

Abstract
Fatal familial insomnia is a genetic prion disease, which is associated with the aspartic acid to asparagine substitution at codon 178 of the prion protein gene. Although the hallmark pathological feature is thalamic and olivary degeneration, there is a patient with an atypical fatal familial insomnia without the hallmark feature. The cause of the pathological variability is unclear. We analysed a Japanese fatal familial insomnia kindred and compared one atypical clinicopathological fatal familial insomnia phenotype case and typical fatal familial insomnia phenotype cases with transmission studies using multiple lines of knock-in mice and with protein misfolding cyclic amplification. We also analysed the transmissibility and the amplification properties of sporadic fatal insomnia. Transmission studies revealed that the typical fatal familial insomnia with thalamic and olivary degeneration showed successful transmission only using knock-in mice expressing human–mouse chimeric prion protein gene. The atypical fatal familial insomnia with spongiform changes showed successful transmission only using knock-in mice expressing bank vole prion protein gene. Two sporadic fatal insomnia cases with thalamic and olivary degeneration showed the same transmissibility as the typical fatal familial insomnia phenotype. Interestingly, one sporadic fatal insomnia case with thalamic/olivary degeneration and spongiform changes showed transmissibility of both the typical and atypical fatal familial insomnia phenotypes. Protein misfolding cyclic amplification could amplify both typical fatal familial insomnia cases and sporadic fatal insomnia cases but not the atypical fatal familial insomnia phenotype or other sporadic Creutzfeldt–Jakob disease subtypes. In addition to clinical findings and neuropathological features, the transmission properties and the amplification properties were different between the typical and atypical fatal familial insomnia phenotypes. It is suggested that two distinct prions were associated with the diversity in the fatal familial insomnia phenotype, and these two prions could also be detected in sporadic fatal insomnia.
Keywords: fatal familial insomnia, sporadic fatal insomnia, knock-in mouse, transmission study, protein misfolding cyclic amplification
Two distinct prions are identified in patients with fatal familial insomnia. A typical fatal familial insomnia with olivary degeneration and an atypical fatal familial insomnia with spongiform changes showed distinct transmission and amplification properties, and these two prions could be detected also in sporadic fatal insomnia. Two distinct prions might be associated with the phenotypic diversity of fatal familial insomnia/sporadic fatal insomnia.
Graphical Abstract
Graphical Abstract.
Introduction
Fatal familial insomnia (FFI) was first described in 1986 in Italy (Lugaresi et al., 1986). This disease was subsequently reported as a genetic prion disease that was linked to the codon 178 point mutation (aspartic acid to asparagine substitution: D178N) of the prion protein gene (PRNP) (Medori et al., 1992). Meanwhile, the same D178N mutation has been reported in a subtype of familial Creutzfeldt-Jakob disease (Goldfarb et al., 1991). Goldfarb et al. revealed that the phenotypic variability among the patients with the D178N mutation depended on a common polymorphism at PRNP codon 129, methionine or valine. The D178N mutation gene coupled with methionine at codon 129 develops FFI, and the D178N mutation gene coupled with valine at codon 129 develops familial Creutzfeldt–Jakob disease (Goldfarb et al., 1992).
The pathological hallmark of the typical FFI phenotype is severe neuronal loss and atrophy in thalamic nuclei and the inferior olivary nucleus. However, atypical FFI cases with predominant lesions in the cerebral cortex, consisting of a spongiform encephalopathy, rather than thalamic degeneration/olivary degeneration (OD), have been reported (McLean et al., 1997; Saitoh et al., 2010). Moreover, FFI cases with conspicuous spongiform changes (SC) in the cerebral cortex in addition to typical thalamic degeneration/OD have been reported in cases with a relatively long clinical course (i.e. >18 months) (Manetto et al., 1992; Parchi et al., 1995, 1998b; Sasaki et al., 2005). Western blot analysis revealed that non-glycosylated bands of the abnormal form of the prion protein (PrPSc) in patients with the D178N mutation had electrophoretic mobility of 19 kDa (type 2) irrespective of the pathological phenotypic variability. Apart from the clear effect on disease duration of the codon 129 genotype on the wild-type PRNP allele (Parchi et al., 1998a, b), the reason for the phenotypic diversity among individuals with the 129M-D178N has not been fully elucidated. Meanwhile, the infectivity of the FFI prion has been examined by transmission studies in animal models (Collinge et al., 1995; Tateishi et al., 1995; Telling et al., 1996; Sasaki et al., 2005).
In this study, we focused on a Japanese FFI kindred with the 129M-D178N genotype showing distinct clinicopathological features, namely atypical FFI without thalamic generation/OD and typical FFI (Saitoh et al., 2010). The mother in the kindred showed the atypical FFI phenotype with rapidly progressing dementia. Histopathological examination and immunohistochemical analysis revealed SC throughout the cerebral cortex and a synaptic pattern of PrP deposition, respectively. The type and glycosylation pattern of accumulated PrPSc were the same as those of typical FFI cases, but a larger amount of PrPSc was detected than in patients with typical FFI. To explore the possible involvement of multiple prions in the phenotypic variability of 129M-D178N, we carried out experimental transmissions of prions from the kindred using multiple lines of knock-in mice. Challenges with knock-in mice expressing bank vole PrP with M109 (Ki-Bank) (Kobayashi et al., 2019b) or knock-in mice expressing human-mouse chimeric PrP (Ki-ChM) (Taguchi et al., 2003) revealed that the transmissibility varied between FFI phenotypes. We also found that typical FFI prions but not the atypical FFI prion could be amplified efficiently with protein misfolding cyclic amplification (PMCA) using recombinant cellular prion protein (PrPC) as a substrate (cell-PMCA). Moreover, we analysed the transmissibility and the amplification property of the thalamic form of sporadic Creutzfeldt–Jakob disease (sCJD-MM2T) because sCJD-MM2T was recognized as sporadic fatal insomnia (sFI) due to the involvement of the same prion as FFI (Mastrianni et al., 1999; Moda et al., 2012).
Materials and methods
Ethics statement
Brain tissues were obtained at autopsy after receiving written informed consent for research use. Animal experiments were performed in strict accordance with the Regulations for Animal Experiments and Related Activities at Tohoku University. The protocol was approved by the Institutional Animal Care and Use Committees of Tohoku University (2017MdA-81).
Sources of prion inocula for transmission experiments
Brain homogenates were prepared from patients with FFI with the 129M/M genotype and from patients with sFI. The diagnosis of Creutzfeldt–Jakob disease, the PRNP genotype and the PrPSc type was confirmed by immunohistochemistry, PRNP sequence analysis and western blot analysis, respectively (Kitamoto et al., 1992, 1993). The subtyping of Creutzfeldt–Jakob disease was carried out according to the classification by Parchi et al. (1999).
Transmission experiments
Knock-in mice carrying human PrP with the 129M/M or 129V/V, Ki-Bank mice and Ki-ChM mice were produced as described previously (Kitamoto et al., 1996, 2002; Asano et al., 2006; Kobayashi et al., 2007, 2019b). Inoculated mice were sacrificed after the onset of clinical disease. One hemisphere of the brain was immediately frozen for western blotting, and the other hemisphere was fixed in 10% buffered formalin for immunohistochemistry.
Immunohistochemistry
Immunohistochemistry of mouse brain tissues was performed as described previously (Kobayashi et al., 2019b). The anti-PrP antiserum PrP-N was used as the primary antibody (Kitamoto et al., 1991). Goat-anti-rabbit immunoglobulin polyclonal antibody labelled with the peroxidase-conjugated dextran polymer, EnVision+ (Dako), was used as the secondary antibody.
Western blotting analysis
PrPSc was extracted from brains with collagenase treatment as described previously with some modifications (Grathwohl et al., 1996). The protease-resistant core of PrPSc of the samples was subjected to SDS–PAGE and western blotting. Anti-PrP monoclonal antibodies 3F4 (Signet, Dedham, MA, USA) or SAF83 (Bertin Pharma) were used as the primary antibodies. Anti-mouse or Anti-rabbit EnVision+ was used as the secondary antibody.
Cell-PMCA procedures
Human brain homogenates from patients with sCJD (MM1, MV1, MM2, MV2, VV1 and VV2) or FFI were seeded. Cell lysates as substrate from FreeStyle 293F cells (Invitrogen) expressing human PrPC with methionine at codon 129 as a substrate or human brain homogenates as seed for cell-PMCA were prepared as previously described (Takeuchi et al., 2016), with some modifications. The cell-PMCA methods are described in detail in the Supplementary material.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Results
Neuropathological findings of the fatal familial insomnia kindred and sFI
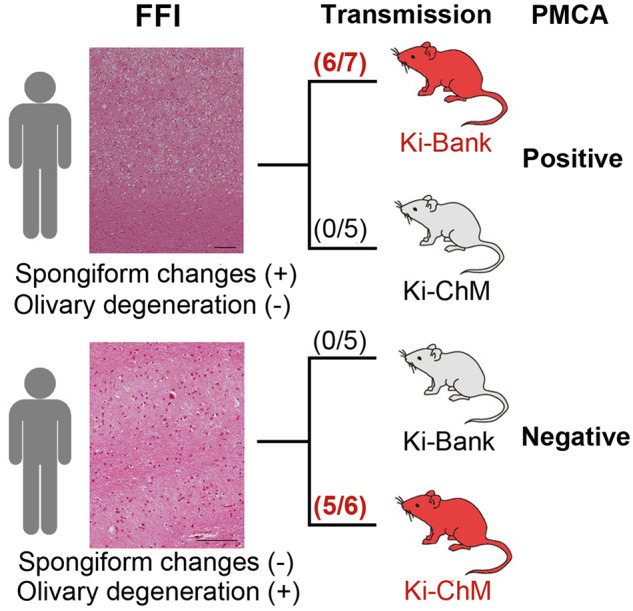
We analysed the neuropathology of the Japanese FFI kindred with the 129M/M genotype. Details of the clinical features and post-mortem studies of the two patients have been described elsewhere (Saitoh et al., 2010), and the clinical details are re-summarized in Table 1. Briefly, FFI-Mother was the biological mother of FFI-Child and she presented atypical phenotypic features of FFI, but FFI-Child showed a typical FFI phenotype. SC throughout the cerebral cortex and synaptic pattern of PrP deposition were observed in the brain of FFI-Mother, whereas neuronal loss and gliosis in the thalamus and inferior olive were not observed (Fig. 1A–C). FFI-Child exhibited a severe neuronal loss in the inferior olive without SC or PrP deposition (Fig. 1D–F). In western blotting with tissues from the frontal cortex, discrimination between FFI-Mother PrPSc and FFI-Child PrPSc was rather difficult because both of them were type 2 PrPSc with a di-glycoform dominant pattern, but the amount of PrPSc from the frontal cortex of FFI-Mother was ∼70 times greater than that of FFI-Child (Supplementary Fig. 1). We also analysed the neuropathological features of two additional Japanese FFI cases, FFI-I37 and FFI-I75 (Table 2). Both FFI-I37 and FFI-I75 showed spotty SC in addition to severe neuronal loss in the inferior olive. It was previously reported that neuropathological findings with a combination of SC and neuronal loss in the inferior olive are observed in patients with typical FFI (Parchi et al., 1995).
Table 1.
Clinical details of the FFI kindreda
| FFI-Mother | FFI-Child | |
|---|---|---|
| Clinical manifestations |
|
|
| Neurological examinations |
|
|
Clinical details of these patients have been reported elsewhere (Saitoh et al., 2010).
Figure 1.
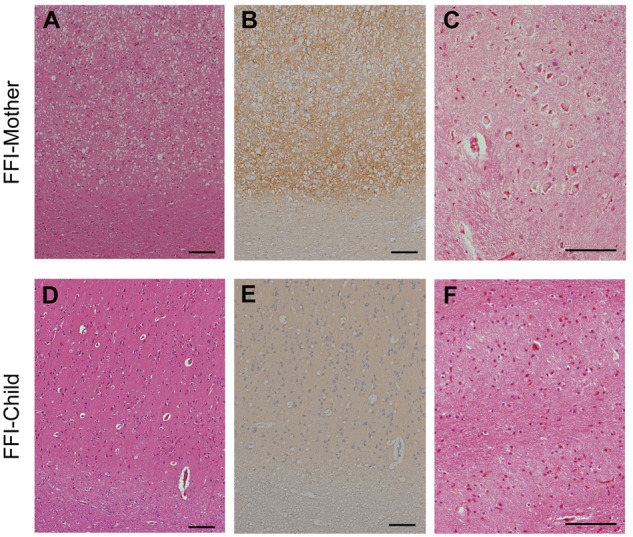
Phenotypic features of FFI-Mother and FFI-Child. (A–F) Neuropathological features of FFI-mother and FFI-child. (A and D) HE stainings. The cerebral cortex showed widespread SC in FFI-Mother but not in FFI-Child. (B and E) Representative microscopic images of PrP immunostaining. The cerebral cortex showed widespread synaptic-type PrP deposition in FFI-Mother. No PrP deposition was observed in FFI-Child. (C and F) HE stainings. Neurons in inferior olive were preserved well in FFI-Mother, whereas inferior OD was observed in FFI-Child. Scale bars = 100 μm.
Table 2.
Histopathological findings in the brains and amplification with cell-PMCA of patients with FFI and sFI
| Case number | PRNP genotype | Histopathology |
Amplification with cell-PMCA b | Reference | |
|---|---|---|---|---|---|
| OD [number of neurons in the olivary nucleus/mm2 (mean ± SEM)] a | Spongiform changes | ||||
| FFI-Mother | 129M/M 219E/E D178N | Negative (55.1 ± 15.9) | +++ | Negative | Saitoh et al. (2010) |
| FFI-Child | Positive (11.8 ± 2.8) | − | Positive | Saitoh et al. (2010) | |
| FFI-I37 | Positive (4.5 ± 6.5) | + | Positive | ||
| FFI-I75 | Positive (0.3 ± 0.8) | + | Positive | ||
| sFI-H91 | Positive (2.2 ± 1.2) | + | Positive | Kawasaki et al. (1997) | |
| sFI-I110 | 129M/M 219E/E | Positive (1.2 ± 1.1) | + | Positive | |
| sFI-T00/06 | Positive (7.0 ± 7.3) | ++ | Positive | Yamashita et al. (2001) | |
| sFI-I48 | Positive (0.6 ± 0.8) | +++ | Positive | Hirose et al. (2006) | |
| sFI-J86 | Positive (5.0 ± 3.9) | − | Positive | ||
| sFI-H10 | Positive (0) | + | Positive | ||
| sFI-H94 | Positive (1.2 ± 1.2) | + | Positive | ||
| sFI-J93 | Positive (2.7 ± 3.1) | + | Positive | ||
| sFI-I43 | Positive (1.8 ± 2.0) | +++ | Positive | ||
| sFI-J99 | Positive (6.8 ± 3.1) | − | Positive | ||
−, absent; +, spotty (lesion is usually within 1 mm2); ++, partial (lesion spreads beyond gyrus or involves all cortical layers); +++, widespread (lesion spreads beyond one lobe).
The average and standard deviation of the number of neurons at each of 5 places/mm2 in the inferior olive.
The cases were described as positive if the brain samples with 10−3-fold dilution could be amplified using this PMCA method.
We also analysed the neuropathology of 10 cases of sFI based on SC and OD (Table 2). Eight of the 10 cases of sFI showed various ranges of SC in the brains, such as spotty (lesion is usually within 1 mm2), partial (lesion spreads beyond gyrus or involves all cortical layers) or widespread (lesion spreads beyond one lobe) in addition to the remarkable atrophy in the inferior olive (Table 2). sFI-I48 was previously described as the MM2-thalamic form with widespread cortical pathology (Hirose et al., 2006), and sFI-T00/06 showing partial SC was previously reported as a thalamic variant of sCJD with severe brain atrophy and plaque-like PrP deposition (Yamashita et al., 2001). These results demonstrated that the combination of SC and OD was also commonly observed in the brains of patients with sFI, as previously reported (Abu-Rumeileh et al., 2018).
Transmission studies of the FFI kindred and sFI
We carried out transmission studies using knock-in mice and inocula from the FFI kindred and three cases of sFI. Although neither FFI-Mother prion nor FFI-Child prion was transmissible to Ki-Hu129M/M mice or Ki-Hu129V/V mice, a difference in the transmissibility was observed between FFI-Mother and FFI-Child (Table 3 and Fig. 2) when Ki-Bank or Ki-ChM mice were used as recipients. FFI-Mother prion was successfully transmitted to Ki-Bank mice (Ki-Bank [FFI-Mother]), and PrP deposition in the brains was observed by immunohistochemistry, but the inoculated Ki-ChM mice did not develop the disease (Fig.2). In contrast, FFI-Child prion was successfully transmitted to Ki-ChM mice (Ki-ChM [FFI-Child]) instead of Ki-Bank mice (Fig. 2). Western blot analysis revealed that the molecular weight of the accumulated PrPSc in the brains of Ki-Bank [FFI-Mother] or Ki-ChM [FFI-Child] was smaller than that of the PrPSc in the Ki-Bank or Ki-ChM mice inoculated with sCJD-MM1 prions, consistent with type 2 PrPSc (Supplementary Fig. 2A). The distinct differences in transmissibility between FFI-Mother and FFI-Child strongly suggested that two different prions were associated with the phenotypic variability among patients with the 129M-D178N mutation.
Table 3.
Transmission of FFI or sFI to multiple knock-in mice lines
| Inoculum a | Knock-in mice line | Attack rate (n/n0) | Mean incubation period (days ± SEM) |
|---|---|---|---|
| FFI-Mother | Ki-Hu129M/M | 0/6 | ∼1037 |
| Ki-Hu129V/V | 0/5 | ∼998 | |
| Ki-ChM | 0/5 | ∼907 | |
| Ki-Bank | 6/7 | 454 ± 104 | |
| FFI-Child | Ki-Hu129M/M | 0/6 | ∼886 |
| Ki-Hu129V/V | 0/6 | ∼767 | |
| Ki-ChM | 5/6 | 502 ± 133 | |
| Ki-Bank | 0/5 | ∼903 | |
| sFI-H91 | Ki-Hu129M/M | 0/3 | ∼806 |
| Ki-Hu129V/V | 0/6 | ∼799 | |
| Ki-ChM | 4/5 | 342 ± 25 | |
| Ki-Bank | 0/6 | ∼803 | |
| sFI-I110 | Ki-Hu129M/M | 0/4 | ∼752 |
| Ki-Hu129V/V | 0/4 | ∼722 | |
| Ki-ChM | 3/6 | 394 ± 23 | |
| Ki-Bank | 0/5 | ∼770 | |
| sFI-T00/06 | Ki-Hu129M/M | 0/6 | ∼907 |
| Ki-Hu129V/V | 0/4 | ∼661 | |
| Ki-ChM | 5/6 | 572 ± 135 | |
| Ki-Bank | 2/7 | 690, 707 |
n: number of positive mice; n0: number of inoculated mice.
Intracerebral inoculation with 20 μl of a 10% (w/v) brain homogenate from either of the patients with FFI or sFI.
Figure 2.
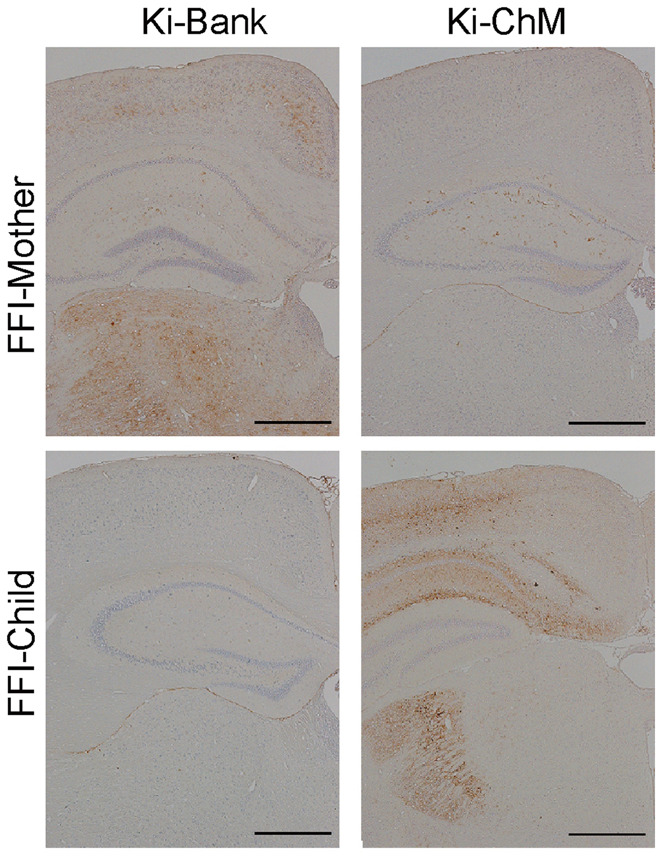
Transmission studies. Immunohistochemical analysis of the brains from the inoculated mice with the FFI kindred. Transmission of FFI-Mother’s sample was successful in Ki-Bank mice but not in Ki-ChM mice. Positive PrP stainings were observed mainly in the cerebral cortex and thalamus of Ki-Bank mice. Transmission of FFI-Child’s sample was successful in Ki-ChM mice but not in Ki-Bank mice. Positive PrP stainings were observed mainly in the cerebral cortex, hippocampus and lateral geniculate body of the Ki-ChM mice. Scale bars = 500 μm.
We analysed the transmission properties of three cases of sFI, such as sFI-H91 (spotty SC) (Kawasaki et al., 1997), sFI-T00/06 (partial SC) and sFI-I110 (spotty SC) (Table 2). The transmissibility of each sFI case is shown in Table 3. Similar to FFI-Child prion, sFI-H91, sFI-I110 and sFI-T00/06 prions transmitted to Ki-ChM mice successfully and type 2 prions were accumulated in the brains of the infected mice (Supplementary Fig. 2B). Interestingly, sFI-T00/06 prion was also transmitted to two of the seven inoculated Ki-Bank mice (Table 3 and Supplementary Fig. 2B), whereas Ki-Bank mice inoculated with brain homogenates from sFI-H91 or sFI-I110 did not develop the disease. Successful transmission of sFI-T00/06 prion to both Ki-Bank mice and Ki-ChM mice correlated with the sFI-T00/06 pathology with a relatively prominent SC in addition to OD, whereas sFI-H91 and sFI-H110 had only spotty SC in a limited area.
Protein misfolding cyclic amplification of FFI and sFI
We performed cell-PMCA of prions from FFI and sFI cases using substrate lysates prepared from 129M human PrP-expressing 293F cells to compare the amplification properties between FFI-Mother prion and FFI-Child prion (Table 2). FFI-Child prion could be amplified significantly, and strong signals were detected with up to a 10−4-fold dilution, whereas a slight amplification was observed only from a 10−2-fold dilution when FFI-Mother prion was used as the seed (Supplementary Fig. 3A). Interestingly, prions from both FFI-I37 and FFI-I75 amplified efficiently even at a 10−4-fold dilution. These results revealed that the amplification efficiency was also different between the atypical FFI prion and typical FFI prions in this cell-PMCA condition.
Since it appeared that OD in a typical FFI phenotype would be coupled with efficient amplification, we also carried out cell-PMCA in the 10 sFI cases. Similar to the typical FFI cases, significant amplification was detected from all 10 cases of sFI (Table 2 and Supplementary Fig. 3B), which revealed that prions associated with OD, namely M2T prions, were specifically amplified. To confirm the reliability for the detection of M2T with cell-PMCA, we examined 115 samples with all subtypes of sCJD and non-Creutzfeldt–Jakob disease. No significant signal of the amplified products from sCJD-MM1, sCJD-MM2C (Supplementary Fig. 3B), other sCJD subtypes without OD (data not shown) or non-Creutzfeldt–Jakob disease (data not shown) was observed.
Discussion
In this study, we have demonstrated that one FFI case with an atypical phenotype is likely caused by a prion strain that is distinct from that associated with typical FFI cases, using two independent methodologies, animal transmission studies and cell-PMCA. Furthermore, it appeared that the atypical prion might exist in sFI.
Reasons why two distinctive prions have detected in fatal familial insomnia or sFI
The transmission properties of FFI or sFI using transgenic mice were demonstrated in several laboratories (Table 4). However, previous studies might have missed the full extent of prion strain diversity in patients carrying the D178N-129M haplotype because the results were obtained by limited transmission experiments using a single transgenic mouse line or only one patient. In this study, we performed transmission studies using multiple knock-in mouse lines (Ki-humanized mice, Ki-Bank mice and Ki-ChM mice) and samples from an FFI kindred carrying the 129M/M genotype showing two distinct clinicopathological features. Notably, we showed that the atypical FFI phenotype without thalamic/olivary is associated with a different prion, indicating that two distinct prions were generated by the same mutation and PRNP haplotype in this kindred. In addition, the successful transmission of one patient with sFI to both Ki-ChM and Ki-Bank mice suggested that two distinct prions were associated with the sporadic form.
Table 4.
Previous reports of the experimental transmission of FFI and sFI using a mouse model
| Inocula | Number of patients (successful transmission) | Recipient | Reference |
|---|---|---|---|
| FFI | 1 | Wild-type NZW mice | Tateishi et al. (1995) |
| FFI | 2 | Tg (human PrP129M) mice | Collinge et al. (1995) |
| FFI | 3a | Tg (human/mouse PrP chimera)b mice | Telling et al. (1996) |
| sFI | 1 | Tg (human/mouse PrP chimera)b mice | Mastrianni et al. (1999) |
| FFI | 1c | Wild-type NZW mice Tg (hamster PrP) mice | Sasaki et al. (2005) |
| sFI | 1d | Ki (human PrP129MM) mice | Moda et al. (2012) |
Three of the four patients with FFI were transmitted to Tg mice. The inoculated Tg mice with one patient with FFI with SC and OD did not develop the disease after 350 days post-inoculation.
The Tg mice have the same PrP sequence of Ki-ChM mice.
This case has widespread SC and OD.
This case has widespread SC; however, OD has not been described.
Experimental transmission using knock-in mice
In the present study, prions from typical FFI or sFI with OD successfully transmitted to Ki-ChM mice expressing human-mouse chimeric PrPC, whereas none of the inoculated Ki-Hu129M/M mice or Ki-Hu129V/V mice developed the disease. To our knowledge, the successful transmission of only one sCJD-MM2T case to Ki-Hu129M/M mice was reported by Moda et al. (2012) (Table 4), which is not consistent with our transmission data in typical FFI or sFI. Because M2T prions may preferentially accumulate in the thalamus, it is possible that the amount of PrPSc in our inoculum from the cerebral cortex might have been insufficient to cause infection. To confirm the infectivity of M2T prion to Ki-Hu129M/M mice, a secondary passage will also be needed in the future. Nevertheless, it was clarified that Ki-ChM mice were more susceptible to M2T prions than Ki-Hu129M/M mice when the same dose of inoculum was used. At present, experimental transmission using Ki mice instead of Tg mice would be suitable for identifying prion strains. Ki mice carrying different PRNP genes but expressing the same level of PrPC allow analysing the susceptibility to each prion strain based only on the length of the incubation period. MMiK prion among patients with dura mater graft-associated Creutzfeldt–Jakob disease with the 129M/M genotype were successfully transmitted to Ki-Hu129V/V mice with a shorter incubation period than Ki-129M/M mice (Kobayashi et al., 2007). The incubation periods of the mice inoculated with MMiK prion were the same as those of mice inoculated with sCJD-VV2 prions, which revealed that MMiK prion was identical to V2 prion (Kobayashi et al., 2009, 2010). It would not be appropriate to use Tg mice to evaluate the susceptibility to prion strains by comparing the length of the incubation period, because the incubation period is significantly affected by the variable expression levels of PrPC in Tg mice. For example, Tg-Bank mice with the M109 genotype in addition to the I109 genotype spontaneously developed the disease, whereas Ki-Bank mice did not (Kobayashi et al., 2019b), which makes Ki-Bank mice more suitable to prove the prion infectivity than Tg-Bank mice. In the present study, typical FFI or sFI prions were successfully transmitted to Ki-ChM mice but not to Ki-Bank mice. However, the atypical FFI case with SC and the sFI case with SC were successfully transmitted to Ki-Bank mice. A previous study reported the successful transmission of a sCJD-MM2 case with bank voles (Nonno et al., 2006). It will be necessary to investigate the presence of OD or SC pathology in this case.
Diagnosis of FFI or sFI according to pathological finding in the inferior olivary nucleus
Neuronal loss or degeneration in the dorsomedial thalamic nucleus or the inferior olivary nucleus has been adopted as neuropathological hallmark for FFI or sFI (Lugaresi et al., 1986; Parchi et al., 1999). However, we consider OD, the most important neuropathological finding for the diagnosis of the disease. Relatively severe neuronal loss in the medial thalamus is also seen in the brains of patients with sCJD-MM1 after a long-term illness (>15 months), while the number of neurons in the inferior olive or the lateral thalamus is largely preserved irrespectively of the disease duration (Kobayashi et al., 2019a). Accordingly, Kobayashi et al. have recently reported that, in cases of sCJD-MM1 with MM2T pathology, neuronal loss in the inferior olivary nucleus rather than in the medial thalamus is more specific for FFI or sFI. Thereby, we propose that the diagnosis of FFI or sFI could be readily achieved based on the neuropathological findings of both thalamic and olivary atrophy. Because cell-PMCA could amplify the M2T prion selectively from the brains even in the presence of co-occurring M1 or M2C prions (or both) (Takeuchi et al., unpublished data), this cell-PMCA may also be useful for the detection of M2T prion in addition to the neuropathological findings in the inferior olivary nucleus.
Addressing unresolved issues in FFI or sFI
By providing evidence for the existence of two distinct prions associated with the pathological variability of patients with FFI, our study helps to explain unresolved issues in FFI. It has been reported that there are patients with FFI who exhibit multiple and mixed clinical symptoms of both FFI and Creutzfeldt–Jakob disease (Zarranz et al., 2005; Synofzik et al., 2009; Chen et al., 2018) and that patients with FFI also show significant neuropathological phenotypic variability (Manetto et al., 1992; McLean et al., 1997; Yamashita et al., 2001; Sasaki et al., 2005). The genotype at codon 129 in the non-mutated allele is known to affect the clinical and pathological features in FFI. It has been reported that significant SC in the cerebral cortex characterize a number of patients with FFI with the 129 methionine/valine genotype because of their longer survival (Parchi et al., 1995, 1998b). However, the coexistence of typical and atypical prions might also be associated with unusual clinical and pathological features in patients with FFI irrespectively of the effect of disease duration.
Furthermore, it is unknown why a lower amount of PrPSc in the thalamus than in other parts of the brain causes remarkable thalamic degeneration in FFI or sFI. It might be difficult to analyse accurately the distribution of M2T prion if a larger amount of atypical prions coexist in the brain. The actual distribution of M2T prion in the brain will only be revealed by a specific quantitative analysis.
It is still uncertain why the distinctive prion strain appeared only in a single case (i.e. the present case), given that, to date, other atypical FFI cases have not been demonstrated. However, it has been reported worldwide that there are large, genetically identified FFI pedigrees in which patients showing heterogeneous phenotypic features have appeared. In the future, it will be necessary to perform a detailed genetic analysis and also search for environmental factors or pathological changes that influence the clinicopathological course in such large FFI pedigrees.
In inherited prion diseases, phenotypic variability has also been reported in Gerstmann–Sträussler–Scheinker disease linked to the P102L mutation. Two distinct pathological phenotypes were observed in patients with the P102L genotype; one with both SC and amyloid plaques and the other without SC (Parchi et al., 1998a; Piccardo et al., 1998). The Gerstmann–Sträussler–Scheinker P102L mutation generates two different types of abnormal PrPSc fragments of 21 and 8 kDa, respectively. The 21-kDa PrPSc fragment was only seen in the brains with SC, whereas the 8-kDa fragment was seen in all cases (Parchi et al., 1998a; Piccardo et al., 1998). Experimental transmission studies revealed different infectivity between 21- and 8-kDa PrPSc in knock-in mice carrying the 101L/L PrP gene (homologous to codon 102 in humans) (Piccardo et al., 2007). Consequently, two distinct prion strains were associated with phenotypic diversity in Gerstmann–Sträussler–Scheinker P102L. The occurrence of multiple prion strains and phenotypic diversity in subjects carrying the same PRNP mutation might be a common feature in inherited prion diseases.
A new classification of sCJD-MM2
In the present study, we have identified a new type 2 prion associated with typical SC and synaptic PrP deposition. SC and synaptic PrP deposition are considered hallmarks of MM1, the most common Creutzfeldt–Jakob disease phenotype, and are currently used by neuropathologists for the identification of MM1 prion. However, here, we showed that the type 2 prion can also be detected in association with typical SC and synaptic PrP deposition and that the transmission properties of these atypical M2 prions were different from those of the MM1 prion. Interestingly, the atypical type 2 prion did not show any large confluent vacuoles or perivacuolar PrP deposition, which are landmarks of sCJD-MM2C.
The classification of sCJD was established according to codon 129 genotyping, PrPSc typing and the pathological findings (Parchi et al., 1999). We propose revising MM2C as MM2C [large vacuolation (lv)] to distinguish it from MM2C ([small vacuolation (sv)] (Table 5). We recently identified an MM2C (sv + lv) case with both widespread SC with synaptic PrP deposition and large confluent vacuoles with perivacuolar PrP staining without OD (Kitamoto et al., unpublished data), suggesting that there are MM2 cases in which M2T, M2C (lv) or M2C (sv) prions exist independently or in combination.
Table 5.
A new classification of sCJD-MM2
| Current classification | Pathological change | PrP immunostain | PMCA | Transmission | Proposed classification | Prion |
|---|---|---|---|---|---|---|
| MM2T | Thalamic degeneration/OD | negative | Positive | Ki-ChM (+) Ki-Bank (−) | MM2T | M2T |
| MM2C | Large vacuolation | Perivacuolar PrP deposits | Negative | Ki-ChM (−) Ki-Bank (+) | MM2C (lv) | M2C (lv) |
| Newly identified | Spongiform changes (small vacuolation) | Synaptic PrP deposits | Negative | Ki-ChM (−) Ki-Bank (+) | MM2Ca (sv) | M2C (sv) |
At the beginning of this study, we detected SC in MM2T patients. However, the SC were mainly in the cerebral cortex. Therefore, an atypical type 2 prion associated with spongiform change is classified as MM2C (sv).
It is still uncertain whether M2C (sv) prion and M2C (lv) prion differ or not. The sCJD-MM2C (lv) was transmitted successfully to Ki-Bank mice (Kobayashi et al., 2019b) but not to Ki-ChM mice (Korth et al., 2003; Kitamoto et al., unpublished data) and neither M2C (sv) prion nor M2C (lv) prion could be amplified by our cell-PMCA. At present, M2C (sv) prion has not been discriminated from M2C (lv) prion according to the transmissibility or amplification properties. However, Cracco et al. (2017) have reported that the biochemical properties of sFI prion were different from those of sCJD-MM2C prion based on a detailed comparative assay of the PrPSc conformer. It would be useful to analyse the biochemical properties of M2C (sv) prion if we can identify a sporadic form of MM2C (sv) without OD.
Supplementary Material
Acknowledgment
We thank H. Kudo, A. Yamazaki, Y. Sasaki and M. Yamamoto for their excellent technical assistance and B. Bell for critical review of the article.
Funding
This study was supported by Grant-in-Aid for Scientific Research from Japan Society for the Promotion of Science (18H02738, 19K22588, and 19KK0213 to T.K. and 18K07490 to A.T.) and a Grant-in-Aid for Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control) from Ministry of Education, Culture, Sports, Science and Technology (T.K.). Research for the development of decontamination/disinfection procedures against various sCJD subgroups has been supported by a Grant-in-Aid from the Research Committee of Surveillance and Infection Control of Prion Disease, the Ministry of Health, Labor and Welfare of Japan (T.K.).
Competing interests
The authors report no competing interests.
Glossary
- FFI
fatal familial insomnia
- lv
large vacuolation
- OD
olivary degeneration
- PMCA
protein misfolding cyclic amplification
- PRNP
human prion protein gene
- PrP
prion protein
- PrPC
normal cellular isoform
- PrPSc
disease-associated isoform
- SC
spongiform changes
- sCJD
sporadic Creutzfeldt–Jakob disease
- sFI
sporadic fatal insomnia
- sv
small vacuolation
References
- Abu-Rumeileh S, Redaelli V, Baiardi S, Mackenzie G, Windl O, Ritchie DL, et al. Sporadic fatal insomnia in Europe: phenotypic features and diagnostic challenges. Ann Neurol 2018; 84: 347–60. [DOI] [PubMed] [Google Scholar]
- Asano M, Mohri S, Ironside JW, Ito M, Tamaoki N, Kitamoto T.. vCJD prion acquires altered virulence through trans-species infection. Biochem Biophys Res Commun 2006; 342: 293–9. [DOI] [PubMed] [Google Scholar]
- Chen S, He S, Shi XH, Shen XJ, Liang KK, Zhao JH, et al. The clinical features in Chinese patients with PRNP D178N mutation. Acta Neurol Scand 2018; 138: 151–5. [DOI] [PubMed] [Google Scholar]
- Collinge J, Palmer MS, Sidle KC, Gowland I, Medori R, Ironside J, et al. Transmission of fatal familial insomnia to laboratory animals. Lancet 1995; 346: 569–70. [DOI] [PubMed] [Google Scholar]
- Cracco L, Notari S, Cali I, Sy MS, Chen SG, Cohen ML, et al. Novel strain properties distinguishing sporadic prion diseases sharing prion protein genotype and prion type. Sci Rep 2017; 7: 38280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb LG, Haltia M, Brown P, Nieto A, Kovanen J, McCombie WR, et al. New mutation in scrapie amyloid precursor gene (at codon 178) in Finnish Creutzfeldt-Jakob kindred. Lancet 1991; 337: 425. [DOI] [PubMed] [Google Scholar]
- Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science 1992; 258: 806–8. [DOI] [PubMed] [Google Scholar]
- Grathwohl KU, Horiuchi M, Ishiguro N, Shinagawa M.. Improvement of PrPSc-detection in mouse spleen early at the preclinical stage of scrapie with collagenase-completed tissue homogenization and Sarkosyl-NaCl extraction of PrPSc. Arch Virol 1996; 141: 1863–74. [DOI] [PubMed] [Google Scholar]
- Hirose K, Iwasaki Y, Izumi M, Yoshida M, Hashizume Y, Kitamoto T, et al. MM2-thalamic-type sporadic Creutzfeldt-Jakob disease with widespread neocortical pathology. Acta Neuropathol 2006; 112: 503–11. [DOI] [PubMed] [Google Scholar]
- Kawasaki K, Wakabayashi K, Kawakami A, Higuchi M, Kitamoto T, Tsuji S, et al. Thalamic form of Creutzfeldt-Jakob disease or fatal insomnia? Report of a sporadic case with normal prion protein genotype. Acta Neuropathol 1997; 93: 317–22. [DOI] [PubMed] [Google Scholar]
- Kitamoto T, Mohri S, Ironside JW, Miyoshi I, Tanaka T, Kitamoto N, et al. Follicular dendritic cell of the knock-in mouse provides a new bioassay for human prions. Biochem Biophys Res Commun 2002; 294: 280–6. [DOI] [PubMed] [Google Scholar]
- Kitamoto T, Muramoto T, Hilbich C, Beyreuther K, Tateishi J.. N-terminal sequence of prion protein is also integrated into kuru plaques in patients with Gerstmann-Straussler syndrome. Brain Res 1991; 545: 319–21. [DOI] [PubMed] [Google Scholar]
- Kitamoto T, Nakamura K, Nakao K, Shibuya S, Shin RW, Gondo Y, et al. Humanized prion protein knock-in by Cre-induced site-specific recombination in the mouse. Biochem Biophys Res Commun 1996; 222: 742–7. [DOI] [PubMed] [Google Scholar]
- Kitamoto T, Ohta M, Doh-Ura K, Hitoshi S, Terao Y, Tateishi J.. Novel missense variants of prion protein in Creutzfeldt-Jakob disease or Gerstmann-Straussler syndrome. Biochem Biophys Res Commun 1993; 191: 709–14. [DOI] [PubMed] [Google Scholar]
- Kitamoto T, Shin RW, Doh-Ura K, Tomokane N, Miyazono M, Muramoto T, et al. Abnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous system in patients with Creutzfeldt-Jakob disease. Am J Pathol 1992; 140: 1285–94. [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Asano M, Mohri S, Kitamoto T.. Cross-sequence transmission of sporadic Creutzfeldt-Jakob disease creates a new prion strain. J Biol Chem 2007; 282: 30022–8. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Asano M, Mohri S, Kitamoto T.. A traceback phenomenon can reveal the origin of prion infection. Neuropathology 2009; 29: 619–24. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Iwasaki Y, Takao M, Saito Y, Iwaki T, Qi Z, et al. A novel combination of prion strain co-occurrence in patients with sporadic Creutzfeldt-Jakob disease. Am J Pathol 2019a; 189: 1276–83. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Matsuura Y, Takeuchi A, Yamada M, Miyoshi I, Mohri S, et al. A domain responsible for spontaneous conversion of bank vole prion protein. Brain Pathol 2019b; 29: 155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Sakuma N, Matsuura Y, Mohri S, Aguzzi A, Kitamoto T.. Experimental verification of a traceback phenomenon in prion infection. J Virol 2010; 84: 3230–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korth C, Kaneko K, Groth D, Heye N, Telling G, Mastrianni J, et al. Abbreviated incubation times for human prions in mice expressing a chimeric mouse-human prion protein transgene. Proc Natl Acad Sci USA 2003; 100: 4784–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugaresi E, Medori R, Montagna P, Baruzzi A, Cortelli P, Lugaresi A, et al. Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N Engl J Med 1986; 315: 997–1003. [DOI] [PubMed] [Google Scholar]
- Manetto V, Medori R, Cortelli P, Montagna P, Tinuper P, Baruzzi A, et al. Fatal familial insomnia: clinical and pathologic study of five new cases. Neurology 1992; 42: 312–9. [DOI] [PubMed] [Google Scholar]
- Mastrianni JA, Nixon R, Layzer R, Telling GC, Han D, DeArmond SJ, et al. Prion protein conformation in a patient with sporadic fatal insomnia. N Engl J Med 1999; 340: 1630–8. [DOI] [PubMed] [Google Scholar]
- McLean CA, Storey E, Gardner RJ, Tannenberg AE, Cervenakova L, Brown P.. The D178N (cis-129M) “fatal familial insomnia” mutation associated with diverse clinicopathologic phenotypes in an Australian kindred. Neurology 1997; 49: 552–8. [DOI] [PubMed] [Google Scholar]
- Medori R, Tritschler HJ, LeBlanc A, Villare F, Manetto V, Chen HY, et al. Fatal familial insomnia, a prion disease with a mutation at codon 178 of the prion protein gene. N Engl J Med 1992; 326: 444–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moda F, Suardi S, Di Fede G, Indaco A, Limido L, Vimercati C, et al. MM2-thalamic Creutzfeldt-Jakob disease: neuropathological, biochemical and transmission studies identify a distinctive prion strain. Brain Pathol 2012; 22: 662–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonno R, Di Bari MA, Cardone F, Vaccari G, Fazzi P, Dell'Omo G, et al. Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog 2006; 2: e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parchi P, Castellani R, Cortelli P, Montagna P, Chen SG, Petersen RB, et al. Regional distribution of protease-resistant prion protein in fatal familial insomnia. Ann Neurol 1995; 38: 21–9. [DOI] [PubMed] [Google Scholar]
- Parchi P, Chen SG, Brown P, Zou W, Capellari S, Budka H, et al. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Sträussler-Scheinker disease. Proc Natl Acad Sci USA 1998a; 95: 8322–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parchi P, Giese A, Capellari S, Brown P, Schulz Schaeffer W, Windl O, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999; 46: 224–33. [PubMed] [Google Scholar]
- Parchi P, Petersen RB, Chen SG, Autilio-Gambetti L, Capellari S, Monari L, et al. Molecular pathology of fatal familial insomnia. Brain Pathol 1998b; 8: 539–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccardo P, Dlouhy SR, Lievens PM, Young K, Bird TD, Nochlin D, et al. Phenotypic variability of Gerstmann-Straussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol 1998; 57: 979–88. [DOI] [PubMed] [Google Scholar]
- Piccardo P, Manson JC, King D, Ghetti B, Barron RM.. Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc Natl Acad Sci USA 2007; 104: 4712–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh Y, Ogawa M, Naito Y, Komatsuzaki Y, Tagaya H, Arima K, et al. Discordant clinicopathologic phenotypes in a Japanese kindred of fatal familial insomnia. Neurology 2010; 74: 86–9. [DOI] [PubMed] [Google Scholar]
- Sasaki K, Doh-Ura K, Wakisaka Y, Tomoda H, Iwaki T.. Fatal familial insomnia with an unusual prion protein deposition pattern: an autopsy report with an experimental transmission study. Neuropathol Appl Neurobiol 2005; 31: 80–7. [DOI] [PubMed] [Google Scholar]
- Synofzik M, Bauer P, Schols L.. Prion mutation D178N with highly variable disease onset and phenotype. J Neurol Neurosurg Psychiatry 2009; 80: 345–6. [DOI] [PubMed] [Google Scholar]
- Taguchi Y, Mohri S, Ironside JW, Muramoto T, Kitamoto T.. Humanized knock-in mice expressing chimeric prion protein showed varied susceptibility to different human prions. Am J Pathol 2003; 163: 2585–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi A, Kobayashi A, Parchi P, Yamada M, Morita M, Uno S, et al. Distinctive properties of plaque-type dura mater graft-associated Creutzfeldt-Jakob disease in cell-protein misfolding cyclic amplification. Lab Invest 2016; 96: 581–7. [DOI] [PubMed] [Google Scholar]
- Tateishi J, Brown P, Kitamoto T, Hoque ZM, Roos R, Wollman R, et al. First experimental transmission of fatal familial insomnia. Nature 1995; 376: 434–5. [DOI] [PubMed] [Google Scholar]
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996; 274: 2079–82. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Yamamoto T, Nishinaka K, Udaka F, Kameyama M, Kitamoto T.. Severe brain atrophy in a case of thalamic variant of sporadic CJD with plaque-like PrP deposition. Neuropathology 2001; 21: 138–43. [DOI] [PubMed] [Google Scholar]
- Zarranz JJ, Digon A, Atares B, Rodriguez-Martinez AB, Arce A, Carrera N, et al. Phenotypic variability in familial prion diseases due to the D178N mutation. J Neurol Neurosurg Psychiatry 2005; 76: 1491–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.