

Abstract
We describe here design, synthesis, and biological evaluation of a series of highly potent HIV-1 protease inhibitors containing stereochemically defined and unprecedented tricyclic furanofuran derivatives as P2 ligands in combination with a variety of sulfonamide derivatives as P2’ ligands. These inhibitors were designed to enhance the ligand-backbone binding and van der Waals interactions in the protease active site. A number of inhibitors containing the new P2 ligand, an aminobenzothiazole as the P2’ ligand and a difluorophenylmethyl as the P1 ligand, displayed very potent enzyme inhibitory potency and also showed excellent antiviral activity against a panel of highly multidrug-resistant HIV-1 variants. The tricyclic P2 ligand has been synthesized efficiently in an optically active form using enzymatic desymmetrization of meso-1,2-(dihydroxymethyl)cyclohex-4-ene as the key step. We determined high-resolution X-ray structures of inhibitor-bound HIV-1 protease. These structures revealed extensive interactions with the backbone atoms of HIV-1 protease and provided molecular insights into the binding properties of these new inhibitors.
Graphical Abstract
INTRODUCTION
HIV-1 protease inhibitors (PIs) are critical components of combined antiretroviral therapy (cART) which dramatically improved the HIV-related mortality and morbidity to patients with HIV-1 infection and AIDS.1,2 PIs block the cleavage of Gag-Pol polyproteins during viral maturation and result in generation of noninfectious virions.3,4 PI-based drugs in general exhibit a relatively high genetic barrier to the emergence of drug-resistant HIV-1 variants compared to non-nucleoside reverse transcriptase inhibitors and integrase strand transfer inhibitors (INSTIs).5-7 The last approved PI, darunavir (1, DRV, Figure 1), is a first-line therapy which is often preferred for cART-naïve HIV-1 infected patients because of its tolerance and a high genetic barrier to the development of drug-resistant viruses.8-10 However, there are reports of failure of DRV-containing regimens and the emergence of darunavir resistant HIV-1 variants.11,12 Furthermore, there are reports that a growing number of HIV/AIDS patients are harboring highly multidrug-resistant HIV-1 variants, and options for treating these patients are limited.13,14 Therefore, the development of potent and structurally novel PIs with broad spectrum antiviral activity are necessary for the future success of cART treatment regimens.
Figure 1.

Structures of HIV-1 PIs 1–4.
Darunavir and its derivatives (2) were designed to promote a network of hydrogen bonding interactions with the backbone atoms at the active site of HIV-1 protease. The backbone binding strategies are likely to slow development of drug resistant HIV-1 variants because of possible reduction of catalytic fitness. Darunavir incorporated the stereochemically defined and conformationally constrained bicyclic polyether, bis-tetrahydrofuran (bis-THF), which makes extensive interactions in the active site.10,15,16 Numerous X-ray crystallographic studies of darunavir-bound HIV-1 protease revealed that darunavir forms a network of hydrogen bonding interactions with the backbone of HIV-1 protease in both S2 and S2’ subsites.17,18 The P2 bis-tetrahydrofuranyl urethane (bis-THF) ligand is an intriguing pharmacophore where both ring oxygens form strong hydrogen bonds with the Asp29 and Asp30 backbone amide NHs. The bicyclic bis-THF ring also engages in van der Waals interaction with active site residues.19,20 In our continuing efforts to optimize both P2 and P2’ structural templates of darunavir, we have recently reported a number of very potent PIs with unprecedented structural features.21-23 In particular, we designed a crown-like tetrahydropyrano-tetrahydrofuran in inhibitor 3 with a bridged methylene group as the P2 ligand to promote additional van der Waals interactions in the active site.24,25 Inhibitors containing such crown-THF ligands are very potent inhibitors and maintained potent antiviral activity against multidrug-resistant HIV-1 variants.25,26 The X-ray structural analysis of crown-THF-derived inhibitors provided molecular insights to further improve ligand-binding site interactions.24,25 Herein, we report a new class of PIs containing cyclohexane fused bis-tetrahydrofuran as the P2 ligands in combination with (R)-hydroxyethylaminesulfonamide isosteres. A number of inhibitors exhibited exceptionally potent enzyme inhibitory and antiviral activity. In particular, inhibitor 4d maintained exceptional antiviral potency against selected multidrug-resistant HIV-1 variants. Our high resolution X-ray structural studies of inhibitor-bound HIV-1 protease revealed important molecular insights into the ligand-binding site interactions responsible for their potent activity. The design and synthesis of new PIs have been an active area of our research. Several potent inhibitors incorporating a variety of heterocyclic P2 ligands for improving interactions at the S2 subsite have been reported.27-30 A number of other design efforts were focused on optimizing both P1 and P2’ moieties to improve potency.27,28,31 Recently, bicyclic piperazine sulfonamide-based inhibitors have been designed where the piperazine NH forms the key interaction with the catalytic aspartic acids of the HIV-1 protease.32,33
RESULTS AND DISCUSSION
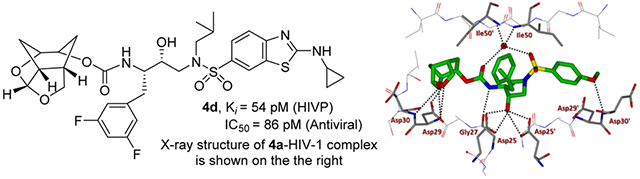
Based upon ligand-binding site interactions of the crown-THF ligand in 3 in the HIV-1 protease active site, we have designed a new tricyclic cyclohexane fused-tetrahydrofuranofuran (Chf-THF) derivative as the P2 ligand shown in inhibitors 4a and 5a (Table 1). This new design may promote better hydrogen bonding interactions with backbone atoms. Furthermore, the new ligand with additional methylene groups around the bis-THF may exhibit better van der Waals interactions in the active site. Our preliminary model of the enantiomeric ligand structure also showed good orientation for hydrogen bonding interactions with backbone amide NHs of Asp29 and Asp30. The conformationally constrained ligand also appeared to engage in additional van der Waals contacts in the S2 subsite compared to the bis-THF ligand to darunavir. While the stereochemistry of the Chf-ligand in inhibitors 4a and 5a appeared optimum, we planned to synthesize both enantiomeric ligands as both enantiomers of bis-THF ligand and crown-THF in inhibitor 3 showed comparable enzyme affinity and ligand binding site interactions. This is due to the fact that the acetal oxygens in both enantiomers are in a similar environment and both ligands can form strong hydrogen bonds with the backbone amide residues in the S2 subsite. Therefore, our plan is to synthesize both enantiomers and gain molecular insights into their ligand-binding site interactions using X-ray structural studies of the inhibitor-HIV-1 protease complex. We also plan to examine various hydroxyethylsulfonamide isosteres containing varying P1 and P2’ ligands. In particular, we plan to investigate an aminobenzothiazole(Abt)-based P2’-ligand which has been showed to promote better hydrogen bonding and van der Waals interactions compared to the 4-aminobenzene-sulfonamide P2’-ligand of darunavir. Structurally, the Abt P2’ ligand forms additional hydrogen bonding and van der Waals interactions in the S2’ subsite. A number of recent inhibitors containing these P2’-ligands exhibited robust antiviral activity against highly multidrug-resistant HIV-1 variants.24,25
Table 1.
HIV-1 Protease Inhibitory and Antiviral Activity of PIs



Ki values represents at least four data points. Standard error in all cases was less than 7%. Darunavir exhibited Ki= 16 pM.
Values are means of at least three experiments. Standard error in all cases was less than 5%. Darunavir exhibited antiviral IC50 = 3.2 nM, saquinavir IC50 = 21 nM.
For our studies, we plan to synthesize our newly designed ligand in a stereodefined manner. Our synthetic strategy for optically active synthesis is shown in Scheme 1. Enantiomeric ligand 6 can be obtained from the functionalized cyclohexane-1,2-diol derivative 7. Structure 7 can be obtained from cyclohexene derivative 8 by asymmetric dihydroxylation reaction.34,35 An optically active aldehyde derivative can be derived conveniently from meso diol derivative 9 by enzymatic desymmetrization as the key reaction. The meso-diol 9 can be derived from commercially available and inexpensive 1,2,3,6-tetrahydrophthalic anhydride 10.
Scheme 1.
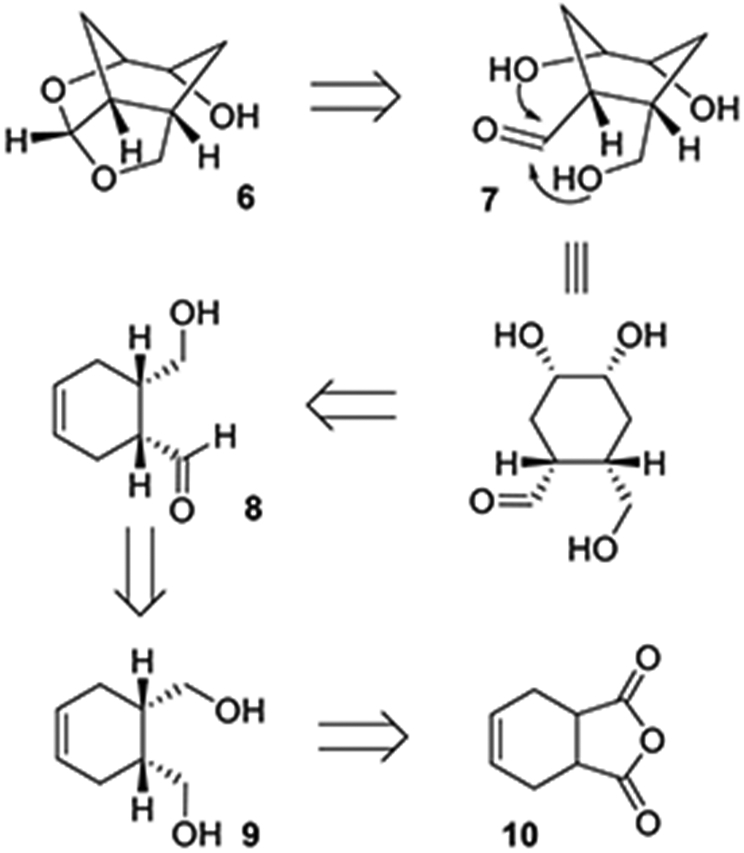
Synthetic Strategy for the Cyclohexane-Fused bis-THF P2 Ligand Structure
Our synthesis of optically active ligand 6 is shown in Scheme 2. meso-1,2,3,6-Tetrahydrophthalic anhydride 10 was reduced by LiAlH4 in THF at 0 °C for 3 h to provide meso-diol derivative 9 in a multigram scale.36 Diol 9 was subjected to enzymatic desymmetrization reaction using porcine pancreatic lipase (PPL) in ethyl acetate at 23 °C for 12 h to provide monoacetate derivative 11 in gram scale in 82% yield and >95% ee, as determined by high-performance liquid chromatography (HPLC) analysis37 (see Supporting Information for further details). Swern oxidation of alcohol 11 provided the corresponding aldehyde which was reacted with trimethylorthoformate in the presence of a catalytic amount of tetrabutylammonium tribromide (TBABr3) at 23 °C for 8 h to provide dimethylacetal derivative 12 in 76% yield over two steps. For diastereoselective dihydroxylation, we carried out Sharpless asymmetric dihydroxylation using AD-mix-β.34 Reaction of 12 with AD-mix-β in a mixture (1:1) of t-butanol and water at 0–23 °C for 24 h resulted in a 1:1 mixture of diastereomeric diol.38 The resulting diol was subjected to saponification using 1 N aqueous NaOH in MeOH at 0–23 °C for 3 h to provide triol derivatives 13 and 14 in 90% yield over two steps. These triol derivatives were separated by silica gel chromatography using 5% MeOH in CH2Cl2 as the eluent. Triol derivative 13 was reacted with a catalytic amount of camphorsulfonic acid (CSA) in CH2Cl2 at 0 °C for 1 h to provide optically active tricyclic ligand alcohol 6 in 82% yield.
Scheme 2.
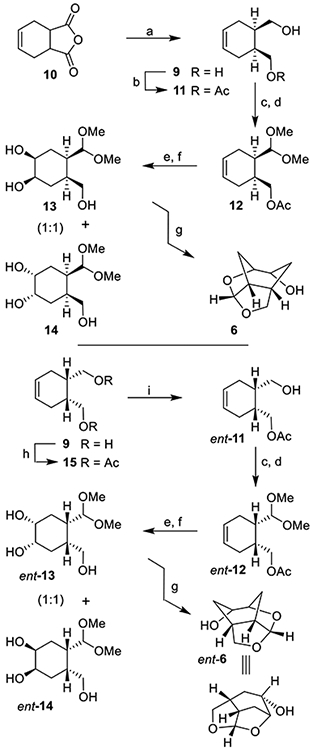
Synthesis of Substituted Tricyclic P2 Ligands 6 and ent-6a
aReagents and conditions. (a) LiAlH4, THF, 0 °C, 3 h (85%); (b) PPL, EtOAc, 23 °C, 12 h (82%); (c) (COCl)2, DMSO, TEA, CH2Cl2, −78 to 0 °C, 1.5 h; (d) CH(OMe)3, TBABr3, MeOH, 23 °C, 8 h (76% over 2-steps for 12; 80% over 2-steps for ent-12); (e) AD mix-β, CH3SO2NH2, t-BuOH/H2O (1:1), 0–23 °C; (f) 1 N NaOH, MeOH, 0–23 °C, 3 h (90% over 2-steps for 13 & 14; 88% over 2-steps for ent-13 & ent-14); (g) CSA, CH2Cl2, 0 °C, 1 h (82% for 6 and 79% for ent-6); (h) Ac2O, Py, DMAP, CH2Cl2, 0–23 °C (98%); (i) PPL, 0.1 M phosphate buffer pH 7, 1 N NaHCO3, 23 °C, 16 h (84%).
Our synthesis of enantiomeric ligand ent-6 from meso-diol 9 is shown in Scheme 2. Diol 9 was converted to diacetate derivative 15 by reaction with acetic anhydride and pyridine in the presence of a catalytic amount of DMAP at 23 °C for 16 h. Exposure of diacetate to PPL in 0.1 M phosphate buffer at pH 7 in the presence of aqueous NaHCO3 at 23 °C for 16 h provided optically active alcohol ent-11 in 84% yield.37,39 Alcohol ent-11 was converted to dimethylacetal ent-12 in 80% yield as described above. Exposure of dimethylacetal ent-12 to sharpless asymmetric dihydroxylation reaction with AD-mix-α afforded a 1:1 mixture of diastereomeric diol.34 Deprotection of the acetate derivative furnished triol derivatives ent-13 and ent-14 which were separated by silica gel chromatography. Treatment of the major triol derivative ent-13 with a catalytic amount of CSA in CH2Cl2 afforded ligand alcohol ent-6 in 79% yield.
The synthesis of the designed PIs was carried out in a two-step sequence involving synthesis of activated carbonates followed by reaction of these carbonates with appropriate hydroxyethylaminesulfonamide isosteres.22,23 The syntheses of various activated carbonates are shown in Scheme 3. Optically active ligand alcohols 6 and ent-6 synthesized above were converted to their respective activated carbonates 16 and ent-16. As shown, reaction of ligand alcohols 6 and ent-6 with 4-nitrophenylchloroformate in the presence of pyridine in CH2Cl2 at 0–23 °C for 12 h provided activated carbonates 16 and ent-16 in 87 and 88% yields, respectively. These carbonates were then converted to urethane derivatives using amines 17–22. The synthesis of various inhibitors containing Chf-THF as the P2 ligands on the hydroxyethylamine sulfonamide isostere is shown in Scheme 4. Reactions of activated carbonate 16 with known22,23 amine derivatives 17–22 in the presence of diisopropylethylamine (DIPEA) in CH3CN at 23 °C for 72 h provided inhibitors 4a–f in good yields (65–86%). Similarly, reactions of carbonate ent-16 with amines 17–22 under similar conditions afforded inhibitors 5a–5f in very good yields (59–87%). The full structures of these inhibitors are shown in Table 1.
Scheme 3.
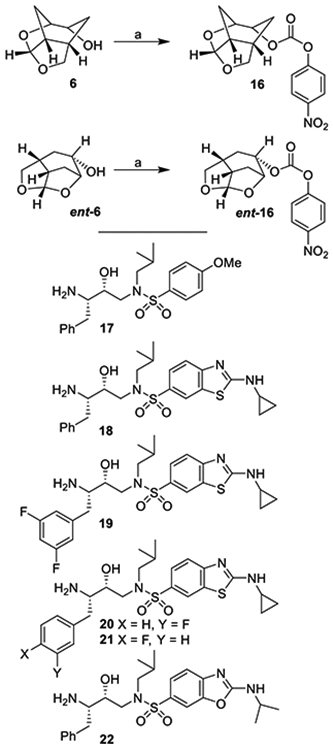
Synthesis of Activated Carbonates 16 and ent-16a
aReagents and Conditions. (a) 4-NO2PhOCOCl, Py, CH2Cl2, 0–23 °C, 8 h (87% for 16 and 88% for ent-16)
Scheme 4.
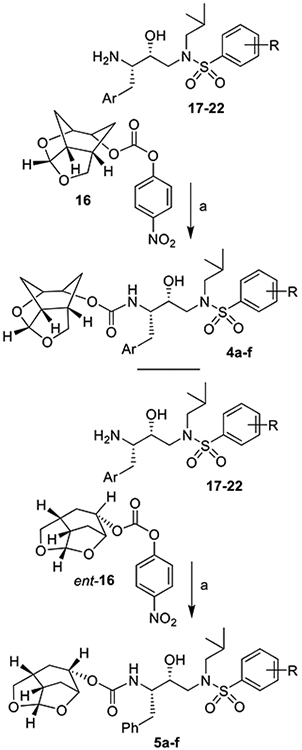
Synthesis of PIs 4a–4f and 5a–fa
aReagents and Conditions. (a) DIPEA, CH3CN, 23 °C, (59–87%)
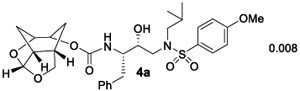
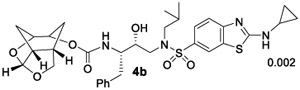
Our preliminary model of the P2 Chf-THF ligand in inhibitor 4a compared to the bis-THF ligand in inhibitor 2 indicated that both oxygens of the new Chf-THF ligand could align optimally with Asp29 and Asp30 backbone amide NHs in the S2 site. In addition, two methylene groups are expected to provide van der Waals interactions surrounding Ile47, Val32, Leu76, and Ile50’ residues in the S2 subsite. Because ligand combinations are critical for overall affinity, we investigated enantiomeric Chf-THF with various P2’-ligands. The results of compounds in HIV-1 protease inhibitory and antiviral assays are shown in Table 1. The assay protocol for HIV-1 inhibition is similar to the reported procedure of Toth and Marshall.40 The protocol for antiviral assay is described previously using MT-2 human T-lymphoid cells exposed to HIV-1LAI.40 To assess the stereochemical effect, we first examined the enantiomeric ligands in combination with a sulfonamide isostere with a 4-methoxy benzene sulfonamide as the P2’-ligand in compounds 4a and 5a. Inhibitor 4a showed very potent HIV-1 protease inhibitory activity with a Ki value of 8 pM. The inhibitor 4a also exhibited a very good antiviral IC50 value of 38 nM (entry 1). Inhibitor 5a with enantiomeric Chf-THF ligand is slightly less potent than 4a. It showed enzyme inhibitory activity with a Ki of 34 pM and antiviral activity with an IC50 value of 72 nM (entry 2). We then examined both enantiomeric ligands in combination with other benzothiazoles and benzoxazoles as the P2’-ligands. Inhibitor 4b showed very potent enzyme Ki of 2 pM, which is nearly 10-fold better than inhibitor 5b with the enantiomeric P2 ligand (entries 3 and 4). However, both inhibitors exhibited very potent antiviral activity with IC50 values of 3.3 and 1.8 nM. Inhibitors 4c and 5c with enantiomeric Chf-THF P2 ligands and benzoxazole P2’ ligands also showed very potent enzyme inhibitors and antiviral activity (entries 5 and 6). Interestingly, inhibitor 5c exhibited over 5-fold improvement of antiviral activity (IC50 = 0.3 nM) compared to inhibitor 4c (IC50 = 1.7 nM).
We then examined inhibitors containing enantiomeric Chf-THF ligands in combination with fluorine-substituted phenylmethyl groups as the P1 ligands. As shown in Table 2, incorporation of a 3,5-difluoro phenylmethyl group as the P1-ligand resulted in inhibitor 4d which displayed enzyme inhibitory Ki of 54 pM and antiviral IC50 value of 86 pM. Inhibitor 4d showed improved antiviral activity compared to the corresponding inhibitor 4b containing a phenylmethyl group as the P1-ligand. This improvement in antiviral activity may be due to increased lipophilicity of inhibitor 4d (clogp 5.4) over inhibitor 4b (clogP 5.1). Inhibitor 5d containing the enantiomeric P2 ligand also showed very potent enzyme Ki value; however, it showed nearly 25-fold reduction of antiviral activity compared to inhibitor 4d with an enantiomeric P2 ligand (entries 1 and 2). In comparison, darunavir and saquinavir exhibited antiviral IC50 values of 3.2 and 21 nM, respectively. We also examined the effect of monofluoro substitution on the P1-ligand. As shown, incorporation of 3-fluorophenylmethyl as the P1 ligand resulted in inhibitors 4e and 5e with only marginal improvement of antiviral activity compared to unsubstituted P1 ligands in inhibitors 4b and 5b (Table 1). Incorporation of 4-fluoro phenylmethyl as the P1 ligand resulted in inhibitors 4f and 5f with improvement of antiviral activity, over 7-fold compared to inhibitor 4b and over 4-fold compared to inhibitor 5b (entry 3, Table 1). Overall, inhibitor 4d with 3,5-bis-fluorines on the P1 ligand showed the best antiviral activity over methoxy sulfonamide and monofluoro derivatives.
Table 2.
HIV-1 Protease Inhibitory and Antiviral Activity of PIs
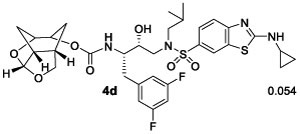
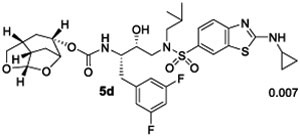
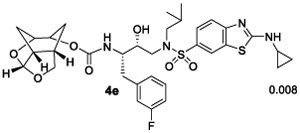
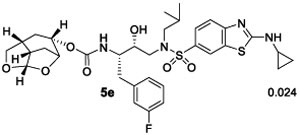

Ki values represents at least four data points. Standard error in all cases was less than 7%. Darunavir exhibited Ki= 16 pM.
Values are means of at least three experiments. Standard error in all cases was less than 5%. Darunavir exhibited antiviral IC50 = 3.2 nM, saquinavir IC50 = 21 nM.
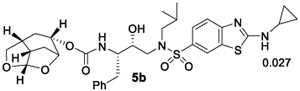
While the cART therapy and treatment guidelines are updated regularly, PI-based drugs are important elements of current cART regimens. In particular, PIs are widely used for the treatment of naïve and experienced HIV/AIDS patients. As mentioned earlier, heavily-ART regimen-experienced HIV/AIDS patients are reported to have drug-failure with the currently available PIs including DRV.11,12 Therefore, the design of new classes of potent PIs with a high genetic barrier to development of drug resistance is important for durable treatment options. In this context, our long-standing objective is to design PIs that can maintain robust potency against a variety of existing multi-PI-resistant HIV-1 variants with better selectivity index and safety profiles. Based upon preliminary antiviral data, we selected two potent inhibitors (4d, antiviral IC50 of 0.086 nM and 5c, antiviral IC50 of 0.3 nM) containing enantiomeric Chf-THF ligands and evaluated their antiviral activity against a panel of highly multidrug-resistant HIV-1 variants that had been selected in vitro with widely used FDA-approved PIs, ATV and DRV. Each of these HIV-1 variants were selected in vitro by propagating HIV-1NL4-3 in the presence of increasing concentrations of each PI (up to 5 μM) in MT-4 cells, as described by us previously.41,42 The results are shown in Table 3. As can be seen, both widely used PI drugs, ATV and DRV, lost activity against these resistant variants. In particular, ATV lost significant potency and is unable to suppress the replication of highly PI-resistant HIV-1 variants examined. DRV showed relatively better results compared to ATV; however, DRV lost 86-fold and 89-fold activity against HIV-1LPV-5 μM and HIV-1DRVRP30 variants. Inhibitor 5c containing the Chf-THF P2 ligand failed to block replication of these highly drug-resistant HIV-1 variants. Inhibitor 4d containing enantiomeric P2-ligand is significantly more potent than DRV and ATV against HIV-1NL4-3 virus. While this inhibitor maintained very good antiviral activity (0.21–4.5 nM) against these highly drug-resistant HIV-1 variants, its fold-change of activity is comparable to DRV. The reason for better overall antiviral activity is possibly due to extensive hydrogen bonding and van der Waals interactions in the S2 and S2’ subsites. Our X-ray crystallographic studies of inhibitors 4a- and 5c-bound HIV-1 protease provided molecular insights into the ligand-binding site interactions responsible for their activity.
Table 3.
| mean IC50 in nM ± SD (fold-change) |
|||||
|---|---|---|---|---|---|
| virus species |
ATV |
DRV |
compound 5c |
compound 4d |
|
| wild-type | HIV-1NL4-3 | 4.0 ± 0.7 | 4.2 ± 0.9 | 38 ± 1 | 0.034 ± 0.026 |
| in vitro HIV-1PRR | HIV-1ATV-5 μM | >1000 (>250) | 26 ± 6(6.1) | 350 ± 30 (9.1) | 0.21 ± 0.07 (6.3) |
| HIV-1LPV-5 μM | 430 ± 10 (107) | 360 ± 50 (86) | >1000 (>26) | 4.3 ± 0.2 (127) | |
| HIV-1DRVRP30 | >1000 (>250) | 370 ± 120 (89) | 640 ± 510 (17) | 4.5 ± 2.5 (133) | |
The amino acid substitutions identified in protease of HIV-1ATV-5 μM, HIV-1LPV-5 μM, and HIV-1DRVRP30 compared to the wild-type HIV-1NL4-3 include L23I/E34Q/K43I/M46I/I50L/G51A/L63P/A71V/V82A/T91A, L10F/V32I/M46I/I47A/A71V/I84V, and L10I/I15V/K20R/L24I/ V32I/M36I/ M46L/L63P/K70Q/V82A/I84V/L89M, respectively.
In vitro HIV-1PRR, in vitro PI-selected HIV-1 variants.
The EC50 (50% effective concentration) values were determined by using MT-4 cells as target cells. MT-4 cells (105/mL) were exposed to 100 TCID50s of each HIV-1, and the inhibition of p24 Gag protein production by each drug was used as an endpoint. Numbers in parentheses represent fold changes in IC50s for each isolate compared to the IC50s for wild-type HIV-1NL4-3. All assays were conducted in duplicate, and the data shown represent mean values (±1 standard deviation) derived from the results of two independent experiments.
We determined the X-ray structures of inhibitors 4a and 5c containing enantiomeric ligands in complex with wild-type HIV-1 protease at a resolution of 1.25 and 1.3 Å, respectively.43 Both complexes crystallized in the orthorhombic space group P21212 with one protease homodimer per asymmetric unit. The inhibitors bind at the active site in two alternate conformations related by 180° rotation with a relative occupancy of 0.7/0.3 for inhibitor 4a and 0.55/0.45 for inhibitor 5c. The overall dimer structure is very similar to the HIV-1 protease–darunavir complex17,18 with an rsmd for superposition of 198 equivalent Cα atoms of 0.14 Å and 0.12 for protease complexes with inhibitors 4a and 5c, respectively. The largest deviation of 0.45 Å in comparison to the protease–darunavir complex occurs at residue Pro39 for 5c complex, while flap residue Phe53’ exhibits a maximum deviation of 0.51 Å in 4a complex. The key interactions of inhibitors 4a and 5c with HIV-1 protease are highlighted in Figures 2 and 3, respectively. Both inhibitors retain all the hydrogen bonds observed between darunavir and the main chain atoms of the HIV-1 protease. The new inhibitors also form a tetracoordinated water-mediated hydrogen bond interaction involving one of the sulfonamide oxygens and carbonyl oxygen with the amide nitrogen of flap residues Ile 50 and Ile 50’ similar to the protease–darunavir complex.44
Figure 2.

Inhibitor 4a-bound HIV-1 protease X-ray structure is shown (pdb code: 6VOD). The inhibitor carbon atoms are shown in green and hydrogen bonds are shown by black dotted lines.
Figure 3.

Inhibitor 5c-bound HIV-1 protease X-ray structure is shown (pdb code: 6VOE). The inhibitor carbon atoms are shown in cyan and hydrogen bonds are shown by black dotted lines.
Inhibitor 5c has a Chf-THF as P2 group and the two acetal oxygens in the bulky Chf-THF form hydrogen bond interactions with the main chain amide of Asp29 and Asp30 similar to the hydrogen bonds formed by the bis-THF oxygens in darunavir. However, unlike the bis-THF of darunavir, the bigger Chf-THF of inhibitors 4a and 5c form a number of additional van der Waals interactions. We compared van der Waals interactions of the enantiomeric Chf-THF ligands in Figure 4. Interestingly, in one conformation, Chf-ligand in compound 5c forms van der Waals contacts with Ile47. The tricyclic core of the Chf-THF ligand forms van der Waals interaction with the side chain atoms of Ile47, the main chain amide of Gly48, and the Cδ1 atom of Ile50’. The two acetal oxygens in the enantiomeric P2 Chf-THF of 4a are shifted by 0.8 Å in comparison to those of 5c. There is an additional hydrogen bond interaction between one of the acetal oxygens and the carboxylate side chain of Asp29. Further, the enantiomeric change allows the 6-member ring of Chf-THF in inhibitor 4a to shift toward Ile84. Because of this shift, the P2 group of 4a form van der Waals interactions with Ile 84 in addition to those with Ile47, Gly48, and Ile50’.
Figure 4.

Side by side comparison of the new Chf-THF moiety of inhibitor 4a (left, green carbon atoms) with the enantiomeric Chf-THF moiety of inhibitor 5c (right, cyan carbon chain) inside the S2 subpocket. Both ligands form extensive van der Waals interactions (Val32, Ile47, Ile50’, and Ile84 for 4a and Val32, Ile47, and Ile50’ for 5c) in the S2 subsite. Also, they are located close to the periphery of the protease active site and form three strong hydrogen bonds in a similar fashion (black dotted lines).
The aminobenzene group at the P2’ of darunavir is replaced by the extended N-isopropylbenzo[d]oxazol-2-amine group in inhibitor 5c. In the protease–darunavir complex, the P2’ amine of darunavir forms direct and water-mediated hydrogen bonds with the two alternate conformations of Asp30’ side chain. In the protease complex with 5c, the single conformation of Asp30’ forms a direct hydrogen bond interaction with the oxazole nitrogen of the inhibitor. In addition, the extended P2’ amine group of inhibitor 5c forms a new hydrogen bond with the carboxylate side chain of Asp30’. Further, the terminal P2’ isopropyl group of 5c forms a C–H⋯ O interaction (2.8–3.2 Å) with the carboxylate side chain of Asp29’ and van der Waals interaction with Ile47’. Asp29’ plays a vital role in the structural integrity of the protease dimer by forming a conserved inter-subunit ion pair with Arg8. The P2’ methoxy group of inhibitor 4a has a similar position as the P2’ aminobenzene of darunavir in the S2’ subsite of protease. The oxygen atom of the P2’ methoxy group in inhibitor 4a forms a hydrogen bond with the main chain amide of Asp30’. The Chf-THF P2-ligand in inhibitor 4a appeared to form enhanced ligand-binding site interactions over the enantiomeric ligand in inhibitor 5c in the HIV-1 protease active site. The combination of the Chf-THF P2-ligand, 3,5-difluorinated P1 ligand and the aminobenzothiazole P2’ ligand resulted in inhibitor 4d which exhibited best antiviral activity and maintained very good antiviral activity against a panel of highly drug-resistant HIV-1 variants.
CONCLUSIONS
In summary, based on X-ray structural data, we have designed a new class of very potent HIV-1 PIs incorporating new polycyclic cylcohexane-fused bis-tetrahydrofuran derivatives as the P2 ligands. The new ligands have been specifically designed to make favorable hydrogen bonding interactions and van der Waals interactions with the residues at the S2-subsite. In general, inhibitors containing enantiomeric ligands have shown very potent activity, particularly inhibitors 4d and 5c. However, inhibitor 4d with a difluoro phenylmethyl P1 ligand and an aminobenzothiazole as the P2’ ligand maintained very potent activity against a panel of highly multidrug-resistant HIV-1 variants. The results show that inhibitor 4d is superior to darunavir and other approved PI drugs. The new polycyclic ligand alcohols were synthesized efficiently in an enantioselective manner using enzymatic desymmetrization of meso-diols as the key steps. Because both enantiomeric ligands exhibited very high enzyme affinity, we determined high resolution X-ray structures of inhibitor-bound HIV-1 protease containing enantiomeric ligands. As it turned out, both oxygens of the enantiomeric tricyclic ligands formed very strong hydrogen bonds with the backbone amide NHs of Asp29 and Asp30 in the S2 subsite. The P2 ligand in inhibitor 4a appears to make better van der Waals interactions compared to the enantiomeric ligands in the S2 site. Incorporation of the Chf-THF P2 ligand, the fluorinated P1 ligand, and the aminobenzothiazole P2’ ligand resulted in inhibitor 4d that showed best antiviral activity. Further optimization of inhibitor properties and ligand-binding site interactions are in progress in our laboratory.
EXPERIMENTAL SECTION
General Methods.
All chemicals and reagents were purchased from commercial suppliers and used without further purification unless otherwise noted. The following reaction solvents were distilled prior to use: dichloromethane from calcium hydride, diethyl ether and tetrahydrofuran from Na/benzophenone, and methanol and ethanol from activated magnesium under argon. All reactions were carried out under an argon atmosphere in either flame or oven-dried (120 °C) glassware. TLC analysis was conducted using glass-backed thin-layer silica gel chromatography plates (60 Å, 250 μm thickness, F-254 indicator). Column chromatography was performed using 230–400 mesh, 60 Å pore diameter silica gel. 1H and 13C NMR spectra were recorded at room temperature on a Bruker AV800, DRX-500 and AV-400. Chemical shifts (δ values) are reported in parts per million and are referenced to the deuterated residual solvent peak. NMR data are reported as δ value (chemical shift, J-value (Hz), integration, where s = singlet, d = doublet, t = triplet, q = quartet, and brs = broad singlet). Optical rotations were recorded on a PerkinElmer 341 polarimeter. HRMS and LRMS spectra were recorded at the Purdue University Department of Chemistry Mass Spectrometry Center. HPLC analysis and purification were done on an Agilent 1100 series instrument using a Chiralcel OZ-3 column of 4.6 mm ID for analysis. The purity of all test compounds was determined by HPLC analysis to be ≥95% pure.
(1R,3aS,7aR)-Octahydro-1,6-epoxyisobenzofuran-5-yl((2S,3R)-3-Hydroxy-4-((N-isobutyl-4-methoxyphenyl)-sulfonamido)-1-phenylbutan-2-yl)carbamate (4a).
To a stirred solution of activated alcohol 16 (15 mg, 0.046 mmol) and isostere 17 (21 mg, 0.051 mmol) in acetonitrile (2 mL) was added DIPEA (40 μL, 0.233 mmol) at 23 °C under an argon atmosphere. The reaction mixture was stirred at 23 °C until completion. Upon completion, solvents were removed under reduced pressure and the crude product was purified by silica gel column chromatography (50% EtOAc in hexane) to give 4a (22 mg, 81%) as an amorphous solid. Rf = 0.3 (70% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3): δ 7.72 (d, J = 8.9 Hz, 2H), 7.30–7.23 (m, 4H), 7.22–7.17 (m, 1H), 6.97 (d, J = 8.9 Hz, 1H), 5.60 (d, J = 3.6 Hz, 1H), 5.02 (d, J = 9.3 Hz, 1H), 4.71 (t, J = 6.2 Hz, 1H), 4.31 (t, J = 5.4 Hz, 1H), 4.10 (m, 1H), 3.87 (m, 1H), 3.86 (s, 3H), 3.82–3.71 (m, 3H), 3.19–3.04 (m, 3H), 2.97 (dd, J = 13.4, 8.3 Hz, 1H), 2.83–2.75 (m, 2H), 2.72 (dt, J = 8.7, 3.9 Hz, 1H), 2.50 (m, 1H), 1.96 (ddd, J = 12.5, 6.0, 4.2 Hz, 1H), 1.91–1.79 (m, 3H), 1.70 (d, J = 12.5 Hz, 1H), 1.28 (m, 1H), 0.91 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 162.8, 155.8, 137.7, 129.9, 129.4, 129.4, 128.3, 126.3, 114.2, 108.7, 76.2, 75.3, 72.4, 58.6, 55.5, 55.1, 53.5, 42.4, 36.0, 34.1, 29.3, 29.0, 27.1, 20.0, 19.8 LRMS-ESI (m/z): 589.2 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C30H41N2O8S, 589.2578; found, 589.2572.
(1R,3aS,7aR)-Octahydro-1,6-epoxyisobenzofuran-5-yl((2S,3R)-4-((2-(Cyclopropylamino)-N-isobutylbenzo[d]-thiazole)-6-sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)-carbamate (4b).
Activated alcohol 16 (8 mg, 0.024 mmol) was treated with isostere amine 18 (14 mg, 0.027 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 4b (12.5 mg, 75%) as an amorphous solid. Rf = 0.15 (80% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3): δ 8.09 (s, 1H), 7.69 (d, J = 8.6 Hz, 1H), 7.55 (d, J = 8.5 Hz, 1H), 7.39–7.15 (m, 5H), 5.59 (d, J = 3.6 Hz, 1H), 5.09 (d, J = 9.7 Hz, 1H), 4.70 (m, 1H), 4.31 (t, J = 5.5 Hz, 1H), 4.09 (t, J = 8.2 Hz, 1H), 3.94–3.71 (m, 4H), 3.21 (dd, J = 15.1, 8.9 Hz, 1H), 3.16–3.07 (m, 2H), 3.02 (dd, J = 13.4, 8.5 Hz, 1H), 2.86–2.68 (m, 4H), 2.50 (m, 1H), 1.99–1.80 (m, 4H), 1.69 (d, J = 12.6 Hz, 1H), 1.26 (m, 1H), 0.97–0.90 (m, 5H), 0.87 (d, J = 6.5 Hz, 3H), 0.81–0.77 (m, 2H); 13C NMR (125 MHz, CDCl3): δ 172.9, 155.8, 155.5, 137.7, 131.1, 130.4, 129.4, 128.3, 126.3, 125.3, 120.8, 118.4, 108.7, 76.2, 75.3, 72.4, 58.7, 55.1, 53.6, 42.4, 36.0, 34.0, 29.6, 29.3, 29.0, 27.2, 26.5, 20.1, 19.8, 7.9. LRMS-ESI (m/z): 671.2 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C33H43N4O7S2, 671.2568; found, 671.2574.
(1R,3aS,7aR)-Octahydro-1,6-epoxyisobenzofuran-5-yl((2S,3R)-3-Hydroxy-4-((N-isobutyl-2-(isopropylamino)benzo-[d]oxazole)-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (4c).
Activated alcohol 16 (10 mg, 0.031 mmol) was treated with isostere amine 22 (16 mg, 0.034 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 4c (17.5 mg, 86%) as an amorphous solid. Rf = 0.4 (5% MeOH/CH2Cl2). 1H NMR (800 MHz, CDCl3): δ 7.71 (d, J = 1.8 Hz, 1H), 7.64 (dd, J = 8.2, 1.8 Hz, 1H), 7.41 (d, J = 8.3 Hz, 1H), 7.31–7.25 (m, 5H), 7.23–7.19 (m, 1H), 5.61 (d, J = 3.6 Hz, 1H), 5.44 (br s, 1H), 5.09 (d, J = 9.3 Hz, 1H), 4.72 (t, J = 6.3 Hz, 1H), 4.33 (t, J = 5.4 Hz, 1H), 4.16–4.09 (m, 2H), 3.91–3.74 (m, 4H), 3.20 (dd, J = 15.1, 8.8 Hz, 1H), 3.15–3.07 (m, 2H), 3.01 (dd, J = 13.4, 8.4 Hz, 1H), 2.85–2.79 (m, 2H), 2.74 (dd, J = 8.8, 4.3 Hz, 1H), 2.52 (m, 1H), 1.97 (dt, J = 12.6, 4.7 Hz, 1H), 1.90–1.85 (m, 2H), 1.81 (br s, 1H), 1.72 (d, J = 12.6 Hz, 1H), 1.38 (d, J = 6.5 Hz, 6H), 1.31–1.27 (m, 1H), 0.94 (d, J = 6.6 Hz, 3H), 0.89 (d, J = 6.6 Hz, 3H); 13C NMR (200 MHz, CDCl3): δ 163.3, 155.9, 147.9, 147.7, 137.8, 130.0, 129.5, 128.4, 126.4, 124.2, 116.0, 108.9, 108.5, 76.4, 75.4, 72.4, 58.8, 55.2, 53.7, 45.7, 42.6, 36.1, 34.2, 29.4, 29.1, 27.3, 23.0, 20.2, 19.9. LRMS-ESI (m/z): 657.2 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C33H45N4O8S, 657.2953; found, 657.2947.
(1R,3aS,7aR)-Octahydro-1,6-epoxyisobenzofuran-5-yl((2S,3R)-4-((2-(Cyclopropylamino)-N-isobutylbenzo[d]-thiazole)-6-sulfonamido)-1-(3,5-difluorophenyl)-3-hydroxybutan-2-yl)carbamate (4d).
Activated alcohol 16 (7 mg, 0.021 mmol) was treated with isostere amine 19 (13 mg, 0.023 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 4d (10 mg, 65%) as an amorphous solid. Rf = 0.1 (70% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3): δ 8.11 (s, 1H), 7.71 (d, J = 10.0 Hz, 1H), 7.57 (d, J = 8.5 Hz, 1H), 6.90 (br s, 1H), 6.80 (d, J = 7.6 Hz, 2H), 6.64 (dd, J = 10.1, 7.7 Hz, 1H), 5.60 (d, J = 3.6 Hz, 1H), 5.13 (d, J = 8.9 Hz, 1H), 4.73 (m, 1H), 4.32 (t, J = 5.2 Hz, 1H), 4.13 (t, J = 8.1 Hz, 1H), 3.96–3.76 (m, 3H), 3.22–3.07 (m, 3H), 3.02 (dd, J = 13.4, 8.4 Hz, 1H), 2.85 (dd, J = 13.4, 6.8 Hz, 1H), 2.81–2.70 (m, 3H), 2.53 (m, 1H), 2.03–1.80 (m, 4H), 1.71 (d, J = 12.6 Hz, 1H), 1.34 (d, J = 16.0 Hz, 1H), 0.97–0.91 (m, 5H), 0.89 (d, J = 6.5 Hz, 3H), 0.82–0.76 (m, 2H); 13C NMR (200 MHz, CDCl3): δ 172.8, 163.5 (d, J = 12.7 Hz), 162.3 (d, J = 12.8 Hz), 155.8, 155.6, 142.2 (m), 131.3, 130.6, 125.4, 121.0, 118.7, 112.4 (d, J = 20.6 Hz), 108.9, 101.9 (t, J = 25.1 Hz), 76.4, 75.3, 72.6, 72.4, 59.0, 55.0, 53.7, 42.5, 35.9, 34.2, 29.7, 29.4, 29.1, 27.4, 26.7, 20.2, 19.9, 8.0. LRMS-ESI (m/z): 707.2 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C33H41F2N4O7S2, 707.2379; found, 707.2385.
(1R,3aS,7aR)-Octahydro-1,6-epoxyisobenzofuran-5-yl((2S,3R)-4-((2-(Cyclopropylamino)-N-isobutylbenzo[d]-thiazole)-6-sulfonamido)-1-(3-fluorophenyl)-3-hydroxybutan-2-yl)carbamate (4e).
Activated alcohol 16 (12 mg, 0.037 mmol) was treated with isostere amine 20 (21 mg, 0.041 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 4e (18 mg, 70%) as an amorphous solid. Rf = 0.2 (80% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3): δ 8.13 (d, J = 1.9 Hz, 1H), 7.74 (dd, J = 8.4, 2.0 Hz, 1H), 7.59 (d, J = 8.9 Hz, 1H), 7.26 (dd, J = 8.1, 6.2 Hz, 1H), 7.18 (br s, 1H), 7.07 (d, J = 7.7 Hz, 1H), 7.00 (m, 1H), 6.92 (t, J = 8.6 Hz, 1H), 5.63 (d, J = 3.7 Hz, 1H), 5.10 (d, J = 9.2 Hz, 1H), 4.75 (m, 1H), 4.35 (t, J = 5.6 Hz, 1H), 4.14 (t, J = 8.3 Hz, 1H), 3.94–3.77 (m, 4H), 3.23 (dd, J = 15.2, 8.4 Hz, 1H), 3.20–3.07 (m, 2H), 3.05 (dd, J = 13.4, 8.3 Hz, 1H), 2.91–2.71 (m, 4H), 2.54 (m, 1H), 2.07–1.85 (m, 4H), 1.73 (d, J = 12.8 Hz, 1H), 1.34 (d, J = 10.0 Hz, 1H), 1.00–0.88 (m, 8H), 0.84–0.80 (m, 2H); 13C NMR (200 MHz, CDCl3): δ 172.9, 163.4 (d, J = 9.4 Hz), 162.2 (d, J = 10.1 Hz), 155.9, 155.5, 140.6 (d, J = 5.9 Hz), 131.2, 130.7, 129.8 (d, J = 8.3 Hz), 125.4 (d, J = 33.8 Hz), 121.0, 118.7, 116.4 (d, J = 21.0 Hz), 113.3 (d, J = 21.0 Hz), 108.9, 76.4, 75.4, 72.5, 72.5, 58.9, 55.1, 53.7, 42.5, 35.9, 34.2, 31.9, 29.7, 29.5, 29.4, 29.4, 29.1, 27.3, 26.7, 20.2, 19.9, 8.0. LRMS-ESI (m/z): 689.2 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C33H42FN4O7S2, 689.2474; found, 689.2466.
(1R,3aS,7aR)-Octahydro-1,6-epoxyisobenzofuran-5-yl((2S,3R)-4-((2-(Cyclopropylamino)-N-isobutylbenzo[d]-thiazole)-6-sulfonamido)-1-(4-fluorophenyl)-3-hydroxybutan-2-yl)carbamate (4f).
Activated alcohol 16 (12 mg, 0.037 mmol) was treated with isostere amine 21 (21 mg, 0.041 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 4f (17.5 mg, 68%) as an amorphous solid. Rf = 0.2 (80% EtOAc/hexanes). 1H NMR (500 MHz, CDCl3): δ 8.10 (d, J = 1.9 Hz, 1H), 7.70 (dd, J = 8.5, 1.9 Hz, 1H), 7.57 (d, J = 8.5 Hz, 1H), 7.25–7.18 (m, 2H), 7.00–6.94 (m, 2H), 6.89 (br s, 1H), 5.60 (d, J = 3.6 Hz, 1H), 5.02 (d, J = 9.0 Hz, 1H), 4.70 (t, J = 6.1 Hz, 1H), 4.33 (t, J = 5.4 Hz, 1H), 4.11 (t, J = 8.2 Hz, 1H), 3.92–3.71 (m, 4H), 3.18 (dd, J = 15.1, 8.2 Hz, 1H), 3.14–3.06 (m, 2H), 3.01 (dd, J = 13.4, 8.5 Hz, 1H), 2.83 (dd, J = 13.4, 6.7 Hz, 1H), 2.79–2.70 (m, 3H), 2.52 (m, 1H), 1.96 (dt, J = 12.5, 5.2 Hz, 1H), 1.92–1.81 (m, 2H), 1.70 (d, J = 12.6 Hz, 1H), 1.29 (d, J = 10.0 Hz, 1H), 0.98–0.90 (m, 5H), 0.88 (d, J = 6.6 Hz, 3H), 0.82–0.77 (m, 2H); 13C NMR (200 MHz, CDCl3): δ 172.9, 162.2, 161.0, 156.0, 133.6, 131.0 (d, J = 8.0 Hz), 130.7, 125.4, 121.0, 118.7, 115.2 (d, J = 21.0 Hz), 108.9, 76.4, 75.4, 72.6, 72.5, 58.9, 55.3, 53.7, 42.6, 35.2, 34.2, 29.7, 29.4, 29.1, 27.3, 26.7, 20.2, 19.9, 8.0. LRMS-ESI (m/z): 689.2 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C33H42FN4O7S2, 689.2474; found, 689.2481.
(1S,3aR,5S,7aS)-Octahydro-1,6-epoxyisobenzofuran-5-yl((2S,3R)-3-Hydroxy-4-((N-isobutyl-4-methoxyphenyl)-sulfonamido)-1-phenylbutan-2-yl)carbamate (5a).
Activated alcohol ent-16 (15 mg, 0.046 mmol) was treated with isostere amine 17 (21 mg, 0.051 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 5a (22 mg, 80%) as an amorphous solid. Rf = 0.3 (70% EtOAc/hexanes). 1H NMR (800 MHz, CDCl3): δ 7.72 (d, J = 8.4 Hz, 2H), 7.33–7.25 (m, 4H), 7.22 (t, J = 7.2 Hz, 1H), 6.98 (d, J = 8.4 Hz, 2H), 5.65 (s, 1H), 5.17 (d, J = 8.8 Hz, 1H), 4.80–4.68 (m, 1H), 4.33 (t, J = 5.4 Hz, 1H), 4.13 (m, 1H), 3.88 (s, 3H), 3.91–3.75 (m, 4H), 3.13–2.91 (m, 4H), 2.87–2.71 (m, 2H), 2.56 (m, 1H), 2.06–1.68 (m, 5H), 1.52 (d, J = 15.8 Hz, 1H), 1.31 (m, 1H), 0.91 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.7 Hz, 3H); 13C NMR (200 MHz, CDCl3): δ 162.9, 156.2, 137.8, 130.1, 129.6, 129.5, 128.5, 126.4, 114.3, 108.9, 76.6, 75.7, 72.6, 72.1, 58.6, 55.6, 55.4, 53.5, 42.7, 35.2, 34.3, 29.7, 29.5, 29.5, 27.2, 20.1, 19.9. LRMS-ESI (m/z): 589.2 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C30H40N2O8SNa, 611.2398; found, 611.2410.
(1S,3aR,5S,7aS)-Octahydro-1,6-epoxyisobenzofuran-5-yl((2S,3R)-4-((2-(Cyclopropylamino)-N-isobutylbenzo[d]-thiazole)-6-sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)-carbamate (5b).
Activated alcohol ent-16 (15 mg, 0.046 mmol) was treated with isostere amine 18 (25 mg, 0.051 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 5b (22 mg, 70%) as an amorphous solid. Rf = 0.2 (80% EtOAc/hexanes). 1H NMR (800 MHz, CDCl3): δ 8.08 (s, 1H), 7.69 (d, J = 8.5 Hz, 1H), 7.57 (d, J = 8.5 Hz, 1H), 7.32–7.26 (m, 5H), 7.24–7.16 (m, 2H), 5.65 (d, J = 3.6 Hz, 1H), 5.19 (d, J = 8.8 Hz, 1H), 4.78 (t, J = 6.1 Hz, 1H), 4.34 (t, J = 5.4 Hz, 1H), 4.13 (t, J = 8.3 Hz, 1H), 3.98–3.81 (m, 4H), 3.15 (dd, J = 15.0, 8.7 Hz, 1H), 3.10–2.95 (m, 3H), 2.85 (dd, J = 13.5, 6.9 Hz, 1H), 2.80–2.70 (m, 2H), 2.55 (m, 1H), 2.05–1.67 (m, 5H), 1.52 (d, J = 15.8 Hz, 1H), 0.97 (d, J = 6.5 Hz, 2H), 0.92 (d, J = 6.6 Hz, 3H), 0.88 (d, J = 6.6 Hz, 3H), 0.81 (m, 2H); 13C NMR (200 MHz, CDCl3): δ 172.98, 156.19, 155.60, 137.81, 131.20, 130.60, 129.62, 128.49, 126.49, 125.40, 120.93, 118.55, 108.88, 76.59, 75.67, 72.67, 72.20, 58.71, 55.48, 53.63, 42.72, 35.24, 34.32, 29.71, 29.50, 27.31, 26.68, 20.18, 19.91, 7.97. LRMS-ESI (m/z): 671.2 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C33H43N4O7S2, 671.2568; found, 671.2563.
(1S,3aR,5S,7aS)-Octahydro-1,6-epoxyisobenzofuran-5-yl((2S,3R)-3-Hydroxy-4-((N-isobutyl-2-(isopropylamino)benzo-[d]oxazole)-6-sulfonamido)-1-phenylbutan-2-yl)carbamate (5c).
Activated alcohol ent-16 (8 mg, 0.024 mmol) was treated with isostere amine 22 (13 mg, 0.027 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 5c (14 mg, 86%) as an amorphous solid. Rf = 0.4 (5% MeOH/CH2Cl2). 1H NMR (800 MHz, CDCl3): δ 7.66 (s, 1H), 7.60 (d, J = 8.2 Hz, 1H), 7.39 (d, J = 8.2 Hz, 1H), 7.30–7.23 (m, 5H), 7.20 (t, J = 7.1 Hz, 1H), 5.63 (d, J = 3.5 Hz, 1H), 5.23 (br s, 1H), 5.07 (d, J = 8.9 Hz, 1H), 4.76 (m, 1H), 4.32 (m, 1H), 4.15–4.05 (m, 2H), 3.87–3.72 (m, 4H), 3.13–2.92 (m, 4H), 2.85–2.67 (m, 2H), 2.53 (m, 1H), 2.03–1.95 (m, 2H), 1.92–1.77 (m, 2H), 1.73 (d, J = 12.5 Hz, 1H), 1.50 (d, J = 15.8 Hz, 1H), 1.36 (d, J = 6.5 Hz, 6H), 0.90 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H); 13C NMR (200 MHz, CDCl3): δ 163.3, 156.2, 147.9, 137.7, 130.1, 129.6, 128.5, 126.5, 124.2, 116.0, 108.9, 108.4, 76.6, 75.7, 72.7, 72.1, 58.7, 56.0, 55.4, 53.7, 45.8, 42.7, 35.3, 34.3, 29.7, 29.5, 27.3, 23.0, 20.2, 19.9. LRMS-ESI (m/z): 657.2 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C33H45N4O8S, 657.2953; found, 657.2948.
(1S,3aR,5S,7aS)-Octahydro-1,6-epoxyisobenzofuran-5-yl((2S,3R)-4-((2-(Cyclopropylamino)-N-isobutylbenzo[d]-thiazole)-6-sulfonamido)-1-(3,5-difluorophenyl)-3-hydroxy-butan-2-yl)carbamate (5d).
Activated alcohol ent-16 (12 mg, 0.037 mmol) was treated with isostere amine 19 (21 mg, 0.041 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 5d (15.5 mg, 59%) as an amorphous solid. Rf = 0.3 (5% MeOH/CH2Cl2). 1H NMR (800 MHz, CDCl3): δ 8.11 (s, 1H), 7.72 (d, J = 8.6 Hz, 1H), 7.59 (d, J = 8.5 Hz, 1H), 6.98 (br s, 1H), 6.87–6.80 (m, 2H), 6.67 (m, 1H), 5.65 (d, J = 3.6 Hz, 1H), 5.25 (d, J = 8.8 Hz, 1H), 4.83–4.73 (m, 1H), 4.36 (t, J = 5.4 Hz, 1H), 4.16–4.05 (m, 2H), 3.95–3.73 (m, 3H), 3.18–2.93 (m, 4H), 2.89 (dd, J = 13.3, 6.9 Hz, 1H), 2.84–2.72 (m, 3H), 2.56 (q, J = 8.2 Hz, 1H), 2.05–1.95 (m, 2H), 1.94–1.83 (m, 2H), 1.76 (d, J = 12.6 Hz, 1H), 1.72 (m, 1H), 1.52 (d, J = 15.9 Hz, 1H), 0.97 (d, J = 6.6 Hz, 2H), 0.94 (d, J = 6.5 Hz, 3H), 0.90 (d, J = 6.9 Hz, 3H), 0.83–0.79 (m, 2H). 13C NMR (200 MHz, CDCl3): δ 173.0, 163.6 (d, J = 11.8 Hz), 162.3 (d, J = 12.1 Hz), 156.2, 155.6, 142.2 (m), 131.3, 130.4, 125.4, 120.9, 118.7, 112.4 (d, J = 20.2 Hz), 108.9, 102.0 (t, J = 24.2 Hz), 76.5, 75.5, 72.7, 72.3, 59.0, 55.3, 53.7, 42.6, 34.8, 34.3, 29.7, 29.5, 27.4, 26.7, 20.2, 19.9, 8.0. LRMS-ESI (m/z): 707.2 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C33H41F2N4O7S2, 707.2379; found, 707.2384.
(1S,3aR,5S,7aS)-Octahydro-1,6-epoxyisobenzofuran-5-yl((2S,3R)-4-((2-(Cyclopropylamino)-N-isobutylbenzo[d]-thiazole)-6-sulfonamido)-1-(3-fluorophenyl)-3-hydroxybutan-2-yl)carbamate (5e).
Activated alcohol ent-16 (4 mg, 0.012 mmol) was treated with isostere amine 20 (7 mg, 0.013 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 5e (7.5 mg, 87%) as an amorphous solid. Rf = 0.2 (80% EtOAc/hexanes). 1H NMR (800 MHz, CDCl3): δ 8.14–8.07 (m, 1H), 7.73–7.65 (m, 1H), 7.56 (dd, J = 8.3, 2.8 Hz, 1H), 7.23 (m, 1H), 7.08–7.00 (m, 2H), 6.98 (d, J = 10.2 Hz, 1H), 6.89 (td, J = 8.8, 3.3 Hz, 1H), 5.63 (d, J = 3.6 Hz, 1H), 5.21 (d, J = 8.8 Hz, 1H), 4.77 (t, J = 6.3 Hz, 1H), 4.32 (t, J = 5.2 Hz, 1H), 4.11 (td, J = 8.9, 8.5, 3.2 Hz, 1H), 4.02–3.73 (m, 4H), 3.15–2.92 (m, 4H), 2.85 (dd, J = 13.3, 6.7 Hz, 1H), 2.79–2.70 (m, 2H), 2.57–2.48 (m, 1H), 2.01–1.93 (m, 2H), 1.89–1.81 (m, 2H), 1.73 (d, J = 12.5 Hz, 1H), 1.50 (d, J = 15.8 Hz, 1H), 0.95–0.84 (m, 8H), 0.81–0.77 (m, 2H); 13C NMR (200 MHz, CDCl3): δ 173.0, 163.4, 162.2, 156.2, 155.6, 140.6, 131.2, 130.6, 130.5, 129.9–125.7 (m), 125.5–125.2 (m), 120.9 (d, J = 8.5 Hz), 118.6, 116.5 (d, J = 22.9 Hz), 113.4 (t, J = 19.0 Hz), 108.9, 76.5 (d, J = 36.1 Hz), 75.5 (d, J = 35.0 Hz), 72.7, 72.3, 58.8, 55.4, 55.2, 53.6, 42.7, 42.5, 34.8, 34.3, 34.2, 29.7, 29.5, 29.4, 29.1, 27.3, 26.7, 20.2, 19.9, 8.0. LRMS-ESI (m/z): 689.2 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C33H42FN4O7S2, 689.2474; found, 689.2481.
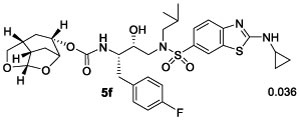
(1S,3aR,5S,7aS)-Octahydro-1,6-epoxyisobenzofuran-5-yl((2S,3R)-4-((2-(Cyclopropylamino)-N-isobutylbenzo[d]-thiazole)-6-sulfonamido)-1-(4-fluorophenyl)-3-hydroxybutan-2-yl)carbamate (5f).
Activated alcohol ent-16 (4 mg, 0.012 mmol) was treated with isostere amine 21 (7 mg, 0.013 mmol) by following the procedure outlined for inhibitor 4a to give inhibitor 5f (6.6 mg, 77%) as an amorphous solid. Rf = 0.2 (80% EtOAc/hexanes). 1H NMR (800 MHz, CDCl3): δ 8.07 (s, 1H), 7.67 (d, J = 8.6 Hz, 1H), 7.56 (d, J = 8.5 Hz, 1H), 7.25–7.20 (m, 2H), 6.98–6.95 (m, 2H), 6.83 (br s, 1H), 5.63 (d, J = 3.5 Hz, 1H), 5.10 (d, J = 9.1 Hz, 1H), 4.74 (m, 1H), 4.32 (t, J = 5.5 Hz, 1H), 4.11 (t, J = 7.9 Hz, 1H), 3.90–3.72 (m, 4H), 3.12 (dd, J = 15.0, 8.5 Hz, 1H), 3.07–3.03 (m, 1H), 3.01–2.96 (m, 2H), 2.91 (dd, J = 14.2, 8.2 Hz, 1H), 2.83 (dd, J = 13.5, 6.8 Hz, 1H), 2.77–2.69 (m, 2H), 2.53 (m, 1H), 2.03–1.95 (m, 2H), 1.89–1.80 (m, 2H), 1.73 (d, J = 12.5 Hz, 1H), 1.48 (d, J = 15.8 Hz, 1H), 0.96–0.93 (m, 2H), 0.91 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.7 Hz, 3H), 0.81–0.78 (m, 2H); 13C NMR (200 MHz, CDCl3): δ 172.9, 162.3, 161.1, 156.1, 155.5, 133.4, 131.2, 131.1–131.0 (m), 130.6, 125.4, 120.9, 118.7, 115.2 (d, J = 20.9 Hz), 108.9, 76.6, 75.5, 72.7, 72.1, 58.8, 55.4, 53.7, 42.7, 34.5, 34.3, 29.7, 29.5, 29.4, 27.3, 26.7, 20.2, 19.9, 8.0. LRMS-ESI (m/z): 689.2 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C33H42FN4O7S2, 689.2474; found, 689.2478.
(1R,3aS,5S,7aR)-Octahydro-1,6-epoxyisobenzofuran-5-ol (6).
To a stirred solution of triol 13 (340 mg, 1.54 mmol) in dichloromethane (16 mL) was added 10-camphorsulfonic acid (36 mg, 0.15 mmol) at 0 °C for 1 h. The crude residue was purified by silica gel column chromatography (40% EtOAc/hexanes) to afford alcohol 6 (198 mg, 82%) as a white amorphous solid. Rf = 0.3 (70% EtOAc/hexanes). + 45.5 (c 0.77, CHCl3). 1H NMR (400 MHz, CDCl3): δ 5.64 (d, J = 4.2 Hz, 1H), 4.30 (t, J = 5.7 Hz, 1H), 4.03–3.87 (m, 3H), 3.21 (d, J = 10.3 Hz, 1H), 2.82 (dt, J = 8.8, 4.4 Hz, 1H), 2.56–2.48 (m, 1H), 1.93 (dt, J = 12.4, 5.0 Hz, 1H), 1.83–1.73 (m, 2H), 1.56 (dt, J = 15.4, 1.1 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 108.6, 76.8, 76.6, 67.5, 42.1, 34.7, 31.8, 29.6. LRMS-ESI (m/z): 157 [M + H]+.
(1S,3aR,7aS)-Octahydro-1,6-epoxyisobenzofuran-5-ol (ent-6).
The title compound ent-6 (62 mg, 79%) was obtained from ent-13 (110 mg, 0.5 mmol) by following the procedure outlined for compound 6. Rf = 0.3 (70% EtOAc/hexanes); = −47.3 (c 0.76, CHCl3). 1H and 13C NMR data is identical with 6.
cis-Cyclohex-4-ene-1,2-dimethanol (9).
To a slurry of lithium aluminum hydride in THF was added cis-4-cyclohexene-1,2-dicarboxylic anhydride 10 (7 g, 46.00 mmol) in THF (300 mL) at 0 °C over 20 min. The reaction mixture was stirred at 0 °C for 3 h and quenched by dropwise addition of methanol over a period of 30 min at 0 °C. The reaction mixture was allowed to warm to room temperature, and aq solution of sodium sulfate was added and stirred at 23 °C overnight. The resulting slurry was filtered, and the solid was rinsed with ethyl acetate. The layers were separated, and the aqueous layer was extracted with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered, and the solvent was removed under reduced pressure to give cis-diol 9 (g, 85%) as liquid. 1H NMR (400 MHz, CDCl3): δ 5.62–5.59 (m, 2H), 3.71 (dd, J = 11.0, 6.5 Hz, 2H), 3.58 (dd, J = 11.1, 3.3 Hz, 2H), 3.27 (br s, 2H), 2.18–1.96 (m, 6H); 13C NMR (101 MHz, CDCl3): δ 125.4, 64.0, 37.7, 26.8. LRMS-ESI (m/z): 143.0 [M + H]+.
((1S,6R)-6-(Hydroxymethyl)cyclohex-3-en-1-yl)methyl Acetate (11).
A mixture of diol 9 (0.5 g, 3.52 mmol) and PPL (porcine pancreatic lipase Sigma type 2, 2.27 g) in ethyl acetate (50 mL) was stirred at 23 °C for 12 h. After completion of diol by TLC, the reaction mixture was filtered and the solvent was removed in vacuo to give a crude residue which was purified by column chromatography over silica gel (30% EtOAc/hexanes) to afford the mono acetate 11 (0.53 g, 82%) along with di acetate (40 mg, 5%) as the minor product. Rf = 0.5 (50% EtOAc/hexanes). + 17.5 (c 2.2, CHCl3), {literature data:2 + 19.0 (c 5.85, CHCl3)}; 1H NMR (400 MHz, CDCl3): δ 5.67–5.57 (m, 2H), 4.17 (dd, J = 11.0, 6.0 Hz, 1H), 3.94 (dd, J = 11.0, 8.0 Hz, 1H), 3.68–3.53 (m, 2H), 2.27–2.20 (m, 1H), 2.17–2.05 (m, 3H), 2.04 (s, 3H), 2.03–1.82 (m, 3H). 13C NMR (101 MHz, CDCl3): δ 171.3, 125.5, 125.0, 64.9, 63.5, 37.1, 33.1, 26.9, 25.9, 20.9. LRMS-ESI (m/z): 185.1 [M + H]+. Spectral data was identical with the reported data.1
((1R,6S)-6-(Hydroxymethyl)cyclohex-3-en-1-yl)methyl Acetate (ent-11).
To a stirred solution of diacetate 15 (18.6 g, 82.30 mmol) in 0.1 M phosphate buffer (242 mL, pH 7) was added PPL (1.86 g, Sigma type II, crude) at 23 °C. NaHCO3 solution (93 mL) (1 N) was added dropwise, and the heterogeneous mixture was stirred for 16 h. The mixture was then filtered through a pad of Celite. The filtrate was extracted with dichloromethane (×3). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and the solvent was evaporated in vacuo. The residue was purified by chromatography over silica gel (30% EtOAc/hexanes) to obtain ent-11 (12.75 g, 84%) as colorless oil. Rf = 0.5 (50% EtOAc/hexanes). [α] = −17.5 (c = 1.43, CHCl3); LRMS-ESI (m/z): 185.1 [M + H]+. 1H and 13C NMR spectral data were identical with 11.
((1S,6R)-6-(Dimethoxymethyl)cyclohex-3-en-1-yl)methyl Acetate (12).
Oxalyl chloride (1.34 mL, 15.21 mmol) in dry CH2Cl2 (60 mL) was cooled to −78 °C under a nitrogen atmosphere. Dimethyl sulfoxide (2.2 mL, 30.43 mmol) was added dropwise. After 15 min, alcohol 11 (1.4 g, 7.60 mmol) in dry CH2Cl2 (20 mL) was added to the reaction mixture via cannula and stirred for 30 min at −78 °C. Then, Et3N (5.3 mL, 38.04 mmol) was added and the mixture stirred for 15 min. Further reaction was carried out at 0 °C. The solvent was concentrated, extracted with EtOAc (2 × 100 mL), and washed with H2O and brine, and the organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (15% EtOAc/hexanes) to afford aldehyde S1 (1.25 g, 90%) as colorless oil. Rf = 0.5 (30% EtOAc/hexanes). − 47.9 (c 0.73, CHCl3); 1H NMR (400 MHz, CDCl3): δ 9.71 (s, 1H), 5.73–5.61 (m, 2H), 4.12–4.03 (m, 2H), 2.65–2.55 (m, 2H), 2.33–2.24 (m, 3H), 2.07–1.96 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 203.2, 170.6, 125.3, 124.7, 64.4, 47.2, 32.7, 26.8, 22.6, 20.6. LRMS-ESI (m/z): 183.0 [M + H]+.
To a stirred solution of above aldehyde (1.25 g, 6.86 mmol) in methanol (20 mL) was added trimethyl orthoformate (7.5 mL, 68.60 mmol) followed by tetrabutylammonium tribromide (66 mg, 0.137 mmol) at 23 °C. The reaction mixture was stirred for 8 h at 23 °C. After this period, the reaction mixture was quenched by the addition of saturated aqueous NH4Cl solution. Methanol was removed under reduced pressure and the reaction mixture was diluted with ethyl acetate. The layers were separated, the aqueous layer was extracted with EtOAc, and combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (10% EtOAc/hexanes) to afford 12 (1.32 g, 84%). Rf = 0.5 (10% EtOAc/hexanes, 3 times). + 5.9 (c 1.2, CHCl3). 1H NMR (400 MHz, CDCl3): δ 5.70–5.54 (m, 2H), 4.33 (dd, J = 8.3, 1.9 Hz, 1H), 4.12 (ddd, J = 10.8, 5.4, 1.8 Hz, 1H), 3.99 (ddd, J = 10.8, 9.0, 1.9 Hz, 1H), 3.36–3.30 (m, 6H), 2.29 (m, 1H), 2.21–1.99 (m, 7H), 1.88 (dddd, J = 16.1, 7.9, 4.3, 2.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 171.1, 125.7, 124.7, 105.0, 64.3, 53.5, 52.5, 37.4, 31.9, 27.5, 24.5, 21.0. LRMS-ESI (m/z): 251.1 [M + Na]+.
((1R,6S)-6-(Dimethoxymethyl)cyclohex-3-en-1-yl)methyl Acetate (ent-12).
The title compound ent-12 (9 g, 80% over two steps) was obtained from ent-11 (9 g, 48.91 mmol) by following the procedure outlined for compound 12. Rf = 0.5 (10% EtOAc/hexanes, 3 times). − 5.5 (c 1.0, CHCl3). 1H and 13C NMR spectral data were identical with 12.
(1R,2S,4R,5S)-4-(Dimethoxymethyl)-5-(hydroxymethyl)-cyclohexane-1,2-diol (13) and (1S,2R,4R,5S)-4-(Dimethoxy-methyl)-5-(hydroxymethyl)cyclohexane-1,2-diol (14).
AD-mix-β (3.0 g) was dissolved in 1:1 tert-butyl alcohol/water (22 mL), and the mixture was stirred for 10 min. MeSO2NH2 (208 mg, 2.19 mmol) was then added, and stirring was continued for a further 10 min. After the mixture was cooled to 0 °C, 12 (500 mg, 2.19 mmol) in t-BuOH (2 mL) was added. The reaction was slowly warmed to ambient temperature and stirred for 24 h. At this time, solid Na2SO3 was added, and the reaction was stirred for an additional 30 min. The reaction was then partitioned between EtOAc/water and the aqueous layer extracted with EtOAc. The combined organic layers were washed brine solution, dried over Na2SO4, filtered, and concentrated under reduced pressure to yield inseparable mixture of diols which were used for the next step with further purification.
The stirred solution of above diol was dissolved in methanol (6 mL), and 1 N NaOH (0.6 mL) at 0 °C was added. The reaction mixture was slowly warmed to ambient temperature and stirred for 3 h. After completion of the starting material, methanol was evaporated and extracted with dichloromethane (3 × 30 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude residue was purified by column chromatography (5% MeOH/CH2Cl2) over silica gel to afford triol 13 (207 mg, 43%) and 14 (227 mg, 47%) as an oily liquids.
Compound 13.
Rf = 0.4 (10% MeOH/CH2Cl2). − 3.2 (c 0.58, CHCl3). 1H NMR (400 MHz, methanol-d4): δ 4.46 (d, J = 8.3 Hz, 1H), 3.82–3.72 (m, 2H), 3.60 (dd, J = 11.4, 4.3 Hz, 1H), 3.55 (m, 1H), 3.36 (s, 3H), 3.35 (s, 3H), 2.00–1.82 (m, 3H), 1.81–1.69 (m, 2H), 1.62 (dt, J = 12.9, 4.3 Hz, 1H); 13C NMR (100 MHz, methanol-d4): δ 104.9, 70.7, 68.4, 62.9, 52.4, 52.1, 39.0, 34.6, 33.2, 27.4. LRMS-ESI (m/z): 243.1 [M + Na]+.
Compound 14.
Rf = 0.2 (10% MeOH/CH2Cl2). − 0.77 (c 3.49, CHCl3). 1H NMR (400 MHz, methanol-d4): δ 4.30 (d, J = 7.8 Hz, 1H), 3.89 (dt, J = 6.0, 3.0 Hz, 1H), 3.77 (dt, J = 10.2, 3.7 Hz, 1H), 3.63 (dd, J = 10.9, 6.0 Hz, 1H), 3.50 (dd, J = 11.0, 8.8 Hz, 1H), 3.33 (s, 3H), 3.31 (s, 3H), 2.26 (ddt, J = 12.0, 8.3, 4.2 Hz, 1H), 2.15–2.07 (m, 1H), 1.87–1.79 (m, 1H), 1.78–1.65 (m, 2H), 1.63–1.51 (m, 1H). 13C NMR (100 MHz, MeOD): δ 105.2, 68.4, 67.3, 60.3, 52.7, 51.7, 35.8, 34.3, 28.8, 28.5. LRMS-ESI (m/z): 243.1 [M + Na]+.
(1S,2R,4S,5R)-4-(Dimethoxymethyl)-5-(hydroxymethyl)-cyclohexane-1,2-diol (ent-13) and (1R,2S,4S,5R)-4-(Dimethoxymethyl)-5-(hydroxymethyl)cyclohexane-1,2-diol (ent-14).
Triol ent-13 (405 mg, 42%) and ent-14 (445 mg, 46%) were synthesized from ent-12 (1 g, 4.38 mmol) by following the procedure outlined for compound 13 and 14.
cis-Cyclohex-4-ene-1,2-diylbis(methylene) Diacetate (15).
To a stirred solution of diol 9 (11.5 g, 80.98 mmol) were added pyridine (26.1 mL, 323.94 mmol), acetic anhydride (15.3 mL, 161.97 mmol), and followed by DMAP (495 mg, 4.05 mmol) at 0 °C. The resulting mixture was stirred at 23 °C overnight. Upon completion, the reaction mixture was quenched with water and extracted with EtOAc (2 × 100 mL). The combined organic layers were dried over NaSO4, filtered, and concentrated under reduced pressure. The crude residue was purified by silica gel column chromatography (20% EtOAc/hexanes) to afford alcohol 15 (18.1 g, 98%). Rf = 0.8 (50% EtOAc/hexanes). 1H NMR (400 MHz, chloroform-d): δ 5.63–5.60 (m, 2H), 4.12–4.07 (m, 2H), 4.04–3.98 (m, 2H), 2.28–2.11 (m, 4H), 2.05 (s, 6H), 1.97–1.89 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 170.9, 125.0, 65.0, 33.6, 26.5, 20.9. LRMS-ESI (m/z): 227.1 [M + H]+.
4-Nitrophenyl ((1R,3aS,7aR)-Octahydro-1,6-epoxyisobenzofuran-5-yl) Carbonate (16).
To a stirred solution of 6 (22 mg, 0.14 mmol) in dichloromethane (1.0 mL) were added pyridine (30 μL, 0.32 mmol) and 4-nitrophenylchloroformate (63 mg, 0.31 mmol) at 0 °C under an argon atmosphere. The reaction mixture was warmed to 23 °C and stirred for 12 h. Upon completion, solvent was removed under reduced pressure. The crude product was purified by silica gel column chromatography (35% EtOAc in hexane) to afford 16 (39.5 mg, 87%) as an amorphous solid. Rf = 0.5 (70% EtOAc/hexanes). + 77.4 (c 0.7, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.27–8.23 (m, 2H), 7.43–7.38 (m, 2H), 5.69 (d, J = 3.6 Hz, 1H), 4.91 (ddt, J = 7.4, 5.0, 1.0 Hz, 1H), 4.62 (t, J = 5.4 Hz, 1H), 4.18 (t, J = 8.3 Hz, 1H), 3.93 (dd, J = 8.8, 2.9 Hz, 1H), 2.82 (dt, J = 8.6, 3.9 Hz, 1H), 2.68–2.59 (m, 1H), 2.11–2.01 (m, 2H), 1.80 (d, J = 12.7 Hz, 1H), 1.71 (d, J = 14.7 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ 155.7, 151.7, 145.3, 125.2, 121.9, 109.1, 77.2, 76.2, 73.9, 42.3, 34.0, 29.2, 28.5. LRMS-ESI (m/z): 344 [M + Na]+.
4-Nitrophenyl ((1S,3aR,7aS)-Octahydro-1,6-epoxyisobenzofuran-5-yl) Carbonate (ent-16).
The title compound ent-16 (100 mg, 88%) was obtained from ent-6 (55 mg, 0.352 mmol) by following the procedure outlined for compound 16. Rf = 0.5 (70% EtOAc/hexanes). − 79.1 (c 0.8, CHCl3). 1H and 13C NMR data are identical with 16.
Determination of X-ray structure of HIV-1 Protease–Inhibitor Complex.
The HIV-1 protease was expressed and purified, as described previously.45 The PR/5c complex was crystallized by the hanging drop vapor diffusion method with a well solution of 1.25 M NaCl and 0.1 M sodium acetate, pH 4.8, while PR/4a crystals were grown with a reservoir solution of 0.65 M NaCl and 0.1 M sodium acetate, pH 6.0. Diffraction data were collected on a single crystal cooled to 90 K at SER-CAT (22-BM beamline), Advanced Photon Source, Argonne National Lab (Chicago, USA) with an X-ray wavelength of 1.0 Å. The two data sets were processed by HKL-200046 to a Rmerge of 5.6 and 7.9%. The complex structures were solved by PHASER47 in CCP4i Suite48-50 using the previously determined isomorphous structure with PDB code 3NU351 as the start model. The PR/5c and PR/4a complexes were refined by SHELX-201452,53 up to 1.3 Å resolution and by REFMAC554 to 1.25 Å resolution. PRODRG-255 and Jligand56 were used to construct inhibitors and the restraints for refinement. COOT57,58 was used for model building. Anisotropic atomic displacement parameters (B factors) were applied for all atoms including solvent molecules. The final refined solvent structure comprised one Na+ ion, two Cl− ions, two acetate ion, and 189 water molecules for PR/5c and one Na+ ion, two Cl− ions, one glycerol, one formic acid, and 222 water molecules for PR/4a. The crystallographic statistics are provided in the Supporting Information section. The coordinates and structure factors of PR/5c and PR/4a were deposited in the Protein Data Bank59 with code 6VOE and 6VOD, respectively.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the National Institutes of Health (Grant AI150466, A.K.G. and Grant AI150461, I.T.W.). X-ray data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22BM at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under contract no. W-31-109-Eng-38. This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (H.M.), and in part by grants for the promotion of AIDS research from the Ministry of Health; grants from Welfare and Labor of Japan (H.M.); grants for the Research Program on HIV/AIDS from the Japan Agency for Medical Research and Development (AMED) under grant numbers JP15fk0410001 and JP16fk0410101 (H.M.); a grant from the National Center for Global Health and Medicine (NCGM) Research Institute (H.M.); and a Grant-in-Aid for Scientific Research (Priority Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu Kagakusho) (H.M.). The authors would like to thank the Purdue University Center for Cancer Research, which supports the shared NMR and mass spectrometry facilities. The authors also thank M.A. for discussion.
ABBREVIATIONS
- ART
antiretroviral therapies
- ATV
atazanavir
- Abt
aminobenzothiazole
- bis-THF
bis-tetrahydrofuran
- Chf-THF
cyclohexane fused bis-tetrahydrofuran
- DIPEA
N,N-diisopropyletylamine
- DRV
darunavir
- PI
protease inhibitor
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00202.
Molecular formula strings and some data (CSV)
Full NMR spectroscopic data for all final compounds X-ray structural data for inhibitors 4a and 5c-bound HIV-1 Protease (PDF)
Accession Codes
The PDB accession codes for X-ray structures of inhibitor 4a and 5c-bound HIV-1 protease are: 6VOD and 6VOE.
The authors declare no competing financial interest.
Contributor Information
Arun K. Ghosh, Department of Chemistry, Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette, Indiana 47907, United States.
Satish Kovela, Department of Chemistry, Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette, Indiana 47907, United States.
Heather L. Osswald, Department of Chemistry, Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette, Indiana 47907, United States
Masayuki Amano, Departments of Infectious Diseases and Hematology, Kumamoto University Graduate School of Biomedical Sciences, Kumamoto 860-8556, Japan.
Manabu Aoki, Department of Medical Technology, Kumamoto Health Science University, Kumamoto 861-5598, Japan; Experimental Retrovirology Section, HIV and AIDS Malignancy Branch, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, United States; Department of Clinical Sciences, Kumamoto University Hospital, Kumamoto 860-8556, Japan.
Johnson Agniswamy, Department of Biology, Georgia State University, Atlanta, Georgia 30303, United States.
Yuan-Fang Wang, Department of Biology, Georgia State University, Atlanta, Georgia 30303, United States.
Irene T. Weber, Department of Biology, Georgia State University, Atlanta, Georgia 30303, United States.
Hiroaki Mitsuya, Department of Refractory Viral Infection, National Center for Global Health and Medicine Research Institute, Tokyo 162-8655, Japan; Experimental Retrovirology Section, HIV and AIDS Malignancy Branch, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, United States; Department of Clinical Sciences, Kumamoto University Hospital, Kumamoto 860-8556, Japan.
REFERENCES
- (1).Lohse N; Hansen A-BE; Gerstoft J; Obel N Improved Survival in HIV-Infected Persons: Consequences and Perspectives. J. Antimicrob. Chemother 2007, 60, 461–463. [DOI] [PubMed] [Google Scholar]
- (2).Montaner JS; Lima VD; Barrios R; Yip B; Wood E; Kerr T; Shannon K; Harrigan PR; Hogg RS; Daly P; Kendall P Association of Highly Active Antiretroviral Therapy Coverage, Population Viral Load, and Yearly New HIV Diagnoses in British Columbia, Canada: A Population-Based Study. Lancet 2010, 376, 532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kohl NE; Emini EA; Schleif WA; Davis LJ; Heimbach JC; Dixon RA; Scolnick EM; Sigal IS Active Human Immunodeficiency Virus Protease is Required for Viral Infectivity. Proc. Natl. Acad. Sci. U.S.A 1988, 85, 4686–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Smith R; Brereton IM; Chai RY; Kent SBH Ionization States of the Catalytic Residues in HIV-1 Protease. Nat. Struct. Mol. Biol 1996, 3, 946–950. [DOI] [PubMed] [Google Scholar]
- (5).Blanco J-L; Varghese V; Rhee S-Y; Gatell JM; Shafer RW HIV-1 Integrase Inhibitor Resistance and Its Clinical Implications. J. Infect. Dis 2011, 203, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Naggie S; Hicks C Protease Inhibitor-Based Antiretroviral Therapy in Treatment-Naive HIV-1-Infected Patients: The Evidence Behind the Options. J. Antimicrob. Chemother 2010, 65, 1094–1099. [DOI] [PubMed] [Google Scholar]
- (7).Koh Y; Nakata H; Maeda K; Ogata H; Bilcer G; Devasamudram T; Kincaid JF; Boross P; Wang Y-F; Tie Y; Volarath P; Gaddis L; Harrison RW; Weber IT; Ghosh AK; Mitsuya H Novel bis-Tetrahydrofuranylurethane-Containing Non-peptidic Protease Inhibitor (PI) UIC-94017 (TMC114) with Potent Activity Against Multi-PI-Resistant Human Immunodeficiency Virus In Vitro. Antimicrob. Agents Chemother 2003, 47, 3123–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).De Meyer S; Azijn H; Surleraux D; Jochmans D; Tahri A; Pauwels R; Wigerinck P; de Béthune M-P TMC114, a Novel Human Immunodeficiency Virus Type 1 Protease Inhibitor Active Against Protease Inhibitor-Resistant Viruses, Including a Broad Range of Clinical Isolates. Antimicrob. Agents Chemother 2005, 49, 2314–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).de Béthune MP; Sekar V; Spinosa-Guzman S; Vanstockem M; De Meyer S; Wigerinck P; Lefebvre E Darunavir (Prezista, TMC114): From Bench to Clinic. Improving Treatment Options for HIV-Infected Patients in Antiviral Drugs: From Basic Discovery through Clinical Trials; John Wiley & Sons, Inc.: New Jersey, 2011; pp 31–45. [Google Scholar]
- (10).Ghosh AK; Chapsal BD Design of the Anti-HIV-1 Protease Inhibitor Darunavir in Introduction to Biological and Small Molecule Drug Research and Development: Theory and Case Studies; Ganellin CR, Jefferis R, Roberts S, Eds.; Elsevier: London, 2013; pp 355–384. [Google Scholar]
- (11).Lambert-Niclot S; Flandre P; Canestri A; Peytavin G; Blanc C; Agher R; Soulié C; Wirden M; Katlama C; Calvez V; Marcelin A-G Factors Associated with the Selection of Mutations Conferring Resistance to Protease Inhibitors (PIs) in PI-experienced Patients Displaying Treatment Failure on Darunavir. Antimicrob. Agents Chemother 2008, 52, 491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).de Meyer S; Vangeneugden T; van Baelen B; de Paepe E; van Marck H; Picchio G; Lefebvre E; de Béthune M-P Resistance Profile of Darunavir: Combined 24-Week Results from the POWER Trials. AIDS Res. Hum. Retroviruses 2008, 24, 379–388. [DOI] [PubMed] [Google Scholar]
- (13).Esté JA; Cihlar T Current Status and Challenges of Antiretroviral Research and Therapy. Antiviral Res. 2010, 85, 25–33. [DOI] [PubMed] [Google Scholar]
- (14).Saylor D; Dickens AM; Sacktor N; Haughey N; Slusher B; Pletnikov M; Mankowski JL; Brown A; Volsky DJ; McArthur JC HIV-Associated Neurocognitive Disorder — Pathogenesis and Prospects for Treatment. Nat. Rev. Neurol 2016, 12, 234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ghosh AK; Ramu Sridhar P; Kumaragurubaran N; Koh Y; Weber IT; Mitsuya H Bis-Tetrahydrofuran: A Privileged Ligand for Darunavir and a New Generation of HIV Protease Inhibitors That Combat Drug Resistance. ChemMedChem 2006, 1, 939–950. [DOI] [PubMed] [Google Scholar]
- (16).Ghosh AK; Anderson DD; Weber IT; Mitsuya H Enhancing Protein Backbone Binding—A Fruitful Concept for Combating Drug-Resistant HIV. Angew. Chem., Int. Ed 2012, 51, 1778–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tie Y; Boross PI; Wang Y-F; Gaddis L; Hussain AK; Leshchenko S; Ghosh AK; Louis JM; Harrison RW; Weber IT High Resolution Crystal Structures of HIV-1 Protease with a Potent Non-peptide Inhibitor (UIC-94017) Active Against Multi-Drug-resistant Clinical Strains. J. Mol. Biol 2004, 338, 341–352. [DOI] [PubMed] [Google Scholar]
- (18).Kovalevsky AY; Liu F; Leshchenko S; Ghosh AK; Louis JM; Harrison RW; Weber IT Ultra-High Resolution Crystal Structure of HIV-1 Protease Mutant Reveals Two Binding Sites for Clinical Inhibitor TMC114. J. Mol. Biol 2006, 363, 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ghosh AK; Chapsal BD Aspartic Acid Proteases as Therapeutic Targets; Ghosh AK, Ed.; Wiley-VCH: Weinheim, Germany, 2010; pp 169–204. [Google Scholar]
- (20).Ghosh AK; Chapsal BD; Weber IT; Mitsuya H Design of HIV Protease Inhibitors Targeting Protein Backbone: An Effective Strategy for Combating Drug Resistance. Acc. Chem. Res 2008, 41, 78–86. [DOI] [PubMed] [Google Scholar]
- (21).Ghosh AK; Osswald HL; Prato G Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J. Med. Chem 2016, 59, 5172–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ghosh AK; Nyalapatla, P. R, Kovela S; Rao KV; Brindisi M; Osswald HL; Amano M; Aoki M; Agniswamy J; Wang Y-F; Weber IT; Mitsuya H Design and Synthesis of Highly Potent HIV-1 Protease Inhibitors Containing Tricyclic Fused Ring Systems as Novel P2 Ligands: Structure–Activity Studies, Biological and X-ray Structural Analysis. J. Med. Chem 2018, 61, 4561–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ghosh AK; Martyr CD; Osswald HL; Sheri VR; Kassekert LA; Chen S; Agniswamy J; Wang Y-F; Hayashi H; Aoki M; Weber IT; Mitsuya H Design of HIV-1 Protease Inhibitors with Amino-bis-Tetrahydrofuran Derivatives as P2-Ligands to Enhance Backbone-Binding Interactions: Synthesis, Biological Evaluation and Protein-Ligand X-ray Studies. J. Med. Chem 2015, 58, 6994–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ghosh AK; Rao KV; Nyalapatla PR; Osswald HL; Martyr CD; Aoki M; Hayashi H; Agniswamy J; Wang Y-F; Bulut H; Das D; Weber IT; Mitsuya H Design and Development of Highly Potent HIV-1 Protease Inhibitors with a Crown-like Oxotricyclic Core as the P2-Ligand to Combat Multidrug-Resistant HIV Variants. J. Med. Chem 2017, 60, 4267–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ghosh AK; Rao KV; Nyalapatla PR; Kovela S; Brindisi M; Osswald HL; Sekhara Reddy B; Agniswamy J; Wang Y-F; Aoki M; Hattori S.-i.; Weber IT; Mitsuya H Design of Highly Potent, Dual Acting and Central Nervous System Penetrating HIV-1 Protease Inhibitors with Excellent Potency against Multidrug-Resistant HIV-1 Variants. ChemMedChem 2018, 13, 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Aoki M; Hayashi H; Rao KV; Das D; Higashi-Kuwata N; Bulut H; Aoki-Ogata H; Takamatsu Y; Yedidi RS; Davis DA; Hattori S.-i.; Nishida N; Hasegawa K; Takamune N; Nyalapatla PR; Osswald HL; Jono H; Saito H; Yarchoan R; Misumi S; Ghosh AK; Mitsuya H A Novel Central Nervous System-Penetrating Protease Inhibitor Overcomes Human Immunodeficiency Virus 1 Resistance with Unprecedented aM to pM potency. eLife 2017, 6, No. e28020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hohlfeld K; Tomassi C; Wegner JK; Kesteleyn B; Linclau B Disubstituted Bis-THF Moieties as New P2 Ligands in Nonpeptidal HIV-1 Protease Inhibitors. ACS Med. Chem. Lett 2011, 2, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hohlfeld K; Wegner JK; Kesteleyn B; Linclau B; Unge J Disubstituted Bis-THF Moieties as New P2 Ligands in Nonpeptidal HIV-1 Protease Inhibitors (II). J. Med. Chem 2015, 58, 4029–4038. [DOI] [PubMed] [Google Scholar]
- (29).Bai X; Yang Z; Zhu M; Dong B; Zhou L; Zhang G; Wang J; Wang Y Design and Synthesis of Potent HIV-1 Protease Inhibitors with (S)-Tetrahydrofuran-Tertiary Amine-Acetamide as P2–Ligand: Structure–Activity Studies and Biological Evaluation. Eur. J. Med. Chem 2017, 137, 30–44. [DOI] [PubMed] [Google Scholar]
- (30).Yang Z-H; Bai X-G; Zhou L; Wang J-X; Liu H-T; Wang Y-C Synthesis and Biological Evaluation of Novel HIV-1 Protease Inhibitors Using Tertiary Amine as P2-Ligands. Bioorg. Med. Chem. Lett 2015, 25, 1880–1883. [DOI] [PubMed] [Google Scholar]
- (31).Yan J; Huang N; Li S; Yang L-M; Xing W; Zheng Y-T; Hu Y Synthesis and Biological Evaluation of Novel Amprenavir-Based P1-Substituted Bi-Aryl Derivatives as Ultra-Potent HIV-1 Protease Inhibitors. Bioorg. Med. Chem. Lett 2012, 22, 1976–1979. [DOI] [PubMed] [Google Scholar]
- (32).Bungard CJ; Williams PD; Ballard JE; Bennett DJ; Beaulieu C; Bahnck-Teets C; Carroll SS; Chang RK; Dubost DC; Fay JF; Diamond TL; Greshock TJ; Hao L; Holloway MK; Felock PJ; Gesell JJ; Su H-P; Manikowski JJ; McKay DJ; Miller M; Min X; Molinaro C; Moradei OM; Nantermet PG; Nadeau C; Sanchez RI; Satyanarayana T; Shipe WD; Singh SK; Truong VL; Vijayasaradhi S; Wiscount CM; Vacca JP; Crane SN; McCauley JA Discovery of MK-8718, an HIV Protease Inhibitor Containing a Novel Morpholine Aspartate Binding Group. ACS Med. Chem. Lett 2016, 7, 702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bungard CJ; Williams PD; Schulz J; Wiscount CM; Holloway MK; Loughran HM; Manikowski JJ; Su H-P; Bennett DJ; Chang L; Chu X-J; Crespo A; Dwyer MP; Keertikar K; Morriello GJ; Stamford AW; Waddell ST; Zhong B; Hu B; Ji T; Diamond TL; Bahnck-Teets C; Carroll SS; Fay JF; Min X; Morris W; Ballard JE; Miller MD; McCauley JA Design and Synthesis of Piperazine Sulfonamide Cores Leading to Highly Potent HIV-1 Protease Inhibitors. ACS Med. Chem. Lett 2017, 8, 1292–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kolb HC; VanNieuwenhze MS; Sharpless KB Catalytic Asymmetric Dihydroxylation. Chem. Rev 1994, 94, 2483–2547. [Google Scholar]
- (35).Jacobsen EN; Marko I; Mungall WS; Schroeder G; Sharpless KB Asymmetric Dihydroxylation via Ligand-accelerated Catalysis. J. Am. Chem. Soc 1988, 110, 1968–1970. [Google Scholar]
- (36).Riva R; Banfi L; Danieli B; Guanti G; Lesma G; Palmisano G Indole Alkaloids. Enantioselective Synthesis of (−)-Alloyohimbane by a Chemoenzymatic Approach. J. Chem. Soc., Chem. Commun 1987, 1987, 299–300. [Google Scholar]
- (37).(a) Danieli B; Lesma G; Mauro M; Palmisano G; Passarella D First Enantioselective Synthesis of (−)-Akagerine by a Chemoenzymatic Approach. J. Org. Chem 1995, 60, 2506–2513. [Google Scholar]; (b) Adams GL; Carroll PJ; Smith AB III Total Synthesis of (+)-Scholarisine A. J. Am. Chem. Soc 2012, 134, 4037–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ghosh AK; Sarkar A Enantioselective Syntheses of (−)-Alloyohimbane and (−)-Yohimbane by an Efficient Enzymatic Desymmetrization Process. Eur. J. Org. Chem 2016, 2016, 6001–6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Dihydroxylation of 12 with OsO4 and NMO resulted in diols 13 and 14 in 1:3 ratio.
- (39).Von Langen DJ; Tolman RL Resolution and Stereo-selective Synthesis of the Herpes Thymidine Kinase Inhibitor L-653180. Tetrahedron: Asymmetry 1997, 8, 677–681. [Google Scholar]
- (40).Toth MV; Marshall GR A Simple, Continuous Fluorometric Assay for HIV Protease. Int. J. Pept. Protein Res 1990, 36, 544–550. [DOI] [PubMed] [Google Scholar]
- (41).Koh Y; Amano M; Towata T; Danish M; Leshchenko-Yashchuk S; Das D; Nakayama M; Tojo Y; Ghosh AK; Mitsuya H In Vitro Selection of Highly Darunavir-Resistant and Replication-Competent HIV-1 Variants by Using a Mixture of Clinical HIV-1 Isolates Resistant to Multiple Conventional Protease Inhibitors. J. Virol 2010, 84, 11961–11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Aoki M; Danish ML; Aoki-Ogata H; Amano M; Ide K; Das D; Koh Y; Mitsuya H Loss of the Protease Dimerization Inhibition Activity of Tipranavir (TPV) and Its Association with the Acquisition of Resistance to TPV by HIV-1. J. Virol 2012, 86, 13384–13396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).For details of X-ray studies, please see Supporting Information.
- (44).Wlodawer A; Vondrasek J Inhibitors of HIV-1 Protease: A Major Success of Structure-assisted Drug Design. Annu. Rev. Biophys. Biomol. Struct 1998, 27, 249–284. [DOI] [PubMed] [Google Scholar]
- (45).Agniswamy J; Kneller DW; Brothers R; Wang Y-F; Harrison RW; Weber IT Highly Drug-Resistant HIV-1 Protease Mutant PRS17 Shows Enhanced Binding to Substrate Analogues. ACS Omega 2019, 4, 8707–8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Otwinowski Z; Minor W Processing of X-ray Diffraction Data Collected in Oscillation Mode In Methods in Enzymology, 276: Macromolecular Crystallography, Part A.; Carter CW Jr., Sweet RM, Eds.; Academic Press: New York, 1997; pp 307–326. [DOI] [PubMed] [Google Scholar]
- (47).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ; Phaser Crystallographic Software. Phasercrystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AGW; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the CCP4 Suite and Current Developments. Acta Crystallogr., Sect. D: Biol. Crystallogr 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Collaborative Computational Project, Number 4. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr 1994, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- (50).Potterton E; Briggs P; Turkenburg M; Dodson E A Graphical User Interface to the CCP4 Program Suite. Acta Crystallogr., Sect. D: Biol. Crystallogr 2003, 59, 1131–1137. [DOI] [PubMed] [Google Scholar]
- (51).Shen C-H; Wang Y-F; Kovalevsky AY; Harrison RW; Weber IT Amprenavir Complexes with HIV-1 Protease and its Drug-Resistant Mutants Altering Hydrophobic Clusters. FEBS J. 2010, 277, 3699–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Sheldrick GM Short History of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr 2008, 64, 112–122. [DOI] [PubMed] [Google Scholar]
- (53).Sheldrick GM Crystal Structure Refinement with SHELXL. Acta Crystallogr. 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Murshudov GN; Vagin AA; Dodson EJ Refinement of Macromolecular Structures by the Maximum-likelihood Method. Acta Crystallogr., Sect. D: Biol. Crystallogr 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- (55).Schüettelkopf AW; van Aalten DMF PRODRG: a Tool for High-Throughput Crystallography of Protein–Ligand Complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr 2004, 60, 1355–1363. [DOI] [PubMed] [Google Scholar]
- (56).Lebedev AA; Young P; Isupov MN; Moroz OV; Vagin AA; Murshudov GN Ligand: A Graphical Tool for the CCP4 Template-restraint Library. Acta Crystallogr., Sect. D: Biol. Crystallogr 2012, 68, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Emsley P; Cowtan K Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (59).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.