

Abstract
Sphingosine-1-phosphate (S1P) produced by sphingosine kinases (SPHK1 and SPHK2) is a signaling molecule involved in cell proliferation and formation of cellular junctions. In this study, we characterized the retinas of Sphk1 knockout (KO) mice by electron microscopy and immunocytochemistry. We also tested cultured Müller glia for their response to S1P. We found that S1P plays an important role in retinal and retinal pigment epithelial (RPE) structural integrity in aging mice. Ultrastructural analysis of Sphk1 KO mouse retinas aged to 15 months or raised with moderate light stress revealed a degenerated outer limiting membrane. This membrane is formed by adherens junctions between neighboring Müller glia and photoreceptor cells. We also show that Sphk1 KO mice have reduced retinal function in mice raised with moderate light stress. In vitro assays revealed that exogenous S1P modulated cytoskeletal rearrangement and increased N-cadherin production in human Müller glia cells. Aged mice also had morphological degeneration of the RPE, as well as increased lipid storage vacuoles and undigested phagosomes reminiscent of RPE in age related macular degeneration. These findings show that SPHK1 and S1P play a vital role in the structural maintenance of the mammalian retina and retinal pigmented epithelium by supporting the formation of adherens junctions.
Keywords: Retina, outer limiting membrane, Sphingosine kinase, sphingosine-1-phosphate, retinal degeneration, N-cadherin
Introduction
The mammalian retina is a highly specialized sensory input tissue that is part of the central nervous system and is organized in laminated layers of neurons. Its proper development and maintenance throughout life is required for accurate vision [1]. Distortion of the integrity of retinal layers is associated with vision loss in many human blinding diseases, including age-related macular degeneration (AMD) and diabetic retinopathy (DR). An important structure of the mammalian retina is the outer limiting membrane (OLM) also known as the external limiting membrane (ELM). It is formed by the apical processes of Müller cells which attach to one another and to the inner segments of the photoreceptor cells via continuous heterotypic tight-like adherens junctions (AJ) [2]. The OLM provides mechanical support and maintains homeostasis of photoreceptors in the outer retinal layer, thus the AJs that are formed in the OLM play a critical role in maintaining the architecture of the retina [1,3–5]. Integrity of the OLM is compromised in many human blinding diseases and the assessment of OLM/ELM structure with optical coherence tomography (OCT) has become a routine diagnostic and prognostic measure for AMD, geographic atrophy, macular hole, diabetic macular edema, retinal detachment, and for acute posterior multifocal placoid pigment epitheliopathy [6–12].
In the retina, the blood-retinal barrier controls the exchange of molecules and fluids between the retina and the circulatory system. In the inner retina, this barrier is formed at the retinal vessels surrounded by pericytes and glial cells, and in the outer retina by a single layer of retinal pigment epithelial (RPE) cells. The tight junctions (TJ) located between endothelial cells in retinal vessels and in RPE cells control entrance of circulating molecules and cells into the retina and thus confers immune privilege to the eye. Like in other epithelial cells, junctions in RPE cells are composed by TJ, AJ, and desmosomes [13].
The sphingolipid sphingosine-1-phosphate (S1P) has emerged as an important signaling molecule for establishing junctional complexes in the vascular and neuronal systems, mainly by activating junctional complexes to form adherens junctions [14]. Sphingolipid signaling is involved in a multitude of cellular activities including cell migration, cellular adhesion, proliferation, and cell death [15–18]. S1P, one of the most studied sphingolipid signaling molecules, is synthesized from sphingosine by two known kinases, sphingosine kinase 1 and 2 (SPHK1 and SPHK2). S1P is the ligand for five G-protein coupled receptors (S1P1-5) and mediates cell growth, adhesion, and migration by activating GTPases like Rac and Rho. S1P can induce the activation and subsequent translocation to the plasma membrane of both Rac and Rho in endothelial cells through activation of the Gi subunit by any of the five S1P receptors [19]. Signaling through the S1P1-5 receptors also releases calcium stores within the cell, aiding in the activation of Rac signaling and the stabilization of adherens proteins [20]. Indeed, S1P signaling via S1P1 is essential during development of flow-competent vessels by the activation of Rac followed by increased vascular endothelial cadherins at cell junctions [19,21]. S1p1 knockout mice die in utero because blood vessels fail to form proper barriers and leak [21]. The same is true in double knockout mice for both sphingosine kinases, resulting in a lack of S1P production and death in utero due to underdeveloped blood vessels. These mice also fail to develop a properly functioning nervous system [22].
Breakdown or loss of the integrity of retinal barriers, either at endothelial junctions, RPE, or at the OLM are associated with major retinal diseases, such as diabetic macular edema, AMD, macular hole, etc. Many of these diseases are age-related. However, the role of S1P and its signaling roles in retinal structural integrity and controlling the permeability barrier have not yet been studied. Here, we studied retinal permeability barrier components in Sphk1 knockout (KO) mice. Sphk1 KO mice are known to have a ~50% reduction in S1P levels in the plasma [22], however the magnitude of S1P reduction in retinal tissue was not known before this study. Our results indicate that S1P generated by SPHK1 is reduced by ~50% in the retina too. S1P is necessary in the maintenance of AJs in the OLM and RPE in aged mice, however, the tight junctions in these tissues were maintained. Using human MIO-M1 Müller glia cells, we further show that S1P signaling in retinal Müller cells is important in activating the Rac1 pathway to increase N-cadherin production and rearrangement of the actin cytoskeleton for building junctional complexes.
Materials and Methods
Animal care
All procedures were performed according to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and the University of Oklahoma Health Sciences Center Guidelines for Animals in Research. Wild-type (WT) and Sphk1 global knockout (KO) mice (BALB/c background) were generated from pigmented mice received from Dr. Richard L. Proia (NIDDK, Bethesda, MD). The mice were born and raised in the Dean A. McGee Eye Institute vivarium and maintained from birth under dim cyclic light (5-10 lux, 12 h on/off, 7 a.m. to 7 p.m. CST) to protect the BALB/c mice from light induced retinal degeneration or stress. All WT and Sphk1 and Sphk2 KO mice in the C57BL/6J background were born and raised in the Dean A. McGee Eye Institute vivarium and maintained from birth under approved cyclic light conditions (30-60 lux, 12 h on/off, 7 a.m. to 7 p.m. CST). All mice were genotyped for the retinal degeneration mutations rd1 and rd8 to ensure they were not present. All procedures, tissue harvests, and methods of euthanasia for mice were reviewed and approved by the OUHSC Institutional Animal Care and Use Committee (OUHSC IACUC). Mice were euthanized by carbon dioxide asphyxiation before harvesting the eye or retinal tissues. Sphk1 and Sphk2 KO mice were gifts from Dr. Richard L. Proia (NIDDK, Bethesda, MD).
Experimental methods for our albino Sphk1 WT and KO mouse models involved rearing in either dim cyclic light, (5-10 lux, 12 h on/off, 7 a.m. to 7 p.m. CST), or bright cyclic light, (100-150 lux, 12 h on/off, 7 a.m. to 7 p.m. CST). The Dean A. McGee Eye Institute vivarium is divided into two separate areas, one allowing bright light exposure (100-150 lux) to mouse colonies and the other allowing dimmed light exposure (5-10 lux) to mainly albino mice who are at risk for environmental retinal degeneration. KO and WT littermates were placed in bright light, after weaning at 21 days of age, and allowed to grow to 3 months and 6 months of age to induce the effects of environmental retinal degeneration in our albino line.
Histology
After euthanasia by carbon dioxide asphyxiation, mouse eyes were enucleated immediately, placed in fixative (Prefer; Anatech LTD, Battle Creek, MI), and embedded in paraffin for light microscope evaluation of retinal structure. Sections of 5 μm were cut along the vertical meridian through the optic nerve and stained with hematoxylin and eosin (H and E). The thickness of the outer nuclear layer (ONL) was measured every 0.2-0.3μm of the retina starting from the optic nerve head (ONH) and moving out to the inferior and superior ora serrata. Measured points were plotted as a spider diagram in GraphPad Prism 7.03. Unstained slides were also obtained to perform immunocytochemistry analysis.
Extraction and analysis of sphingolipids
Animals were anesthetized intraperitoneally with ketamine (100 mg/kg body weight) and xylazine (5 mg/kg body weight) and perfused with 1× PBS via the left ventricle of the heart. The eyes were enucleated, and the retinas were dissected out and flash frozen in liquid nitrogen. Five to six retinas were combined together to make one pooled sample (n=1) with a total of an n=4 used for analysis. Plasma from each individual animal was used as an independent sample. Extraction and LC/MS were performed using established procedures at Virginia Commonwealth University, as published earlier [23–25].
Transmission Electron Microscopy (TEM)
Animals were anesthetized, 7-9 hours after light onset in the animal facility, with ketamine (100 mg/kg body weight) and xylazine (5 mg/kg body weight). Mice were then perfused via the left ventricle of the heart with 2% electron microscopy (EM) grade paraformaldehyde and 2% EM grade glutaraldehyde made in cacodylate buffer (final concentration of 0.1M cacodylate salt with 0.1 μM CaCl2 at pH 7.3). The eyes were carefully dissected out of the skull and cauterized on the superior side of the globe. They were then placed immediately into the same fixative and incubated on an orbital shaker overnight at room temperature. The fix was carefully washed out with three cacodylate buffer rinses, and a secondary fixation of 1% osmium tetroxide made in 0.1 M cacodylate buffer was applied for 90 minutes at room temperature. The osmium was then washed out, and the samples were bisected from the superior to the inferior side of the globe. These two halves were then dehydrated through an ethanol gradient and finally propylene oxide. After dehydration, the samples were infiltrated with an epon-araldite mixture and cured with heat. Blocks were sectioned at 100 nm on a Leica UC6 Ultracut and put on 75 Cu mesh grids coated with formvar. These were stained with Sato’s lead and uranyl acetate to enhance contrast and then imaged on a Hitachi H-7600 transmission electron microscope at the Oklahoma Medical Research Foundation.
Retinal flat mounts
After euthanasia, the eyes were enucleated and submerged in 4% PFA. After 30 minutes the eyes were dissected and the retina was removed. The retinas were permeablized in 1% Triton X-100 overnight at 4°C. The following day the retinas were probed with primary antibodies and incubated overnight at 4°C. The samples were then washed and counterstained with secondary antibodies. The samples were then flat-mounted on a slide using micro-scissors to cut the retina into a “four-leaf clover” so that it lays flat. These were then coverslipped in 50% glycerol mounting media and imaged on an Olympus FV1200 confocal microscope. Antibodies: 10 μg/ml Armenian hamster anti-CD31 (Developmental Studies Hybridoma Bank [DSHB], 2H8 was deposited to the DSHB by Bogen, S.A. [DSHB Hybridoma Product 2H8]); 1:100 guinea pig anti-NG2 (a gift from Dr. William B. Stallcup, Sanford-Burham Medical Research Institute, La Jolla, CA); 1:200 mouse anti- alpha smooth muscle actin conjugated to Cy3 (Sigma-Aldrich).
Retinopathy of Prematurity Model (ROP)
Pregnant dams were monitored for the birth of pups. At P7, the mother and pups were placed in an oxygen chamber with 80% concentration of oxygen. The mothers were replaced every other day with nursing surrogates. At P12, the pups were removed back to an ambient oxygen concentration and placed with a new surrogate mother. At P17, the pups were euthanized by carbon dioxide followed by decapitation. The eyes were enucleated, and the retinas were dissected for immunohistochemistry. Retinas were stained for CD31 and flat-mounted for image acquisition on an Olympus MVX microscope. Multiple images were stitched together in Photoshop and then analyzed for areas of obliteration and neovascularization as described previously [26].
Electroretinography (ERG)
Flash ERGs were recorded with the Diagnosys Espion E2 ERG system (Diagnosys LLC, Lowell, MA). Mice were maintained in total darkness overnight and prepared for ERG recording under dim red light. They were anesthetized via intraperitoneal injection of ketamine (100 mg/kg body weight) and xylazine (5 mg/kg body weight). One drop of 10% (v/v) phenylephrine was applied to the cornea to dilate the pupil, and one drop of 0.5% (v/v) proparacaine HC1 was applied for local anesthesia. Mice were kept on a heating pad at 37°C during recordings. A gold electrode was placed on the cornea, a reference electrode was positioned in the mouth, a ground electrode was placed in the tail, and mice were placed inside a Ganzfeld illuminating sphere. Responses to light were differentially amplified, averaged, and stored. For the assessment of rod photoreceptor function (scotopic ERG), five strobe flash stimuli were presented at flash intensities at −2.3, −1.3, 0.7, 2.7, and 3.3 log candela (cd)·s/m2. The amplitude of the a-wave was measured from the pre-stimulus baseline to the a-wave trough. The amplitude of the b-wave was measured from the trough of the a-wave to the peak of the b-wave. For the evaluation of cone function (photopic ERG), a strobe flash stimulus (3.3 log cd·s/m2) was presented to dilated, light-adapted (5 minutes at 1.7 log cd·s/m2) mice. Photopic, blue and green cone, and 3, 10, 20, and 30 Hz flickers were used for cone functional analysis. The amplitude of the cone b-wave was measured from the trough of the a-wave to the peak of the b-wave.
Cell culture
Moorfields/Institute of Ophthalmology-Müller 1 (MIO-M1) cells [27], provided by G. Astrid Limb (Institute of Ophthalmology, University College London, London, UK) were seeded in 10 cm plates at 1×106 cells/plate and maintained in 1:1 DMEM/F12 (Gibco) supplemented with 10% fetal bovine serum (Gibco), 0.05 mM glutamate (ThermoFisher), and 1% penicillin/streptomycin antibiotics (ThermoFisher) [28]. Whole cell extracts were prepared using octyl-glucoside lysis buffer with protease inhibitors (Roche). Protein concentrations were determined by BCA assay (Thermo).
Cell culture treatments
S1P: S1P was prepared according to manufacturer’s recommendation (Avanti). It was resuspended in 4 mg/mL fatty-acid free BSA. Cells were treated with either 0.5 μM or 1 μM of this S1P solution.
BSA: 4 mg/mL stock of fatty-acid free BSA (Sigma-Aldrich) in FBS free DMEM/F12 media. Cells were treated with an equal concentration of BSA as in 0.5 μM and 1 μM S1P that was given to each cell culture well of a 6-well plate.
ML141: Cdc42/Rac1 GTPase inhibitor (EMD Millipore, 217708) was dissolved in 100% DMSO. Cells were treated with 10 μM ML141 inhibitor diluted in DMSO, and controls were treated with the same volume of DMSO.
All treatments were performed one hour after serum starvation, and the cells remained in serum-free media for the duration of the treatment.
Immunohisto- and Immunocytochemistry
Paraffin slides were deparaffinized and then antigens were retrieved in 1× citrate buffer heated for 10 minutes in a 90 °C water bath. The slides were then allowed to return to room temperature for 20 minutes. The sections were blocked for one hour at room temperature with 10% horse serum (Jackson ImmunoResearch Laboratories). Primary antibodies were then applied and incubated overnight at 4 °C in a humid chamber. (Primary antibodies: glutamine synthase −2 μg/mL from Millipore MAB302; N-cadherin – 10 μg/mL from Developmental Studies Hybridoma Bank [DSHB] MNCD2, MNCD2 was deposited to the DSHB by Takeichi, M. / Matsunami, H. [DSHB Hybridoma Product MNCD2]; peanut agglutinin [PNA] – 50 μg/mL from Vector Labs RL-1072; glial fibrillary acidic protein [GFAP] 5 μg/mL– Dako 70182. All primary antibodies were diluted in 1× phosphate buffered solution [PBS]). The following day the slides were rinsed with 1× PBS and secondary antibodies were applied. (PNA was added at this step because it was conjugated to rhodamine.) (Secondary antibodies: Alexa Fluor donkey anti-rat 488 6 μg/mL; Alexa Fluor donkey anti-mouse 647 3 μg/mL; Alexa Fluor donkey anti-rabbit 568 6 μg/mL, from Jackson ImmunoResearch Laboratories). The incubation for the secondary antibodies was two hours at room temperature, and samples were then washed and briefly stained with 1 μg/mL 4’,6-Diamidine-2’-phenylindole dihydrochloride (DAPI). The samples were mounted with Prolong Diamond mounting media (Invitrogen), and a coverslip was applied. Slides were imaged on an Olympus FV1200 Confocal microscope.
Immunocytochemistry for MIO-M1 cells was performed following a similar protocol. Cells were grown to 80% confluence on No1.5 coverslips. They were fixed with 2% paraformaldehyde for 15 minutes. The coverslips were blocked with 2% BSA with 0.01% Triton X-100 for one hour at room temperature. N-cadherin antibody, 10 μg/mL made in 2% BSA, was applied and the coverslips were incubated overnight at 4 °C in a humid chamber. The coverslips were then washed with PBS, and secondary antibody was added (6 μg/mL Alexa Fluor donkey anti-rat 488 and 1 U/100μL phalloidin-594 [Invitrogen, A12381]). The samples were incubated with secondary antibodies for one hour at room temperature. The samples were washed and then stained with DAPI, washed again and finally coverslipped in Prolong Diamond mounting media. Cells were imaged on an Olympus FV1200 Confocal microscope or DeltaVision OMX super-resolution imaging system at the Oklahoma Medical Research Foundation.
Immunoprecipitation
Activated Rac1 detection was performed using an Activated Rac1/Cdc42 Pulldown Assay (EMD Millipore, Rac1/Cdc42 Activation Assay Kit 17-441). MIO-M1 cells were grown to 85-90% confluency, serum-starved for one hour, and then treated with 0.5 μM S1P, or the equivalent volume of BSA as a vehicle control, for 12 hours. Positive and negative control cells received no treatment of BSA or S1P. All cells were lysed with ice-cold Mg2+ Lysis buffer with protease inhibitors (Roche). Positive and negative control cell extracts for Rac1 activation received GTPγS (100 μM) or GDP (1 mM), respectively. Samples were then incubated for 30 minutes. Activated Rac1/Cdc42 was pulled down with PAK-1-PBD agarose beads (activated GTP-Rac1 binds to the PAK-1 GTPase binding domain). Immunoprecipitated proteins were then resuspended in Laemmli buffer, separated by SDS-PAGE, transferred to nitrocellulose membrans, and probed by western blot with anti-Rac1 and anti-Cdc42 antibodies. Rac1 and Cdc42 were visualized using the appropriate HRP-conjugated secondary antibody. Images of the gel were quantified in FIJI/Image-J [29] by measuring the intensity of the bands and calculating the area under the intensity histogram.
Western blot
MIO-M1 whole cell extracts were prepared via 30 min incubation in octyl glucoside lysis buffer. Protein concentration was determined via BCA assay. Samples were then diluted in Laemmli buffer and 30 μg denatured protein was separted on 4-20% Tris-glycine gradient gels (Novex) using SDS-PAGE. Proteins were transferred to nitrocellulose membranes (Bio-Rad) andstained with anti-N-cadherin rat monoclonal (DSHB MNCD2; 1 μg/mL) and vimentin mouse monoclonal (Abeam, ab8978; 1:1000) antibodies. Membranes were visualized using the appropriate HRP-conjugated secondary antibody and quantified using the Kodak In Vivo FX Pro imaging system (Carestream Health Inc., Rochester, NY).
Statistics
All statistics were done using GraphPad Prism version 7.03 for Windows, GraphPad Software, La Jolla California USA. Column data were assessed for normality by a Shapiro-Wilk normality test to determine whether to use two-tailed t-tests or Mann-Whitney two-tailed tests (α=0.05). Grouped data was evaluated by two-way ANOVA (α=0.05) followed by Bonferroni’s correction for multiple comparisons.
Results
Sphingolipid composition in Sphk1 KO mouse retinas
Structure, function, and retinal composition of sphingolipids in Sphk1 KO mice have not previously been studied. We obtained Sphk1 global knockout (Sphk1 KO) mice from Dr. Richard L. Proia (NIDDK, Bethesda, MD). The mice were on C57BL/6 pigmented background [22]. We generated an albino line of these mice by 7 generation backcrossing with BALB/c mice. Both pigmented and albino mice are used in this study. We used LC/MS to measure retinal and plasma sphingolipid species from adult albino mice raised in standard dim light conditions. Previous studies have shown that Sphk1 KO mice have about a 50% reduction in plasma concentrations of S1P [22]; we tested our newly generated BALB/c background Sphk1 KO mice and found that plasma S1P was reduced by 60% and dihydroS1P (dhS1P) was reduced by 75% (Figure 1 A, p<0.0001, n=5). We found that the retinas of Sphk1 KO mice had a significant increase in sphingosine (Sph) concentration when compared to the littermate wild type (WT) mice (Figure 1 B, p=0.01, n=4). The retinal concentrations of S1P and dhS1P were measured by combining 5-6 retinas to make one pooled sample, for a total of n=4 individual pooled samples. We found that S1P was reduced by 50% (Figure 1 B, p=0.0004, n=4) and dhS1P was reduced by 40% (Figure 1 B, p=0.002, n=4). Ceramide (Cer), monohexosylceramides (HexCer), and sphingomyelin (SM) were also analyzed. Total Cer and SM concentrations were significantly increased in retinas from Sphk1 KO mice compared to the WT mice (Table 1 and Figure 1 C, n=4, *p=0.04 and *p=0.02 respectively). The overall composition of these sphingolipids, as calculated by mole %, remained unchanged for all species (Supplemental Figure 1 A–C). These findings show that the deletion of Sphk1 affects the retinal composition of sphingolipids by reducing S1P and accumulating Sph, Cer, and SM.
Figure 1.
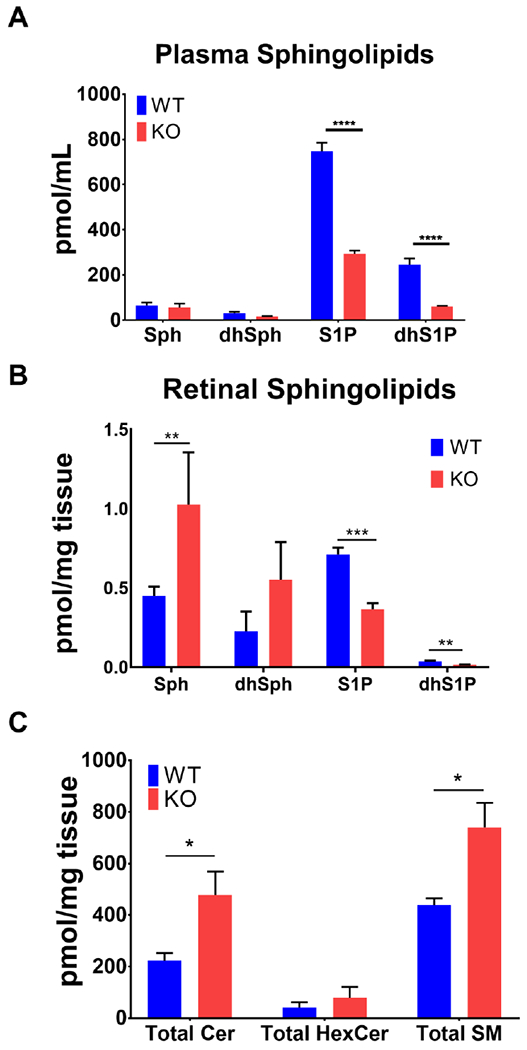
Sphk1 KO mouse retinas have increased sphingosine, ceramide, and sphingomyelin profiles, while there is a significant loss of circulating S1P and dhS1P. Retinas and plasma were harvested from six-month-old mice raised at 5-10 lux and analyzed by LC/MS for pmol/mg wet weight tissue or mL of plasma. A) Circulating S1P (p<0.0001) and dhS1P (p=0.0002) are reduced in the Sphk1 KO mice. B) Significant increases in sphingosine (Sph, p=0.01) were measured in Sphk1 KO mouse retinas. S1P (p=0.0004) and dhS1P (p=0.002) were significantly reduced in retinal tissue from the Sphk1 KO mice. C) Total ceramides (Cer, p=0.02) and sphingomyelin (SM, p=0.04) were significantly increased in Sphk1 KO retinas. For all, two-tailed Student’s t-test, α=0.05, error bars are SEM.
Table 1). Ceramide, HexCer, and SM species from WT and Sphk1 KO mouse retinas.
Retinas were analyzed by LC/MS for individual species of ceramides, HexCer, and sphingomyelin; pmol/mg wet weight tissue.
| Sphingolipid Species | Ceramides (pmol/mg tissue) | Monohexosylceramides (pmol/mg tissue) | Sphingomyelin (pmol/mg tissue) | |||
|---|---|---|---|---|---|---|
| WT | KO | WT | KO | WT | KO | |
| 14:0 | 1.4 ± 0.28 | 2.9 ± 0.69 | 0.30 ± 0.00* | 0.50 ± 0.070* | 7.0 ± 1.2 | 9.0 ± 0.71 |
| 16:0 | 68 ± 11 | 150 ± 34 | 1.9 ± 0.13* | 4.4 ± 0.75* | 120 ± 12 | 170 ± 23 |
| 18:1 | 2.2 ± 0.41 | 6.4 ± 1.8 | 0.03 ± 0.030* | 0.10 ± 0.00* | 25 ± 4.1* | 57 ± 8.1* |
| 18:0 | 79 ± 5.1* | 160 ± 31* | 6.0 ± 1.8 | 13 ± 5.8 | 93 ± 6.7 | 170 ± 40 |
| 20:0 | 27 ± 8.1 | 59 ± 14 | 2.3 ± 0.70 | 3.8 ± 1.2 | 75 ± 11 | 140 ± 30 |
| 22:0 | 16 ± 3.4* | 34 ± 5.7* | 7.3 ± 3.9 | 11 ± 6.6 | 59 ± 4.6* | 96 ± 14* |
| 24:1 | 21 ± 1.7* | 44 ± 8.5* | 12 ± 7.4 | 26 ± 19 | 37 ± 5.1 | 62 ± 13 |
| 24:0 | 6.6 ± 0.56* | 16 ± 3.7* | 11 ± 6.0 | 19 ± 11 | 21 ± 3.5 | 37 ± 8.8 |
| 26:1 | 0.25 ± 0.90 | 0.40 ± 0.11 | 0.45 ± 0.25 | 0.68 ± 0.38 | 0.65 ± 0.090* | 1.4 ± 0.27* |
| 26:0 | 1.4 ± 0.88 | 1.8 ± 0.72 | 0.20 ± 0.070 | 0.18 ± 0.050 | 0.45 ± 0.030 | 0.75 ± 0.13 |
| Total | 220 ± 30* | 480 ± 91* | 41 ± 20 | 79 ± 43 | 440 ± 26* | 740 ± 96* |
Data presented as mean ± SEM, two-tailed Student’s t-test n=4,
p= >0.05-0.01.
Ultrastructural analysis of Sphk1 KO retinas
An advantage to studying mice from the BALB/c genetic background is the ability to stress the retina with light in a titratable manner. These mice are normally housed in dim light conditions (5-10 lux) to protect the retina from stress induced degeneration. However, raising BALB/c background mice at ambient light conditions (100-150 lux) adds stress to the retina, though not enough to initiate degeneration. We analyzed the retinal structure of Sphk1 KO and WT mice raised in dim and ambient light conditions by light microscopy in paraffin sections stained by hematoxylin and eosin and did not observe gross morphological differences nor detect any significant differences in outer nuclear layer thickness, indicating no loss of photoreceptors (Supplemental Figure 2A and B). To elucidate any possible morphological differences between Sphk1 KO and WT mice, we conducted ultrastructural studies using transmission electron microscopy (TEM) on mice raised in the dim and ambient light conditions. Interestingly, we observed several unique phenotypic characteristics in ambient light-raised Sphk1 KO mice at 6 months of age: in Sphk1 KO mice, the outer segments of the photoreceptors were disorganized and many were displaced into the inner segment region of the tissue (Figure 2 B, arrows, n=3) when compared to WT retinas (Figure 2 A, n=3). The integrity of the OLM was also compromised in Sphk1 KO mouse retinas (Figure 2 a and b, n=3). The OLM is made up of adherens junctions between neighboring Müller glia and by photoreceptors and Müller glia [30]. In TEM micrographs, these are electron-dense lines that run perpendicular to the cilia body of the photoreceptors. Sphk1 KO animals lost this structure, and the electron-dense junctions appeared punctate and parallel to the cilia body (Figure 2 A and B, and enlargements a and b). However, in 6-month-old WT mice and Sphk1 KO mice raised in 5-10 lux light, no morphological differences were found at the OLM or the inner and outer photoreceptor segments (Figure 2 C and D, and enlargements c and d, n=3). We then investigated if age would affect the morphology of the Sphk1 KO mouse retinas from mice raised at 5-10 lux. Sphk1 WT mice aged to 15 months at 5-10 lux maintained their ordered retinal structure and OLM integrity (Figure 2 E and e). However, retinas from 15-month-old Sphk1 KO mice raised at 5-10 lux showed a collapse of photoreceptor architecture, with inner and outer photoreceptor segments losing all stratification (Figure 2 F). These mice also had complete OLM loss (Figure 2 f), very similar to 6-month-old KO mice raised in ambient light. These results suggest that SPHK1 may be necessary for the maintenance of retinal structure and integrity as a function of light stress and with age.
Figure 2.
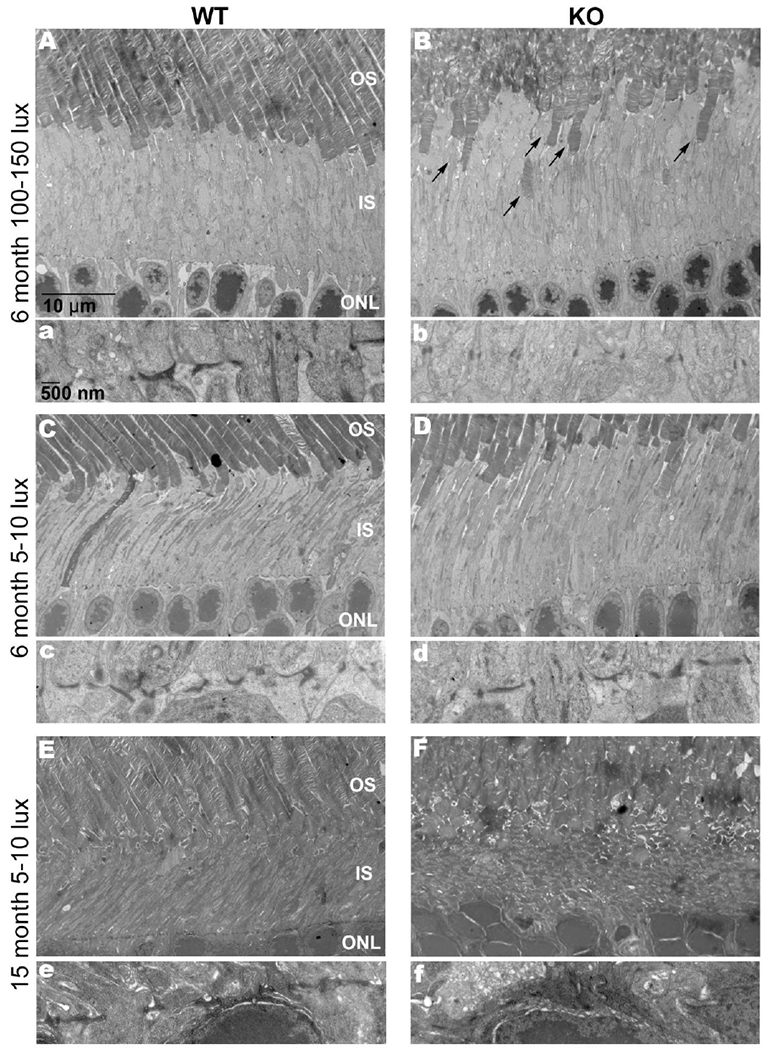
Sphk1 KO results in a loss of the OLM and structural organization of the outer and inner segments of the photoreceptors. Retinas were collected 7-9 hours after light onset and an analyzed by transmission electron microscopy. A) Retina from WT mouse raised at 100-150 lux for 6 months. Enlargement a) Shows broad electron-dense bands that make up the OLM composed of adherens proteins. B) Retina from KO mouse raised at 100-150 lux for 6 months. Outer segments become displaced (arrows). Enlargement b) shows small punctate adhesions of the OLM. C) Retina from WT mouse raised at 5-10 lux conditions for 6 months. Enlargement c) shows the OLM. D) Retina from KO mouse raised at 5-10 lux conditions for 6 months. Enlargement d) shows an intact OLM comparable to the WT retina. E) Retina from WT mouse raised at 5-10 lux conditions for 15 months. Enlargement e) shows an intact OLM at 15 months of age. F) Retina from KO mouse raised at 5-10 lux conditions for 15 months. The retinal architecture has become extremely disorganized. The inner and outer photoreceptor segments have collapsed and the OLM is absent. Enlargement e) shows an almost complete loss of adherens junctions that comprise the OLM. Representative images from n=3 mice for each light condition and time point. All scale bars are the same for each group unless otherwise noted.
Ultrastructural analysis of RPE in Sphk1 KO mice
The retinal pigmented epithelium (RPE) is composed of a layer of epithelial cells that help nourish the neural retina. The RPE is responsible for absorbing scattered light and recycling lipids, proteins, and metabolites needed for visual transduction [31–33]. To recycle components of photoreceptors, the RPE phagocytoses the most distal part of the outer segments each day after the presumptive onset of light [34]. Complications with the RPE are attributed to a number of vision loss related diseases [31]. We wanted to determine if Sphk1 deletion affected RPE structure. TEM images showed only subtle Sphk1 KO-dependent changes in the RPE of 6-month-old mice raised at 100-150 lux (Figure 3 A and B). Adherens junctions remained intact between adjacent RPE cells. The adherens junctions can be observed by first identifying the electron-dense tight junctions at the apical side of the RPE (black arrows in Figure 3), then the adherens junction can be traced through the cell body (white arrows, Figure 3). Sphk1 KO mouse RPE showed an increased number of lipid-filled vacuoles. These appear as monochromatic grey vacuoles inside the cell (asterisks, Figure 3 B). There was also a noticeable increase in multivesicular phagosomes within these cells (arrowhead, Figure 3 B). No differences in RPE morphology were observed between WT and Sphk1 KO mice raised at 5-10 lux for 6 months (Figure 3 C and D). However, in Sphk1 KO mice raised at 5-10 lux and aged to 15 months, the RPE underwent severe morphological changes (Figure 3 E and F). WT mouse RPE maintained adherens junctions between cells (white arrow, Figure 3 E), however, Sphk1 KO mouse RPE failed to maintain adherens junctions between neighboring cells (Figure 3 F). This loss led to the formation of large membrane-filled spaces between the RPE cells (double dagger, Figure 3 F). The basal membrane folds of the RPE also became highly disordered, leading to the formation of large membrane-filled vacuoles throughout the cell body. Large membrane-filled phagosomes were also observed (arrowhead, Figure 3 F), which were indicative of delayed and dysfunctional phagocytosis and lysosomal digestion. Interestingly, in Sphk1 KO mice raised with mild light stress or at an advanced age, the RPE showed signs of increased lipid storage problems and increased membrane-filled phagosomes, which is suggestive of RPE dysfunction. In preliminary assays, retinas from Sphk1 KO and WT mice raised to 6 months in dim light conditions were labelled with fluorescent anti-rhodopsin antibody. These mice were either subjected to dark adaptation overnight and harvested at 0900, or subjected to ambient light for 2 hours after overnight dark adaptation and harvested at 1100. These mice showed no obvious difference in phagolysosomal activity, though further investigation with mice raised in various light conditions and for longer periods is needed (Supplemental Figure 3).
Figure 3.
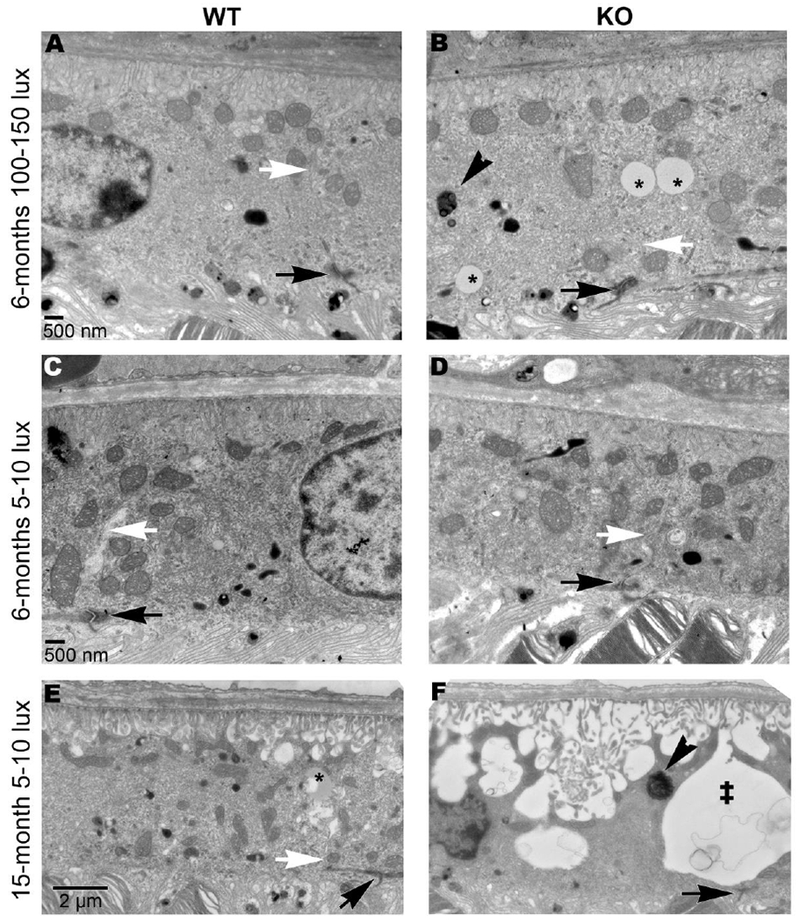
Sphk1 KO results in increased lipid storage vacuoles and adherens junction loss between adjoining RPE cells. A) RPE from a WT mouse raised at 100-150 lux for 6 months. Tight junctions (black arrow) remain intact, as do the adherens junctions along the cell body where the junction of cells can be traced (white arrow). B) RPE from a Sphk1 KO mouse raised at 100-150 lux for 6 months. The tight junctions (black arrow) and adherens junctions (white arrow) remain intact. However, there was a distinct increase in vacuoles containing lipids (asterisks) in these cells. An increase of electron-dense multivesicular phagosomes was also observed (black arrowhead). C and D) RPE from WT (C) and KO (D) mouse raised at 5-10 lux for 6 months. RPE from both genotypes have similar morphology with no change in tight junctions (black arrow) or adherens junctions (white arrow). E) RPE from WT mouse raised at 5-10 lux for 15 months. Adherens junctions remain intact. Lipid storage vesicles can be observed but were fewer in number (asterisks) compared to the KO mouse. F) RPE from KO mouse raised at 5-10 lux for 15 months. Tight junctions (black arrow) remain between RPE cells, but adherens junctions are lost, resulting in large membrane-filled gaps (‡) between RPE cells. Electron dense phagosomes (black arrowhead) are observed throughout the RPE. The basal membrane invaginations of the RPE are also highly disrupted and result in large open and membrane filled vacuoles in the cell. Representative images from n=3 mice. All scale bars are the same for each row unless otherwise noted.
Structural phenotype of Sphk1 KO retina on pigmented (C57BL/6J) background and comparison with Sphk2 KO.
As albino mice are sensitive to environmental light, and as we found differences in retinal phenotypes in albino Sphk1 KO mice raised in different light conditions, we further verified whether this retinal structural pathology is indeed due to Sphk1 deletion by subjecting Sphk1 KO mice on pigmented (C57BL/6J) background for ultrastructural analyses. One-year-old WT mice exhibited normal morphology of the retina (Figure 4 A-i). The OLM appeared as broad, electron-dense structures that orient perpendicular to the ONL (Figure 4 A-ii). The RPE also appeared healthy and had tightly sealed cell borders (Figure 4 A-iii). Like the Sphk1 KO mice in the BALB/c background, the pigmented Sphk1 KO line had a disruption of the OLM. The inner segments were also unorganized and had lost stratification (Figure 4 B-i and B-ii). While the RPE maintained some adherens junctions (white arrow, Figure 4 B-iii), there was a loss of junctions along the entire border. This can be observed in Figure 4 B-iii (double dagger) above and below the white arrow, where the space between neighboring RPE cells had swelled and filled with debris. The RPE of Sphk1 KO mice in the C57BL/6J background showed a considerable number of lingering phagosomes full of outer segment membranes compared to the WT mouse (asterisks, Figure 4 B-iv).
Figure 4.
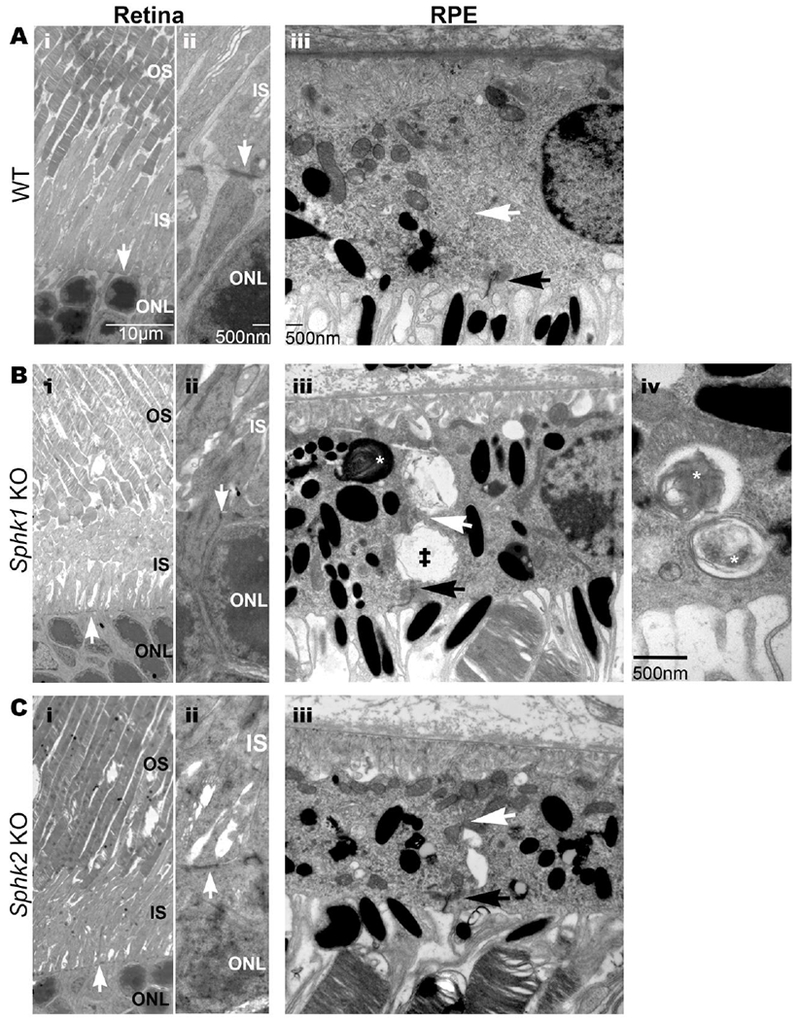
Loss of adherens junctions at the OLM and in the RPE is specific to Sphk1 KO and not Sphk2 KO. Electron microscopy was performed to compare WT, Sphk1 KO, and Sphk2 KO mouse retinas and RPE in 1-year-old mice in a C57B/6J background. A-i) WT mouse retinas show structured inner and outer segments of the photoreceptors. White arrow indicates the OLM. A-ii) Enlargement of the OLM shows broad electron-dense adherens contacts in WT mice (white arrow). A-iii) The RPE have intact tight junctions (black arrow) and adherens junctions (white arrow). B-i and -ii) Sphk1 KO mouse retinas had disordered photoreceptors starting with a collapse of the inner segments. The OLM had a loss of adherens junctions and an overall loss of the membrane. Electron dense adherens junctions (white arrow) were short punctate connections compared to the WT OLM. B-iii) The RPE have normal tight junctions (black arrow). Some adherens junctions remain (white arrow). However, degradation of these junctions is observed between RPE cells (double daggar). These areas are observed as large open spaces between two cells fdled with membrane debris. B-iv) Sphk1 KO mouse RPE cells also had an increase of phagosomes (asterisk). The enlargement shows that these phagosomes are full of undigested membranes. C-i and -ii) Sphk2 KO mice RPE have a similar phenotype as the WT mouse retinas. C-iii) Sphk2 KO mouse RPE close resembled WT RPE. The tight junction (black arrow) denotes the border of two RPE cells and the adherens junctions (white arrow) can be traced the length of the cell. Representative images from n=3 mice. All scale bars are the same for each column unless otherwise noted.
We wanted to determine if the loss of adherens junctions in the OLM and the RPE would also be observed in Sphk2 KO mice. While Sphk1 KO mice have been shown to have reduced S1P in serum/plasma and some tissues, Sphk2 KO mice have increased levels of S1P in serum/plasma [35–37,22]. We obtained Sphk2 KO mice from Dr. Richard Proia (NIDDK/NIH) in C57BL/6J background and analyzed their retinas at one year of age. As shown in Figure 4, retinal structure in Sphk2 KO mice remained unremarkable with a complete OLM and well-stratified inner and outer segments (Figure 4 C-i and C-ii). The RPE also remained healthy with intact cell borders and no increase in phagosomes (Figure 4 C-iii). These results suggest a specific role of SPHK1 in the maintenance of structural integrity of mammalian retina and RPE cells with aging.
Survey of endothelial junctions in Sphk1 KO retinas
Our data thus far suggests that the loss of Sphk1 and reduction of S1P levels compromised the AJs in retinal neuronal junctions. Though there is no report of endothelial barrier or vascular defect in Sphk1 KO mice retinas, we investigated retinal vascular integrity in aged (1-year-old) albino mice. As shown in Figure 5, we did not find any major defects in the vascular system (arteries, vessels, and capillaries) in the retinas of Sphk1 KO mice (Figure 5 A a-h). We also tested for any difference in the neovascular response of these mice in a retinopathy of prematurity (ROP) model by subjecting the newborn mice to 80% oxygen saturation. We did not observe any differences in neovascularization capabilities between the wild type and the Sphk1 KO mice (Figure 5 B and C). It should be noted that circulatory or plasma levels of S1P are decreased 60% in Sphk1 KO mice (Figure 1 A), which appears to be sufficient to form and maintain the endothelial junctions.
Figure 5.
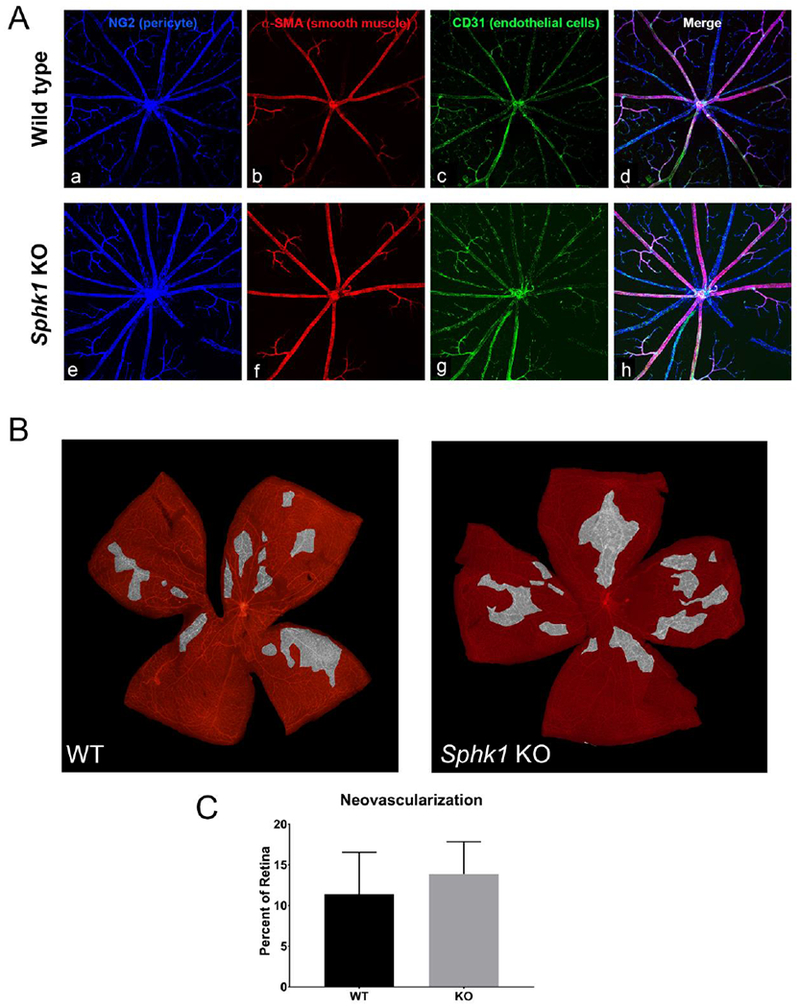
Vascular adherens junctions and neovascular potential are not affected by a deletion of Sphk1. A) Flat mounted retina from WT (a-d) and Sphk1 KO (e-h) mice were probed for pericyte coverage (a and e), smooth muscle cell coverage of arterioles (b and f), and endothelial cells (c and g). No differences were observed. B) Retinopathy of prematurity model for neovascularization showed no difference between WT or Sphk1 KO mouse retinas. Areas highlighted in grey have a high density of NV tufts, formed in response to the hypoxic environment after the mice are returned to normoxia for 5 days. C) Quantification of NV tuft areas after ROP show no major difference in NV between Sphk1 KO or WT. (n=7, two-tailed Student’s t-test, α=0.05, error bars are SD).
Immunohistochemical analysis of Sphk1 KO retinas
To further investigate the loss of the OLM as shown in Figure 6, we probed paraffin-embedded eyes from WT and Sphk1 KO 6-month-old mice raised at 100-150 lux and 15-month-old mice raised at 5-10 lux with a panel of antibodies against adherens junction proteins. N-cadherin staining was diminished at the OLM in KO mice raised for 6 months in 100-150 lux light conditions and KO mice raised for 15 months in 5-10 lux light conditions (Figure 6 A). The OLM in KO mice had large gaps where no N-cadherin staining was observed (arrows Figure 6 B). To determine if the loss of the OLM was due to Müller cell gliosis activation, we probed samples for glial fibrillary acidic protein (GFAP). Upon Müller cell activation, GFAP is upregulated throughout the entire cell and produces both cytokines and chemokines that increase inflammatory responses [1] which could account for the loss of the OLM. However, GFAP staining remained minimal and unchanged between KO and WT animals in all conditions tested (Figure 6 C). This indicated that the loss of the OLM is not due to activation of the Müller glia. Müller cells were also evaluated for glutamine synthetase (GS) to determine if there was a loss of the Müller cell population in KO retinas (Figure 6 D). While no substantial loss of Müller cells in KO mice was observed, the apical side of the Müller cell processes stained with less intensity than those in WT mice (Figure 6 E, all images were captured with exact same imaging parameters). There was no loss of GS staining observed at the basal Müller cell process that makes up the inner limiting membrane, which remained intact and unchanged between WT and KO animals (Figure 6 F). The reduction of GS staining at the apical Müller end-feet further confirms that there was a loss of the normal cellular architecture at the OLM and inner segments of the retina in Sphk1 KO mice.
Figure 6.
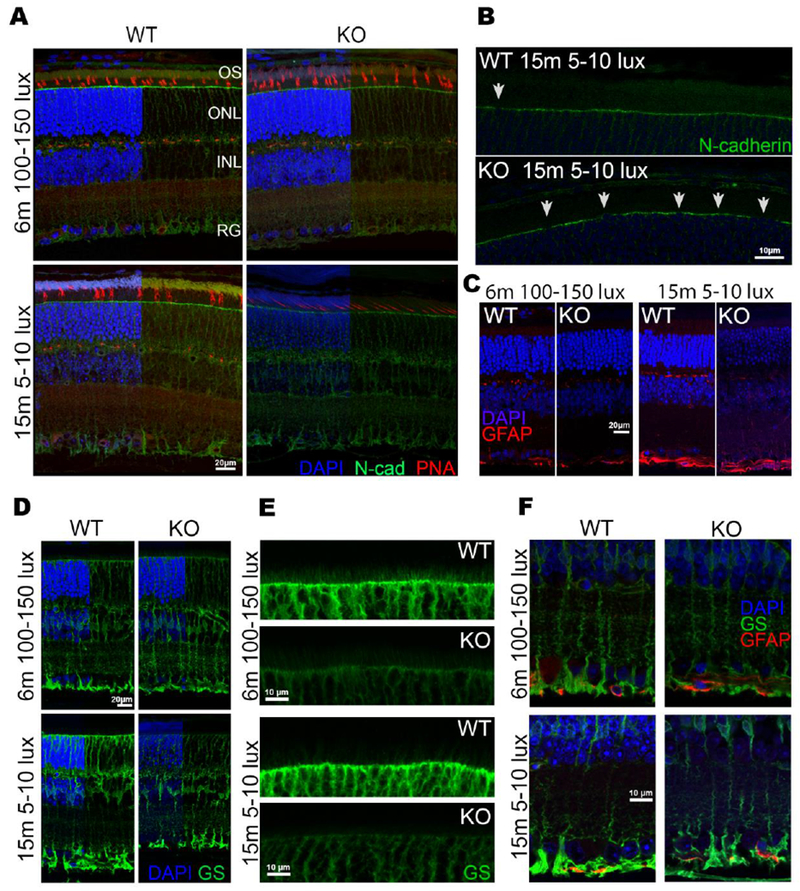
Sphk1 KO mouse retina shows a loss of N-cadherin at the OLM. A) Retinas from WT and KO mice raised in both the 5-10 lux and 100-150 lux light conditions were stained for the cone outer sheath with peanut agglutinin (PNA, red) and show no change between WT and KO mice. Retinas from both WT and KO mice were also stained for N-Cadherin. Gaps in N-cadherin coverage are seen in KO retinas at the OLM. B) Magnification (2× zoom) of N-cadherin staining at the OLM in 15-month-old mice raised at 5-10 lux. There is a significant loss of the normally continuous coverage of N-cadherin (white arrowheads) at the OLM in KO mice. C) Retinas stained for glial fibrillary acidic protein (GFAP) to detect glial activation of Müller cells show that they do not become activated, and thus are not the reason for the loss of the OLM. D) Müller glia stained for glutamine synthetase (GS) show no overall loss of Müller glia cell numbers, but a loss of staining at the apical side can be detected. E) 2× magnification of the apical Müller feet that make up the OLM shows a loss of staining and villous structures that reach into the inner segments in Sphk1 KO mice. F) Basal end feet of the Müller glia that make up the inner limiting membrane remain unaffected in the KO mouse retina. Representative images from n=3 mice.
Retinal function in Sphk1 KO mice
Because we detected a loss of the OLM by TEM and immunohistological analysis in Sphk1 KO mice, we wanted to see if there was also a loss of function in the retina. WT and Sphk1 global KO mice raised in bright light (100-150 lux) or dim light (5-10 lux) conditions were analyzed for retinal function by scotopic and photopic ERG at 3 months and 6 months of age. We found that 3-month-old Sphk1 KO mice raised in 100-150 lux light showed a significant a-wave reduction at flashes of 400 and 2000 cd.s/m2 (Figure 7 A, WT n=11, KO n=13; two-way ANOVA with Bonferroni’s correction for multiple comparisons; *p=0.03; **p=0.008). The a-wave is an indicator of photoreceptor cell hyperpolarization; reduction of a-wave amplitude indicates failure of photoreceptor signaling and loss of visual function. B-wave amplitudes in 3-month-old 100-150 lux Sphk1 KO mice were significantly reduced at 2000 cd.s/m2 flash intensity (Figure 7 B; WT n=11, KO n=13; two-way ANOVA with Bonferroni’s correction for multiple comparisons; *p=0.007). The b-wave is a measurement of post-synaptic depolarization of various cells after the initial hyperpolarization of the photoreceptor, and is commonly used as a clinical measure of retinal function. Similar results were seen in 6-month-old mice raised at 100-150 lux. Here we saw a significant reduction in a-wave amplitude in KO mice at 400 cd.s/m2 (Figure 6 C; WT n=9, KO n=10, two-way ANOVA with Bonferroni’s correction for multiple comparisons; *p=0.03). In 6-month-old mice raised at 100-150 lux, we found that the b-wave responses were not significantly different between KO and WT mice (Figure 7 D). Mice raised at 5-10 lux showed little difference between WT and Sphk1 KO mice in scotopic ERG response at either 3- or 6-months of age (Figure 7 E–H). Photopic ERG, which is used to measure cone photoreceptor function, indicated no differences between WT and Sphk1 KO mice at 3- or 6-months of age when mice were raised in either light condition (Supplemental Figure 4).
Figure 7.
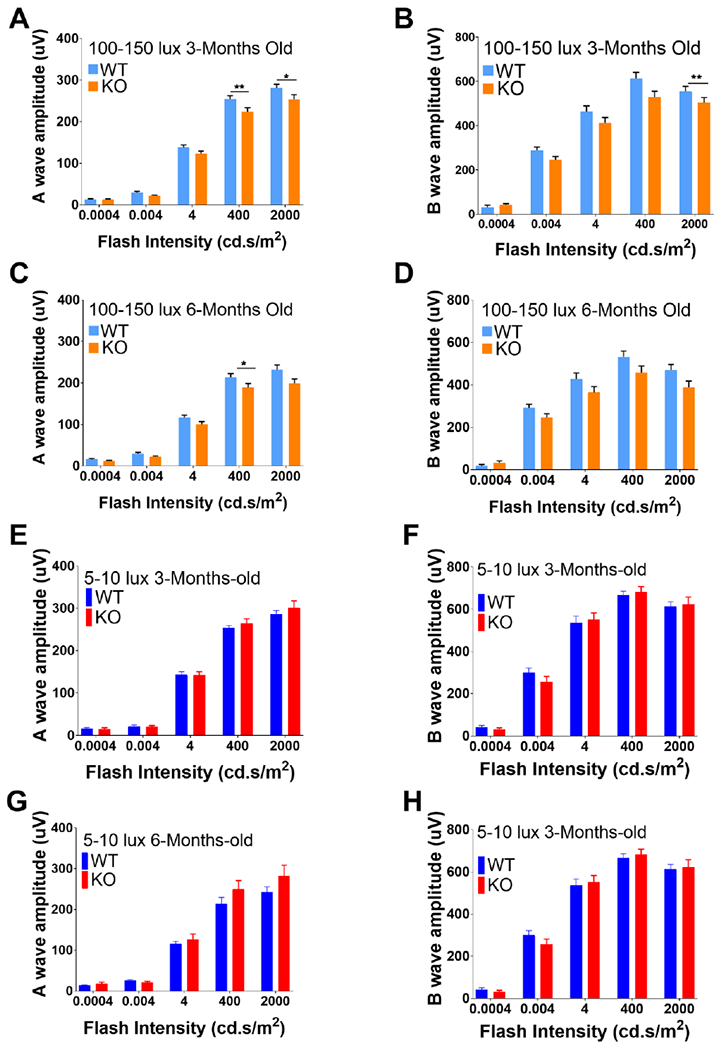
Scotopic ERG and ONL quantification for WT and Sphk1 KO mice. ERG responses for scotopic function were recorded for five flash stimuli at intensities of 0.0004, 0.004, 4, 400 and 2000 cd.sec/m2. A-D) Animals raised in 100-150 lux light conditions. A) A-wave responses in 3-month-old WT and KO mice raised (WT n=11, KO n=13; *p=0.03; **p=0.008). B) B-wave responses in 3-month-old mice (WT n=11, KO n=13 B; *p=0.007). C) A-wave responses in 6-month-old animals (WT n=9, KO n=10, *p=0.03). D) B-wave responses in 6-month-old animals (A-D: WT n=9, KO n=10.) E-H) Animals raised in 5-10 lux light conditions. E) A-wave responses for 3-month-old mice raised in dim light. F) B-wave responses for 3-month-old mice raised in dim light. G) A-wave responses for 6-month-old mice raised in dim light. H) B-wave responses for 6-month-old mice raised in dim light (E-H n=5). Two-way ANOVA with Bonferroni’s correction for multiple comparisons, α=0.05. All error bars are SEM.
Effects of S1P in human Müller glia cells
Because there was a loss of N-cadherin at the OLM in Sphk1 KO retinas, we wanted to test if S1P could induce N-cadherin production in Müller cells. We used the human-derived MIO-M1 Müller cell culture model to test the effects of S1P on Müller glia. MIO-M1 cells were grown on glass coverslips and treated with 0.5 μM of S1P in fatty acid-free BSA after one hour of serum starvation [27]. At 30 minutes, the S1P-treated cells increased in size compared to cells treated with an equivalent volume of BSA control (Figure 8 A). By 4 hours, the cells treated with S1P were larger in size and had made extensive contacts with adjoining cells, unlike the BSA-treated cells (Figure 8 B). At 12 hours after the addition of S1P or BSA, N-cadherins were noticeably increased in the S1P-treated cells, with large concentrations at the filipodia (Figure 8 C). Using super-resolution structured illumination microscopy, we observed an increase of both actin filaments and N-cadherin puncta in S1P-treated cells (both 0.5 μM and 1 μM) (Figure 8 D). Especially interesting was that N-cadherin puncta were observed on actin-positive processes extending outside of the cells (Figure 8 D enlargements, arrows). These N-cadherin puncta were not seen in BSA controls, indicating that extracellular S1P signaling could also be needed to localize N-cadherin to specific environments in the cell.
Figure 8.

Human cultured Müller glia (MIO-M1) cells respond to S1P by increased actin assembly and increased N-cadherin production. A) MIO-M1 cells treated with 0.5 μM S1P and BSA that serves as a control. S1P, after 30 min of treatment, increases cell size and actin assembly. B) 4 hours after treatment, BSA treated cells remained spindly with small contacts between neighboring cells, while S1P-treated cells had broad contact with other cells. Representative images from n=3 replicates. C) N-cadherin was increased in MIO-M1 cells 12 hours after S1P treatment. Representative images from n=3 replicates. D) Structured illumination microscopy of MIO-M1 cells showed an increase of N-cadherin puncta in S1P treated cells. Villous actin positive protrusions from the cells were increased in S1P treated cells. Actin positive protrusions in S1P treated cells were decorated with N-cadherin puncta (arrows) seen in the enlargements below. N-cadherin decoration was not seen in the BSA treated cells. Representative images from n=3 replicates. E) Western blot of MIO-M1 cells probed for N-cadherin. There is a significant increase of N-cadherin after treatment with 1 uM S1P for 12 hours (3 replicates, normalized to vimentin loading control, two-tailed Student’s t-test, α=0.05, p=0.02, error bars SD).
N-cadherin was also measured by western blots in MI0-M1 cells 12 hours after treatment with 1 μM of S1P or an equal volume of BSA as a control. A significant increase in N-cadherin was seen after S1P treatment (Figure 8 E, 3 replicates, two-tailed Student’s t-test, α=0.050, p=0.02). These data show that S1P can signal Müller glia to increase the production of N-cadherins and dynamically change the actin cytoskeleton, allowing the cells to make broad contacts with one another. In preliminary assays to determine differences in retinal N-cadherin, we analyzed N-cadherin levels from the retinas of 6-month-old Sphk1 KO and WT mice raised in dim light conditions by western blot. We found no differences in N-cadherin levels, however, further investigation using mice raised in various light conditions and for longer periods of time is necessary (Supplemental Figure 5).
S1P activates RAC1 and CDC42 in MIO-M1 cells
A downstream target of S1P receptors 1, 2, and 3 is RAC1, a GTPase that is responsible for signaling actin polymerization and that can upregulate cadherin production once activated [38]. To determine if S1P activates RAC1 in Müller glia, we treated MIO-M1 cells with 0.5 μM S1P or an equal volume of BSA as a control. The cells were harvested and used for immunoprecipitation. The p21-activating kinases (PAK) are downstream effectors of both RAC1 and CDC42 (also a GTPase with similar function to RAC1) once they have been activated by GTP. PAK-1 agarose beads were used to bind GTP-bound RAC1 or CDC42. The bead complexes were then probed by western blot for RAC1 or CDC42. After a 12-hour incubation with 0.5 μM S1P, we observed a significant increase in the activated form of RAC1. Increased band intensity was seen in western blots for the S1P treated cells. Semi-quantification of the blots in FIJI confirmed the increase in activated RAC1 (Figure 9 A, 2 replicates on the same blot, two-tailed t-test, α=0.050, p=0.04). A substantial increase of activated CDC42 was also observed by western blot and after semi-quantification using FIJI in the S1P treated cells (Figure 9 B, 2 separate replicates).
Figure 9.
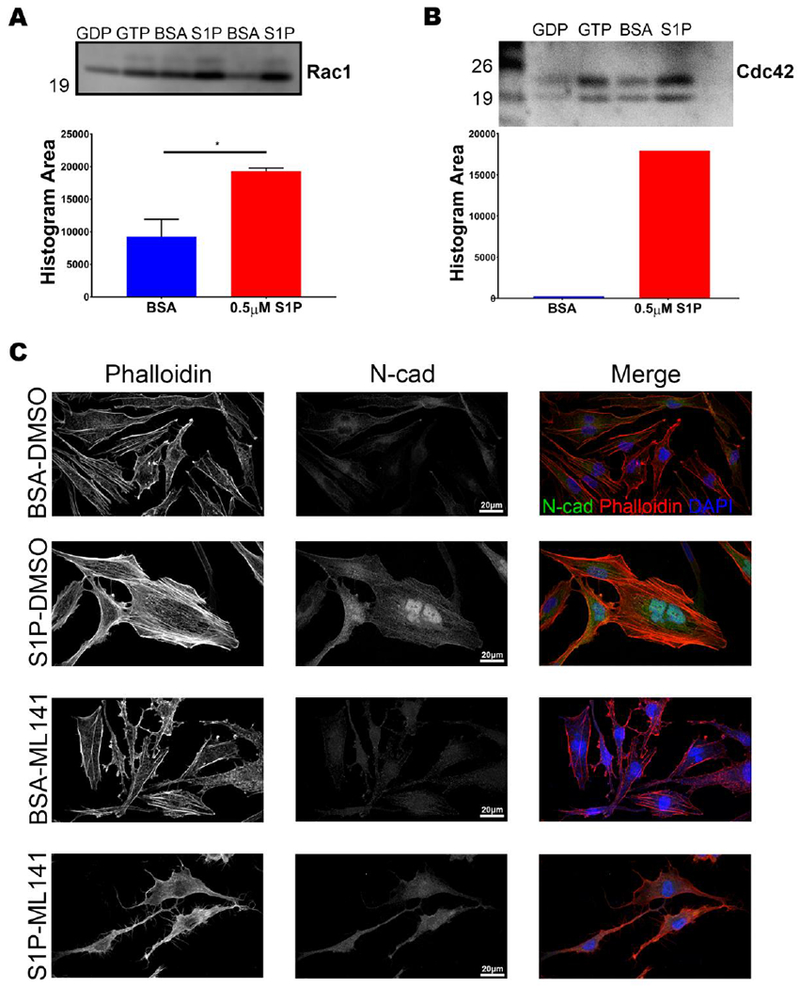
S1P activates RAC1 and CDC42 in MIO-M1 cells. MIO-M1 cells were treated with 0.5 μM S1P or the equivalent volume of BSA and incubated for 12 hours. Immunoprecipitation of GTP bound RAC1 and CDC42 was performed on MIO-M1 cells using PAK1-agarose beads. These pull-downs were then analyzed by western blot for RAC1 and CDC42. A) The blot shows GDP negative control, GTP positive control, and bands for RAC1 after treatments. Quantification of band intensity in FIJI showed that S1P treatment significantly increased activated RAC1 in MIO-M1 cells (2 replicates on the same blot for quantification, two-tailed Student’s t-test, α=0.05, p=0.04, error bars SD). B) Increased levels of active CDC42 were detected in the S1P treated cells. The blot shows GDP negative control, GTP positive control, and bands for CDC42. Band intensity in FIJI showed that S1P treatment increased activated Rac1 in MIO-M1 cells (2 replicates). C) MIO-M1 cells were treated with either control BSA or 0.5 μM S1P in the presence of DMSO (vehicle control) or ML141, an inhibitor of RAC1/CDC42. S1P in the presence of DMSO results in actin assembly and increased N-cadherin staining. However, S1P in the presence of ML141 results in a loss of actin assembly and N-cadherin production, yielding cells that resemble BSA-treated cells. Representative images from n=3 replicates.
To further understand if S1P acts through the RAC1 signaling pathway to induce the observed changes in the cytoskeleton and N-cadherin in cultured Müller glia, we used the inhibitor ML141 to block the activation of RAC1 and CDC42. BSA-only and S1P-treated MIO-M1 cells, treated with DMSO as a vehicle control, responded exactly as previously shown. The cells had an increase of N-cadherin, swelling of the cell, and an increase in actin filaments when treated with 0.5 μM of S1P. However, when 0.5 μM S1P-treated cells were also treated with 10 μM ML141, this response was abrogated. This resulted in MIO-M1 cells exhibiting the same spindly shape as seen in BSA-only-treated cells (Figure 9 C). These results highlight that S1P can activate RAC1 and CDC42, leading to an increase in N-cadherin and dynamic changes to the actin cytoskeleton in MIO-M1 cells.
Discussion
In this study, we have shown an essential role of S1P signaling in maintaining retinal structure, especially through stabilization of adherens junctions at the OLM. Our findings in Sphk1 KO mice, showing age and stress-related disruption of the OLM, are very intriguing and provide the first evidence of a role of lipid signaling in maintaining retinal structural homeostasis in aging and age-related diseases. We also show that the loss of Sphk1 leads to reduced photoreceptor function (Figure 7). Sphk1 expression in mammalian retinas increases significantly under conditions of retinal stress, along with an increase in retinal S1P levels in light damage models, suggesting its role in maintaining the stressed retina [24]. Studies have shown that reduced expression of Sphk1 increases oxidative stress in the aging brain [39,40]. The aged and light-stressed (mice raised at 100-150 lux) phenotypes observed in our Sphk1 KO mice may be a result of increased oxidative stress and reduced ability of SPHK2 to compensate for the lack of SPHK1.
A hallmark of S1P signaling is the regulation of adherens proteins. In vascular endothelial cells, S1P signals through the S1P1 receptor, which activates RAC to form adherens junctions in between cells and make flow-competent blood vessels [41,21,42]. In Sphk1 KO mice, the circulatory or plasma S1P levels are reduced by 60% (Figure 1 A) and there are no reports of any vascular or endothelial junctional defects in these mice. In our analysis, we did not see any gross morphological abnormalities in adult Sphk1 KO mouse retinal vasculature (Figure 5). We also induced vascular obliteration and neovascularization in neonatal Sphk1 KO mice and failed to determine any variation from the wild-type littermates (Figure 5). This clearly suggests that endothelial junctional integrity is not compromised in Sphk1 KO mice. However, Sphk1 deletion could affect adherens junctions made by either neurons or glial cells. Notably, we found a reduction of N-cadherin labeling at the OLM in aging and light-stressed Sphk1 KO retinas (Figure 6). We also showed that Müller glia cells in culture respond to exogenous S1P by increasing N-cadherin production (Figure 8), and that addition of S1P stimulates RAC1 and CDC42 (Figure 8), thereby indicating the role of S1P in mobilizing the actin cytoskeleton for the formation of adherens junctions between the apical end feet of Müller glia cells and photoreceptor inner segments. N-cadherin has been shown previously to be essential for development of the eye [43–45]. While the role of cadherins has been broadly studied in development, they also play an important role in the mature retina to maintain the necessary architecture required for proper vision [46,3]. Our observation that S1P stimulates CDC42 in MIO-M1 cells is important because CDC42 has been shown to locate PALS1 to junctions in epithelial cells and is needed to generate their apical polarity [47]. Interestingly, Sphk1 KO mice exhibit similar OLM degeneration and photoreceptor and RPE cell morphological abnormalities as Pals1 KO mice [46]. It should be noted that the results seen in MIO-M1 cells are from S1P exogenously added to cell culture media, while the observed in vivo results arise from endogenous or exogenous S1P on Müller glia and surrounding retinal cell types such as photoreceptors.
The mechanisms of S1P intra- and paracellular signaling, the exact roles of S1P receptors, and the cell types which generate S1P in the retina are unknown. Previous studies have shown that cultured glia from the retina are susceptible to S1P-mediated actions [48,49]. The addition of S1P induces an increase in intracellular calcium and enhanced cell migration [48]. Cultured Müller glia migration is disrupted when SPHK1 or S1P3 is inhibited [49]. Evaluation of each individual S1P receptor is vital to our understanding of the biologically active receptors in the retina. In preliminary studies using electron microscopy, we observed no remarkable morphological changes in global S1p3 KO or retinal S1p1 conditional KO mice (Chx10 Cre) with respect to junctional complexes in the OLM or RPE at one year of age (Mandal Lab, unpublished data). A further investigation is therefore necessary using in vitro cultures of various retinal cell types to isolate and identify the sources of S1P and the actions of S1P receptors in maintaining junctional complexes in the retina and RPE.
Though development of the retina and RPE are not affected by the loss of Sphk1, it is apparent that aging and cellular stress are major factors in the observed phenotype. In the RPE, we observed increased lipid storage vacuoles (Figures 3 and 4). This finding is interesting considering that lipid metabolism and storage are connected to the formation of drusen in the aging RPE. Drusen are associated with macular degeneration and are filled with lipid and protein debris [50]. We also observed increased numbers of multivesicular and membrane-rich phagosomes in Sphk1 KO mouse RPE (Figures 3 and 4). Many of these phagosomes contained undigested membranes that characteristically resemble the outer segments of the photoreceptors that under normal conditions are digested and recycled. The work here shows that SPHK1 and S1P are critical for proper RPE structure and function in aging mice.
The ability to produce S1P keeps ceramide, sphingosine, and sphingomyelin species balanced, however, all of these sphingolipids were increased in Sphk1 KO mouse retinas. In the RPE, the accumulation of these sphingolipid species could contribute to increases in lipid-filled vacuoles and phagocytosis dysfunction, similar to that seen in other sphingolipidoses [51]. In the neural retina, the accumulation of ceramides could lead to the observed phenotypes in our Sphk1 KO mice. However, TEM micrographs of acid ceramidase (Asah1) knockout mice, which are deficient in ceramide degradation, show no OLM loss at one year of age (Mandal Lab, unpublished data). These mice have elevated ceramides starting at an early age but show no loss of AJs in the retina and no loss of the OLM. While we cannot rule out that increased ceramides and sphingosine can induce cell death contributing to loss of retinal function or OLM degeneration, our data shows no changes in outer nuclear layer thickness (Supplemental figure 2) and no major changes in the structure of Müller glia by both GS and GFAP staining (Figure 6), which does not indicate that cell death is the reason for the loss of AJs and the OLM in the retina. Our results indicate that adherens junction disruption at the OLM and the RPE is associated with SPHK1 but not with SPHK2, as the OLM and RPE AJs remained intact in aged Sphk2 KO mice (Figure 4). Sphk2 KO mice are known to have increased plasma S1P, which can be attributed to compensatory increases in Sphk1 expression as a response to the loss of Sphk2 [37]. This suggests that normal or high levels of S1P in the plasma are as important for barrier formation in the RPE and OLM as they are for vascular AJ formation.
In conclusion, our findings show the importance of SPHK1 in the maintenance of neural retinal and RPE structural integrity with aging and cellular stress. Specifically, we show that SPHK1 is needed to build and maintain the adherens junctions between RPE cells and the OLM in the retina. The interaction between Müller glia and photoreceptors is critical for the polarity of the retina and forming the fully functional structure of the tissue. We also show retinal functional deficits in the absence of SPHK1. The phenotype seen in Sphk1 KO mice, in both the retina and the RPE, has similar morphological characteristics to many human retinal diseases such as AMD, geographic atrophy, macular hole, diabetic macular edema, retinal detachment, and acute posterior multifocal placoid pigment epitheliopathy. SPHK1-mediated interaction of neuronal and glial cells in the retina implies that S1P could play a bigger role in glia-neuron interaction throughout the nervous system and neurovascular coupling. Further investigation of the role of S1P and its synthesis by glial and neural cell lines is required for understanding these interactions and its role in normal development, aging, and diseases associated with neuronal tissue.
Supplementary Material
Acknowledgments:
We would like to thank the services of the Imaging Core and all the hard work done by the animal facility at the Dean McGee Eye Institute. We also want to thank Ben Fowler and Julie Crane at the Oklahoma Medical Research Foundation for their help with the super-resolution microscopy and use of the Imaging Facility to do our TEM work; Dr. Koushik Mondal from Ophthalmology, UTHSC for his help in animal experiments; Dr. Jeremy Allegood, Operational Director of Lipidomics, Virginia Commonwealth University School of Medicine, Richmond, VA 23298-5048; and VCU Lipidomics/Metabolomics Core. , the NIH-NCI Cancer Center Support Grant P30 CA016059 to the VCU Massey Cancer Center, as well as a shared resource grant (S10RR031535) from the National Institutes of Health in all manuscripts of which data generated by the VCU Lipidomics/Metabolomics Core is included as well as maintain NIH compliance for NCBI registration of manuscripts that utilize data derived from the VCU Lipidomics/Metabolomics core.
Funding: NAM: NIH grants EY022071, EY025256, EY021725. Foundation Fighting Blindness and Research to Prevent Blindness, USA.
JLW: T32EY023202
VCU Lipidomics/Metabolomics Core: NIH grants P30 CA016059 and S10 RR031535.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of interest: The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Vecino E, Rodriguez FD, Ruzafa N, Pereiro X, Sharma SC (2016) Glia-neuron interactions in the mammalian retina. Progress in retinal and eye research 51:1–40. doi: 10.1016/j.preteyeres.2015.06.003 [DOI] [PubMed] [Google Scholar]
- 2.van de Pavert SA, Kantardzhieva A, Malysheva A, Meuleman J, Versteeg I, Levelt C, Klooster J, Geiger S, Seeliger MW, Rashbass P, Le Bivic A, Wijnholds J (2004) Crumbs homologue 1 is required for maintenance of photoreceptor cell polarization and adhesion during light exposure. J Cell Sci 117 (Pt 18):4169–4177. doi: 10.1242/jcs.01301 [DOI] [PubMed] [Google Scholar]
- 3.van Rossum AG, Aartsen WM, Meuleman J, Klooster J, Malysheva A, Versteeg I, Arsanto JP, Le Bivic A, Wijnholds J (2006) Pals1/Mpp5 is required for correct localization of Crb1 at the subapical region in polarized Müller glia cells. Hum Mol Genet 15 (18):2659–2672. doi: 10.1093/hmg/ddl194 [DOI] [PubMed] [Google Scholar]
- 4.Stuck MW, Conley SM, Naash MI (2012) Defects in the outer limiting membrane are associated with rosette development in the Nrl−/− retina. PloS one 7 (3):e32484. doi: 10.1371/journal.pone.0032484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abramoff MD, Garvin MK, Sonka M (2010) Retinal imaging and image analysis. IEEE Rev Biomed Eng 3:169–208. doi: 10.1109/RBME.2010.2084567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oishi A, Hata M, Shimozono M, Mandai M, Nishida A, Kurimoto Y (2010) The significance of external limiting membrane status for visual acuity in age-related macular degeneration. American journal of ophthalmology 150 (1):27–32 e21. doi: 10.1016/j.ajo.2010.02.012 [DOI] [PubMed] [Google Scholar]
- 7.Wolf-Schnurrbusch UE, Enzmann V, Brinkmann CK, Wolf S (2008) Morphologic changes in patients with geographic atrophy assessed with a novel spectral OCT-SLO combination. Investigative ophthalmology & visual science 49 (7):3095–3099. doi: 10.1167/iovs.07-1460 [DOI] [PubMed] [Google Scholar]
- 8.Landa G, Gentile RC, Garcia PM, Muldoon TO, Rosen RB (2012) External limiting membrane and visual outcome in macular hole repair: spectral domain OCT analysis. Eye (Lond) 26 (1):61–69. doi: 10.1038/eye.2011.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen X, Zhang L, Sohn EH, Lee K, Niemeijer M, Chen J, Sonka M, Abramoff MD (2012) Quantification of external limiting membrane disruption caused by diabetic macular edema from SD-OCT. Investigative ophthalmology & visual science 53 (13):8042–8048. doi: 10.1167/iovs.12-10083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scarinci F, Shaarawy A, Narala R, Jampol LM, Fawzi AA (2016) Loss of External Limiting Membrane Integrity Predicts Progression of Hydroxychloroquine Retinal Toxicity after Drug Discontinuation. Retina 36 (10): 1951–1957. doi: 10.1097/IAE.0000000000001217 [DOI] [PubMed] [Google Scholar]
- 11.Narala R, Scarinci F, Shaarawy A, Simonett JM, Flaxel CJ, Fawzi AA (2016) Longitudinal Quantitative Evaluation of Photoreceptor Volume Following Repair of Macula-Off Retinal Detachment. Retina 36 (8): 1432–1438. doi: 10.1097/IAE.0000000000000971 [DOI] [PubMed] [Google Scholar]
- 12.Scarinci F, Fawzi AA, Shaarawy A, Simonett JM, Jampol LM (2017) Longitudinal Quantitative Evaluation of Outer Retinal Lesions in Acute Posterior Multifocal Placoid Pigment Epitheliopathy Using Optical Coherence Tomography. Retina 37 (5):851–857. doi: 10.1097/IAE.0000000000001245 [DOI] [PubMed] [Google Scholar]
- 13.Williams DS, Arikawa K, Paallysaho T (1990) Cytoskeletal components of the adherens junctions between the photoreceptors and the supportive Müller cells. J Comp Neurol 295 (1): 155–164. doi: 10.1002/cne.902950113 [DOI] [PubMed] [Google Scholar]
- 14.Paik JH, Chae S, Lee MJ, Thangada S, Hla T (2001) Sphingosine 1-phosphate-induced endothelial cell migration requires the expression of EDG-1 and EDG-3 receptors and Rho-dependent activation of alpha vbeta3- and beta1-containing integrins. The Journal of biological chemistry 276 (15):11830–11837. doi: 10.1074/jbc.M009422200 [DOI] [PubMed] [Google Scholar]
- 15.Futerman AH, Hannun YA (2004) The complex life of simple sphingolipids. EMBO reports 5 (8):777–782. doi: 10.1038/sj.embor.7400208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagahashi M, Ramachandran S, Kim EY, Allegood JC, Rashid OM, Yamada A, Zhao R, Milstien S, Zhou H, Spiegel S, Takabe K (2012) Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer research 72 (3):726–735. doi: 10.1158/0008-5472.CAN-11-2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang H, Buckley NE, Gibson K, Spiegel S (1990) Sphingosine stimulates cellular proliferation via a protein kinase C-independent pathway. The Journal of biological chemistry 265 (1):76–81 [PubMed] [Google Scholar]
- 18.Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL (2005) Essential role for sphingosine kinases in neural and vascular development. Molecular and cellular biology 25 (24): 11113–11121. doi: 10.1128/MCB.25.24.11113-11121.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha’afi RI, Hla T (1999) Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 99 (3):301–312 [DOI] [PubMed] [Google Scholar]
- 20.Mehta D, Konstantoulaki M, Ahmmed GU, Malik AB (2005) Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. The Journal of biological chemistry 280 (17): 17320–17328. doi: 10.1074/jbc.M411674200 [DOI] [PubMed] [Google Scholar]
- 21.Jung B, Obinata H, Galvani S, Mendelson K, Ding BS, Skoura A, Kinzel B, Brinkmann V, Rafii S, Evans T, Hla T (2012) Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Developmental cell 23 (3):600–610. doi: 10.1016/j.devcel.2012.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allende ML, Sasaki T, Kawai H, Olivera A, Mi Y, van Echten-Deckert G, Hajdu R, Rosenbach M, Keohane CA, Mandala S, Spiegel S, Proia RL (2004) Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. The Journal of biological chemistry 279 (50):52487–52492. doi: 10.1074/jbc.M406512200 [DOI] [PubMed] [Google Scholar]
- 23.Qi H, Priyadarsini S, Nicholas SE, Sarker-Nag A, Allegood J, Chalfant CE, Mandal NA, Karamichos D (2017) Analysis of sphingolipids in human corneal fibroblasts from normal and keratoconus patients. Journal of lipid research 58 (4):636–648. doi: 10.1194/jlr.M067264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H, Tran JT, Eckerd A, Huynh TP, Elliott MH, Brush RS, Mandal NA (2013) Inhibition of de novo ceramide biosynthesis by FTY720 protects rat retina from light-induced degeneration. Journal of lipid research 54 (6): 1616–1629. doi: 10.1194/jlr.M035048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stiles M, Qi H, Sun E, Tan J, Porter H, Allegood J, Chalfant CE, Yasumura D, Matthes MT, LaVail MM, Mandal NA (2016) Sphingolipid profile alters in retinal dystrophic P23H-1 rats and systemic FTY720 can delay retinal degeneration. Journal of lipid research 57 (5):818–831. doi: 10.1194/jlr.M063719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Connor KM, Krah NM, Dennison RJ, Aderman CM, Chen J, Guerin KI, Sapieha P, Stahl A, Willett KL, Smith LE (2009) Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nature protocols 4 (11): 1565–1573. doi: 10.1038/nprot.2009.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Limb GA, Salt TE, Munro PM, Moss SE, Khaw PT (2002) In vitro characterization of a spontaneously immortalized human Müller cell line (MIO-M1). Investigative ophthalmology & visual science 43 (3):864–869 [PubMed] [Google Scholar]
- 28.Wu M, Yang S, Elliott MH, Fu D, Wilson K, Zhang J, Du M, Chen J, Lyons T (2012) Oxidative and endoplasmic reticulum stresses mediate apoptosis induced by modified LDL in human retinal Müller cells. Investigative ophthalmology & visual science 53 (8):4595–4604. doi: 10.1167/iovs.12-9910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9 (7):676–682. doi: 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mehalow AK, Kameya S, Smith RS, Hawes NL, Denegre JM, Young JA, Bechtold L, Haider NB, Tepass U, Heckenlively JR, Chang B, Naggert JK, Nishina PM (2003) CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum Mol Genet 12 (17):2179–2189. doi: 10.1093/hmg/ddg232 [DOI] [PubMed] [Google Scholar]
- 31.Wright CB, Redmond TM, Nickerson JM (2015) A History of the Classical Visual Cycle. Prog Mol Biol Transl Sci 134:433–448. doi: 10.1016/bs.pmbts.2015.06.009 [DOI] [PubMed] [Google Scholar]
- 32.Kevany BM, Palczewski K (2010) Phagocytosis of retinal rod and cone photoreceptors. Physiology (Bethesda) 25 (1):8–15. doi: 10.1152/physiol.00038.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young RW, Bok D (1969) Participation of the retinal pigment epithelium in the rod outer segment renewal process. J Cell Biol 42 (2):392–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.LaVail MM (1976) Rod outer segment disk shedding in rat retina: relationship to cyclic lighting. Science 194 (4269):1071–1074 [DOI] [PubMed] [Google Scholar]
- 35.Sensken SC, Bode C, Nagarajan M, Peest U, Pabst O, Graler MH (2010) Redistribution of sphingosine 1-phosphate by sphingosine kinase 2 contributes to lymphopenia. J Immunol 184 (8):4133–4142. doi: 10.4049/jimmunol.0903358 [DOI] [PubMed] [Google Scholar]
- 36.Zemann B, Kinzel B, Müller M, Reuschel R, Mechtcheriakova D, Urtz N, Bornancin F, Baumruker T, Billich A (2006) Sphingosine kinase type 2 is essential for lymphopenia induced by the immunomodulatory drug FTY720. Blood 107 (4): 1454–1458. doi: 10.1182/blood-2005-07-2628 [DOI] [PubMed] [Google Scholar]
- 37.Kharel Y, Raje M, Gao M, Gellett AM, Tomsig JL, Lynch KR, Santos WL (2012) Sphingosine kinase type 2 inhibition elevates circulating sphingosine 1-phosphate. The Biochemical journal 447 (1): 149–157. doi: 10.1042/BJ20120609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mendelson K, Evans T, Hla T (2014) Sphingosine 1-phosphate signalling. Development 141 (1):5–9. doi: 10.1242/dev.094805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pyszko JA, Strosznajder JB (2014) The key role of sphingosine kinases in the molecular mechanism of neuronal cell survival and death in an experimental model of Parkinson’s disease. Folia Neuropathol 52 (3):260–269 [DOI] [PubMed] [Google Scholar]
- 40.Pchejetski D, Kunduzova O, Dayon A, Calise D, Seguelas MH, Leducq N, Seif I, Parini A, Cuvillier O (2007) Oxidative stress-dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A-associated cardiac cell apoptosis. Circ Res 100 (1):41–49. doi: 10.1161/01.RES.0000253900.66640.34 [DOI] [PubMed] [Google Scholar]
- 41.Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, Hobson JP, Rosenfeldt HM, Nava VE, Chae SS, Lee MJ, Liu CH, Hla T, Spiegel S, Proia RL (2000) Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. The Journal of clinical investigation 106 (8):951–961. doi: 10.1172/JCI10905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paik JH, Skoura A, Chae SS, Cowan AE, Han DK, Proia RL, Hla T (2004) Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes & development 18 (19):2392–2403. doi: 10.1101/gad.1227804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malicki J, Jo H, Pujic Z (2003) Zebrafish N-cadherin, encoded by the glass onion locus, plays an essential role in retinal patterning. Dev Biol 259 (1):95–108 [DOI] [PubMed] [Google Scholar]
- 44.Pujic Z, Malicki J (2001) Mutation of the zebrafish glass onion locus causes early cell-nonautonomous loss of neuroepithelial integrity followed by severe neuronal patterning defects in the retina. Dev Biol 234 (2):454–469. doi: 10.1006/dbio.2001.0251 [DOI] [PubMed] [Google Scholar]
- 45.Lele Z, Folchert A, Concha M, Rauch GJ, Geisler R, Rosa F, Wilson SW, Hammerschmidt M, Bally-Cuif L (2002) parachute/n-cadherin is required for morphogenesis and maintained integrity of the zebrafish neural tube. Development 129 (14):3281–3294 [DOI] [PubMed] [Google Scholar]
- 46.Park B, Alves CH, Lundvig DM, Tanimoto Ν, Beck SC, Huber G, Richard F, Klooster J, Andlauer TF, Swindell EC, Jamrich M, Le Bivic A, Seeliger MW, Wijnholds J (2011) PALS1 is essential for retinal pigment epithelium structure and neural retina stratification. J Neurosci 31 (47):17230–17241. doi: 10.1523/JNEUROSCI.4430-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wells CD, Fawcett JP, Traweger A, Yamanaka Y, Goudreault M, Elder K, Kulkarni S, Gish G, Virag C, Lim C, Colwill K, Starostine A, Metalnikov P, Pawson T (2006) A Rich1/Amot complex regulates the Cdc42 GTPase and apical-polarity proteins in epithelial cells. Cell 125 (3):535–548. doi: 10.1016/j.cell.2006.02.045 [DOI] [PubMed] [Google Scholar]
- 48.Esche M, Hirrlinger PG, Rillich K, Yafai Y, Pannicke T, Reichenbach A, Weick M (2010) Signalling of sphingosine-1-phosphate in Müller glial cells via the S1P/EDG-family of G-protein-coupled receptors. Neurosci Lett 480 (2): 101–105. doi: 10.1016/j.neulet.2010.06.014 [DOI] [PubMed] [Google Scholar]
- 49.Simon MV, Prado Spalm FH, Politi LE, Rotstein NP (2015) Sphingosine-1-Phosphate Is a Crucial Signal for Migration of Retina Muller Glial Cells. Investigative ophthalmology & visual science 56 (10):5808–5815. doi: 10.1167/iovs.14-16195 [DOI] [PubMed] [Google Scholar]
- 50.Wang L, Clark ME, Crossman DK, Kojima K, Messinger JD, Mobley JA, Curcio CA (2010) Abundant lipid and protein components of drusen. PloS one 5 (4):e10329. doi: 10.1371/journal.pone.0010329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen H, Chan AY, Stone DU, Mandal NA (2014) Beyond the cherry-red spot: Ocular manifestations of sphingolipid-mediated neurodegenerative and inflammatory disorders. Survey of ophthalmology 59 (1):64–76. doi: 10.1016/j.survophthal.2013.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.