

Abstract
Plasma amyloid-β peptide concentration has recently been shown to have high accuracy to predict amyloid-β plaque burden in the brain. These amyloid-β plasma markers will allow wider screening of the population and simplify and reduce screening costs for therapeutic trials in Alzheimer’s disease. The aim of this study was to determine how longitudinal changes in blood amyloid-β track with changes in brain amyloid-β. Australian Imaging, Biomarker and Lifestyle study participants with a minimum of two assessments were evaluated (111 cognitively normal, 7 mild cognitively impaired, 15 participants with Alzheimer’s disease). Amyloid-β burden in the brain was evaluated through PET and was expressed in Centiloids. Total protein amyloid-β 42/40 plasma ratios were determined using ABtest® assays. We applied our method for obtaining natural history trajectories from short term data to measures of total protein amyloid-β 42/40 plasma ratios and PET amyloid-β. The natural history trajectory of total protein amyloid-β 42/40 plasma ratios appears to approximately mirror that of PET amyloid-β, with both spanning decades. Rates of change of 7.9% and 8.8%, were observed for total protein amyloid-β 42/40 plasma ratios and PET amyloid-β, respectively. The trajectory of plasma amyloid-β preceded that of brain amyloid-β by a median value of 6 years (significant at 88% confidence interval). These findings, showing the tight association between changes in plasma and brain amyloid-β, support the use of plasma total protein amyloid-β 42/40 plasma ratios as a surrogate marker of brain amyloid-β. Also, that plasma total protein amyloid-β 42/40 plasma ratios has potential utility in monitoring trial participants, and as an outcome measure.
Keywords: plasma amyloid, Alzheimer’s disease, amyloid imaging, aging
Total protein amyloid β42/40 plasma ratios determined by the ABtest® assay were able to effectively map and track longitudinal amyloid PET accumulation. This adds to the body of information that blood-based biomarkers hold utility for diagnosis, prognosis, monitoring and, thus, treatment in the field of Alzheimer’s.
Graphical Abstract
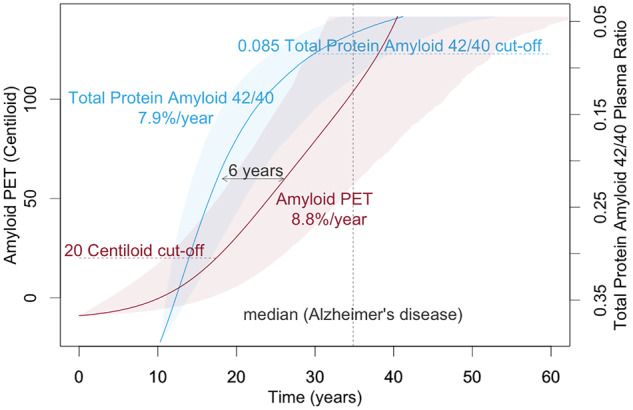
Graphical Abstract.
Introduction
Recently proposed frameworks suggest that a triad of biomarkers (Burnham et al., 2019a, b; Jack et al., 2019; van Maurik et al., 2019), namely amyloid-β (Aβ; A), tau (T) and neurodegeneration ([N]), are able to biologically classify Alzheimer’s disease even at preclinical disease stages (Jack et al., 2018). These biomarkers have predominantly been quantified using imaging techniques (PET, MRI) or CSF levels. PET imaging is relatively expensive and has limited availability; CSF requires lumbar puncture and is considered invasive by many. Therefore, alternative approaches are desirable for widespread population screening.
A number of reports now demonstrate the accuracy with which plasma levels estimate both the insoluble Aβ via PET imaging and the soluble Aβ via CSF measures. Plasma Aβ has reported area under the receiver operating curve values of 90% for discriminating between brain Aβ positives and negatives (Fandos et al., 2017; Ovod et al., 2017; Nakamura et al., 2018). In regard to the Aβ (A), tau (T) and neurodegeneration ([N]) framework (Jack et al., 2018), recent reports show that plasma pTau can discriminate between controls and Alzheimer’s disease, achieving area under the receiver operating curve values of 80% (Kovacs et al., 2017; Fossati et al., 2019). Along the same line, neurofilament light has emerged as an accurate plasma measure for Alzheimer’s disease progression and has been shown to discriminate between controls and Alzheimer’s disease, suggesting that it could be used as a robust marker of neurodegeneration (Kovacs et al., 2017; Lewczuk et al., 2018; Mattsson et al., 2019).
Given the reported accuracies of the plasma biomarkers for quantifying Aβ, Tau and neurodegeneration as well as the ease and access for utilizing blood for screening, the Aβ (A), tau (T) and neurodegeneration ([N]) framework for biologically classifying Alzheimer’s disease now appears achievable using a blood-based screening test.
Whilst threshold and correlation analyses on cross-sectional data look promising for such screening tests, the dynamic nature of plasma levels and their relationship with imaging and CSF biomarkers need to be further elucidated to fully understand the value such tests may have for monitoring progression as well as potential outcome measures in clinical trials. This contribution aims to evaluate longitudinal data to understand the natural history of Aβ as measured in both plasma and by PET imaging.
Materials and methods
Australian Imaging, Biomarker and Lifestyle cohort
The Australian Imaging, Biomarker and Lifestyle (AIBL) cohort study of ageing combines data from neuroimaging, biomarkers, lifestyle, clinical and neuropsychological assessments. Two study centres in Melbourne, VIC, and Perth, WA, Australia, recruit mild cognitively impaired (MCI) individuals and individuals with Alzheimer’s disease from primary care physicians or tertiary Memory Disorders Clinics. Cognitively healthy normal controls (CN) were recruited through advertisement or from spouses of participants in the study. Exclusion criteria were a history of non-Alzheimer’s disease dementia, Parkinson’s disease, schizophrenia, bipolar disorder, obstructive sleep apnoea, serious head injury, current depression (geriatric depression score >5 out of 15), cancer in the past 2 years (with the exception of basal-cell skin carcinoma), symptomatic stroke, uncontrolled diabetes or current regular alcohol use. Between 3 November 2006 and 30 October 2008, AIBL recruited 1112 eligible volunteers, who were aged 60 years or older and fluent in English. An enrichment cohort of 86 patients with Alzheimer’s disease, 124 MCI and 389 CN was recruited by AIBL between 30 March 201 and 29 June 2015. At baseline, the AIBL study participants were an average of 72 years of age, the study consisted of 58% women, and 36% were apoliporotein E-ε4 carriers. The institutional ethics committees of Austin Health, St Vincent’s Health, Hollywood Private Hospital and Edith Cowan University approved the AIBL study, and all volunteers gave written informed consent before participating.
A subset of the AIBL cohort, who had undergone at least two evaluations of both Aβ PET and total protein Aβ42/Aβ40 plasma ratios using the ABtest® assays (Araclon Biotech, Zaragoza, Spain), was utilized in this study. Furthermore, individuals considered to be Aβ negative [<20 Centiloids (CL)] and non-accumulators (longitudinal rates of change <0.0 CL/year) based on PET evaluations who were not exhibiting pathology of disease were excluded from this study (Villemagne et al., 2013). Thus, data from 133 participants (111 CN, 7 MCI, 15 Alzheimer’s disease) who were an average of 72 years of age, consisted of 54% males and 32% apoliporotein E-ε4 carriers were evaluated.
PET Aβ evaluation
AIBL Aβ PET studies consisted of a 20-min acquisition starting 50 min after the injection of 370 MBq of 11C-Pittsburgh compound-B. For the semi-quantitative analysis, PET images were spatially normalized with CapAIBL® using an adaptive atlas (Bourgeat et al., 2015). The spatially normalized PET images were then converted into CL (Bourgeat et al., 2018) using the standard CL cortical and whole cerebellum masks (Bourgeat et al., 2018). Images were then corrected to the standard CL SPM8 approach as previously described (Bourgeat et al., 2018). The abnormal threshold for levels of brain Aβ in AIBL participants was set as ˃20 CL (equivalent to 1.44 standardized uptake value ratio using the cerebellar cortex as reference region and 1.25 standardized uptake value ratio using the whole cerebellum as a reference region) (Rowe et al., 2018).
Plasma Aβ evaluation
Plasma samples were obtained using Ethylenediaminetetraacetic acid (EDTA) as anticoagulant and following AIBL procedures (Rembach et al., 2014) and were conserved at −70°C until analysis without undergoing any extra freezing/thaw cycles. Total protein Aβ42/Aβ40 plasma ratios were determined using ABtest® assays, two validated colorimetric assays based in the sandwich enzyme-linked immunosorbent assay technique, as described elsewhere (Pérez-Grijalba et al., 2016). The analyses were always performed in a coded manner to ensure that the knowledge of other participant evaluations was not available to the operator. The abnormal threshold for the ratio of plasma Aβ in AIBL participants was set as ˂0.085. This threshold was based upon receiver operating characteristics using the Youden index for plasma Aβ against brain Aβ positives and negatives (Doecke et al., 2020).
Statistical analyses
AIBL participants (n = 133) with at least 18 months of follow-up evaluations for Aβ who were considered to be accumulating Aβ (rate of deposition >0.0 CL/year; Villemagne et al., 2013) were included in this study. All analyses were performed in the R environment (R Development Core Team, 2017). Note, analyses were replicated in a subset of these participants (n = 112) with at least 3 years of follow-up testing, the findings were not altered.
Baseline differences between CN, MCI and Alzheimer’s disease were assessed with one-way t-tests for continuous data (age, amyloid PET, plasma amyloid, rate of change in amyloid measures), χ2 testing for categorized data (sex, years of education, disease classification, number of follow-ups) and Kruskal–Wallis testing for non-normally distributed data (length of follow-up).
Natural history of deposition for both plasma and PET Aβ was evaluated using the four-step procedure described previously (Villemagne et al., 2013; Budgeon et al., 2017). Briefly, the four-step procedure comprises (i) estimating the mean and slope of each individuals’ Aβ using linear models (refer to Fig. 1A and D), (ii) fitting a polynomial to the estimated means and slopes across all individuals (refer to Fig. 1B and E), (iii) integrating the reciprocal of the fitted polynomial (refer to Fig. 1C and F) and (iv) inverting the function to obtain the natural history trajectory (refer to Fig. 2). Confidence intervals for the natural history curves were created using the bootstrapping procedure described previously (Budgeon et al., 2017). Axes were anchored by taking the mean of the values exhibited by individuals at the extremes of each measure. The curves were anchored using the median values exhibited by the mild Alzheimer’s disease participants.
Figure 1.

Graphical representations of the first three steps of the data analysis method for Ab PET (A, C, E) and plasma AbTP42/40 ratio (B, D, F). Step 1 (A and B) represents the raw data and fitted linear models for each individual; Step 2 (C and D) represents plots of the individuals’ slopes versus their mean observed values and the fitted cubic polynomial to this data; and Step 3 (E and F) represents the integral of the fitted cubic polynomial. Green points are CN, blue points are MCI, orange points are Alzheimer’s disease and black points are outliers. TP42/40 = total protein amyloid-β 42/40 plasma ratios.
Figure 2.
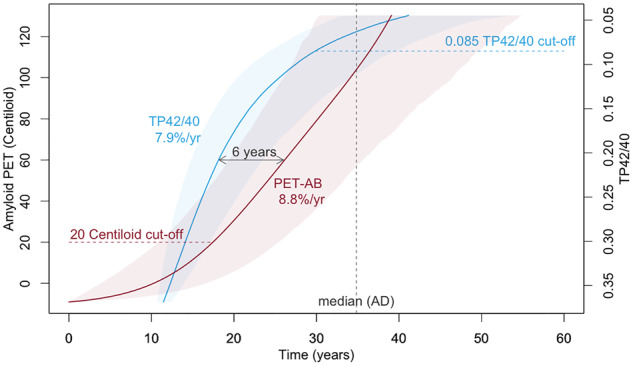
The natural history of brain Aβ deposition and inverted plasma Aβ changes anchored by median levels observed in mild Alzheimer’s disease participants.
Data availability
A subset of the AIBL data including images is shared through the LONI Image and Data Archive (http://adni.loni.usc.edu), a secure research data repository. Applications for access to the entirety of the AIBL data can be made via application through the AIBL website (https://aibl.csiro.au/).
Results
Demographics
The Alzheimer’s disease participants had significantly less education than CN, and MCI and Alzheimer’s disease had significantly less follow-up than CN (Table 1).
Table 1.
Demographics
| Participant demographics summary | CN | MCI | Alzheimer’s disease | Total |
|---|---|---|---|---|
| Number of participants, N | 111 | 7 | 15 | 133 |
| Male gender, N (%) | 63 (65.8) | 4 (57.1) | 5 (33.3) | 72 (54.1) |
| Age (years), mean (SD) | 71.8 (6.3) | 75.6 (7.9) | 74.9 (9.1) | 72.3 (6.8) |
| Years of education, mean (SD) | 13.4 (2.7) | 11.9 (2.8) | 12.5 (3.0) | 13.2 (2.8) |
| APOE ε4 carriers, N (%) | 30 (27.0) | 3 (42.9) | 9 (60.0) | 42 (31.6) |
| Years of follow-up, mean (SD) | 4.1 (0.8) | 2.1 (1.2) | 2.6 (1.4) | 3.9 (1.1) |
| Number of data collections, median (range) | 4 (2–4) | 2 (2–3) | 2 (2–4) | 4 (2–4) |
| Centiloid scores at baseline, mean (SD) | 17.9 (31.0) | 62.6 (55.8) | 87.3 (39.5) | 28.2 (40.8) |
| TP42/40 at baseline, mean (SD) | 0.10 (0.05) | 0.09 (0.03) | 0.10 (0.10) | 0.10 (0.06) |
| Rate of change in TP42/40 per year, mean (SD) | 0.007 (0.01) | 0.003 (0.01) | 0.008 (0.01) | −0.01 (0.01) |
Bold fonts indicate statistically significant differences from the cognitively normal subgroup.
APOE = apoliporotein E; TP42/40 = total protein amyloid-β 42/40 plasma ratios.
Baseline amyloid evaluations
MCI and Alzheimer’s disease participants had significantly higher levels of brain Aβ than the CN. No differences were observed for the plasma Aβ.
Natural history of brain Aβ
Analyses of the present cohort indicated that brain Aβ followed a trajectory of exponential growth spanning 40 years. The abnormal threshold of 20 CL was reached 17 years prior to typical levels seen in mild Alzheimer’s disease, and the fast, dynamic part of trajectory was associated with an 8.8% change per year (Fig. 2).
Natural history of plasma Aβ
Analyses indicated that plasma Aβ followed a trajectory of logarithmic growth spanning 34 years. The abnormal threshold of total protein amyloid-β 42/40 plasma ratios 0.085 was reached 5 years prior to typical levels seen in mild Alzheimer’s disease, and the fast, dynamic part of trajectory was associated with an 7.9% change per year (Fig. 2).
Comparison of natural history curves
Inverting the trajectory for the plasma Aβ and overlaying on the brain Aβ trajectory, anchored at the median levels observed in the mild Alzheimer’s disease participants, indicated that the trajectory for plasma amyloid preceded that of brain amyloid by a median value of 6 years; however, this was not statistically significant with 95% confidence (Fig. 2). It should be noted that 88% confidence was observed in this temporal difference.
Discussion
Our findings indicate that the natural history of plasma Aβ changes preceded that of brain Aβ deposition by 6 years. This temporal difference in the longitudinal trajectories of brain and plasma Aβ was observed with 88% confidence. However, it should be noted that we were under-powered to observe a statistical difference with 95% confidence. The figure also suggests that we were not able to observe an early inflection point, representing a change from steady state to a dynamic change, in plasma Aβ with the observed trajectory starting at a very fast dynamic rate of (7.9%/year). Conversely, for brain Aβ, we were able to observe an initial inflection point but not a second inflection point indicating a plateauing of the deposition. These observations also lend to the hypothesis that changes in plasma Aβ precede changes in brain Aβ. Increasing both the sample size and the sample range, to capture higher levels of brain Aβ (more abnormal) and higher ratios of plasma Aβ (more normal), in future studies would further our knowledge, understanding and confidence in these findings.
The differences observed in the shape and timing of the different trajectories seen between brain and plasma Aβ, as previously reported between imaging and CSF (Villemagne, 2013; Toledo 2013), are indicative of the different pools of Aβ, a more soluble easily diffusible pool captured in biofluid assays and a larger, insoluble pool of Aβ captured by imaging (Roberts et al., 2017). This explains the non-linear relationship between Aβ measured in biofluids and by imaging. The changes in these pools likely reflect the same process of Aβ aggregation and accumulation, translating into a decrease in Aβ in biofluids and an increase in the imaging signal.
Given the correlation between brain and plasma Aβ, ethical concerns should also be considered in obtaining and revealing blood-based levels of Aβ. It is likely that some of the same considerations regarding the appropriate use of Aβ imaging (Johnson et al., 2013) would also be applicable here.
It is noted that, whilst the longitudinal comparisons of the trajectories indicate that plasma Aβ precedes that of brain Aβ, this is contradicted when they are considered as dichotomous diagnostic biomarkers, with brain Aβ reaching abnormal levels 12 years before that of plasma Aβ. The utility of brain and plasma Aβ for screening is currently based upon thresholds of 20 CL and a ratio of 0.085, respectively. If the ratios were to change to increase sensitivity or specificity, then the difference in timing, reported as 12 years here, would also change. The results presented here suggest that, if the sensitivity of the plasma biomarkers is sufficient, then a higher ratio capturing earlier changes in the plasma Aβ signal may better serve the screening process.
There are some limitations in the present study. First, the number of participants included in the assessments is somewhat small, especially in the Alzheimer’s disease group, warranting validation in a larger independent cohort. Second, the participants were volunteers who were not randomly selected from the community and were generally well educated and had high scores on cognitive tests; thus, these findings might only be valid in similar cohorts and this limitation precludes the generalization of the findings to the general population. Third, AIBL’s astringent selection criteria, which excluded individuals with other neurodegenerative conditions (such as dementia with Lewy bodies or frontotemporal lobar degeneration) as well as other metabolic co-morbidities, might have influenced the trajectories of Aβ in blood and brain reported. Finally, the 20 CL (1.25 standardized uptake value ratio using the whole cerebellum as a reference region) threshold used is equivalent to a 11C-Pittsburgh compound-B 1.44 standardized uptake value ratio using the cerebellar cortex as reference region. This is slightly higher than the threshold of 1.40 standardized uptake value ratio using the cerebellar cortex as reference region which is usually applied. This, along with the use of only two points to establish the rates of Aβ accumulation, and the small number of Alzheimer’s disease patients, may explain the shorter time to reach mild Alzheimer’s disease levels and the lack of a plateau.
Findings indicate that longitudinal changes in plasma Aβ match and potentially precede changes in brain Aβ. Therefore, plasma Aβ may be a suitable screening tool for recruitment to clinical trials and may be able to identify individuals at risk to develop Alzheimer’s disease even earlier than with PET. The use of plasma as a screening tool could significantly reduce the cost of trial recruitment, allowing for larger clinical trials in the attempt to find disease-modifying therapies. Furthermore, the results presented here provide strong evidence that plasma Aβ has a strong dynamic range that is likely useful for monitoring, and even as an outcome measure in prodromal and preclinical trials.
The ABtest® assay was able to effectively map and track brain Aβ accumulation. The results presented here add to the body of information that blood-based biomarkers hold utility for diagnosis, prognosis, monitoring and, thus, treatment in the field of Alzheimer’s.
Acknowledgements
We thank the participants who took part in the study and their families.
Funding
Core funding for the AIBL study was provided by the Commonwealth Scientific and Industrial Research Organization (CSIRO) Flagship Collaboration Fund and the Science and Industry Endowment Fund (SIEF) in partnership with the Cooperative Research Centre (CRC) for Mental Health, Edith Cowan University (ECU), Mental Health Research institute (MHRI), Alzheimer’s Australia (AA), National Ageing Research Institute (NARI), Austin Health, Macquarie University, CogState Ltd, Hollywood Private Hospital and Sir Charles Gairdner Hospital. The study also received funding from the National Health and Medical Research Council (NHMRC; 1156891), Dementia Collaborative Research Centres program (DCRC) and McCusker Alzheimer’s Research Foundation and operational infrastructure support from the Government of Victoria.
Competing interests
N.F., V.P.-G., M.S. and P.P. are full-time employees of Araclon Biotech. No other authors report any relevant conflicts of interest.
Glossary
- Aβ =
amyloid-β
- AIBL =
The Australian Imaging, Biomarker and Lifestyle
- CL =
Centiloids
- CN =
cognitively healthy normal controls
- MCI =
mild cognitively impaired
References
- Bourgeat P, Doré V, Fripp J, Ames D, Masters CL, Salvado O, et al. Implementing the centiloid transformation for 11C-PiB and β-amyloid 18F-PET tracers using CapAIBL. NeuroImage 2018; 183: 387–93. [DOI] [PubMed] [Google Scholar]
- Bourgeat P, Villemagne VL, Dore V, Brown B, Macaulay SL, Martins R, et al. Comparison of MR-less PiB SUVR quantification methods. Neurobiol Aging 2015; 36: S159–66. [DOI] [PubMed] [Google Scholar]
- Budgeon CA, Murray K, Turlach BA, Baker S, Villemagne VL, Burnham SC.. Constructing longitudinal disease progression curves using sparse, short‐term individual data with an application to Alzheimer’s disease. Stat Med 2017; 36: 2720–34. [DOI] [PubMed] [Google Scholar]
- Burnham SC, Coloma P, Li Q-X, Collins S, Savage G, Laws S, et al. Application of the NIA-AA research framework: towards a biological definition of Alzheimer’s disease using cerebrospinal fluid biomarkers in the AIBL study. J Prev Alzheimer’s Dis 2019. a; 6: 248–55. [DOI] [PubMed] [Google Scholar]
- Burnham SC, Loi SM, Doecke J, Fedyashov V, Dore V, Villemagne VL, et al. The dawn of robust individualised risk models for dementia. Lancet Neurol 2019. b; 18: 985–7. [DOI] [PubMed] [Google Scholar]
- Doecke J, Virginia P-G, Fandos N, Fowler C, Villemagne V, Masters C, et al. Total Aβ42/Aβ40 ratio in plasma predict amyloid-PET status, independent of clinical AD diagnosis. Neurology 2020; 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fandos N, Pérez-Grijalba V, Pesini P, Olmos S, Bossa M, Villemagne VL, et al. AIBL Research Group. Plasma amyloid β 42/40 ratios as biomarkers for amyloid β cerebral deposition in cognitively normal individuals. Alzheimers Dement 2017; 8: 179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossati S, Cejudo JR, Debure L, Pirraglia E, Sone JY, Li Y, et al. Plasma tau complements CSF tau and P-tau in the diagnosis of Alzheimer’s disease. Alzheimers Dement 2019; 11: 483–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018; 14: 535–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Therneau TM, Weigand SD, Wiste HJ, Knopman DS, Vemuri P, et al. Prevalence of biologically vs clinically defined Alzheimer spectrum entities using the National Institute on Aging–Alzheimer’s Association research framework. JAMA Neurol 2019; 76: 1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Minoshima S, Bohnen NI, Donohoe KJ, Foster NL, Herscovitch P, et al. Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. J Nucl Med 2013; 54: 476–90. [DOI] [PubMed] [Google Scholar]
- Kovacs GG, Andreasson U, Liman V, Regelsberger G, Lutz MI, Danics K, et al. Plasma and cerebrospinal fluid tau and neurofilament concentrations in rapidly progressive neurological syndromes: a neuropathology‐based cohort. Eur J Neurol 2017; 24: 1326–e77. [DOI] [PubMed] [Google Scholar]
- Lewczuk P, Ermann N, Andreasson U, Schultheis C, Podhorna J, Spitzer P, et al. Plasma neurofilament light as a potential biomarker of neurodegeneration in Alzheimer’s disease. Alzheimers Res Ther 2018; 10: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Cullen NC, Andreasson U, Zetterberg H, Blennow K.. Association between longitudinal plasma neurofilament light and neurodegeneration in patients with Alzheimer disease. JAMA Neurol 2019; 76: 791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Doré V, et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018; 554: 249–54. [DOI] [PubMed] [Google Scholar]
- Ovod V, Ramsey KN, Mawuenyega KG, Bollinger JG, Hicks T, Schneider T, et al. Amyloid β concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimers Dement 2017; 13: 841–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Grijalba V, Fandos N, Canudas J, Insua D, Casabona D, Lacosta AM, et al. Validation of immunoassay-based tools for the comprehensive quantification of Aβ 40 and Aβ 42 peptides in plasma. J Alzheimers Disease 2016; 54: 751–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. Vienna; 2017.
- Rembach A, Faux NG, Watt AD, Pertile KK, Rumble RL, Trounson BO, et al. Changes in plasma amyloid beta in a longitudinal study of aging and Alzheimer’s disease. Alzheimers Dement 2014; 10: 53–61. [DOI] [PubMed] [Google Scholar]
- Roberts BR, Lind M, Wagen AZ, Rembach A, Frugier T, Li Q-X, et al. Biochemically-defined pools of amyloid-β in sporadic Alzheimer’s disease: correlation with amyloid PET. Brain 2017; 140: 1486–98. [DOI] [PubMed] [Google Scholar]
- Rowe C, Amadoru S, Dore V, McLean C, Hinton F, Shepherd C, et al. Correlation of amyloid PET in Centiloid units with neuropathological findings in Alzheimer’s disease. J Nucl Med 2018; 59 (Suppl 1): 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Maurik IS, Vos SJ, Bos I, Bouwman FH, Teunissen CE, Scheltens P, et al. Biomarker-based prognosis for people with mild cognitive impairment (ABIDE): a modelling study. Lancet Neurol 2019; 18: 1034–44. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol 2013; 12: 357–67. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
A subset of the AIBL data including images is shared through the LONI Image and Data Archive (http://adni.loni.usc.edu), a secure research data repository. Applications for access to the entirety of the AIBL data can be made via application through the AIBL website (https://aibl.csiro.au/).