

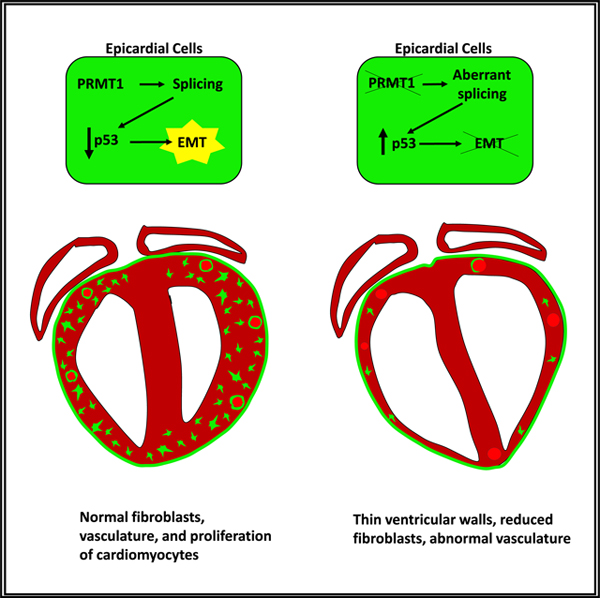
SUMMARY
Epicardial cells are cardiac progenitors that give rise to the majority of cardiac fibroblasts, coronary smooth muscle cells, and pericytes during development. An integral phase of epicardial fate transition is epithelial-to-mesenchymal transition (EMT) that confers motility. We uncover an essential role for the protein arginine methyltransferase 1 (PRMT1) in epicardial invasion and differentiation. Using scRNA-seq, we show that epicardial-specific deletion of Prmt1 reduced matrix and ribosomal gene expression in epicardial-derived cell lineages. PRMT1 regulates splicing of Mdm4, which is a key controller of p53 stability. Loss of PRMT1 leads to accumulation of p53 that enhances Slug degradation and blocks EMT. During heart development, the PRMT1-p53 pathway is required for epicardial invasion and formation of epicardial-derived lineages: cardiac fibroblasts, coronary smooth muscle cells, and pericytes. Consequently, this pathway modulates ventricular morphogenesis and coronary vessel formation. Altogether, our study reveals molecular mechanisms involving the PRMT1-p53 pathway and establish its roles in heart development.
In Brief
Jackson-Weaver et al. show that PRMT1 drives epicardial invasion and differentiation in heart development. PRMT1 regulates splicing of Mdm4 and decreases p53 stability, which enhances Slug degradation to block epicardial EMT. The PRMT1-p53 axis is required for epicardial invasion and formation of epicardial-derived lineages during development.
Graphical Abstract
INTRODUCTION
Epicardial cells represent an important progenitor population in the heart. During heart development, epicardial cells undergo epithelial-to-mesenchymal transition (EMT) to invade the developing muscle wall, giving rise to the majority of cardiac fibroblasts, coronary vascular smooth muscle cells (cVSMCs), and pericytes (von Gise and Pu, 2012). At the same time, these epicardial-derived cells are signaling centers that modulate myocardial growth and coronary vessel formation (Olivey and Svensson, 2010; Pé rez-Pomares and de la Pompa, 2011). The epicardial EMT is a cellular program in which cells lose their epithelial cell morphology and become motile and invasive (Lamouille et al., 2014). EMT is initiated by a network of signaling pathways, including transforming growth factor β (TGF-β), platelet-derived growth factor (PDGF), and Wnt signaling, which converge on key transcription factors such as Snail and Slug to achieve transcriptional reprogramming that leads to morphological changes and acquisition of migratory and invasive propensity (Lamouille et al., 2014). Nonetheless, the molecular mechanisms of epicardial cell fate transition are not fully understood.
Protein arginine methyltransferases (PRMTs) are a class of enzymes that methylate arginine residues on histones and non-histone proteins. PRMT1 is the major PRMT, is responsible for 75% of arginine methylation activity in mammalian cells (Bedford and Clarke, 2009), and is documented to regulate signal transduction, epigenetic regulation, and DNA repair (Blanc and Richard, 2017; Xu et al., 2013). The physiological functions of PRMT1 are increasingly understood because of roles in embryonic development, such as craniofacial morphogenesis and neural development, and in diseases such as inflammatory conditions and cancer (Gou et al., 2018; Scaglione et al., 2018; Yang and Bedford, 2013; Zhang et al., 2018a).
Here we show that PRMT1 drives epicardial differentiation and invasion during cardiac development and pinpoint p53 as a previously unappreciated mediator of PRMT1 activity. We first showed roles of PRMT1 in epicardial fate transition using singlecell RNA sequencing (scRNA-seq). Further investigation demonstrated that loss of PRMT1 leads to p53 accumulation, increasing p53-mediated degradation of Slug to block epicardial EMT. This PRMT1-p53 axis regulates the transcriptional reprogramming required for epicardial EMT and the acquisition of motility. During heart development, the PRMT1-p53 pathway is required for the formation of epicardial-derived mesenchymal lineages and supports ventricular morphogenesis and coronary vessel formation.
RESULTS
PRMT1 Drives Epicardial EMT and Invasion
Epicardial cells undergo cell fate transition into cardiac fibroblasts, cVSMCs, and pericytes through a process of EMT (von Gise and Pu, 2012). To study the role of PRMT1 in epicardial EMT, we used a cell line established from embryonic ventricular epicardial cells, MEC1 (Li et al., 2011). First, to characterize the ability of MEC1 cells to undergo EMT, we treated cells with TGF-b, a robust inducer of EMT (Lamouille et al., 2014). TGF-β treatment induced a progressive change characteristic of EMT, with cells losing their cobblestone morphology and acquiring a spindle shape (Figure 1A) and with a stepwise decrease in circularity (roundness) (Figure 1B). Expression of epithelial-associated proteins decreased, whereas mesenchymal-associated proteins increased (Figures 1C and S1A). The epicardial identity marker Wilms Tumor 1 (WT1) (Martínez-Estrada et al., 2010) was unaffected (Figures 1C and S1A), suggesting that EMT precedes the loss of epicardial identity. Silencing Prmt1 prevented TGF-β-induced loss of epicardial marker E-cadherin and gain of mesenchymal markers fibronectin and Slug. However, the increased expression of Snail, which is closely related to Slug, was unaffected (Figures 1D and S1B). Notably, Slug is critical in mouse epicardial EMT (Takeichi et al., 2013), but Snail is dispensable (Casanova et al., 2013). MEC1 cells produced a dense assemblage of fibronectin fibers in response to TGF-β-induced EMT, but Prmt1 depletion prevented fibronectin deposition to the extracellular matrix (ECM) (Figure 1E). Prmt1 depletion also inhibited actin stress fiber formation, a feature of EMT that facilitates motility (Haynes et al., 2011) (Figure 1F). An important outcome of EMT is the development of migratory and invasive propensity. We showed that Prmt1 knockdown significantly decreased MEC1 cell migration (Figures 1G and 1H), reduced baseline invasion, and prevented TGF-β-induced invasion (Figures 1I and 1J). Altogether, these results indicate that PRMT1 is required for epicardial EMT and its functional consequences, including migration and invasion.
Figure 1. PRMT1 Is Required for Epicardial Migration and Invasion.
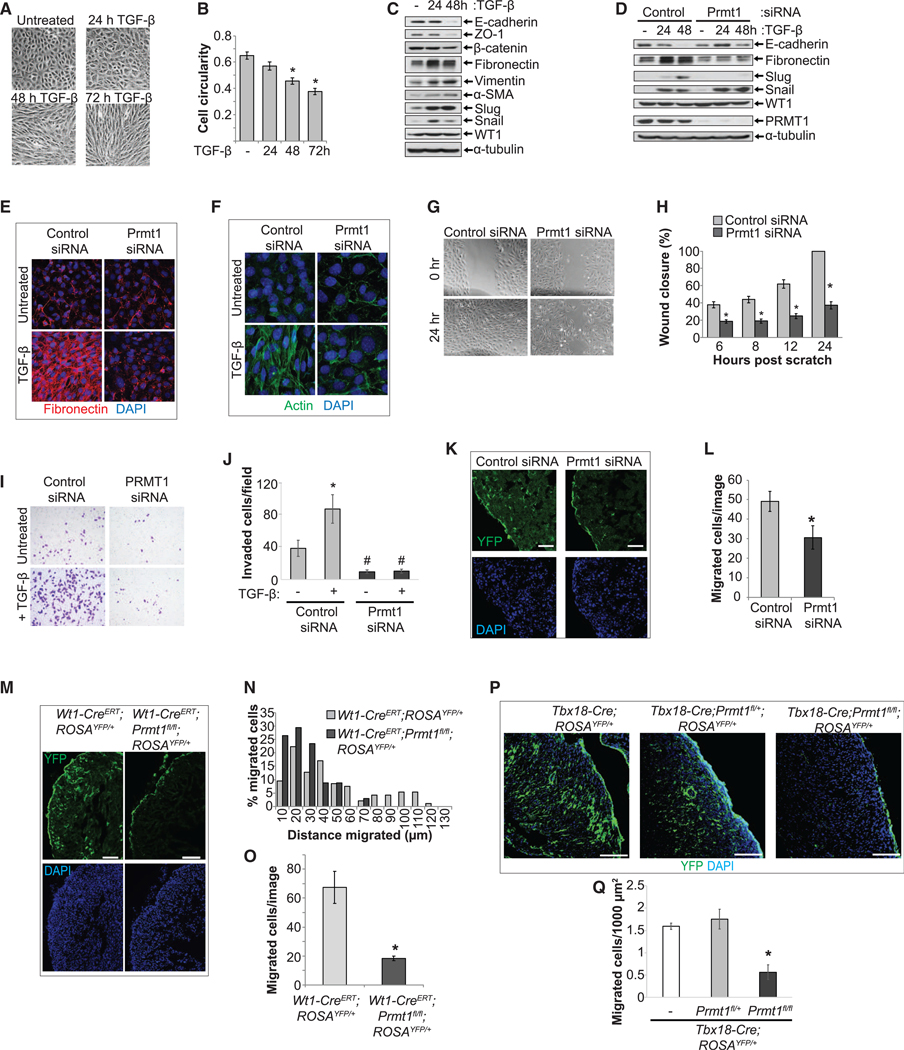
(A and B) TGF-β induced characteristic EMT changes in epicardial MEC1 cells. Phase contrast images shown in (A). Cell circularity is quantified in (B).
(C) Epicardial cells that underwent EMT exhibited decreased epithelial markers and increased mesenchymal markers. MEC1 cells lysates were analyzed by SDSPAGE and blotted for the indicated proteins.
(D) Depleting Prmt1 prevented TGF-β-induced changes in EMT marker proteins E-cadherin, fibronectin, and Slug. Snail and WT1 were unaffected.
(E) Depleting Prmt1 prevented fibronectin matrix formation. Confocal images for fibronectin immunostaining.
(F) Depleting Prmt1 blocked actin stress fiber formation. Confocal images of actin fibers visualized by phalloidin.
(G and H) Prmt1 depletion reduced epicardial cell migration. Phase contrast images of MEC1 cells at 0 or 24 h after scratching are shown in (G). The percentage of wound closure is quantified in (H). *p < 0.05 versus control siRNA.
(I and J) Prmt1 depletion prevented epicardial cell invasion. MEC1 cells that invaded are imaged in (I) and quantified in (J). *p < 0.05 versus untreated, #p < 0.05 versus control siRNA.
(K and L) Prmt1 knockdown significantly decreased the number of YFP-labeled epicardial-derived cells that migrated into myocardium. Wt1-CreERT;ROSAYFP/+ embryonic hearts dissected at E12.5 were incubated with control or Prmt1 siRNA and then cultured in the presence of TGF-β. YFP was visualized by immunostaining and confocal imaging (K). The left ventricular wall (LV) is shown. Scale bar, 25 μm. Migrated cells per image are quantified in (L). *p < 0.05 versus control siRNA.
(M–O) Epicardial-specific deletion of Prmt1 using Wt1-CreERT led to inhibition of epicardial invasion. YFP is visualized in E15.5 hearts in (M). The LV is shown. Scale bar, 25 μm. The histogram in (N) shows the percentage of migrated cells at increasing distances from the epicardium. Data were pooled from 3 hearts per genotype. Migrated cells per image are quantified in (O). *p < 0.05 versus Wt1-CreERT;ROSAYFP/+.
(P and Q) Tbx18-Cre-driven deletion of Prmt1 in epicardial cells suppressed invasion. YFP is visualized in P0 hearts in (P). The LV is shown. Scale bar, 100 μm. Migrated cells per 1,000 mm2 are quantified in (Q). *p < 0.05 versus Tbx18-Cre;ROSAYFP/+.
PRMT1 is highly expressed and highly active in epicardial cells compared with differentiated cardiac fibroblasts (Figure S1C). To determine whether endogenous PRMT1 is required for epicardial EMT, we knocked down Prmt1 in embryonic epicardium using ex vivo culture of embryonic day (E) 12.5 hearts from Wt1-CreERT;ROSAYFP/+ mice, which allows the tracing of epicardial-derived cells yellow fluorescent protein (YFP) (Srinivas et al., 2001; Zhou and Pu, 2012). We observed that numerous epicardial-derived YFP+ cells penetrated into the myocardium in control small interfering RNA (siRNA)-treated hearts, indicative of epicardial invasion, but that in the Prmt1-silenced group, migrated cells were significantly reduced (Figures 1K, 1L, S1D, and S1E).
To determine whether PRMT1 is required for epicardial invasion in vivo, we generated Wt1-CreERT;Prmt1fl/fl;ROSAYFP/+ mice to delete Prmt1 in the epicardium and its cellular derivatives and concomitantly label epicardial-derived cells with YFP. Loss of Prmt1 significantly altered the profile of epicardial invasion (Figures 1M–1O). In a plot of cumulative numbers of migrating cells at increasing distances into the myocardium, epicardial-derived cells in the control group invaded as far as 120 μm at E15.5. But in the Prmt1-deficient group, they accumulated proximal to the epicardium (Figure 1N). The Wt1-CreERT;Prmt1fl/fl;ROSAYFP/+ embryos died between E16 and E18, potentially from tamoxifen toxicity and developmental defects. We next used a constitutive Tbx18-Cre to delete Prmt1 in epicardial-derived cells (Figures S1F and S1G) (Christoffels et al., 2009). Tbx18-Cre;Prmt1fl/fl;ROSAYFP/+ pups died between postnatal day (P) 0 and P7. At P0, Prmt1-deficient hearts had 65% fewer YFP-labeled epicardial-derived cells (Figures 1P and 1Q). Prmt1 partial deletion (Tbx18-Cre;Prmt1fl/+) did not cause significant effects (Figures 1P, 1Q, and S1G). Altogether, these results demonstrate that PRMT1 is required for epicardial EMT and invasion during cardiac development.
Loss of Prmt1 Impairs the Formation of Epicardial-Derived Cardiac Fibroblasts, cVSMCs, and Pericytes
Epicardial invasion is coupled with cell fate transition into cardiac fibroblasts, cVSMCs, and pericytes (von Gise and Pu, 2012). To determine whether Prmt1 deficiency impairs the formation of these epicardial-derived cell lineages, we quantified each cell type with lineage-specific markers. Tbx18-Cre was shown to have leaky expression in cardiomyocytes (CMs) when crossed with the ROSALacZ reporter (Christoffels et al., 2009). We crossed Tbx18-Cre with a weaker ROSAYFP reporter, in which YFP+ CMs were observed sporadically (20%) in the ventricular septum, but not in the ventricular free walls. Therefore, ventricular free walls were used for quantification (Figure S2A). For littermate controls, we used Prmt1 heterozygous deletion (Tbx18Cre;Prmt1fl/+) mice for most experiments, based on the data that Tbx18-Cre;Prmt1fl/+ did not show noticeable differences in PRMT1 expression or cardiac phenotype when compared with Tbx18-Cre hearts (Figures S1E and S2D–S2G).
First, we assessed the number of epicardial-derived cardiac fibroblasts using the fibroblast marker Vimentin (Figure S2B). In Prmt1-deficient hearts, Vimentin+ cells declined by 71% (Figures 2A, 2B, and S2H). This decrease in cardiac fibroblasts also reduced the expression of ECM, affecting genes that encode matrix components and remodeling proteins (Figure 2C). Next, we investigated whether Prmt1 deficiency impairs the formation of epicardial-derived cVSMCs using smooth muscle marker αSMA (Figure S2C). In Prmt1-deficient hearts at P0, αSMA+ cells were significantly reduced and the cardiac expression of αSMA was dramatically decreased (Figures 2D–2F). The αSMA+ cells were almost completely ablated in the apical region of the heart (Figures 2D and S2I), but in the basal region within 500 mm of the valves, most coronary vessels displayed a smooth muscle layer (Figures 2G and 2H). We found that 57% of αSMA+ cells in the basal region were YFP−, suggesting that non-epicardial sources compensated for cVSMC formation in the basal region (Arima et al., 2012; Chen et al., 2016). Despite the compensatory attempt, we noted incomplete perivascular coverage of cVSMCs and minimal or absent medial and adventitial layers (Figures 2G–2I and S2J). We then assessed whether Prmt1 deficiency impairs pericyte formation using pericyte marker NG2. Prmt1 deletion resulted in >40% loss of pericyte in the ventricular walls and decreased NG2 expression in the whole heart (Figures 2J–2L). Pericyte numbers in the septum were not affected (Figure S2K). In addition, in the control hearts, 90% of NG2+ cells in the ventricular walls were YFP+, similar to previous findings (Figures 2M and 2N) (Volz et al., 2015). Altogether, these data show that epicardial-specific deletion of Prmt1 impairs the formation of epicardial-derived cardiac fibroblasts, cVSMCs, and pericytes.
Figure 2. Epicardial Prmt1 Deletion Impairs the Formation of Cardiac Fibroblasts, cVSMCs, and Pericytes.
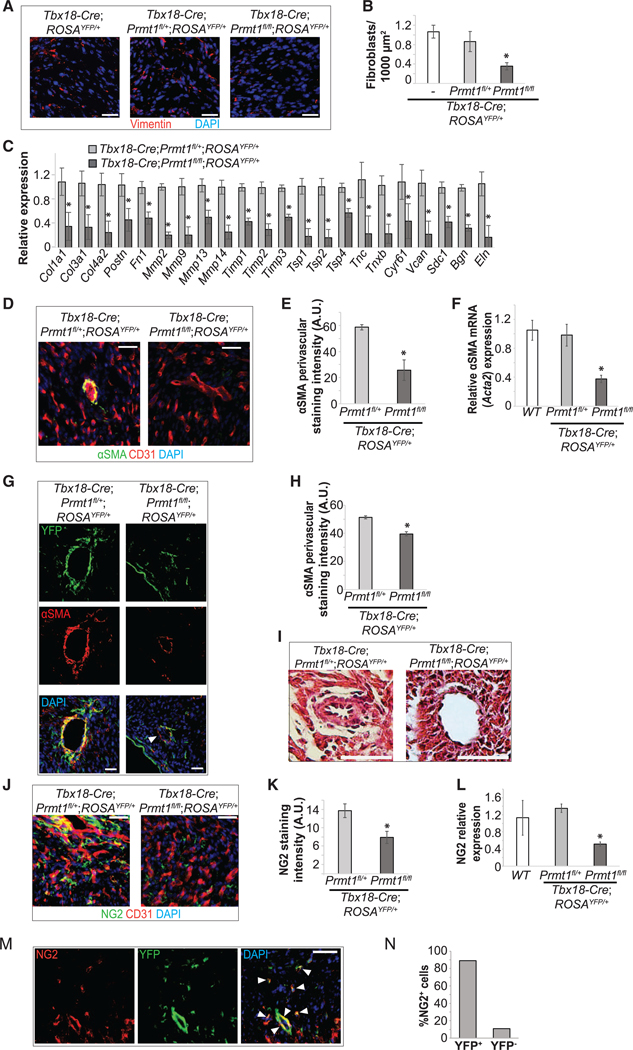
(A and B) Number of cardiac fibroblasts was reduced by epicardial-specific Prmt1 deletion. Cardiac fibroblasts revealed by Vimentin staining in P0 ventricular walls are shown in (A) and quantified in (B). *p < 0.05 versus Tbx18-Cre;ROSAYFP/+.
(C) Epicardial-specific Prmt1 deletion reduced ECM gene expression. The mRNA was analyzed by qRT-PCR in whole hearts from P0 mice. *p < 0.05 versus Tbx18-Cre;Prmt1fl/+;ROSAYFP/+.
(D–F) cVSMC formation was reduced by epicardial-specific Prmt1 deletion. cVSMCs were revealed by aSMA immunostaining in P0 hearts and ECs were revealed by CD31, as shown in (D). Scale bar, 25 mm. Perivascular aSMA staining intensity in large vessels of the ventricles wall is quantified in (E). *p < 0.05 versus Tbx18Cre;Prmt1fl/+;ROSAYFP/+. αSMA mRNA was analyzed in P0 whole-heart lysates by qRT-PCR, as shown in (F). *p < 0.05 versus Tbx18-Cre;Prmt1fl/+;ROSAYFP/+.
(G and H) In the basal region of the heart, cVSMC coverage was only moderately reduced after epicardial Prmt1 deletion. αSMA is immunostained in P0 hearts in
(G). The arrow indicates a αSMA+;YFP cell, suggesting non-epicardial origin. Scale bar, 25 μm. Perivascular αSMA staining intensity is quantified in (H). *p < 0.05 versus Tbx18-Cre;Prmt1fl/+;ROSAYFP/+.
(I) Medial and adventitial morphology was absent after epicardial Prmt1 deletion, shown by H&E staining of P0 hearts.
(J–L) Pericyte formation was reduced by epicardial Prmt1 deletion. Coronary pericytes were revealed by NG2+ immunostaining in P0 hearts, as shown in (J). NG2 staining intensity is quantified in (K). *p < 0.05 versus Tbx18-Cre;Prmt1fl/+;ROSAYFP/+. NG2 mRNA was analyzed in P0 whole-heart lysates by qRT-PCR, as shown in (L). *p < 0.05 versus Tbx18-Cre;Prmt1fl/+;ROSAYFP/+.
(M and N) Most NG2+ pericytes were epicardial derived. NG2 and YFP co-immunostaining in P0 Tbx18-Cre;Prmt1fl/+;ROSAYFP/+ hearts shows significant overlap in (M) (arrows). Scale bar, 50 μm. The percentage of NG2+ cells that were YFP+ or YFP are quantified in (N).
PRMT1-Regulated Epicardial Function Is Essential for Coronary Formation and Ventricular Morphogenesis
Epicardial-derived cardiac mesenchymal cells are critical for coronary vessel development and support myocardial growth (Cavallero et al., 2015; Tian and Morrisey, 2012). In Prmt1-deficient hearts (Tbx18-Cre;Prmt1fl/fl;ROSAYFP/+), we observed consistent coronary vessel dilation at P0 (Figures 3A and 3B). A similar phenotype was observed in Wt1-CreERT;Prmt1fl/fl embryos at E15.5, illustrated by a significant increase of vessel diameter, without altering vessel density (Figures 3C–3E).
Figure 3. Epicardial-Specific Prmt1 Deletion Impairs Coronary Formation and Cardiac Morphogenesis.
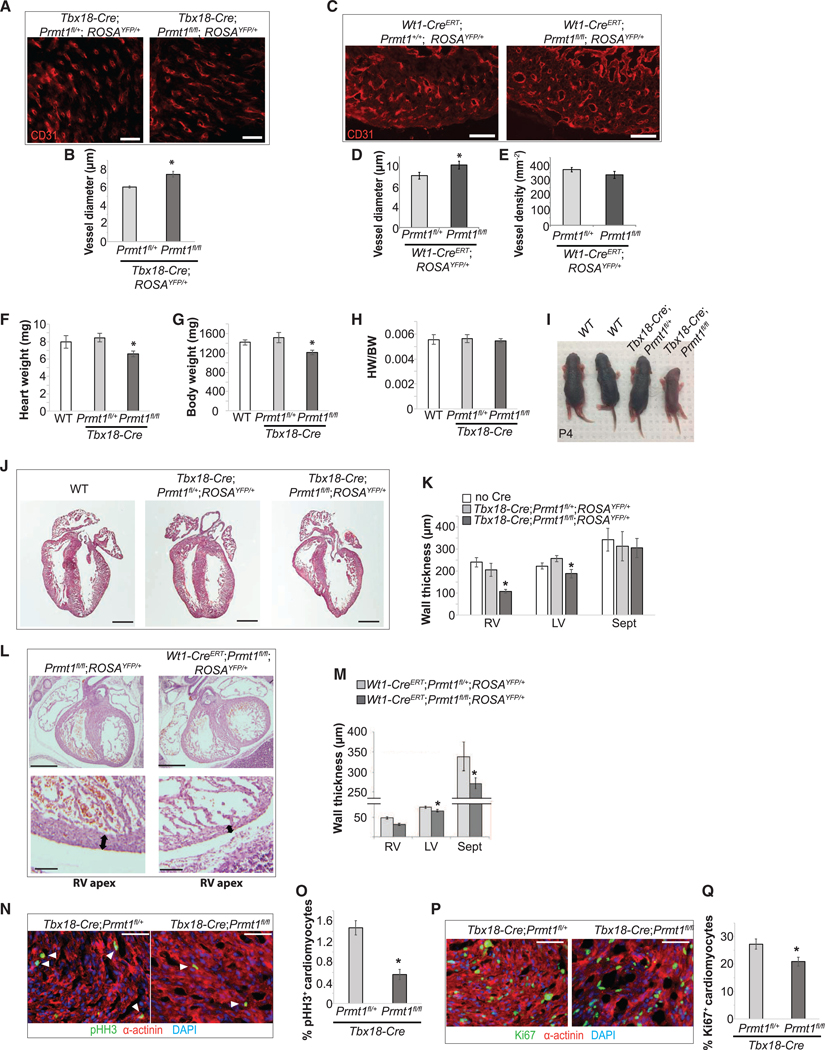
(A and B) Epicardial Prmt1 deletion driven by Tbx18-Cre led to coronary vessel dilation. Coronary vessels were revealed by CD31 immunostaining in P0 hearts in (A). Scale bar, 25 μm. Vessel diameter is quantified in (B). *p < 0.05 versus Tbx18-Cre;Prmt1fl/+;ROSAYFP/+.
(C–E) Epicardial Prmt1 deletion using Wt1-CreERT led to coronary vessel dilation. Coronary vessels were revealed by CD31 in E15.5 hearts, as shown in (C). Scale bar, 25 μm. Vessel diameter is quantified in (D), and density is quantified in (E). *p < 0.05 versus Wt1-CreERT;Prmt1fl/+;ROSAYFP/+.
(F–I) Epicardial Prmt1 deficiency reduced heart weight and body weight. Heart weight, body weight, and heart weight-to-body weight ratio (HW/BW) at P0 are compared in (F), (G), and (H), respectively. *p < 0.05 versus Tbx18-Cre;Prmt1fl/+;ROSAYFP/+. Representative pups at P4 are shown in (I).
(J) and (K) Epicardial Prmt1 deletion led to thinner ventricular walls. H&E staining of P0 hearts is shown in (J). Scale bar, 500 μm. Thicknesses of the right ventricular wall (RV), LV, and septum (Sept) are quantified in (K). *p < 0.05 versus Tbx18-Cre;Prmt1fl/+;ROSAYFP/+.
(L and M) Thin ventricular wall was apparent after epicardial Prmt1 deletion using Wt1-CreERT. H&E staining of E15.5 hearts is shown in (L). Arrows indicate thinner walls in the RV apex. Wall thickness is quantified in (M). *p < 0.05 versus Wt1-CreERT;Prmt1fl/+;ROSAYFP/+.
(N–Q) Epicardial Prmt1 deletion reduced CM proliferation. Proliferating CMs in P0 hearts were revealed by pH3 (N) and Ki67 (P) and are quantified in (O) and (Q), respectively. α-actinin labels CMs. *p < 0.05 versus Tbx18-Cre;Prmt1fl/+;ROSAYFP/+.
To investigate the impact of epicardial Prmt1 deficiency on myocardial growth, we first examined ventricular morphology. The Tbx18-Cre;Prmt1fl/fl mutant pups were smaller than littermates at P0; the mutant hearts were also smaller (n = 14) (Figures 3F–3I). Prmt1-deficient hearts exhibited hypoplasia with thinner ventricular walls in both chambers (Figures 3J and 3K). In Wt1CreERT;Prmt1fl/fl hearts, the thinning of ventricular walls was most dramatic in the apical region at E15.5 (Figures 3L and 3M). We further quantified myocyte proliferation using markers for cycling cells during interphase (Ki67) and mitosis (phosphohistone H3, pH3). Prmt1-deficient hearts showed a significant reduction of Ki67+ and pH3+ myocytes, indicating decreased myocyte proliferation (Figures 3N–3Q).
These findings demonstrate that epicardial-specific deletion of Prmt1 impairs cardiac mesenchymal cell formation, which subsequently compromises coronary development and ventricular morphogenesis.
Loss of Prmt1 Alters the Transcriptional Profile in Epicardial-Derived Cell Lineages at E12.5
To further investigate the role of PRMT1 in epicardial cell fate transition, we used scRNA-seq analysis to profile the cardiac transcriptome at the initiation stage of epicardial invasion. We pooled hearts from control (Tbx18-Cre;ROSAYFP/+) or Prmt1 CKO (conditional knockout) (Tbx18-Cre;Prmt1fl/fl;ROSAYFP/+) E12.5 embryos and profiled the transcriptomes of 6,330 single cells from the control group and 7,181 from the mutant group (Figure 4A). Cells were clustered based on transcriptional similarities using an unsupervised canonical correlation analysis (Butler et al., 2018) and visualized using t-distributed stochastic neighbor embedding (tSNE) (van der Maaten, 2008, 2014). We identified 23 distinct clusters and assigned identities to each cluster using the expression pattern of cluster-enriched genes. In total, we observed 2 clusters of epicardial (Epi-1 and Epi-2) cells (Wt1+/Tbx1+/ Cebpb+/Krt18+), 1 fibroblast-like cells (Tcf21+/Fn1+), 4 smooth muscle/pericyte-like cells (Cnn1+/Acta2+/Tagln2+/Tagln+), 1 neural crest cell-derived cells (Msx1+/Twist+/Sox9+), 6 CMs (all Myh6+Nppa+, four ventricular Myl2+/Myl3+, and two atrial Myl1+/ Myl4+), 4 endothelial cells (ECs) (Klf2+/Pecam1+/Cdh5+), and 5 hemopoietic cells (Hba-a1+ red blood, Fcer1g+ myeloid, and Ctla2a+ lymphoid) (Figures 4B, 4C, and S3A–S3F) (Xiao et al., 2018). Our analysis indicated that murine heart at E12.5 is composed of 30% CMs (6% atrial myocytes and 24% ventricular myocytes) and 7.6% ECs (Figure 4D). These data reveal the cell-type heterogeneity of the murine heart at E12.5 and identify epicardial-derived lineages based on transcriptional markers.
Figure 4. scRNA-Seq Analysis of Embryonic Heart at E12.5.
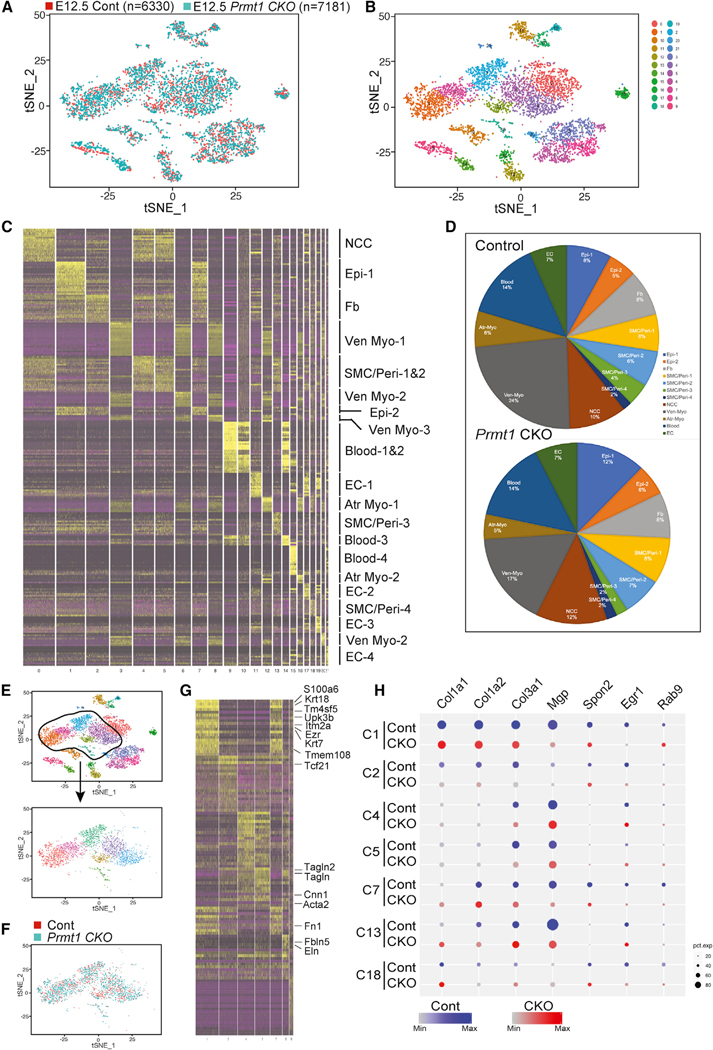
(A and B) Cellular composition of E12.5 hearts was visualized using tSNE. Two control Tbx18-Cre;ROSAYFP/+ or seven mutant Tbx18-Cre;Prmt1fl/fl;ROSAYFP/+ hearts were pooled for single-cell isolation, barcoding, and RNA-seq. Individual single-cell transcriptomes were colored by experimental group in (A) and cluster identity in (B).
(C) Heatmap illustrating genes most highly enriched in each cluster to predict distinct cell populations. NCC, neural crest-derived cell; Epi, epicardial cell; VenMyo, ventricular myocyte; SMC/Peri, SMC/pericyte-like cell; Atr Myo, atrial myocyte.
(D) Pie chart depicting cellular composition in each group.
(E–H) Epi, Fb, and SMC/Peri clusters were isolated by subsetting in (E) and clustering in (F). A heatmap is generated in (G). The dot blot in (H) illustrates downregulation of matrix genes in Epi-1 (C1), Epi-2 (C7), and SMA/Peri-3 (C13), but not Fb (C2) or SMA/Peri-1/2/4 (C4/5/18) clusters. Dot size depicts the percentage of cells that express individual genes, and the color gradient shows the expression level (H). p < 0.05 for all genes illustrated. C, cluster; Cont, Tbx18Cre;ROSAYFP/+; CKO, Tbx18-Cre;Prmt1fl/fl;ROSAYFP/+.
We then focused on epicardial and epicardial-derived progeny clusters (Figure 4E). Epi-1 and Epi-2 clusters expressed characteristic epicardial genes. The Epi-1 cluster expressed prospective epicardial progenitor genes Tm4sf5, Tmem108, S100a6, Ezr, and Itm2a; the mesothelial gene Upk3b; and epithelial genes Krt18 and Krt7 (Figure 4G) (Bochmann et al., 2010; Xiao et al., 2018). Compared with Epi-1, Epi-2 clusters expressed lower levels of the epicardial progenitor and mesothelial/epithelial genes but exhibited a subset of fibroblast markers such as Spon2 and Tcf21, indicating an epicardial-to-fibroblast transitional phenotype. We also identified four smooth muscle cell (SMC)/pericyte-like clusters. Prmt1 CKO hearts showed a moderate decrease in SMA/Peri-3 (Figure 4D) and a subtle increase in the other three SMC/pericyte clusters. The SMA/Peri-3 population shared the SMC/pericyte progenitor genes Cnn1, Tagln, and Tagln2 but was distinct in the high expression of Fbln5 and Eln (Figures 4F and 4G). We examined gene expression within each cluster (Table S1) and revealed that Prmt1 deletion downregulated genes in ECM, focal adhesion, and regulation of cell shape in the Epi-1, Epi-2, and SMA/Peri-3 clusters, suggesting that cells in these clusters contributed to decreased ECM formation (Figures 4H and S3G–S3M). The cardiac fibroblast-like cell (Fb) cluster also showed significant alterations in ribosome and splicing genes in Prmt1 CKO hearts (Figure S3I). These findings demonstrated that epicardial-specific deletion of Prmt1 alters the expression of genes related to matrix formation and splicing at E12.5.
PRMT1 Regulates EMT via p53-Slug Pathway
To investigate the molecular mechanism underlying PRMT1regulated epicardial invasion, we surveyed master transcription factors that drive EMT (Xu et al., 2009). In epicardial EMT, we observed prominent induction of Snail and Slug in contrast to other EMT transcription factors and Prmt1-depletion impaired Slug induction (Figures 1C, 1D, and S4A–S4E). Slug induction during epicardial EMT occurred mainly at the protein level, in contrast to Snail (Figures 5A–5D, S4F, and S4G). Because Slug protein is regulated by p53-targeted degradation in cancer cells (Wang et al., 2009), the role of p53 in epicardial cells was evaluated. We first identified that Slug recruitment by p53 is regulated by TGF-β and proteasomal degradation (Figures 5E, 5F, and S4H). We then observed a modest but significant increase of p53 protein levels (1.6-fold) in Prmt1-depleted MEC1 cells (Figures 5G, 5H, and S4I). To test whether a modest increase in p53 expression is sufficient to block EMT, we increased p53 levels by 2-fold using Nutlin-3, an inhibitor that stabilizes p53 (Figures 5I and 5J) (Vassilev et al., 2004). This increase in p53 levels prevented the induction of Slug and preserved epicardial morphology despite TGF-β treatment (Figures 5I and 5K), phenocopying the effects of Prmt1 depletion. Furthermore, a low-dose Nutlin-3 treatment of embryonic hearts decreased epicardial invasion without increasing cell death (Figures 5L and 5M). These data indicate that a modest increase in p53 level is sufficient to inhibit epicardial EMT and invasion.
Figure 5. Silencing PRMT1 Increases the p53 Level to Reduce Slug Induction.
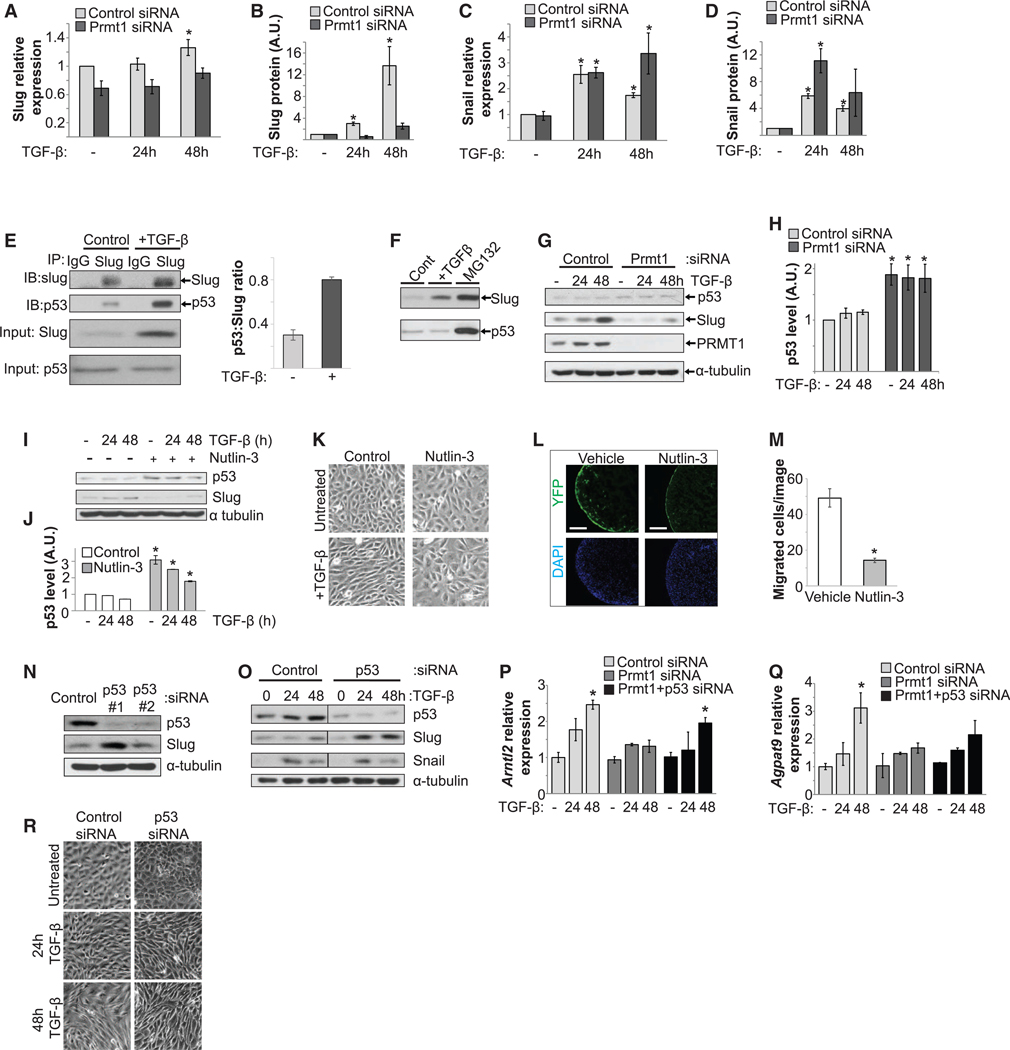
(A–D) Slug induction during epicardial EMT occurred mainly at the protein level, in contrast to Snail. TGF-β induced a modest increase in mRNA levels of Slug (A) but a dramatic increase in protein expression (B), in contrast to a dramatic increase in both mRNA (C) and protein levels (D) for Snail. mRNA was measured by qRT-PCR, and protein expression was quantified by immunoblot from multiple assays. *p < 0.05 versus untreated.
(E) Slug interacted with p53 in MEC1 cells. Co-immunoprecipitation (coIP) using anti-Slug antibody or immunoglobulin G (IgG) control was blotted for Slug andp53, and quantification for the p53:slug ratio. IB, immunoblot. IP, immunoprecipitation. The input images were the same as the first two columns of (F) from the same assay.
(F) Slug and p53 were degraded by the proteasome. The protein levels of Slug and p53 were increased by MG132, which blocks proteasomal degradation.
(G and H) Silencing Prmt1 increased p53 protein levels. p53 is blotted in (G), and the blot is quantified in (H). *p < 0.05 versus control siRNA.
(I and J) Enhancing p53 levels with Nutlin-3 reduced TGF-β-induced Slug in MEC1 cells, as assessed by immunoblotting in (I) and quantified in (J). *p < 0.05 versus control.
(K) Nutlin-3 prevented EMT morphological change, as assessed by phase contrast microscopy.
(L and M) Nutlin-3 decreased epicardial invasion. Hearts dissected from E12.5 Wt1-CreERT;ROSAYFP/+ embryos were incubated with vehicle or Nutlin-3 in ex vivo culture. YFP-labeled epicardial-derived cells in the myocardium are visualized in (L) and quantified in (M). The left ventricle is shown. *p < 0.05 versus vehicle.
(N) Depletion of p53 increased Slug levels. MEC1 cells transfected with control or two independent p53 siRNAs were assessed.
(O) Depletion of p53 with siRNA 1 enhanced Slug induction, but not Snail, during epicardial EMT.
(P and Q) PRMT1-p53 pathway regulated Slug target gene induction. mRNA levels of Slug target genes Arntl2 (P) and Agpat9 (Q) were analyzed by qRT-PCR. *p < 0.05 versus control siRNA.
(R) Depletion of p53 accelerated EMT morphological change, as assessed by phase contrast microscopy.
Next, we determined whether endogenous p53 regulates EMT. In unstimulated cells, robust p53 knockdown elicited a dramatic increase in Slug levels and modest knockdown elicited a subtle increase (Figures 5N and S4J). In TGF-β-stimulated cells, p53 knockdown enhanced Slug induction to facilitate EMT (Figures 5O and S4K). We further tested pro-invasive Slug target genes Arntl2 and Agpat9 in MEC1 cells (Mistry et al., 2014). TGF-β-induced upregulation of Arntl2 and Agpat9 was blocked by Prmt1 knockdown and rescued in Prmt1 and p53 double-knockdown (Figures 5P and 5Q), providing evidence that experimentally locates p53 downstream of PRMT1. The depletion of p53 also accelerated EMT-associated morphological changes (Figure 5R). Altogether, these data indicate that p53 regulates epicardial EMT and invasion downstream of PRMT1.
PRMT1RegulatesEpicardial EMT and Invasion Partly via p53
To assess whether PRMT1-regulated epicardial EMT depends on p53, we compared gene expression during EMT in control, Prmt1-depleted, and Prmt1/p53 double-depleted MEC1 cells by RNA sequencing (RNA-seq) analysis (Figure 6A). In control epicardial cells, TGF-β caused downregulation of genes that define epithelial properties and upregulation of genes that confer mesenchymal features. We observed upregulation of 589 genes and downregulation of 762 genes in TGF-β-induced epicardial EMT. In the Prmt1-depleted cells, 40% of these changes (545 genes) were blocked, including 44.3% of the upregulated genes and 37.2% of the downregulated genes. When Prmt1 and p53 were both depleted, changes in 36.8% (201 of 545 genes) of the Prmt1-responsive genes were restored (Figure 6B). One of the major categories regulated by PRMT1-p53 includes genes regulating cell migration and protrusion (Figure 6C).
Figure 6. PRMT1 Silencing Prevents EMT through p53 and Alternative Splicing.
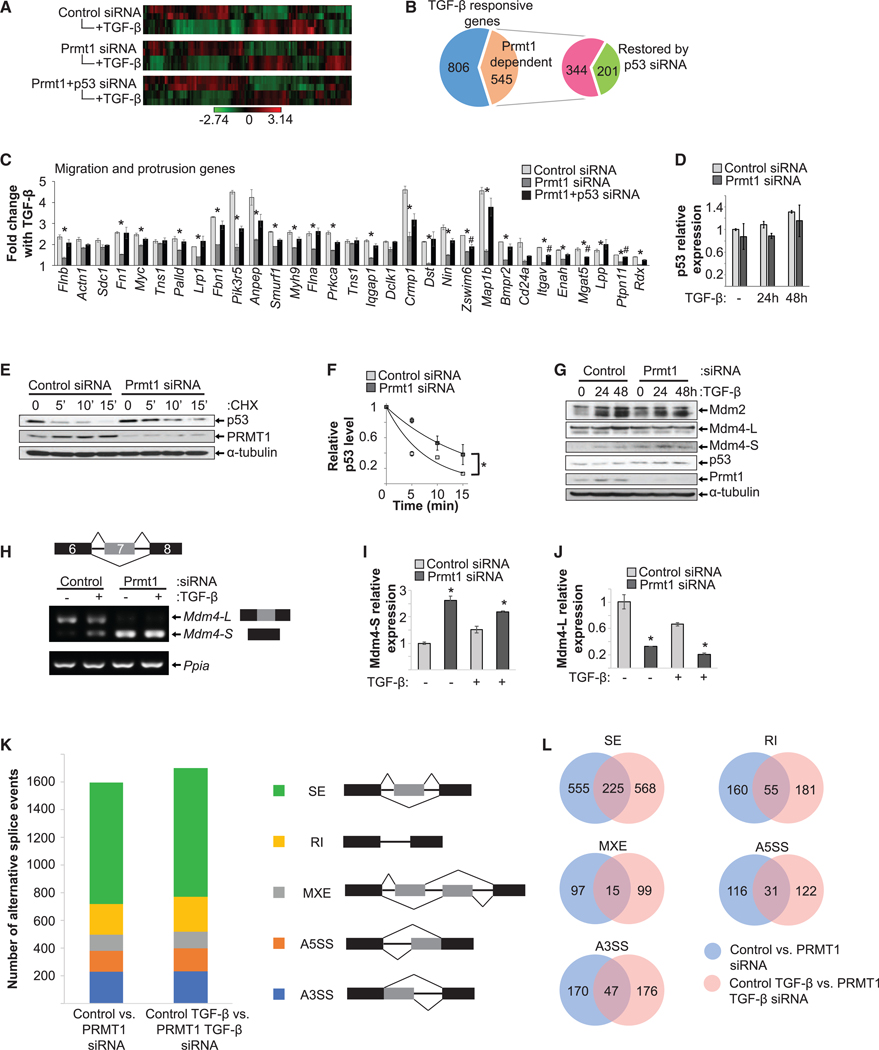
(A–C) RNA-seq analysis of transcriptional regulation by the PRMT1-p53 axis during epicardial EMT. (A) Heatmap of differentially regulated genes in control, Prmt1 siRNA, or Prmt1+p53 siRNA groups ± TGF-β treatment (n = 2 per group). (B) Plot of the number of TGF-β-responsive (upregulated or downregulated) genes that depend on PRMT1-p53 expression. Of the 545 PRMT1-dependent genes, combined Prmt1 and p53 siRNA restored 201 genes. (C) Migration and protrusion genes were significantly reduced by Prmt1 siRNA and restored by combined Prmt1 and p53 siRNA, revealed by Ingenuity Pathway Analysis (IPA). *p < 0.05 versus control siRNA, #p < 0.05 versus control siRNA.
(D) Prmt1 silencing did not change p53 mRNA expression.
(E and F) Silencing Prmt1 increased p53 half-life from ~4 to ~10 min. CHX chase was used to measure the p53 degradation rate in MEC1 cells. Densitometry measurements of p53 levels in (E) was plotted against time and fitted to exponential curves to calculate half-life in (F). *p < 0.05.
(G) Silencing Prmt1 did not affect protein levels of Mdm2 or full-length Mdm4 (Mdm4-L) but increased Mdm4 short form (Mdm4-S) in MEC1 cells.
(H–J) Prmt1 silencing increased Mdm4-S and reduced Mdm4-L levels, assessed by semi-qPCR in (H) and RT-PCR in (I)(short form) and (J) (long form). Ppia was used as the loading control. *p < 0.05 versus control siRNA.
(K and L) Quantification of splicing events altered by Prmt1 depletion. The number of splicing events between control and Prmt1 siRNA-treated cells is compared in (K). Overlap between alternatively spliced genes in the two comparisons is shown in (L), separated by types of alternative splicing. A3SS, alternative 3’ splice site; A5SS, alternative 5’ splice site; MXE, mutually exclusive exon; RI, retained intron; SE, skipped exon.
To understand how PRMT1 increases p53 protein expression, we first examined the mRNA levels of p53 and showed that Prmt1 depletion did not change p53 mRNA levels in MEC1 cells (Figure 6D). Next, p53 protein stability was assessed by cycloheximide (CHX) chase, in which new protein synthesis was blocked by CHX to measure the degradation rate of existing proteins. After Prmt1 depletion, p53 half-life was increased by 2.5-fold (Figures 6E and 6F). We next examined key regulators of p53 stability during heart development, the E3 ligases Mdm2 and Mdm4 (Grier et al., 2006). Prmt1 depletion did not alter protein expression of Mdm2 or full-length Mdm4, but it increased the levels of a short form of Mdm4 in MEC1 cells (Figures 6G and S5A). Mdm4 splicing in MEC1 cells was then examined. The transcript of a Mdm4 short form that skips exon 7 was significantly increased with a concomitant decrease of the long form (Figures 6H–6J). This short form of Mdm4 is defective in p53 binding, acting in a dominant negative fashion to block the long form from binding to p53 and thus stabilizing p53 (Bardot et al., 2015). To further assess whether Prmt1 depletion altered splicing on a larger scale, we analyzed RNAseq data with rMATS (replicate multivariate analysis of transcript splicing) (Shen et al., 2014) and identified that Prmt1 depletion caused ~1,600 alternative splicing events, including alternative 3’ splice sites, alternative 5’ splice sites, mutually exclusive exons, retained introns, and skipped exons (Figures 6K and 6L), indicating that PRMT1 regulates multiple splicing events.
PRMT1-p53 Pathway Regulates Epicardial Invasion and Differentiation In Vivo
To determine the physiological significance of the PRMT1-p53 axis, we assessed its role in epicardial invasion and differentiation in vivo. The Prmt1-deficient epicardial cells expressed higher levels of p53 and lower levels of Slug, echoing findings in MEC1 cells (Figures 5G; 7A, arrowhead; and S6A). By deleting one allele of p53, the enhanced p53 level was reduced to the basal level in the Tbx18-Cre;Prmt1fl/fl;p53fl/+ group (Figure 7A). We then examined epicardial invasion and epicardial-derived cardiac fibroblast formation. The number of Vimentin+ fibroblasts in Tbx18Cre;Prmt1fl/fl;p53fl/+ hearts was restored to a level similar to that of the Tbx18-Cre;Prmt1fl/fl control (Figures 7B and 7C). We measured invasion using the distance traveled by epicardial-derived cardiac fibroblasts. Fibroblasts localized primarily within 75 μm of the peripheral epicardium in Prmt1-deficient mice, compared with a range of 150 μm in control group and the Prmt1-deficient group with reduced p53 (Figure 7D). Restoration of cardiac fibroblast number and invasion also reestablished ECM expression (Figure 7E). Next, we assessed cVSMC formation. In the apical region, where Prmt1 deficiency almost completely ablated αSMA+ cells, reducing p53 in Prmt1-deficient epicardial cells restored migration to the perivascular region and rescued the number of αSMA+ cells by 68% (Figures 7F and 7G). However, some αSMA+ cells in Tbx18-Cre;Prmt1fl/fl;p53fl/+ mouse hearts fail to form a compact layer around vascular ECs (Figure 7F, arrowhead). Subsequently, the medial and adventitial morphology was not fully restored (Figure S6B), indicating that p53 reduction was not sufficient to restore cVSMC function. We next examined pericyte formation. Reducing p53 in Prmt1-deficient epicardial cells rescued pericyte formation to 85% of control levels, and these pericytes localized to the perivascular region(Figures 7H and 7I).
Figure 7. Reducing p53 Restores the Formation of Epicardial-Derived Lineages and Ventricular Morphology.
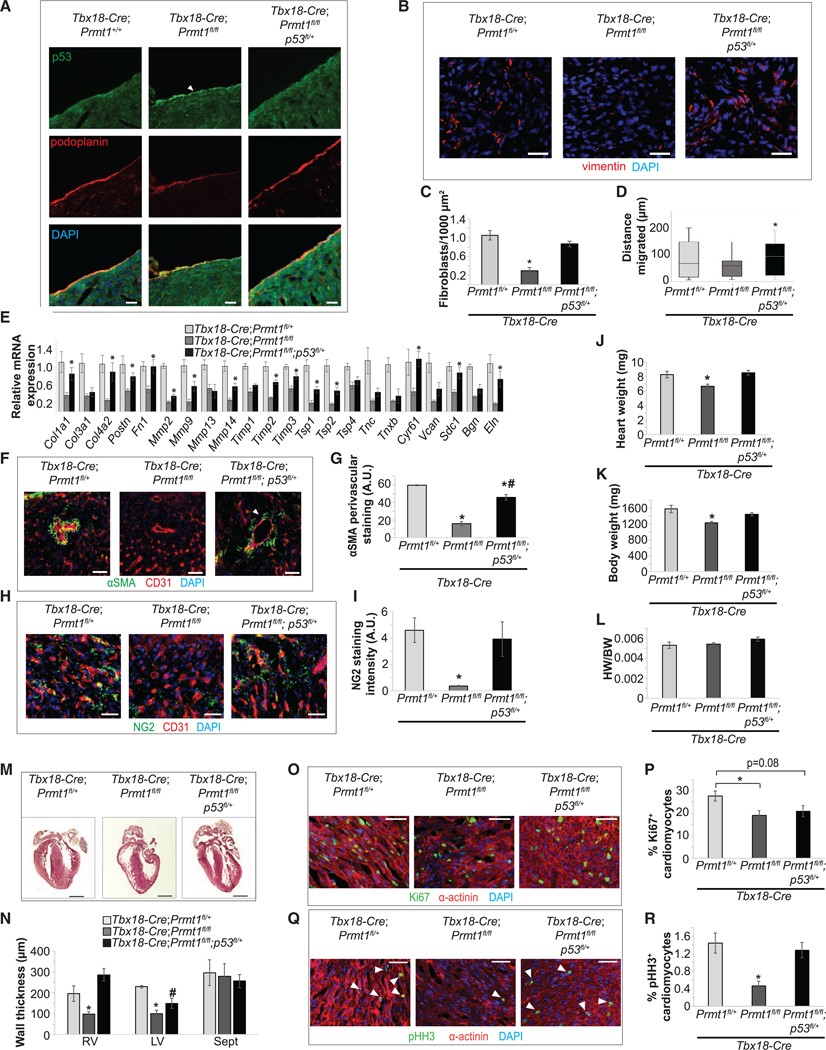
(A) Epicardial Prmt1 deletion increased p53 expression and p53 partial deletion reduced p53 levels to basal levels in P0 hearts. Podoplanin labels the epicardium (arrow).
(B–D) Reducing p53 restored epicardial invasion and cardiac fibroblast formation. Cardiac fibroblasts in P0 hearts stained with vimentin are shown in (B) and quantified in (C). Scale bar, 25 μm. *p < 0.05 versus Tbx18-Cre;Prmt1fl/+. The distance of cardiac fibroblasts from the epicardium as a measure of epicardial-derived cell invasion is quantified in (D). *p < 0.05 versus Tbx18-Cre;Prmt1fl/fl.
(E) Reducing p53 restored most ECM gene expression. mRNA collected from P0 whole hearts was assessed by qRT-PCR. *p < 0.05 versus Tbx18-Cre;Prmt1fl/fl. (F and G) Reducing p53 restored cVSMC formation in P0 hearts. aSMA labels cVSMC and CD31 labels ECs in (F). Perivascular aSMA staining intensity is quantified in (G). *p < 0.05 versus Tbx18-Cre;Prmt1fl/+, #p < 0.05 versus Tbx18-Cre;Prmt1fl/fl.
(H and I) Reducing p53 restored pericyte formation in P0 hearts. NG2 labels pericytes in (H). NG2 staining intensity is quantified in (I). *p < 0.05 versus Tbx18Cre;Prmt1fl/+.
(J–L) Reducing p53 rescued the reduction in heart weight because of Prmt1 deletion. Heart weight, body weight, and HW/BW were examined at P0 in (J), (K), and (L), respectively. *p < 0.05 versus Tbx18-Cre;Prmt1fl/+.
(M and N) Reducing p53 rescued the thinning of ventricular walls. H&E staining of P0 hearts is shown in (M). Scale bar, 500 μm. Thicknesses of the RV, LV, and Sept are quantified in (N). *p < 0.05 versus Tbx18-Cre;Prmt1fl/+, #p < 0.05 versus Tbx18-Cre;Prmt1fl/fl.
(O–R) Reducing p53 restored CM proliferation. Ki67 (O) and pH3 (Q) staining was used to assess CM proliferation in P0 hearts and is quantified in (P) and (R), respectively. a-actinin labels CMs. *p < 0.05 versus Tbx18-Cre;Prmt1fl/+.
We further determined whether reducing p53 expression rescues the abnormal cardiac morphology in Prmt1-deficient heart. The Tbx18-Cre;Prmt1fl/fl;p53fl/+ mutant pups displayed normal heart and body weights at P0 (Figures 7J–7L) and survive to adulthood without obvious abnormality. Reducing p53 rescued the hypoplastic phenotype caused by Prmt1 deficiency, because Tbx18-Cre;Prmt1fl/fl;p53fl/+ mice displayed ventricular wall thickness comparable to controls (Figures 7M and 7N). Reducing p53 also restored myocardial expansion, because the percentages of Ki67+ and pH3+ myocytes in Tbx18-Cre;Prmt1fl/fl;p53fl/+ mice were comparable to controls (Figures 7O–7R).
These findings showed that reducing p53 in Prmt1-deficient epicardial cells largely rescues epicardial invasion; restores the formation of cardiac fibroblasts, cVSMCs, and pericytes; and rescues ventricular morphogenesis.
DISCUSSION
We identified a PRMT1-p53 pathway that drives epicardial invasion and differentiation. In epicardial cells, depletion of PRMT1 causes a significant change in alternative splicing, including Mdm4 splicing, which stabilizes p53. The accumulation of p53 in turn decreases Slug levels and blocks epicardial EMT and invasion. We further show that during embryonic heart development, epicardial-specific deletion of Prmt1 causes p53 accumulation, disrupts epicardial invasion, and impairs the formation of epicardial cell-derived cardiac mesenchymal lineages, including cardiac fibroblasts, cVSMCs, and pericytes. By reducing epicardial p53 in vivo, the disrupted invasion, impaired cardiac mesenchymal cell formation, and cardiac morphological defects were largely rescued.
Epicardial cells are multipotent progenitors that give rise to cardiac fibroblasts, cVSMCs, and pericytes during heart development. Epicardial EMT is an integral part of epicardial progenitor differentiation and confers motility; however, the sequence and timing of these events remain unclear. Our scRNA-seq analysis of E12.5 hearts demonstrates that epicardial fate commitment is initiated prior to epicardial invasion. The epicardial progenitor population (Epi-1) was identified by the expression of mesothelial/epithelial genes and prospective epicardial progenitor genes Tm4sf5, Tmem108, S100a6, Ezr, and Itm2a (Bochmann et al., 2010; Xiao et al., 2018), but experimental validation is needed to define these prospective markers and their physiological roles. We noted an increase in the Epi-1 population in the Prmt1 mutant hearts (12%) compared with controls (8%). Whether PRMT1 regulates epicardial progenitor awaits further assessment. Fibroblasts and SMC/pericyte lineage cells were identified before they invaded the myocardium. In Prmt1 mutant hearts, a SMC/pericyte-like cluster marked by high expression of Fbln5 and Eln showed a 49% reduction (Figures 4F and 4G). Fbln5 and Eln encode ECM proteins Fibulin-5 and Elastin, which are abundantly expressed in large vessels and indispensable for elastic lamina formation (Li et al., 1998; Yanagisawa et al., 2002). This information is in line with our data that Prmt1 deficiency impaired medial and adventitial morphology in coronary vessels.
Previous works showed that epicardial differentiation uses distinct signaling pathways. For example, loss of PDGFRα in epicardial cells disrupts cardiac fibroblast formation without altering cVSMC formation (Smith et al., 2011), whereas loss of myocardin-related transcription factors disrupts epicardial-derived pericyte formation (Trembley et al., 2015). Our study indicates a requirement for PRMT1 in epicardial motility and the formation of all three epicardial-derived lineages. Whether PRMT1 functions upstream of or acts in concert with other factors in epicardial fate commitment awaits further investigation.
We also revealed a role for PRMT1 in alternative splicing. Splicing factors are among the earliest characterized substrates for PRMT1. For example, PRMT1 methylates the heterogeneous nuclear ribonucleoprotein (hnRNP) components SAF-A and FMRP to regulate their activities (Blackwell et al., 2010; Guo et al., 2014; Herrmann et al., 2004; Zhou et al., 2017). We show that PRMT1 regulates Mdm4 splicing and link PRMT1-regulated alternative splicing to p53 stability in the control of EMT and cell invasion.
We previously reported that in mammary epithelial cells, PRMT1 regulates EMT through the TGF-b/Smad pathway via Smad7 methylation (Katsuno et al., 2018). Others also documented PRMT1 functions in cancer cell EMT through various mechanisms (Avasarala et al., 2015; Chen et al., 2019; Federico et al., 2019; Gao et al., 2016; Wei et al., 2019; Zhang et al., 2018b). Here, in epicardial cells, PRMT1 regulates epicardial EMT through a distinct pathway without affecting TGF-β-induced Smad3 signaling (Figures S6C–S6F). Instead, PRMT1 drives the p53-Slug axis to control epicardial EMT and invasion, a pathway that is also relevant in cancer such as mesothelioma.
In summary, we have identified a PRMT1-p53 pathway that promotes epicardial EMT, invasion, and cell fate transition to facilitate the formation of cardiac mesenchymal cells and support cardiac morphogenesis. These findings should shed light on the molecular basis of congenital heart disease and open up the possibility of using the PRMT1-p53 pathway as a potential therapeutic avenue.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jian Xu (xujian@usc.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All sequencing data that support the findings of this study have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO). RNA-seq data are accessible through the GEO Series accession number GSE122200. scRNA-seq data are accessible through GSE144271. All other relevant data are available from the corresponding author on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal care and experimentation were performed in accordance with University of Southern California Institutional Animal Care and Use Committee (IACUC)-approved protocols, and following guidelines from the Institute for Laboratory Animal Research.
Prmt1fl/fl (Yu et al., 2009), Tbx18-Cre (Christoffels et al., 2009) and WT1-CreERT (Jax 010912) mice was used for epicardial-specific deletion of PRMT1. For WT1-CreERT (Zhou et al., 2008; Zhou and Pu, 2012), tamoxifen (100 mg/kg) was injected at E9.5 to induced Cre expression and embryos were collected at E15.5.
METHOD DETAILS
Chemicals and antibodies
TGF-β was purchased from HumanZyme (Chicago, IL). MG132, Cycloheximide, and crystal violet were purchased from Sigma-Aldrich (St. Louis, MO). Nutlin-3 was purchased from Santa Cruz Biotechnology. HRP-conjugated Secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA). Fluorescent-conjugated secondary antibodies and fluorescent-conjugated phalloidin were purchased from Thermo Fisher Scientific (Waltham MA).
Ex vivo embryo thorax culture
epicardial migration was assessed using a modified ex vivo culture protocol from Shen et al. (2015). To trace epicardial cells that invaded into the myocardium, Wt1-CreERT;ROSAYFP/+ mice was used, in which an inducible epicardial-specific Cre activates a ROSAYFP reporter to label epicardial-derived cells (Soriano, 1999; Srinivas et al., 2001; Zhou et al., 2008). YFP expression was induced in epicardial cells from Wt1-CreERT;ROSAYFP/+ embryos by injecting the pregnant mother with tamoxifen on E9.5. Embryos were harvested on E12.5, when epicardial invasion initiates (Figure S1D), and the head and abdomen dissected away, exposing the heart to the medium. Embryos were then transfected with control or Prmt1 siRNA for 15 hours using RNAiMax (Invitrogen) in 1:1 DMEM:M199. SiRNA transfection using this method primarily targeted surface epicardium with ~50% efficiency (Figure S1E). Media was then changed to 0.1% FBS, 1:1 DMEM:M199 supplemented with 2 ng/ml bFGF and 10 ng/ml TGF-β and cultured for an additional 48 hr. Embryos were then fixed in 4% PFA overnight, washed in PBS, and cryopreserved in 30% sucrose in PBS overnight. Embryos were embedded in OCT and sectioned at 10 μm. Sections were blocked in 10% horse serum and 0.1% Triton X-100 in PBS for 1 hour. Primary antibodies diluted in 10% horse serum in PBS were applied overnight at 4°C. Secondary antibodies and DAPI diluted in 10% horse serum in PBS were applied for 1 hr at room temperature. The number of YFP positive cells penetrated into the myocardium were quantified in at least 3 images per heart from 3 embryos for each condition.
Immunofluorescence staining
Tissue was dissected and washed briefly in ice cold PBS, followed by overnight fixation at 4°C in 4% paraformaldehyde. Next, tissues were washed in ice cold PBS and incubated with 30% sucrose in PBS overnight at 4°C for cryopreservation. Tissue was then embedded in Optimal Cutting Temperature medium (Sakura) and frozen with dry ice. Tissue was then sectioned in a cryostat at −21°C at 10 μm section thickness, and sections were affixed to a positively charged glass slide. Tissue sections were blocked for 1 hour in 10% goat serum in PBS +0.1% Triton X-100. Next, sections were incubated in primary antibodies overnight at 4°C. Antibodies and dilutions used were: PRMT1 (1:100, Cell Signaling Technology Cat# 2449, RRID:AB_2237696), GFP (1:200, Molecular Probes Cat# A-6455, RRID:AB_221570), CD31 (1:50, Santa Cruz Biotechnology Cat# sc-1506-R, RRID:AB_831096), Vimentin (1:100, BD Biosciences Cat# 550513, RRID:AB_393716), NG2 (1:100, Millipore Cat# AB5320, RRID:AB_11213678), Ki67 (1:200, Abcam Cat# ab16667, RRID:AB_302459), pHH3 (1:200, Millipore Cat# 06–570, RRID:AB_310177), α-actinin (1:50, Sigma-Aldrich Cat# A7732, RRID:AB_2221571), Fibronectin (1:200, Sigma-Aldrich Cat# F3648, RRID:AB_476976), a-SMA (1:200, Millipore Cat# 113200, RRID:AB_262054), and Podoplanin (1:200, Thermo Fisher Scientific Cat# 14–5381-85, RRID:AB_1210507). Stained sections were imaged on a Leica DMI 3000B. Images were analyzed using ImageJ (ImageJ, RRID:SCR_003070).
Cell culture and siRNA transfection
MEC1 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum (FBS). For siRNA transfection assays in 6-well plates, MEC1 cells were plated at 0.5 to 2.5 × 105 cells per well and transfected with RNAiMax (Invitrogen). Four to six hours after transfection, cells were transferred to fresh medium containing 10% FBS.
Cell circularity (C) was calculated using the formula C=4πA/P2 where A and P are the area and perimeter of the cell, respectively. Area and perimeter were measured using ImageJ.
SDS-PAGE, immunoblotting, and immunoprecipitation
Cells were lysed in lysis buffer (50 mM Tris-HCl pH 7.5, 250 mM NaCl, 2mM EDTA, 0.1% NP-40, 10% glycerol, and protease inhibitor cocktail). Proteins were quantified using Bio-Rad protein assay (Bio-Rad Laboratories), and 20–80 μg of protein was separated by SDS-PAGE on a 10% acrylamide gel run at 80–110V. Proteins were then transferred to 0.45 mm PVDF membrane at 100V for 3 hours. Membranes were blocked in TBS, 0.1% Tween 20, and 5% powdered milk (blocking solution) for 1 h, followed by overnight incubation with primary antibody diluted in TBS, 0.1% Tween 20, and 3% BSA, and 1 h incubation with HRP-conjugated secondary antibody diluted at 1:5,000. Immunoreactive protein was detected using ECL (GE Healthcare) and HyBlot CL film (Denville Scientific). For immunoprecipitation, cells were lysed in lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 2mM EDTA, 0.1% NP-40, 10% glycerol, and protease inhibitor cocktail) and pre-cleared with IgG and protein A/G Sepharose beads. Primary antibody was added and incubated overnight at 4°C. Immune complexes were then precipitated with protein A/G Sepharose beads and separated by SDS-PAGE followed by immunoblotting. Antibodies and dilutions were: PRMT1 (1:1000, Cell Signaling Technology Cat# 2449, RRID:AB_2237696), Asymmetric di-methyl arginine (1:1000, Cell Signaling Technology Cat# 13522, RRID:AB_2665370), α-tubulin (1:2000, Santa Cruz Biotechnology Cat# sc-8035, RRID:AB_628408), α-SMA (1:2000, Millipore Cat# 113200, RRID:AB_262054), E-cadherin (1:1000, Cell Signaling Technology Cat# 3195, RRID:AB_2291471), ZO-1 (1:1000, Cell Signaling Technology Cat# 8193, RRID:AB_10898025), β-catenin (1:2000, Cell Signaling Technology Cat# 9582, RRID:AB_823447), Fibronectin (1:2000, Sigma-Aldrich Cat# F3648, RRID:AB_476976), Vimentin (1:1000, BD Biosciences Cat# 550513, RRID:AB_393716), Slug (1:1000, Cell Signaling Technology Cat# 9585, RRID:AB_2239535), Snail (1:1000, Cell Signaling Technology Cat# 3879, RRID:AB_2255011), Zeb1 (1:1000, Cell Signaling Technology Cat# 3396, RRID:AB_1904164), Twist1 (1:1000, Abcam Cat# ab50581, RRID:AB_883292), Smad2 (1:1000, Cell Signaling Technology Cat# 5339, RRID:AB_10626777), phospho-Smad2 (1:1000, Cell Signaling Technology Cat# 3108, RRID:AB_490941), Smad3 (1:1000, Cell Signaling Technology Cat# 9523, RRID:AB_2193182), phospho-Smad3 (1:1000, Cell Signaling Technology Cat# 9520, RRID:AB_2193207), p53 (1:1000, Santa Cruz Biotechnology Cat# sc-6243, RRID:AB_653753), MDM2 (1:1000, Santa Cruz Biotechnology Cat# sc-965, RRID:AB_627920), MDM4 (1:1000, Millipore Cat# 04–1556, RRID:AB_10562655), WT-1 (1:1000, Cell Marque Corp Cat# 348M-94, RRID:AB_1161120). For immunoprecipitation, antibodies and dilutions were: p53 (1:1000, Cell Signaling Technology Cat# 2524, RRID:AB_331743) and Slug (1:500, Abcam Cat# ab51772, RRID:AB_1142919).
Quantitative RT-PCR
To quantify mRNA expression, MEC1 cells were transfected with Control or Prmt1 siRNA as above and treated with TGF-β (2 ng/ml) for a two-day time course (0, 24 hr, and 48 hr) or a short time course (0, 3 hr, and 6 hr) and RNA was isolated with Trizol (Invitrogen) and used as a template for reverse transcriptase (iScript RT supermix, Bio-Rad). mRNAs were quantified by real-time PCR with IQ Sybr Green Supermix (Bio-Rad), and normalized against RPL19 or Ppia mRNA. Relative changes in expression were calculated using the DDCt method. Primer sequences are listed in Table S2.
Semiquantitative RT-PCR
MEC1 cells were transfected with Control or Prmt1 siRNA as above and treated with TGF-β (2 ng/ml) for a two-day time course (0, 24 hr, and 48 hr) and RNA was isolated with Trizol (Invitrogen) and used as a template for reverse transcriptase (iScript RT supermix, Bio-Rad). For PCR reactions, 10 μL GoTaq Green Master Mix 2X, 1 μL of forward and reverse primers, 0.5 μL cDNA, 7 μL H2O, and 0.5 μL 25 mM MgCl2. Cycling protocol: 95°C for 2 minutes, 35 cycles of denaturation at 95°C for 25 s, annealing at 55°C for 35 s, elongation at 72°C for 70 s, final elongation of 72°C for 5 minutes and 4°C holding temperature. The primer sequences are found in Table S2.
RNaseq analysis
Total RNA from MEC1 cells was extracted using TRIzol (Thermo Fisher Scientific). Total RNA was enriched for poly(A) RNA and fragmented for library construction. RNA fragments were reverse transcribed followed by A-tailing and index adaptor ligation, then denatured and amplified on cBot. Library analysis and quality checks were performed on a LabChip GX. Sequencing was performed on a HiSeq 4000 (Illumina) with paired end reads of 100 bp. Analysis was performed using Partek Flow software (Partek Inc.). First, reads were first trimmed to remove adaptor sequences and then mapped to the mouse genome (mm10) using TopHat2 (Kim et al., 2013). Aligned reads were then quantified using Partek E/M. Differential expressed genes in each condition were identified using GSA. For gene ontology and pathway analysis, genes that were significantly up- or downregulated were compiled and analyzed using Ingenuity Pathway Analysis (QIAGEN Inc.). For alternate splice analysis, RNaseq reads were aligned using STAR aligner, mapped to GENCODE M19 mouse genome, and analyzed using rMATS.
Single cell RNA-sequencing and analysis
Embryonic mouse hearts were dissected at E12.5. For control group, YFP-positive Tbx18-Cre;ROSAYFP/+ hearts were pooled from multiple litters and subjected to 15~30 minutes of digestion in TrypLE solution (Thermo Fisher Scientific) on gentle rotation. For CKO group, embryos were subjected to fast-genotyping and then YFP-positive, Tbx18-Cre;Prmt1fl/fl;ROSAYFP/+ hearts were pooled from multiple litters for digestion. The embryo harvest and heart digestion for cell isolation dissociation were performed at the same day, and then single cell barcoding was performed simultaneously to minimize variation. Cells were counted and viability was confirmed to be > 85% before loading onto the 10X Genomics Chromium Controller. Libraries were generated with the 10× Chromium Single Cell 3’ v3 reagent kit according to the manufacturer’s instructions and sequenced on NOVAseq6000 S2. Following sequencing, raw fastq files were first converted to expression matrix using the 10× Genomics Cell Ranger software (http://www.10Xgenomics.com). Subsequently, expression matrix file from each experimental group was imported into R studio (Seurat package, version 2.3.4), where log normalization and scaling were performed (Satija et al., 2015). Batch effects were corrected by regressing out the number of molecules per cell and the percentage of mitochondria reads using the RegressOut function (Seurat package). Principle components analysis (PCA) was performed and significant PCs were used as input for graph-based clustering. Two-dimensional visualization of the multi-dimensional data was performed with t-distributed stochastic neighbor embedding (tSNE). Heatmap illustrating genes most highly enriched in each cluster was generated, with each column representing a gene and each row representing average expression level of that gene in each cluster. Distinct cell populations were predicted by the most enriched gene. Differential expression of gene in individual clusters was analyzed using the likelihood-ratio test for single cell feature expression. P value for differential expression analysis was adjusted based on Bonferroni correction using all genes in the dataset, which is also known as a false discovery rate (FDR) adjusted p value (McDavid et al., 2013). Differentially expressed genes in individual clusters were imported into Metascape (Metascape.org) for gene ontology (GO) analysis.
Migration and invasion assays
MEC1 cells were transfected with siRNA as above and cultured for 48 hours. Confluent monolayers were scratched with a pipette tip to create a cell-free area. Distance across the area was measured at time 0 as baseline and at 6, 8, 12, and 24 hours after scratching. Invasion was measured using a modified Boyden chamber assay (Lamouille and Derynck, 2007). Control or Prmt1 siRNA transfection of MEC1 cells was performed as above. 0.25×105 cells with or without 48 hours of treatment with 2 ng/ml TGF-β were added to Matrigel-coated inserts (BioCoat Matrigel Invasion Chamber; Becton Dickinson) in serum-free DMEM. These were then placed in companion plates with DMEM + 10% FBS for 24 h. Non-migrated cells in the top of the chamber were removed, and remaining cells were fixed with 4% PFA in PBS and stained for 30 mins in crystal violet (0.05% in PBS). Stained cells were counted on an Olympus IX70 microscope.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are means ± SEM; comparisons between groups were performed using two-tailed t test or 1 way ANOVA with Bonferroni post hoc test for comparisons of more than two groups. p53 degradation experiment was analyzed by 2-way ANOVA with Bonferroni post hoc test. All experiments were independently repeated at least three times and representative images are shown. SDS-PAGE quantification and statistical results are shown in supplemental figures.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal mix anti-Asymmetric Dimethylarginine | Cell Signaling | Cat# 13522; RRID:AB_2665370 |
| Mouse monoclonal anti-α-smooth muscle actin | Millipore | Cat# 113200; RRID:AB_262054 |
| Mouse monoclonal anti-α-actinin | Sigma | Cat# A7732; RRID:AB_2221571 |
| Rabbit monoclonal E-cadherin | Cell Signaling | Cat# 3195; RRID:AB_2291471 |
| Rabbit monoclonal anti-ZO-1 | Cell Signaling | Cat# 8193; RRID:AB_10898025 |
| Rabbit monoclonal anti-B-catenin | Cell Signaling | Cat# 9582; RRID:AB_823447 |
| Rabbit polyclonal anti-Fibronectin | Sigma | Cat# F3648; RRID:AB_476976 |
| Mouse monoclonal anti-Vimentin | BD Biosciences | Cat# 550513; RRID:AB_393716 |
| Rabbit monoclonal anti-Slug | Cell Signaling | Cat# 9585; RRID:AB_2239535 |
| Mouse monoclonal anti-Slug | Abcam | Cat# Ab51772; RRID:AB_1142919 |
| Rabbit monoclonal anti-Snail | Cell Signaling | Cat# 3879; RRID:AB_2255011 |
| Mouse monoclonal anti-WT-1 | Cell Marque | Cat# 348M-94; RRID:AB_1161120 |
| Mouse monoclonal anti-α-tubulin | Santa Cruz | Cat# Sc-8035; RRID:AB_628408 |
| Rabbit polyclonal anti-PRMT1 | Cell Signaling | Cat# 2449; RRID:AB_2237696 |
| Rabbit polyclonal anti-GFP | Invitrogen | Cat# A6455; RRID:AB_221570 |
| Rabbit polyclonal anti-ZEB1 | Cell Signaling | Cat# 3396; RRID:AB_1904164 |
| Rabbit polyclonal anti-Twist | Abcam | Cat# Ab50581; RRID:AB_883292 |
| Rabbit monoclonal anti-Smad2 | Cell Signaling | Cat# 5339; RRID:AB_10626777 |
| Rabbit monoclonal anti-phospho-Smad2 | Cell Signaling | Cat# 3108; RRID:AB_490941 |
| Rabbit monoclonal anti-Smad3 | Cell Signaling | Cat# 9523; RRID:AB_2193182 |
| Rabbit monoclonal anti-phospho-Smad3 | Cell Signaling | Cat# 9520; RRID:AB_2193207 |
| Mouse monoclonal anti-p53 | Cell Signaling | Cat# 2524; RRID:AB_331743 |
| Rabbit polyclonal anti-p53 | Sana Cruz | Cat# sc-6243; RRID:AB_653753 |
| Mouse monoclonal anti-MDM2 | Sana Cruz | Cat# sc-965; RRID:AB_627920 |
| Mouse monoclonal anti-MDM4 | Millipore | Cat# 04–1556; RRID:AB_10562655 |
| Goat polyclonal anti-CD31 | Sana Cruz | Cat# sc-1506-R; RRID:AB_831096 |
| Rabbit polyclonal anti-NG2 | Millipore | Cat# AB5320; RRID:AB_11213678 |
| Rabbit polyclonal anti-Ki67 | Abcam | Cat# ab16667; RRID:AB_302459 |
| Rabbit polyclonal anti-pHH3 | Millipore | Cat# 06–570; RRID:AB_310177 |
| Hamster monoclonal anti-Podoplanin | eBioscience | Cat# 14–5381-85; RRID:AB_1210507 |
| HRP secondary antibodies | Jackson Immunoresearch | various |
| AlexaFluor secondary antibodies | Thermo Fischer | various |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TRIzol | Thermo Fisher | Cat# 15596026 |
| TGFβ−1 | HumanZyme | Cat# HZ-1011 |
| AlexaFluor 488 Phalloidin | Invitrogen | Cat# A12379 |
| Crystal Violet | Sigma | Cat# C0775 |
| MG-132 | Sigma | Cat# 474787 |
| Cycloheximide | Sigma | Cat# 01810 |
| Nutlin-3 | Santa Cruz | Cat# sc-45061 |
| Tamoxifen | Sigma | Cat# T5648 |
| O.C.T. compound | Electron Microscopy Sciences | Cat# 62550–01 |
| Triton X-100 | Sigma | Cat# X100 |
| DAPI | Sigma | Cat# D9542 |
| Lipofectamine RNAiMAX | Thermo Fisher | Cat# 13778500 |
| Paraformadehyde | Sigma | Cat# P6148 |
| BioRad Protein Assay | Bio-Rad | Cat# 5000006 |
| ECL developing reagent | GE Life Sciences | Cat# RPN2106 |
| Protein A Sepharose beads | Sigma | Cat# P3391 |
| Protein G Sepharose beads | Sigma | Cat# P3296 |
| bFGF | Sigma | Cat# 341583 |
| Critical Commercial Assays | ||
| iScript RT supermix | Bio-Rad | Cat# 1708841 |
| iQ Sybr green supermix | Bio-Rad | Cat# 1708884 |
| GoTaq green master mix | Promega | Cat# M7122 |
| Deposited Data | ||
| Raw and analyzed RNaseq data | This paper | GEO: GSE122200 |
| Raw and analyzed scRNaseq data | This paper | GEO: GSE144271 |
| Experimental Models: Cell Lines | ||
| MEC1 mouse epicardial cell line | Li et al., 2011 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Prmt1tm1Rchd | Yu et al., 2009 | RRID:MGI:3848228 |
| Mouse: Tbx18:Cre | Cai et al., 2008 | N/A |
| Mouse: Gt(ROSA)26Sortm1(EYFP)Cos | Soriano, 1999 | RRID:IMSR_JAX:006148 |
| Mouse: Wt1tm2(cre/ERT2)Wtp/J | Zhou et al., 2008 | RRID:IMSR_JAX:010912 |
| Oligonucleotides | ||
| Primers for PCR and qPCR, see Table S1 | Primer Bank and this paper | N/A |
| PRMT1 siRNA | QIAGEN | Cat# SI00441861 and SI02663493 |
| p53 siRNA | QIAGEN | Cat# SI01456511and SI01456525 |
| Negative control siRNA | QIAGEN | Cat# 1022076 |
| Software and Algorithms | ||
| Partek Flow | Partek Inc. | N/A |
| rMATS | Kim et al., 2013 | N/A |
Highlights.
PRMT1 drives epicardial EMT and invasion in vitro and in vivo
PRMT1-p53 pathway controls the formation of epicardial-derived lineages
PRMT1-p53 regulates ventricular morphogenesis and coronary vessel formation
PRMT1 modulates alternative splicing of Mdm4 and decreases p53 stability
ACKNOWLEDGMENTS
We thank Drs. Michael Stallcup and Ching-Ling Lien for their thoughtful input and Dr. Bridget Samuels for editing. This research was supported by an American Heart Association postdoctoral fellowship (15POST22730019 to O.J.-W.), National Institutes of Health Interdisciplinary Research Training Award (5T90DE021982–07 to O.J.-W.), American Heart Association Scientist Development Grant (11SDG5220002 to J.X.), American Heart Association Beginning Grant-in-Aid (16BGIA26540000 to J.X.), Rose Hills Foundation research fellowship (to J.X.), STOP CANCER Research Career Development Award (to J.X.), and USC School of Dentistry seed grant (to J.X.) and start-up fund (to J.X.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107739.
REFERENCES
- Arima Y, Miyagawa-Tomita S, Maeda K, Asai R, Seya D, Minoux M, Rijli FM, Nishiyama K, Kim KS, Uchijima Y, et al. (2012). Preotic neural crest cells contribute to coronary artery smooth muscle involving endothelin signalling. Nat. Commun. 3, 1267. [DOI] [PubMed] [Google Scholar]
- Avasarala S, Van Scoyk M, Karuppusamy Rathinam MK, Zerayesus S, Zhao X, Zhang W, Pergande MR, Borgia JA, DeGregori J, Port JD, et al. (2015). PRMT1 Is a Novel Regulator of Epithelial-Mesenchymal-Transition in Non-small Cell Lung Cancer. J. Biol. Chem. 290, 13479–13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardot B, Bouarich-Bourimi R, Leemput J, Lejour V, Hamon A, Plancke L, Jochemsen AG, Simeonova I, Fang M, and Toledo F. (2015). Mice engineered for an obligatory Mdm4 exon skipping express higher levels of the Mdm4-S isoform but exhibit increased p53 activity. Oncogene 34, 2943–2948. [DOI] [PubMed] [Google Scholar]
- Bedford MT, and Clarke SG (2009). Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell E, Zhang X, and Ceman S. (2010). Arginines of the RGG box regulate FMRP association with polyribosomes and mRNA. Hum. Mol. Genet. 19, 1314–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc RS, and Richard S. (2017). Arginine Methylation: The Coming of Age. Mol. Cell 65, 8–24. [DOI] [PubMed] [Google Scholar]
- Bochmann L, Sarathchandra P, Mori F, Lara-Pezzi E, Lazzaro D, and Rosenthal N. (2010). Revealing new mouse epicardial cell markers through transcriptomics. PLoS ONE 5, e11429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C-L, Martin J, Sun Y, Li C, Lianchun W, Kunfu O, et al. (2008). A myocardial lineage derives from Tbx18 epicardial cells. Nature 454, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova JC, Travisano S, and de la Pompa JL (2013). Epithelial-to-mesenchymal transition in epicardium is independent of Snail1. Genesis 51, 32–40. [DOI] [PubMed] [Google Scholar]
- Cavallero S, Shen H, Yi C, Lien CL, Kumar SR, and Sucov HM (2015). CXCL12 Signaling Is Essential for Maturation of the Ventricular Coronary Endothelial Plexus and Establishment of Functional Coronary Circulation. Dev. Cell 33, 469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Zhang H, Liu Y, Adams S, Eilken H, Stehling M, Corada M, Dejana E, Zhou B, and Adams RH (2016). Endothelial cells are progenitors of cardiac pericytes and vascular smooth muscle cells. Nat. Commun. 7, 12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YY, Peng XF, Liu GY, Liu JS, Sun L, Liu H, Xiao L, and He LY (2019). Protein arginine methyltranferase-1 induces ER stress and epithelial-mesenchymal transition in renal tubular epithelial cells and contributes to diabetic nephropathy. Biochim. Biophys. Acta Mol. Basis Dis. 1865, 2563–2575. [DOI] [PubMed] [Google Scholar]
- Christoffels VM, Grieskamp T, Norden J, Mommersteeg MT, Rudat C, and Kispert A. (2009). Tbx18 and the fate of epicardial progenitors. Nature 458, E8–E9, discussion E9–E10. [DOI] [PubMed] [Google Scholar]
- Federico A, Sepe R, Cozzolino F, Piccolo C, Iannone C, Iacobucci I, Pucci P, Monti M, and Fusco A. (2019). The complex CBX7-PRMT1 has a critical role in regulating E-cadherin gene expression and cell migration. Biochim. Biophys. Acta. Gene Regul. Mech. 1862, 509–521. [DOI] [PubMed] [Google Scholar]
- Gao Y, Zhao Y, Zhang J, Lu Y, Liu X, Geng P, Huang B, Zhang Y, and Lu J. (2016). The dual function of PRMT1 in modulating epithelial-mesenchymal transition and cellular senescence in breast cancer cells through regulation of ZEB1. Sci. Rep. 6, 19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou Y, Li J, Jackson-Weaver O, Wu J, Zhang T, Gupta R, Cho I, Ho TV, Chen Y, Li M, et al. (2018). Protein Arginine Methyltransferase PRMT1 Is Essential for Palatogenesis. J. Dent. Res. 97, 1510–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grier JD, Xiong S, Elizondo-Fraire AC, Parant JM, and Lozano G. (2006). Tissue-specific differences of p53 inhibition by Mdm2 and Mdm4. Mol. Cell. Biol. 26, 192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo A, Gu H, Zhou J, Mulhern D, Wang Y, Lee KA, Yang V, Aguiar M, Kornhauser J, Jia X, et al. (2014). Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell. Proteomics 13, 372–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes J, Srivastava J, Madson N, Wittmann T, and Barber DL (2011). Dynamic actin remodeling during epithelial-mesenchymal transition depends on increased moesin expression. Mol. Biol. Cell 22, 4750–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann F, Bossert M, Schwander A, Akgün E, and Fackelmayer FO (2004). Arginine methylation of scaffold attachment factor A by heterogeneous nuclear ribonucleoprotein particle-associated PRMT1. J. Biol. Chem. 279, 48774–48779. [DOI] [PubMed] [Google Scholar]
- Katsuno Y, Qin J, Oses-Prieto J, Wang H, Jackson-Weaver O, Zhang T, Lamouille S, Wu J, Burlingame A, Xu J, and Derynck R. (2018). Arginine methylation of SMAD7 by PRMT1 in TGF-β-induced epithelial-mesenchymal transition and epithelial stem-cell generation. J. Biol. Chem. 293, 13059–13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, and Derynck R. (2007). Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 178, 437–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, Xu J, and Derynck R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, and Keating MT (1998). Elastin is an essential determinant of arterial morphogenesis. Nature 393, 276–280. [DOI] [PubMed] [Google Scholar]
- Li P, Cavallero S, Gu Y, Chen TH, Hughes J, Hassan AB, Brüning JC, Pashmforoush M, and Sucov HM (2011). IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development 138, 1795–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Estrada OM, Lettice LA, Essafi A, Guadix JA, Slight J, Velecela V, Hall E, Reichmann J, Devenney PS, Hohenstein P, et al. (2010). Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nat. Genet. 42, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDavid A, Finak G, Chattopadyay PK, Dominguez M, Lamoreaux L, Ma SS, Roederer M, and Gottardo R. (2013). Data exploration, quality control and testing in single-cell qPCR-based gene expression experiments. Bioinformatics 29, 461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry DS, Chen Y, Wang Y, Zhang K, and Sen GL (2014). SNAI2 controls the undifferentiated state of human epidermal progenitor cells. Stem Cells 32, 3209–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivey HE, and Svensson EC (2010). Epicardial-myocardial signaling directing coronary vasculogenesis. Circ. Res. 106, 818–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Pomares JM, and de la Pompa JL (2011). Signaling during epicardium and coronary vessel development. Circ. Res. 109, 1429–1442. [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A. (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglione A, Patzig J, Liang J, Frawley R, Bok J, Mela A, Yattah C, Zhang J, Teo SX, Zhou T, et al. (2018). PRMT5-mediated regulation of developmental myelination. Nat. Commun. 9, 2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y. (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 111, E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Cavallero S, Estrada KD, Sandovici I, Kumar SR, Makita T, Lien CL, Constancia M, and Sucov HM (2015). Extracardiac control of embryonic cardiomyocyte proliferation and ventricular wall expansion. Cardiovasc. Res. 105, 271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CL, Baek ST, Sung CY, and Tallquist MD (2011). Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ. Res. 108, e15–e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70–71. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, and Costantini F. (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi M, Nimura K, Mori M, Nakagami H, and Kaneda Y. (2013). The transcription factors Tbx18 and Wt1 control the epicardial epithelial-mesenchymal transition through bi-directional regulation of Slug in murine primary epicardial cells. PLoS ONE 8, e57829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, and Morrisey EE (2012). Importance of myocyte-nonmyocyte interactions in cardiac development and disease. Circ. Res. 110, 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trembley MA, Velasquez LS, de Mesy Bentley KL, and Small EM (2015). Myocardin-related transcription factors control the motility of epicardium-derived cells and the maturation of coronary vessels. Development 142, 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L, and Hinton G. (2008). Visualizing Data using t-SNE. Journal of Machine Learning Research 9, 2579–2605. [Google Scholar]
- van der Maaten L. (2014). Accelerating t-SNE using Tree-Based Algorithms. Journal of Machine Learning Research 15, 3221–3245. [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. (2004). In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848. [DOI] [PubMed] [Google Scholar]
- Volz KS, Jacobs AH, Chen HI, Poduri A, McKay AS, Riordan DP, Kofler N, Kitajewski J, Weissman I, and Red-Horse K. (2015). Pericytes are progenitors for coronary artery smooth muscle. eLife 4, e10036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gise A, and Pu WT (2012). Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circ. Res. 110, 1628–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SP, Wang WL, Chang YL, Wu CT, Chao YC, Kao SH, Yuan A, Lin CW, Yang SC, Chan WK, et al. (2009). p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat. Cell Biol. 11, 694–704. [DOI] [PubMed] [Google Scholar]
- Wei H, Liu Y, Min J, Zhang Y, Wang J, Zhou M, Xiong E, Yu G, Zhou H, He J, et al. (2019). Protein arginine methyltransferase 1 promotes epithelial-mesenchymal transition via TGF-b1/Smad pathway in hepatic carcinoma cells. Neoplasma 66, 918–929. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Hill MC, Zhang M, Martin TJ, Morikawa Y, Wang S, Moise AR, Wythe JD, and Martin JF (2018). Hippo Signaling Plays an Essential Role in Cell State Transitions during Cardiac Fibroblast Development. Dev. Cell 45, 153–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lamouille S, and Derynck R. (2009). TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 19, 156–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wang AH, Oses-Prieto J, Makhijani K, Katsuno Y, Pei M, Yan L, Zheng YG, Burlingame A, Brückner K, and Derynck R. (2013). Arginine Methylation Initiates BMP-Induced Smad Signaling. Mol. Cell 51, 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa H, Davis EC, Starcher BC, Ouchi T, Yanagisawa M, Richardson JA, and Olson EN (2002). Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature 415, 168–171. [DOI] [PubMed] [Google Scholar]
- Yang Y, and Bedford MT (2013). Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 13, 37–50. [DOI] [PubMed] [Google Scholar]
- Yu Z, Chen T, Hé bert J, Li E, and Richard S. (2009). A mouse PRMT1 null allele defines an essential role for arginine methylation in genome maintenance and cell proliferation. Mol. Cell. Biol. 29, 2982–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Wu J, Ungvijanpunya N, Jackson-Weaver O, Gou Y, Feng J, Ho TV, Shen Y, Liu J, Richard S, et al. (2018a). Smad6 Methylation Represses NFκB Activation and Periodontal Inflammation. J. Dent. Res. 97, 810–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang D, Zhang M, Wei H, Lu Y, Sun Y, Zhou M, Gu S, Feng W, Wang H, et al. (2018b). Protein arginine methyltransferase 1 coordinates the epithelial-mesenchymal transition/proliferation dichotomy in gastric cancer cells. Exp. Cell Res. 362, 43–50. [DOI] [PubMed] [Google Scholar]
- Zhou B, and Pu WT (2012). Genetic Cre-loxP assessment of epicardial cell fate using Wt1-driven Cre alleles. Circ. Res. 111, e276–e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Ma Q, Rajagopal S, Wu SM, Domian I, Rivera-Feliciano J, Jiang D, von Gise A, Ikeda S, Chien KR, and Pu WT (2008). Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 454, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou LT, Ye SH, Yang HX, Zhou YT, Zhao QH, Sun WW, Gao MM, Yi YH, and Long YS (2017). A novel role of fragile X mental retardation protein in pre-mRNA alternative splicing through RNA-binding protein 14. Neuroscience 349, 64–75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data that support the findings of this study have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO). RNA-seq data are accessible through the GEO Series accession number GSE122200. scRNA-seq data are accessible through GSE144271. All other relevant data are available from the corresponding author on request.