

Abstract
Purpose
N-methyl-N-nitrosourea (MNU) is an alkylating toxicant with potent mutagenic ability. This study was designed to induce apoptosis in lens epithelial cells (LECs) and corneal endothelial cells (CECs) via MNU administration. We sought to build ocular disease models of cataract and corneal endothelial decompensation.
Methods
MNU was delivered into the intraperitoneal cavities of neonatal rats and the anterior chambers of adult rabbits. The MNU-treated animals were then subjected to a series of functional and morphological analyses at various time points.
Results
MNU treatment induced pervasive apoptosis of LECs and CECs. These effects were dose and time dependent. Mature cataracts were found in neonatal rats 3 weeks after MNU treatment. Histological analysis revealed that MNU toxicity induced swelling, vacuolation, and liquefaction in lens fibers of MNU-treated rats. Pentacam examination showed that the average density of rat lens increased significantly after MNU administration. Terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) analysis showed pervasive apoptotic staining in the lenses of MNU-treated rats. In rabbit eyes, intracameral treatment with MNU induced corneal edema and significantly increased central corneal thickness, which peaked at P14. Morphological and immunohistochemical analysis showed that CECs were effectively ablated in the MNU-treated rabbits. The expression of 8-OHdG increased significantly in the cornea of MNU-treated rabbits, compared with vehicle-treated controls.
Conclusions
MNU is sufficient to induce ocular cell apoptosis in animal models. These models of MNU-induced cataract and corneal endothelial decompensation represent valuable tools for efforts to develop relevant therapies.
Keywords: N-methyl-N-nitrosourea, cataract, corneal endothelial decompensation, apoptosis
N-methyl-N-nitrosourea (MNU) is a direct-acting alkylating agent that increases the frequency of mutations in DNA.1,2 When these mutations are left unrepaired, the accumulated damage increases cancer risk. MNU has been used extensively for the induction of mammary cancer in rats.3,4 When DNA damage is severe, MNU acts as a cell-disrupting agent and causes cell death in susceptible organs and tissues by inducing apoptosis.5 The photoreceptor cells in mammals are highly sensitive to MNU toxicity. MNU can induce excessive DNA damage in the nucleus and activate apoptosis in photoreceptors.6,7 In the vertebrate eye, apoptosis is associated with ocular disease affecting the retina, lens, and cornea.8–10 Previous ophthalmological studies established the capacity of MNU to induce retinopathy.6,11,12 However, the effects of MNU in other ocular tissues have rarely been investigated. MNU may induce apoptosis in other ocular cell types, thereby generating ophthalmological disease models for experimental use.
Cataract is the leading cause of blindness among elderly individuals and accounts for approximately half of all forms of vision loss.13,14 The pathogenesis of cataract is associated with risk factors, such as oxidative damage, metabolic dysfunction, and exposure to a variety of physical or chemical agents.15,16 Terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) assay analysis of surgically isolated capsular epithelium has revealed that various types of cataract are associated with differing levels of lens epithelial cell (LEC) apoptosis.9,17–24 To reduce the need for cataract surgery, alternative medical therapies are urgently required to delay cataract formation. Chemically induced and genetic animal models of cataract are currently used in therapeutic trials.25 However, genetic animal models are costly, and transgenic mice with different mutations may share the same phenotype.26,27 On the other hand, chemically induced animal models require a lengthy period for the development of pathology and carry the risk of animal mortality.28,29 It is unlikely that a single animal model will suffice for replicating the entire process of cataract formation, as it occurs in humans.25,26 Therefore, it is necessary to establish a model with suitable expedience, ease of use, and reproducibility to mimic the pathophysiological process of cataract formation and to test anticataract therapies.
With the rate of cataract surgery growing exponentially,14 cataract surgery, typically phacoemulsification, is the most common cause of corneal endothelial decompensation.30–35 Corneal endothelium decompensation occurs as a surgical complication in 1 to 2% of cataract surgeries.30,36,37 Cornea edema after cataract surgery was the predominant indication for penetrating keratoplasty in North America (28.0%) and ranked second in Europe (20.6%), Australia (21.1%), the Middle East (13.6%), Asia (15.5%), and South America (18.6%).35 In previous studies, staining for 8-hydroxy-2-deoxyguanosine (8-OHdG), a marker of oxidative stress, has revealed that the free radicals produced by phacoemulsification cause damage to the corneal endothelium.38 Cataract surgery is the most common surgical cause of toxic anterior segment syndrome (TASS), which also results in severe damage to the corneal endothelium.33,39 Unlike the corneal epithelium, which can self-renew, human corneal endothelial cells (CECs) are quiescent in vivo.40 Damage to human CECs is associated with corneal endothelial decompensation, which causes blurred vision and discomfort, or even severe pain. Thus far, the only definitive treatment for corneal endothelial decompensation has been corneal transplantation. However, the shortage of donor corneas is challenging, and researchers have sought to develop potential alternatives using animal models.30,41 Several approaches are used to produce corneal endothelial decompensation in animal eyes, including mechanical injury,42–45 the intracameral injection of magnetic foreign particles or toxic chemicals,46,47 transcorneal freezing,48 phacoemulsification,38 and alkali burns.49 Although these approaches are proven to be effective in destroying CECs, they are technically difficult, inconsistently reproducible, and time consuming. Moreover, they may cause unwanted complications, such as anterior chamber inflammation, high intraocular pressure, corneal neovascularization, and intraocular damage. Some of these approaches are not effective for mimicking the corneal endothelial decompensation caused by phacoemulsification or TASS. Mammals are typically used to produce models of corneal endothelial decompensation. Previous studies have shown that the regenerative capacity of the corneal endothelium in cats and monkeys is analogous to that in humans.50,51 However, the use of cats and mentally developed animals, such as monkeys, are not recommended for use as animal models because of ethical reasons and economic issues. Rabbits and rodents, on the other hand, are easier to maintain in animal facilities. Although the rodent eye is too small for complex corneal operations, the rabbit eyeball is large enough for surgery and subsequent evaluation.50–55 Although the corneal endothelium of rabbits has potent regenerative ability, sufficiently severe injury to the rabbit endothelium can result in irreversible corneal decompensation.50,56 Selecting the most appropriate model would allow us to better determine the effectiveness of therapies designed to protect CECs from decompensation, and provide a stronger scientific rationale for future testing of these therapies in human patients.
Previous studies have shown that MNU is toxic to the LECs of neonatal Sprague-Dawley rats.28,29,57–60 The intraperitoneal injection of MNU induces lens pathologies, such as swelling, vacuolation, and fiber liquefaction. These changes mimic the pathology of cataract. However, the specific dose-effect mechanism of the MNU-induced cataract has not been verified. Several factors can influence the pathological process of MNU-induced cataract. The rate of development and severity of MNU-induced lens pathologies are closely related to the dose injected and the time that has elapsed since administration.29,57 Researchers tend to study MNU-induced retinal degeneration in mouse models, as well as cataract changes in Sprague-Dawley rats.4 Variation among studies in the dose and timing of MNU injections and in the animal model selected may hinder further standardization of the MNU-induced cataract model. Therefore, further study is required to characterize the MNU-induced cataract model. In particular, MNU administration is effective to induce corneal edema. Previous study showed that an intravitreal injection of MNU (6 mg/kg) caused corneal edema in adult rabbits.61 However, the mechanism underlying corneal edema remains to be explored. We hypothesized that MNU may cause damage to the corneal endothelium, which would subsequently give rise to corneal edema. With the experimental protocol outlined below, we sought to induce cataract and decompensation of the corneal endothelium via MNU administration. We examine the specific characteristics of MNU-induced pathology and evaluate various modeling parameters, including onset, progression, and optimal dosing. We found that MNU can act as a pluripotent toxicant to induce an ocular disease model. The data obtained should enrich our knowledges of MNU toxicity and its potential use in the field of ophthalmological research. The animal models generated should be valuable for future therapeutic trials.
Materials and Methods
Animal Procedures
All experiments were conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. All procedures regarding the use and the handling of animals were approved by the Institutional Animal Care and Use Committee of the General Hospital of Chinese PLA. Neonatal Sprague-Dawley rats (5 male and 5 female pups per mother, born within 24 hours) were obtained, together with their maternal rats, from the same breeder (SPF Biotechnology Co, Ltd., Beijing, China). The rats were maintained under specified pathogen-free (SPF1) conditions (room temperature 22 ± 2 deg Celsius (°C), relative humidity 60 ± 10%, 12-hour light/dark cycle). The rats were housed in 590 × 400 × 200-mm cages (Zhongke Animal Tec, ZX210001; Beijing, China) with food and water available ad libitum. The illumination intensity in cages was set below 60 lux. Adult New Zealand rabbits (2.0–2.5 kg, 12 months old, male) were purchased from Beijing Jinmuyang Laboratory Animal Breeding Co, Ltd. and housed under SPF1 conditions. Rabbits were allowed to acclimatize for 1 week before being used for experiments.
Chemicals and Dose Formulation
MNU (Fluorochem Ltd., Derbyshire, UK) was kept at −20°C in the dark. MNU was dissolved in physiologic saline containing 0.05% acetic acid just before use. MNU (10 mg/mL) was administered via an intraperitoneal (IP) injection at a dose of 50, 60, or 70 mg/kg body weight (BW) to induce lens damage. Vehicle-treated controls received an IP injection of a volume of 0.05% acetic acid equivalent to that administered to MNU-treated animals. To induce decompensation of the corneal endothelium, MNU was dissolved in a mixture of 30% dimethylsulfoxide (DMSO; Serva, Heidelberg, Germany) and 70% phosphate-buffered saline (PBS). This solution was further diluted with PBS to achieve the desired concentration of MNU.61 Rabbits received an intracameral injection of MNU at a dose of 2.0, 2.5, or 3.0 mg/kg BW; 150 µl of MNU solution or vehicle (DMSO + PBS mixture) was injected into the anterior chamber through the corneal limbus with a sterile 30-gauge steel needle.
Weight and Blood Samples
MNU-treated rats were examined daily for the appearance of clinical symptoms. Rats were weighed at P1, P3, P7, P14, and P21 after MNU treatment. At P21, blood samples were obtained by puncturing the aortaventralis to evaluate general health status. Samples were analyzed with an automatic analyzer (MEK-6450; Nihon Kohden, Rosbach, Germany) according to guidelines provided by the Institutional Animal Care and Use Committee of the National Beijing Center for Drug Safety Evaluation and Research. The following blood chemistry measurements were obtained: creatinine, blood urea nitrogen, albumin, alanine aminotransferase, aspartate aminotransferase, glucose, bilirubin, total protein, and calcium chloride.
Lens Opacity and Classification
The pupils of rats were dilated with 0.5% tropicamide solution (Santen Oy, Japan). Rat eyes were examined under a slit-lamp microscope (Opto-Sil, Optoprobe, UK), at P1, P3, P7, P14, and P21 days after MNU treatment. Cataract stage was graded according to a previously described method62 (Table). All observations were conducted by the same double-blinded investigator.
Table.
The Cataract Grading Scale
| Grade | Findings on Slit-Lamp Microscopy of the Lens |
|---|---|
| Grade 0 | Normal transparent lens |
| Grade 1 | Initial signs of posterior subcapsular or nuclear opacity involving tiny scatters |
| Grade 2 | Slight nuclear opacity with swollen fibers or posterior subcapsular scattering foci |
| Grade 3 | Diffuse nuclear opacity with cortical scattering |
| Grade 4 | Partial nuclear opacity |
| Grade 5 | Nuclear opacity without lens cortex |
| Grade 6 | Mature cataract of the entire lens |
Measurements of Lens Density and Thickness
The pupils of rats were dilated with 0.5% tropicamide solution (Santen Oy). Lens density and thickness were measured with a Pentacam anterior segment analyzer (Pentacam 70700; Oculus, Germany). The enhanced dynamic mode with 15 pictures / 0.3 seconds was used to visualize the lens. Lens density and thickness measurements were measured in vertical linear planes. Centrally vertical lines were drawn from the anterior lens capsule, the anterior subcapsular cortex, the anterior subcortical nucleus, the nucleus (highest point), the nucleus (lowest point), the posterior cortex, and the posterior capsule on the apical axis of the cornea. After lines were drawn, lens density and thickness were calculated by the Pentacam analyzer.
Morphological Examination of Lens Sections
Rats were euthanized with an overdose of anesthesia with pentobarbital sodium 100 mg/kg (Beijing Huaye Huayu Chemical Co., Ltd., Beijing, China) at P1, P3, P7, P14, and P21. Eye cups were enucleated and fixed overnight in FAS eyeball fixative, then embedded in paraffin, sectioned at 4-µm thickness, and stained with hematoxylin and eosin (H&E). H&E-stained slides were scanned with a high-resolution digital slide scanner (NanoZoomer 2.0 digital slide scanner; Hamamatsu Photonics, Hamamatsu, Japan) to obtain digital images that could be opened in color mode. The morphology, size, and nuclear staining of each lens section was evaluated.
For examination under the transmission electron microscope, lenses were prefixed in 2.5% glutaraldehyde solution, then postfixed in 2% OsO4 and processed for Luveak-812 embedding. Thin sections (80-nm thickness) were stained with toluidine blue to locate areas for imaging with electron microscopy. Ultrathin sections stained with uranyl acetate and lead citrate were examined with a Hitachi H-600 electron microscope (Hi-tachi, Tokyo, Japan).
TUNEL Assay
TUNEL staining was performed using a one-step apoptosis assay kit (Beyotime Institute of Biotechnology, China). Sections of lens and cornea tissue were incubated with proteinase K (100 µg/mL), rinsed, incubated in 3% H2O2, permeabilized with 0.5% Triton X-100, rinsed again, and incubated in a TUNEL reaction mixture. Sections were rinsed and visualized using Converter-POD with 0.03% 3,3’-diaminobenzidine (DAB), then mounted onto gelatin-coated slides. The slides were air-dried overnight at room temperature, and nuclei were stained with 40,6-diamidino-2-phenylindole (DAPI). The slides were examined under the confocal scanning laser microscope (SP8, Leica, Germany). Apoptotic index (AI) was calculated as the number of apoptotic cells / total cell count.
Expression of Crystalline Antioxidants and Enzymes
Each lens was extracted to measure levels of antioxidants and enzymes. Total superoxide dismutase (SOD) activity was measured with the nitroblue tetrazolium (NBT) method using the detection kit according to the manufacturer's instructions (Category number: S0109; Beyotime Institute of Biotechnology). Mn-SOD activity was measured using the Mn-SOD Assay Kit with WST-8 (Category number: S0103, Beyotime Institute of Biotechnology). SOD activity was expressed as U/mg protein. Reduced glutathione (GSH) was measured with the DTNB method, which was performed with a GSH assay kit (Beyotime Biotechnology). Wavelength was set at 405 nm and calculated according to a standard curve, then normalized to the protein level measured with the bicinchoninic acid (BCA) method, using a BCA protein assay kit from Beyotime Biotechnology. The results are expressed as µmol/g protein. The content of malondialdehyde (MDA) was determined by thiobarbituric acid reaction chronometry with reference to the MDA assay kit from Beyotime Biotechnology. The intensity of the resulting pink color was read at 532 nm. Levels of lipid peroxide are expressed as nmol of MDA formed per mg protein.
Intraocular Pressure and Anterior Segment Examination
Rabbits received an intracameral injection of MNU, at a dose of 2.0, 2.5, or 3.0 mg/kg (n = 8 per group). Rabbits were subjected to tonometry examinations, slit-lamp photography, and anterior segment optical coherence tomography (AS-OCT) at P1, P3, P7, P14, P21, and P28. Intraocular pressure (IOP) was measured with a TONO-PEN AVIA, which was designed according to the Mackay–Marg principle to yield measurements that are more accurate and less influenced by corneal thickness than those obtained with other devices63,64 (Reichert, Inc., Depew, NY). Ten measurements per eye are completed within 15 seconds, and the LCD sensor will automatically display the IOP value and statistical confidence interval. The anterior segment of MNU administered rabbits was evaluated by slit-lamp microscope (Opto-Sil; Optoprobe, UK). An AS-OCT device (Visante OCT 1000, Carl Zeiss) was used to measure central corneal thickness (CCT). For measurements of IOP and CCT, the average of three measurements was recorded.
In vivo Confocal Microscopy
Samples were examined with in vivo confocal microscopy (IVCM; HRT III; Heidelberg Engineering, Heidelberg, Germany) at P1, P3, P7, P14, P21, and P28. The laser source used in the Rostock Cornea Module was a diode laser with wavelength of 670 nm. Images consisted of 384 × 384 pixels covering an area of 400 × 400 µm with transverse optical resolution of approximately 1 µm/pixel (Heidelberg Engineering). For all eyes examined, 50 to 100 IVCM images parallel to the corneal epithelium, stroma, and endothelium were obtained.
Histologic Analysis of Cornea Sections
Rabbits from each group were euthanized by sodium pentobarbital overdose (Beijing Huaye Huayu Chemical Co., Ltd.), at P1, P3, P7, P14, P21, and P28, respectively. Eyes were enucleated, and the corneas were excised into quarters. One quarter of the corneal specimen was placed endothelial side up, and one drop of 0.25% trypan blue was applied for 1 minute. Alizarin red S (1%; pH 4.2) was then applied for 3 minutes. The cornea samples were examined under an inverted light microscope (BX51, Olympus, Japan). Another quarter of each cornea was fixed in 10% neutral buffered formalin. The formalin-fixed tissues were routinely processed for paraffin embedding, and 4-µm thick sections were cut for H&E staining and immunohistochemistry.
Immunohistochemistry
After samples were deparaffinized and rehydrated, antigen retrieval was performed. After blocking endogenous peroxidase activity and blocking with serum, samples were incubated with primary antibody (8-OHDG, Abcam, ab48508, 1:100) overnight at 4°C. Slides were washed three times with PBS (pH 7.4; 5 minutes each) and incubated with secondary antibody (goat anti-mouse, Servicebio, GB23301, 1:200) for 50 minutes at room temperature. Slides were dried, and fresh prepared DAB chromogenic reagent (Servicebio, G1211) was used for marking purpose. Reaction time was determined by observation under microscopy until the nucleus appeared brown-yellow in color. Finally, sections were washed with distilled water (5 minutes each) and mounted on slides. The results of H&E staining and immunohistochemical analysis were visualized with a high-resolution digital slide scanner (NanoZoomer 2.0 digital slide scanner; Hamamatsu Photonics, Hamamatsu, Japan). Another quarter of each cornea was embedded in optimal cutting temperature (OCT) compound (SAKURA, Japan). Sections of 6-µm thickness were cut in the sagittal plane with a Microm HM550 cryostat (Microm, Walldorf, Germany). The sections were placed on polylysine-coated glass slides (TBD, China) for immunofluorescence staining. After fixation with 4% polyformaldehyde (Solarbio Beijing, China), the slides were washed for 5 minutes (×3) in PBS (pH 7.2–7.4), and 200 mL 10% donkey serum (Solarbio Beijing) in PBS was dropped on the cover glass (1 hour, 4°C). The slides were then incubated overnight at 4°C with primary antibody (anti-alpha 1 sodium potassium ATPase antibody, Abcam, ab7671, 1:200). After washing with PBS, immuno-reactivity was revealed by incubation for 1 hour using a mixture of secondary antibodies linked to fluorophores: Alexa Fluor 488-conjugated goat anti-mouse IgG H+L (ZSGB-BIO, ZF-0512, 1:100). Nuclei were stained with DAPI. Fluorescent images were acquired with a confocal scanning laser microscope (SP8, Leica, Germany).
Electron Microscopy Scanning of Corneal Specimens
Corneal sections were examined with electron microscopy at P1, P3, P7, P14, P21, and P28, respectively. First, globes were immersed in fixative solution containing 2.5% glutaraldehyde in 0.1 M sodium cacodylate (pH 7.4; Electron Microscopy Sciences, Hatfield, PA) overnight at 4°C. For the final fixation step, corneas were excised from the globes and immersed in 1% osmium tetroxide (FMB, Singapore) at 22°C for 2 hours. After corneas were dehydrated in serial dilutions of ethanol, they were dried in a critical-point dryer (BALTEC, Balzers, Liechtenstein), and mounted on stubs. Samples were then sputter-coated with a 10 nm-thick layer of gold (BALTEC) and examined with a scanning electron microscope (FEI; Holland).
Statistical Analysis
All data are expressed as mean ± standard error of the mean (SEM). Statistical analysis of the data was subject to 1-way ANOVA analysis, followed by Duncan’s post hoc test and 2-way ANOVA analysis followed by Tukey's multiple-comparison test. The χ2 test was applied for categorical variables. The one-sample t-test was followed by the Mann-Whitney test. The Kolmogorov-Smirnov test was used to verify the distribution of the data among groups. All statistical calculations were carried out with GraphPadPrism version 7 software. P < 0.05 was considered statistically significant.
Results
General Condition of MNU-Treated Rats
Seven days after the intraperitoneal injection of MNU)P7(, toxic effects on hair follicle cells resulted in hair thinning.4 At P14, MNU cytotoxicity caused more severe clinical signs, such as loss of fur, pale skin, and decreased body weight. At P21, these clinical symptoms were alleviated, and body weight gain returned to normal. At P21, gross observation revealed eyeball opacity. These data relating to MNU-treated rats are summarized in Figures 1A, 1B. The results of blood analysis showed that 70-mg/kg MNU-treated animals and controls had similar expression levels of Glu (2.25 ± 0.31 mmol/L vs. 3.47 ± 0.45 mmol/L, respectively), ALT (6.42 ± 0.50 U/L vs. 12.08 ± 2.30 U/L, respectively), BUN (25.48 ± 0.1408 mmol/L vs. 25.64 ± 0.23 mmol/L, respectively), AST (33.29 ± 11.15 U/L vs. 33 ± 5.45 U/L, respectively), CREA (7.04 ± 0.45 µmol/L vs. 7.24 ± 0.58 µmol/L, respectively), and ALP (110.5 ± 31.84 U/L vs. 149.7 ± 29.21 U/L, respectively; P > 0.05, n = 10). Compared with normal animals, those in the 70-mg/kg MNU group had decreased expression of total protein (TP; 10.51 ± 2.50 vs. 29.16 ± 4.62 g/L, P < 0.01, n = 10), TG (0.17 ± 0.02 vs. 0.55 ± 0.11 mmol/L, P < 0.001, n = 10), and TCHO (0.41 ± 0.11 vs. 1.09 ± 0.18 mmol/L, P < 0.01, n = 10; Fig. 1C).
Figure 1.

(A) The mean body weights of the rats in the 50 / 60 / 70-mg/kg groups were significantly lower compared with those of the control group at P14 and P21. (B) Body weight gain (growth rate) in the 70-mg/kg MNU-treated pups tended to be lower than that in the 50- and 60-mg/kg MNU groups and control group at P14. At P21, body weight gain tended to recover (2-way analysis of variance [ANOVA] followed by Tukey's multiple-comparisons test, ****P < 0.0001, * P < 0.05, n = 20, for differences compared with control; all values represent mean ± SEM). (C) GLU, glucose; ALT, alanine aminotransferase; BUN, blood urea nitrogen; AST, aspartate transaminase; CREA, creatinine; ALP, alkaline phosphatase; TG, triglyceride; TP, total protein; TCHO, total cholesterol. No significant effects on blood levels of GLU, ALT, AST, ALP, BUN, and CREA were observed in MNU-treated rats. Decreases in TP, TG, and TCHO were observed in MNU-treated rats (one-sample t-test followed by Mann-Whitney U test, *** P < 0.001** P < 0.01 for between-group differences; n =10, all values represent mean ± SEM).
MNU-Induced Lens Opacification
The eyes of MNU-treated rats were examined under slit-lamp microscopy. In the 70-mg/kg group, posterior subcapsular lens opacity was observed at P3. The severity of lenticular opacity progressed over time. At P7, overt lenticular opacity was found in 10% of rats (2/20). At P14, overt lenticular opacity was found in 45% of rats (9/20). At P21, the incidence of overt lenticular opacity increased to 100% (20/20). Lenticular opacity most commonly arose bilaterally as nuclear cataract. There was no gender difference with regard to the incidence and severity of lens pathology. The MNU-induced changes in lenticular opacification are presented in Figure 2A. At P21, lenticular changes were rare in the 50-mg/kg and 60-mg/kg groups, except that 10% (2/20) of rats in the 50-mg/kg group and 20% (4/20) of the rats in the 60-mg/kg had small spots of posterior subcapsular opacity. The severity of MNU-induced lenticular damage was quantified with the cataract index (Fig. 2B). Cataract index scores were significantly higher in the 70-mg/kg group, compared with the 60- and 50-mg/kg MNU and control groups (P < 0.0001, n = 20). Cataract index scores in the 60-mg/kg and 50-mg/kg groups were not significantly different from those of the control group (P > 0.05, n = 20), suggesting that a dose of 70 mg/kg effectively induced lenticular opacity in rat eyes (Fig. 2C).
Figure 2.

(A) The lenses of experimental animals were examined under a slit-lamp microscope (magnification ×20). In rats from the 70-mg/kg group, the first sign of opacity appeared as posterior subcapsular opacity 3 days after MNU administration. Opacity size subsequently increased, with mature cataracts observed at P21. No lenticular abnormalities were observed in the 50- or 60-mg/kg MNU groups at P21. Lenses in the control group remained transparent. Diffuse, diffusion illumination; slit, direct illumination. (B) The severity of the lenticular damage induced by various doses of MNU was quantified with the cataract index. At P21, in the 70-mg/kg group, the incidence of overt lenticular opacity increased to 100% (20/20). Abnormal lenticular changes were rarely seen in the 50-mg/kg and 60-mg/kg groups in the days leading up to P21, except that 10% (2/20)/20% (4/20) of the 50/60 mg/kg MNU-treated rats had small posterior subcapsular cataracts. (C) The cataract index in the 70-mg/kg group was significantly higher than that observed in the 60 mg/kg MNU group, the 50 mg/kg MNU group, and the control group. The cataract index was similar in the 60-mg/kg group, the 50-mg/kg group, and the control group (1-way ANOVA followed by Dunn's multiple-comparisons test, ****P < 0.0001, compared with the 60-mg/kg group, the 50-mg/kg MNU group, and the control group; n = 20; all values represent mean ± SEM).
MNU induced Changes in Lens Density and Thickness
At P3, lens density in the MNU-treated groups was not significantly different from that in the control group (50 / 60 / 70 mg/kg MNU vs. control: 8.05 ± 0.45 / 8.4 ± 0.4 / 8.6 ± 1.3 vs. 7.75 ± 0.75%, P > 0.05, n =10; Figs. 3A, 3B). Mean density values in the 70-mg/kg group began to increase at P7 and continued to increase until P21 (70 mg/kg MNU vs. 50/60 mg/kg MNU and control: P7: 13 ± 0 vs. 8.0 ± 0.1 / 8.5 ± 0.8/7.9 ± 0.1%, P < 0.05; P14: 15.65 ± 2.15 vs. 8.1 ± 0 / 8.2 ± 0/8.1 ± 0.3%, P < 0.005; P21: 47.35 ± 4.25 vs. 8.0 ± 0 / 8.15 ± 0.35 / 7.95 ± 0.55 %, P < 0.0001, n = 10; see Fig. 3A,B). From P1 to P14, lens thickness in the MNU-treated groups was not significantly different from that in the control group (P > 0.05, n =10). Mean thickness in the 70-mg/kg group was significantly higher than that in the control group and 50- and 60-mg/kg groups at P21 (70 mg/kg MNU vs. 50/60 mg/kg MNU and control: 5340 ± 180 vs. 4486 ± 195.5 / 4438 ± 132.5 / 4431 ± 199.0 µm, P < 0.01, n = 10; Fig. 3C). Conversely, mean density and thickness in the 50- and 60-mg/kg groups were not significantly different from values observed in the control group at any time point (P > 0.05, n = 10, Fig. 3C).
Figure 3.

(A) Lens of rat anterior segment were analyzed with the Pentacam system. (B) At P3. lens density of all MNU-treated groups was not significantly different from that of the control group. In the 70-mg/kg group, mean density began to increase at P7 and continued to increase until P21. (C) From P1 to P14, lens thickness in all MNU-treated groups was not significantly different from that in the control group. Mean thickness in the 70-mg/kg group was significantly higher than that in the control group and the 50- and 60-mg/kg groups at P21. Mean density and thickness in the 50- and 60-mg/kg groups were not significantly different from those in controls at all time-points (2-way ANOVA followed by Tukey's multiple-comparisons test, **** P < 0.0001,** P < 0.01* P < 0.05; n = 10; all values represent mean ± SEM.)
MNU Induced Changes in Lens Histology
Lens sections were examined under the microscope. The MNU-treated rats, especially these in the 70-mg/kg group, had decreased numbers of nuclear germinative zone LECs, from P3 onward. At P7, several swollen, vacuolated, and liquefied fibers were observed in the 70-mg/kg group, and the number of LECs in the 70-mg/kg group decreased substantially. At P14, lens fiber vacuolation had increased, with the capsules of affected lenses becoming extremely thin. In the 70-mg/kg group, all the LECs disappeared, and the lens capsules became very thin. At P21, the MNU-induced lesions extended to the lens nucleus. Atypical nuclei and calcified, degenerative fibers in liquefied lenses were commonly seen in the 70-mg/kg group (Fig. 4, Fig. 5A). These changes corresponded to a diagnosis of mature cataract.50 In contrast, eyes in the control group did not show evidence of lenticular changes. In the 60-mg/kg and 50-mg/kg groups, the extent of lens damage was decreased, and the affected area was limited to subcapsular cortex, without associated nuclear abnormalities (see Fig. 5A). In the 70-mg/kg group, the results of electron microscopy showed that LECs had shrunken cytoplasm with nuclear chromatin condensation and dysmorphism. The subepithelial fibers of LECs were swollen and disorganized. The mitochondria of affected LECs were significantly decreased in number and in size. Conversely, the LECs from control group animals had numerous mitochondria with well-defined nuclei (Fig. 5B).
Figure 4.

Lens sections with H&E staining were examined under microscopy. In the 70 mg/kg MNU-treated group, the number of nuclear LECs in the germinative zone had decreased at P3. At P7, sparse swollen, vacuolated, and liquefied fibers were observed in the 70-mg/kg group, and the number of LECs in the 70-mg/kg group had decreased remarkably. At P14, lens fiber vacuolation had expanded further, and the lens capsules in these rats had become extremely thin, and all the LECs had disappeared. At P21, atypical nuclei and calcified and degenerative fibers were detected in the lens tissue. At P21, the regenerated epithelial cells had accumulated under the lens capsule. In the 50- and 60-mg/kg MNU-treated groups, lens damage was much milder; the affected area was free of atypical nuclei and restricted to the subcapsular cortex. In the control rats, no lenticular abnormalities were found. Scale bar = 50 µm.
Figure 5.
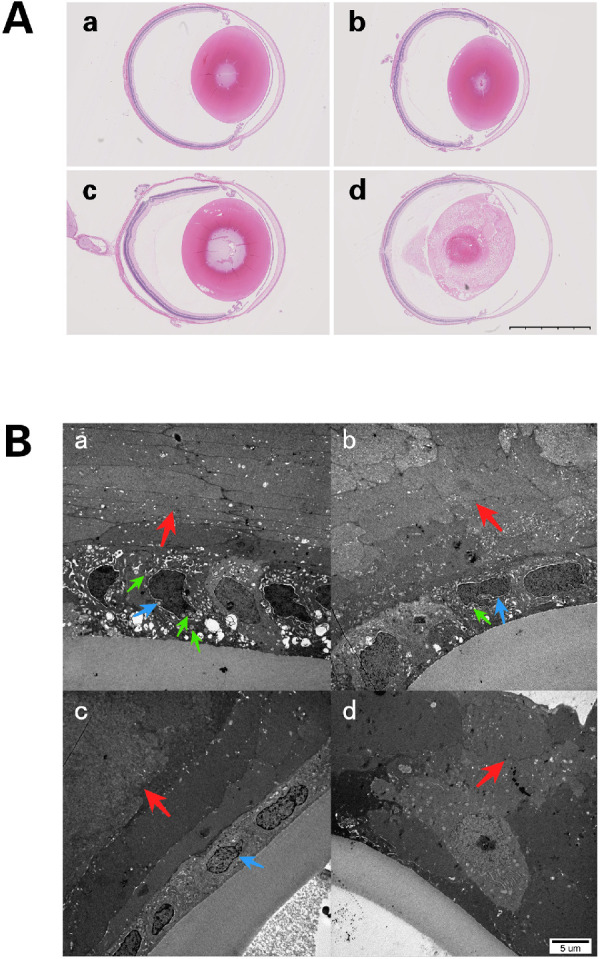
(A) Lens sections with H&E staining at P21. (a) Lenses from the control group were normal in appearance, (b) lenses from the 50-mg/kg group were nearly normal in appearance. (c) Liquefied fibers were found in the posterior pole in rats from the 60-mg/kg group. (d) Mature cataract formation in lenses from the 70-mg/kg group. Scale bar = 2.5 mm. (B) Transmission electron microscopy examination at P14. (a) Samples from the control group show normal LECs with normal nuclei (blue arrow), abundant mitochondria (green arrow) in the cytoplasm, and organized fibers (red arrow). (b) In the 50-mg/kg group, LECs have slightly shrunken cytoplasm and nuclei (blue arrow) with limited numbers of mitochondria (green arrow) and fibers showing slight swelling and lack of organization (red arrow). (c) In the 60-mg/kg group, LECs show deeply shrunken cytoplasm with condensation and dysmorphism of the nuclear chromatin (blue arrow). The subepithelial fibers of these LECs were swollen and disorganized (red arrow). The number of mitochondria in these LECs decreased markedly. (d) In the 70-mg/kg group, the LECs have disappeared, and the fibers are markedly swollen and disorganized (red arrow).
MNU-Induced Apoptosis in LECs
TUNEL staining was performed to count apoptotic cells in the lenses of MNU-treated rats at P3. Photomicrographs of TUNEL-positive cells in lens sections are shown in Figure 6A. Few TUNEL-positive cells were observed in the lenses of animals in the control group. The lens epithelium of MNU-treated rats had abundant TUNEL-positive cells. The AI in the 50-mg/kg group was not significantly different from that in the control group (3.250 ± 0.250 vs. 2.313 ± 0.210, P > 0.05, n = 6). The AI of the 60- and 70-mg/kg groups were significantly higher than that of the control group (5.000 ± 0.267 / 43.750 ± 1.191 vs. 2.313 ± 0.210, P < 0.05, n = 6; Fig. 6B). The apoptotic LECs were located closer to the equator of the lens in animals treated with lower doses of MNU. These findings suggested that MNU treatment induced LEC apoptosis in a dose-dependent manner.
Figure 6.

(A) Confocal photomicrographs of terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL)-positive cells in the lens epithelium at P3 (red: apoptosis; blue: DAPI). (B) Number of TUNEL-positive cells in each group. Few TUNEL-positive cells can be seen in lenses from the control group. Massive TUNEL-positive cells were found in the lens epithelium of MNU-treated rats. The AI of the 50-mg/kg group was not significantly different from that of the control group. The AI of the 60- and 70-mg/kg groups was significantly higher than that of the control group. (C) Total superoxide dismutase (SOD) activity in the lenses of each animal group. Treatment with 70 mg/kg MNU caused a significant reduction in total SOD, compared to levels observed in the control group. (D) Mn-SOD activity in lens tissue from each. Treatment with 70 mg/kg MNU caused a significant reduction in Mn-SOD levels, compared to those observed in the control group. (E) The level of reduced glutathione (GSH) in lenses from each group. Treatment with 70 mg/kg MNU caused a significant reduction in GSH, compared to levels observed in the control group. (F) Levels of malondialdehyde (MDA), a marker of lipid peroxidation, in lenticular samples from each group. The MDA concentration was significantly higher in the 70-mg/kg group than in the control group (1-way ANOVA followed by Dunnett's multiple-comparisons test, ****P < 0.0001, *** P < 0.001, ** P < 0.01 for comparisons with control group; n = 3; all values represent mean ± SEM).
Levels of Crystalline Antioxidants and Enzymes
The levels of antioxidants and enzymes in the lenses of MNU-treated rats were examined. The mean SOD activity in the 70-mg/kg group was significantly lower than that in the control group (3.65 ± 0.79 vs. 25.55 ± 0.79 U/mg; P < 0.0001, n = 6; Fig. 6C). Moreover, total SOD activity levels in the 60-mg group (16.68 ± 1.08 U/mg protein) and the 50-mg group (20.97 ± 0.87 U/mg protein) were also significantly lower than levels measured in the control group (P < 0.05, n = 6; see Fig. 6C). Similarly, mean Mn-SOD activity levels in the 70-mg/kg group were significantly lower than in the control group (0.7083 ± 0.07115 vs. 2.613 ± 0.1071 U/mg; P < 0.0001, n = 6; Fig. 6D). Moreover, Mn-SOD activity levels in the 60-mg group (2.183 ± 0.06922 U/mg protein) and the 50-mg group (2.298 ± 0.03351 U/mg protein) were also significantly lower than in the control group (P < 0.05, n = 6, Fig. 6D). MNU treatment caused a significant reduction in GSH levels in the 70-mg/kg group, compared to the control group (2.265 ± 0.1901 vs. 8.210 ± 0.04726 µmol/g protein, P < 0.0001, n = 6; Fig. 6E). However, this decrease was less noticeable in the 60-mg/kg group (7.287 ± 0.08939 µmol/g protein, P < 0.001, n = 6) and the 50-mg/kg group (7.658 ± 0.1310 µmol/g protein, P < 0.05, n = 6). MDA levels were significantly higher in the 70-mg/kg group than in the control group (0.48 ± 0.02 vs. 0.21 ± 0.02 nmol/mg protein, P < 0.001, n = 6; Fig. 6F). MDA levels in the 60-mg/kg group (0.32 ± 0.01 nmol/mg protein) were significantly higher than in the control group (P < 0.01, n = 6; see Fig. 6F). MDA levels in the 50-mg/kg group (0.26 ± 0.02 nmol/mg protein) were not significantly different from those in the control group (P > 0.05, n = 6; see Fig. 6F). These results suggest that MNU may affect crystalline antioxidants and enzymes and cause oxidative stress.
Effects of MNU Treatment on IOP
There was no significant difference between pre- vs. post-injection IOP times in any group (pre-injection: control / MNU 2.0 mg/kg / MNU 2.5 mg/kg / MNU 3.0 mg/kg: 14.0 ± 1.0 / 13.5 ± 1.5 / 12.5 ± 0.5 / 14.0 ± 0 vs. post-injection: control / MNU2.0mg/kg / MNU2.5mg/kg / MNU3.0mg/kg: 13.92 ± 0.4167 / 10.0 ± 0.3651 / 13.33 ± 0.7149 / 14.92 ± 1.186 mm Hg, P > 0.05; n = 8). IOP was slightly lower in the 2.0-mg/kg group, compared with the control group, at all time points after MNU treatment, but the difference was not significant (P > 0.05, n = 8; Fig. 7A).
Figure 7.

(A) MNU-treated rabbits were subjected to ocular examination. There was no significant difference among groups in IOP before injection or between pre-injection and post-injection IOP. There was no significant difference in post-injection IOP between MNU-treated groups and the control group. IOP was slightly lower in the 2.0-mg/kg group, compared with the control group, at all time-points after MNU treatment, but these differences were not significant (2-way ANOVA followed by Tukey's multiple-comparisons test, P > 0.05; n = 8; all values represent mean ± SEM). (B) The anterior segment was evaluated by slit-lamp microscopy (magnification ×16). In vehicle-treated rabbits, slight corneal edema was noted at P1 but resolved by P7. Corneal edema, inflammatory infiltration, and mydriasis mimicking TASS occurred in all MNU-treated rabbits, from P1 onward. Corneal pathology was more severe in the 3.0-mg/kg group than in the 2.0- and 2.5-mg/kg groups. Irreversible corneal edema was observed in all rabbits treated with 2.5 and 3.0 mg/kg, throughout the study period. Hyphema was observed in the 2.0- and 2.5-mg/kg groups.
Examination of the Anterior Segment
Effects of MNU treatment on the anterior segment in rabbits were evaluated by slit-lamp microscopy (Fig. 7B). In vehicle-treated rabbits, slight corneal edema was noted at P1 but resolved by P7. Corneal edema, inflammatory infiltration, and mydriasis (Pupil diameter: MNU/control:8.42 ± 0.34 mm vs. 2.65 ± 0.07 mm; P < 0.0001) mimicking TASS occurred in all MNU-treated rabbits, from P1 until the end of the study. The corneal pathology in the 3.0-mg/kg group was more severe than that in the 2.0- and 2.5-mg/kg groups. Irreversible corneal edema was found in all rabbits treated with 3.0 mg/kg or 2.5 mg/kg MNU throughout the study period. No sign of iridocorneal adhesion, hypopyon, or corneal neovascularization was found in any of the MNU-treated animals. Epithelial bullae were found in four cases (3 cases in the 3.0-mg/kg group at P14; and 1 case in the 2.5-mg/kg group at P21). Corneal dissolution occurred in one rabbit in the 3.0-mg/kg group at P28. Corneal scarring was observed in two cases (1 case in the 3.0-mg/kg group at P28; and 1 case in the 2.5-mg/kg group at P28). Hyphema was observed in six cases after MNU injection (4 cases in the 2.0-mg/kg group at P3; 2 cases in the 2.5-mg/kg group at P3). AS-OCT examination showed remarkable corneal edema in the 3.0-mg/kg group from P1 onward, with progression until P28 (Fig. 8A). Between P3 and P28, CCT was significantly greater in the 3.0-mg/kg MNU group, compared with the other three groups (3.0 mg/kg vs. 2.5 mg/kg MNU / 2.0 mg/kg MNU / control: P3: 1.55 ± 0.23 vs. 0.74 ± 0.00/0.73 ± 0.01 / 0.61 ± 0.01 mm; P7: 2.06 ± 0.22 vs. 0.92 ± 0.02 / 0.60 ± 0.00 / 0.51 ± 0.07 mm; P14: 3.21 ± 0.19 vs. 1.29 ± 0.01 / 0.59 ± 0.00 / 0.39 ± 0.02 mm; P21: 2.73 ± 0.08 vs. 1.29 ± 0.10 / 0.63 ± 0.06 / 0.38 ± 0.02 mm; P28: 1.89 ± 0.06 vs. 1.37 ± 0.02 / 0.62 ± 0.03 / 0.35 ± 0.02 mm; P < 0.0001; n = 8; Fig. 8B). CCT in the 2.5-mg/kg MNU group was not significantly different from that in the control group at P3. However, CCT in the 2.5-mg/kg MNU group increased at P7, with values significantly higher than those in the control group between P7 and P28. These findings suggest that treatment with MNU at a dose of 3.0 mg/kg or 2.5 mg/kg alters CCT in rabbit eyes. Corneal pathology was most severe in the 3.0-mg/kg group. Comparison of the 2.0-mg/kg MNU group and the control group revealed no significant change in CCT throughout the follow-up period (P > 0.05, n = 8), suggesting that MNU treatment at a dose of 2.0 mg/kg failed to induce any change in CCT.
Figure 8.

(A) AS-OCT examination revealed remarkable corneal edema in rabbits from the 3.0-mg/kg group, from P1 onward. The edema progressed over time until P28. (B) CCT was significantly higher in the 3-mg/kg group, compared with the other 3 groups, during the period from P3 to P28, with levels peaking at P14. CCT values in the 2.5-mg/kg group were not significantly different from those in the control group at P3. However, CCT increased in the 2.5-mg/kg group at P7; values were significantly higher than those observed in the control group at P7 and P28. These findings suggest that treatment with MNU at 3.0 mg/kg or 2.5 mg/kg induced stable alterations in CCT in rabbit eyes. Corneal pathology was more severe in the 3.0-mg/kg group (2-way ANOVA followed by Tukey's multiple-comparison test, **** P < 0.0001** P < 0.01; n = 8; all values represent mean ± SEM). Scale bar = 100 and 50 µm.
In vivo Confocal Microscopy
Rabbit corneas were examined under in vivo confocal microscopy (IVCM; Fig. 9). In the control group, normal CECs were found in the endothelial cell layer, which was characterized by uniformly sized hexagonal cells with low-reflection discernible cell borders. CECs in the 2.5- and 3.0-mg/kg groups disappeared from P1 to P28. In the 2.0-mg/kg MNU group, highly reflective, irregular, sparse CECs with diffusely distributed dark shadows were found in the endothelial cell layer at P1. In the 2.0-mg/kg MNU group, CECs were not found in the endothelial cell layer at P3, P7, P14, or P21. Conversely, numerous regenerated CECs were observed in the 2.0-mg/kg MNU group at P28. In the control group, clear images of CECs were obtained by IVCM at all time points.
Figure 9.

Representative IVCM images for experimental animals. In the control group, normal CECs were found in the endothelial cell layer, which was characterized by uniformly sized cells with a predominantly hexagonal shape, with low-reflection discernible cell borders. From P1 to P28, CECs were absent in the 2.5- and 3.0-mg/kg groups. In the 2.0-mg/kg MNU group, the endothelial cell layer had highly reflective, irregular, sparse CECs with diffusely distributed dark shadows. CECs were not found in the endothelial cell layer of the 2.0 mg/kg MNU group at P3, P7, P14, or P21. Numerous regenerated CECs were observed in the 2.0 mg/kg MNU group at P28. Scale bar = 100 µm.
Morphological Examination
As shown in the cornea sections with H&E staining, CECs were absent from eyes in the 2.5- and 3.0-mg/kg groups. There was no evidence of CEC regeneration in eyes from the 2.5- and 3.0-mg/kg groups. Corneal sections in the 2.0-mg/kg group had no CECs at P1 or P7. However, CEC regeneration was observed at P14 and continued until P28. In the 3.0-mg/kg group, evident damage was observed in Descemet's membrane (DM) throughout the follow-up period. DM remained intact in the 2.0- and 2.5mg/kg groups. The severity of DM damage differed significantly among the 3.0-mg/kg MNU group and the groups of animals treated with other doses of MNU (Fig. 10). The staining of CECs with alizarin red S and trypan blue showed complete ablation of the corneal endothelial layer in the 2.5- and 3.0-mg/kg groups throughout the follow-up period. No regeneration of CECs was observed in the 2.5- or 3.0-mg/kg groups. The corneal endothelial layer was also ablated in eyes from the 2.0-mg/kg group, from P1 to P21. Small clusters of cells thought to be regenerating CECs were found in peripheral cornea in the 2.0-mg/kg group at P28. Normal CECs were seen at all time points in the control group (Fig. 11A). Immunofluorescence staining results for the expression of Na+/K+-ATPase in the corneal endothelial layer were negative in the 2.5- and 3.0-mg/kg groups throughout the entire follow-up period. Na+/K+-ATPase expression was negative at P1, P3, and P7 after MNU dosing in the 2.0-mg/kg group, whereas Na+/K+-ATPase expression appeared at P14, P21, and P28 after MNU injection (Fig. 11B).
Figure 10.
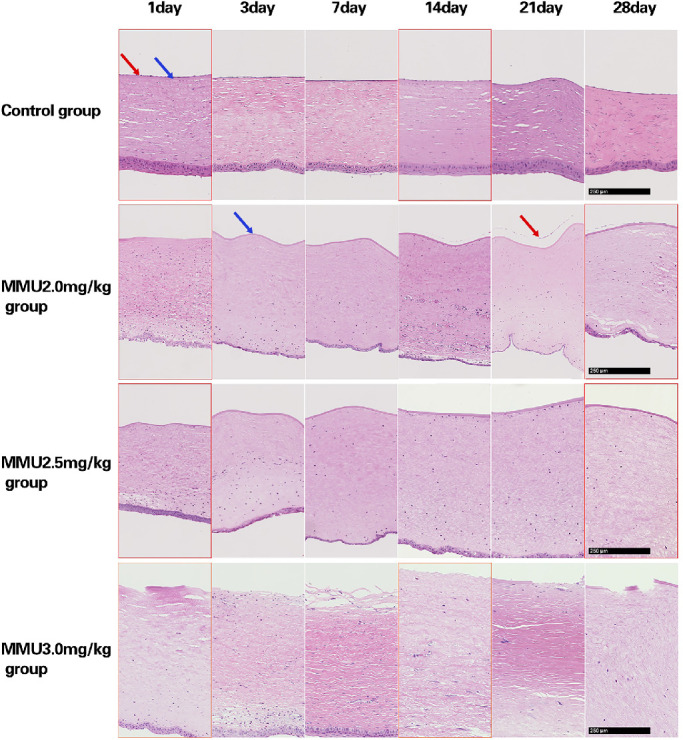
Cornea sections with H&E staining showed a lack of endothelial cells in the 2.5- and 3.0-mg/kg MNU groups, at all time points. In the 3.0 mg/kg MNU group, defects in Descemet's membrane (DM) were detected at all time points. DM appeared to be preserved in the 2.5- and 2.0- mg/kg MNU group. In the 2.0-mg/kg MNU group, CEC regeneration was observed from day 14 after MNU injection. In the control group, normal CECs were observed at all time-points. Scale bar = 250 µm.
Figure 11.

(A) Staining CECs with alizarin red S and trypan blue revealed complete ablation of the corneal endothelial layer during the follow-up period in the 2.5- and 3.0-mg/kg groups. In the 2.0-mg/kg group, small clusters of CECs (likely regenerated) were noted in the periphery at P28. In the control group, normal CECs were observed at all time points. Scale bar = 50 µm. (B) Staining for Na+/K+-ATPase (green) reveals a lack of Na+/K+-ATPase expression in the 2.5- and 3.0-mg/kg groups throughout the study period. In the 2.0-mg/kg MNU group, Na+/K+-ATPase staining was negative on P1, P3, and P7, but positive from P14 to P28. In the control group, Na+/K+-ATPase staining was positive at all time points (green: Na+/K+-ATPase; blue: DAPI). Scale bar = 250 µm.
Immunohistochemistry and TUNEL
The expression of 8-OHdG in the corneas of experimental animals was examined at P3 (Fig. 12A). The immunohistochemical expression of 8-OHdG was significantly higher in the 3.0-mg/kg group than in the control group (0.04133 ± 0.001542 vs. 0.007 ± 0.0005164 IOD/area, P < 0.0001, n = 3). Immunohistochemical staining for 8-OHdG in the 2.0- and 2.5-mg/kg groups was not significantly different from that in the control group (0.01 ± 0.00 / 0.01 ± 0.00 vs. 0.01 ± 0.00 IOD / area, P > 0.05, n = 3). Although TUNEL staining revealed few apoptotic cells in corneal sections of control group, numerous apoptotic cells throughout all corneal layers were observed in MNU-treated rabbits at P3 (Fig. 12B). The AI of the 2.0 / 2.5 / 3.0-mg/kg MNU groups was significantly higher than in the control group (2.5/3.0-mg/kg MNU vs. control: 17.25 ± 1.461 / 39.25 ± 1.436 vs.2.250 ± 0.3660, P < 0.0001; MNU 2.0 vs. control: 7.750 ± 0.750 / 32.250 ± 0.3660, P < 0.01, n = 3).
Figure 12.

(A) Immunohistochemical staining for 8-OHdG was significantly stronger in the 3.0-mg/kg group than in the control group. Staining for 8-OHdG was similar among the 2.0-mg group, the 2.5-mg/kg group, and the control group. Scale bar = 500 µm. (B) TUNEL staining revealed a limited number of apoptotic cells in corneal sections from animals in the control group. MNU-treated rabbits had numerous apoptotic cells throughout all corneal layers )red: apoptosis; blue: DAPI(. The AI of the 3.0-mg/kg group was significantly higher than that of the 2.0- and 2.5-mg/kg groups (1-way ANOVA followed by Dunnett's multiple-comparisons test, ****P < 0.0001, ** P < 0.01 in comparison with the control group; n = 3; all values represent mean ± SEM). Scale bar = 250 and 100 µm.
Scanning Electron Microscopy
SEM showed that the CECs were attached to DM in the control group. However, no CECs were found in the corneas of animals from the 2.5- or 3.0-mg/kg groups during follow-up. In the 2.0-mg/kg group, CECs were completely absent from P1 to P7. Islands of CEC regeneration were found in the 2.0-mg/kg group between P14 and P28.As shown in the SEM images, the DM was detached from the posterior surface of the cornea in the 3.0-mg/kg group. DM was intact in the 2.0- and 2.5-mg/kg groups. (Fig. 13).
Figure 13.

Scanning electron microscopy (SEM) showed that CECs were completely absent in the2.5- and 3.0-mg/kg MNU groups at all time-points. In the 2.0 mg/kg MNU group, islands of regenerated CECs (red arrow) were noted between P14 and P28. CECs (blue arrow) remained attached to DM in the control group. Serial SEM revealed severe damage to DM (green arrow) in the 3.0-mg/kg MNU group. Scale bar = 100 µm.
Discussion
MNU is an alkylating agent that interacts directly with DNA. Some types of cells with the capacity for rapid division, such as hematopoietic cells, brain neurons, and photoreceptors, are highly sensitive to MNU, and excessive DNA damage may result in apoptosis.3 MNU toxicity can activate poly (ADP-ribose) polymerase (PARP) in retina. MNU induces DNA adduct formation in photoreceptor cell nuclei, and increased PARP activity leads to the inactivation of NF-kB and JNK/AP-1, which downregulates Bcl-2, upregulates Bax, and activates the caspase-dependent apoptosis cascade.65,66 In this study, we induced apoptosis in ocular LECs and CECs via MNU administration to establish animal models of cataract and corneal endothelial decompensation.
MNU toxicity targets the LECs of neonatal rats. The development of a wide spectrum of MNU lesions depends on the method by which MNU is administered, dose, animal age, and species.3 Cataract models are typically established by administering MNU IP to neonatal Sprague-Dawley rats.29,57,58 The IP injection of MNU does not require anesthesia and is relatively feasible and safe. After IP treatment, MNU has an in vivo half-life of approximately 30 minutes.67 MNU decomposes at physiological pH, producing cyanate ion, which then reacts with proteins by carbamylation.68 This reaction with serum proteins may be responsible for the toxic effects of MNU on the epithelial cells of the lens. The IP injection of MNU at a high dose (100 mg/kg) can cause severe cataract in neonatal rats.57 However, neonatal rats are vulnerable to high doses of MNU. Moreover, retinal dysplasia may occur when MNU is administered to neonatal rats.69,70 In Sprague-Dawley rats, the critical time window for lens maturation is 12 to 16 days after birth.71 The growing lens epithelium may be more susceptible to MNU than developed lens epithelium. Thus, the dose of MNU and treatment time window should be optimized. Herein, we show that a single IP injection of 70-mg/kg MNU induced nuclear cataract in 15-day-old Sprague-Dawley rats within 3 weeks. No obvious complications were seen in these animals. The timing and dosing of MNU described above, therefore, seem to be safe and reliable for inducing rapid cataract formation in neonatal rats. Cataract index and density are critical indicators for monitoring the severity of cataractous pathology. In MNU-treated rats, changes in cataract index score and lens density are associated with pathological changes in lens sections. Lens sections revealed that the eyeballs were quite different in size. Ocular sections were cut along a vertical line parallel to the optic axis. Thus, the size of the eye was closely associated with the position of the incision.72 If the distance between the visual axis and the cutting line was small, the eye appeared larger in sections. Notably, because it is difficult to fix eye specimens and slice ocular sections in a standardized fashion, the eyes appeared to differ in size when subjected to histological analysis. Generally, diabetic animals are prone to the development of cataract, and cataracts have been produced experimentally through treatment with diabetogenic agents.73,74 Although MNU is reported to induce pancreatic damage,75,76 no blood glucose abnormality was observed in any of the animals included in this study. Thus, MNU-induced cataractous pathology may not be related to diabetic condition. No retinal damage was observed in neonatal rats after MNU treatment (data not shown). The weaning period of rats (20 days postnatal) is just at the ending stage of retinal differentiation. It is reported that the retina is resistant to MNU when it is administered close to weaning.72 MNU treatment did not induce toxic effects during retinal differentiation but did cause apoptosis in mature photoreceptor cells.77
LEC apoptosis is closely correlated with cataractogenesis.18,19 Different pathogenic factors may activate the apoptotic cascade in LECs, thus destroying epithelial control of lens fiber homeostasis and transparency and ultimately initiating cataractogenesis.9,17–22 MNU induces selective 7-methyldeoxyguanosine DNA adduct formation in LEC nuclei, which subsequently leads to cell death via an apoptotic mechanism.6,57 In the present study, abundant TUNEL-positive cells were found in the lens epithelium of MNU-treated rats. Moreover, the location of the TUNEL-positive cells appeared to shift in a dose-dependent manner (more central in animals treated with lower MNU doses compared to the animals treated with the highest MNU dose). It is highly possible that TUNEL-positive cells with self-renewal capacity originate in the equatorial zone, which is the LEC germinative zone. With increasing doses of MNU, TUNEL-positive cells may spread throughout the lens. This observation is consistent with space- and time-dependent MNU-induced retinal degeneration.78 Transmission electron microscopy revealed typical nuclear chromatin condensation and the gradual loss of mitochondria and LECs. Mitochondria modulate LEC function by controlling cell metabolism and apoptotic signaling.79 Moreover, remarkable TUNEL staining was seen in LECs in the 70-mg/kg MNU group. The gradual loss of LECs may cause metabolic dysfunction and damage to lens fibers, which subsequently leads to lens opacification.
Previous studies have shown that oxidative stress contributes strongly to cataract formation.80 Oxidative stress mainly arises from the strong cellular oxidizing potential of excess reactive oxygen species (ROS).81,82 ROS, generated by the leakage of superoxide ions from the respiratory chain after perturbations of mitochondrial structure and function, are countered by antioxidative systems. SOD is an important enzyme that protects the lens and LECs from oxidative damage by preventing the production of superoxide in mitochondria.83–85 There are three isoforms of SOD: cytosolic-Cu/Zn SOD (SOD1), mitochondrial superoxide dismutase Mn-SOD (SOD2), and extracellular secreted enzyme (SOD3), which is anchored to proteoglycans and the cell surface.86 All three isoforms are capable of converting O2- to H2O2, which is detoxified into H2O by either glutathione peroxidase or catalase.85,87,88 SOD2 is a major component of ROS metabolism in the mitochondrial matrix.89–91 Studies have verified that SOD2-deficient cells have extensive mitochondrial damage, cytochrome C leakage, caspase 3 activation, and increased apoptotic cell death when challenged with O2-. Thus, mitochondrial enzyme (SOD2) deficiency plays an important role in the initiation of apoptosis in the lens epithelium.85,92,93 Consistent with previous studies, our results confirmed that MNU causes excessive oxidative injury, leading to decreased levels of SOD2, subsequent mitochondrial damage, and, finally, LEC apoptosis. GSH is also a critical and primary lenticular antioxidant. The loss of GSH from the nuclear region is associated with reduced numbers of LECs, followed by age-related nuclear (ARN) cataract formation.25,94,95 The level of MDA, a reliable marker of oxidative stress, increases significantly.96 Oxidative stress and damage contribute to MNU-induced cataract. MNU-induced cataract formation in rats may serve as an animal model for noncongenital cataracts, especially the ARN cataract. Previous studies have shown that MNU induces epithelial-mesenchymal transition (EMT)-like phenotype changes in ocular and breast epithelial cells.97,98 These changes are mediated by methylation damage-induced activation of DNA-dependent protein kinase (DNA-PK).99 EMT is central to fibrotic posterior capsule opacification in cases of anterior subcapsular cataract and posterior polar cataract.100 EMT yields myofibroblasts that contribute to the formation of fibrotic tissue, which impairs tissue homeostasis and functionality.101 The EMT may be common to fibrotic forms of cataract and cancer metastasis. Additional experiments are needed to verify whether MNU induces lens epithelial cells to undergo EMT and, thus, loss of the epithelial phenotype and acquisition of mesenchymal markers as well as migratory capacity.
Our study used a novel approach to create an animal model of corneal endothelial decompensation mimicking phacoemulsification or TASS damage. To the best of our knowledge, this is the first study demonstrating that the intracameral injection of MNU is an effective and safe approach to induce corneal endothelial decompensation in mammalian eyes. We have studied the efficacy and safety of MNU treatment. We found that both 2.5 mg/kg and 3.0 mg/kg of MNU effectively induced corneal endothelial decompensation in rabbit eyes, with few complications. A dose of 3.0 mg/kg MNU may cause severe edema, whereas a dose of 2.5 mg/kg may cause relatively mild edema. CCT increased by 5.5- and 3-fold in the 3.0-mg/kg and 2.5-mg/kg group, respectively. Conversely, the intracameral injection of vehicle failed to produce any keratopathy or loss of CECs, suggesting that the aforementioned corneal pathologies are unique to MNU toxicity rather than simply caused by intracameral injection.
The intracameral injection is a relatively safe approach for the delivery of MNU to rabbit eyes. IVCM and histological analyses showed that corneal endothelial decompensation was induced efficiently in MNU-treated rabbits (2.5 and 3.0-mg/kg groups). In MNU-treated rabbits, the severity of corneal damage and swelling were dose dependent. CEC regeneration was detected in the 2.0-mg/kg MNU group from P14 onward. Conversely, CEC regeneration was not observed until the end of the follow-up periods in the 2.5- and 3.0-mg/kg MNU groups. The CEC regeneration observed in the 2.0-mg/kg MNU group may be ascribed to the compensatory function of CECs. Because the CECs of rabbits are regeneration cells, they can self-renew after injury. DM damage was found in the 3.0-mg/kg MNU group, whereas DM remained intact in the 2.5 and 2.0-mg/kg MNU groups. The increase in swelling observed was caused by the destruction of DM; in the presence of a normal DM, the rate of swelling is lower, and the healing of CECs is rapid and complete.102 CECs and DM, which is a product of endothelial secretion, have a bidirectional relationship. Corneal endothelial health relies on the intact DM, and healthy endothelial cells are necessary for the production of a normal extracellular matrix.103 Impairments of DM may collectively contribute to corneal endothelial decompensation.
MNU-induced corneal endothelial decompensation lasts for 28 days, which is significantly longer than in other models of corneal endothelial decompensation. For instance, in the transcorneal freezing rabbit model, CEC regeneration takes place 3 to 7 days after modeling, depending on the scale of the initial injury.102 In the corneal endothelium erasure rabbit model, the healing of rabbit corneal endothelium takes place 2 days after modeling.104,105 The injection of benzalkonium bromide into the anterior rabbit eye can cause corneal edema and corneal neovascularization, with corneal edema degrading 2 weeks after modeling.106 During the follow-up period, average IOP was lower in the 2.0- and 2.5-mg/kg groups than in the 3.0-mg/kg group. Previous studies have shown that decreased IOP is closely correlated with hyphema. If IOP decreases to a level lower than episcleral venous pressure, blood may reflux into the anterior chamber and cause hyphema.107,108
In summary, MNU acts as a pluripotent toxicant to induce LEC and CEC apoptosis, which results in cataract formation and corneal endothelial decompensation. The MNU dosage and the parameters of animal models are systemically characterized in this study. In particular, this study demonstrates that the intracameral injection of MNU is an effective and safe approach to induce corneal endothelial decompensation in rabbit eyes. These findings may promote the standardization and acceptance of MNU-induced animal models. The animal models generated may be valuable for future therapeutic trials.
Acknowledgments
Supported by 2017YFA0103204 National Key R&D Program of China (No: 2017YFA0103204) and the National Natural Science Foundation of China (No. 81770887; 81600767; and 81670830).
The authors alone are responsible for the content and writing of the paper.
Disclosure: Y. Qu, None; R. Li, None; X. Li, None; Q. Yang, None; J. Chen, None; Y. Dong, None; W. Xiao, None; S. Zheng, None; L. Wang, None; Y. Tao, None; Y. Huang, None
References
- 1. Kyrtopoulos SA. DNA adducts in humans after exposure to methylating agents. Mutat Res. 1998; 405: 135–143. [DOI] [PubMed] [Google Scholar]
- 2. Newbold RF, Warren W, Medcalf AS, Amos J.. Mutagenicity of carcinogenic methylating agents is associated with a specific DNA modification. Nature. 1980; 283: 596–599. [DOI] [PubMed] [Google Scholar]
- 3. Faustino-Rocha AI, Ferreira R, Oliveira PA, Gama A, Ginja M.. N-Methyl-N-nitrosourea as a mammary carcinogenic agent. Tumour Biol. 2015; 36: 9095–9117. [DOI] [PubMed] [Google Scholar]
- 4. Tsubura A, Lai YC, Miki H, et al.. Review: Animal models of N-Methyl-N-nitrosourea-induced mammary cancer and retinal degeneration with special emphasis on therapeutic trials. In Vivo. 2011; 25: 11–22. [PubMed] [Google Scholar]
- 5. Wijnhoven SW, van Steeg H.. Transgenic and knockout mice for DNA repair functions in carcinogenesis and mutagenesis. Toxicology. 2003; 193: 171–187. [DOI] [PubMed] [Google Scholar]
- 6. Yoshizawa K, Tsubura A.. [Characteristics of N-methyl-N-nitrosourea-induced retinal degeneration in animals and application for the therapy of human retinitis pigmentosa]. Nippon Ganka Gakkai Zasshi 2005; 109: 327–333. [PubMed] [Google Scholar]
- 7. Nakajima M, Nambu H, Shikata N, Senzaki H, Miki H, Tsubura A. Pigmentary degeneration induced by N-methyl-N-nitrosourea and the fate of pigment epithelial cells in the rat retina. Pathol Int. 1996; 46;874–882. [DOI] [PubMed] [Google Scholar]
- 8. Lolley RN. The rd gene defect triggers programmed rod cell death. The Proctor Lecture. Invest Ophthalmol Vis Sci. 1994; 35: 4182–4191. [PubMed] [Google Scholar]
- 9. Li DW, Spector A.. Hydrogen peroxide-induced expression of the proto-oncogenes, c-jun, c-fos and c-myc in rabbit lens epithelial cells. Mol Cell Biochem. 1997; 173: 59–69. [DOI] [PubMed] [Google Scholar]
- 10. Wilson SE. Stimulus-specific and cell type-specific cascades: emerging principles relating to control of apoptosis in the eye. Exp Eye Res. 1999; 69: 255–266. [DOI] [PubMed] [Google Scholar]
- 11. Schaller JP, Wyman M, Weisbrode SE, Olsen RG.. Induction of retinal degeneration in cats by methylnitrosourea and ketamine hydrochloride. Vet Pathol. 1981; 18: 239–247. [DOI] [PubMed] [Google Scholar]
- 12. Ogino H, Ito M, Matsumoto K, et al.. Retinal degeneration induced by N-methyl-N-nitrosourea and detection of 7-methyldeoxyguanosine in the rat retina. Toxicol Pathol. 1993; 21: 21–25. [DOI] [PubMed] [Google Scholar]
- 13. Pararajasegaram R. VISION 2020-the right to sight: from strategies to action. Am J Ophthalmol. 1999; 128: 359–360. [DOI] [PubMed] [Google Scholar]
- 14. Pascolini D, Mariotti SP.. Global estimates of visual impairment: 2010. Br J Ophthalmol. 2012; 96: 614–618. [DOI] [PubMed] [Google Scholar]
- 15. Jaanus SD. Drug-related cataract. Optom Clin. 1991; 1: 143–157. [PubMed] [Google Scholar]
- 16. Yang CX, Yan H, Ding TB.. Hydrogen saline prevents selenite-induced cataract in rats. Mol Vis. 2013; 19: 1684–1693. [PMC free article] [PubMed] [Google Scholar]
- 17. Li WC, Kuszak JR, Wang GM, Wu ZQ, Spector A. Calcimycin-induced lens epithelial cell apoptosis contributes to cataract formation. Exp Eye Res. 1995; 61: 91–98. [DOI] [PubMed] [Google Scholar]
- 18. Li WC, Spector A.. Lens epithelial cell apoptosis is an early event in the development of UVB-induced cataract. Free Radic Biol Med. 1996; 20: 301–311. [DOI] [PubMed] [Google Scholar]
- 19. Li WC, Kuszak JR, Dunn K, et al.. Lens epithelial cell apoptosis appears to be a common cellular basis for non-congenital cataract development in humans and animals. J Cell Biol. 1995; 130: 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen F, Chen C, Song X.. [The ultrastructure of human and mouse cataractous lens epithelial cell apoptosis]. Zhonghua Yan Ke Za Zhi. 2000; 36: 107–108, 110. [PubMed] [Google Scholar]
- 21. Tamada Y, Fukiage C, Nakamura Y, Azuma M, Kim YH, Shearer TR. Evidence for apoptosis in the selenite rat model of cataract. Biochem Biophys Res Commun. 2000; 275;300–306. [DOI] [PubMed] [Google Scholar]
- 22. Pandya U, Saini MK, Jin GF, Awasthi S, Bodley BF, Awasthi YC. Dietary curcumin prevents ocular toxicity of naphthalene in rats. Toxicol Lett. 2000; 115: 195–204. [DOI] [PubMed] [Google Scholar]
- 23. Balaram M, Tung WH, Kuszak JR, Ayaki M, Shinohara T, Chylack LT Jr. Noncontact specular microscopy of human lens epithelium. Invest Ophthalmol Vis Sci. 2000; 41: 474–481. [PubMed] [Google Scholar]
- 24. Guggenmoos-Holzmann I, Engel B, Henke V, Naumann GO.. Cell density of human lens epithelium in women higher than in men. Invest Ophthalmol Vis Sci. 1989; 30: 330–332. [PubMed] [Google Scholar]
- 25. Lim JC, Umapathy A, Donaldson PJ.. Tools to fight the cataract epidemic: a review of experimental animal models that mimic age related nuclear cataract. Exp Eye Res. 2016; 145: 432–443. [DOI] [PubMed] [Google Scholar]
- 26. Gelatt KN, Das ND.. Animal models for inherited cataracts: a review. Curr Eye Res. 1984; 3: 765–778. [DOI] [PubMed] [Google Scholar]
- 27. Smith RS, Sundberg JP, Linder CC.. Mouse mutations as models for studying cataracts. Pathobiology. 1997; 65: 146–154. [DOI] [PubMed] [Google Scholar]
- 28. Roy B, Fujimoto N, Watanabe H, Ito A.. Induction of cataract in methylnitrosourea treated Fischer (F344) rats. Hiroshima J Med Sci. 1989; 38: 95–98. [PubMed] [Google Scholar]
- 29. Kiuchi K, Yoshizawa K, Moriguchi K, Tsubura A.. Rapid induction of cataract by a single intraperitoneal administration of N-methyl-N-nitrosourea in 15-day-old Sprague-Dawley (Jcl: SD) rats. Exp Toxicol Pathol. 2002; 54: 181–186. [DOI] [PubMed] [Google Scholar]
- 30. Feizi S. Corneal endothelial cell dysfunction: etiologies and management. Ther Adv Ophthalmol. 2018; 10: 2515841418815802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morrison LK, Waltman SR.. Management of pseudophakic bullous keratopathy. Ophthalmic Surg. 1989; 20: 205–210. [PubMed] [Google Scholar]
- 32. Claesson M, Armitage WJ, Stenevi U. Corneal oedema after cataract surgery: predisposing factors and corneal graft outcome. Acta Ophthalmologica Scandinavica. 2010; 87: 154–159. [DOI] [PubMed] [Google Scholar]
- 33. Sharma N, Singhal D, Nair SP, Sahay P, Sreeshankar SS, Maharana PK. Corneal edema after phacoemulsification. Indian J Ophthalmol. 2017; 65: 1381–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xie L, Song Z, Zhao J, Shi W, Wang F.. Indications for penetrating keratoplasty in north China. Cornea. 2007; 26: 1070–1073. [DOI] [PubMed] [Google Scholar]
- 35. Matthaei M, Sandhaeger H, Hermel M, et al.. Changing indications in penetrating keratoplasty: a systematic review of 34 years of global reporting. Transplantation. 2017; 101: 1387–1399. [DOI] [PubMed] [Google Scholar]
- 36. Ljubimov AV, Atilano SR, Garner MH, Maguen E, Nesburn AB, Kenney MC. Extracellular matrix and Na+,K+-ATPase in human corneas following cataract surgery: comparison with bullous keratopathy and Fuchs' dystrophy corneas. Cornea. 2002; 21: 74–80. [DOI] [PubMed] [Google Scholar]
- 37. Kenney MC, Kenney MC, Zorapapel N, Atilano S, Chwa M, Ljubimov A, Brown D. Insulin-like growth factor-I (IGF-I) and transforming growth factor-beta (TGF-beta) modulate tenascin-C and fibrillin-1 in bullous keratopathy stromal cells in vitro. Exp Eye Res. 2003; 77: 537–546. [DOI] [PubMed] [Google Scholar]
- 38. Igarashi T, Ohsawa I, Kobayashi M, et al.. Hydrogen prevents corneal endothelial damage in phacoemulsification cataract surgery. Sci Rep. 2016; 6: 31190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mamalis N, Edelhauser HF, Dawson DG, Chew J, LeBoyer RM, Wener L. Toxic anterior segment syndrome. J Cataract Refract Surg. 2006; 32: 324–333. [DOI] [PubMed] [Google Scholar]
- 40. Koizumi N, Okumura N, Kinoshita S.. Development of new therapeutic modalities for corneal endothelial disease focused on the proliferation of corneal endothelial cells using animal models. Exp Eye Res. 2012; 95: 60–67. [DOI] [PubMed] [Google Scholar]
- 41. Tan DT, Dart JK, Holland EJ, Kinoshita S. Corneal transplantation. Lancet. 2012; 379: 1749–1761. [DOI] [PubMed] [Google Scholar]
- 42. Maurice DM, Giardini AA.. Swelling of the cornea in vivo after the destruction of its limiting layers. Br J Ophthalmol. 1951; 35: 791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stocker FW. The endothelium of the cornea and its clinical implications. Trans Am Ophthalmol Soc. 1953; 51: 669–786. [PMC free article] [PubMed] [Google Scholar]
- 44. Morton PL, Ormsby HL, Basu PK.. Healing of endothelium and Descemet's membrane of rabbit cornea. Am J Ophthalmol. 1958; 46: 62–67. [DOI] [PubMed] [Google Scholar]
- 45. McDonald JE. The effect of endothelial curettement on corneal wound healing: experimental study. Am J Ophthalmol. 1961; 51: 930–941. [DOI] [PubMed] [Google Scholar]
- 46. Cogan DG. A new method for studying endothelial regeneration. Ophthalmologica. 1949; 118: 440–443. [DOI] [PubMed] [Google Scholar]
- 47. Hayashi T, Yamagami S, Tanaka K, et al.. A mouse model of allogeneic corneal endothelial cell transplantation. Cornea. 2008; 27: 699–705. [DOI] [PubMed] [Google Scholar]
- 48. Han SB, Ang H, Balehosur D, et al.. A mouse model of corneal endothelial decompensation using cryoinjury. Mol Vis. 2013; 19: 1222–1230. [PMC free article] [PubMed] [Google Scholar]
- 49. Chung JH, Fagerholm P.. Endothelial healing in rabbit corneal alkali wounds. Acta Ophthalmol (Copenh). 1987; 65: 648–656. [DOI] [PubMed] [Google Scholar]
- 50. Van Horn DL, Sendele DD, Seideman S, Buco PJ.. Regenerative capacity of the corneal endothelium in rabbit and cat. Invest Ophthalmol Vis Sci. 1977; 16: 597–613. [PubMed] [Google Scholar]
- 51. Van Horn DL, Hyndiuk RA.. Endothelial wound repair in primate cornea. Exp Eye Res. 1975; 21: 113–124. [DOI] [PubMed] [Google Scholar]
- 52. Ilmonen M, Lehtosalo JI, Virtanen J, Uusitalo H, Palkama A.. Initial healing of the posterior corneal surface following perforating trauma in guinea pig: a scanning electron microscope study. Acta Ophthalmol (Copenh). 1984; 62: 787–795. [DOI] [PubMed] [Google Scholar]
- 53. Ling TL, Vannas A, Holden BA.. Long-term changes in corneal endothelial morphology following wounding in the cat. Invest Ophthalmol Vis Sci. 1988; 29: 1407–1412. [PubMed] [Google Scholar]
- 54. Tuft SJ, Williams KA, Coster DJ.. Endothelial repair in the rat cornea. Invest Ophthalmol Vis Sci. 1986; 27: 1199–1204. [PubMed] [Google Scholar]
- 55. Maurice D, Perlman M.. Permanent destruction of the corneal endothelium in rabbits. Invest Ophthalmol Vis Sci. 1977; 16: 646–649. [PubMed] [Google Scholar]
- 56. Joyce NC, Navon SE, Roy S, Zieske JD.. Expression of cell cycle-associated proteins in human and rabbit corneal endothelium in situ. Invest Ophthalmol Vis Sci. 1996; 37: 1566–1575. [PubMed] [Google Scholar]
- 57. Yoshizawa K, Oishi Y, Nambu H, et al.. Cataractogenesis in neonatal Sprague-Dawley rats by N-methyl-N-nitrosourea. Toxicol Pathol. 2000; 28: 555–564. [DOI] [PubMed] [Google Scholar]
- 58. Emoto Y, Yoshizawa K, Hamazaki K, et al.. Mead acid supplementation does not rescue rats from cataract and retinal degeneration induced by N-methyl-N-nitrosourea. J Toxicol Pathol. 2015; 28: 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Miyazono Y, Harada K, Sugiyama K, et al.. Toxicological characterization of N-methyl-N-nitrosourea-induced cataract in rats by LC/MS-based metabonomic analysis. J Appl Toxicol. 2011; 31: 655–662. [DOI] [PubMed] [Google Scholar]
- 60. Miki K, Yoshizawa K, Uehara N, Yuri T, Matsuoka Y, Tsubura A. PARP inhibitors accelerate N-methyl-N-nitrosourea-induced cataractogenesis in Sprague-Dawley rats. In Vivo. 2007; 21: 739–744. [PubMed] [Google Scholar]
- 61. Rosch S, Werner C, Muller F, Walter P.. Photoreceptor degeneration by intravitreal injection of N-methyl-N-nitrosourea (MNU) in rabbits: a pilot study. Graefes Arch Clin Exp Ophthalmol. 2017; 255: 317–331. [DOI] [PubMed] [Google Scholar]
- 62. Hiraoka T, Clark JI.. Inhibition of lens opacification during the early stages of cataract formation. Investigative ophthalmology & visual science. Nov 1995; 36(12): 2550–2555. [PubMed] [Google Scholar]
- 63. Mok KH, Wong CS, Lee VW. Tono-Pen tonometer and corneal thickness. Eye (London, England). 1999; 13: 35–37. [DOI] [PubMed] [Google Scholar]
- 64. Sharma R, Majumdar S, Sobti A, Arora T, Agarwal T, Dada T. Comparison of Tono-Pen AVIA intraocular pressure measurements performed at limbus with central corneal Tono-Pen AVIA intraocular pressure. Cornea. Jul. 2013; 32(7): 943–946. [DOI] [PubMed] [Google Scholar]
- 65. Petrin D, Baker A, Coupland SG, et al.. Structural and functional protection of photoreceptors from MNU-induced retinal degeneration by the X-linked inhibitor of apoptosis. Invest Ophthalmol Vis Sci. 2003; 44: 2757–2763. [DOI] [PubMed] [Google Scholar]
- 66. Oka T, Nakajima T, Tamada Y, Shearer TR, Azuma M.. Contribution of calpains to photoreceptor cell death in N-methyl-N-nitrosourea-treated rats. Exp Neurol. 2007; 204: 39–48. [DOI] [PubMed] [Google Scholar]
- 67. Bosland MC. Chemical and hormonal induction of prostate cancer in animal models. Urol Oncol. 1996; 2: 103–110. [DOI] [PubMed] [Google Scholar]
- 68. Knox P. Carcinogenic nitrosamides and cell cultures. Nature. 1976; 259: 671–673. [DOI] [PubMed] [Google Scholar]
- 69. Yoshizawa K, Oishi Y, Nambu H, et al.. Cataractogenesis in neonatal Sprague-Dawley rats by N-methyl-N-nitrosourea. Toxicol Pathol. 2000; 28: 555–564 [DOI] [PubMed] [Google Scholar]
- 70. Nambu H, Taomoto M, Ogura E, Tsubura A.. Time-specific action of N-methyl-N-nitrosourea in the occurrence of retinal dysplasia and retinal degeneration in neonatal mice. Pathol Int. 2010; 48: 199–205. [DOI] [PubMed] [Google Scholar]
- 71. Groth-Vasselli B, Farnworth PN.. A critical maturation period in neonatal-rat-lens development. Exp Eye Res. 1986; 43: 1057–1066. [DOI] [PubMed] [Google Scholar]
- 72. Nambu H, Yoshizawa K, Yang J, et al.. Age-specific and dose-dependent retinal dysplasia and degeneration in- duced by a single intraperitoneal administration of N- methyl-N-nitrosourea to rats. J Toxicol Pathol. 1998; 11: 127–131, 10.3791/57861. [DOI] [Google Scholar]
- 73. Ishida H, Mitamura T, Takahashi Y, et al.. Cataract development induced by repeated oral dosing with FK506 (tacrolimus) in adult rats. Toxicology. 1997; 123: 167–175. [DOI] [PubMed] [Google Scholar]
- 74. White JH, Cinotti AA.. Streptozotocin-produced cataracts in rats. Invest Ophthalmol. 1972; 11: 56–57. [PubMed] [Google Scholar]
- 75. Murthy A, Vawter G, Petersen R.. Ocular lesions and neoplasms in Wistar rats after a single injection of N-methyl-N-nitrosourea. Toxicol Lett. 1979; 4: 439–447. [Google Scholar]
- 76. Niedermüller H. Age dependency of DNA repair in rats after DNA damage by carcinogens. Mech Ageing Dev. 1982; 19: 259–271. [DOI] [PubMed] [Google Scholar]
- 77. Nambu H, Taomoto M, Ogura E, Tsubura A.. Time-specific action of N-methyl-N-nitrosourea in the occurrence of retinal dysplasia and retinal degeneration in neonatal mice. Pathol Int. 1998; 48: 199–205. [DOI] [PubMed] [Google Scholar]
- 78. Tao Y, Chen T, Fang W, et al.. The temporal topography of the N-Methyl-N-nitrosourea induced photoreceptor degeneration in mouse retina. Sci Rep. 2015; 5: 18612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kubota M, Shui YB, Liu M, et al.. Mitochondrial oxygen metabolism in primary human lens epithelial cells: association with age, diabetes and glaucoma. Free Radic Biol Med. 2016; 97: 513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Beebe DC, Holekamp NM, Shui YB.. Oxidative damage and the prevention of age-related cataracts. Ophthalmic Res. 2010; 44: 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wright E Jr, Scism-Bacon JL, Glass LC.. Oxidative stress in type 2 diabetes: the role of fasting and postprandial glycaemia. Int J Clin Pract. 2006; 60: 308–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Reddy PH. Amyloid precursor protein-mediated free radicals and oxidative damage: implications for the development and progression of Alzheimer's disease. J Neurochem. 2006; 96: 1–13. [DOI] [PubMed] [Google Scholar]
- 83. Manikandan R, Thiagarajan R, Beulaja S, Sudhandiran G, Arumugam M.. Effect of curcumin on selenite-induced cataractogenesis in Wistar rat pups. Curr Eye Res. 2010; 35: 122–129. [DOI] [PubMed] [Google Scholar]
- 84. Periyasamy P, Shinohara T. Age-related cataracts: role of unfolded protein response, Ca(2+) mobilization, epigenetic DNA modifications, and loss of Nrf2/Keap1 dependent cytoprotection. Prog Retin Eye Res. 2017; 60: 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Reddy VN, Kasahara E, Hiraoka M, Lin LR, Ho YS.. Effects of variation in superoxide dismutases (SOD) on oxidative stress and apoptosis in lens epithelium. Exp Eye Res. 2004; 79: 859–868. [DOI] [PubMed] [Google Scholar]
- 86. Carlsson LM, Jonsson J, Edlund T, Marklund SL.. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc Natl Acad Sci U S A. 1995; 92: 6264–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003; 552: 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ohsawa I, Ishikawa M, Takahashi K, et al.. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med. 2007; 13: 688–694. [DOI] [PubMed] [Google Scholar]
- 89. Zelko IN, Mariani TJ, Folz RJ.. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002; 33: 337–349. [DOI] [PubMed] [Google Scholar]
- 90. Pani G, Koch OR, Galeotti T.. The p53-p66shc-Manganese Superoxide Dismutase (MnSOD) network: a mitochondrial intrigue to generate reactive oxygen species. Int J Biochem Cell Biol. 2009; 41: 1002–1005. [DOI] [PubMed] [Google Scholar]
- 91. Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011; 15: 1583–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Liu L, Keefe DL.. Cytoplasm mediates both development and oxidation-induced apoptotic cell death in mouse zygotes. Biol Reprod. 2000; 62: 1828–1834. [DOI] [PubMed] [Google Scholar]
- 93. Karbowski M, Kurono C, Wozniak M, et al.. Free radical-induced megamitochondria formation and apoptosis. Free Radic Biol Med. 1999; 26: 396–409. [DOI] [PubMed] [Google Scholar]
- 94. Reddy VN. Glutathione and its function in the lens–an overview. Exp Eye Res. 1990; 50: 771–778. [DOI] [PubMed] [Google Scholar]
- 95. Giblin FJ. Glutathione: a vital lens antioxidant. J Ocul Pharmacol Ther. 2000; 16: 121–135. [DOI] [PubMed] [Google Scholar]
- 96. Babizhayev MA, Deyev AI, Linberg LF.. Lipid peroxidation as a possible cause of cataract. Mech Ageing Dev. 1988; 44: 69–89. [DOI] [PubMed] [Google Scholar]
- 97. Anandi L, Chakravarty V, Ashiq KA, Bodakuntla S, Lahiri M.. DNA-dependent protein kinase plays a central role in transformation of breast epithelial cells following alkylation damage. J Cell Sci. 2017; 130: 3749–3763. [DOI] [PubMed] [Google Scholar]
- 98. Ul Quraish R, Sudou N, Nomura-Komoike K, Sato F, Fujieda H. p27(KIP1) loss promotes proliferation and phagocytosis but prevents epithelial-mesenchymal transition in RPE cells after photoreceptor damage. Mol Vis. 2016; 22: 1103–1121. [PMC free article] [PubMed] [Google Scholar]
- 99. Mohapatra P, Ranjan Satapathy S, Das D, Siddhart S, Choudhuri T, Nath Kundu C. Resveratrol mediated cell death in cigarette smoke transformed breast epithelial cells is through induction of p21Waf1/Cip1 and inhibition of long patch base excision repair pathway. Toxicol Appl Pharmacol. 2014; 275: 221–231. [DOI] [PubMed] [Google Scholar]
- 100. Lovicu FJ, Shin EH, McAvoy JW.. Fibrosis in the lens. Sprouty regulation of TGFβ-signaling prevents lens EMT leading to cataract. Exp Eye Res. 2016; 142: 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Shirai K, Tanaka SI, Lovicu FJ, Saika S.. The murine lens: a model to investigate in vivo epithelial-mesenchymal transition. Dev Dyn. 2018; 247: 340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Minkowski JS, Bartels SP, Delori FC, Lee SR, Kenyon KR, Neufeld AH. Corneal endothelial function and structure following cryo-injury in the rabbit. Invest Ophthalmol Vis Sci. 1984; 25: 1416–1425. [PubMed] [Google Scholar]
- 103. Ali M, Raghunathan V, Li JY, Murphy CJ, Thomasy SM.. Biomechanical relationships between the corneal endothelium and Descemet's membrane. Exp Eye Res. 2016; 152: 57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Matsuda M, Sawa M, Edelhauser HF, Bartels SP, Neufeld AH, Kenyon KR. Cellular migration and morphology in corneal endothelial wound repair. Invest Ophthalmol Vis Sci. 1985; 26: 443–449. [PubMed] [Google Scholar]
- 105. Chi HH, Teng CC, Katzin HM.. Healing process in the mechanical denudation of the corneal endothelium. Am J Ophthalmol. 1960; 49: 693–703. [PubMed] [Google Scholar]
- 106. Maurice D, Perlman M. Permanent destruction of the corneal endothelium in rabbits. Invest Ophthalmol Vis Sci. 1977; 16: 646–649. [PubMed] [Google Scholar]
- 107. Ahuja Y, Malihi M, Sit AJ.. Delayed-onset symptomatic hyphema after ab interno trabeculotomy surgery. Am J Ophthalmol. 2012; 154: 476–480.e472. [DOI] [PubMed] [Google Scholar]
- 108. Kassam F, Stechschulte AC, Stiles MC, Buhrmann R, Damji KF.. Delayed spontaneous hyphemas after Ab interno trabeculectomy surgery for glaucoma. J Glaucoma. 2014; 23: 660–661. [DOI] [PubMed] [Google Scholar]