

Abstract
Dementia severity can be quantitatively described by the latent dementia phenotype ‘δ’ and its various composite ‘homologues’. We have explored δ’s blood-based protein biomarkers in the Texas Alzheimer’s Research and Care Consortium. However, it would be convenient to replicate them in the Alzheimer’s Disease Neuroimaging Initiative. To that end, we have engineered a δ homologue from the observed cognitive performance measures common to both projects [i.e. ‘d:Texas Alzheimer’s Research and Care Consortium to Alzheimer’s Disease Neuroimaging Initiative’ (dT2A)]. In this analysis, we confirm 13/22 serum proteins as partial mediators of age’s effect on dementia severity as measured by dT2A in the Texas Alzheimer’s Research and Care Consortium and then replicate 4/13 in the Alzheimer’s Disease Neuroimaging Initiative’s plasma data. The replicated mediators of age-specific effects on dementia severity are adiponectin, follicle-stimulating hormone, pancreatic polypeptide and resistin. In their aggregate, the 13 confirmed age-specific mediators suggest that ‘cognitive frailty’ pays a role in dementia severity as measured by δ. We provide both discriminant and concordant support for that hypothesis. Weight, calculated low-density lipoprotein and body mass index are partial mediators of age’s effect in the Texas Alzheimer’s Research and Care Consortium. Biomarkers related to other disease processes (e.g. cerebrospinal fluid Alzheimer’s disease-specific biomarkers in the Alzheimer’s Disease Neuroimaging Initiative) are not. It now appears that dementia severity is the sum of multiple independent processes impacting δ. Each may have a unique set of mediating biomarkers. Age’s unique effect appears to be at least partially mediated through proteins related to frailty. Age-specific mediation effects can be replicated across cohorts and biofluids. These proteins may offer targets for the remediation of age-specific cognitive decline (aka ‘senility’), help distinguish it from other determinants of dementia severity and/or provide clues to the biology of Aging Proper.
Keywords: aging, cognition, dementia, g, intelligence
We confirm 13 proteins as mediators of age’s unique association with ‘δ’ (a dementia-specific phenotype) (path a) and replicate four across samples and biofluids (i.e. serum–plasma). Our results suggest that the dementing effect of Aging Proper (i.e. ‘senility’) is mediated by frailty and not via Alzheimer’s disease-specific neurodegeneration.
Graphical Abstract
Introduction
Dementia’s essential feature is a disruption of the ‘cognitive correlates of functional status’ (Royall et al., 2007). The assessment of those correlates can be approached by confirmatory factor analysis in a structural equation model framework (Royall et al., 2012). Functional status appears to be linked to cognitive performance through Spearman’s general intelligence factor g rather than through domain-specific cognitive abilities (Spearman, 1904; Royall and Palmer, 2014). Using bifactor confirmatory factor analysis we can parse g into two orthogonal (unrelated) fractions: (i) the psychometric correlates of functional status (i.e. ‘δ’, for ‘dementia’) and (ii) g', i.e. residual variance in g that is empirically unrelated to instrumental activities of daily living. This approach divorces functionally salient cognitive impairment from cognitive impairment per se.
The latent variable δ can be reified as a composite ‘d-score’ and applied to individuals as an omnibus dementia severity metric, i.e. a dementia-specific phenotype. As g is thought to contribute to all cognitive measures, it has proven feasible to construct δ from a wide range of measures/batteries. So many batteries are available that we distinguish each embodiment as a δ ‘homologue’. In genetics, a homologue is a gene descended from an ancestral gene in the same species and preserves the original’s function.
All validated δ homologues exhibit strong associations with dementia severity (e.g. as measured by the Clinical Dementia Rating Scale ‘Sum of Boxes’; Hughes et al., 1982) and achieve high areas under the receiver operating characteristic curve (ROC) for the discrimination of various dementias from normal controls (NC). Moreover, δ appears to be agnostic to dementia’s aetiology. Although it has a high area under the receiver operating characteristic curve to discriminate all-cause dementia from NC and cases of mild cognitive impairment (MCI) (Gavett et al., 2015), δ cannot distinguish any two dementing conditions (John et al., 2016).
We have been studying δ homologues and their biomarkers in the Texas Alzheimer’s Research and Care Consortium (TARCC). TARCC is a large (N ≅ 3500), well-characterized, ethnically diverse convenience sample with annual longitudinal follow-up (Waring et al., 2008). Age, APOE ε4 and depressive symptoms are independently associated with δ and may exert their dementing effects through it. Each of their associations is partially mediated by largely nonoverlapping panels of serum protein biomarkers suggesting that they reflect independent dementing processes (Royall et al., 2016, 2017a, b).
Age’s effect was mediated by 22 proteins (Royall et al., 2016). Several were ‘somatomedins’ including insulin-like growth factor 1 (IGF-1) and IGF-binding protein 2 (IGF-BP2), which had the largest effect. In their aggregate, they implicate ‘cognitive frailty’ as the cause of age-specific functionally salient cognitive impairment (i.e. ‘senility’). Frailty has traditionally been conceived as a strictly physical problem (Fried et al., 2001). Its cognitive aspects are largely unexplored. However, we have been able to show that a frailty index, comprising physical indicators, mediates the majority (51%) of age’s effect on δ in a population-based cohort of elderly Mexican-Americans (Palmer and Royall, 2019). Thus, cognitive frailty may be a dementing condition.
Although each risk factor’s association with δ is statistically weak to moderate, five proteins rationally selected by our method fully attenuate their 9-fold aggregate 5-year MCI conversion risk (Royall and Palmer, 2019a). This suggests first that the protein mediators selected by our methods may offer treatment targets for individual dementia risks (i.e. by a personalized treatment approach) but second that these risk factors are independent dementia-specific processes and do not contribute to a single dementing illness (e.g. Alzheimer’s disease) by any final common pathway.
Regardless, our findings await confirmation in other cohorts. To that end, we have developed the ability to replicate TARCC’s biomarker findings in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). ADNI is a second large, well-characterized convenience sample created to test biomarker findings of relevance to Alzheimer’s disease. As it happens, TARCC’s methods were largely predicated on ADNI’s. Both studies share a common subset of cognitive measures and a panel of ≅100 blood-based biomarkers by a common vendor.
The ‘TARCC to ADNI’ δ homologue (dT2A) was engineered from a common set of cognitive performance measures (Royall et al., 2019). In TARCC, dT2A has been reported (i) to have excellent fit, (ii) to exhibit factor equivalence across random subsets of the sample, (iii) to be strongly correlated with dementia severity as measured by the Clinical Dementia Rating Scale and (iv) to exhibit an area under the receiver operating characteristic curve of 0.981 (0.976–0.985) for the discrimination between Alzheimer’s disease (AD) and NC. In ADNI, dT2A also had excellent fit, correlated (r = 0.96) with Clinical Dementia Rating Scale ‘Sum of Boxes’ (P < 0.001) and achieved an area under the receiver operating characteristic curve of 1.0 (0.995–1.00) for the discrimination of Alzheimer’s disease from NC.
Both datasets have limitations that may hinder replications. All δ homologues ‘target’ a measure of instrumental activities of daily living. TARCC used Lawton and Brody’s instrumental activities of daily living index (IADL) (Lawton and Brody, 1969), but ADNI uses the Functional Assessment Questionnaire (FAQ) (Pfeffer et al., 1982). While their biomarker panels were obtained from a common vendor, TARCC measures them in serum, while ADNI measures them in plasma. Some proteins on each study’s panel are not available on the other’s, and technical problems prevent the analysis of certain proteins in either sample.
On the other hand, each study has unique strengths. ADNI provides access to both structural neuroimaging and functional neuroimaging and so-called Alzheimer’s disease-specific cerebrospinal fluid (CSF) biomarkers [e.g. amyloid beta 1–42 (Aβ1–42), total tau (t-tau) and phosphorylated tau (p-tau18)]. TARCC is an ethnically diverse cohort. Thirty-six percent of its participants are Mexican-Americans (MA).
We propose to replicate the previously reported mediation effects of 22 age-related serum proteins in ADNI. This will involve confirmations within TARCC across two δ homologues with minimally overlapping cognitive batteries and replications across studies and two biofluids. We will also test several new mediators to provide both discriminant and concordant support for the hypothesis that age’s unique effect on dementia severity as measured by δ is mediated via frailty and not by plausible alternative aetiologies (e.g. diabetes mellitus or Alzheimer’s disease). We predict that weight and body mass index (BMI) will mediate age’s effect, but neither haemoglobin A1c (HgbA1c) nor Alzheimer’s disease-specific CSF biomarkers (in ADNI) (etc.). We also predict that treatment with acetylcholinesterase inhibitors will mediate age’s effect, as it has been reported to lower serum resistin levels (Satapathy et al., 2011), which have in turn been found to be elevated in Alzheimer’s disease patients (Kizilarslanoğlu et al., 2015), confirmed to show a dose-dependent association with clinical diagnoses in TARCC (Royall and Palmer, 2019b) and have been identified by us as a mediator of both age’s and depression’s independent effects on δ (in mutually adjusted models; Royall et al., 2016, 2017b).
Materials and methods
Subjects
This is a secondary analysis of data collected by TARCC and ADNI. Informed consent was obtained from all participants (or their legally authorized proxies) before data collection, and both studies were approved by their respective Institutional Review Boards. Because ADNI has few Hispanic subjects, and because ethnicity moderates the association between multiple serum proteins and δ in TARCC (Royall and Palmer, 2015, 2016), we restricted this analysis to non-Hispanic whites (NHW).
Texas Alzheimer’s Research and Care Consortium
TARCC is a longitudinally followed convenience sample of elderly volunteers recruited from five Texas medical schools (Waring et al., 2008). Each participant underwent a standardized annual examination that included a medical evaluation, neuropsychological testing and clinical interview. Categorical clinical diagnoses of ‘Alzheimer’s disease’, ‘MCI’ and ‘NC’ were established through consensus. The diagnosis of Alzheimer’s disease was based on National Institute for Neurological Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association criteria (McKhann et al., 1984). Consensus-based clinical diagnoses of ‘MCI’ were based on all available clinical data. Although TARCC is an ethnically diverse cohort, only NHW participants (N = 2551) were included in this analysis.
Alzheimer’s Disease Neuroimaging Initiative
ADNI data were obtained from the ADNI database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public–private partnership, led by Principal Investigator Michael W. Weiner, MD (Weiner and Veitch, 2015). The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early Alzheimer’s disease.
The initial 5-year study, ADNI-1, enrolled cognitively normal, MCI and Alzheimer’s disease cases. Subsequent studies (ADNI-GO and ADNI-2) added early- and late-MCI cohorts. ADNI has provided a framework for similar initiatives worldwide, including TARCC. Only ADNI’s NHW participants were included (N = 1668).
Clinical variables
dT2A
dT2A’s construction and validation have been recently reported (Royall et al., 2019). Its cognitive indicators were limited to observed measures that are common to both TARCC and ADNI, including the Boston Naming Test (BNT) (Kaplan et al., 1983), Category Fluency (Animals) (Morris et al., 1989), Logical Memory I and II (Wechsler, 1997), the Mini–Mental State Examination (Folstein et al., 1975), and Trail-Making Test Part B (Reitan, 1958).
dT2A’s target indicators
In TARCC, we used informant-rated instrumental activities of daily living (Lawton and Brody, 1969) as dT2A‘s target indicator. The Functional Assessment Questionnaire (Pfeffer et al., 1982) was used in ADNI. It is commonly used in dementia evaluations (Juva et al., 1997; Teng et al., 2010). Other investigators have employed Functional Assessment Questionnaire as the target of a δ homologue (Gavett et al., 2015; John et al., 2016).
Observed clinical measures
Observed clinical measures are often used as covariates or to provide external validation. The following measures are available in both TARCC and ADNI.
Self (informant)-reported age and gender are self-explanatory. Education was coded as a continuous measure of subject/informant reported years of formal education.
The Clinical Dementia Rating Scale ‘Sum of Boxes’ (Hughes et al., 1982): the Clinical Dementia Rating Scale is used to evaluate dementia severity. The rating assesses the patient’s cognitive ability to function in six domains—memory, orientation, judgement and problem solving, community affairs, home and hobbies and personal care. Information is collected during an interview with the patient and their caregiver (15 min).
Geriatric depression scale (GDS): depressive symptoms were assessed in both studies by the GDS (Sheikh and Yesavage, 1986; Maixner et al., 1995). GDS scores range from 0 to 30. Higher scores are worse. The GDS is valid in demented persons (Burke et al., 1989).
Apolipoprotein E genotyping
APOEε4 burden was coded zero—two, based on the number of ε4 alleles. TARCC’s APOE genotyping was conducted by the Ballantyne Lab at the Baylor College of Medicine in Houston Texas using standard polymerase chain reaction methods (Koch et al., 2002). ADNI’s APOE genotyping was performed on DNA extracted from peripheral blood cells and processed by the University of Pennsylvania AD Biofluid Bank Laboratory, as previously described (Saykin et al., 2010).
Mediating variables
Blood-based protein biomarkers from both studies were tested as mediators of age’s association with dT2A. Both studies obtained a highly reduplicative panel of blood-based protein biomarkers (N = 120) via a multiplexed immunoassay (i.e. the human multi-analyte profile) processed in by a common vendor [i.e. Rules-Based Medicine (RBM) of Austin, TX, USA]. A complete listing of the biomarkers offered by the human multi-analyte profile panel is available at http://www.myriadrbm.com/products-services/humanmap-services/humanmap/ (4 December 2019, date last accessed).
Blood-based biomarker methods
All RBM analyses were run in duplicate, and data were discarded when the duplicate values differed by >5%. All values recorded by RBM as ‘LOW’ were recorded and analysed. If >50% of the samples for a given analyte were recorded as ‘LOW’, all readings for that analyte were dropped. If <50% of the analytes were recorded as ‘LOW’, the LOW values were recorded as the least detectable dose divided by two. Some proteins in the human multi-analyte profile panel are not available to TARCC, ADNI or both.
Raw biomarker data from both studies were inspected to ascertain their normality. Data points beyond 3.0 standard deviations about the mean were labelled as ‘outliers’ and deleted. Logarithmic transformation was used to normalize highly skewed distributions. The data were then standardized, within each dataset, to a mean of zero and unit variance.
Other mediators
Other variables of interest were tested as potential mediators of age’s association with dT2A. Some (e.g. BMI) were chosen to provide construct validity for the hypothesis that age’s effect on dementia severity is mediated through cognitive ‘frailty’. Others (e.g. HgbA1c) were chosen to provide discriminant validity. Some variables were selected from TARCC while others were available only in ADNI.
Texas Alzheimer’s Research and Care Consortium mediators
Serum cholesterol, C-peptide, homocysteine, HgbA1c, high-density lipoprotein, low-density lipoprotein (calculated), lipoprotein-associated phospholipase A2 and triglycerides were obtained from the Ballantyne Lab. HgbA1c was measured in whole blood by the turbidimetric inhibition immunoassay. Homocysteine was measured in serum using the recombinant enzymatic cycling assay (i.e. Roche Hitachi 911).
Height and weight were obtained at all TARCC sites, and BMI was calculated from those variables. The presence of diabetes mellitus, hyperlipidaemia and/or hypertension were obtained from the subject or their informant and coded dichotomously. The duration of smoking exposure was obtained from the subject or their informant and coded continuously.
Alzheimer’s Disease Neuroimaging Initiative mediators
CNS structural and functional neuroimaging biomarkers and certain ‘Alzheimer’s disease-specific’ biomarkers measured in CSF were tested as potential mediators of age’s association with dT2A in ADNI.
Imaging biomarkers
Magnetic resonance imaging data
The participants underwent a standardized 1.5-T MRI protocol (www.loni.ucla.edu/ADNI/Research/Cores/index.shtml (4 December 2019, date last accessed)), which included 2 T1-weighted MRI scans using a sagittal volumetric magnetization prepared rapid gradient echo sequence with the following acquisition parameters: echo time 4 ms, repetition time 9 ms, flip angle 8° and acquisition matrix size 256 × 256 × 166 in the x-, y- and z-dimensions with a nominal voxel size of 0.94 × 0.94 × 1.2 mm3. Only one of the magnetization prepared rapid gradient echo sets was used for the analysis. The ADNI MRI quality control centre at the Mayo Clinic selected the magnetization prepared rapid gradient echo image with higher quality and corrected for system-specific image artefacts, as described in Jack et al. (2008). Further details are described in Wyman et al. (2013).
18F-Fluorodeoxyglucose positron emission tomography imaging data
All fluorodeoxyglucose PET scans were acquired using ADNI’s standardized fluorodeoxyglucose PET acquisition protocols. Raw PET data were uploaded to the University of Michigan for preprocessing to correct for differences in the PET scanners used across ADNI sites. During preprocessing, each of the 5-min emission frames acquired in every scan were co-registered and then averaged to the first frame. The image was then reoriented such that the anterior–posterior axis of the subject ran parallel to the anterior commissure–posterior commissure line and interpolated onto a uniform 60 × 160 × 96 voxel image grid, with 1.5-mm cubic voxels (http://adni.loni.usc.edu/methods/pet-analysis/pre-processing (4 December 2019, date last accessed)). Finally, a subject-specific mask was applied for intensity normalization (where average in the mask was one). Further details are described in Jagust et al. (2010).
Cerebrospinal fluid biomarkers
CSF was collected from the lower spine by lumbar puncture after an overnight fast as described in the ADNI procedures manual (www.adni-info.org). In brief, CSF was transferred in polypropylene tubes on dry ice within 1 h after collection and shipped overnight to the ADNI Biomarker Core laboratory at the University of Pennsylvania Medical Center. Aβ1–42, t-tau and p-tau18 were measured in each aliquot using the multiplex xMAP Luminex platform (Luminex Corp, Austin, TX, USA) with Innogenetics (INNO-BIA AlzBio3; Ghent, Belgium) immunoassay kit-based reagents. Full details of ADNI baseline CSF biomarker measurements are provided by Shaw et al. (2009).
Statistical analyses
These analyses were conducted in the NHW subset of TARCC’s most recent dataset (N = 2551) and in a combined sample of ADNI-1, ADNI-2 and ADNI-GO data (N = 1668).
The analysis was performed using Analysis of Moment Structures software (Arbuckle, 2006). The maximum likelihood estimator was chosen for these models. Covariances between the residuals were allowed to be estimated if they were significant and improved model fit.
Analysis sequence
We used the ‘TARCC to ADNI’ δ homologue (dT2A) as previously described (Royall et al., 2019). dT2A was constructed from baseline data using unadjusted indicators.
First, we constructed Multiple Indicators Multiple Causes (MIMIC) models (Muthén, 1979) of age’s effect on cognitive performance. By that approach, the latent dT2A construct acted as a mediator of age’s direct effects on dT2A’s observed indicators. The MIMIC model thereby distinguishes age’s direct effects on individual cognitive measures from its δ-specific indirect effects on all of them. Only age’s δ-related effects are likely to be disabling and therefore potentially ‘dementing’.
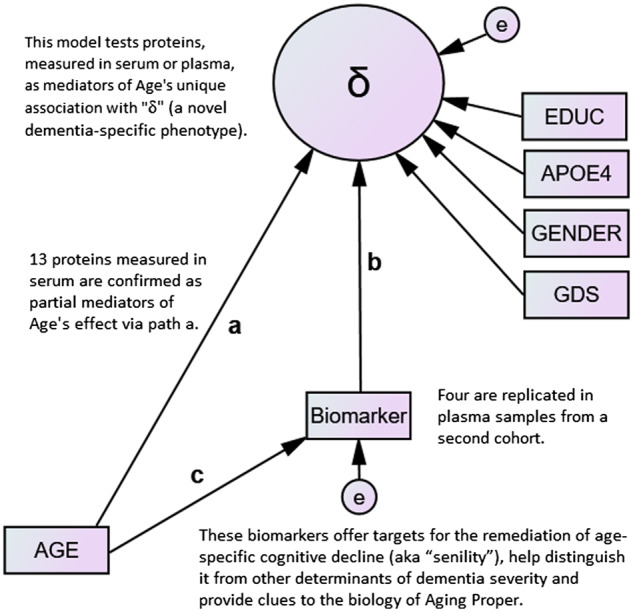
Next, we converted age’s association with dT2A in the MIMIC model into the ‘direct’ effect (i.e. ‘path a’) of a mediation model by introducing an observed biomarker (Fig. 1). Path ‘a’ represents age’s direct association with dT2A. Path ‘b’ represents the proposed mediator’s independent effect on dT2A. Because these models were hypothesis driven, no Bonferroni correction was applied. Path ‘c’ represents age’s effect on the proposed mediator. When both paths b and c are significant, the mediator’s effect on age’s direct association can be calculated by MacKinnon’s method (MacKinnon, 1994).
Figure 1.
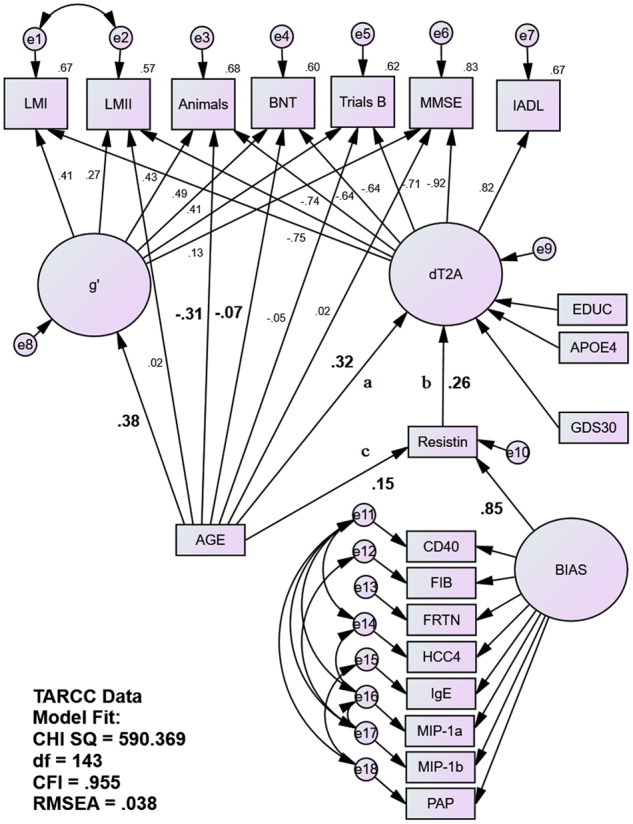
Fully adjusted MIMIC mediation model of serum resistin as a mediator of age’s unique association with the latent dT2A homologue in TARCC. Age’s association with the bifactor dT2A δ homologue is being partially mediated by serum resistin in TARCC’s NHW sample (N = 2251) independently of age’s direct effects on individual cognitive performance measures. Gender had no significant independent association with dT2A and was omitted from the model. AGE is correlated to EDUC, APOE4, GENDER and GDS (paths not shown). EDUC is also correlated with e8. Statistically significant structural paths of interest are in bold. Animals = animal naming; BNT = Boston Naming Test; CHI SQ = chi square; CD40 = cluster of differentiation 40; CFI = comparative fit index; EDUC = education (years); FIB = fibrinogen; FRTN = ferritin; HCC4 = human C–C motif chemokine-4; IADL = instrumental activities of daily living; IgE = immunoglobulin E; LMI = Wechsler Logical Memory immediate recall; LMII = Wechsler Logical Memory delayed recall; MIP-1a = macrophage inflammatory protein 1 alpha; MIP-1b = macrophage inflammatory protein 1 beta; MMSE = Mini–Mental State Examination; PAP = prostatic acid phosphatase; RMSEA = root-mean-square evaluative assessment; Trails B = Trail-Making Test Part B.
Age’s effect by path a and indirect effect via paths c–b were adjusted for APOE ε4 burden, education, gender and depressive symptoms (i.e. variables previously shown to exert age-independent effects on δ). We restricted this analysis to NHW only. Gender had no significant effect on δ in TARCC’s NHW, and so it was omitted from TARCC’s model (to improve fit).
TARCC’s RBM biomarkers are known to exhibit significant batch effects. In the past, we have adjusted each TARCC biomarker with dichotomous dummy variables coding batch. However, in this analysis, batch effects were assumed to be a source of ‘systematic’ error and were adjusted by the introduction of a latent ‘BIAS’ variable, indicated by the proposed mediator and multiple other proteins (i.e. cluster of differentiation 40, fibrinogen, ferritin, human C–C motif chemokine-4, immunoglobulin E, macrophage inflammatory protein 1α, macrophage inflammatory protein 1β and prostatic acid phosphatase). The latter biomarkers were chosen for their lack of associations with either age or δ in TARCC (when measured in serum; Royall et al., 2016). The same proteins, measured in plasma, were used as indicators of a BIAS construct in ADNI. The BIAS variable should also account for biofluid-specific measurement bias across studies, as that too is a systematic source of variance across all protein biomarkers. The BIAS construct and its indicators were dropped from models that tested non-protein mediators.
Missing data
We used full information maximum likelihood methods to address missing data. Full information maximum likelihood uses the entire observed data matrix to estimate parameters with missing data. In contrast to listwise or pairwise deletion, full information maximum likelihood yields unbiased parameter estimates and preserves the overall power of the analysis (Schafer and Graham, 2002; Graham, 2009).
Fit indices
The validity of structural models was assessed using two common test statistics. A non-significant chi square signifies that the data are consistent with the model (Bollen and Long, 1993). The ratio of the chi square to the degrees of freedom in the model is also of interest. A chi square/degrees of freedom ratio of <5.0 suggests an adequate fit to the data (Wheaton et al., 1977). The comparative fit index, with values ranging from 0 to 1, compares the specified model with a model of no change (Bentler, 1990). Comparative fit index values <0.95 suggest model misspecification. Values ≥0.95 indicate adequate to excellent fit. A root-mean-square error of approximation of ≤0.05 indicates a close fit to the data, with models <0.05 considering as ‘good’ fit and models up to 0.08 considering as ‘acceptable’ (Browne and Cudeck, 1993). All three fit statistics should be simultaneously considered to assess the adequacy of the models to the data.
Data availability
The data underlying the results presented in the study are available from TARCC and ADNI. Requests for TARCC data should be made to http://www.txalzresearch.org/research/ (4 December 2019, date last accessed). Requests for ADNI data should be made to adni.loni.usc.edu.
Results
Descriptive statistics is presented for two cohorts in Table 1 and by diagnoses in Table 2 (TARCC) and Table 3 (ADNI). The cohorts differed significantly on all measures. ADNI has a relatively high fraction of MCI cases, which were recruited explicitly into ADNI-2 and ADNI-GO. TARCC has a much higher prevalence of Mexican-American participants.
Table 1.
Descriptive statistics by sample (raw scores except where indicated)
| TARCC NHW (N = 2251) | ADNI NHW (N = 1668) | |
|---|---|---|
| Alzheimer’s disease cases, n (%) | 1100 (49.7) | 342 (19.7) |
| MCI cases, n (%) | 395 (17.8) | 978 (56.3) |
| NC, n (%) | 718 (32.4) | 417 (24.8) |
| Gender, female, n (%) | 1288 (57.2) | 919 (55.1) |
| Mean (SD) | Mean (SD/d1) | |
| Age | 73.1 (8.96) | 73.8 (7.2/0.09**) |
| Education | 15.1(2.78) | 15.9 (2.9/0.28**) |
| MMSE | 25.2 (5.04) | 27.2 (2.7/0.49**) |
| Animals | 14.4 (6.04) | 17.2 (5.9/0.47**) |
| BNTa | 9.2 (4.19) | 26.1 (4.51/b) |
| CDR-SB | 3.1 (3.59) | 1.6 (1.8/0.53**) |
| GDS | 4.9 (4.10) | 1.4 (1.4/1.14**) |
| LMI | 7.7 (4.43) | 9.3 (4.8/0.35**) |
| LMII | 7.5 (4.75) | 7.1 (5.3/0.08**) |
| Trails B | 8.0 (4.08) | 122.2 (75.8/c) |
d1 = Cohen’s d versus TARCC. Animals = animal naming; BNT = Boston Naming Test; CDR-SB = Clinical Dementia Rating scale ‘Sum of Boxes’; LMI = Wechsler Logical Memory immediate recall; LMII = Wechsler Logical Memory delayed recall; MA = Mexican-American; MMSE = Mini–Mental State Examination; SD = standard deviation; Trails B = Trail-Making Test Part B.
Scaled scores.
TARCC uses 30-item BNT, and ADNI uses 60-item BNT.
TARCC uses scale scores, and ADNI uses in seconds.
**P < 0.001.
Table 2.
Descriptive statistics by diagnosis (TARCC)
| TARCC total NHW sample (N = 2251), mean (SD) | TARCC NHW Alzheimer’s disease (N = 1100), mean (SD) | TARCC NHW MCI (N = 395), mean (SD) | TARCC NHW controls (N = 718), mean (SD) | |
|---|---|---|---|---|
| Gender, female (%) | 57.2 | 54.8 | 51.9 | 63.9 |
| Age | 73.1 (8.97) | 75.46 (8.51) | 72.4 (8.54) | 69.8 (8.81) |
| Education | 15.1 (2.79) | 14.75 (2.95) | 14.9 (2.59) | 15.6 (2.54) |
| MMSE | 25.2 (5.04) | 21.68 (4.87) | 27.5 (2.15) | 29.3 (0.97) |
| Animals | 14.3 (6.05) | 10.40 (4.28) | 15.8 (5.52) | 19.6 (3.90) |
| BNTa | 9.2 (4.19) | 6.88 (3.55) | 10.0 (3.43) | 12.4 (3.11) |
| CDR-SB | 3.1 (3.59) | 5.82 (3.29) | 1.3 (0.98) | 0.0 (0.08) |
| GDS30 | 4.9 (4.08) | 5.59 (4.20) | 5.6 (4.43) | 3.3 (3.21) |
| LMI | 7.7 (4.43 ) | 4.42 (2.65) | 8.3 (3.01) | 12.3 (2.68) |
| LMII | 7.5 (4.75) | 3.88 (2.53) | 8.2 (3.21) | 12.6 (2.71) |
| Trails B (s) | 8.0 (4.08) | 5.34 (3.37) | 9.1 (3.03) | 11.3 (2.53) |
Animals = animal naming; BNT = Boston Naming Test; CDR-SB = Clinical Dementia Rating scale ‘Sum of Boxes’; LMI = Wechsler Logical Memory immediate recall; LMII = Wechsler Logical Memory delayed recall; MA = Mexican-American; MMSE = Mini–Mental State Examination; SD = standard deviation; Trails B = Trail-Making Test Part B.
TARCC uses 30-item BNT.
Table 3.
Descriptive statistics by diagnosis (ADNI)
| ADNI total NHW sample (N = 1668), mean (SD) | ADNI NHW Alzheimer’s disease (N = 342), mean (SD) | ADNI NHW MCI a (N = 978), mean (SD) | ADNI NHW controls (N = 417), mean (SD) | |
|---|---|---|---|---|
| Gender, female (%) | 55.1 | 44.7 | 42.8 | 49.9 |
| Age | 73.8 (7.19) | 75.03 (7.79) | 72.91 (7.42) | 74.76 (5.73) |
| Education | 15.91 (2.86) | 15.18 (2.99) | 16.00 (2.82) | 16.28 (2.73) |
| MMSE | 27.17(2.67) | 23.22 (2.07) | 27.75 (1.81) | 29.07 (1.12) |
| Animals | 17.15 (5.93) | 12.25 (4.98) | 17.39 (5.22) | 20.60 (5.50) |
| BNTb | 25.97 (4.51) | 22.24 (6.05) | 26.43 (3.68) | 27.94 (2.66) |
| CDR-SB | 1.64 (1.79) | 4.39 (1.67) | 1.36 (0.95 | 0.03 (0.13) |
| GDS30 | 1.42 (1.40) | 1.65 (1.44) | 1.63 (1.41) | 0.75 (1.12) |
| LMI | 9.28 (4.83) | 4.08 (2.80) | 9.10 (3.91) | 13.98 (3.25) |
| LMII | 7.07 (5.33) | 1.37 (1.89) | 6.46 (4.10) | 13.18 (3.33) |
| Trails B (s) | 122.23 (75.78) | 191.46 (89.69) | 113.61 (65.42) | 85.68 (43.18) |
Animals = animal naming; BNT = Boston Naming Test; CDR-SB = Clinical Dementia Rating scale ‘Sum of Boxes’; LMI = Wechsler Logical Memory immediate recall; LMII = Wechsler Logical Memory delayed recall; MA = Mexican-American; MMSE = Mini–Mental State Examination; SCI = subjective cognitive impairment; SD = standard deviation; Trails B = Trail-Making Test Part B.
All subtypes and SCI.
ADNI uses 60-item BNT.
Technical issues precluded the testing of 6/22 proteins in ADNI. We can assume that >50% of those analytes’ samples were reported to be ‘LOW’ by RBM. We had no access to the unprocessed biomarker data and could not determine how many samples were removed from each cohort because their duplicate values differed by >5%, or because they were assessed as ‘outliers’ prior to analysis. However, an upper bound for these issues can be estimated by the number of cases with missing data when biomarker data were available and cannot have been more than 123/886 = 13.9% (i.e. for the unconfirmed biomarker glutathione S-transferase in TARCC). No data were missing in ADNI’s (N = 566) samples. The eight discordant confirmed biomarkers, i.e. angiopoetin-2N, creatinine kinase-MB, epidermal growth factor receptor 1, FAS, plasminogen activator inhibitor type 1, serum amyloid protein (SAP), thyroxine-binding globulin, and von Willebrand factor had no more than 15/886 = 1.7% missing values (i.e. for creatinine kinase-MB) in TARCC.
Age was significantly associated with dT2A in both cohorts (TARCC: r = 0.36, P < 0.001); ADNI: r = 0.16, P < 0.001) (by unadjusted path a). All mediation models had acceptable fit (e.g. resistin; Figs 1 and 2). Significant mediation effects were found for 13/22 of TARCC’s serum proteins (Table 4). Of the 16 potential mediators available in ADNI, significant mediation effects were found for four including adiponectin (APN; Z = 2.32, P = 0.02; 12.0%), FSH (FHS; Z = 2.21, P = 0.03; 8.2%), pancreatic polypeptide (PPP; Z = 2.00, P = 0.05; 14.2%) and resistin (Z = 2.01, P = 0.04; 8.3%). All mediation effects in both cohorts were partial, ranging from 3.3% (creatinine kinase-MB in TARCC) to 32.8% (IGF-binding protein 2 in TARCC). Near significant mediation effects were observed for plasma complement 3 (Z = −1.82, P = 0.07) and thyroxine-binding globulin (Z = −1.83, P = 0.07) in ADNI.
Table 4.
ADNI replication of age’s mediators on dT2A in TARCC
| Biomarker | TARCC (serum) | ADNI (plasma) |
|---|---|---|
| APN | r = 0.33 (P < 0.001) | r = 0.14 (P < 0.001) |
| Z = 2.50 (P = 0.01) | Z = 2.32 (P = 0.02) | |
| 8.8% | 12.0% | |
| ANG-2 | r = 0.33 (P < 0.001) | r = 0.16 (P < 0.001) |
| Z = 3.52 (P < 0.001) | Z = −1.01 (P = 0.31) | |
| 9.6% | c | |
| C3 | r = 0.36 (P < 0.001) | r = 0.17 (P < 0.001) |
| Z = −0.17 (P = 0.87) | Z = −1.82 (P = 0.07) | |
| b | b, c | |
| CK-MB | r = 0.35 (P < 0.001) | r = 0.15 (P < 0.001) |
| Z = 1.93 (P = 0.05) | Z = 0.42 (P = 0.68) | |
| 3.3% | c | |
| EGFR | r = 0.26 (P < 0.001) | r = 0.17 (P < 0.001) |
| Z = 5.20 (P < 0.001) | Z = −1.09 (P = 0.28) | |
| 26.9% | C | |
| FAS | r = 0.35 (P < 0.001) | r = 0.15 (P < 0.001) |
| Z = 2.37 (P = 0.02) | Z = 0.80 (P = 0.42) | |
| 4.3% | c | |
| FSH | r = 0.34 (P < 0.001) | r = 0.14 (P < 0.001) |
| Z = 2.77 (P = 0.006) | Z = 2.21 (P = 0.03) | |
| 7.1% | 8.2% | |
| GST | r = 0.37 (P < 0.001) | r = 0.15 (P < 0.001) |
| Z = −0.38 (P = 0.70) | Z = 1.40 (P = 0.16) | |
| b | b | |
| G-CSF | – | – |
| IGF-1 | r = 0.40 (P < 0.001) | r = 0.15 (P < 0.001) |
| Z = 0.20 (P = 0.84) | ||
| Z = −1.35 (P = 0.18) | ||
| c | c | |
| IGF-BP2 | r = 0.23 (P < 0.001) | – |
| Z = 5.80 (P < 0.001) | ||
| 32.8% | ||
| IL-5 | r = 0.34 (P < 0.001) | – |
| Z = 1.59 (P = 0.11) | ||
| b | ||
| MyG | r = 0.38 (P < 0.001) | r = 0.16 (P < 0.001) |
| Z = −1.53 (P = 0.13) | Z = −0.04 (P = 0.99) | |
| c | c | |
| PP | r = 0.32 (P < 0.001) | r = 0.13 (P < 0.001) |
| Z = 2.96 (P =0.003) | Z = 2.00 (P = 0.05) | |
| 10.2% | 14.2% | |
| PAI-1 | r = 0.32 (P < 0.001) | r = 0.16 (P < 0.001) |
| Z = 3.64 (P < 0.001) | Z = −1.57 (P = 0.12) | |
| 12.5% | b | |
| PDGF | r = 0.36 (P < 0.001) | – |
| Z = P 0.48 (P = 0.63) | ||
| c | ||
| Progesterone | r = 0.36 (P < 0.001) | – |
| Z = 0.51 (P = 0.61) | ||
| Resistin | r = 0.32 (P < 0.001) | r = 0.14 (P = 0.001) |
| Z = 4.02 (P < 0.001) | Z = 2.01 (P = 0.04) | |
| 10.7% | 8.3% | |
| S100b | r = 0.37 (P < 0.001) | – |
| Z = −1.25 (P = 0.21) | ||
| b | ||
| SAP | r = 0.32 (P < 0.001) | r = 0.16 (P < 0.001) |
| Z = 3.35 (P < 0.001) | Z = −0.86 (P = 0.39) | |
| 12.3% | b | |
| TBG | r = 0.34 (P < 0.001) | r = 0.17 (P < 0.001) |
| Z = 2.22 (P = 0.03) | Z = −1.83 (P = 0.07) | |
| 5.4% | b | |
| vWF | r = 0.34 (P < 0.001) | r = 0.16 (P < 0.001) |
| Z = 1.82 (P = 0.07) | Z = 0.10 (P = 0.92) | |
| 6.3% | c |
b: path b (mediator to dT2A) is significant at P < 0.05; c: path c (age to mediator) is significant at P < 0.05. ANG-2 = angiopoetin-2N; C3 = complement 3; CK-MB = creatinine kinase-MB; EGFR = epidermal growth factor receptor 1; FSH = follicle-stimulating hormone; G-CSF = granulocyte colony-stimulating factor; GST = glutathione S-transferase; IGF-BP2 = IGF-binding protein 2; IL-5 = interleukin 5; MyG = myoglobin; PAI-1 = plasminogen activator inhibitor type 1; PDGF = platelet-derived growth factor; PP = pancreatic polypeptide; S100b = S100 calcium-binding protein B; TBG = thyroxine-binding globulin; vWF = von Willebrand factor.
Figure 2.
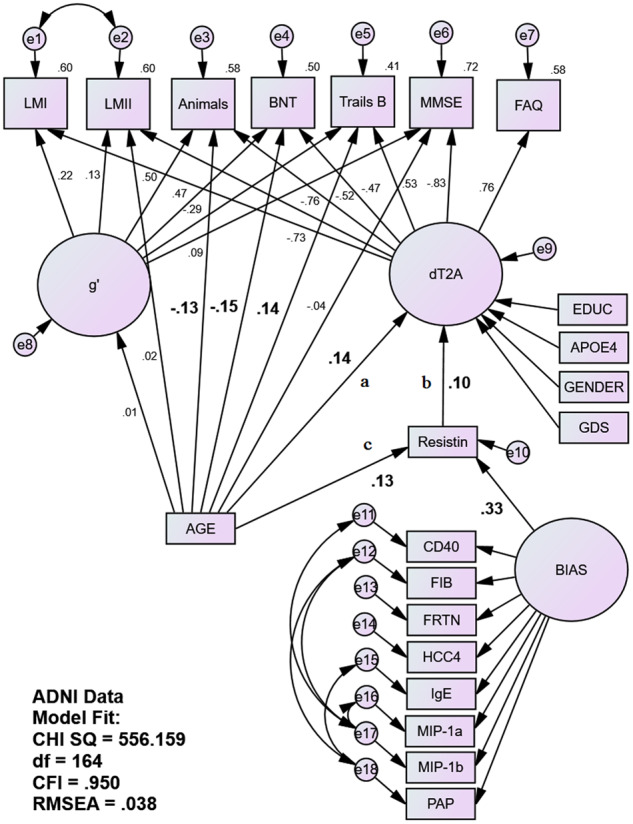
Fully adjusted MIMIC mediation model of plasma resistin as a mediator of age’s unique association with the latent dT2A homologue in in ADNI. Age’s association with the bifactor dT2A δ homologue is being partially mediated by plasma resistin in ADNI’s NHW sample (N = 1668) independently of age’s direct effects on individual cognitive performance measures. AGE is correlated to EDUC, APOE4, GENDER and GDS (paths not shown). EDUC is also correlated with e8. Statistically significant structural paths of interest are in bold. Animals = animal naming; BNT = Boston Naming Test; CHI SQ = chi square; CD40 = cluster of differentiation 40; CFI = comparative fit index; EDUC = education (years); FAQ = Functional Abilities Questionnaire; FIB = fibrinogen; FRTN = ferritin; HCC4 = human C–C motif chemokine-4; IgE = immunoglobulin E; LMI = Wechsler Logical Memory immediate recall; LMII = Wechsler Logical Memory delayed recall; MIP-1a = macrophage inflammatory protein 1 alpha; MIP-1b = macrophage inflammatory protein 1 beta; MMSE = Mini–Mental State Examination; PAP = prostatic acid phosphatase; RMSEA = root-mean-square evaluative assessment; Trails B = Trail-Making Test Part B.
ADNI also confirmed that 6/22 proteins were not likely to be mediators of senile cognitive decline in TARCC, at least in NHW. In every case, both models agreed which path (b or c) remained significant and which did not. In half of the cases (3/6), the proposed mediator was related to age but not to δ (in NHW) (i.e. complement 3, glutathione S-transferase and thyroxine-binding globulin). IGF-1, myoglobin and von Willebrand factor were related to δ but not age (in NHW). Thus, the TARCC and ADNI models agreed with regard to 8/16 testable proteins.
Table 5 presents additional mediation effects. In TARCC, age’s association with dT2A was not mediated by serum cholesterol, C-peptide, diabetes mellitus, homocysteine, HgbA1c, hyperlipidaemia, hypertension, lipoprotein-associated phospholipase A2, serum triglycerides or years of smoking history. It was partially mediated by high-density lipoprotein C (Z = 2.38, P = 0.02; 3.5%), low-density lipoprotein (calculated) (Z = −2.24, P = 0.03; 3.4%), weight (Z = 3.62, P < 0.001; 5.1%) and BMI (Z = 4.28, P < 0.001; 5.4%). Age’s effect was also partially mediated by both treatment by galantamine (Z = 2.51, P = 0.01; 1.0%), which has been associated with change in resistin levels (Satapathy et al., 2011), and treatment by any acetylcholinesterase inhibitors (Z = 7.76, P < 0.001; 8.5%).
Table 5.
Other mediators of age’s effect in TARCC and ADNI
| Mediator | TARCC | ADNI |
|---|---|---|
| Hyperlipidaemia | r = 0.36 (P < 0.001) | |
| Z = −0.97 (P = 0.33) | ||
| Hypertension | r = 0.37 (P < 0.001) | |
| Z = −1.29 (P = 0.20)c | ||
| Diabetes mellitus | r = 0.36 (P < 0.001) | |
| Z = 0.87 (P = 0.38) | ||
| Years smoked | r = 0.36 (P < 0.001) | |
| Z = 0.15 (P = 0.88) | ||
| Cholesterol | r = 0.37 (P < 0.001) | |
| Z = −0.97 (P = 0.33)c | ||
| Triglycerides | r = 0.36 (P < 0.001) | |
| Z = 0.17 (P = 0.87) | ||
| HDL-c | r = 0.35 (P < 0.001) | |
| Z = 2.38 (P = 0.02) | ||
| 3.5% | ||
| LDL-c | r = 0.37 (P < 0.001) | |
| Z = −2.24 (P = 0.03) | ||
| 3.4% | ||
| LpPLA2 | r = 0.35 (P < 0.001) | |
| Z = 1.66 (P = 0.10) | ||
| b, c | ||
| HCY | r = 0.35 (P < 0.001) | |
| Z = 1.58 (P = 0.11) | ||
| c | ||
| C-peptide | r = 0.37 (P < 0.001) | |
| Z = −0.78 (P = 0.44) | ||
| b | ||
| HgbA1c | r = 0.36 (P < 0.001) | |
| Z = −0.49 (P = 0.62) | ||
| Weight | r = 0.34 (P < 0.001) | |
| Z = 3.62 (P < 0.001) | ||
| 5.1% | ||
| BMI | r = 0.34 (P < 0.001) | |
| Z = 4.28 (P < 0.001) | ||
| 5.4% | ||
| On galantamine | r = 0.36 (P < 0.001) | |
| Z = 2.51 (P = 0.01) | ||
| 1.0% | ||
| On any AChEI | r = 0.33 (P < 0.001) | |
| Z = 7.76 (P < 0.001) | ||
| 8.5% | ||
| CSF Aβ1–42 | r = 0.16 (P < 0.001) | |
| Z = −0.33 (P = 0.74) | ||
| b | ||
| CSF t-tau | r = 0.14 (P < 0.001) | |
| Z = 0.92 (P = 0.36) | ||
| b | ||
| CSF p-tau18 | r = 0.15 (P < 0.001) | |
| Z = 0.16 (P = 0.88) | ||
| b | ||
| CSF t-tau/Aβ1–42 | r = 0.17 (P < 0.001) | |
| Z = −1.00 (P = 0.87) | ||
| b | ||
| AV45 PET | r = 0.06 (P = 0.052) | |
| Z = 7.12 (P < 0.001) | ||
| 55.8% | ||
| FDG PET | r = 0.08 (P = 0.009) | |
| Z = 4.83 (P < 0.001) | ||
| 47.4% | ||
| Whole brain | r = 0.05 (P = 0.14) | |
| Z = 10.39 (P < 0.001) | ||
| 66.0% | ||
| Ventricular size | r = 0.05 (P = 0.13) | |
| Z = 8.88 (P < 0.001) | ||
| 70.5% | ||
| Hippocampal volume | r = −0.10 (P < 0.001) | |
| Z = 14.47 (P < 0.001) | ||
| 100% |
b: path b (mediator to dT2A) is significant at P < 0.05; c: path c (age to mediator) is significant at P < 0.05. AChEI = acetylcholine esterase inhibitors; FDG = fluorodeoxyglucose; LDL-c = low-density lioprotein (calculated); LpPLA2 = lipoprotein-associated phospholipase A2; HCY = homocysteine; HDL-c, high-density lipoprotein C.
Age’s association with dT2A in ADNI was not mediated by CSF t-tau, p-tau18, Aβ1–42 or the t-tau/Aβ1–42 ratio but was severely attenuated by whole brain atrophy (Z = 10.39, P < 0.001; 66.0%), hippocampal atrophy, ventricular size (Z = 8.88, P < 0.001; 70.5%), fluorodeoxyglucose (Z = 4.83, P < 0.001; 47.4%) and AV45 PET (Z = 7.12, P < 0.001; 55.8%). SAP mediated age’s effect in serum (TARCC; Z = 3.35, P < 0.001; 12.3%) but not in plasma (ADNI; Z = −0.86, P = 0.39).
Discussion
We have replicated age-specific mediation effects (or the lack thereof) for eight blood-based biomarkers across two independent cohorts representing convenience samples with differing case mixes. Our analysis also produced confirmations of 13/22 mediation effects in TARCC across two δ homologues with few common indicators. Finally, the present analysis replicates the mediation effects of four protein biomarkers across biofluids.
In TARCC, we confirm 13 of the 22 previously reported mediation effects. Our original reports were exploratory analyses of an ad hoc panel in Bonferroni corrected models. The current replications are now hypothesis driven and relate to a second δ homologue. Moreover, the MIMIC model better distinguishes age’s δ-specific effect on cognitive performance from its other effects on individual measures. Age’s δ-independent effects on animal naming, Boston Naming Test and Trail-Making Test Part B were also confirmed. However, only age’s δ-specific effect is likely to be functionally salient and it is precisely that effect, which is being mediated by the biomarkers.
As in our earlier report, all 13 confirmed proteins were partial mediators of age’s effect on δ. Once again, IGF-binding protein 2 emerged with the strongest effect. Unfortunately, technical issues prevented a test of that biomarker in ADNI. However, the effect of IGF-1 could be neither confirmed in TARCC nor replicated in ADNI. IGF-1 was associated with age by path c in both cohorts but with δ in neither. This does not preclude age-specific effects on other organs or on less functionally salient cognitive domains and/or measures.
There was considerable concordance across the cohorts. We replicated 8/16 testable effects (or the lack thereof). In 8/8 discordant cases, we replicated at least one of the intervening paths (e.g. paths b or c). All the discordant dyads included a confirmed mediation effect in TARCC. No protein measured in plasma was found to be a mediator in ADNI but not in TARCC. In every case where mediation was not confirmed in TARCC, the ADNI model was concordant. Moreover, both models agreed on which paths were significant and which were not. The 2/8 discordant dyads involved near significant plasma effects in ADNI. These findings lend credibility to TARCC’s serum findings and suggest that the measurement of these proteins in serum may be more clinically meaningful and generalizable than their measurement in plasma.
Several issues may have contributed to our failure to achieve confirmations in some of the previously reported effects. The current analysis involves a new δ homologue with a minimally overlapping set of indicators. We also limited these analyses to NHW. This reduced our sample size and may have undermined some replications. Our use of the MIMIC model has narrowed the focus of this analysis to age’s unique effect on δ. Our use of the BIAS variable or our selection of its indicators may have inadequately accounted for batch effects. Finally, our earlier models targeted a d-composite. That may have introduced a reification bias relative to the latent constructs being targeted in the present work.
Regardless, we have replicated four of TARCC’s 13 confirmed mediation effects across cohorts and biofluids. Such replications are difficult to achieve in the opinion of many experts (Committee on the Review of Omics-Based Tests for Predicting Patient Outcomes in Clinical Trials et al., 2012; O’Bryant et al., 2015). However, we may have been advantaged by our use of a latent variable approach, which has not been previously applied to blood-based protein biomarkers.
The replicated proteins were APN, follicle-stimulating hormone (FSH), PPP and resistin. APN and resistin are so-called ‘apidokines’ associated with adipocytes and their related secretome (Arai et al., 2019). We recently demonstrated that APN’s effect on cognitive performance to be fully mediated via δ (in TARCC; Benavente et al., 2019). In addition, APN, resistin and PPP have each been previously associated with ‘Alzheimer’s disease’ (Kiliaan et al., 2014). However, 20% of so-called ‘Alzheimer’s disease’ cases do not exhibit amyloidosis by PET (Witte et al., 2014; Degenhardt et al., 2016; Landau et al., 2016). It cannot be assumed that APN’s, resistin’s and PPP’s associations with δ in the current analysis are mediated by Alzheimer’s disease pathology. δ is ‘agnostic’ to dementia’s aetiology (Gavett et al., 2015; John et al., 2016).
Instead, APN resistin and PPP have been shown to be mediators of age’s unique effect on dementia severity. Alzheimer’s disease-specific CSF biomarkers were not. The latter biomarkers impacted δ independently of age’s effect. This suggests that the replicated biomarkers’ associations with clinical ‘Alzheimer’s disease’ in other studies could have been attributable to age’s effect (i.e. senility) rather than Alzheimer’s disease’s.
Resistin is associated with resistance to oral hypoglycemics in diabetes mellitus (Acquarone et al., 2019). However, we did not confirm either serum insulin (Royall et al., 2016) or HgbA1c (in the present analysis) as mediators of age’s effect. Resistin must be acting here through other mechanisms.
Resistin levels have been shown to be elevated in Alzheimer’s disease (Kizilarslanoğlu et al., 2015) and to rise progressively in MCI and Alzheimer’s disease relative to NC (in TARCC; Royall and Palmer, 2019b). However, resistin has been shown here to be a mediator of age’s effect. Its association with clinical dementia in other studies should not be attributed to Alzheimer’s disease in the absence of a formal test of mediation via an Alzheimer’s disease-specific biomarker.
Ironically perhaps, serum resistin levels can be shown to be modulated by treatment with acetylcholinesterase inhibitors (Satapathy et al., 2011). This opens the door to possibly modulation of δ by acetylcholinesterase inhibitors in demented persons with elevated resistin levels. Our results suggest that these can be found among the fraction of demented persons with advanced age and/or depressive symptoms (Royall et al., 2017b). Serum resistin levels fully attenuate the GDS’ nearly 3-fold prospective 5-year MCI conversion is risk in an age-adjusted TARCC model (Royall and Palmer, 2019a). We have recently proposed a method to select individuals who might revert to non-demented states after the correction of any prespecified δ-related condition or risk factor (Royall and Palmer, 2019b). Demented cases selected by that approach on the basis of their depressive symptoms have demonstrably higher serum resistin levels, as we predict similarly selected age-specific dementias would be.
Resistin, APN and PPP have been proposed as biomarkers of weight loss and frailty in older persons (Cardoso et al., 2018). We have confirmed weight and BMI as mediators of age’s effect on δ (in TARCC). In contrast, alternative disease-specific conditions and biomarkers did not mediate that effect, notably HgbA1c, homocysteine and lipoprotein-associated phospholipase A2 (an ischaemic vasculopathy-associated risk factor).
Alzheimer’s disease-specific CSF biomarkers were also not found to be mediators of age’s effect. Given that CSF concentrations of Aβ1–42, t-tau and p-tau18 have been shown to provide high sensitivity and specificity for diagnosing Alzheimer’s disease, predict conversion from MCI to a diagnosis of probable Alzheimer’s disease and identify non-demented elderly persons likely to progress (Blennow and Zetterberg, 2009), the failure of these analytes to exhibit mediation effects helps distinguish Alzheimer’s disease from the dementing effects of Aging Proper (i.e. senility) under consideration here. This is consistent with the relative paucity of Alzheimer’s disease neuropathology among centenarians and the oldest old (von Guten et al., 2010).
Regardless, age’s association with δ has been shown to be fully attenuated by a latent variable indicated by Alzheimer’s disease pathology in autopsy-proven Alzheimer’s disease cases of the National Alzheimer’s Coordinating Center’s Unified Dataset (at a mean age of 81.6 ± 10.6 years; Gavett et al., 2016). However, Alzheimer’s disease neuropathology was ‘inversely related to age’ in that sample (r = −0.49, P < 0.001), as was NIA-Reagan AD staging (National Institute on Aging, 1997). Thus, it is more properly stated that the ‘lack of AD pathology’ mediates age’s association with δ in the National Alzheimer’s Coordinating Center.
Although CSF Aβ1–42, t-tau, p-tau18 and the t-tau:Aβ1–42 ratio were related to δ by path b, none were related to age by path c, and so they cannot mediate age’s effect on dementia severity. These biomarkers may be sensitive to clinical and pre-clinical Alzheimer’s disease and contribute to its dementing effects, but Alzheimer’s disease may not be entirely responsible for an individual’s d-score and, therefore, for conversion risk (Ritchie et al., 2017). Age’s independent effect on δ might instead manifest as an erosion of the ‘cognitive reserve’ that has been proposed to explain the empirically weak relationships between many Alzheimer’s disease-specific biomarkers (including autopsy findings) and measures of dementia severity (Stern, 2012). Our structural equation model approach has the potential to distinguish each compartment of δ’s variance and to identify its unique biomarker profile.
In contrast to the CSF biomarkers, CNS amyloidosis by AV45 florbetapir PET was a strong mediator of age’s association with δ. PiB-PET was available in only a small minority of ADNI participants and so could not be tested. There are subtle differences in the distributions of these radioligands. Florbetapir is more strongly associated with Aβ1–40 than with Aβ1–42 (Beach et al., 2018). Aβ1–40 is associated with diffuse plaque while the latter is more closely associated with NP. AV45’s mediation effect in the absence of effects via CSF Alzheimer’s disease-specific biomarkers might be explained by an Aβ1–40 amyloidosis rather than Aβ1–42 and NP formation.
In fact, extreme old age is associated with both a diminished NP burden and a reduced distribution of neurofibrillary tangles, limited largely to the mesiotemporal region, i.e. ‘primary age-related tauopathy’ (Crary et al., 2014). As a result, most primary age-related tauopathy cases present at lower Braak stages (Besser et al., 2019). Such a restricted distribution is unlikely to impact general cognition and hence g and δ, suggesting that the reserve of demented primary age-related tauopathy cases may be eroded by some process other than primary age-related tauopathy-related neurofibrillary tangle formation.
The age-specific AV45 signal might also be influenced by CNS amylin deposition (Jackson et al., 2013). Amylin (i.e. insular amyloid polypeptide) is secreted by the pancreas and contributes to the pancreatic amyloidosis of type 2 diabetes. AV45 labels pancreatic amylin deposits (Templin et al., 2018). A genome-wide association study (GWAS) using AV45 as an endophenotype (presumably of ‘Alzheimer’s disease’) found single-nucleotide polymorphisms in the insular amyloid polypeptide gene to be predictive ‘of CNS amyloisdosis’ (Roostaei et al., 2017). Amylin deposits can be demonstrated in the brains of demented persons with AODM, and even in the absence of that condition (Fawver et al., 2014).
Plasma SAP levels are associated with Alzheimer’s disease in Han Chinese (Cheng et al., 2018). However, that finding was age-adjusted and may not be relevant to Aging Proper. However, elevated plasma SAP concentrations are also associated with cognitive impairment in centenarians (Nybo et al., 1998). SAP was associated with δ in both TARCC and ADNI but was found to be a mediator of age’s effect in TARCC’s serum data only.
SAP is co-localized with a variety of amyloids, including both Aβ1–42 and amylin. It co-localizes with florbetapir both in and outside the CNS (Wagner et al., 2018). SAP is thought to accelerate Aβ1–42 deposition in NP (Hamazaki, 1995; Tennent et al., 1995). However, it ‘interferes’ with insular amyloid polypeptide deposition in amylin deposits (Gao et al., 2015). In TARCC’s data, SAP is associated with lower d-scores (a salutary effect). Age’s adverse effect is mediated by ‘lower’ serum SAP levels in advancing age but not higher levels.
Plasma PPP levels are also predictive of CNS amyloidosis, both by PiB-PET (Kiddle et al., 2012) and by AV45 (Voyle et al., 2015). PPP levels rise exponentially with age (Brimnes et al., 1997) and are associated with anorexia and weight loss in older persons (Moss et al., 2012). High plasma PPP levels are associated with both weight loss and MCI and moderate the association of diabetes with that diagnosis (Roberts et al., 2015).
In summary, we cannot be certain that the CNS AV45 PET signal is specific to Aβ1–42 and therefore that AV45’s mediation effect involves that aggregate. Age’s effect on δ could be mediated instead by Aβ1–40 in the absence of NP formation or by CNS amylin deposition, accelerated by falling SAP levels, in parallel with rising PPP and in the presence of PPP-induced anorexia and weight loss. Since we are addressing age-specific contribution to dementia severity, neither scenario would conflict with AV45’s co-localization at autopsy with NP ‘in age-adjusted’ models (Clark et al., 2012).
We also found important mediation effects by other less Alzheimer’s disease-specific structural and functional imaging modalities (e.g. fluorodeoxyglucose PET and whole brain atrophy by voxel-based morphometry). These were consistently stronger mediators than the blood-based protein biomarkers. An earlier ADNI analysis has associated CSF Aβ1–42 and t-tau concentrations with the same structural features (Tosun et al., 2010). However, as that analysis was age-adjusted, it cannot speak to the effects of Aging Proper. Brain atrophy’s mediation of age’s unique effect on δ is likely to be independent of CSF Aβ1–42 and t-tau concentrations, as they were not themselves mediators.
Low high-density lipoprotein C and high low-density lipoprotein (calculated) were found to be partial mediators of age’s effect on δ. They might offer potentially modifiable risk factors for age-specific cognitive decline and frailty (Ramsay et al., 2015). Both were found to be associated with clinical ‘Alzheimer’s disease’ in an early TARCC report (Warren et al., 2012). However, those effects were age-adjusted once again. The present results suggest that high-density lipoprotein C and low-density lipoprotein (calculated) may additionally affect δ by an ‘age-specific’ mechanism. C-peptide, diabetes mellitus and LpLA2 were not mediators, but they were associated with dementia severity by path b, suggesting that they may be involved in ‘age-independent’ dementing processes.
Finally, FSH is associated with frailty in older men participating in the European Male Aging Study (Tajar et al., 2011) and the Concord Health and Ageing in Men Project (Travison et al., 2011). FSH’s unadjusted effect on frailty in the Concord Health and Ageing in Men Project is fully attenuated by adjustment for age.
In summary, several blood-based protein biomarkers related to frailty have been shown to mediate age-specific differences in dementia severity as measured by δ. The 13 proteins have been confirmed by a second δ homologue in TARCC (serum). Four have been replicated across cohorts and biofluids. These may offer targets for the remediation of age-specific cognitive decline (aka ‘senility’), help distinguish it from other determinants of dementia severity and/or provide clues to the biology of Aging Proper.
Acknowledgments
TARCC had no role in study design, data analysis, decision to publish or preparation of the article. However, TARCC investigators contributed to the design and implementation of TARCC and/or provided data. Texas Alzheimer’s Research and Care Consortium List of Investigators are as follows: Baylor College of Medicine: Valory Pavlik, PhD, Paul Massman, PhD, Eveleen Darby, MA/MS, Monica Rodriguear, MA, and Aisha Khaleeq Ansari, MD; Texas Tech University Health Sciences Center: John C. DeToledo, MD, Hemachandra Reddy, PhD, Henrick Wilms, MD, PhD, Kim Johnson, PhD, and Victoria Perez; University of North Texas Health Science Center: Thomas Fairchild, PhD, Janice Knebl, DO, Sid E. O’Bryant, PhD, James R. Hall, PhD, Leigh Johnson, PhD, Robert C. Barber, PhD, Douglas Mains, DrPH, and Lisa Alvarez; University of Texas Southwestern Medical Center: Munro Cullum, PhD, Roger Rosenberg, MD, Benjamin Williams, MD, PhD, Mary Quiceno, MD, Joan Reisch, PhD, Linda S. Hynan, PhD, Ryan Huebinger, PhD, Janet Smith, and Trung Nguyen, MD, PhD; University of Texas Health Science Center—San Antonio: Donald Royall, MD, Raymond Palmer, PhD, and Marsha Polk; Texas A&M University Health Science Center: Alan Stevens, PhD, and Marcia Ory, PhD/MPH; University of Texas at Austin/Dell Medical School: David Paydarfar, MD, John Bertelson, MD, Martin Woon, PhD, and Gayle Ayres, DO; Alyssa Aguirre LCSW; and University of North Carolina: Kirk C. Wilhelmsen, MD, PhD, Jeffrey L. Tilson, PhD. ADNI had no role in study design, data collection and analysis, decision to publish or preparation of the article. However, ADNI investigators contributed to the design and implementation of ADNI and/or provided data. A list of relevant ADNI investigators can be found below, in the Appendix. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf (4 December 2004, date last accessed).
Funding
This work was supported by the Julia and Vann Buren Parr endowment for the study of Alzheimer’s disease. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the article. Some data used in the preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). Data collection and sharing for this project was funded by the ADNI (National Institutes of Health Grant U01 AG024904) and Department of Defense ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging and the National Institute of Biomedical Imaging and Bioengineering and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai, Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche, Ltd., and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer, Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Some data used in the preparation of this article were obtained from the TARCC, which is supported by funding provided by the Darrell K Royal Texas Alzheimer’s Initiative, directed by the Texas Council on Alzheimer’s Disease and Related Disorders.
Competing interests
Dr. D.R.R. has disclosed his co-invention of δ, its homologues and orthologues to the University of Texas Health Science Center at San Antonio (UTHSCSA), which has filed patent application 2012.039.US1.HSCS and provisional patents 61/603,226 and 61/671,858 relating to the latent variable d’s construction and biomarkers. Dr. R.F.P. has disclosed his co-invention of δ, its homologues and orthologues to UTHSCSA.
Glossary
- ADNI
Alzheimer’s Disease Neuroimaging Initiative
- APN
adiponectin
- APOE
apolipoprotein E
- BMI
body mass index
- CSF c
erebrospinal fluid
- GDS
geriatric depression scale
- IGF-1
insulin-like growth factor-1
- LDD l
east detectable dose
- MCI
mild cognitive impairment
- MIMIC
Multiple Indicators Multiple Causes
- MRI
magnetic resonance imaging
- NC
normal controls
- NHW
non-Hispanic white
- PET
positron emission tomography
- PPP
pancreatic polypeptide
- RBM R
ules-Based Medicine
- SAP
serum amyloid protein
- TARCC
Texas Alzheimer’s Research and Care Consortium
Appendix: ADNI collaborators
I. ADNI I, GO, II and III
Part A: leadership and infrastructure
Principal Investigator
Michael W. Weiner, MD, UC San Francisco
ATRI PI and Director of Coordinating Center Clinical Core
Paul Aisen, MD, University of Southern California
Executive Committee
Michael Weiner, MD, UC San Francisco
Paul Aisen, MD, University of Southern California
Ronald Petersen, MD, PhD, Mayo Clinic, Rochester
Clifford R. Jack, Jr., MD, Mayo Clinic, Rochester
William Jagust, MD, UC Berkeley
John Q. Trojanowki, MD, PhD, U Pennsylvania
Arthur W. Toga, PhD, USC
Laurel Beckett, PhD, UC Davis
Robert C. Green, MD, MPH Brigham and Women’s Hospital/Harvard Medical School
Andrew J. Saykin, PsyD, Indiana University
John Morris, MD, Washington University St. Louis
Leslie M. Shaw, University of Pennsylvania
ADNI External Advisory Board
Zaven Khachaturian, PhD, Prevent Alzheimer’s Disease 2020 (Chair)
Greg Sorensen, MD, Siemens
Maria Carrillo, PhD, Alzheimer’s Association
Lew Kuller, MD, University of Pittsburgh
Marc Raichle, MD, Washington University St. Louis
Steven Paul, MD, Cornell University
Peter Davies, MD, Albert Einstein College of Medicine of Yeshiva University
Howard Fillit, MD, AD Drug Discovery Foundation
Franz Hefti, PhD, Acumen Pharmaceuticals
David Holtzman, MD, Washington University St. Louis
M. Marcel Mesulam, MD, Northwestern University
William Potter, MD, National Institute of Mental Health
Peter Snyder, PhD, Brown University
Data and Publications Committee
Robert C. Green, MD, MPH BWH/HMS (Chair)
Resource Allocation Review Committee
Tom Montine, MD, PhD, University of Washington (Chair)
Clinical Core Leaders
Ronald Petersen, MD, PhD, Mayo Clinic, Rochester (Core PI)
Paul Aisen, MD, University of Southern California
Clinical Informatics and Operations
Gustavo Jimenez, MBS, USC
Michael Donohue, PhD, USC
Devon Gessert, BS, USC
Kelly Harless, BA, USC
Jennifer Salazar, MBS, USC
Yuliana Cabrera, BS, USC
Sarah Walter, MSc, USC
Lindsey Hergesheimer, BS, USC
Biostatistics Core Leaders and Key Personnel
Laurel Beckett, PhD, UC Davis (Core PI)
Danielle Harvey, PhD, UC Davis
Michael Donohue, PhD, UC San Diego
Biomarkers Core Leaders and Key Personnel
Leslie M. Shaw, PhD, UPenn School of Medicine
John Q. Trojanowki, MD, PhD, UPenn School of Medicine
Virginia Lee, PhD, MBA, UPenn School of Medicine
Magdalena Korecka, PhD, UPenn School of Medicine
Michal Figurski, PhD, UPenn School of Medicine
Initial Concept Planning & Development
Michael W. Weiner, MD, UC San Francisco
Lean Thal, MD, UC San Diego
Zaven Khachaturian, PhD, Prevent Alzheimer’s Disease 2020
Early Project Proposal Development
Leon Thal, MD, UC San Diego
Neil Buckholtz, National Institute on Aging
Michael W. Weiner, MD, UC San Francisco
Peter J. Snyder, PhD, Brown University
William Potter, MD, National Institute of Mental Health
Steven Paul, MD, Cornell University
Marilyn Albert, PhD, Johns Hopkins University
Richard Frank, MD, PhD, Richard Frank Consulting
Zaven Khachaturian, PhD, Prevent Alzheimer’s Disease 2020
Part B: investigators by site
Oregon Health & Science University
Joseph Quinn, MD
Lisa C. Silbert, MD
Betty Lind, BS
Jeffrey A. Kaye, MD, A.—Past Investigator
Raina Carter, BA—Past Investigator
Sara Dolen, BS—Past Investigator
University of Southern California
Lon S. Schneider, MD
Sonia Pawluczyk, MD
Mauricio Becerra, BS
Liberty Teodoro, RN
Bryan M. Spann, DO, PhD—Past Investigator
University of California—San Diego
James Brewer, MD, PhD
Helen Vanderswag, RN
Adam Fleisher, MD—Past Investigator
University of Michigan
Jaimie Ziolkowski, MA, BS, TLLP
Judith L. Heidebrink, MD, MS
Joanne L. Lord, LPN, BA, CCRC—Past
Investigator
Mayo Clinic, Rochester
Ronald Petersen, MD, PhD
Sara S. Mason, RN
Colleen S. Albers, RN
David Knopman, MD
Kris Johnson, RN—Past Investigator
Baylor College of Medicine
Javier Villanueva-Meyer, MD
Valory Pavlik, PhD
Nathaniel Pacini, MA
Ashley Lamb, MA
Joseph S. Kass, MD, LD, FAAN
Rachelle S. Doody, MD, PhD—Past Investigator
Victoria Shibley, MS—Past Investigator
Munir Chowdhury, MBBS, MS—Past Investigator
Susan Rountree, MD—Past Investigator
Mimi Dang, MD—Past Investigator
Columbia University Medical Center
Yaakov Stern, PhD
Lawrence S. Honig, MD, PhD
Karen L. Bell, MD
Randy Yeh, MD
Washington University, St. Louis
Beau Ances, MD, PhD, MSc
John C. Morris, MD
David Winkfield, BS
Maria Carroll, RN, MSN, GCNS-BC
Angela Oliver, RN, BSN, MSG
Mary L. Creech, RN, MSW—Past Investigator
Mark A. Mintun, MD—Past Investigator
Stacy Schneider, APRN, BC, GNP—Past
Investigator
University of Alabama—Birmingham
Daniel Marson, JD, PhD
David Geldmacher, MD
Marissa Natelson Love, MD
Randall Griffith, PhD, ABPP—Past Investigator
David Clark, MD—Past Investigator
John Brockington, MD—Past Investigator
Mount Sinai School of Medicine
Hillel Grossman, MD
Effie Mitsis, PhD—Past Investigator
Rush University Medical Center
Raj C. Shah, MD
Melissa Lamar, PhD
Patricia Samuels
Wien Center
Ranjan Duara, MD
Maria T. Greig-Custo, MD
Rosemarie Rodriguez, PhD
Johns Hopkins University
Marilyn Albert, PhD
Chiadi Onyike, MD
Daniel D’Agostino II, BS
StepMohammed O. Sheikh, MD
Jamika Singleton-Garvin, CCRP
Anaztasia Ulysse
Mrunalini Gaikwad
Duke University Medical Center
P. Murali Doraiswamy, MBBS, FRCP
Jeffrey R. Petrella, MD
Olga James, MD
Salvador Borges-Neto, MD
Terence Z. Wong, MD—Past Investigator
Edward Coleman—Past Investigator
University of Pennsylvania
Jason H. Karlawish, MD
David A. Wolk, MD
Sanjeev Vaishnavi, MD
Christopher M. Clark, MD—Past Investigator
Steven E. Arnold, MD—Past Investigator
University of Kentucky
Charles D. Smith, MD
Greg Jicha, MD
Peter Hardy, PhD
Riham El Khouli, MD
Elizabeth Oates, MD
Gary Conrad, MD
University of Pittsburgh
Oscar L. Lopez, MD
MaryAnn Oakley, MA
Donna M. Simpson, CRNP, MPH
University of Rochester Medical Center
Anton P. Porsteinsson, MD
Kim Martin, RN
Nancy Kowalksi, MS, RNC
Melanie Keltz, RN
Bonnie S. Goldstein, MS, NP—Past Investigator
Kelly M. Makino, BS—Past Investigator
M. Saleem Ismail, MD—Past Investigator
Connie Brand, RN—Past Investigator
University of California Irvine IMIND
Gaby Thai, MD
Aimee Pierce, MD
Beatriz Yanez, RN
Elizabeth Sosa, PhD
Megan Witbracht, PhD
University of Texas Southwestern Medical School
Kyle Womack, MD
Dana Mathews, MD, PhD
Mary Quiceno, MD
Emory University
Allan I. Levey, MD, PhD
James J. Lah, MD, PhD
Janet S. Cellar, DNP, PMHCNS-BC
University of Kansas, Medical Center
Jeffrey M. Burns, MD
Russell H. Swerdlow, MD
William M. Brooks, PhD
University of California, Los Angeles
Ellen Woo, PhD
Daniel H.S. Silverman, MD, PhD
Edmond Teng, MD, PhD
Sarah Kremen, MD
Liana Apostolova, MD—Past Investigator
Kathleen Tingus, PhD—Past Investigator
Po H. Lu, PsyD—Past Investigator
George Bartzokis, MD—Past Investigator
Mayo Clinic, Jacksonville
Neill R Graff-Radford, MBBCH, FRCP (London)
Francine Parfitt, MSH, CCRC
Kim Poki-Walker, BA
Indiana University
Martin R. Farlow, MD
Ann Marie Hake, MD
Brandy R. Matthews, MD—Past Investigator
Jared R. Brosch, MD
Scott Herring, RN, CCRC
Yale University School of Medicine
Christopher H. van Dyck, MD
Richard E. Carson, PhD
Pradeep Varma, MD
McGill Univ., Montreal-Jewish General Hospital
Howard Chertkow, MD
Howard Bergman, MD
Chris Hosein, MEdhanie Kielb, BS—Past Investigator
Sunnybrook Health Sciences, Ontario
Sandra Black, MD, FRCPC
Bojana Stefanovic, PhD
Chris (Chinthaka) Heyn, BSC, PhD, MD, FRCPC
U.B.C. Clinic for AD & Related Disorders
Ging-Yuek Robin Hsiung, MD, MHSc, FRCPC
Benita Mudge, BS
Vesna Sossi, PhD
Howard Feldman, MD, FRCPC—Past Investigator
Michele Assaly, MA—Past Investigator
Cognitive Neurology—St. Joseph's, Ontario
Elizabeth Finger, MD
Stephen Pasternack, MD, PhD
William Pavlosky, MD
Irina Rachinsky, MD—Past Investigator
Dick Drost, PhD—Past Investigator
Andrew Kertesz, MD—Past Investigator
Cleveland Clinic Lou Ruvo Center for Brain Health
Charles Bernick, MD, MPH
Donna Munic, PhD
Northwestern University
Marek-Marsel Mesulam, MD
Emily Rogalski, PhD
Kristine Lipowski, MA
Sandra Weintraub, PhD
Borna Bonakdarpour, MD
Diana Kerwin, MD—Past Investigator
Chuang-Kuo Wu, MD, PhD—Past Investigator
Nancy Johnson, PhD—Past Investigator
Premiere Research Inst (Palm Beach Neurology)
Carl Sadowsky, MD
Teresa Villena, MD
Georgetown University Medical Center
Raymond Scott Turner, MD, PhD
Kathleen Johnson, NP
Brigid Reynolds, NP
Brigham and Women's Hospital
Reisa A. Sperling, MD
Keith A. Johnson, MD
Gad A. Marshall, MD
Stanford University
Jerome Yesavage, MD
Joy L. Taylor, PhD
Steven Chao, MD, PhD
Barton Lane, MD—Past Investigator
Allyson Rosen, PhD—Past Investigator
Jared Tinklenberg, MD—Past Investigator
Banner Sun Health Research Institute
Edward Zamrini, MD
Christine M. Belden, PsyD
Sherye A. Sirrel, CCRC
Boston University
Neil Kowall, MD
Ronald Killiany, PhD
Andrew E. Budson, MD
Alexander Norbash, MD—Past Investigator
Patricia Lynn Johnson, BA—Past Investigator
Howard University
Thomas O. Obisesan, MD, MPH
Ntekim E. Oyonumo, MD, PhD
Joanne Allard, PhD
Olu Ogunlana, BPharm
Case Western Reserve University
Alan Lerner, MD
Paula Ogrocki, PhD
Curtis Tatsuoka, PhD
Parianne Fatica, BA, CCRC
University of California, Davis—Sacramento
Evan Fletcher, PhD
Pauline Maillard, PhD
John Olichney, MD
Charles DeCarli, MD
Owen Carmichael, PhD—Past Investigator
Neurological Care of CNY
Smita Kittur, MD—Past Investigator
Parkwood Institute
Michael Borrie, MB ChB
T-Y Lee, PhD
Dr Rob Bartha, PhD
University of Wisconsin
Sterling Johnson, PhD
Sanjay Asthana, MD
Cynthia M. Carlsson, MD, MS
Banner Alzheimer's Institute
Pierre Tariot, MD
Anna Burke, MD
Joel Hetelle, BS
Kathryn DeMarco, BS
Nadira Trncic, MD, PhD, CCRC—Past Investigator
Adam Fleisher, MD—Past Investigator
Stephanie Reeder, BA—Past Investigator
Dent Neurologic Institute
Vernice Bates, MD
Horacio Capote, MD
Michelle Rainka, PharmD, CCRP
Ohio State University
Douglas W. Scharre, MD
Maria Kataki, MD, PhD
Rawan Tarawneh, MD
Albany Medical College
Earl A. Zimmerman, MD
Dzintra Celmins, MD
David Hart, MD
Hartford Hospital, Olin Neuropsychiatry Research Center
Godfrey D. Pearlson, MD
Karen Blank, MD
Karen Anderson, RN
Dartmouth-Hitchcock Medical Center
Laura A. Flashman, PhD
Marc Seltzer, MD
Mary L. Hynes, RN, MPH
Robert B. Santulli, MD—Past Investigator
Wake Forest University Health Sciences
Kaycee M. Sink, MD, MAS
Mia Yang, MD
Akiva Mintz, MD, PhD
Rhode Island Hospital
Brian R. Ott, MD
Geoffrey Tremont, PhD
Lori A. Daiello, Pharm.D, ScM
Butler Hospital
Stephen Salloway, MD, MS
Paul Malloy, PhD
Stephen Correia, PhD
Athena Lee, PhD
UC San Francisco
Howard J. Rosen, MD
Bruce L. Miller, MD
David Perry, MD
Medical University South Carolina
Jacobo Mintzer, MD, MBA
Kenneth Spicer, MD, PhD
David Bachman, MD
St. Joseph’s Health Care
Elizabeth Finger, MD
Stephen Pasternak, MD
Irina Rachinsky, MD
John Rogers, MD
Andrew Kertesz, MD—Past Investigator
Dick Drost, MD—Past Investigator
Nathan Kline Institute
Nunzio Pomara, MD
Raymundo Hernando, MD
Antero Sarrael, MD
University of Iowa College of Medicine
Delwyn D. Miller, PharmD, MD
Karen Ekstam Smith, RN
Hristina Koleva, MD
Ki Won Nam, MD
Hyungsub Shim, MD
Susan K. Schultz, MD—Past Investigator
Cornell University
Norman Relkin, MD, PhD
Gloria Chiang, MD
Michael Lin, MD
Lisa Ravdin, PhD
University of South Florida: USF Health Byrd Alzheimer’s Institute
Amanda Smith, MD
Christi Leach, MD
Balebail Ashok Raj, MD—Past Investigator
Kristin Fargher, MD—Past Investigator
Courtney Bodge, PhD
References
- Acquarone E, Monacelli F, Borghi R, Nencioni A, Odetti P.. Resistin: a reappraisal. Mech Ageing Dev 2019; 178: 46–63. [DOI] [PubMed] [Google Scholar]
- Arai Y, Kamide K, Hirose N.. Adipokines and aging: findings from centenarians and the very old. Front Endocrinol (Lausanne) 2019; 10: 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbuckle JL. Analysis of Moment Structures—AMOS (version 7.0) (computer program). Chicago: SPSS; 2006.
- Beach TG, Maarouf CL, Intorcia A, Sue LI, Serrano GE, Lu M, Joshi A, Pontecorvo MJ, . Antemortem-postmortem correlation of Florbetapir (18F) PET amyloid imaging with quantitative biochemical measures of Aβ42 but not Aβ40. J Alzheimers Dis 2018; 61: 1509–16. [DOI] [PubMed] [Google Scholar]
- Benavente KS, Palmer R, Royall DR.. Serum adiponectin is related to dementia. J Gerontol A: Biol Sci Med Sci 2019, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentler PM. Comparative fit indexes in structural models. Psychol Bull 1990; 107: 238–46. [DOI] [PubMed] [Google Scholar]
- Besser LM, Mock C, Teylan MA, Hassenstab J, Kukull WA, Crary JF.. Differences in cognitive impairment in primary age-related tauopathy versus Alzheimer disease. J Neuropathol Exp Neurol 2019; 78: 219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow K, Zetterberg H.. Cerebrospinal fluid biomarkers for Alzheimer's disease. J Alzheimers Dis 2009; 18: 413–7. [DOI] [PubMed] [Google Scholar]
- Bollen KA, Long JS.. Testing structural equation models. Sage Publications, Thousand Oaks, CA, 1993. [Google Scholar]
- Wheaton B, Muthén B, Alwin DF, Summer GF.. Assessing reliability and stability in panel models In:Heise DR, editor. Sociology methodology. San Francisco, CA: Jossey-Bass; 1977. [Google Scholar]
- Browne M, Cudeck R.. Alternative ways of assessing model fit In: Bollen KA, Long JS, editors. Testing structural equation models. Thousand Oaks, CA: Sage Publications; 1993. p. 136–62. [Google Scholar]
- Brimnes DM, Rasmussen BK, Hilsted L, Jensen R, Hilsted J.. Basal serum pancreatic polypeptide is dependent on age and gender in an adult population. Scand J Clin Lab Invest 1997; 57: 695–702. [DOI] [PubMed] [Google Scholar]
- Burke WJ, Houston MJ, Boust SC, Roccaforte WH.. Use of the geriatric depression scale in dementia of the Alzheimer type. J Am Geriatr Soc 1989; 37: 856–60. [DOI] [PubMed] [Google Scholar]
- Cardoso AL, Fernandes A, Aguilar-Pimentel JA, de Angelis MH, Guedes JR, Brito MA, . Towards frailty biomarkers: candidates from genes and pathways regulated in aging and age-related diseases. Ageing Res Rev 2018; 47: 214–77. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Yin J, Yuan H, Jin C, Zhang F, Wang Z, . Blood-derived plasma protein biomarkers for Alzheimer’s disease in Han Chinese. Front Aging Neurosci 2018; 10: 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark CM, Pontecorvo MJ, Beach TG, Bedell BJ, Coleman RE, Doraiswamy PM, . Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-β plaques: a prospective cohort study. Lancet Neurol 2012; 11: 669–78. [DOI] [PubMed] [Google Scholar]
- Committee on the Review of Omics-Based Tests for Predicting Patient Outcomes in Clinical Trials, Board on Health Care Services, Board on Health Sciences Policy, Institute of Medicine, Micheel CM, Nass SJ, Omenn GS, editors. Evolution of translational omics: lessons learned and the path forward. Washington, DC: National Academies Press (US; ); 2012. [PubMed] [Google Scholar]
- Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, . Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014; 128: 755–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt EK, Witte MM, Case MG, Yu P, Henley DB, Hochstetler HM, . Florbetapir F18 PET amyloid neuroimaging and characteristics in patients with mild and moderate Alzheimer dementia. Psychosomatics 2016; 57: 208–16. [DOI] [PubMed] [Google Scholar]
- Fawver JN, Ghiwot Y, Koola C, Carrera W, Rodriguez-Rivera J, Hernandez C, . Islet amyloid polypeptide (IAPP): a second amyloid in Alzheimer's disease. Curr Alzheimer Res 2014; 11: 928–40. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR.. Mini-mental state: a practical method for grading the cognitive state of patients for the clinician. J Psychiatry Res 1975; 12: 189–98. [DOI] [PubMed] [Google Scholar]
- Fried LP, Tangen CM, Walston J, Newman AB, Hirsch C, Gottdiener J.. Frailty in older adults: evidence for a phenotype. J Gerontol A: Biol Sci Med Sci 2001; 56: M146–M156. [DOI] [PubMed] [Google Scholar]
- Gao M, Estel K, Seeliger J, Friedrich RP, Dogan S, Wanker EE, . Modulation of human IAPP fibrillation: cosolutes, crowders and chaperones. Phys Chem Chem Phys 2015; 17: 8338–48. [DOI] [PubMed] [Google Scholar]
- Gavett BE, John SE, Gurnani AS, Bussell CA, Saurman JL.. The role of Alzheimer’s and cerebrovascular pathology in mediating the effects of age, race, and apolipoprotein E genotype on dementia severity in pathologically confirmed Alzheimer’s disease. J Alzheimers Dis 2016; 49: 531–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavett BE, Vudy V, Jeffrey M, John SE, Gurnani A, Adams J.. The δ latent dementia phenotype in the NACC UDS: cross-validation and extension. Neuropsychology 2015; 29: 344–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham JW. Missing data analysis: making it work in the real world. Annu Rev Psychol 2009; 6: 549–76. [DOI] [PubMed] [Google Scholar]
- Hamazaki H. Amyloid P component promotes aggregation of Alzheimer’s beta-amyloid peptide. Biochem Biophys Res Commun 1995; 211: 349–53. [DOI] [PubMed] [Google Scholar]
- Hughes CP, Berg L, Danziger WL, Coben LA, Martin RA.. New clinical scale for the staging of dementia. Br J Psychiatry 1982; 140: 566–72. [DOI] [PubMed] [Google Scholar]
- Jack CR, Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, . The Alzheimer's Disease Neuroimaging Initiative (ADNI): MRI methods. J Mag Reson Imaging 2008; 27: 685–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson K, Barisone GA, Diaz E, Jin LW, DeCarli C, Despa F.. Amylin deposition in the brain: a second amyloid in Alzheimer disease? Ann Neurol 2013; 74: 517–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust WJ, Bandy D, Chen K, Foster NL, Landau SM, Mathis CA, . The Alzheimer’s disease neuroimaging initiative positron emission tomography core. Alz Dement 2010; 6: 221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John SE, Gurnani AS, Bussell C, Saurman JL, Griffin JW, Gavett BE.. The effectiveness and unique contribution of neuropsychological tests and the δ latent phenotype in the differential diagnosis of dementia in the uniform data set. Neuropsychology 2016; 30: 946–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juva K, Mäkelä M, Erkinjuntti T, Sulkava R, Yukoski R, Valvanne J, . Functional assessment scales in detecting dementia. Age Ageing 1997; 26: 393–400. [DOI] [PubMed] [Google Scholar]
- Kaplan EF, Goodglass H, Weintraub S.. The Boston Naming Test: experimental edition. 2nd edn Philadelphia: Lea & Febiger; 1983. [Google Scholar]
- Kiddle SJ, Thambisetty M, Simmons A, Riddoch-Contreras J, Hye A, Westman E, . Plasma based markers of [11C] PiB-PET brain amyloid burden. PLoS One 2012; 7: e44260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiliaan AJ, Arnoldussen IA, Gustafson DR.. Adipokines: a link between obesity and dementia? Lancet Neurol 2014; 13: 913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizilarslanoğlu MC, Kara Ö, Yeşil Y, Kuyumcu ME, Öztürk ZA, Cankurtaran M.. Alzheimer disease, inflammation, and novel inflammatory marker: resistin. Turk J Med Sci 2015; 45: 1040–6. [PubMed] [Google Scholar]
- Koch W, Ehrenhaft A, Griesser K, Pfeufer A, Müller J, Schömig A, . TaqMan systems for genotyping of disease‐related polymorphisms present in the gene encoding apolipoprotein E. Clin Chem Lab Med 2002; 40: 1123. [DOI] [PubMed] [Google Scholar]
- Landau SM, Horng A, Fero A, Jagust WJ, Alzheimer's Disease Neuroimaging Initiative. Amyloid negativity in patients with clinically diagnosed Alzheimer disease and MCI. Neurology 2016; 86: 1377–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawton MP, Brody EM.. Assessment of older people: self-maintaining and instrumental activities of daily living. Gerontologist 1969; 9: 179–86. [PubMed] [Google Scholar]
- MacKinnon D. Analysis of mediating variables in prevention and intervention research. In: Czarees A, Beatty L, editors. Scientific methods for prevention intervention research. NIDA Research Monograph 139; 1994. p. 137–53. [PubMed]
- Maixner SM, Burke WJ, Roccaforte WH, Wengel SP, Potter JF.. A comparison of two depression scales in a geriatric assessment clinic. Am J Geriatr Psychiatr 1995; 3: 60–7. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM.. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group. Neurology 1984; 34: 939–44. [DOI] [PubMed] [Google Scholar]
- Morris JC, Heyman A, Mohs RC, Hughes JP, vanBelle G, Fullenbaum G, . The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer’s disease. Neurology 1989; 39: 1159–65. [DOI] [PubMed] [Google Scholar]
- Moss C, Dhillo WS, Frost G, Hickson M.. Gastrointestinal hormones: the regulation of appetite and the anorexia of ageing. J Hum Nutr Diet 2012; 25: 3–15. [DOI] [PubMed] [Google Scholar]
- Muthén BO. A structural model with latent variables. J Am Stat Assoc 1979; 74: 807–11. [Google Scholar]
- National Institute on Aging. Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging 1997; 18(4 Suppl): S1–2. [PubMed] [Google Scholar]
- Nybo M, Olsen H, Jeune B, Andersen-Ranberg K, Holm Nielsen E, Svehag SE.. Increased plasma concentration of serum amyloid P component in centenarians with impaired cognitive performance. Dement Geriatr Cogn Disord 1998; 9: 126–9. [DOI] [PubMed] [Google Scholar]
- O’Bryant SE, Gupta V, Henriksen K, Edwards M, Jeromin A, Lista S, . Guidelines for the standardization of preanalytic variables for blood-based biomarker studies in Alzheimer's disease research. Alzheimers Dement 2015; 11: 549–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer RF, Royall DR.. Frailty mediates senility in Mexican-Americans. J Frailty Aging 2019; 8: S21. [Google Scholar]
- Pfeffer RI, Kurosaki TT, Harrah CH, Chance JM, Filos S.. Measurement of functional activities in older adults in the community. J Gerontol 1982; 37: 323–9. [DOI] [PubMed] [Google Scholar]
- Ramsay SE, Arianayagam DS, Whincup PH, Lennon LT, Cryer J, Papacosta AO, . Cardiovascular risk profile and frailty in a population-based study of older British men. Heart 2015; 101: 616–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitan RM. Validity of the Trail Making test as an indicator of organic brain damage. Percept Mot Skills 1958; 8: 271–6. [Google Scholar]
- Ritchie C, Smailagic N, Noel-Storr AH, Ukoumunne O, Ladds EC, Martin S.. CSF tau and the CSF tau/ABeta ratio for the diagnosis of Alzheimer's disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst Rev 2017; 3: CD010803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RO, Aakre JA, Cha RH, Kremers WK, Mielke MM, Velgos SN, . Association of pancreatic polypeptide with mild cognitive impairment varies by APOE ε4. Front Aging Neurosci 2015; 7: 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roostaei T, Nazeri A, Felsky D, De Jager PL, Schneider JA, Pollock BG, . Genome-wide interaction study of brain beta-amyloid burden and cognitive impairment in Alzheimer's disease. Mol Psychiatry 2017; 22: 287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royall DR, Al-Rubaye S, Bishnoi R, Palmer RF.. Few serum proteins mediate APOE’s association with dementia. PLoS One 2017a; 12: e0172268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royall DR, Al-Rubaye S, Bishnoi R, Palmer RF.. Serum protein mediators of depression’s association with dementia. PLoS One 2017b; 12: e0175790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royall DR, Al-Rubaye S, Bishnoi R, Palmer RF.. Serum protein mediators of dementia and Aging Proper. Aging 2016; 8: 3241–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royall DR, Lauterbach EC, Kaufer DI, Malloy P, Coburn KL, Black KJ.. The cognitive correlates of functional status: a review from the Committee on Research of the American Neuropsychiatric Association. J Neuropsychiatry Clin Neurosci 2007; 19: 249–65. [DOI] [PubMed] [Google Scholar]
- Royall DR, Palmer RF.. δ-Related biomarkers mediate multiple AD conversion risks and offer targets for intervention. J Gerontol Ser A: Med Sci 2019a, in press. [DOI] [PubMed] [Google Scholar]
- Royall DR, Palmer RF.. “Executive functions” cannot be distinguished from general intelligence: two variations on a single theme within a symphony of latent variance. Frontiers Behav Neurosci 2014; 8: 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royall DR, Palmer RF.. Ethnicity moderates dementia’s biomarkers. J Alzheimers Dis 2015; 43: 275–87. [DOI] [PubMed] [Google Scholar]
- Royall DR, Palmer RF.. Thrombopoeitin is associated with δ’s intercept, and only in non-Hispanic whites. Alzheimers Dement (Amst) 2016; 3: 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royall DR, Palmer RF.. Selection for depression-specific dementia cases with replication in two cohorts. PLoS One 2019b; 14: e0216413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royall DR, Palmer RF, Alzheimer’s Disease Neuroimaging Initiative. A δ homolog for dementia case finding with replication in the Alzheimer's Disease Neuroimaging Initiative. J Alzheimers Dis 2019; 67: 67–79. [DOI] [PubMed] [Google Scholar]
- Royall DR, Palmer RF, O’Bryant SE.. Validation of a latent variable representing the dementing process. J Alzheimers Dis 2012; 30: 639–49. [DOI] [PubMed] [Google Scholar]
- Satapathy SK, Ochani M, Dancho M, Hudson LK, Rosas-Ballina M, Valdes-Ferrer SI, . Galantamine alleviates inflammation and other obesity-associated complications in high-fat diet-fed mice. Mol Med 2011; 17: 599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saykin AJ, Shen L, Foroud TM, Potkin SG, Swaminathan S, Kim S, . Alzheimer’s Disease Neuroimaging Initiative biomarkers as quantitative phenotypes: genetics core aims, progress, and plans. Alz Dement 2010; 6: 265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer JL, Graham JW.. Missing data: our view of the state of the art. Psychol Methods 2002; 7: 147–77. 2002. [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, . Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009; 65: 403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh JI, Yesavage JA.. Geriatric Depression Scale (GDS): recent evidence and development of a shorter version. Clin Gerontologist 1986; 5: 165–73. [Google Scholar]
- Spearman C. General intelligence, objectively determined and measured. Am J Psychol 1904; 15: 201–93. [Google Scholar]
- Stern Y. Cognitive reserve in ageing and Alzheimer's disease. Lancet Neurol 2012; 11: 1006–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajar A, O'Connell MD, Mitnitski AB, O'Neill TW, Searle SD, Huhtaniemi IT, . Frailty in relation to variations in hormone levels of the hypothalamic-pituitary-testicular axis in older men: results from the European male aging study. J Am Geriatr Soc 2011; 59: 814–21. [DOI] [PubMed] [Google Scholar]
- Templin AT, Meier DT, Willard JR, Wolden-Hanson T, Conway K, Lin YG, . Use of the PET ligand florbetapir for in vivo imaging of pancreatic islet amyloid deposits in hIAPP transgenic mice. Diabetologia 2018; 61: 2215–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng E, Becker BW, Woo E, Cummings JL, Lu PH.. Subtle deficits in instrumental activities of daily living in subtypes of mild cognitive impairment. Dement Geriatr Cogn Disord 2010; 30: 189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennent GA, Lovat LB, Pepys MB.. Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. Proc Natl Acad Sci USA 1995; 92: 4299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosun D, Schuff N, Truran-Sacrey D, Shaw LM, Trojanowski JQ, Aisen P, . Relations between brain tissue loss, CSF biomarkers, and the ApoE genetic profile: a longitudinal MRI study. Neurobiol Aging 2010; 31: 1340–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travison TG, Nguyen A-H, Naganathan V, Stanaway FF, Blyth FM, Cumming RG, . Changes in reproductive hormone concentrations predict the prevalence and progression of the frailty syndrome in older men: the concord health and ageing in men project. J Clin Endocrinol Metab 2011; 96: 2464–74. [DOI] [PubMed] [Google Scholar]
- von Guten A, Ebbing K, Imhof A, Giannakopoulos P, Kövari E.. Brain aging in the oldest‐old. Curr Gerontol Geriatr Res 2010. pii:358531. doi: 10.1155/2010/358531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voyle N, Baker D, Burnham SC, Covin A, Zhang Z, Sangurdekar DP. Blood protein markers of neocortical amyloid-β burden: a candidate study using SOMAscan technology. J Alzheimers Dis 2015; 46: 947–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner T, Page J, Burniston M, Skillen A, Ross JC, Manwani R, . Extracardiac 18F-florbetapir imaging in patients with systemic amyloidosis: more than hearts and minds. Eur J Nucl Med Mol Imaging 2018; 45: 1129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waring S, O’Bryant SE, Reisch JS, Diaz-Arrastia R, Knebl J, Doody R, . The Texas Alzheimer's Research Consortium longitudinal research cohort: study design and baseline characteristics. Texas Public Health Journal 2008; 60: 9–13. [Google Scholar]
- Warren MW, Weiner LS, Texas MF, Research Care Consortium A.. Lipids and adipokines as risk factors for Alzheimer’s disease. J Alzheimers Dis 2012; 29: 151–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler D, Wechsler memory scale—third edition. San Antonio, TX: The Psychological Corporation; 1997. [Google Scholar]
- Weiner MW, Veitch DP.. Introduction to special issue: overview of Alzheimer's Disease Neuroimaging Initiative. Alzheimers Dement 2015; 11: 730–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte MM, Trzepacz P, Case M, Yu P, Hochstetler H, Quinlivan M, . Association between clinical measures and florbetapir F18 PET neuroimaging in mild or moderate Alzheimer's disease dementia. J Neuropsychiatry Clin Neuzrosci 2014; 26: 214–20. [DOI] [PubMed] [Google Scholar]
- Wyman BT, Harvey DJ, Crawford K, Bernstein MA, Carmichael O, Cole PE, . Standardization of analysis sets for reporting results from ADNI MRI data. Alzheimers Dement 2013; 9: 332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data underlying the results presented in the study are available from TARCC and ADNI. Requests for TARCC data should be made to http://www.txalzresearch.org/research/ (4 December 2019, date last accessed). Requests for ADNI data should be made to adni.loni.usc.edu.