would result in a realistic estimation of the exposure to SSOS (E 1450) from its use as a food additive according to Annex II for infants above 16 weeks of age and toddlers.
3.5. Proposed revision to existing EU Specifications for SSOS (E 1450)
The Panel considered that the maximum limits in the EU specifications for toxic elements should be established based on actual levels measured in the food additive. Therefore, if the European Commission decides to revise the current limits in the EU specifications to more appropriate values, the estimations of toxic elements intake as described below could be considered.
The interested party proposed maximum limits for toxic elements (< 0.05 (As), < 0.01 (Cd), < 0.03 (Pb) and < 0.05 (Hg) mg/kg) based on the lowest technologically achievable levels which were consistent with the analytical data, expressed as ‘less than values’ given above, provided for 27 commercial samples of the food additive (i.e. lowest achievable levels declared equal to the highest ‘less than values’ reported). Of note, the interested party declared that these analytical data and maximum levels proposed are specifically intended only for food for infants below 16 weeks of age (a clarification letter was provided by Starch Europe on 29 October 2019).
The Panel agreed to consider these proposed values as a starting point to characterise the risk of exposure to toxic elements derived from the consumption of the food additive. The potential exposure to these toxic elements can be calculated by assuming contamination of the additive may be up to the maximum limits (0.05 (As), 0.01 (Cd), 0.03 (Pb) and 0.05 (Hg) mg/kg), as proposed for the revision of the EU specifications, and then by calculation pro rata to the dietary exposure to the food additive itself.
With regard to the dietary exposure of the food additive, the Panel considered the refined estimated exposure assessment scenario based on maximum use levels (95th percentile) for infants below 16 weeks of age (5200 mg/kg bw per day, see Table 4), for E 1450 from carry‐over into infant formulae (100 mg/kg bw per day, see Table 5) and for toddlers (2,579 mg/kg bw per day, see Table 6). The above‐mentioned proposed maximum limits for toxic elements (0.05 (As), 0.01 (Cd), 0.03 (Pb) and 0.05 (Hg) mg/kg), combined with the estimated intakes of E 1450 (5,200, 100 and 2,579 mg/kg bw per day) could result in an exposure which can be compared with the following reference points, or health‐based guidance values (HBGVs), for the four toxic elements: a BMDL01 of 0.3–8 μg/kg bw per day for arsenic (EFSA CONTAM Panel, 2009), a total weekly intake (TWI) of 2.5 μg/kg bw for cadmium (EFSA CONTAM Panel, 2009), a BMDL01 of 0.5 μg/kg bw per day for lead (EFSA CONTAM Panel, 2010) and a TWI of 4 μg/kg bw for mercury (EFSA CONTAM Panel, 2012).
The outcome of such an exercise illustrates the health impact that would result if the proposed maximum limits for toxic elements were to be used: for arsenic and lead, the MOS/MOE could be as low as 1.2 and 2.3, respectively (see Table 8). For cadmium and mercury, the exhaustion of their HBGVs could be up to 15% and 46%, respectively.
Table 8.
Exposure to toxic elements based on the maximum limits for toxic elements in SSOS (E1450) for use in food for infants below 16 weeks of age as proposed by interested party (Documentation provided to EFSA n. 1)
| Exposure to the additive (mg/kg bw per day) | MOS/MOE for As | MOS/MOE for Pb | % of the TWI for Cd | % of the TWI for Hg |
|---|---|---|---|---|
| 0.05 mg/kg | 0.03 mg/kg | 0.01 mg/kg | 0.05 mg/kg | |
| 5200 (Table 4) | 1.2–31 | 3.2 | 15 | 46 |
| 100 (Table 5) | 60–1,600 | 166.7 | 0.28 | 0.9 |
| 2579a (Table 6) | 2.3–62.5 | 6.5 | 7.2 | 22.4 |
In EFSA ANS Panel (2017), the highest dietary exposure to modified starches E 1404‐1451 for the population above 16 weeks was 3,053 mg/kg bw per day (toddlers, brand‐loyal refined exposure assessment scenario). Therefore, the dietary exposure to E 1450 (see Table 6) for this population group was considered in the current assessment.
The Panel observed that if these maximum levels were to be applied, i.e. 0.05 (As), 0.01 (Cd), 0.03 (Pb) and 0.05 (Hg) mg/kg), this would mean that the performance of the analytical method applied should guarantee a limit of quantification (LOQ) of two‐fifths of the maximum level, i.e. 0.02 (As), 0.004 (Cd), 0.012 (Pb) and 0.02 (Hg) mg/kg, as in accordance to the provisions of Commission Regulation (EC) No 333/200719 for toxic elements in food. These LOQ values, especially for cadmium, may be technically difficult to be achieved with the analytical techniques commonly applied for the measurement of toxic element (e.g. ICP‐MS). Of note, E 1450 is a sodium‐containing food additive and sodium inhibits ionisation efficiency in the ICP‐MS; thus, a lower sensitivity would be expected.
In addition, the Panel pointed out that it should be checked whether these limits are technologically achievable also in the food additive intended for food for population groups other than infants below 16 weeks of age, according to what declared by the interested party.
The Panel also noted that there is some uncertainty on the exact concentration of the toxic elements reported for the 27 production batches, as the analytical data provided on the samples are expressed as ‘less than a reporting level’, which may be assumed referring to the LOQs of the analytical measurements, and not as measured values (with the exception for three samples in the lead determination). Therefore, the Panel decided to perform also an estimate of the exposure to toxic elements, derived from the food additive, considering the highest ‘less than value’ reported per each toxic element multiplied by an ‘uncertainty’ factor (UF) of 10 in order to cover uncertainties, such as representativeness, homogeneity and analytical measurement uncertainty. The resulting calculated concentration values of toxic elements (i.e. 0.5 (As), 0.1 (Cd), 0.3 (Pb), 0.5 (Hg) mg/kg), combined with the estimated intakes of E 1450 (5,200, 100 and 2,579 mg/kg bw per day), were then compared with the corresponding reference points or HBGVs for the four toxic elements (a BMDL01 of 0.3–8 μg/kg bw per day for arsenic (EFSA CONTAM Panel, 2009), a TWI of 2.5 μg/kg bw for cadmium (EFSA CONTAM Panel, 2009), a BMDL01 of 0.5 μg/kg bw per day for lead (EFSA CONTAM Panel, 2010) and a TWI of 4 μg/kg bw for mercury (EFSA CONTAM Panel, 2012)).
The health impact that would result if the highest ‘less than values’ reported, multiplied by UF of 10, for example, were to be used as maximum levels are as follows: for arsenic and lead, the MOS/MOE could be as low as 0.12 and 0.32, respectively (see Table 9). For cadmium and mercury, the TWIs are exceeded by factors up to 1.5 (Cd) to 4.6 (Hg).
Table 9.
Exposure to toxic elements considering the highest ‘less than value’, multiplied by an UF of 10, reported per each toxic element in 27 batches of E 1450 (Documentation provided to EFSA n. 1)
| Exposure to the additive (mg/kg bw per day) | MOS/MOE for As | MOS/MOE for Pb | % of the TWI for Cd | % of the TWI for Hg |
|---|---|---|---|---|
| 0.5 mg/kg | 0.3 mg/kg | 0.1 mg/kg | 0.5 mg/kg | |
| 5200 (Table 4) | 0.12–3.1 | 0.32 | 146 | 455 |
| 100 (Table 5) | 6–160 | 17 | 2.8 | 8.8 |
| 2579a(Table 6) | 0.23–6.3 | 0.65 | 72 | 224 |
In EFSA ANS Panel (2017), the highest dietary exposure to modified starches E 1404‐1451 for the population above 16 weeks was 3,053 mg/kg bw per day (toddlers, brand‐loyal refined exposure assessment scenario). Therefore, the dietary exposure to E 1450 (see Table 6) for this population group was considered in the current assessment.
The resulting figures show, in both exposure scenarios described (i.e. using the maximum levels as proposed by the interested party or using the highest reported ‘less than value’ multiplied by UF of 10), that the exposure to toxic elements from the consumption of E 1450 is not marginal. Therefore, this supports the Panel recommendation to substantially decrease the current maximum limits set for arsenic, lead and mercury and to introduce a maximum limit for cadmium.
The Panel emphasises that the choice of the magnitude of an acceptable MOS/MOE or an acceptable level of exceedance of the TWI from one source only as a basis to conclude on the maximum limits for toxic elements in the specifications is in the remit of risk management. However, the Panel noted that the MOS/MOE for arsenic and lead is very low, considering that for lead, the reference point is based on perturbation of intellectual development in children, (who have the highest exposure), and for arsenic, the reference point is based on carcinogenicity.
With regard to the maximum limit for octenylsuccinic acid residue, the Panel considered the proposal by the interested party to maintain the value currently set in the EU Reg. 213/2012, i.e. not more than 0.3%, to be adequate as it reflects the analytical levels measured in the food additive (Documentation provided to EFSA n. 1).
With regard to the maximum limit for sulfur dioxide, the proposal by the interested party is to lower the limit to not more than 10 mg/kg according to the analytical results obtained from the analysis of commercial samples of the food additive. The Panel agreed with this proposal and pointed out that sulfur dioxide is an authorised food additive (E 220) with a maximum permitted level (MPL) up to 2,000 mg/kg. In addition, under EU Reg. 1169/2011 on the provisions of food labelling information to consumers, sulfur dioxide and sulfites are considered allergens. For prepacked foods, their presence in a food or beverage must be indicated on the label, by its full name, when the level exceeds 10 mg/kg or 10 mg/L (expressed as SO2). Therefore, the presence of sulfur dioxide in E 1450 as impurity, within the proposed maximum limit, does not constitute a safety concern.
The interested party declared that the proposed maximum limits both for octenylsuccinic acid residue and sulfur dioxide are specifically intended for food for infants below 16 weeks of age (like the specification proposals for toxic elements). Therefore, the Panel reiterated that it should be checked whether these limits can be technologically achievable also in food for other population groups.
The Panel noted also that the use of ingredients for infant formula and follow‐on formula is regulated in the EU by Regulation 2016/127 on infant formulae and follow‐on formulae, clearly stating (Annex II) that ‘The use of ingredients containing gluten shall be prohibited8 ’. Therefore, the food additive SSOS (E 1450) should not contain gluten (only in infant formula and follow‐on formula, in accordance with Regulation 2016/127).
Overall, based on the data provided by interested parties in response to EFSA call for data16 and the relative above considerations, the FAF Panel recommends the following revisions of the existing EU Specifications for SSOS (E 1450) as outlined in Table 10.
Table 10.
Proposal for a revised version of the existing EU Specifications for SSOS (E 1450)
| Commission Regulation (EU) No 231/2012 | Comment/justification for revision | |
|---|---|---|
| Definition | Starch sodium octenyl succinate is the sodium salt of starch esterified with octenylsuccinic anhydride | Unchanged |
| Synonym | SSOS | Unchanged |
| Description | White or nearly white powder or granules or (if pregelatinised) flakes, amorphous powder or coarse particles | Unchanged |
| Microscopic observation | Passes test (if not pregelatinised) | Unchanged |
| Iodine staining | Passes test (dark blue to light red colour) | Unchanged |
| Loss on drying |
Not more than 15.0% for cereal starch Not more than 21.0% for potato starch Not more than 18.0% for other starches |
Unchanged |
| Octenylsuccinyl groups | Not more than 3% (on an anhydrous basis) | Unchanged |
| Octenylsuccinic acid residue | Not more than 0.3% (on an anhydrous basis) | Unchangeda |
| Sulfur dioxide | Lowered based on the available analytical resultsa | |
| Arsenic | Lowered based on the available analytical resultsa | |
| Lead | Lowered based on the available analytical resultsa | |
| Mercury | Lowered based on the available analytical resultsa | |
| Cadmium | Included based on the available analytical resultsa | |
| Gluten | Gluten free, only in infant formula and follow‐on formula, in accordance with Commission Delegated Regulation (EU) 2016/127/EC of 25 September 2015 | Included according to Commission Delegated Regulation (EU) 2016/127/EC |
| Microbiological criteria introduced for reason of harmonisation | ||
| Aerobic plate count | < 100 CFU/g | Included based on the available informationa |
| Yeasts | < 100 CFU/g | Included based on the available informationa |
| Salmonella spp | Negative in 375 g | Included based on the available informationa |
| E. coli | Negative in 10 g | Included based on the available informationa |
| Cronobacter (Enterobacter) sakazakii | Negative in 10 g | Included based on the available informationa |
CFU: colony‐forming unit.
The Panel noted that the interested party has submitted data and proposals of SSOS (E 1450) samples specifically intended for food for infants below 16 weeks of age (a clarification letter was provided by Starch Europe on 29 October 2019).
3.6. Biological and Toxicological data
3.6.1. Absorption, distribution, metabolism and excretion studies
The following text (in italics) is from the opinion published in 2017 (EFSA ANS Panel, 2017). New information and assessments related to the specific age group below 16 weeks of age are added in the following paragraphs.
In vitro study
The in vitro digestibility of OSA‐modified starch by porcine pancreatic and human salivary α‐amylase, a fungal (Aspergillus niger) glucoamylase and a barley β‐amylase was compared with that of the corresponding unmodified starch from which it was prepared (NSCC, 1984; cited in JECFA, 2015 ). The digestibility of OSA‐modified starch, measured by the rate of production of reducing substances, ranged from 83% to 98% of that of its corresponding native starch. It was suggested that the slight differences in the rate of digestibility were likely due to those anhydroglucose units in the starch substituted with OSA (about 1 in 50) inhibiting the hydrolysis of the α1‐4 and α1‐6 bonds. The in vitro enzyme digestibility of OSA‐modified starch was comparable to that reported for other modified food starches.
In vivo studies
The excretion of OSA and its related metabolites was analysed in 17 hospitalised infants and children (aged 2 months–6 years) fed one of three commercial hydrolysed protein formulas containing OSA‐modified starch 21 for an unspecified duration (Kelley, 1991 ). Random or 24‐h urine samples were collected, and urinary metabolites were identified using gas chromatography–mass spectrometry (GC–MS). In addition, plasma samples were collected from five patients and analysed for free fatty acids and organic acids. The results indicated that between 10% and 25% of the OSA hydrolysed from ingested OSA‐modified starch was absorbed and excreted in the urine. The average amount of OSA absorbed was estimated to be approximately 50–70 mg/kg bw. The principal compounds identified in the urine were OSA and at least nine metabolites that appeared to be produced from the oxidation of OSA by a combination of microsomal and mitochondrial or peroxisomal processes. The levels of OSA detected in the urine ranged from 121 to 1,353 mg/g creatinine, whereas urinary levels of OSA‐related metabolites ranged from 73 to 2,168 mg/g creatinine. In the plasma, measurable concentrations of OSA (9.5–57.9 μmol/L) were detected, but no other related metabolites were detected at concentrations higher than 1 μg/mL. Based on the molecular weight and mass fragmentation of the nine identified metabolites associated with the excretion of OSA, the author proposed that OSA is metabolised in infants by a combination of ω‐, ω‐1 and β‐oxidation steps, similar to valproic acid.
One hundred and seven female healthy term infants (aged 2–16 days), comprising 55 infants administered a milk‐based formula containing OSA‐modified starch (concentration not specified) and 52 administered a milk‐based formula containing distarch phosphate modified tapioca starch (control), were fed for 120 days ad libitum (MJNR, 1994; cited in JECFA, 2015 ). Urine samples collected on day 90 were analysed for OSA and related metabolites. In the infants consuming OSA‐modified starch, urinary OSA levels ranged from 0 to 1,398.6 μg/mg creatinine (mean of 546.1 μg/mg creatinine). The concentration of 1,2,9‐non-4‐enetricarboxylate, a metabolite of OSA, ranged from 0 to 865.5 μg/mg creatinine (mean of 343.8 μg/mg creatinine).
3.6.1.1. Absorption, distribution, metabolism and excretion studies in animals
A study in juvenile rats is described by the JECFA (2015). The JECFA report did not mention the method used to measure the parent compound and the metabolites. According to JECFA, the rats were divided into three groups (n = 4) from which group 1 received a proprietary formula, not containing OSA20 or SSOS (E 1450),21 as control; the second group (group 2) received a proprietary formula (formula 1) to which OSA was added (0.72 mg/mL) and the third group formula 2 which contained SSOS (E 1450) with an OSA content of 0.42 mg/ml (0.58 fold less OSA than in group 2). The animals received the formula once by oral application (not further specified). By comparison of the OSA and metabolites excreted in urine after administration of OSA as such or after administration of SSOS, it could be shown that 35% of the dose is excreted in urine when OSA is added to formula and that 19% of the OSA‐dose is excreted in the urine when SSOS (E 1450) is added (see Table 11). The result indicates that OSA is split off from SSOS (E 1450) and that the amount of OSA which is split off accounted to 54.3%.
Table 11.
Metabolite urinary excretion in rats following the administration of a formula added with OSA (formula 1) and to a formula containing SSOS (E 1450) (formula 2), from JECFA (2015)
| OSAa | SSOS (E 1450)b | |
|---|---|---|
| Dose (μmol/kg bw) | 120 | 69.1 |
| Urinary excretion | μmol/24 h | μmol/24 h |
| Tricarboxylic acid derivate of OSA | 3.13 ± 1.19 | 1.06 ± 0.07 |
| 7‐hydroxyacetyl succinate | 0.71 ± 0.21 | 0.13 ± 0.03 |
| 6‐hydroxyacetyl succinate | 2.03 ± 0.95 | 0.48 ± 0.18 |
| 1,2,7‐hept‐4‐enetricarboxylate | 0.39 ± 0.13 | 0.17 ± 0.04 |
| OSA | 0.1 ± 0.09 | 0.03 ± 0.01 |
| Excretion of OSA and metabolites (% of dose) | 35 ± 12 | 19 ± 2 |
OSA: octenyl succinic acid; SSOS: starch sodium octenyl succinate; bw: body weight.
28% (w/v) suspension of proprietary formula 1 to which OSA (0.72 mg/mL) was added.
Suspension of proprietary formula 2 containing SSOS (E 1450), with an OSA content of 0.42 mg/mL.
Absorption, distribution, metabolism and excretion studies in humans
In a study in 17 infants and children (age 2 months to 6 years), receiving SSOS (E 1450) (reported as OSA‐modified cornstarch) containing formulas, OSA and OSA metabolites were determined in the urine (Kelley, 1991), see summary above from EFSA ANS Panel (2017). The ratio of OSA metabolites to OSA was not different when infants up to 4 months were compared to infants above 4 months (up to 4 months: mean 2.4, range 0.35–4.8; above 4 months: mean 1.9, range 0.31–4.7; calculations by the Panel), indicating no metabolic differences. The publication does not give the dose which the infants received nor was the sampling scheme the same for all children (24 h collection in six infants; sampling at random in 11 infants). In this publication, the excretion of glutarate and ketoglutarate in urine was in about half of the infants above the normal values. Because of the lack of information and the interference with the concomitant treatment of the infants, it was not possible for the Panel to draw any conclusions on the association between the intake of SSOS (E 1450) and the elevated excretion of glutarate and ketoglutarate. Overall, from the rat study, the conclusion can be drawn that OSA is split off from SSOS (E 1450) and metabolised to the same metabolites as found in urine after OSA administration. In humans, Kelley (1991) proposes that OSA is split off from SSOS (E 1450) (reported as OSA‐modified cornstarch) and then metabolised to several metabolites whereby metabolites are produced by shortening the side chain after omega oxidation by cycles of beta oxidation. In the study MJNR, 1994 (cited in JECFA, 2015), the excretion of OSA and of its metabolite 1,2,9‐non‐4‐enetricarboxylate in urine indicates also that OSA is split off from SSOS (E 1450) and metabolites are formed by omega oxidation. Comparison among different species and data comparing young and old population were not available.
Gut microbiome
In general, when starches are reaching the caecum and colon, they are fermented by the gut microbiome into short‐chain fatty acids (SCFA). The SCFA (acetate, propionate, butyrate) are further metabolised after absorption and have multiple actions in diverse organs where they are introduced into the general metabolism (Hu et al., 2018). They have been reported to have a wide array of beneficial effects (den Besten et al., 2015a,b; Canfora et al., 2015; Gonzalez‐Bermudez et al., 2018). Information specific for SSOS (E 1450) was not available.
An interplay between microbiome and the diet has been described (den Besten et al., 2013). In infants, it is known that the microbiome depends on the mode of delivery (vaginal or caesarean), and also on the feeding, e.g. more diverse microbiomes in formula‐fed infants have been found compared to breast‐fed infants. A recent narrative review (Chong et al., 2018) confirmed these factors and expanded them by mentioning age, diet, host genetics, antibiotic usage and the birth environment of the infants e.g. neonatal intensive care unit (NICU). In both publications, the authors conclude that more data are required for a better understanding of the interaction between the factors. The Panel noted that changes in the composition of the gut microbiota without measuring a specific health outcome are difficult to interpret.
3.6.2. Short‐term and subchronic toxicity
The following information from short‐term and subchronic toxicity studies was reviewed by the ANS Panel in the context of the re‐evaluation of the safety of modified starches (EFSA ANS Panel, 2017):
‘In a 8‐week study, groups of 12 weanling albino rats (6/sex) were maintained on diets containing 64% carbohydrate ingredients consisting of 29% cellulose, with the remaining 35% consisting of starch sodium octenyl succinate, or corn starch as a control (…) Rats fed the substituted starch showed a slightly slower growth rate than control rats fed corn starch. The decreased growth rate was associated with decreased food consumption. Efficiency of food utilisation was not affected by the test compound.
Rats (Charles River) received a diet containing 6%, 12% or 30% starch sodium octenyl succinate (plus cornstarch at a 30% level of the diet) or 30% cornstarch (Buttolph and Newberne, 1980) (…) The animals were allowed to mate twice. The F1b generation was maintained on the same test diet as the parents and used for the study (…) Twenty animals from the 30% starch sodium octenyl succinate and control groups were killed at 30 days post‐weaning, and the remainder of the animals killed 90 days post‐weaning (…) There was no significant effect on growth rate. Serum chemistry and haematology were within normal levels and showed no compound‐related effects. Urine chemistry showed higher concentrations of urinary calcium and magnesium in females but not in males. Relative organ weight data showed a trend for increased liver and kidney weight with increased concentration of the substituted starch in the diet. There was an increased caecal weight in the animals fed 30% starch sodium octenyl succinate in both sexes after 30 days, but this was only observed in females after 90 days on the test diet. The only significant histological finding was an incidence of corticomedullary mineralisation in the kidneys. The effect was more severe in females than in males, and occurred in animals fed either the modified or unmodified starch.
In a 90‐day study (Unilever, 1984; cited in JECFA, 2015 ), groups of 10 male and 10 female Colworth−Wistar rats (…) the control diets for each of the OSA‐modified test groups contained unmodified starch. The modified starch diets provided approximately 37,000 mg/kg bw per day of OSA‐modified starch (…)
There were no differences in body weight gain, feed consumption, plasma chemistry measurements or urine analysis parameters when comparing animals on the test diets (OSA‐modified starch) with those on the corresponding basal control diets (…). Similarly, no significant test article‐related changes in liver, kidney or caecum weights were observed when comparing the animals on the test diets with those on the corresponding basal control diets. (…) Increased kidney weights were observed (…) considered to be related to corticomedullary nephrocalcinosis. All female animals fed the ‘in‐house’ purified diets exhibited corticomedullary nephrosclerosis, and it was noted that the inclusion of OSA‐modified starch did not influence its severity. Similar effects were not apparent in male rats.
The authors concluded that the inclusion of OSA‐modified starch in the diet of rats for 90 days at a concentration of 30% (approximately 37,000 mg/kg bw per day) did not adversely affect any parameter examined, when compared with the control unmodified starch (Unilever, 1984; cited in JECFA, 2015). The Panel agreed with this conclusion’.
3.6.3. Post‐natal studies
In the evaluation of the ANS Panel in 2017 (EFSA ANS Panel (2017)) and in JECFA (2015), a study in pups of Beagle dogs (age 5–9 days) which were dosed with 0 (water control), 5,000 or 10,000 mg/kg bw per day of SSOS (E 1450)21 or 5,000 or 10,000 mg/kg bw per day of a control starch for 6 weeks was described. There were no significant differences in blood chemistry, haematology or urine parameters among groups. No deaths, gross lesions or histological findings were attributable to the treatment. The full study report of this study was not available for evaluation by the FAF Panel.
In the former evaluation (EFSA ANS Panel, 2017) a 3‐week dietary toxicity study of SSOS (E 1450) in farm piglets was briefly described; however, the study report was not available. The Panel received the full study report (Documentation provided to EFSA n. 3). The results of this study were also published by Mahadevan et al. (2014).
The study was performed according to Good laboratory practice (GLP), FDA (2006), EMA (2009) and ICH (2010). The objective of this study was to evaluate the safety of SSOS (E 1450) after 3 weeks of dietary administration to farm piglets starting 2 days after birth and evaluate the impact of SSOS (E 1450) on their growth. The pig was selected for use in this study because of similarity of the digestive system between neonatal swine and human infants. The RoB Risk of Bias score (RoB) was rated as tier 1, which indicating a low risk of bias (see Appendix C).
Domestic Yorkshire cross‐bred piglets (n = 6/sex per group; body weight males 1.7–2.5 and females 1.4–2.6 kg) were administered 500 mL/kg bw per day of milk containing 0, 2, 4 or 20 g SSOS (E 1450) per litre (equivalent to 0, 1,000, 2,000 or 10,000 mg/kg bw per day) from post‐natal day (PND) 2 for 3 weeks. The highest dose was chosen to represent up to the regulatory limit in the EU. To ensure that the total caloric intake was similar among groups, accounting for the decreased digestibility of SSOS (E 1450), the control, low‐dose, mid‐dose and high‐dose groups also received amioca powder (control article) at levels of 8,000, 7,200, 6,400 or 0 mg/kg bw per day, respectively. SSOS (E 1450) and amioca were added to a commercially available milk replacer. The energetic value of the formulation was assumed to be 60 kcal/L in all test groups. The test formulations were offered orally via a feeding bowl at a dose volume of 500 mL/kg bw per day, six times per day (~ 83.33 mL/kg bw per dose, 3.25 h between doses). Animals were randomly assigned by sex using body weight to the treatment groups. The first feeding of the formulation started on lactation day 2; day of arrival of the animals at the test facility. All animals survived to scheduled necropsy on day 21, and there were no compound‐related changes in clinical observations during the study except for a decrease in growth of the male piglets of the high‐dose group. In female animals’ significant effect on body weight gain were not observed; only one female showed a lower body weight gain in the high‐dose group). However, the Panel noted that four out of six male animals had a decrease in the body weight gain at the high‐dose and one out of six male animals in the mid‐dose group. This effect was not statistically significant; however, the change in body weight compared to the control over the entire study (study day 1–21) was approximately 30%. The feed intake of the male piglets of the high‐dose group was decreased when compared to the control group; the difference was statistically significant on study day 20. The Panel noted that it was unclear whether reduced palatability alone was responsible of the reduced feed intake observed in the high‐dose male group. No dose‐related effects on haematology, clinical chemistry and urine analysis were observed on study day 8 and 21. Faecal samples were negative for parasites. Organ weights were obtained for the brain, heart, kidney, large intestine (caecum, colon, rectum), liver, small intestine (duodenum, jejunum and ileum) and spleen. The relative liver and kidney weights of the male piglets of the high‐dose group were statistically significantly decreased (approximately 15%). No macroscopical effects were observed at necropsy. Microscopical examination of the organs listed above, as well as eye, including optic nerve; gall bladder; stomach; gross lesions; lung with bronchi; pancreas; and Peyer's patch did not show treatment‐related effects. While the authors argued that the effect on liver and kidney weight could be secondary to decreased growth/body weight gain and/or incidental, the Panel considered the effect on relative organ weight as treatment‐related, although not adverse as no histopathological changes were observed. The authors concluded that administration of SSOS (E 1450) in the diet for a 3‐week period after birth was well tolerated in piglets and that exposure to SSOS (E 1450) did not produce any definitive compound‐related effects and considered 10,000 mg/kg bw per day, the highest dose tested as the NOAEL. The Panel performed a benchmark dose modelling (BMD) on body weight gain in males according to the guidance of the Scientific Committee (EFSA Scientific Committee, 2017b) and concluded that based on the reported data, no dose response could be identified.
In summary, in the animal studies evaluated by the ANS Panel (EFSA ANS Panel, 2017), no indication of significant toxic effects of SSOS were observed. However, the FAF Panel considered that the 8‐week study in weanling rats and the 90‐day rat study were not appropriate for the evaluation of SSOS as a food additive in food for infants below 16 weeks of age. In the study in pups of Beagle dogs up to 10,000 mg SSOS/kg bw per day for 6 weeks effects on body weight and food consumption were not described. The full study report was not available to the Panel and, therefore, a reference point could not be derived from this study.
The results of the post‐natal study in piglets were considered by the FAF Panel as the most suitable animal data for the evaluation of SSOS as food additive in food for infants below 16 weeks of age. However, due to the absence of effects in female animals and a lack of a dose‐response in the effect on body weights of male piglets, the Panel could not identify a reference point for the hazard characterisation of SSOS based on the data from this study.
3.6.4. Clinical studies
The following six clinical studies were submitted by the interested business operators in response to the call for data launched by EFSA: Ahrens et al. (2018); Borschel and Kajzer (2011) (documentation provided to EFSA n. 3); Borschel et al. (2013) (full study report provided by the interested business operators, documentation provided to EFSA n. 7); Burks et al. (2008) (full study report provided by the interested business operators, documentation provided to EFSA n. 8 and 9); Fleddermann et al. (2014) and Scalabrin et al. (2009) (full study report provided by the interested business operators, documentation provided to EFSA n. 10).
The Panel noted that according to the study reports or the publications on the studies, the studies were not aimed to investigate the influence of SSOS (E 1450) on the development of weight or on the tolerability.
The reviewers gave identical RoB scores for all studies (see Figure 3 and Appendix D for further details). Only minor inconsistencies on the scoring for some of the questions/elements were noted and clarified. Five of the studies were allocated to tier 3 (high risk of bias) (Ahrens et al., 2018; Borschel and Kajzer, 2011; Borschel et al., 2013; Burks et al., 2008; Scalabrin et al., 2009). The study of Fleddermann et al. (2014) was allocated to tier 2 (moderate risk of bias). Studies allocated to a RoB tier 3 could only be used as supporting evidence and are briefly described here. The elements considered for RoB appraisal by the reviewers are summarised in Figure 3.
Figure 3.
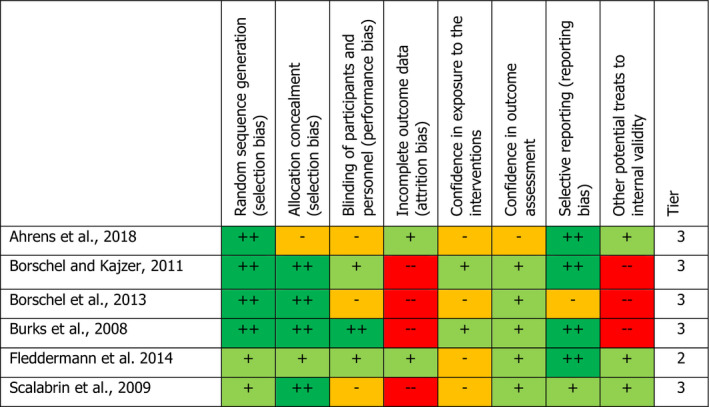
- Definitely low risk of bias (++), Probably low risk of bias (+), Probably high risk of bias (−), Definitely high risk of bias (−−).
The aim of the study of Ahrens et al. ( 2018 ); was described as follows: ‘A high protein content of non‐hydrolysed infant formula exceeding metabolic requirements can induce rapid weight gain and obesity. Hydrolyzed formulas with too low protein (LP) content may result in inadequate growth. The aim of this study was to investigate non inferiority of partial and extensively hydrolysed formulas (pHF, eHF) with lower hydrolysed protein content than conventionally, regularly used formulas, with or without synbiotics for normal growth of healthy term infants’. Four hundred and two infants were randomised to four treatment groups; LP‐formula containing partially hydrolysed proteins (1.9 g protein/100 kcal) with or without synbiotics, LP‐eHF formula with synbiotics or formula with regular protein content (2.3 g protein/100 Kcal). The primary endpoint was the average daily weight gain over 84 days. The trial was set up as a non‐inferiority trial with a non‐inferiority margin of ‐ 3.5 g/day, resulting in a total weight difference of ‐ 294 g as being equivalent with the control. In the publication and the supplementary information, the content of the formulas is given without mentioning SSOS (E 1450) as one of the components of the formula. According to the information from the interested business operators, the SSOS (E 1450) content was ■■■■■ in the control formula and ■■■■■ in the experimental formulae (Specialised Nutrition Europe, clarification letter dated 19 September 2019). All tested formulas showed non‐inferiority compared with the control. The main deficiencies of the study were that the composition in proteins content differed between, on the method of measurement and on the loss of participants above 20% which is seen as invalidating study results (Genaidy et al., 2007). The study was allocated to tier 3 (high risk of bias).
In the study report from Borschel and Kajzer (2011) (documentation provided to EFSA n. 3), it is not mentioned that SSOS (E 1450) was used but only starch. In a communication (Abbott, communication dated March 2019), it was reported that SSOS (E 1450) in two of the formulations used (EF‐1 and EF‐2) in this study was 1.9 g/L (as fed). The study was a randomised, multicentre, controlled, double‐blinded, parallel group study. One hundred and sixty‐eight healthy term infants between 0 and 8 days of age were randomised to receive ad libitum one of the three study formulas, control product without SSOS (E 1450) and one product containing SSOS (E 1450). ‘The purpose of this study was to evaluate the tolerance of infants fed experimental casein hydrolysate‐based formulas containing alternate CHO sources, ■■■■■ compared to a commercially available casein hydrolysate, based formula. The primary endpoint was MRSC (mean rank stool consistency) from Study Visit 1 to Study Visit 3 (approximately 28 days of age) (cited from the study report).’ Further outcomes included weight and height and adverse event monitoring. The main deficiency of the study was the loss of participants above 20% (25% in EF‐1 and 22% in EF‐2) which is seen as invalidating study results (Genaidy et al., 2007) and the lack of quantitative information on the content of the different ingredients in the formula, in particular of SSOS (E 1450) in EF‐1 and EF‐2 in the study report. The study was allocated to tier 3 (high risk of bias).
The main objective of the study by Borschel et al. ( 2013 ) was described as follows: ‘The present study was designed to assess the growth of healthy term infants fed an amino acid ‐based formula versus an extensively hydrolyzed casein‐based formula in a randomized controlled trial that met robust criteria proposed for evaluating the ability of a formula to support growth of infants.’ In the publication, it is declared that the extensively hydrolysed casein‐based formula contained 1.6% SSOS (E 1450). This extensively hydrolysed casein‐based formula was compared to an amino acid‐based formula which besides the difference in the source of proteins also contained different sources for the fat content and no SSOS. The randomised, double‐blind parallel group study was performed in 213 infants entering the study between 0 and 9 days old; 107 infants were randomised to the amino acid‐based formula whereas 106 infants were randomised to the formula containing 1.6% SSOS (E 1450). The study duration was 112 days. The formulas were given ad libitum and were the sole source of nutrition throughout the study. The primary endpoint was weight gain between 14 and 112 days of age. Secondary endpoints included length, head circumference, study formula intake, daily stool number, MRSC and serum albumin concentration. Seventy‐nine infants (37%) dropped out, with similar demographic characteristics between groups. The numbers of infants who finished the study early because of intolerance symptoms were similar. The group receiving SSOS (E 1450) had a significantly greater number of daily stools and average MRSC. This finding may be explained by the differences in protein composition between the two study formulae. No statistically significant differences between groups in weight, length, head circumference or mean serum albumin concentration were noted. The Panel noted that 40 of the 106 infants randomised to the formula containing 1.6% SSOS (E 1450) dropped out from the study, 21 of them because of intolerance which was not further specified. From the 66 infants which completed the study, no adverse events were noted, the SSOS (E 1450) consumption being a mean of 2.35 g/kg bw per day over 112 days. The main deficiencies of the study were the loss of participants above 20% which is seen as invalidating study results (Genaidy et al., 2007) and the difference in nutritional composition others than SSOS (E 1450) between the formulae. The study was allocated to tier 3 (high risk of bias).
In the study by Burks et al. ( 2008 ) for which a report was made available by interested business operators, the aim is described as follows: ‘The primary objective of this study was to compare weight gain from 14 to 120 days in term infants fed one of two study formulas: 1) a commercially available casein hydrolysate formula (Control formula or CF); or 2) an experimental amino acid‐based formula (Test Formula, or TF). Secondary objectives were to evaluate length gain and head circumference gain; formula intake acceptance; and tolerance; and incidence of adverse events among infants consuming the study formulas. An additional secondary objective was to verify that the resulting plasma amino acid profile of infants fed the test formula was similar to that of infants fed the control formula and to published values of breastfed infants.’ In the study report, the composition of the two formulas is given. However, it is not mentioned whether the formulas contained SSOS (E 1450). The Panel was later informed by the interested business operators that both formulas contained SSOS (E 1450) with a content of ■■■■■ (control formula) and ■■■■■ (experimental formula) without further documentation (Specialised Nutrition Europe, clarification letter dated 6 December 2019). A difference in length increase and final length (0.113 ± 0.003 cm/day in control group vs. 0.107 ± 0.003 cm in the experimental group, p = 0.030; 64.3 ± 0.4 cm in control group vs. 63.4 ± 0.4 in the experimental group) was noted, these differences are not biologically relevant. The main deficiencies of the study were that the composition concerning the source for proteins/amino acids differed between the formulas and loss of participants above 20% which is seen as invalidating study results (Genaidy et al., 2007). The study was allocated to tier 3 (high risk of bias).
The Panel noted that the study from Burks et al., 2008 included a trial assessing the hypoallergenicity of infants formulae, this part of the study was considered not relevant for the assessment of SSOS (E 1450) and was not considered further.
The objective of the study by Fleddermann et al., 2014 ; was the following: ‘In the present study we compared growth and blood biochemistry of infants fed a modified infant formula with reduced protein content and rich in ALAB (α‐lactalbumin) as well as added LC‐PUFA (long chain polyunsaturated fatty acid) to a formula with standard protein content and without LC‐PUFA, and a reference group of breastfed infants. The randomized controlled trial aimed to assess the suitability of a reduced‐protein ALAB and LC‐PUFA containing formula focusing on growth velocity, adverse events, markers of fatty acids and protein status and energetic efficiency in infants until the age of 10 days.’ In the study publication, the composition of the two formulas is given. However, it is not mentioned whether the formulas contained SSOS (E 1450). The Panel was later informed by the interested business operators that both formulas contained SSOS with a content of ■■■■■ without further documentation (Specialised Nutrition Europe, clarification letter dated 19 September 2019). In the method section, a primary endpoint is not mentioned; however, the statistical consideration was based on weight gain. Two hundred and thirteen infants were randomised to the two treatments and the duration of the study was from 1 month to 4 months. Weight gain was not statistically different between the intervention and the control group. The main deficiencies of the study were that the composition of proteins and fat differed between the formulas and that the information on the content of SSOS (E1450) is not given in the publication. The study was allocated to tier 2 (moderate risk of bias) and provides limited evidence for the safe use of SSOS in a relatively low concentration of ■■■■■.
The primary objective of the study of Scalabrin et al. ( 2009 ) was described as follows: ‘The primary objective was to compare the rate of weight gain (g/day) from 14 to 120 days of age in healthy, term infants fed 1 of 3 study formulas. Secondary objectives were to compare formula groups with respect to other growth parameters (length and head circumference [HC] growth rates), formula intake and tolerance, adverse events (AEs), blood lipids, allergic sensitization, and antibody response to immunizations commonly given to infants.’ In the study report, the composition of the two formulas is given However, whereas the content of carbohydrate is given, it is not specified whether one or more of the formulas contained SSOS (E 1450). The study tested three different formulas in 276 infants (14 days old), from which 220 infants completed the study after 120 days of exposure. The study was planned to assess the influence on growth and on the tolerance of adding Lactobacillus rhamnosus to a hydrolysed infant formula. According to an information of the interested business operators, two of the formulas contained SSOS (E 1450).The three formulas were as follows: EHF, a formula without Lactobacillus rhamnosus, but with ■■■■■; EHF‐LGG, a formula with Lactobacillus rhamnosus, and with ■■■■■ and PHF‐LGG, a formula with Lactobacillus rhamnosus, without SSOS (E 1450) (Specialised Nutrition Europe, clarification letter dated 6 December 2019). The study was not intended to investigate the tolerability of SSOS (E1450). Hence, the control group in the study received ■■■■■. In evaluating the data of the study, the Panel considered PHF‐LGG, a formula with Lactobacillus rhamnosus, without SSOS (E 1450) as the control group and the EHF‐LGG group, a formula with Lactobacillus rhamnosus, and with ■■■■■ as the experimental group. No difference in weight, length and head circumference was observed. A difference was noted in stool frequency with lower frequency in the group receiving the formula with Lactobacillus rhamnosus, without SSOS (E 1450) (p < 0.05) and in firmness of stools with firmer stools in the group receiving the formula with Lactobacillus rhamnosus, without SSOS (E 1450) (p < 0.005). It is to be noted that in the group receiving the formula containing SSOS (E 1450) relatively more infants dropped out (33%) than in the group receiving the formula containing no SSOS (E 1450) (21%), which may indicate a reduced tolerability of the formula with ■■■■■. The main deficiencies of the study were that the composition concerning the lack of information on SSOS in the study report and loss of participants above 20% (21% EHF group, 26% PHF‐LGG group, 33% EHF‐LGG group), which is seen as invalidating study results (Genaidy et al., 2007). The study was allocated to tier 3 (high risk of bias).
The levels in the formulas given as interventions in the clinical trials described above were calculated by the Panel, please refer to Appendix E for a detailed description on how the calculations were performed.
3.6.5. Post‐market monitoring
■■■■■
■■■■■
■■■■■
■■■■■
-
■■■■■
■■■■■
No reports in the scientific and medical literature were identified during the reporting period by Abbott Nutrition (Documentation provided to EFSA by a SNE Member).
■■■■■
■■■■■
No additional cases of adverse reactions were found by a literature search submitted by the interested business operators (Documentation provided to EFSA n. 3).
3.7. Discussion
This assessment is a follow‐up addressing data gaps previously identified during the re‐evaluation for SSOS (E 1450) and the safety in the special subpopulation of infants below 16 weeks of age (EFSA ANS Panel, 2017), see also Appendix B.
The process involved the publication of a dedicated call for data, allowing all interested business operators to provide the requested information for completing the assessment and to confirm that the additive is used in FC 13.1.5.1 and is also present in infant formula (13.1.1) as a carry‐over resulting from the authorised use in accordance with Annex III to Regulation (EC) No 1333/2008. Further consideration on the use as food additive in food according to FC 13.1.5.1 and FC 13.1.5.2 (dietary foods for babies and young children for special medical purposes as defined in Directive 1999/21/EC) in infants above 16 weeks of age and young children up to 3 years was also requested. The data submitted comprise technical information on impurities of the additive, formulation examples for products on the market, use levels in relevant infant formulae, toxicity data such as a report on a 3‐week dietary study on piglets, clinical data and post‐marketing surveillance reports. No adequate data were submitted by the interested business operators which can serve as the basis to assess the safety of SSOS (E 1450) in the use according to FC 13.1.5.1 and 13.1.5.2 for uses in food for infants above 16 weeks and young children.
The Panel considered it feasible to amend a number of specifications based on the analytical evidence submitted in response to the call for data. This refers to lowering existing limits for SO2, toxic elements, as well as introducing new specifications for cadmium and microbiological criteria for the food additive.
A study in juvenile rats, described in JECFA (2015), showed that the relative absorption of OSA (bound and unbound) from SSOS (E 1450) resulted in 53% (mean of excretion of four metabolites and the parent compound OSA), indicating that in the rat at least half of the SSOS (E 1450) is hydrolysed to OSA.
In infants between 2 months and 5 years, the ratio of OSA metabolites to OSA was not different when infants up to 4 months were compared to infants above 4 months indicating no metabolic differences. The metabolites are excreted via urine. In general, when starches are reaching the caecum and colon, they are fermented by the gut microbiome into SCFA. The SCFA (acetate, propionate, butyrate) are further metabolised after absorption and have multiple actions in diverse organs where they are introduced into the general metabolism (Hu et al., 2018). The publication of Kelley (1991) reported that the excretion of glutarate and ketoglutarate in urine was in about half of the infants above the normal values. However, when analysing the data, there was no indication that infants up to 4 months were more prone to have elevated levels. No further quantitative information is available on the fate of SSOS (E 1450) in infants up to 4 months as compared to older infants, adolescents or adults. Comparison among different species and data comparing young and old population were not available.
An interplay between microbiome and the diet has been described (den Besten et al., 2013). In infants, it is known that the microbiome depends on the mode of delivery and also on the feeding, e.g. more diverse microbiomes in formula‐fed infants have been found compared to breast feed infants. A recent narrative review (Chong et al., 2018) confirmed these factors and expanded them by mentioning age, diet, host genetics, antibiotic usage and the birth environment of the infants (e.g. NICU). In both publications, the authors conclude that more data are required for a better understanding of the interaction between the factors and what is necessary to maintain intestinal homoeostasis in terms of microbiome in the different population groups. The Panel noted that changes in the composition of the gut microbiota without measuring a specific health outcome are difficult to interpret.
In the animal studies evaluated by the ANS Panel (EFSA ANS Panel, 2017), no indication of significant toxic effects of SSOS was observed. However, the FAF Panel considered that the 8‐week study in weanling rats and the 90‐day rat study were not appropriate for the evaluation of SSOS as a food additive in food for infants below 16 weeks of age. In the study in pups of Beagle dogs up to 10,000 mg SSOS/kg bw per day for 6 weeks, effects on body weight and food consumption were not described. The full study report was not available to the Panel and, therefore, reference point could not be derived from this study.
The results of the post‐natal study in piglets were considered by the FAF Panel as the most suitable animal data for the evaluation of SSOS as food additive in food for infants below 16 weeks of age. However, due to the absence of effects in female animals and a lack of a dose‐response in the effect on body weights of male piglets, the Panel could not identify a reference point for the hazard characterisation of SSOS based on the data from this study.
Further to the call for data, five references and one study report from clinical trials conducted in infants below 16 weeks of age were submitted by interested business operators. Following requests for additional information, original study reports were made available for three of the published clinical trials, so that for four of them, the assessment could be based on the full study reports.
However, in only one study, the content of SSOS (E 1450) in the formula was clearly stated and could be calculated to be 1.6%; in the remaining studies, no information concerning the content of SSOS (E 1450) in the formulae was found, and in the study reports, no exact information on the SSOS (E 1450) content was given. The Panel was later provided with the information about the content of SSOS (E 1450) in the studies.
The Panel noted that even the primary full study reports did not contain information on the content of SSOS (E 1450) in the formulae and that the SSOS (E 1450) levels provided by interested business operators in 2019 were not accompanied by certificates, which could be a confirmation of the SSOS levels. The lack of information on the level of SSOS (E 1450) can be explained by the fact that none of the studies was planned to investigate the influence of SSOS (E 1450) on body weight and its tolerability in infants below 16 weeks of age. Hence, in none of the studies, the composition of the formula of the control group was identical to the formula containing the SSOS (E1450). Additionally, in some of the studies, the SSOS (E1450) was contained in the formula of the control group in nearly the same concentration as in the formula of the experimental group.
The six studies were appraised for their risk of bias applying an assessment tool modified from the OHAT RoB tool (NTP‐OHAT, 2015, 2019). The elements considered for the appraisal are described in the Appendix D to this opinion, as well as the decision rule for assigning the studies to tiers of reliability. Five of the studies were allocated to tier 3 (high risk of bias) (Ahrens et al., 2018; Borschel and Kajzer, 2011; Borschel et al., 2013; Burks et al., 2008; Scalabrin et al., 2009). The study of Fleddermann et al. (2014) was allocated to tier 2 (moderate risk of bias). Concerning the outcome of the assessment of RoB of the clinical studies, it is general agreement that studies allocated to tier 3 can only be used as supportive evidence. Insofar, the study of Fleddermann et al. (2014), allocated to tier 2 would be the only study which could serve as study providing main evidence.
In a case report series from 1991, no adverse effects specific to the SSOS (E 1450) in children were identified. Specific information about post‐marketing monitoring data for formula with high SSOS (E 1450) content (1.6% and higher) is not available.
In EFSA ANS Panel opinion (2017), the Panel concluded that the available data did not allow for an adequate assessment of the safety of the use of SSOS (E 1450) in ‘dietary foods for special medical purposes and special formulae for infants’ (food category 13.1.5.1) and in foods belonging to food category 13.1.5.2, in infants and young children consuming these foods at the presently authorised maximum use levels of 20,000 or 50,000 mg/kg, respectively. After the call for data, no studies and no data were submitted by the interested business operators.
Dietary exposure to SSOS (E 1450) from its use as a food additive was assessed based on (1) MPLs set out in the EU legislation (defined as the regulatory maximum level exposure assessment scenario) and (2) the reported use levels (defined as the refined exposure assessment scenario), (3) the levels in the formulas given as interventions in the clinical trials considered in this assessment.
Both scenarios (1) and (2) are based on the recommended consumption levels from SC Guidance (EFSA Scientific Committee, 2017a) which recommends values of 200 and 260 mL formula18/kg bw per day as conservative mean and high‐level consumption values for 14–27 days old infants.
For infants below 16 weeks of age consuming FSMP (FC 13.1.5.1), mean exposure in the regulatory maximum level exposure assessment scenario was estimated at 4,000 mg/kg bw per day while at the high level was estimated at 5,200 mg/kg bw per day. As the maximum level reported by industry was equal to the MPL of 20,000 mg/kg, exposure estimates are the same for the refined scenario based on maximum levels of use provided by the interested business operators. For the scenario using the mean of the reported use levels from industry, exposure estimates were of 1,676 mg/kg bw per day at the mean and 2,179 mg/kg bw per day at the high level of consumption.
Exposure to SSOS (E 1450) estimated for the clinical trials described above were ■■■■■, ■■■■■ in the Borschel and Kajzer (2011) study, the average intake of SSOS was between 262 and 362 mg/kg bw per day depending on the formulae and the day of the study; ■■■■■ ■■■■■
For infants below 16 weeks of age consuming infant formulae which could contain SSOS (E 1450) from carry‐over (FC 13.1.1), in the regulatory maximum level exposure assessment scenario, mean exposure was estimated at 220 mg/kg bw per day while at the high level was estimated at 286 mg/kg bw per day. For the scenario using the maximum level provided by industry, mean exposure was estimated at 77 mg/kg bw per day while at the high level was estimated at 100 mg/kg bw per day. The scenario using the mean of the reported use levels from industry, mean exposure was estimated 12 mg/kg bw per day at the mean and 16 mg/kg bw per day at the high level.
The Panel emphasised that the refined exposure estimates are based on information provided on the reported level of use of SSOS (E 1450). If actual practice changes, these refined estimates may no longer be representative and should be updated.
In conclusion, when considering the available information to set a reference point, studies in healthy infants would be the preferred data source. However, most of them (five of the six studies provided) had low internal validity, reflected in the high risk of bias (tier 3). The study with a moderate risk of bias (tier 2) had very low content of SSOS in the formulae used in both study arms of ■■■■■. In addition, in all the studies, the composition of the control formula, used without SSOS (E 1450), differed from the composition of the experimental formula with SSOS (E 1450). The Panel explored the possibility to compare the studies concerning growth with non‐randomised comparisons (i.e. comparisons to historical controls, such as comparisons to growth reference charts the data of which are of observational nature), but considered that the available data on these growth reference charts were not sufficiently informative for the European population. Hence, the Panel concluded that a reference point could not be derived from the clinical studies. The Panel considered whether the results from the piglet study could be used for identifying a reference point but had to note that the uncertainty surrounding the results precludes deriving a reference point from this study.
On the other hand, both data sources did not clearly indicate an adverse effect due to SSOS. In the former evaluation, the ANS Panel concluded that there was no safety concern for the use of modified starches as food additives at the reported uses and use levels and that there was no need for a numerical ADI for the general population (EFSA ANS Panel, 2017). Based on this and the newly available data, the FAF Panel considered that for exposure to SSOS of infants below 16 weeks, there is no indication for a concern when within the range reported in the clinical studies (up to 2,725 mg/kg bw per day).
When extrapolating the conclusion above to the safety assessment of the food additive when used in FCs 13.1.5.1 and 13.1.5.2 in food for infants above 16 weeks of age and young children, the Panel considered that for exposure to SSOS of infants above 16 weeks and young children, there is no indication for a concern when within the range reported in the clinical studies (up to 2,725 mg/kg bw per day).
4. Conclusions
Due to the low internal validity of the clinical studies, the Panel concluded that a reference point could not be derived from them. The Panel noted that the uncertainty surrounding the results of the piglet study precludes deriving a reference point from this study.
On the other hand, both data sources did not clearly indicate an adverse effect due to SSOS. Given the available data, the Panel concluded that at use levels of SSOS in food for infants below 16 weeks within the range reported in the clinical studies (up to 2,725 mg/kg bw per day), there is no indication for safety concern and reiterated the conclusion of the ANS Panel (EFSA ANS Panel, 2017) that there was no need for a numerical ADI.
When extrapolating the conclusion above to the safety assessment of the food additive when used in FCs 13.1.5.1 and 13.1.5.2 in food for infants above 16 weeks of age and young children, the Panel considered that there is no indication for safety concern also for these uses within the range reported in the clinical studies.
The Panel noted that at the reported use levels, the estimates of exposure could exceed the higher end of the exposure in the clinical trials.
Having considered the analytical data submitted by the interested business operators, the Panel concluded that these data support a revision of the existing specifications.
5. Recommendation
The Panel recommends that:
The European Commission considers revising the MPL of SSOS (E 1450) in FCs 13.1.5.1 and 13.1.5.2.
The European Commission to revise the current specifications for the food additive SSOS (E 1450) in line with the proposals made in Table 10.
6. Documentation as provided to EFSA
Starch Europe, 2019. Submission of data in response to the call for technical and toxicological data on starch sodium octenyl succinate (E 1450) for uses as a food additive in foods for all population groups including infants below 16 weeks of age submitted on January 2019.
SNE (Specialised Nutrition Europe), 2019. Response from Specialised Nutrition Europe of EFSA call for data on starch sodium octenyl succinate (E 1450) authorised as a food additive in support of the risk assessment for infants below 16 weeks of age. January 2019.
Abbott Nutrition Research & Development, 2019. Submission of data in response to the call for technical and toxicological data on starch sodium octenyl succinate (E 1450) for uses as a food additive in foods for all population groups including infants below 16 weeks of age submitted on January and February 2019.
Mead Johnson, 2019. Submission of data in response to the call for technical and toxicological data on starch sodium octenyl succinate (E 1450) for uses as a food additive in foods for all population groups including infants below 16 weeks of age. January 2019.
HiPP, 2019. Submission of data in response to the call for technical and toxicological data on starch sodium octenyl succinate (E 1450) for uses as a food additive in foods for all population groups including infants below 16 weeks of age. January 2019.
United Pharmaceuticals, 2019. Submission of data in response to the call for technical and toxicological data on starch sodium octenyl succinate (E 1450) for uses as a food additive in foods for all population groups including infants below 16 weeks of age. January 2019.
SNE (Specialised Nutrition Europe), 2020. Final report of Borschel, MW 2001. Growth of infants fed an elemental medical food ‐ A masked, randomized, parallel growth study of healthy term infants fed an elemental medical food or a protein hydrolysate formula. Submitted on January 2020.
SNE (Specialised Nutrition Europe), 2020. Final report of Berseth, 2006. The effects on growth and development of an elemental formula fed to term infants. Submitted on January 2020.
SNE (Specialised Nutrition Europe), 2020. Final report of Scalabrin, 2006. Double‐blind, placebo‐controlled trial of an amino acid‐based formula versus Neocate® in children with cow's milk allergy. Submitted on January 2020.
SNE (Specialised Nutrition Europe), 2020. Final report of Scalabrin, 2008. The effects on growth and tolerance of healthy, term infants fed hydrolyzed formulas with Lactobacillus rhamnosus GG. Submitted on January 2020.
Abbreviations
- ADI
acceptable daily intake
- AEs
Adverse events
- ALAB
alpha‐lactalbumin
- ANS Panel
EFSA Panel on Food Additives and Nutrient Sources added to Food
- AOAC
Association of Official Analytical Chemists
- As
Arsenic
- BMD
benchmark dose
- BMDL
lower confidence limit of the benchmark dose
- bw
body weight
- CAS
Chemical Abstract Service
- Cd
Cadmium
- CF
control formula
- CFU
Colony forming unit
- CHO
Carbohydrates
- CONTAM Panel
EFSA Panel on Contaminants in the Food Chain
- DG Santé
Directorate General for Health and Food safety
- E 1450
SSOS (starch sodium octenyl succinate)
- EF
Experimental formula
- e(E)HF
extensively hydrolysed formula
- EMA
European Medicines Agency
- EU
European Union
- FAF Panel
EFSA Panel on Food Additives and Flavourings
- FAO/WHO
Food and Drug Organisation/World Health Organisation
- FC
Food category
- FDA
Food and drug administration
- FSMP
Food for special medical purposes
- GLP
Good laboratory practice
- HBGVs
Health based guidance values
- HC
head circumference
- Hg
Mercury
- ICH
International Conference on Harmonisation
- ICP‐MS
inductively coupled plasma mass spectrometry
- JECFA
Joint FAO/WHO Expert Committee on Food Additives
- LC
long chain
- LOQ
Limit of quantification
- LP
Low protein
- Mintel GNPD
Mintel's Global New Products Database
- MOE
margin of exposure
- MOS
margin of safety
- MPL
maximum permitted levels
- MRSC
mean rank stool consistency
- NICU
Neonatal intensive care unit
- NOAEL
no‐observed‐adverse‐effect level
- OHAT
Office of Health Assessment and Translation
- OSA
octenyl succinic acid
- Pb
Lead
- p(P)HF
partially hydrolysed formula
- PND
postnatal day
- PUFA
Polyunsaturated fatty acids
- RoB
Risk of bias
- SC
Scientific Committee of EFSA
- SCFA
short‐chain fatty acids
- SNE
Specialised Nutrition Europe
- SSOS
starch sodium octenyl succinate
- TAMC
total anaerobic microbial count
- TF
test formula
- TWI
total weekly intake
- TYMC
total combined yeast and mould count
- UF
Uncertainty factor
- WHO
World Health Organization
Appendix A – Summary of the reported use levels (mg/kg or mg/L as appropriate) of food additive (E 1450) provided by industry
1.
| Food category number | Food category name | E‐number | MPL | Number of samples | Mean of typical usage levels | Minimum of typical usage levels | Maximum of typical usage levels | Maximum usage level | Comments, restrictions | Provided by |
|---|---|---|---|---|---|---|---|---|---|---|
| 13.1.1 | Infant formulae as defined by Commission Directive Commission Delegated Regulation (EU) 2016/127 | E 1450 | 1,100 (as carry‐over) | 12 | ■■■■■ | ■■■■■ | ■■■■■ | – | ■■■■■ | |
| 13.1.5.1 | Dietary foods for infants for special medical purposes and special formulae for infants | E 1450 with thickeners | 20,000 | 1 | ■■■■■ | ■■■■■ | ■■■■■ | – | ■■■■■ | |
| 13.1.5.1 | Dietary foods for infants for special medical purposes and special formulae for infants | E 1450 | 20,000 | 2 | ■■■■■ | ■■■■■ | ■■■■■ | – | ■■■■■ | |
| 13.1.5.1 | Dietary foods for infants for special medical purposes and special formulae for infants | E 1450 with thickeners | 20,000 | 2 | ■■■■■ | ■■■■■ | ■■■■■ | – | ■■■■■ | |
| 13.1.5.1 | Dietary foods for infants for special medical purposes and special formulae for infants | E 1450 | 20,000 | 5 | ■■■■■ | ■■■■■ | ■■■■■ | – | ■■■■■ | |
| 13.1.5.1 | Dietary foods for infants for special medical purposes and special formulae for infants | E 1450 | 20,000 | 1 | ■■■■■ | ■■■■■ | ■■■■■ | – | ■■■■■ |
Appendix B – Data requested in the call for data (Call for technical and toxicological data on starch sodium octenyl succinate (E 1450) for uses as a food additive in foods for all population groups including infants below 16 weeks of age.22
1.
| Kind of data | Data requested in the call for data | Responses from interested business operators | Comment |
|---|---|---|---|
| A. Information regarding the follow‐up of the conclusions and the recommendations of the EFSA ANS Panel opinion on the safety starch sodium octenyl succinate (E 1450) as a food additive | |||
| 1.Technical data | Analytical data on current levels of current levels of lead, mercury and arsenic in commercial samples of the food additive | No analytical data and no proposal for specifications for use in food for other population groups than infants below the age of 16 weeks were provided | No analytical data and no proposal for specifications for use in food for other population groups than infants below the age of 16 weeks were provided as requested in part A.1 of the call for data (clarification letter by Starch Europe dated 29 October 2019) |
| The lowest technologically achievable level for lead, mercury, cadmium and arsenic in order to adequately define their maximum limits in the specifications | |||
| The lowest technologically achievable level for sulfur dioxide | |||
| The lowest technologically achievable level for octenylsuccinic acid residue | No analytical data and no proposal for specifications for use in food for other population groups than infants below the age of 16 weeks were provided | Specifications unchanged on the basis of the received data, see Section B.1 | |
| Because of both the botanical origin and the polysaccharidic nature of starch sodium octenyl succinate (E 1450), it can be a substrate of microbiological contamination. Data should be provided demonstrating the absence of Salmonella spp. and Escherichia coli and on the lowest total aerobic microbial count (TAMC) and total combined yeast and mould count (TYMC) that can be reached | No analytical data and no proposal for specifications for use in food for other population groups than infants below the age of 16 weeks were provided | No analytical data and no proposal for specifications for use in food for other population groups than infants below the age of 16 weeks were provided as requested in part A.1 of the call for data (clarification letter by Starch Europe dated 29 October 2019) | |
| 2. Toxicological data | According to the conclusions and recommendations in the Scientific opinion on the re‐evaluation of starch sodium octenyl succinate (E 1450) as a food additive by the EFSA ANS Panel published in 2017, the generation of additional data to assess the potential health effects of starch sodium octenyl succinate (E 1450) when used as a food additive in ‘dietary foods for infants for special medical purposes and special formulae for infants’ (Food category 13.1.5.1) and in ‘dietary foods for babies and young children for special medical purposes as defined in Directive 1999/21/EC’ (Food category 13.1.5.2) was recommended. These requirements will be addressed as outlined in section B.2 | No data submitted | When extrapolating the conclusion for the uses in FC 13.1.5.1 for infants below 16 weeks of age to the FCs 13.1.5.1 and 13.1.5.2 in food for infants above 16 weeks of age and young children, the Panel considered that there is no safety concern also for these uses within the range reported in the clinical studies |
| 3. Literature search | Literature searches should be conducted relevant for the safety evaluation of starch sodium octenyl succinate (E 1450) for all uses in foods for all population groups from 02/05/2017 up to the date of the data submission, as described in the Guidance for submission for food additive evaluations (see its section 5.3) | Received | Assessed, further follow‐up |
| B. Information required for the risk assessment of starch sodium octenyl succinate (E 1450) for uses in foods for infants below 16 weeks of age | |||
| 1. Technical data | Information on the levels of use of starch sodium octenyl succinate (E 1450) in special formulae for infants of that age under special medical conditions (FC 13.1.5.1) and in infant formulae (FC 13.1.1) when added as food additive in nutrients used in these formulae | Received | Assessed, no further follow‐up |
| Information on the fate and the reaction products of starch sodium octenyl succinate (E 1450) in special formulae for infants of that age under special medical conditions (FC 13.1.5.1) and in infant formulae (FC 13.1.1) when added as food additive in nutrients used in these formulae | Received | Assessed, no further follow‐up | |
|
Information on particular specification requirements for identity and purity of starch sodium octenyl succinate (E 1450) (e.g. with respect content in lead and other heavy metals, sulfur oxide and octenylsuccinic acid) when used in special formulae for infants of that age under special medical conditions (FC 13.1.5.1) and when added in infant formulae (FC 13.1.1) as food additive in nutrients used in these formulae. Analytical data on toxic elements in the final special formulae for infants below 16 weeks of age need to be provided when no legal limit. Analytical data on toxic elements in the final special formulae for infants below 16 weeks of age need to be provided when no legal limit In addition, data should be provided demonstrating the absence of Cronobacter (Enterobacter) sakazakii in the food additive |
Received | Assessed, no further follow‐up | |
| Toxicological data | The full report of the randomised, multicentre, double‐blind, good clinical practice (GCP)‐compliant trial (Borschel and Kajzer, 2011) | Received | Assessed, no further follow‐up |
| The full report of the repeated dose study in neonatal piglets (Mahadevan et al., 2014) | Received | Assessed, no further follow‐up | |
| Updated post‐marketing surveillance report since 2016 on undesired and adverse reactions (e.g. flatulence, gastrointestinal discomfort, changes of stool frequencies and consistency and diarrhoea), indicating the ages and other relevant data of the exposed infants and young children and the use level of starch sodium octenyl succinate (E 1450) in the marketed products, where the FSMPs are already in use | Received | Assessed, no further follow‐up | |
| Published and unpublished case reports (e.g. available nutrivigilance data) on undesired and adverse effects, including e.g. flatulence, gastrointestinal discomfort, changes of stool frequencies and consistency and diarrhoea, associated with the oral administration of starch sodium octenyl succinate in any form to infants and young children | Received | Assessed, no further follow‐up | |
| 3. Literature searches | Literature searches should be conducted relevant for the safety evaluation of starch sodium octenyl succinate (E 1450) when used in foods for infants below 16 weeks of age up to the date of the data submission, as described in the Guidance for submission for food additive evaluations (its Section 5.3) | Received | Assessed, no further follow‐up |
Appendix C – Risk of bias/Internal validity for Experimental Animal Studies (modified from NTP‐OHAT, 2015, 2019)
1.
Decision rules
The ratings of the key and non‐key questions (++, +, –, −−) will be integrated to classify the studies in tiers from 1 to 3 corresponding to decreasing levels of internal validity.
Tier 1:
All the key questions are scored +/++
AND
No more than one non‐key question is scored –
AND
No non‐key question is scored −−
Tier 2:
All the other combinations not falling under tier 1 or 3
Tier 3:
Any question is scored −−
OR
More than one key question is scored –
Outcome of the RoB assessment
Assessed study: A 3‐week dietary toxicity study of octenyl succinic anhydride (OSA) in farm piglet. Abbott Nutrition, study 126‐658; Study report 21 November 2012, Study completion 26 November 2012 (Documentation provided to EFSA n. 3).
Outcome of the assessment:
| Question | Domain of bias | Rating (++, +, −, −−) |
Rating of the study Reviewer 1 |
Rating of the study Reviewer 2 |
|---|---|---|---|---|
|
1. Was the administered dose or exposure level adequately randomised? Key question |
Selection | ++ If the method is described and it is adequate |
++ The method is described, and it is adequate |
++ Standard, by weight, measured value randomisation procedure |
| + If the authors only indicate that randomisation was done but do not describe the method | ||||
| – No mentioning of randomisation | ||||
| −− Direct evidence of no randomisation | ||||
| 2. Was allocation to the study groups adequately concealed? | Selection | ++ Properly concealed and described how concealment was performed |
++ Properly concealed and described how concealment was performed |
++ Animal number to be used in the Provantis data collection system assigned to each animal |
| + Mentioning that concealment was performed; + is also appropriate if non‐concealment does not influence the outcome | ||||
| − If non‐concealment does influence the outcome (measurements with a subjective part (e.g. preparation of fat pads, observation of behaviour)) | ||||
| −− If non‐concealment does influence the outcome to a very important part (subjective measurements) | ||||
|
3. Were experimental conditions identical across study groups? Key question |
Performance | ++ Experimental conditions described and identical across study groups (feeding, water supply, bedding, day/night cycle; temperature; humidity) |
++ Experimental conditions described and identical across study groups (feeding, water supply, bedding, day/night cycle; temperature; humidity) |
++ According to the Study Director minor deviations did not impact the study results |
| + Incomplete description of experimental conditions; + is also appropriate if lack of information does not influence the outcome | ||||
| − If lack of information does influence the outcome | ||||
| −− If factors clearly indicate that treatment conditions were different does influence the outcome to a very important part | ||||
|
4. Was the research personnel blinded to the study group? Key question |
Performance | ++ If there is direct evidence that the research personnel did not know what group animals were allocated to, and it is unlikely that they could have broken the blinding of allocation |
+ Not reported and lack of adequate allocation concealment would not appreciably affect the allocation of animals to different study groups (e.g. methods used which do not have a subjective component) |
+ Not reported, unless answer to question 2 implies blinding. Specifically, the blinding of the pathologist is not mentioned. However, no macroscopic or microscopic treatment‐related findings were reported. Therefore, this should not be a major issue |
| + If not reported and lack of adequate allocation concealment would not appreciably affect the allocation of animals to different study groups (e.g. methods used which do not have a subjective component) | ||||
| − If not reported and lack of adequate allocation concealment would appreciably affect the allocation of animals to different study groups (e.g. methods used which have a subjective component) | ||||
| −− If there is direct evidence that it was possible for the research personnel to know what group animals were allocated to, or it is likely that they could have broken the blinding of allocation | ||||
| 5. Were outcome data complete without attrition or exclusion from analysis? | Attrition/exclusion |
++ There is direct evidence that loss of animals was adequately addressed, and reasons were documented when animals were removed from a study OR Missing data have been imputed using appropriate methods (ensuring that characteristics of animals are not significantly different from animals retained in the analysis) |
++ Direct evidence that loss of animals was adequately addressed, and reasons were documented when animals were removed from a study |
++ |
|
+ There is indirect evidence that loss of animals was adequately addressed, and reasons were documented when animals were removed from a study OR It is deemed that the proportion lost would not appreciably bias results. This would include reports of no statistical differences in characteristics of animals removed from the study from those remaining in the study OR There is insufficient information provided about loss of animals (record ‘NR’ as basis for answer), but it is considered that this does not have an impact on the validity of the study | ||||
|
− There is indirect evidence that loss of animals was unacceptably large and not adequately addressed (e.g. if unexplained loss is equal or more than 25%) OR There is insufficient information provided about loss of animals (record ‘NR’ as basis for answer) and it is suspected that this would have an impact on the validity of the study Note: Unexplained inconsistencies between materials and methods and results sections (e.g. inconsistencies in the numbers of animals in different groups) could be an example of indirect evidence | ||||
| −− There is direct evidence that loss of animals was unacceptably large and not adequately addressed | ||||
|
6. Can we be confident in the exposure characterization? Key question |
Detection | ++ There is direct evidence that the substance was sufficiently described and consistently administered (e.g. with the same method and timeframe) across treatment groups. |
++ There is direct evidence that the substance was sufficiently described and consistently administered (e.g. with the same method and timeframe) across treatment groups. |
++ Analytical report in Annex B |
|
+ There is indirect evidence that the substance was sufficiently described and consistently administered (i.e. with the same method and time frame) across treatment groups. OR There is insufficient information provided about description and administration of the substance (record ‘NR’ as basis for answer), but it is considered that this does not have an impact on the validity of the study. | ||||
|
− There is indirect evidence that the substance was not sufficiently described and was not consistently administered (e.g. with the same method and timeframes) across groups. OR There is insufficient information provided about description and administration of the substance (record ‘NR’ as basis for answer) and it is suspected that this has an impact on the validity of the study. | ||||
| −− There is direct evidence that the substance was not sufficiently described and/or was not consistently administered (e.g. with the same method and time frames) across groups. | ||||
| 7. Can we be confident in the outcome assessment? Key question | Detection | |||
|
Element 1 Was the outcome assessed at the same length of time (i.e. day and/or time of day) after initial exposure in all study groups? (remember to take into consideration the endpoints assignments) Element 2 Was a reliable and sensitive animal model used for investigating the test compound and selected end points? Element 3 Was the number of animals per dose group appropriate? Element 4 Was the number of animals per sex in each cage appropriate for the study type and animal model? Element 5 Was the timing and duration of administration of the test compound appropriate? Element 6 Were reliable and sensitive test methods used for investigating the selected end points? Element 7 Were the measurements collected at suitable time points in order to generate sensitive, valid and reliable data? |
++ There is direct evidence + It is deemed that deviation would not appreciably bias results. OR There is insufficient information provided, but it is considered that this does not have an impact on the validity of the study. − There is insufficient information provided (record ‘NR’ as basis for answer) and it is suspected that this has an impact on the validity of the study. −− There is direct evidence for a deviation Decision rules for the final assessment of 7 if element 4 −− results in −− if element 4 − results in − if more than 3 elements ++ and the remainder + or − (exception element 4) results in ++, if less than 3 elements ++ and the remainder + or − (exception element 4) results in + if more than 3 elements + and the remainder −− (exception element 4) results in + if less than 3 elements + and the remainder + or − (exception element 4) results in − if more than 3 elements − (excluding element 4) results in − if more than 3 elements − (including element 4) results in −− if less than 3 elements (excluding element 4) −− results in − if less than 3 elements (including element 3) −− results in −− if more than 3 elements −− results in −− |
++ All elements: ++ Animal number was low but acceptable |
++ All elements: ++ Animal number was low but acceptable |
|
| 8. Were all outcomes measured according to the methodology section reported? | Selective reporting |
++ There is direct evidence that all of the study's measured outcomes (apical and intermediate) outlined in the protocol, methods, abstract, and/or introduction that are relevant for the evaluation have been reported This would include outcomes reported with sufficient detail to be included in meta‐analysis or fully tabulated during data extraction and analyses had been planned in advance |
++ | ++ |
|
+ There is indirect evidence that all of the study's measured outcomes (apical and intermediate) outlined in the protocol, methods, abstract and/or introduction that are relevant for the evaluation have been reported. This would include outcomes reported with insufficient detail such as only reporting that results were statistically significant (or not) OR Analyses that had not been planned in advance (i.e. retrospective unplanned subgroup analyses) are clearly indicated as such and it is deemed that the unplanned analyses were appropriate and selective reporting would not appreciably bias results (e.g. appropriate analyses of an unexpected effect) OR There is insufficient information provided about selective outcome reporting (record ‘NR’ as basis for answer) but it is considered that this does not have an impact on the validity of the study. | ||||
|
− There is indirect evidence that all of the study's measured outcomes (apical and intermediate) outlined in the protocol, methods, abstract and/or introduction that are relevant for the evaluation have not been reported OR There is indirect evidence that unplanned analyses were included that may appreciably bias results OR There is insufficient information provided about selective outcome reporting (record ‘NR’ as basis for answer) and it is suspected that this has an impact on the validity of the study Note: Unexplained inconsistencies between materials and methods and results/abstract or summary sections (e.g. inconsistencies in the numbers of animals in different groups) could be an example of indirect evidence | ||||
|
−− There is direct evidence that not all of the study's measured outcomes (apical and intermediate) outlined in the protocol, methods, abstract and/or introduction that are relevant for the evaluation have not been reported In addition to not reporting outcomes, this would include reporting outcomes based on composite score without individual outcome components or outcomes reported using measurements, analysis methods or subsets of the data that were not prespecified or reporting outcomes not prespecified, or that unplanned analyses were included that would appreciably bias results |
||||
| 9. Were statistical methods appropriate? | Other sources of bias | ++ There is direct evidence that the statistical methods seem appropriate and were clearly reported (adequate treatment of multiple testing) |
++ There is direct evidence that all of the study's measured outcomes (apical and intermediate) outlined in the protocol, methods, abstract and/or introduction that are relevant for the evaluation have been reported This would include outcomes reported with sufficient detail to be included in meta‐analysis or fully tabulated during data extraction and analyses had been planned in advance |
++ Methods are clearly reported and seem to be acceptable |
|
+ Statistical methods were not clearly reported but it may be inferred from other information that they were appropriate OR There is insufficient information provided about statistical methods (record ‘NR’ as basis for answer), but it is considered that this does not have an impact on the validity of the study. | ||||
|
− Statistical methods were not clearly reported but it may be inferred from other information that they were not appropriate OR There is insufficient information provided about statistical methods (record ‘NR’ as basis for answer) and it is suspected that this has an impact on the validity of the study | ||||
| −− There is direct evidence that the statistical methods applied were inappropriate |
Summary of the RoB
| ++ | + | – | – | |
|---|---|---|---|---|
| Key question (No) |
1, 3, 6, 7 1, 3, 6, 7 |
4, 8, 9 4, 8, 9 |
– | – |
| Non‐key question (No) |
2 2 |
5 5 |
– | – |
| Outcome/Tier |
1 1 |
|||
Appendix D – Risk of bias/Internal validity for the clinical studies (modified from to NTP‐OHAT, 2015, 2019)
1.
Decision rules
The ratings of the key and non‐key questions (++, +, −, −−) will be integrated to classify the studies in tiers from 1 to 3 corresponding to decreasing levels of internal validity.
Tier 1:
All the key questions are scored +/++
AND
No more than one non‐key question is scored –
AND
No non‐key question is scored −
Tier 2:
All the other combinations not falling under tier 1 or 3
Tier 3:
Any question is scored – −
OR
More than one key question is scored –
Elements considered in the assessment
| Question | Rating | Explanation for expert judgment |
|---|---|---|
|
1. Was the administered dose or exposure level adequately randomised? Key question |
++ | There is direct evidence that subjects (or clusters) were allocated to any study group including controls using a method with a random component. Acceptable methods of randomisation include referring to a random number table, using a computer random number generator, coin tossing, shuffling cards or envelopes, throwing dice, or drawing of lots (Higgins and Green, 2011). Restricted randomisation (e.g. blocked randomisation) to ensure particular allocation ratios will be considered low risk of bias. Similarly, stratified randomisation and minimisation approaches that attempt to minimise imbalance between groups on significant prognostic factors (e.g. body weight) will be considered acceptable |
| + |
There is indirect evidence that subjects (or clusters) were allocated to study groups using a method with a random component (i.e. authors state that allocation was random, without description of the method used) OR It is deemed that allocation without a clearly random component during the study would not appreciably bias results. For example, approaches such as biased coin or urn randomisation, replacement randomisation, mixed randomisation and maximal randomisation may require consultation with a statistician to determine risk‐of‐bias rating (Higgins and Green, 2011) |
|
| NR | There is insufficient information provided about how subjects (or clusters) were allocated to study groups | |
| − |
There is indirect evidence that subjects (or clusters) were allocated to study groups using a method with a non‐random component NOTE: Non‐random allocation methods may be systematic but have the potential to allow participants or researchers to anticipate the allocation to study groups. Such ‘quasi‐random’ methods include alternation, assignment based on date of birth, case record number, or date of presentation to study |
|
| −− | There is direct evidence that subjects (or clusters) were allocated to study groups using a non‐random method including judgement of the clinician, preference of the participant, the results of a laboratory test or a series of tests, or availability of the intervention (Higgins and Green, 2011) | |
| 2. Was the allocation to study groups adequately concealed? | ++ | There is direct evidence that at the time of recruitment the research personnel and subjects did not know what study group subjects were allocated to, and it is unlikely that they could have broken the blinding of allocation until after assignment was complete and irrevocable. Acceptable methods used to ensure allocation concealment include central allocation (including telephone, web‐based and pharmacy‐controlled randomisation); sequentially numbered drug containers of identical appearance; sequentially numbered, opaque, sealed envelopes; or equivalent methods |
| + |
There is indirect evidence that the research personnel and subjects did not know what study group subjects were allocated to and it is unlikely that they could have broken the blinding of allocation until after recruitment was complete and irrevocable OR It is deemed that lack of adequate allocation concealment would not appreciably bias results (e.g. some crossover designs) |
|
| NR | There is insufficient information provided about allocation to study groups | |
| − |
There is indirect evidence that at the time of recruitment it was possible for the research personnel and subjects to know what study group subjects were allocated to, or it is likely that they could have broken the blinding of allocation before assignment was complete and irrevocable NOTE: Inadequate methods include using an open random allocation schedule (e.g. a list of random numbers); assignment envelopes used without appropriate safeguards (e.g. if envelopes were unsealed or non‐opaque or not sequentially numbered); alternation or rotation; date of birth; case record number; or any other explicitly unconcealed procedure. For example, if the use of assignment envelopes is described, but it remains unclear whether envelopes were sequentially numbered, opaque and sealed |
|
| −− | There is direct evidence that at the time of recruitment it was possible for the research personnel and subjects to know what study group subjects were allocated to, or it is likely that they could have broken the blinding of allocation before recruitment was complete | |
| 3. Were the research personnel and human subjects blinded to the study group during the study? | ++ | There is direct evidence that the subjects and research personnel were adequately blinded to study group, AND it is unlikely that they could have broken the blinding during the study. Methods used to ensure blinding include central allocation; sequentially numbered drug containers of identical appearance; sequentially numbered, opaque, sealed envelopes; or equivalent methods |
| + |
There is indirect evidence that the subjects and research personnel were adequately blinded to study group, AND it is unlikely that they could have broken the blinding during the study OR There is direct evidence for no blinding during the study (including where it was not possible to implement) AND it is deemed that no blinding would appreciably bias results BUT bias minimising measures have been adequately implemented OR It is deemed that lack of adequate blinding or no blinding during the study would not appreciably bias results (e.g. controls unlikely to behave differently for factors other than sodium intake) (e.g. cross‐over) |
|
| NR | There is insufficient information provided about blinding to study group during the study (including possible breaking and minimising measures) | |
| − | There is indirect evidence that it was possible for research personnel or subjects to infer the study group AND it is deemed that lack of adequate blinding or no blinding during the study would appreciably bias results (e.g. no comparable treatment of controls, including not comparable exposure to factors other than the interventions of interest; differential behaviour) AND no bias minimising measures have been adequately implemented | |
| −− | There is direct evidence for lack of adequate blinding of the study group (including no blinding or incomplete blinding) of research personnel and subjects AND it is deemed that lack of adequate blinding or no blinding during the study would appreciably bias results (e.g. no comparable treatment of controls, including not comparable exposure to factors other than the interventions of interest, differential behaviour) AND no bias minimising measures have been adequately implemented | |
|
4. Were outcome data complete without attrition or exclusion from analysis? Key question |
++ |
There is direct evidence that there was no loss of subjects during the study and outcome data were complete OR Loss of subjects (i.e. incomplete outcome data) was adequately addressed and reasons were documented when human subjects were removed from a study or analyses. Review authors should be confident that the participants included in the analysis are exactly those who were randomised into the trial. Acceptable handling of subject attrition includes: very few missing outcome data (e.g. less than 10% in each group (Genaidy et al., 2007)) AND reasons for missing subjects unlikely to be related to outcome (for survival data, censoring unlikely to be introducing bias) AND missing outcome data balanced in numbers across study groups, with similar reasons for missing data across groups (i.e. unlikely to be related to exposure) OR Analyses (such as intention‐to‐treat analysis) in which missing data have been imputed using appropriate methods (ensuring that the characteristics of subjects lost to follow up or with unavailable records are described in identical way and are not significantly different from those of the study participants) NOTE: Participants randomised but subsequently found not to be eligible need not always be considered as having missing outcome data) (Higgins and Green, 2011) |
| + |
There is indirect evidence that loss of subjects (i.e. incomplete outcome data) was adequately addressed and reasons were documented when human subjects were removed from a study OR It is deemed that the proportion lost to follow‐up would not appreciably bias results (e.g. less than 20% in each group in parallel studies (Genaidy et al., 2007)). This would include reports of no statistical differences in characteristics of subjects lost to follow up or with unavailable records from those of the study participants. Generally, the higher the ratio of participants with missing data to participants with events, the greater potential there is for bias. For studies with a long duration of follow‐up, some withdrawals for such reasons are inevitable NB: For crossover designs, this may be less of an issue |
|
| NR | There is insufficient information provided about numbers of subjects lost to follow‐up | |
| − | There is indirect evidence that loss of subjects (i.e. incomplete outcome data) was unacceptably large (e.g. greater than 20% in each group in parallel studies (Genaidy et al., 2007)) and not adequately addressed | |
| −− | There is direct evidence that loss of subjects (i.e. incomplete outcome data) was unacceptably large and not adequately addressed (e.g. greater than 20% in each group in parallel studies (Genaidy et al., 2007)). Unacceptable handling of subject attrition includes: reason for missing outcome data likely to be related to true outcome, with either imbalance in numbers or reasons for missing data across study groups (i.e. likely to be related to the exposure); or potentially inappropriate application of imputation | |
|
5. Can we be confident in the exposure characterisation? Key question |
++ |
There is direct evidence that the exposure (including compliance with the treatment, if applicable) was independently characterised AND that exposure was consistently administered (i.e. with the same method and time frame) across treatment groups |
| + |
There is indirect evidence that the exposure (including compliance with the treatment, if applicable) was independently characterised AND there is indirect evidence that exposure was consistently administered (i.e. with the same method and time‐frame) across treatment groups |
|
| NR | There is insufficient information provided to judge the exposure characterisation | |
| − |
There is indirect evidence that the exposure (including compliance with the treatment, if applicable) was assessed using poorly validated methods (e.g. FFQs, spot urine etc.) OR There is indirect evidence that the exposure assessment was probably biased |
|
| −− |
There is direct evidence that the exposure (including compliance with the treatment, if applicable) was assessed using poorly validated methods (e.g. FFQs, spot urine etc.) OR There is direct evidence that the exposure assessment was biased |
|
|
6. Can we be confident in the outcome assessment? Key question |
++ |
There is direct evidence that the outcome was assessed using well‐established methods (e.g. for office BP: according to a clearly described methodology, including e.g. repeated measurements per visit, trained technician, resting period before each measurement) AND There is direct evidence that the outcome assessors were adequately blinded to the study group, and it is unlikely that they could have broken the blinding prior to reporting outcomes |
| + |
There is indirect evidence that the outcome was assessed using acceptable methods (i.e. deemed valid and reliable but not the gold standard) OR it is deemed that the outcome assessment methods used would not appreciably bias results AND There is indirect evidence that the outcome assessors were adequately blinded to the study group, and it is unlikely that they could have broken the blinding before reporting outcomes OR it is deemed that lack of adequate blinding of outcome assessors would not appreciably bias results |
|
| NR | There is insufficient information provided about blinding of outcome assessors or the method of measurement | |
| − |
There is indirect evidence that the outcome assessment method is an unacceptable method OR There is indirect evidence that it was possible for outcome assessors to infer the study group before reporting outcomes |
|
| −− |
There is direct evidence that the outcome assessment method is an unacceptable method OR There is direct evidence for lack of adequate blinding of outcome assessors (including study subjects if home BP is the outcome), including no blinding or incomplete blinding |
|
| 7. Were all measured outcomes reported? | ++ | There is direct evidence that all of the study's measured outcomes (primary and secondary) outlined in the protocol, methods, abstract and/or introduction (that are relevant for the evaluation) have been reported |
| + |
There is indirect evidence that all of the study's measured outcomes (primary and secondary) outlined in the methods, abstract, and/or introduction (that are relevant for the evaluation) have been reported OR Analyses that had not been planned in advance (i.e. retrospective unplanned subgroup analyses) are clearly indicated as such and it is deemed that the unplanned analyses were appropriate and selective reporting would not appreciably bias results (e.g. appropriate analyses of an unexpected effect). This would include outcomes reported with insufficient detail such as only reporting that results were statistically significant (or not) |
|
| NR | There is insufficient information provided about selective outcome reporting | |
| − |
There is indirect evidence that all of the study's measured outcomes (primary and secondary) outlined in the methods, abstract and/or introduction (that are relevant for the evaluation) have not been reported OR There is indirect evidence that unplanned analyses were included that may appreciably bias result |
|
| −− | There is direct evidence that all of the study's measured outcomes (primary and secondary) outlined in the methods, abstract and/or introduction (that are relevant for the evaluation) have not been reported. In addition to not reporting outcomes, this would include reporting outcomes based on composite score without individual outcome components or outcomes reported using measurements, analysis methods or subsets of the data (e.g. subscales) that were not prespecified or reporting outcomes not prespecified, or that unplanned analyses were included that would appreciably bias results | |
|
8. Were there no other potential threats to internal validity? NOTE: Baseline characteristics should be appraised only if Q1 (randomisation) was rated with ++/+ and Q2 (allocation concealment) was rated with ++/+/NR |
++ |
There is evidence that variables, other than the exposure and outcome, did not differ between groups during the course of the intervention in a way that could bias results AND, in case randomisation is rated ‘probably low’/’definitely low’ RoB and allocation concealment is rated ‘probably low’/‘definitely low’ RoB or ‘not reported’: There is no evidence of differences in baseline characteristics OR There is no information on both BUT no concern |
| + |
1. There is evidence that variables, other than the exposure and outcome, differed between groups during the course of the intervention AND it is deemed that these differences would not appreciably bias results (no concern or adequately addressed by analysis) AND, in case randomisation is rated ‘probably low’/’definitely low’ RoB and allocation concealment is rated ‘probably low’/’definitely low’ RoB or ‘not reported’: There is evidence that reported variables differed between groups at baseline AND It is deemed that these differences would not appreciably bias results (no concern or adequately addressed by analysis) OR 2. There is evidence that variables, other than the exposure and outcome, did not differ between groups during the course of the intervention in a way that could bias results AND, in case randomisation is rated ‘probably low’/’definitely low’ RoB and allocation concealment is rated ‘probably low’/’definitely low’ RoB or ‘not reported’: There is evidence that reported variables differed between groups at baseline AND It is deemed that these differences would not appreciably bias results (no concern or adequately addressed by analysis) OR 3. There is evidence that variables, other than the exposure and outcome, differed between groups during the course of the intervention. AND It is deemed that these differences would not appreciably bias results (no concern or adequately addressed by analysis) AND, in case randomisation is rated ‘probably low’/’definitely low’ RoB and allocation concealment is rated ‘probably low’/’definitely low’ RoB or ‘not reported’: There is no evidence of differences in baseline characteristics OR There is no information BUT no concern |
|
| − |
There is no information on baseline characteristics AND/OR there is no information about differences between groups during the course of the intervention AND There is concern |
|
| −− |
There is evidence that variables, other than the exposure and outcome, differed between groups during the course of the intervention AND It is deemed that these differences appreciably biased results (there is concern (e.g. not adequately addressed by analysis)) OR, in case randomisation is rated ‘probably low’/’definitely low’ RoB and allocation concealment is rated ‘probably low’/’definitely low’ RoB or ‘not reported’: There is evidence that reported variables differed between groups at baseline AND It is deemed that these differences appreciably biased results (there is concern (e.g. not adequately addressed by analysis)) |
Appendix E – Estimated exposure levels in the clinical trial
1.
| Study (authors, year) | Preparation | Consumed amount (mg/kg bw per day) | |
|---|---|---|---|
| Mean or range* | High level | ||
| Borschel and Kajzer (2011) (documentation provided to EFSA n. 3) | EF‐1 | 351.2 | |
| EF‐2 | 341.6 | ||
| Borschel et al. (2013) | 2,142.5–2,724.9 | ||
| Ahrens et al. (2018) | Control | ■■■■■ | |
| LPpHF | ■■■■■ | ||
| LPpHF + Syn | ■■■■■ | ||
| LPeHF + Syn | ■■■■■ | ||
| Burks et al. (2008) | Control | ■■■■■ | |
| Experimental | ■■■■■ | ||
| Fleddermann et al. (2014) | Control | ■■■■■ | ■■■■■ |
| Experimental | ■■■■■ | ■■■■■ | |
| Scalabrin et al. (2009)** | Preparation 1 | ■■■■■ | ■■■■■ |
| Preparation 2 | ■■■■■ | ■■■■■ | |
A range is given in cases where consumption could be calculated for several periods in the study otherwise means are calculated.
No consumption data were given in this study and the EFSA default consumption data were used and mean and high level exposure was calculated.
Suggested citation: EFSA FAF Panel (EFSA Panel on Food Additives and Flavourings) , Younes M, Aquilina G, Castle L, Engel K‐H, Fowler P, Frutos Fernandez MJ, Fürst P, Gürtler R, Husøy T, Manco M, Mennes W, Moldeus P, Passamonti S, Shah R, Waalkens‐Berendsen I, Wölfle D, Wright M, Dusemund B, Mortensen A, Turck D, Barmaz S, Rincon AM, Smeraldi C, Tard A, Vianello G and Gundert‐Remy U, 2020. Opinion on the re‐evaluation of starch sodium octenyl succinate (E 1450) as a food additive in foods for infants below 16 weeks of age and the follow‐up of its re‐evaluation as a food additive for uses in foods for all population groups. EFSA Journal 2020;18(8):5874, 60 pp. 10.2903/j.efsa.2020.5874
Requestor: European Commission
Question number: EFSA‐Q‐2018‐00102
Panel members: Maged Younes, Gabriele Aquilina, Laurence Castle, Karl‐Heinz Engel, Paul Fowler, Maria Jose Frutos Fernandez, Peter Fürst, Rainer Gürtler, Ursula Gundert‐Remy, Trine Husøy, Melania Manco, Wim Mennes, Peter Moldeus, Sabina Passamonti, Romina Shah, Ine Waalkens‐Berendsen, Matthew Wright and Detlef Wölfle.
Note: The full opinion will be published in accordance with Article 8 of Regulation (EU) No 257/2010 once the decision on confidentiality will be received from the European Commission.
* This opinion was first adopted by the FAF Panel on 25 September 2019, as reflected in the minutes of the FAF Plenary Meeting of 24‐26 September 2019: http://www.efsa.europa.eu/sites/default/files/event/190924-m.pdf. However, the adopted version was withdrawn prior to publication (see https://www.efsa.europa.eu/sites/default/files/event/191112-m.pdf) and the current version was adopted by the FAF Panel on 30 June 2020.
Adopted: 30 June 2020
Notes
Regulation (EC) No 1333/2008 of the European Parliament and of the Council of 16 December 2008 on food additives (Text with EEA relevance). OJ L 354, 31.12.2008, p. 16–33.
Regulation (EU) No 609/2013 of the European Parliament and of the Council of 12 June 2013 on food intended for infants and young children, food for special medical purposes, and total diet replacement for weight control and repealing Council Directive 92/52/EEC, Commission Directives 96/8/EC, 1999/21/EC, 2006/125/EC and 2006/141/EC, Directive 2009/39/EC of the European Parliament and of the Council and Commission Regulations (EC) No 41/2009 and (EC) No 953/2009. OJ L 181, 29.6.2013, p. 35–56.
Commission Regulation (EU) No 257/2010 of 25 March 2010 setting up a programme for the re‐evaluation of approved food additives in accordance with Regulation (EC) No 1333/2008 of the European Parliament and of the Council on food additives. OJ L 80, 26.3.2010, p. 19–27.
See Section 1.1.3.
Regulation (EC) No 178/2002 of the European Parliament and of the Council of 28 January 2002 laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety. OJ L 31, 1.2.2002, p. 1–24.
Commission Delegated Regulation (EU) 2016/127 supplementing Regulation (EU) No 609/2013 of the European Parliament and of the Council as regards the specific compositional and information requirements for infant formula and follow‐on formula and as regards requirements on information relating to infant and young child feeding. OJ: JOL_2016_025_R_0001, p. 1–29.
The same statement is mentioned in the Commission delegated Regulation of 25 September 2015 that will apply for infant and follow‐on formulae from 22 February 2020, except for formulae manufactured from protein hydrolysates (applicable from 22 February 2021).
Editorial addition.
Call for technical and toxicological data on starch sodium octenyl succinate (E 1450) for uses as a food additive in foods for all population groups including infants below 16 weeks of age. Available from: http://www.efsa.europa.eu/en/consultations/call/180718-4.
Missing Cyprus, Luxembourg and Malta.
Commission Regulation (EU) No 231/2012 of 9 March 2012 laying down specifications for food additives listed in Annexes II and III to Regulation (EC) No 1333/2008 of the European Parliament and of the Council. OJ L 83, 22.3.2012, p. 1–295.
Taken from: https://pubs.rsc.org/image/article/2015/ra/c5ra10849g/c5ra10849g-s3_hi-res.gif. Accessed on 29 August 2019.
Call for technical and toxicological data on starch sodium octenyl succinate (E 1450) for uses as a food additive in foods for all population groups including infants below 16 weeks of age. Available online: www.efsa.europa.eu/en/consultations/call/180718-4
Call for food additives usage level and/or concentration data in food and beverages intended for human consumption. Published: 12 October 2015. Available online: https://www.efsa.europa.eu/en/data/call/151012
http://www.gnpd.com/sinatra/home/URLHASH; accessed on 22/6/2020
Editorial.
Commission Regulation (EU) No 333/2007 of 28 March 2007 laying down the methods of sampling and analysis for the official control of the levels of lead, cadmium, mercury, inorganic tin, 3‐MCPD and benzo(a)pyrene in foodstuffs. OJ L 88, 29.3.2007, p. 29–38.
The hydrolysis product of OSA (Octenyl Succinic Acid) ‐modified starch, from JECFA, 2015).
Name used by JECFA (2015): OSA (Octenyl Succinic Acid)‐modified starch.
Available online: http://www.efsa.europa.eu/en/consultations/call/180718-4) and responses from interested parties
References
- Ahrens B, Hellmuth C, Haiden N, Olbertz D, Hamelmann E, Vusurovic M, Fleddermann M, Roehle R, Knoll A, Koletzko B, Wahn U and Beyer K, 2018. Hydrolyzed formula with reduced protein content supports adequate growth: a randomized controlled noninferiority trial. Journal of Pediatric Gastroenterology and Nutrition, 66, 822–830. 10.1097/MPG.0000000000001853 [DOI] [PubMed] [Google Scholar]
- den Besten G, van Eunen K, Vanema K, Groen AK, Reijngoud DJ and Bakker BM, 2013. The role of short‐chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. Journal of Lipid Research, 54, 2325–2340. 10.1194/jlr.R036012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Besten G, Gerding A, van Dijk TH, Ciapaite J, Bleeker A, van Eunen K, Havinga R, Groen AK, Reijngoud DJ and Bakker BM, 2015a. Protection against the metabolic syndrome by guar gum‐derived short‐chain fatty acids depends on peroxisome proliferator‐activated receptor γ and glucagon‐like peptide‐1. PLoS ONE, 10, e0136364 10.1371/journal.pone.0136364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Besten G, Bleeker A, Gerding A, van Eunen K, Havinga R, van Dijk TH, Oosterveer MH, Jonker JW, Groen AK, Reijngoud DJ and Bakker BM, 2015b. Short‐chain fatty acids protect against high‐fat diet‐induced obesity via a PPARγ‐dependent switch from lipogenesis to fat oxidation. Diabetes, 64, 2398–2408. 10.2337/db14-1213 [DOI] [PubMed] [Google Scholar]
- Borschel MW, Ziegler EE, Wedig RT and Oliver JS, 2013. Growth of healthy term infants fed an extensively hydrolyzed casein‐based or free amino acid‐based infant formula: a randomized, double‐blind, controlled trial. Clinical Pediatrics, 52, 910–917. 10.1177/0009922813492883 [DOI] [PubMed] [Google Scholar]
- Burks W, Jones SM, Berseth CL, Harris C, Sampson HA and Scalabrin DM, 2008. Hypoallergenicity and effects on growth and tolerance of a new amino acid‐based formula with docosahexaenoic acid and arachidonic acid. Journal of Pediatrics, 153, 266–271. 10.1016/j.jpeds.2008.02.043 [DOI] [PubMed] [Google Scholar]
- Canfora EE, Jocken JW and Blaak EE, 2015. Short‐chain fatty acids in control of body weight and insulin sensitivity. National Reviews. Endocrinology, 11, 577–591. 10.1038/nrendo.2015.128 [DOI] [PubMed] [Google Scholar]
- Chong CYL, Bloomfield FH and O'Sullivan JM, 2018. Factors affecting gastrointestinal microbiome development in neonates. Nutrients, 10, 274 10.3390/nu10030274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA (European Food Safety Authority), 2007. Opinion of the Scientific Committee related to uncertainties in dietary exposure assessment. EFSA Journal 2007;5(1):438, 54 pp. 10.2903/j.efsa.2007.438 [DOI] [Google Scholar]
- EFSA ANS Panel (EFSA Panel on Food Additives and Nutrient Sources added to Food), Mortensen A, Aguilar F, Crebelli R, Di Domenico A, Dusemund B, Frutos MJ, Galtier P, Gott D, Gundert‐Remy U, Lambré C, Leblanc J‐C, Lindtner O, Moldeus P, Mosesso P, Parent‐Massin D, Oskarsson A, Stankovic I, Waalkens‐Berendsen I, Wright M, Younes M, Tobback P, Horvath Z, Tasiopoulou S and Woutersen RA, 2017. Scientific Opinion on the re‐evaluation of oxidised starch (E 1404), monostarch phosphate (E 1410), distarch phosphate (E 1412), phosphated distarch phosphate (E 1413), acetylated distarch phosphate (E 1414), acetylated starch (E 1420), acetylated distarch adipate (E 1422), hydroxypropyl starch (E 1440), hydroxypropyl distarch phosphate (E 1442), starch sodium octenyl succinate (E 1450), acetylated oxidised starch (E 1451) and starch aluminium octenyl succinate (E 1452) as food additives. EFSA Journal 2017;15(10):4911, 96 pp. 10.2903/j.efsa.2017.4911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA CONTAM Panel (Panel on Contaminants in the Food Chain), 2009. Scientific opinion on Cadmium in food. EFSA Journal 2009;7(3):980, 139 pp. 10.2903/j.efsa.2009.980 [DOI] [Google Scholar]
- EFSA CONTAM Panel (Panel on Contaminants in the Food Chain), 2010. Scientific opinion on Lead in food. EFSA Journal 2010;8(4):1570, 151 pp. 10.2903/j.efsa.2010.1570 [DOI] [Google Scholar]
- EFSA CONTAM Panel (Panel on Contaminants in the Food Chain), 2012. Scientific Opinion on the risk for public health related to the presence of mercury and methylmercury in food. EFSA Journal 2012;10(12):2985, 15 pp. 10.2903/j.efsa.2012.2985 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2009. Guidance of the Scientific Committee on transparency in the scientific aspects of risk assessment carried out by EFSA. Part 2: general principles. EFSA Journal 2009;7(11):1051, 22 pp. 10.2903/j.efsa.2009.1051 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2012. Guidance on selected default values to be used by the EFSA Scientific Committee, Scientific Panels and Units in the absence of actual measured data. EFSA Journal 2012;10(3):2579, 32 pp. 10.2903/j.efsa.2012.2579 [DOI] [Google Scholar]
- EFSA Scientific Committee , 2017a. Guidance on the risk assessment of substances present in food intended for infants below 16 weeks of age. EFSA Journal 2017;15(5):4849, 58 pp. 10.2903/j.efsa. 2017.4849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2017b. Update: guidance on the use of the benchmark dose approach in risk assessment. EFSA Journal 2017;15(1):4658, 41 pp. 10.2903/j.efsa.2017.4658 [DOI] [Google Scholar]
- EMA (European Medicines Agency), 2009. ICH guideline M3(R2) on non‐clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. EMA/CPMP/ICH/286/1995. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-m-3-r2-non-clinical-safety-studies-conduct-human-clinical-trials-marketing-authorization_en.pdf
- FDA (Food and Drug Administration), 2006. Guidance for Industry Nonclinical Safety Evaluation of Pediatric Drug Products. Available online: https://www.fda.gov/media/119658/download
- Fleddermann M, Demmelmair H, Grote V, Nikolic T, Trisic B and Koletzko B, 2014. Infant formula composition affects energetic efficiency for growth: the BeMIM study, a randomized controlled trial. Clinical Nutrition, 33, 588–595. 10.1016/j.clnu.2013.12.007 [DOI] [PubMed] [Google Scholar]
- Genaidy AM, Lemasters GK, Lockey J, Succop P, Deddens J, Sobeih T and Dunning K, 2007. An epidemiological appraisal instrument ‐ a tool for evaluation of epidemiological studies. Ergonomics, 50, 920–960. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Bermudez CA, Lopez‐Nicolas R, Peso‐Echarri P, Frontela‐Saseta C and Martinez‐Gracia C, 2018. Effects of different thickening agents on infant gut microbiota. Food and Function, 9, 1768. [DOI] [PubMed] [Google Scholar]
- Higgins JPT and Green S, 2011. Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 [updated March 2011]: http://handbook-5-1.cochrane.org/
- Hu J, Lin S, Zheng B and Cheun PCK, 2018. Short‐chain fatty acids in control of energy metabolism. Critical Reviews in Food Science and Nutrition, 58, 1243–1249. 10.1080/10408398.2016.1245650 [DOI] [PubMed] [Google Scholar]
- ICH (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use), 2010. Guidance on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals, ICH, M3 (R2). Available online: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidisciplinary/M3_R2/Step4/M3_R2__Guideline.pdf [PubMed]
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 2000. Guidelines for the preparation of toxicological working papers for the Joint FAO/WHO Expert Committee on Food Additives. Geneva, Switzerland.
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 2014. Evaluation of certain food additives: 79th report of the Joint FAO/WHO Expert Committee on Food Additives. WHO technical report series No 990.
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 2015. Safety evaluation of certain food additives. Octenyl succinic acid (OSA)‐modified starch. WHO food additives series No 70.
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 2016a. Compendium of food additives specifications prepared by the meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA), 82nd meeting 2016. FAO JECFA Monographs No 19.
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 2016b. Evaluation of certain food additives (82nd report of the Joint FAO/WHO Expert Committee on Food Additives). WHO technical report series No 1000.
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 2017. Residue Monograph prepared by the meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA), 82nd meeting 2016. WHO food additives series No 70.
- JECFA (Joint FAO/WHO Expert Committee on Food Additives), 2018. Compendium of food additives specifications prepared by the meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA), 86th meeting 2018. FAO JECFA Monographs No 22. Available online: http://www.fao.org/documents/card/en/c/CA2330EN
- Johns PW, Jacobs WA, Hroncich MM and Vurma M, 2014. Determination of Octenyl succinic acid in nutritional products. Food Analysis Methods, 8, 807–814. 10.1007/s12161-014-9945-0 [DOI] [Google Scholar]
- Kelley RL, 1991. Octenylsuccinic aciduria in children fed protein‐hydrolysate formulas containing modified cornstarch. Pediatric Research, 30, 564–569. 10.1203/00006450-199112000-00015 [DOI] [PubMed] [Google Scholar]
- Mahadevan B, Thorsrud BA, Brorby GP and Ferguson HE, 2014. A 3‐week dietary safety study of octenyl succinic anhydride (OSA)‐modified starch in neonatal farm piglets. Food and Chemistry Toxicology, 72, 83–89. 10.1016/j.fct.2014.07.009 [DOI] [PubMed] [Google Scholar]
- NTP‐OHAT , 2015. Handbook for conducting a literature‐based health assessment using OHAT approach for systematic review and evidence integration. Available online: http://ntp.niehs.nih.gov/ntp/ohat/pubs/handbookjan2015_508.pdf
- NTP‐OHAT , 2019. Handbook for conduction a literature‐based health assessment using OHAT approach for systematic review and evidence integration (4 March, 2019). Available online: https://ntp.niehs.nih.gov/ntp/ohat/pubs/handbookmarch2019_508.pdf
- Scalabrin DM, Johnston WH, Hoffman DR, P'Pool VL, Harris CL and Mitmesser SH, 2009. Growth and tolerance of healthy term infants receiving hydrolyzed infant formulas supplemented with Lactobacillus rhamnosus GG: randomized, double‐blind, controlled trial Clinical Pediatrics (Phila), 48, 734–744. 10.1177/0009922809332682 [DOI] [PubMed] [Google Scholar]