

Abstract
Astrocytes are glial cells of the central nervous system that become reactive under conditions of stress. The functional properties of reactive astrocytes depend on their stimulus that induces the upregulation of specific genes. Reactive astrocytes are a neuropathological feature of prion disorders; however, their role in the disease pathogenesis is not well understood. Here, we describe our studies of one polarization state of reactive astrocytes, termed A1 astrocytes, in the frontal cortex region of 35 human sporadic Creutzfeldt–Jakob disease brains encompassing a range of molecular sub-types. Examination of two mRNA markers of A1 astrocytes, C3 and GBP2, revealed a strong linear correlation between the two following their log-normalization (P = 0.0011). Both markers were found upregulated in the sporadic Creutzfeldt–Jakob disease brain compared with age-matched control tissues (P = 0.0029 and 0.0002, for C3log and GBP2log, respectively), and stratifying samples based on codon 129 genotype revealed that C3log is highest in homozygous methionine and lowest in homozygous valine patients, which followed a linear trend (P = 0.027). Upon assessing other disease parameters, a significant positive correlation was found between GBP2log and disease duration (P = 0.031). These findings provide evidence for a divergence in the astrocytic environment amongst patients with sporadic Creutzfeldt–Jakob disease based on molecular sub-type parameters of disease. While more research will be needed to determine the global changes in the genomic profiles and resulting functional properties of reactive astrocytes in disease, considering the evidence demonstrating that A1 astrocytes harbour neurotoxic properties, the changes seen in C3log and GBP2log in the current study may reflect differences in pathogenic mechanisms amongst the sporadic Creutzfeldt–Jakob disease sub-types associated with the A1 polarization state.
Keywords: prion, astrocyte, inflammation, Creutzfeldt–Jakob disease, microglia
Two RNA markers of A1 astrocytes were assessed in 35 sporadic Creutzfeldt–Jakob disease brain tissues that represented various molecular sub-type parameters. Following log-normalization, we show that the A1 astrocyte markers C3log and GBP2log stratify to disease parameters: C3log stratifies to codon 129 genotype and GBP2log positively correlates to disease duration.
Graphical Abstract
Graphical Abstract.
Introduction
Prion disorders are a group of transmissible, incurable neurodegenerative conditions that affect humans and animals. These diseases are caused by ‘prions’ (Prusiner, 1982) that are composed predominantly, if not entirely of PrPSc, which is a misfolded isoform of the normal cellular prion protein, PrPC. The ability of PrPSc to auto-catalytically propagate, by inducing the template-driven misfolding of PrPC, is the central pathogenic event in disease. Amplification of PrPSc with resultant accumulation results in neuronal loss in the CNS, as well as widespread vacuolation and reactive astrogliosis and microgliosis through unresolved mechanisms.
In humans, around 85% of Creutzfeldt–Jakob disease (CJD) cases are sporadic CJD (sCJD) with an unknown aetiology. An important genetic component in sCJD is the genotype [either valine (V) or methionine (M)] at codon 129 in the human prion protein gene with homozygous genotypes [homozygous methionine (MM) or homozygous valine] being over-represented compared with the heterogeneous methionine/valine genotype after standardization to their frequency in the normal population (Palmer et al., 1991; Alperovitch et al., 1999; Parchi et al., 1999). Historically, sCJD-associated PrPSc was distinguished from PrPC by its resistance to proteinase K digestion with different banding patterns of the glycosylated and unglycosylated isoforms of proteinase K-resistant PrP (especially mobility of the unglycosylated band) constituting identifiable ‘molecular types’ in the brains of such patients (Collinge et al., 1996; Parchi et al., 1996; Hill et al., 2003). The presence of proteinase K-resistant PrP types suggests that different conformations of PrPSc exist and, in combination with codon 129 genotype, defines various molecular sub-types of sCJD correlating with features of disease including disease duration and clinical presentation (Collinge et al., 1996; Parchi et al., 1999; Hill et al., 2003; Bishop et al., 2010). The correspondence of distinct clinical features with molecular sub-types suggests that divergent pathogenic mechanisms exist, although the neurobiological mechanisms underpinning these associations are not well understood.
Astrocytes are important glial cells of the CNS that become reactive in states of stress, causing them to adopt protective or damaging properties (reviewed in Liddelow and Barres, 2017). Two states of polarization have been observed for reactive astrocytes whereby specific activators cause the upregulation of certain genes (Zamanian et al., 2012), the combination of which is considered to dictate the cell’s functional phenotype. This has been thoroughly characterized for one of these states, termed A1 reactive astrocytes, that harbour highly neurotoxic properties (Liddelow et al., 2017). While more research is needed to determine the extent of these neurotoxic properties in disease models, the observation that A1-specific astrocyte markers C3 and GBP2 co-localize with astrocytes in the frontal cortex of sCJD brain (Hartmann et al., 2019) confirms that they are present in disease; however, it is not known whether these cells preferentially occur in disease of a certain molecular sub-type.
In the current study, we measured mRNA expression of the aforementioned A1 markers in frontal cortex samples from a cohort of 35 patients with sCJD , encompassing various molecular sub-types, and eight healthy age-matched controls. In further support of C3 and GBP2 as specific markers of A1 astrocytes, we report a strong correlation between the two in human brain and confirm both are elevated in sCJD. Critically, we found that C3log stratified to codon 129 genotype. Moreover, we report C3log and GBP2log expression levels associated with glycotype within codon 129 groups and observed a significant positive correlation between GBP2log and disease duration of patients with sCJD. These differences in the expression of the A1 astrocyte markers suggest that the reactive astrocyte environment differs between the molecular sub-types in sCJD. While further research into the genetic and functional properties of A1 astrocytes will provide important insight into the nature of these changes, their reported neurotoxic properties (Liddelow et al., 2017) suggest that their differential expression may contribute to divergences in prion disease pathogenesis amongst the molecular sub-types.
Materials and methods
Patient selection
The use of human tissue in this study was with the approval of La Trobe Human Research Ethics Committee (HEC18004), and all consent was obtained according to the Declaration of Helsinki. sCJD brain tissues were obtained from the Australian National CJD Registry, and control brain tissues were obtained from the Victorian Brain Bank. Frozen tissue was from the frontal cortex. For sCJD samples, cases for analysis were chosen from research-consented tissues after the consideration of disease duration of the patient, proteinase K-resistant PrP glycotype (Types 1–3) and codon 129 genotype, determined as described previously (Lewis et al., 2005). Diagnostic tests that may indicate prion disease ante-mortem include the detection of 14-3-3 protein in the CSF, an abnormal electroencephalogram showing periodic sharp wave activity and brain magnetic resonance imaging showing increased T2 signal in the basal ganglia and/or cerebral cortex. Whenever available, the following additional information was collected: age, gender, post-mortem delay, years of storage and results of ante-mortem clinical assessment for suspected CJD reviewed as per established diagnostic criteria (Will, 1999; Will et al., 2000). The control brain tissues used in this study were chosen according to the following criteria: (i) a non-neurological cause of death as reported from the Coroner or Full Medical Cause of Death Certificate written by the donor’s General Practitioner, (ii) no known neurological/psychiatric history in the donor’s general medical history, obtained from their treating doctor and (iii) absence of amyloid-β and tau (proteins found aggregated in the brain in many neurodegenerative proteinopathies, including Alzheimer’s disease) deposition in brain tissue sections following immunostaining to detect these proteins. The sCJD and control cohorts had an M:F gender ratio of 1:1.5 and 1:0.6, respectively.
RNA isolation from human brain
Extraction of RNA from human brain tissues was performed using the QIAzol lysis buffer and the RNeasy RNA isolation kit (Qiagen) according to the manufacturer’s instructions with the following adjustments: 5PRIME phase lock gel light tubes were used for phase separation to physically separate the aqueous from the interphase and the organic phase. The isolated aqueous phase was diluted 2:1:1 with chloroform and RNA-free water in fresh phase lock tubes, vortexed and centrifuged again. This was repeated for two chloroform extractions in total. After phase separation, tubes were checked to ensure the gel separating the phases resolved below the aqueous phase. In cases where the density of the sample was greater than the gel, RNA-free water was injected under the gel to reduce the density of the aqueous phase; the sample was then vortexed and centrifuged again. RNA was isolated in 60 μl of RNA-free water and purity and concentration determined using NanoDrop.
Reverse transcriptase and real-time qualitative PCR
cDNA was produced using the high-capacity cDNA reverse transcriptase kit, gene primers and fast advanced master mix by PCR under the following conditions: 25°C for 10 min, 37°C for 120 min, 85°C for 5 min and then held at 4°C. Quantitative reserve transcription polymerase chain reaction (RT-qPCR; Taqman Fast Advanced Master Mix; Applied Biosystems) was performed using the following Taqman gene expression array primers (Life Technologies): RPLP0: Hs00420895_gH, C3: Hs00163811_m1 and GBP2: Hs00894837_m1. Samples were processed using a ViiA7 Real-Time PCR System (Life Technologies) whereby each biological replicate was run in triplicate and normalized to the house-keeping gene RPLP0. Normalization of Ct values of each gene and determination of fold differences in gene expression (normalized to control samples) were calculated by the −2ΔΔCt method. QuantStudio™ Real-Time PCR software (Applied Biosystems) was used to analyse the data.
Statistical analysis
All data were assessed for outliers (ROUT Q = 1%) and normality (Shapiro–Wilk test). Pearson r correlation analysis was performed to determine the relationship between two normally distributed variables. Disease duration data was non-normally distributed and contained statistical outliers and so Spearman r correlation was used to assess this dataset. Statistical differences of mRNA between groups were examined using two-tailed Student’s t-test or one-way Analysis of Variance (ANOVA) with a multiple comparisons test for linear trend. All statistical analyses were performed using GraphPad Prism with a statistical criterion of 0.05.
Data availability
Upon reasonable request, the data that support the findings of this study are available from the corresponding author.
Results
Assessment of the relationship between two markers of A1 astrocytes, C3 and GBP2, in human brain
Patient information and molecular sub-type analysis of the 35 pathologically confirmed subjects with sCJD used in this study are shown in Table 1. The outcome (when available) of clinical investigations indicative of CJD is also summarized here. Eight normal brains were used as controls, which were age-matched to the disease cohort (Supplementary Fig. 1). Patient information of the control cohort is shown in Table 2.
Table 1.
Summary of sCJD patient information: sCJD PrPres glycotype 1–3, grouped by codon 129 genotype
| Codon 129 genotype | Glycotype | n | Sex (M, F) | Age (years), median (range) | Disease duration (months), median (range) | Typical magnetic resonance imaginga | Positive 14-3-3a | Typical electro encephalograma | PMDb (days), median (range) | Storage (years), median (range) |
|---|---|---|---|---|---|---|---|---|---|---|
| MM | 1 | 6 | 1, 5 | 72 (58–81) | 2.2 (1–3) | 5/5 | 5/5 | 2/5 | 1.5 (0–3) | 11.5 (5–12) |
| MM | 2 | 6 | 2, 4 | 64 (60–82) | 4.6 (1.5–9.6) | 5/5 | 6/6 | 4/5 | 2 | 11.5 (9–13) |
| MM | 3 | 6 | 4, 2 | 56.5 (47–85) | 8.4 (2–21) | 4/4 | 3/5 | 1/4 | 1 (0–2) | 11.5 (8–15) |
| MV | 1 | 2 | 1, 1 | 65.5 (58–73) | 1.8 (1.5–2.1) | 2/2 | 2/2 | 1/2 | 2 (1–3) | 14 (11–17) |
| MV | 2 | 4 | 2, 2 | 67 (63–74) | 2.7 (2–8) | 4/4 | 3/4 | 0/4 | 3 | 11.5 (7–13) |
| MV | 3 | 5 | 1, 4 | 67 (50–75) | 11.5 (6–40.5) | 4/4 | 3/3 | 1/3 | 2.5 (2–6) | 12 (10–15) |
| VV | 3 | 6 | 3, 3 | 71 (59–78) | 4.5 (2–6.5) | 6/6 | 3/3 | 1/5 | 4 (2–6) | 11 (6–15) |
| Total | 35 | 14, 21 | 67 (47–85) | 3.5 (1–40.5) | 30/30 | 25/28 | 10/28 | 2 (0–6) | 12 (5–17) | |
Disease duration and age are represented as median (range). Wherever performed, outcome of ante-mortem clinical testing results suggestive of CJD are provided. Variables relating to tissue handling are included as PMD and storage time of tissues prior to experimental use.
Values indicate the number of positive outcomes/total number of results available.
Value indicates number reported, where information is available.
PMD, post-mortem delay; F, female; M, male; VV, homozygous valine; PrPres, proteinase K-resistant PrP; MV, heterozygous methionine/valine.
Table 2.
Summary of control donor information: donor information, including cause of death of control cases
| Diagnosis | Sex | Age (years) | Cause of death | PMD (days) | Storage (years) |
|---|---|---|---|---|---|
| Control | M | 75 | Abdominal aortic aneurysm | 1 | 12 |
| Control | M | 73 | Ischaemic heart disease | 0 | 14 |
| Control | M | 77 | Acute posterolateral myocardial infarction | 2 | 15 |
| Control | M | 48 | Cardiac failure | 2 | 13 |
| Control | M | 57 | Intra-abdominal haemorrhage | 0 | 11 |
| Control | F | 60 | Pancreatic cancer | 1 | 1 |
| Control | F | 68 | Pulmonary thromboembolism | 2 | 14 |
| Control | F | 81 | Metastatic endometrial stromal sarcoma | 1 | 6 |
| Median (range) | 70 (48–81) | 1 (0–2) | 12.5 (1–15) | ||
Variables relating to tissue handling are included as PMD and storage time of tissues prior to experimental use.
PMD, post-mortem delay; F, female; M, male.
The utility of C3 and GBP2 to serve as markers of A1 astrocytes in human brain has been confirmed previously (Liddelow et al., 2017; Hartmann et al., 2019). The translated proteins co-localize with astrocytes in the frontal cortex of human prion-diseased brain (Hartmann et al., 2019). Using fluorescence in situ hybridization, C3 selectively co-localizes with astrocytes and its mRNA is elevated in a range of neurodegenerative diseases associated with A1 astrocyte reactivity (Liddelow et al., 2017). As specific markers for the same cell population and given their low basal expression in the CNS under normal conditions (Fagerberg et al., 2014), it is expected that C3 and GBP2 correlate within diseased and healthy human brain, although this has not been confirmed. Therefore, we first performed correlation analysis between C3 and GBP2 using all brain tissues. Data were non-normally distributed and hence log-normalization was performed for results of all samples. Assessment of the correlation between C3log and GBP2log was found to be strongly significant (Fig. 1, n = 43, P = 0.0011) suggesting that either may be used to describe the A1 phenotype but does not exclude some degree of complementarity. Accordingly, the A1 astrocyte markers were subsequently assessed in the sCJD cohort with respect to the molecular sub-types of disease.
Figure 1.
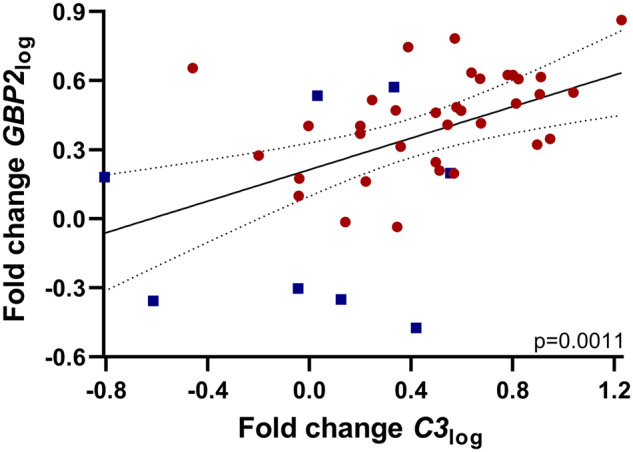
Correlation analysis of two mRNA markers of A1 astrocytes, GBP2 and C3, following log-transformation. Symbols represent the following: ■, control; ●, sCJD frontal cortex brain tissues. GBP2log and C3log significantly correlate (Pearson r test, P = 0.0011). Graph shows line of best fit, and dotted lines show the 95% confidence interval (95% Confidence Interval = 0.2111–0.6827, r = 0.4810); n = 43.
C3 log and GBP2log stratify to molecular sub-type parameters in sCJD brains
The expression of C3log and GBP2log was examined in both sCJD and control brain tissues. C3log was significantly elevated in sCJD brain tissues compared with control counterparts (Fig. 2A, P = 0.0029). Using the sCJD samples, these data were then divided into groups according to the genotype at codon 129 of the patient. Following this stratification to genotype, a significant difference was found in C3log expression between the groups that exhibited a linear trend whereby MM samples had the highest and homozygous valine had the lowest C3log expression (Fig. 2B, P = 0.027). Likewise, GBP2log was also elevated in sCJD brain compared with control tissues (Fig. 2C, P = 0.0002). Although a similar trend was observed, unlike C3log, GBP2log stratification according to codon 129 did not reach significance (Fig. 2D). The expression of both markers in sCJD tissues was subsequently subdivided according to the proteinase K-resistant PrP glycotype and the codon 129 genotype (Fig. 3A and C). While no significant differences were identified amongst the groups, Type 3 patients of both MM and heterozygous methionine/valine genotypes exhibited the highest C3log and GBP2log mean values compared with Type 1 and Type 2 counterparts. Type 1 cases in both MM and heterozygous methionine/valine typically have shorter disease durations than Type 3 counterparts (Lewis et al., 2005); hence, correlation analysis was next performed between C3log or GBP2log and disease duration (Fig. 3B and D). A significant positive correlation was found between disease duration and GBP2log (Fig. 3D, P = 0.031). No significant correlation was observed between disease duration and C3log (Fig. 3B). Neither C3log nor GBP2log correlated with other patient variables (gender, age, post-mortem delay or years of tissue storage) in either the control or sCJD groups (Supplementary Figs 2 and 3).
Figure 2.
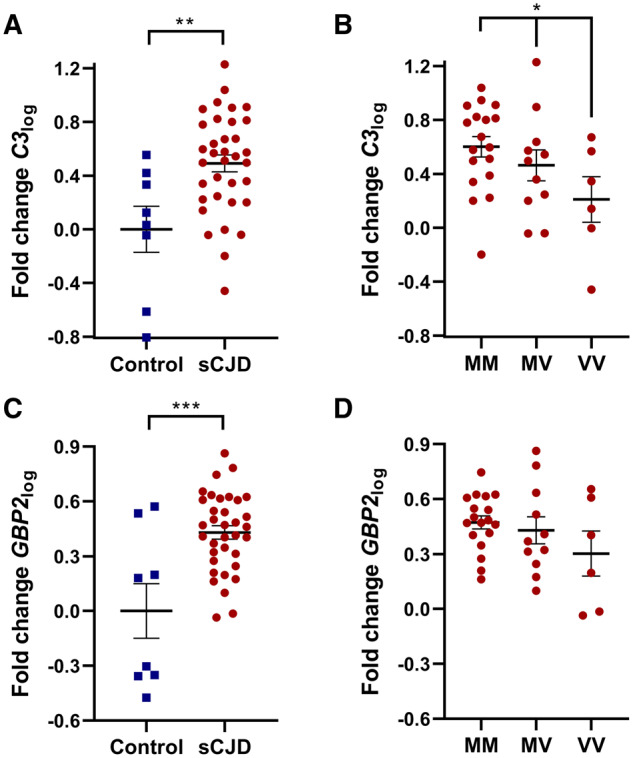
Expression of reactive A1 astrocyte markers in sCJD and control frontal cortex brain tissues and stratification to codon 129 genotype in sCJD. Symbols represent the following: ■, control; ●, sCJD brain tissues. (A) C3log was significantly elevated in sCJD brain tissues compared with control samples (Student’s t-test, n = 8,35, P = 0.0029) and (B) in the sCJD tissues C3log expression stratified according to codon 129 genotype (one-way ANOVA with a multiple comparisons test for linear trend, n = 18,11,6, P = 0.027). (C) GBP2log was significantly elevated in sCJD brain tissues compared with control samples (Student’s t-test, n = 8,35, P = 0.0002). (D) Stratifying GBP2log expression to codon 129 genotype was not statistically significant (one-way ANOVA with a multiple comparisons test for linear trend, n = 18,11,6).
Figure 3.

Expression of A1 markers in sCJD frontal cortex brain tissues based on codon 129 and PrPres glycotype and correlations to disease duration. (A) C3log expression grouped by PrPres glycotype and genotype at codon 129 (n = 6,6,6,2,4,5,6) and (B) C3log did not correlate with disease duration (Spearman r correlation, n = 35). Graph shows line of best fit, and dotted lines show the 95% confidence interval (95% Confidence Interval =−0.2817 to −0.4002, r = 0.06707). (C) GBP2log expression grouped based on PrPres glycotype and genotype at codon 129 (n = 6,6,6,2,4,5,6) and (D) GBP2log correlated with disease duration (Spearman r correlation, n = 35). Graph shows line of best fit, and dotted lines show the 95% confidence interval (95% Confidence Interval = 0.02573–0.6286, r = 0.3648). PrPres, proteinase K-resistant PrP.
Discussion
Reactive A1 astrocytes have been reported to exhibit powerful neurotoxic properties (Liddelow et al., 2017). These cells are present in sCJD brain (Hartmann et al., 2019), as well as brains from a range of other neurodegenerative conditions (Liddelow et al., 2017). In a transgenic rodent model of Parkinson’s disease, ablating the cytokines that stimulate their production results in neuroprotection (Yun et al., 2018). Likewise, an A1 profile is observed in a tauopathy mouse model (Shi et al., 2017). These studies have led to merited discussion on A1 astrocytes as a therapeutic target for neurodegenerative disorders that are associated with their activation [reviewed in Liddelow and Barres (2017)].
It is well established that sCJD is phenotypically highly heterogeneous, with several groups reporting clinical correlation with molecular sub-type parameters of disease (Collinge et al., 1996; Parchi et al., 1999; Hill et al., 2003; Bishop et al., 2010). It is not known whether any given sub-type is particularly vulnerable to produce an A1 environment. In this study, we confirm that two markers of A1 astrocytes are significantly elevated in sCJD brain and that they associate with disease parameters: C3log stratifies to codon 129 genotype while GBP2log positively correlates to disease duration.
In rodents, A1 astrocytes can be induced by factors released from activated microglia following exposure to the proinflammatory molecule lipopolysaccharide (Zamanian et al., 2012; Liddelow et al., 2017). Hence, the generation of A1 astrocytes is intimately related to inflammation. Their stratification to codon 129 status in sCJD suggests that disease associated with the 129MM genotype exhibits enhanced microglial and inflammatory responses. This is consistent with another study showing that microglial (CD11b, CD68) and proinflammatory (Tumor Necrosis Factor α) markers are elevated in frontal cortex sCJD tissues of 129MM compared with 129 homozygous valine (Llorens et al., 2014). However, the relevance of the codon 129 genotype to A1 astrocytes may also be related to astrocyte-specific PrP propagation. Astrocytes are amenable hosts for prion replication and astrocytes derived from human stem cells host prion replication most efficiently when the infecting inoculum (derived from human sCJD brain) has the same codon 129 genotype as the stem cells (Krejciova et al., 2017). Given that the astrocyte-specific marker, Glial Fibrillary Acidic Protein, appears in concert with PrPSc deposition in human sCJD brain tissues (Muhleisen et al., 1995), this could indicate that astrocyte-specific propagation is relevant to 129MM patients. Additional assessment of these A1 astrocyte markers and PrPSc deposition across brain regions that have differential disease pathology amongst the sCJD sub-types would provide insight into these notions.
The significant positive correlation of GBP2log to disease duration suggests that A1 astrocytes may not solely be necessary for sCJD-associated neurotoxicity. This is consistent with a recent study that reported transgenic mice lacking the factors that activate A1 astrocytes do not exhibit neuroprotection following prion exposure (Hartmann et al., 2019). Collectively, these findings support the concept that microglia are neuroprotective in disease. The role of microglia in prion disease pathogenesis has long been a point of interest, with contrasting reports of their activation as enhancing or inhibiting disease progression in various animal models of disease [reviewed in Aguzzi and Zhu (2017)]. Several comprehensive in vivo and ex vivo studies using transgenic mice show a protective role of microglia associated with prion clearance (Falsig et al., 2008; Zhu et al., 2016). Hence, it is possible in sCJD, A1 astrocytes may be produced from activated microglia as a by-product or result of significant and/or prolonged neuroprotective actions. The progressive accumulation of these astrocytes in patients based on the duration of illness could ultimately mean that microglia can play both protective and damaging roles in disease, whereby their initial capacity to be protective may switch to damaging neurotoxic properties over time with corresponding generation of A1 astrocytes. Such considerations prompt the need for further analysis of A1 astrocytes in larger cohorts of sCJD tissues as well as better models to determine the precise nature of their generation over disease course. This is particularly relevant given that, despite the majority of patients having short disease durations (Table 1), 5–10% of CJD cases have duration of illness 24 months (Brown et al., 1984). Hence, comprehensive modelling of the role of A1 astrocytes and disease duration requires large patient numbers.
While there are numerous studies detailing the detrimental effects of A1 astrocytes and the utility of C3 and GBP2 to identify them, the genetic and phenotypic changes that astrocytes undergo upon activation are not well understood. Hence, there may be unknown nuances to the capacity of C3 and GBP2 to serve as markers for A1 astrocytes. For example, possibly, multiple polarization states exist that share a degree of overlap in genomic profiles. Alternatively, the markers themselves may undergo expression changes in other brain morbidities. Such notions may explain the variation in the abilities of the two markers to stratify to disease sub-types in the current study. However, the positive correlation between the two markers in control and sCJD brain, and their co-localization with astrocytes in sCJD brain (Hartmann et al., 2019) is evidence for C3 and GBP2 reporting a population of astrocytes that are equivalent or highly similar. Regardless, further investigations into reactive astrocytes and their activator/s will provide important additional insight into their role in disease and markers to identify them.
The results of this study may contribute to our understanding across the range of broadly related neurodegenerative conditions. Pathogenic mechanisms are shared amongst the different proteins that aggregate in the brain causing neurodegenerative disorders, including significant heterogeneity in the clinical and biochemical features of the diseases (reviewed in Ugalde et al., 2016; Walker 2016). In the context of A1 astrocytes, it is possible that their involvement in the range of neurodegenerative disorders depends on patient-specific disease parameters. Phenotyping of disease, including molecular sub-typing, could represent an important paradigm for determining the best strategies to target A1 astrocytes, with the identification of patient groups who would most benefit from therapies aimed at neurotoxic astrocytes across a range of neurodegenerative conditions.
Supplementary Material
Acknowledgements
The authors wish to thank the Australian National CJD Registry, the CJD Support Group Network, the friends, families and healthcare professionals who support these organizations. Control tissues were received from the Victorian Brain Bank, supported by The Florey Institute of Neuroscience and Mental Health and The Alfred and the Victorian Forensic Institute of Medicine and funded in part by Parkinson’s Victoria, MND Victoria, FightMND and Yulgilbar Foundation.
Funding
This project was funded by the CJD Support Group Network Memorial Award 2019 in memory of Catherine and Michael Heagerty (to C.L.U.) and the National Health and Medical Research Council (NHMRC; APP1132604 to A.F.H.). S.J.C. is supported in part by a National Health and Medical Research Council Practitioner Fellowship (APP1105784).
Competing interests
The authors report no competing interests.
Glossary
- CJD
Creutzfeldt–Jakob disease
- MM
homozygous methionine
- sCJD
sporadic Creutzfeldt–Jakob disease
References
- Aguzzi A, Zhu C.. Microglia in prion diseases. J Clin Invest 2017; 127: 3230–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alperovitch A, Zerr I, Pocchiari M, Mitrova E, Cuesta JDP, Hegyi I, et al. Codon 129 prion protein genotype and sporadic Creutzfeldt-Jakob disease. Lancet 1999; 353: 1673–4. [DOI] [PubMed] [Google Scholar]
- Bishop MT, Will RG, Manson JC.. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci USA 2010; 107: 12005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown P, Rodgers-Johnson P, Cathala FO, Gibbs CJ, Gajdusek DC.. Creutzfeldt-Jakob disease of long duration: clinicopathological characteristics, transmissibility, and differential diagnosis. Ann Neurol 1984; 16: 295–304. [DOI] [PubMed] [Google Scholar]
- Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF.. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 1996; 383: 685–90. [DOI] [PubMed] [Google Scholar]
- Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics 2014; 13: 397–406., [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsig J, Julius C, Margalith I, Schwarz P, Heppner FL, Aguzzi A, et al. A versatile prion replication assay in organotypic brain slices. Nat Neurosci 2008; 11: 109–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann K, Sepulveda-Falla D, Rose IVL, Madore C, Muth C, Matschke J, et al. Complement 3(+)-astrocytes are highly abundant in prion diseases, but their abolishment led to an accelerated disease course and early dysregulation of microglia. Acta Neuropathol Commun 2019; 7: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AF, Joiner S, Wadsworth JDF, Sidle KCL, Bell JE, Budka H, et al. Molecular classification of sporadic Creutzfeldt-Jakob disease. Brain 2003; 126: 1333–46. [DOI] [PubMed] [Google Scholar]
- Krejciova Z, Alibhai J, Zhao C, Krencik R, Rzechorzek NM, Ullian EM, et al. Human stem cell-derived astrocytes replicate human prions in a PRNP genotype-dependent manner. J Exp Med 2017; 214: 3481–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis V, Hill AF, Klug GM, Boyd A, Masters CL, Collins SJ. Australian sporadic CJD analysis supports endogenous determinants of molecular-clinical profiles. Neurology 2005; 65: 113–18 [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Barres BA.. Reactive astrocytes: production, function, and therapeutic potential. Immunity 2017; 46: 957–67. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017; 541: 481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens F, López-González I, Thüne K, Carmona M, Zafar S, Andréoletti O, et al. Subtype and regional-specific neuroinflammation in sporadic Creutzfeldt-Jakob disease. Front Aging Neurosci 2014; 6: 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhleisen H, Gehrmann J, Meyermann R.. Reactive microglia in Creutzfeldt-Jakob disease. Neuropathol Appl Neurobiol 1995; 21: 505–17. [DOI] [PubMed] [Google Scholar]
- Palmer MS, Dryden AJ, Hughes JT, Collinge J.. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature 1991; 352: 340–2. [DOI] [PubMed] [Google Scholar]
- Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 1996; 39: 767–78. [DOI] [PubMed] [Google Scholar]
- Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999; 46: 224–33. [PubMed] [Google Scholar]
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216: 136–44. [DOI] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. ; Alzheimer’s Disease Neuroimaging Initiative. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017; 549: 523–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugalde CL, Finkelstein DI, Lawson VA, Hill AF.. Pathogenic mechanisms of prion protein, amyloid-β and α-synuclein misfolding: the prion concept and neurotoxicity of protein oligomers. J Neurochem 2016; 139: 162–80. [DOI] [PubMed] [Google Scholar]
- Walker LC. Proteopathic strains and the heterogeneity of neurodegenerative diseases. Annu Rev Genet 2016; 50: 329–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will RG. Prion related disorders. J R Coll Physicians Lond 1999; 33: 311–5. [PMC free article] [PubMed] [Google Scholar]
- Will RG, Zeidler M, Stewart GE, Macleod MA, Ironside JW, Cousens SN, et al. Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol 2000; 47: 575–82. [PubMed] [Google Scholar]
- Yun SP, Kam T-I, Panicker N, Kim S, Oh Y, Park J-S, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med 2018; 24: 931–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, et al. Genomic analysis of reactive astrogliosis. J Neurosci 2012; 32: 6391–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Herrmann US, Falsig J, Abakumova I, Nuvolone M, Schwarz P, et al. A neuroprotective role for microglia in prion diseases. J Exp Med 2016; 213: 1047–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Upon reasonable request, the data that support the findings of this study are available from the corresponding author.