

Abstract
Importance:
As the second leading cause of death in the world, stroke has a large societal impact. Nearly one-third of ischemic strokes are lacunar strokes (LS), or small subcortical infarcts. While smaller in size they represent large problems, leaving many patients disabled and demented. Little is known about the underlying etiology of LS, slowing the development of novel therapeutics.
Observations:
When the term lacune was described in the 1800s, its underlying pathophysiological basis was obscure. In the 1960s, C. Miller Fisher performed autopsy studies that showed that vessels supplying lacunes displayed segmental arteriolar disorganization, characterized by vessel enlargement, hemorrhage, and fibrinoid deposition. For these pathological changes, he coined the term lipohyalinosis. Apart from these early descriptions of LS and lipohyalinosis, few attempts have been made to reconcile this pathological description with modern mechanisms of cerebral small vessel disease (cSVD).
Conclusions and Relevance:
During the last six years, progress has been made in understanding the clinical mechanisms, imaging characteristics, and genetic basis of LS. Questions persist regarding the order of events related to the initiation and progression of cSVD, how LS is related to other sequelae of cSVD, and if LS is part of a systemic disease process. These advances prompt a re-assessment of the current understanding of the etiology of LS and cSVD. The development of targeted therapies depends on a complete understanding of these mechanisms.
Historical Background
Stroke is the second leading cause of death in the world and the leading cause of disability in the United States.1 Among ischemic strokes there are different subtypes, including large artery atherosclerosis, cardioembolism, and cerebral small vessel disease (cSVD). While cSVD has several clinical and radiographic manifestations, lacunar stroke (LS) is prototypical and accounts for 20–30% of ischemic strokes.2,3 Clinically, LS can manifest with several syndromes depending on lesion location.4 Silent LS are found in 20–50% of healthy elderly people.5 LS are particularly burdensome given a 20% recurrence rate, 25% 5-year mortality, and associated morbidities such as vascular cognitive impairment.3
LS, appropriately named given their propensity to form cavities (lacunes), were first described in 1838 (Figure 1).3 More recently, the STandards for ReportIng Vascular changes on nEuroimaging (STRIVE) definitions were developed to standardize terms that describe imaging sequelae of cSVD, including recent small subcortical infarcts, lacunes, white matter hyperintensities (WMH), perivascular spaces, microbleeds, and brain atrophy.6 LS encompasses two terms: recent small subcortical infarcts, defined as recent infarctions in the territory of one perforating arteriole with imaging features or clinical symptoms consistent with occurrence in the previous few weeks, and lacunes, defined as round or ovoid, subcortical, fluid-filled cavities of between 3 mm and about 15 mm in diameter consistent with previous acute small subcortical infarcts or hemorrhages in the territory of one perforating arteriole.6 While the focus of this review is LS, it is important to appreciate other pathologies that result from cSVD as there are inter-related mechanisms and treatment implications. For example, many intracerebral hemorrhages (ICH) likely result from similar vessel pathology,7 suggesting that LS patients may be at higher bleeding risk on antithrombotic medications.
Figure 1. Cerebral small vessel disease (cSVD) histopathological section, lacunar stroke gross section, and lacunar stroke computed tomography (CT) images.
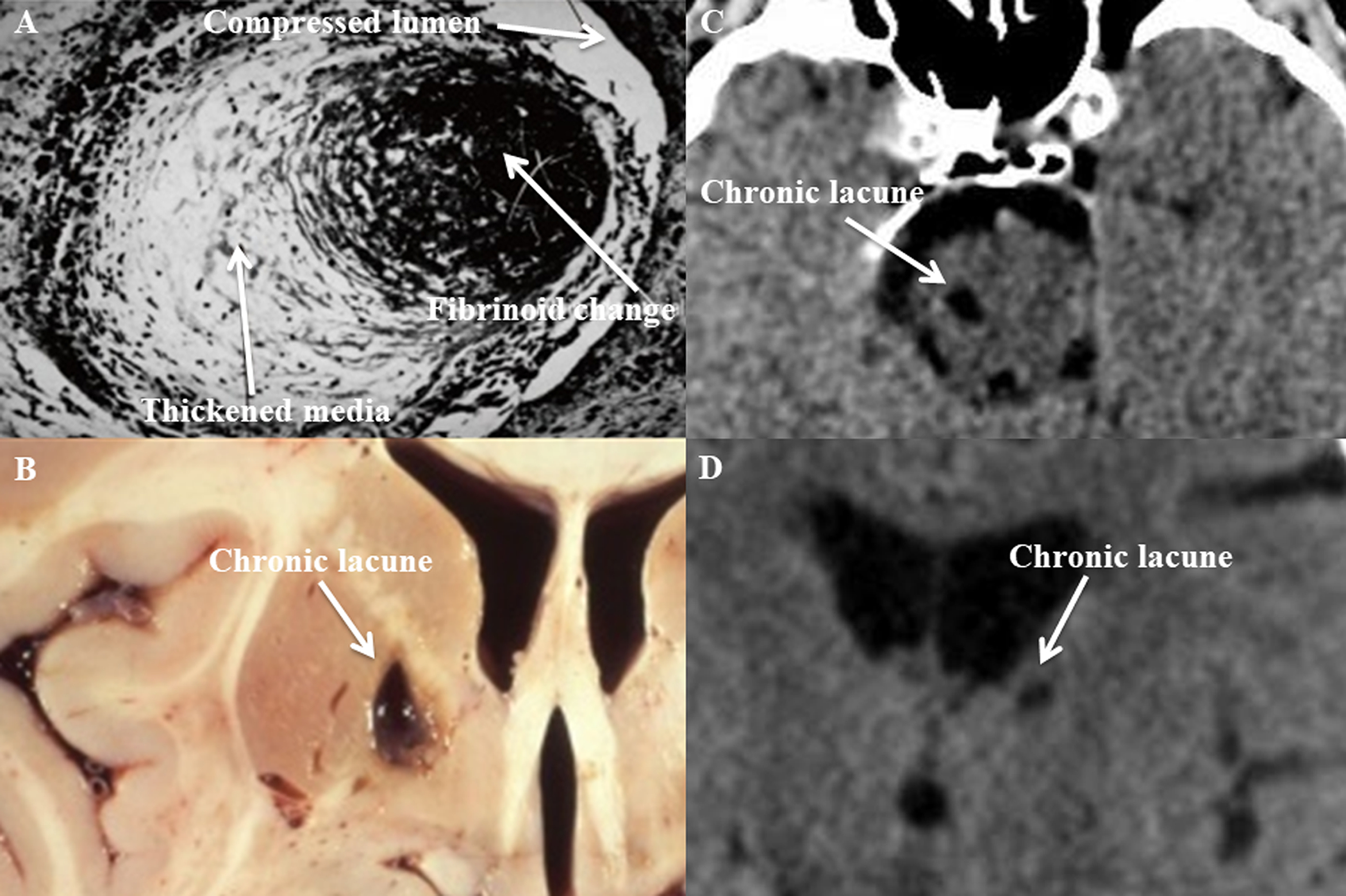
Photomicrograph of cSVD affecting a small penetrating artery (A), showing thickened media and fibrinoid necrosis (Gift from Dr. C. Miller Fisher). Photograph of a cavity secondary to a chronic lacune in the medial basal ganglia (B) found at autopsy.2 CT images showing hypodensities secondary to chronic lacunes involving the right paramedian pons (C) and left thalamus (D).
In the 1960s, Fisher performed numerous pathological studies showing most LS are found distal to occlusive lesions of small perforating arteries.4 He used the term lipohyalinosis to describe what he believed was a unique hypertensive cerebral vasculopathy characterized by fibrinoid vessel wall necrosis and segmental arteriolar disorganization. There were three prominent, often overlapping findings: vessel enlargement, hemorrhage, and fibrinoid deposition.4 Modern classification schemes for sporadic non-amyloid cSVD pathology vary and overlap, including atherosclerosis, microatheroma, arteriosclerosis, arteriolosclerosis, and lipohyalinosis.8 cSVD affects a range of vessel sizes, from 40–900 μm in diameter.7 Arteriosclerosis and atherosclerosis affect vessels of 200–800 μm, arteriolosclerosis causes concentric vessel wall thickening in arteries of 40–150 μm, and lipohyalinosis affects vessels of 40–300 μm.8
The focus of this review will be LS caused by sporadic non-amyloid cSVD, and we limit discussion of other imaging markers of cSVD. Aside from thrombolysis for acute LS, there has been limited progress in the targeted treatment of this condition compared to other stroke subtypes. These pathologies were traditionally described as resulting from chronic hypertension; despite today’s anti-hypertensive agents, LS due to cSVD persists, and its etiology is likely multifactorial.2 This review discusses progress in understanding the pathogenesis of LS, its imaging characteristics, and its genetic basis. We explore connections between LS and systemic small vessel disease. Lastly, we describe new models and approaches that can be used to study LS and cSVD.
Search Strategy and Selection Criteria
References in the remainder of this review were identified by searches of PubMed from 2012–2017, and references from relevant articles. The search terms “lipohyalinosis AND lacune” (2 total hits, 0 relevant), “small vessel disease AND lacune” (27,6), “genetics AND lacune” (4,1), “models AND lacune” (10,1), “lipohyalinosis AND lacunar stroke” (12,1), “small vessel disease AND lacunar stroke” (289,59), “genetics AND lacunar stroke” (85,4), “models AND lacunar stroke” (172,6) were used. Articles were independently screened by 2 authors for appropriateness and relevance to the topic. There were no language restrictions.
Clinical Correlations and Underlying Mechanisms
Recent studies suggest that not all LS are attributable to cSVD (Figure 2). Branch atheromatous disease is one proposed etiology involving large artery disease that obstructs the orifices of penetrators.2 One study showed that a quarter of LS patients had large artery abnormalities on vessel imaging, emphasizing the importance of vessel imaging.9 Furthermore, a small portion of LS may occur from artery-to-artery embolism. A population-based study showed a relationship between calcium content in the carotid siphon and silent LS.10 Another study evaluated patients with acute stroke by TEE for aortic atheroma, and LS increased the odds of having aortic plaques.11
Figure 2. Etiology and clinical phenotype of lacunar stroke (LS).
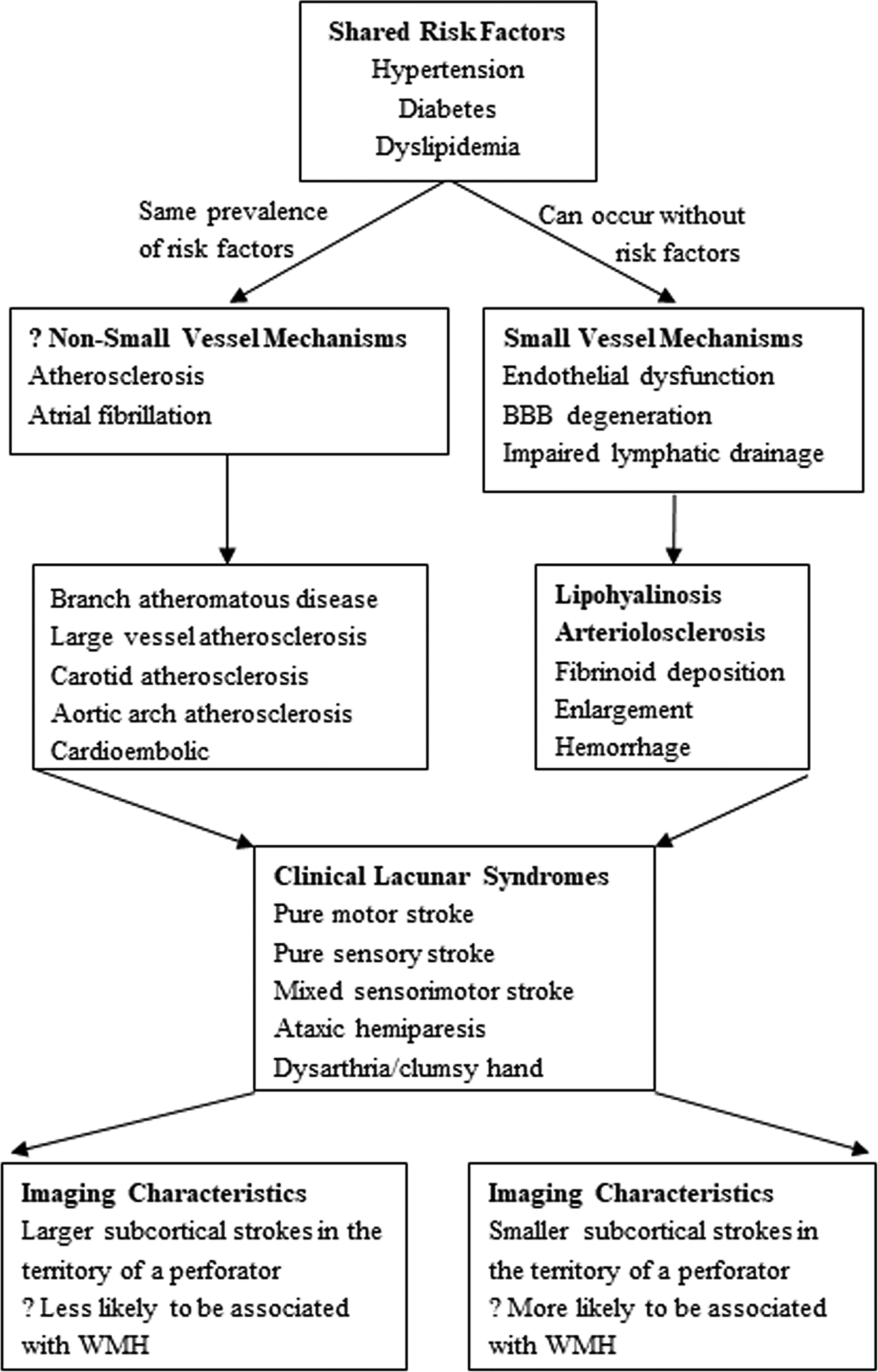
LS results most commonly from small vessel mechanisms, but alternative mechanisms are feasible. Shared risk factors make many studies difficult to interpret. While the clinical lacunar syndromes are identical, there may be subtle differences in imaging characteristics but meaningful differences in treatment approaches based on etiology. BBB (blood brain barrier), WMH (white matter hyperintensity).
Cardioembolism may also account for some rare cases of LS. In one study, 11% of patients with symptomatic LS were found to have a possible carotid or cardiac source. LS in the basal ganglia were slightly larger and were three times more likely to have a possible embolic source, but no other risk factors differed with stroke size, shape, or location.12 Another study examined patients with atrial fibrillation who had either recent LS or non-lacunar stroke. Chronic lacunes were an independent predictor of the incident infarct pattern, suggesting that LS might be caused by intrinsic cSVD despite the presence of concomitant atrial fibrillation.13 These associations may be due to shared risk factors, such as hypertension, dyslipidemia, and diabetes mellitus (DM).
Recent work in cSVD-related LS has focused on elucidating two pathogenic mechanisms: endothelial dysfunction and blood-brain-barrier (BBB) disruption.2 While several mechanisms are involved in the sequelae of cSVD, endothelial dysfunction may be the most important for LS. The endothelium regulates vessel tone, regulates fibrinolysis/coagulation, participates in inflammation, and is involved in angiogenesis. Its dysfunction reflects a shift towards a vasoconstrictive, procoagulation, proinflammatory, and proliferative balance.14 The details of this shift remain largely unknown and likely result from a complex interplay of aging, oxidative stress, mechanical stress, genetic predisposition, and other risk factors such as hypertension. The endothelium incurs structural and functional damage, becoming leaky and inflamed. Further damage leads to impaired autoregulation, where the vessel is unable vasodilate to maintain perfusion.15 The vessel wall thickens as connective tissue replaces normal wall layers with ultimate lumen narrowing, thrombosis, and occlusion.2 A recent study examining the endothelium of small penetrating arteries showed that thrombomodulin, a marker of endothelial dysfunction, was elevated in brains with cSVD compared to aged controls.16 Vascular endothelial growth factor has also been examined as a contributor to cSVD proliferation. In one recent study, however, it was not associated with cSVD severity.17 Recently, brains with pathologic evidence of cSVD were utilized to show an impaired vasodilator function of white matter penetrating arterioles compared to pial arterioles. Furthermore, areas of white matter injury were associated with decreased mature oligodendrocytes, suggesting impaired myelination.18
In addition to endothelial dysfunction, BBB degradation is thought to play a critical role in cSVD and LS. There is also overlap with the pathogenesis of WMH, as edema that develops surrounding leaky vessels can cause gliosis and appear as WMH on imaging. The BBB is composed of inter-related structures, including endothelial cells joined by tight junctions, basement membranes, associated perivascular spaces, pericytes, glia limitans, and astrocyte end feet.15 In one study to assess BBB permeability, patients with nondisabling LS or cortical strokes (controls) underwent contrast-enhanced magnetic resonance imaging (MRI) acutely and then again in 3 years. Poor functional outcome was associated with increased basal ganglia BBB permeability.19 More recently the same group demonstrated further evidence that BBB leakage mediates cSVD, and the severity of leakage was associated with hypertension.20 Another study confirmed global cSVD burden is associated with increased BBB degradation in both the acute LS and non-ischemic areas.21
Improved Understanding through Imaging
Recent advances in ultrasonography, such as transcranial Doppler (TCD), have provided insight into understanding LS and cSVD. One TCD parameter, the pulsatility index (PI), reflects distal cerebral vascular resistance and is posited to be a surrogate marker of cSVD. In a study of patients with acute LS who underwent TCD and MRI, PI was associated with infarct volume.22 An additional parameter assessable by TCD is dynamic cerebral autoregulation. A small study of unilateral LS patients showed that even though strokes were unilateral, there were bilateral impairments in autoregulation even 6 months after stroke,23 suggesting the presence of global pathology.
Computed Tomography (CT) studies also shed light on the underlying pathophysiology and role of hypoperfusion in LS and cSVD. In a study using CT perfusion, acute LS showed a smaller perfusion deficit compared to cortical strokes,9 supporting a focal small vessel pathology. However, another study showed that hypoperfusion of white matter remote to acute infarcts was associated with LS compared to other stoke subtypes, further supporting a global pathology.24 In a comparison of WMH to contralateral white matter, one study showed WMH had decreased cerebral blood flow (CBF) and decreased vascular reactivity.25 Another study compared baseline and 4-year follow-up MRIs in patients with LS; baseline WMH volumes were associated with decline in CBF over time, but baseline CBF was not associated with progression of WMH or LS.26 However, another study of minor strokes compared MRIs at baseline to those after 18 months, and regions with low baseline CBF were associated with the development of new WMH.27 There are conflicting data about whether or not lower CBF predates WMH and LS development,28 but this hypothesis is favored.
Advancements in MRI technology have dramatically improved the clinical phenotyping of LS. For example, since the advent of MRI, heritability estimates for lacunar stroke were markedly increased compared to prior studies using CT and clinical exam.29 Furthermore, MRI has increased the ability to detect cSVD markers, including cerebral microinfarcts (CMI).30 CMI, at 0.2–3 mm in size, are smaller than lacunes at 3–15 mm. Another distinction between CMI and LS is their presence throughout the brain including the cortex. While deep CMI are likely secondary to non-amyloid cSVD,31,32 cortical CMI are more heterogeneous in etiology as they are related to cerebral amyloid angiopathy,31,32 non-amyloid cSVD and LS,30 and intracranial stenosis and large cortical infarcts.30 New techniques are also being developed, including overlapping small vessel templates on MRI images. One study categorized first, second, and third-order branch infarcts, and as vessel branch order increased (smaller vessel), the size of LS decreased.33
Longitudinal changes can be seen in many cSVD MRI surrogates, including LS, demonstrating these involve dynamic processes.34 Recent studies showed that some acute LS can become undetectable on repeat imaging35 and some WMH can regress after minor stroke.36 Several imaging studies show a relationship between LS and WMH (Figure 3). A study using advanced MRI techniques identified incident LS and showed they have a predilection for the edge of WMH, supporting the concept of a WMH “penumbra”.37 However, another study of incident LS showed that their main axes and planes aligned with the orientation of perforating arteries and not with fiber tracts.38 This supports the presumed vascular etiology, despite the predilection for forming near the edge of existing WMH.
Figure 3. Lacunar stroke (LS) and white matter hyperintensity (WMH) proximity.
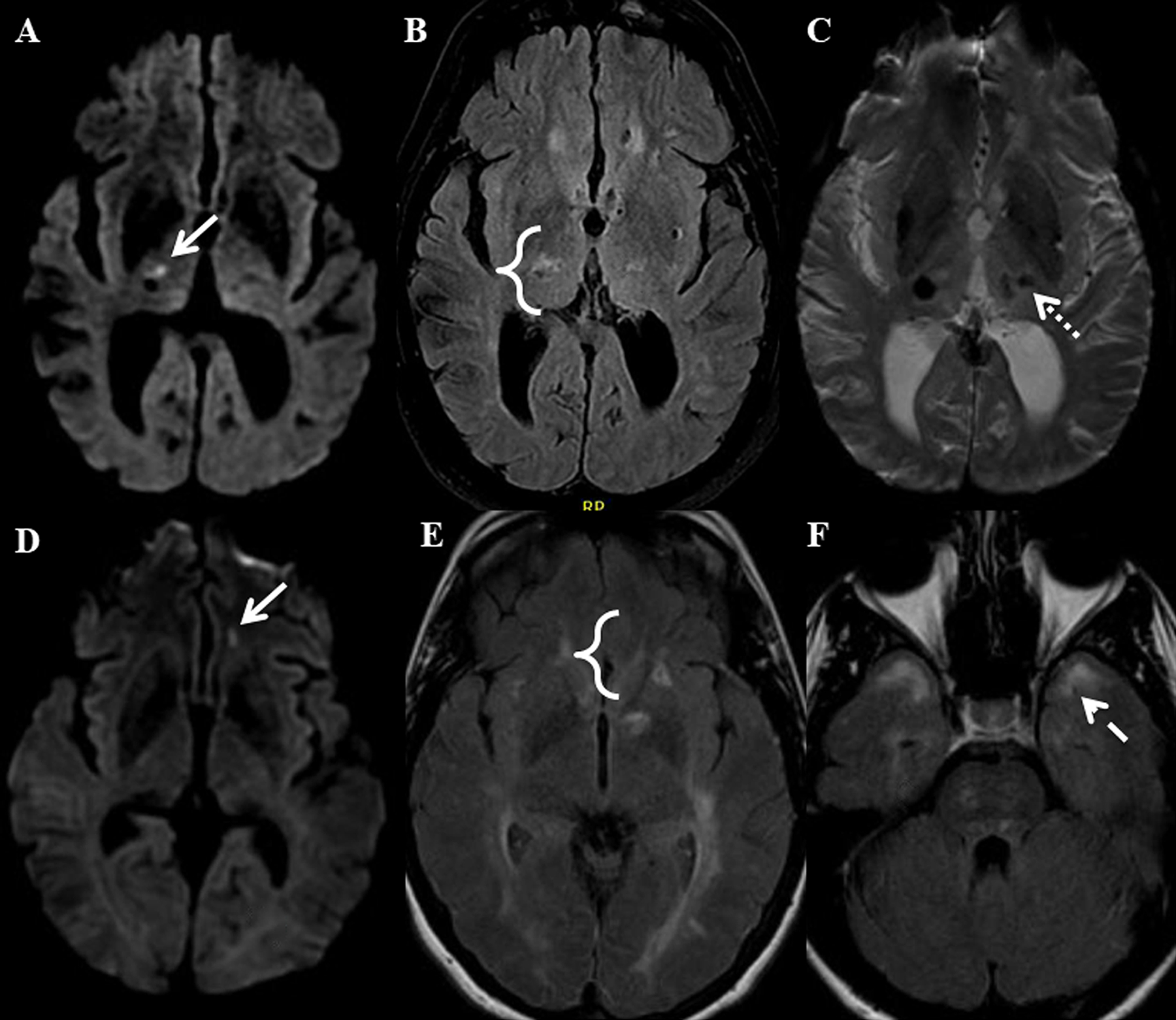
Panels A-C show the brain of a patient with severe sporadic hypertension-related cerebral small vessel disease (cSVD). An acute LS (arrow) is shown in the right thalamus on DWI imaging (A). FLAIR imaging (B) shows there is an adjacent WMH (bracket). SWI imaging (C) shows there is also a microhemorrhage in the left thalamus (dotted arrow). Panels D-F show the brain of a patient with CADASIL. An acute LS (arrow) is shown in the left orbitofrontal white matter on DWI imaging (D). FLAIR imaging (E) shows there is an adjacent WMH (bracket). WMH in the anterior temporal lobe (dashed arrow) is also present on FLAIR imaging (F).
Advancements in Understanding Genetics
Although LS is a multifactorial disease, there are considerable differences in susceptibility to risk factors when comparing individuals. Family history of vascular disease is an independent risk factor for LS, and twin and family studies show that cSVD has a genetic basis.39 In addition, there are several single gene disorders that precipitate familial cSVD, which accounts for 1–5% of all strokes.39 Although the contribution of individual genes in developing sporadic cSVD is unknown, a more complete understanding of familial cSVD may provide insight into its pathogenesis. Mutations resulting in familial cSVD are highly penetrant and involve NOTCH3, HTRA1, TREX1, GLA, COL4A1, COL4A2, FOXC1, and PITX2.40 Single nucleotide polymorphisms (SNPs) in COL4A2 have been associated with cSVD-related sequalae,41 and variants of COL3A1 have been shown to influence LS recurrence and mortality.42 Mutations in COL4A1 have also been identified in patients with ICH.43 In another analysis, loci in COL4A2 and HTRA1 were found to be associated with both LS and deep ICH, supporting a common mechanism.44
Although early genetic studies of stroke have many of the same pitfalls as those of other multifactorial diseases, some important genes were implicated using candidate gene studies. These employ the use of SNP analysis in genes that are suspected to play a role in the disease. A meta-analysis showed the ACE I/D polymorphism was associated with LS, although larger studies have not replicated this finding.45 Several studies examined MTHFR C677T, showing it is also associated with the development of cSVD.46 ApoE has also been studied in elderly individuals, where ApoE4 carriers were found to have significantly more cSVD and increased progression of WMH on subsequent MRIs. This may be due to increased Aβ deposition,47 though other studies suggest ApoE4 acts through other mechanisms.48 Interestingly, a recent study has shown a correlation between autosomal dominant Alzheimer’s disease and increased WMH, even before expected symptom onset.49
Genome wide association studies (GWAS) represent a powerful non-hypothesis based approach that allows the simultaneous genotyping of more than one million polymorphisms across the genome. In one study of early-onset LS patients, two subtypes were defined: isolated LS and multiple LS with leukoaraiosis.29 There was significant heritability of these subtypes, suggesting they may be distinct entities. A subsequent study showed that oxidative phosphorylation pathways were associated with multiple LS with leukoaraiosis but not isolated LS.50 In another study, the rs12445022 SNP was found to be associated with LS.51 A study examining WMH volume, instead of LS specifically, found several associated SNPs involving the genes TRIM65 and TRIM47.52 Yet another study of WMH volume found further associated SNPs, including some in genes implicated in Alzheimer’s disease and ICH.53
One potential drawback to GWAS is that these studies are underpowered to detect rare variants. Whole-exome sequencing (WES) using next-generation technology allows for the rapid sequencing of entire coding sequences. A study in the Chinese Han population showed that SNPs in C1ORF156and XYLB were related to ischemic stroke.54 The former variant may carry some importance in patients without hypertension or DM, while the latter may be important in patients with hypertension but without DM. Another small WES study identified two genes, CSN3 and HLA-DPB1, that were unique to patients with LS; CSN3 has also been implicated in coronary disease and DM.55 In another WES study, a gene implicated in Charcot-Marie-Tooth disease, SH3TC1, was related to WMH.56 The genetics of LS will be advanced by the Small Vessel and Lacunar Project, an ongoing large, prospective study utilizing next generation sequencing.57
Relationship to Systemic Small Vessel Disease
Growing evidence supports a connection between cSVD and other systemic disease processes. As with the theory that some LS are secondary to emboli, it is important to recognize that the associations discussed here may be due to shared risk factors. One study compared cerebral, carotid, and brachial vasoreactivity in patients with recent LS. Patients with LS had more severely impaired cerebral vasoreactivity one month after stroke; the only extracranial vasoreactivity measure that correlated with brain parenchymal abnormalities was brachial endothelial-independent vasodilation.58 Systemic arterial stiffness has also been evaluated for an association with cSVD. Arterial stiffness can be approximated by ankle-brachial index (ABI), pulse wave velocity (PWV, aortic [aPWV], brachial-ankle [baPWV], and carotid-femoral), cardio-ankle vascular index (CAVI), and augmentation index. One study comparing these parameters for different stroke subtypes showed that ABI was lower in large artery atherosclerosis compared to LS and controls, baPWV was higher in both large artery atherosclerosis and LS compared to controls, and CAVI increased in the order of controls, LS, and large artery atherosclerosis.59 A different study showed patients with ABI<0.90 and >1.4 were almost four times more likely to have silent LS.60 In a population-based study, higher aPWV was associated with larger WMH volumes.61 Silent LS were associated with second peak systolic blood pressure wall tension and end diastolic wall tension independently of arterial stiffness in yet another study.62
Additional research has focused on comparing cSVD to pathology in the small vessels of other organs, including the kidneys and retina. There are anatomic and functional similarities as all are small, short vessels that arise from arteries under high pressure. A recent meta-analysis explored this relationship and found an association between worsening renal impairment and asymptomatic cSVD but not symptomatic LS, after controlling for hypertension.63 However, in a cohort from the Rotterdam Study, worsening renal function correlated with higher prevalence of LS.64 Two studies of first-ever LS patients showed worsening renal function was associated with increasing overall burden of cSVD.65,66 In addition to kidney disease, retinal disease may also be related to cSVD, especially in patients with DM. Although there are conflicting results,67 one study of patients without DM who had either acute ICH or LS showed that both were associated with retinal disease.68 More investigation is needed to better understand these complex relationships. It is surprising that cSVD would only affect the cerebral vasculature as Fisher speculated, but a definitive link to other organs has been elusive.
Challenges and Opportunities
In summary, not all clinical and imaging defined LS result from cSVD, and this distinction is important for patient treatment approaches and future research. Through mechanisms related to aging, oxidative stress, mechanical stress, genetic predisposition, and other risk factors such as hypertension, the endothelium in cSVD becomes damaged, both in structure and function. The BBB degrades allowing leakage of blood contents, contributing to gliosis and the formation of WMH. Autoregulation also fails which decreases blood flow as vessels cannot appropriately dilate. Focal narrowing due to thickening of vessel walls further contributes to decreased flow; ischemia may lead to WMH and, when areas of focal narrowing become completely occluded, LS. There are conflicting data about whether or not lower CBF predates WMH and LS development, but this stands to reason mechanistically.
While we limit discussion of other sequelae of cSVD, it is clear there is a dynamic interaction between LS, WMH, and CMI. Incident LS can occur in a WMH “penumbra”, and high order vessel disease may be involved in CMI. Perhaps flow-limiting lesions affecting lower-order branched vessels contribute to hypoperfusion distally. When subsequent branches develop worsening pathology and/or when perfusion decreases further in these already vulnerable areas, LS occur and WMH progress. These processes are dynamic, where LS can become undetectable and WMH can regress, occurring through a balance of blood flow changes, focal cSVD changes, and parenchymal repair processes (Figure 4).
Figure 4.
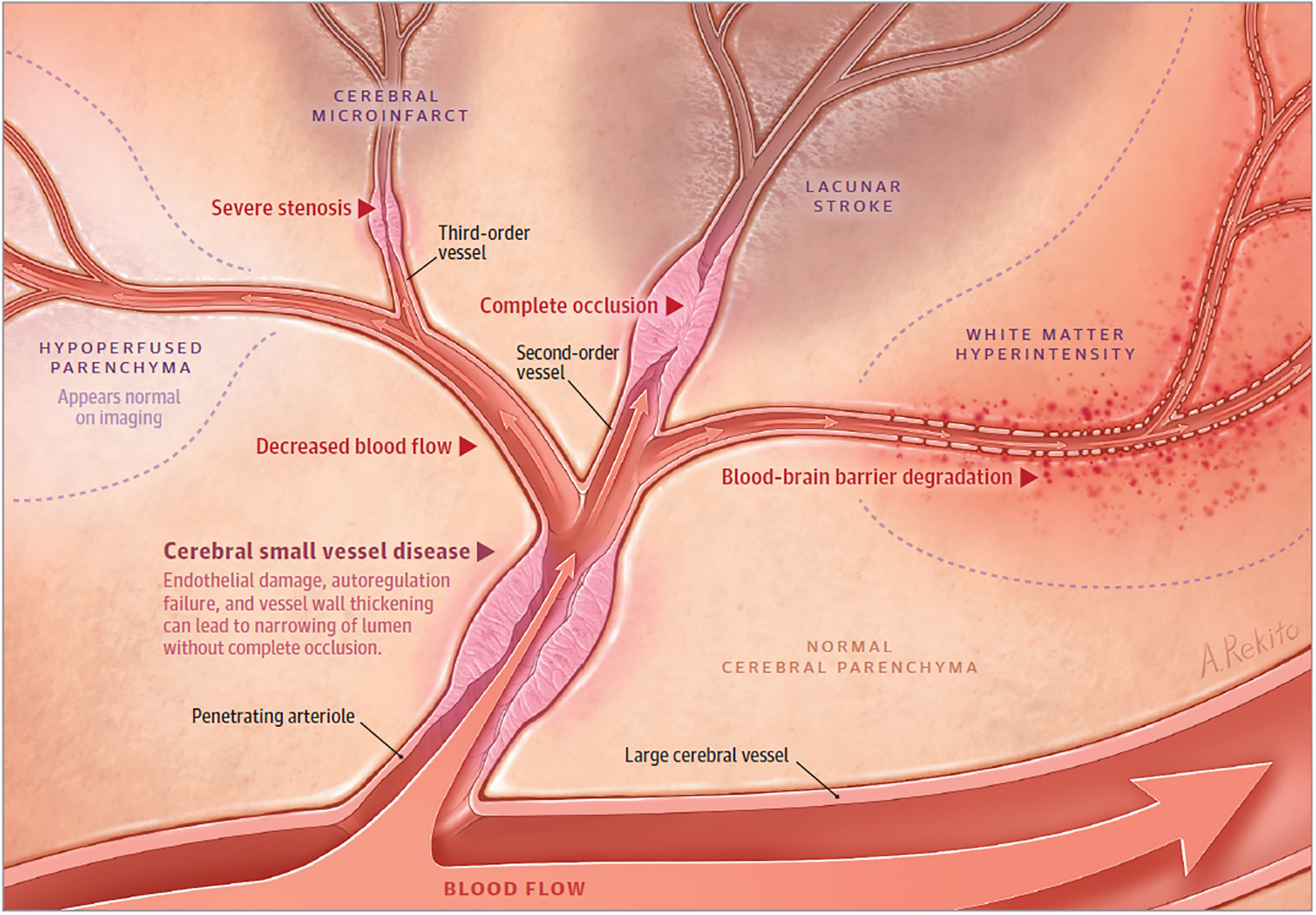
Originating from a large vessel, a penetrating arteriole is shown with narrowing and impaired autoregulation from cerebral small vessel disease. Downstream, there is hypoperfused parenchyma that appears normal on imaging. In the setting of occluded cerebral small vessel disease in a second-order vessel, lacunar stroke occurs. In higher-order vessels with occluded cerebral small vessel disease, smaller lacunar strokes and cerebral microinfarcts occur. Areas of blood-brain barrier degradation with decreased cerebral blood flow result in white matter hyperintensity. Lacunar strokes align with penetrating vessels, but they also have a predilection to form at the edge of white matter hyperintensities. Hypoperfused parenchyma can progress to lacunar stroke and white matter hyperintensity, although these sequelae of cerebral small vessel disease can also improve over time. The dynamic nature of lacunar stroke likely results from a balance of blood flow changes, focal cerebral small vessel disease changes, and parenchymal repair processes.
Several knowledge gaps need to be addressed. Future studies should focus on the initiation and progression of both the vessel and parenchymal pathology. What are the relative roles of aging, oxidative stress, mechanical stress, genetic predisposition, and other vascular risk factors? Autopsy studies comparing present day cSVD pathology to the lipohyalinosis of Fisher’s time may yield insight as hypertension is better controlled today. Are fibrinoid deposition and necrosis as prevalent as they once were? Has cSVD evolved over time? Furthermore, the time dependent order of events needs to be confirmed in more controlled experiments. Is there evidence of these injury mechanisms before BBB degradation and endothelial dysfunction? Does this cause decreased blood flow and ischemia? Does functional damage precede the pathologic vessel thickening? While progress has been made understanding endothelial dysfunction, future work on the role of media and adventitial dysfunction, including mechanisms of bleeding and perivascular leakage, may prove fruitful. Recent advances in mapping the brain vasculome may generate new hypotheses.69 Detailed gene and protein expression profiles for endothelium and perivascular cells may show how function is regulated at different levels of the vascular tree and how homeostasis is affected by co-morbidities and disease. Questions about the best therapeutics persist and may offer further insight into mechanism. Are phosphodiesterase inhibitors more effective than other antiplatelet agents? Does the presence of headache or augmentation of CBF after their use predict effectiveness? Do statins have a salutary effect on the endothelium in cSVD? Even more importantly, what are new therapeutic targets?
For many of these basic questions, experimental models may enhance our understanding. However, they should be chosen carefully as none perfectly mimics the human condition; consideration of several models in complementary fashion may be worthwhile. A recent analysis discussed three main categories of cSVD models: chronically hypertensive animals, chronically hypoperfused animals, and animals that undergo targeted small focal infarcts.70 One chronically hypertensive model is the stroke-pone Spontaneously Hypertensive Rat,71 which shares several features with the human condition such as BBB degradation and impaired vascular reactivity. Bilateral carotid artery occlusion is the most common model of hypoperfusion, which ultimately causes white matter injury.72 For targeted focal infarcts, laser coagulation models and stereotactic injection of vasoconstrictors, such as endothelin-173 or N5-(1-iminoethyl)-L-ornithine74 can be used. These focal infarcts can be targeted to the subcortical white matter, creating lesions that induce a phenotype of hemiparesis similar to human LS.74 Transgenic mouse models are also utilized; COL4A1/2 models replicate cSVD,43 while Notch models exhibit WMH with minimal vessel pathology.40 Lastly, in vitro models of oligodendrocytes and blood brain barrier also exist.75
While experimental models will provide the opportunity to ask certain questions that cannot be tested in humans, advances in the clinical science of this field continue to move forward. Translation and reverse translation will be important for future research efforts. Recent progress in understanding these disease processes at the clinical level has been made through advancements in imaging and genetics. Currently, there are few targeted treatments for LS. In the future, therapeutics aimed at targeting cSVD mechanisms and white matter injury and repair are urgently needed.
Table 1. Recent genetic studies of lacunar stroke (LS) and cerebral small vessel disease (cSVD).
SNP (single nucleotide polymorphism), GWAS (genome wide association study), WES (whole exome sequencing), ICH (intracerebral hemorrhage), WMH (white matter hyperintensity).
| Study Type | Related Gene | Biomarker Studied | SNP | Locus |
|---|---|---|---|---|
| Single Gene43 | COL4A1 | LS, ICH | rs515201 | 13q34 |
| Single Gene44 | COL4A2 | LS, ICH | rs4771674 | 13q34 |
| Single Gene42 | COL3A1 | LS | rs1800255 | 2q32.2 |
| Single Gene44 | HTRA1 | LS, ICH | rs79043147 | 10q26.13 |
| Single Gene45 | ACE | LS | rs464994 | 17q23.3 |
| Single Gene46 | MTHFR | LS, WMH | rs1801133 | 1p36.22 |
| Single Gene47 | APOE | WMH | rs429358 | 19q13.32 |
| GWAS51 | ZCCH14 | LS, WHM | rs12445022 | 16q24.2 |
| GWAS52 | TRIM65 | WMH | rs3744028 | 17q25 |
| GWAS52 | TRIM47 | WMH | rs1055129 | 17q25 |
| GWAS52 | PMF1 | WMH | rs1052053 | 1q22 |
| GWAS53 | SH3PXD2A | WMH | rs12357919 | 10q24.33 |
| GWAS53 | HAAO | WMH | rs11679640 | 2p21 |
| GWAS53 | PMF1-BGLAP | WMH | rs2984613 | 1q22 |
| GWAS53 | EFEMP1 | WMH | rs78857879 | 2p16.1 |
| WES54 | C1ORF156 | Stroke | rs1048177 | 1q24.2 |
| WES54 | XYLB | Stroke | rs17118 | 3p21.3 |
| WES55 | CSN3 | LS | N/A | 4q13.3 |
| WES55 | HLA-DPB1 | LS | N/A | 6p21.32 |
| WES56 | SH3TC1 | WMH | N/A | 4p16.1 |
Acknowledgements:
We wish to acknowledge Stephen M Greenberg, MD, PhD1 for providing comments to help guide this work. All conflict of interest disclosure information for all authors is accurate, complete, and up-to-date. RWR is supported by R25 NS065743. EHL is supported by P01 NS055104, R01 NS099620, R01 AG055559, and R01 NS093415. Each author had full access to the review and takes responsibility for the integrity the analysis.
References
- 1.Regenhardt RW, Das AS, Stapleton CJ, et al. Blood pressure and penumbral sustenance in stroke from large vessel occlusion. Front Neurol. 2017;8(JUL). doi: 10.3389/fneur.2017.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caplan LR. Lacunar Infarction and Small Vessel Disease: Pathology and Pathophysiology. J Stroke. 2015;17(1):2. doi: 10.5853/jos.2015.17.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Norrving B Long-term prognosis after lacunar infarction. Lancet Neurol. 2003;2(4):238–245. http://www.ncbi.nlm.nih.gov/pubmed/12849212. Accessed August 11, 2017. [DOI] [PubMed] [Google Scholar]
- 4.Fisher CM. The arterial lesions underlying lacunes. Acta Neuropathol. 1968;12(1):1–15. [DOI] [PubMed] [Google Scholar]
- 5.Vermeer SE, Longstreth WT, Koudstaal PJ. Silent brain infarcts: a systematic review. Lancet Neurol. 2007;6(7):611–619. doi: 10.1016/S1474-4422(07)70170-9. [DOI] [PubMed] [Google Scholar]
- 6.Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013;12(8):822–838. doi: 10.1016/S1474-4422(13)70124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Charidimou A, Pantoni L, Love S. The concept of sporadic cerebral small vessel disease : A road map on key definitions and current concepts. Int J Stroke. 2016;11(1):6–18. doi: 10.1177/1747493015607485. [DOI] [PubMed] [Google Scholar]
- 8.Thal DR, Grinberg LT, Attems J. Vascular dementia: different forms of vessel disorders contribute to the development of dementia in the elderly brain. Exp Gerontol. 2012;47(11):816–824. doi: 10.1016/j.exger.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan MYQ, Singhal S, Ma H, et al. Examining Subcortical Infarcts in the Era of Acute Multimodality CT Imaging. Front Neurol. 2016;7:220. doi: 10.3389/fneur.2016.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Del Brutto OH, Mera RM, Gillman J, Ha J, Zambrano M. Calcifications in the carotid siphon correlate with silent cerebral small vessel disease in community-dwelling older adults: A population-based study in rural Ecuador. Geriatr Gerontol Int. 2016;16(9):1063–1067. doi: 10.1111/ggi.12599. [DOI] [PubMed] [Google Scholar]
- 11.Song T-J, Kim YD, Yoo J, et al. Association between Aortic Atheroma and Cerebral Small Vessel Disease in Patients with Ischemic Stroke. J Stroke. 2016;18(3):312–320. doi: 10.5853/jos.2016.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Del Bene A, Makin SDJ, Doubal FN, Inzitari D, Wardlaw JM. Variation in Risk Factors for Recent Small Subcortical Infarcts With Infarct Size, Shape, and Location. Stroke. 2013;44(11):3000–3006. doi: 10.1161/STROKEAHA.113.002227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park Y, Chung P-W, Kim YB, et al. Small Deep Infarction in Patients with Atrial Fibrillation: Evidence of Lacunar Pathogenesis. Cerebrovasc Dis 2013;36(3):205–210. doi: 10.1159/000353736. [DOI] [PubMed] [Google Scholar]
- 14.Poggesi A, Pasi M, Pescini F, Pantoni L, Inzitari D. Circulating biologic markers of endothelial dysfunction in cerebral small vessel disease: A review. J Cereb Blood Flow Metab. 2016;36(1):72–94. doi: 10.1038/jcbfm.2015.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wardlaw JM, Smith C, Dichgans M. Mechanisms of sporadic cerebral small vessel disease: insights from neuroimaging. Lancet Neurol. 2013;12(5):483–497. doi: 10.1016/S1474-4422(13)70060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giwa MO, Williams J, Elderfield K, et al. Neuropathologic evidence of endothelial changes in cerebral small vessel disease. Neurology. 2012;78(3):167–174. doi: 10.1212/WNL.0b013e3182407968. [DOI] [PubMed] [Google Scholar]
- 17.Ahmed-Jushuf F, Jiwa NS, Arwani AS, et al. Age-dependent expression of VEGFR2 in deep brain arteries in small vessel disease, CADASIL, and healthy brains. Neurobiol Aging. 2016;42:110–115. doi: 10.1016/j.neurobiolaging.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Bagi Z, Brandner DD, Le P, et al. Vasodilator Dysfunction and Oligodendrocyte Dysmaturation in Aging White Matter. Ann Neurol. 2017;83(1):142–152. doi: 10.1002/ana.25129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wardlaw JM, Doubal FN, Valdes-Hernandez M, et al. Blood-Brain Barrier Permeability and Long-Term Clinical and Imaging Outcomes in Cerebral Small Vessel Disease. Stroke. 2013;44(2):525–527. doi: 10.1161/STROKEAHA.112.669994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muñoz Maniega S, Chappell FM, Valdés Hernández MC, et al. Integrity of normal-appearing white matter: Influence of age, visible lesion burden and hypertension in patients with small-vessel disease. J Cereb Blood Flow Metab. 2017;37(2):644–656. doi: 10.1177/0271678X16635657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arba F, Leigh R, Inzitari D, et al. Blood–brain barrier leakage increases with small vessel disease in acute ischemic stroke. Neurology. 2017;89(21):2143–2150. doi: 10.1212/WNL.0000000000004677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim Y, Lee H, An S-A, et al. The Effect of Pulsatility Index on Infarct Volume in Acute Lacunar Stroke. Yonsei Med J. 2016;57(4):950. doi: 10.3349/ymj.2016.57.4.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo Z-N, Xing Y, Wang S, Ma H, Liu J, Yang Y. Characteristics of dynamic cerebral autoregulation in cerebral small vessel disease: Diffuse and sustained. Sci Rep 2015;5:15269. doi: 10.1038/srep15269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bivard A, Cheng X, Lin L-T, et al. Global White Matter Hypoperfusion on CT Predicts Larger Infarcts and Hemorrhagic Transformation after Acute Ischemia. CNS Neurosci Ther 2016;22(3):238–243. doi: 10.1111/cns.12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sam K, Crawley AP, Poublanc J, et al. Vascular Dysfunction in Leukoaraiosis. Am J Neuroradiol 2016;37(12):2258–2264. doi: 10.3174/ajnr.A4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Veen PH, Muller M, Vincken KL, et al. Longitudinal Relationship Between Cerebral Small-Vessel Disease and Cerebral Blood Flow: The Second Manifestations of Arterial Disease-Magnetic Resonance Study. Stroke. 2015;46(5):1233–1238. doi: 10.1161/STROKEAHA.114.008030. [DOI] [PubMed] [Google Scholar]
- 27.Bernbaum M, Menon BK, Fick G, et al. Reduced blood flow in normal white matter predicts development of leukoaraiosis. J Cereb Blood Flow Metab. 2015;35(10):1610–1615. doi: 10.1038/jcbfm.2015.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi Y, Thrippleton MJ, Makin SD, et al. Cerebral blood flow in small vessel disease: A systematic review and meta-analysis. J Cereb Blood Flow Metab. 2016;36(10):1653–1667. doi: 10.1177/0271678X16662891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Traylor M, Bevan S, Baron J-C, Hassan A, Lewis CM, Markus HS. Genetic Architecture of Lacunar Stroke. Stroke. 2015;46(9):2407–2412. doi: 10.1161/STROKEAHA.115.009485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hilal S, Sikking E, Shaik MA, et al. Cortical cerebral microinfarcts on 3T MRI. Neurology. 2016;87(15):1583–1590. doi: 10.1212/WNL.0000000000003110. [DOI] [PubMed] [Google Scholar]
- 31.Arvanitakis Z, Capuano AW, Leurgans SE, Buchman AS, Bennett DA, Schneider JA. The Relationship of Cerebral Vessel Pathology to Brain Microinfarcts. Brain Pathol. 2017;27(1):77–85. doi: 10.1111/bpa.12365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith EE, Schneider JA, Wardlaw JM, Greenberg SM. Cerebral microinfarcts: the invisible lesions. Lancet Neurol. 2012;11(3):272–282. doi: 10.1016/S1474-4422(11)70307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phan TG, van der Voort S, Beare R, et al. Dimensions of Subcortical Infarcts Associated with First- to Third-Order Branches of the Basal Ganglia Arteries. Cerebrovasc Dis 2013;35(3):262–267. doi: 10.1159/000348310. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt R, Seiler S, Loitfelder M. Longitudinal change of small-vessel disease-related brain abnormalities. J Cereb Blood Flow Metab. 2016;36(1):26–39. doi: 10.1038/jcbfm.2015.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi Y, Wardlaw JM. Update on cerebral small vessel disease: a dynamic whole-brain disease. BMJ. 2016;1(3):83–92. doi: 10.1136/svn-2016-000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wardlaw JM, Chappell FM, Valdés Hernández MDC, et al. White matter hyperintensity reduction and outcomes after minor stroke. Neurology. 2017;89(10):1003–1010. doi: 10.1212/WNL.0000000000004328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duering M, Csanadi E, Gesierich B, et al. Incident lacunes preferentially localize to the edge of white matter hyperintensities: insights into the pathophysiology of cerebral small vessel disease. Brain. 2013;136(9):2717–2726. doi: 10.1093/brain/awt184. [DOI] [PubMed] [Google Scholar]
- 38.Gesierich B, Duchesnay E, Jouvent E, et al. Features and Determinants of Lacune Shape. Stroke. 2016;47(5):1258–1264. doi: 10.1161/STROKEAHA.116.012779. [DOI] [PubMed] [Google Scholar]
- 39.Choi JC. Genetics of Cerebral Small Vessel Disease. J Stroke. 2015;17(1):7. doi: 10.5853/jos.2015.17.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joutel A, Faraci FM. Cerebral Small Vessel Disease: Insights and Opportunities From Mouse Models of Collagen IV-Related Small Vessel Disease and Cerebral Autosomal Dominant Arteriopathy With Subcortical Infarcts and Leukoencephalopathy. Stroke. 2014;45(4):1215–1221. doi: 10.1161/STROKEAHA.113.002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rannikmae K, Davies G, Thomson PA, et al. Common variation in COL4A1/COL4A2 is associated with sporadic cerebral small vessel disease. Neurology. 2015;84(9):918–926. doi: 10.1212/WNL.0000000000001309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lv W, Lin Y, Song W, et al. Variants of COL3A1 Are Associated with the Risk of Stroke Recurrence and Prognosis in the Chinese Population: a Prospective Study. J Mol Neurosci. 2014;53(2):196–203. doi: 10.1007/s12031-014-0283-x. [DOI] [PubMed] [Google Scholar]
- 43.Weng Y-C, Sonni A, Labelle-Dumais C, et al. COL4A1 mutations in patients with sporadic late-onset intracerebral hemorrhage. Ann Neurol. 2012;71(4):470–477. doi: 10.1002/ana.22682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rannikmäe K, Sivakumaran V, Millar H, et al. COL4A2 is associated with lacunar ischemic stroke and deep ICH: Meta-analyses among 21,500 cases and 40,600 controls. Neurology. 2017;89(17):1829–1839. doi: 10.1212/WNL.0000000000004560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao J, Qin X, Li S, Zeng Z. Association between the ACE I/D polymorphism and risk of ischemic stroke: An updated meta-analysis of 47,026 subjects from 105 case–control studies. J Neurol Sci. 2014;345(1–2):37–47. doi: 10.1016/j.jns.2014.07.023. [DOI] [PubMed] [Google Scholar]
- 46.Rutten-Jacobs LCA, Traylor M, Adib-Samii P, et al. Association of MTHFR C677T Genotype With Ischemic Stroke Is Confined to Cerebral Small Vessel Disease Subtype. Stroke. 2016;47(3):STROKEAHA.115.011545. doi: 10.1161/STROKEAHA.115.011545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo X, Jiaerken Y, Yu X, et al. Associations between APOE genotype and cerebral small-vessel disease: a longitudinal study. Oncotarget. 2017;8(27):44477–44489. doi: 10.18632/oncotarget.17724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim HJ, Ye BS, Yoon CW, et al. Effects of APOE ɛ4 on brain amyloid, lacunar infarcts, and white matter lesions: a study among patients with subcortical vascular cognitive impairment. Neurobiol Aging. 2013;34(11):2482–2487. doi: 10.1016/j.neurobiolaging.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 49.Lee S, Viqar F, Zimmerman ME, et al. White matter hyperintensities are a core feature of Alzheimer’s disease: Evidence from the dominantly inherited Alzheimer network. Ann Neurol. 2016;79(6):929–939. doi: 10.1002/ana.24647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Traylor M, Anderson CD, Hurford R, Bevan S, Markus HS. Oxidative phosphorylation and lacunar stroke. Neurology. 2016;86(2):141–145. doi: 10.1212/WNL.0000000000002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Traylor M, Malik R, Nalls MA, et al. Genetic variation at 16q24.2 is associated with small vessel stroke. Ann Neurol. 2017;81(3):383–394. doi: 10.1002/ana.24840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lopez LM, Hill WD, Harris SE, et al. Genes From a Translational Analysis Support a Multifactorial Nature of White Matter Hyperintensities. Stroke. 2015;46(2):341–347. doi: 10.1161/STROKEAHA.114.007649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Verhaaren BFJ, Debette S, Bis JC, et al. Multiethnic Genome-Wide Association Study of Cerebral White Matter Hyperintensities on MRICLINICAL PERSPECTIVE. Circ Cardiovasc Genet 2015;8(2):398–409. doi: 10.1161/CIRCGENETICS.114.000858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, Tong Y, Zhang Y, et al. Two Novel Susceptibility SNPs for Ischemic Stroke Using Exome Sequencing in Chinese Han Population. Mol Neurobiol. 2014;49(2):852–862. doi: 10.1007/s12035-013-8561-0. [DOI] [PubMed] [Google Scholar]
- 55.Cole JW, Stine OC, Liu X, et al. Rare variants in ischemic stroke: an exome pilot study. PLoS One. 2012;7(4):e35591. doi: 10.1371/journal.pone.0035591 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fornage M, Jian X, Chouraki V, et al. Whole exome sequence analysis of white matter hyperintensities on cranial MRI. Alzheimer’s Dement. 2015;11(7):P278–P279. doi: 10.1016/j.jalz.2015.07.371. [DOI] [Google Scholar]
- 57.Bersano A, Zuffardi O, Pantoni L, et al. Next Generation Sequencing for Systematic Assessment of Genetics of Small-Vessel Disease and Lacunar Stroke. J Stroke Cerebrovasc Dis. 2015;24(4):759–765. doi: 10.1016/j.jstrokecerebrovasdis.2014.10.019. [DOI] [PubMed] [Google Scholar]
- 58.Deplanque D, Lavallee PC, Labreuche J, et al. Cerebral and Extracerebral Vasoreactivity in Symptomatic Lacunar Stroke Patients: A Case-Control Study. Int J Stroke. 2013;8(6):413–421. doi: 10.1111/j.1747-4949.2011.00755.x. [DOI] [PubMed] [Google Scholar]
- 59.Saji N, Kimura K, Yagita Y, Kawarai T, Shimizu H, Kita Y. Comparison of arteriosclerotic indicators in patients with ischemic stroke: ankle–brachial index, brachial–ankle pulse wave velocity and cardio–ankle vascular index. Hypertens Res 2015;38(5):323–328. doi: 10.1038/hr.2015.8. [DOI] [PubMed] [Google Scholar]
- 60.Del Brutto OH, Sedler MJ, Mera RM, et al. The Association of Ankle-Brachial Index with Silent Cerebral Small Vessel Disease: Results of the Atahualpa Project. Int J Stroke. 2015;10(4):589–593. doi: 10.1111/ijs.12450. [DOI] [PubMed] [Google Scholar]
- 61.Poels MMF, Zaccai K, Verwoert GC, et al. Arterial Stiffness and Cerebral Small Vessel Disease: The Rotterdam Scan Study. Stroke. 2012;43(10):2637–2642. doi: 10.1161/STROKEAHA.111.642264. [DOI] [PubMed] [Google Scholar]
- 62.Okada Y, Kohara K, Ochi M, et al. Mechanical Stresses, Arterial Stiffness, and Brain Small Vessel Diseases. Stroke. 2014;45(11):3287–3292. doi: 10.1161/STROKEAHA.114.006539. [DOI] [PubMed] [Google Scholar]
- 63.Makin SDJ, Cook FAB, Dennis MS, Wardlaw JM. Cerebral Small Vessel Disease and Renal Function: Systematic Review and Meta-Analysis. Cerebrovasc Dis 2015;39(1):39–52. doi: 10.1159/000369777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akoudad S, Sedaghat S, Hofman A, et al. Kidney Function and Cerebral Small Vessel Disease in the General Population. Int J Stroke. 2015;10(4):603–608. doi: 10.1111/ijs.12465. [DOI] [PubMed] [Google Scholar]
- 65.Xiao L, Lan W, Sun W, et al. Chronic Kidney Disease in Patients With Lacunar Stroke. Stroke. 2015;46(8):2081–2086. doi: 10.1161/STROKEAHA.114.008155. [DOI] [PubMed] [Google Scholar]
- 66.Yang S, Cai J, Lu R, Wu J, Zhang M, Zhou X. Association Between Serum Cystatin C Level and Total Magnetic Resonance Imaging Burden of Cerebral Small Vessel Disease in Patients With Acute Lacunar Stroke. J Stroke Cerebrovasc Dis 2017;26(1):186–191. doi: 10.1016/j.jstrokecerebrovasdis.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 67.Umemura T, Kawamura T, Hotta N. Pathogenesis and neuroimaging of cerebral large and small vessel disease in type 2 diabetes: A possible link between cerebral and retinal microvascular abnormalities. J Diabetes Investig. 2017;8(2):134–148. doi: 10.1111/jdi.12545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gobron C, Erginay A, Massin P, et al. Microvascular retinal abnormalities in acute intracerebral haemorrhage and lacunar infarction. Rev Neurol (Paris). 2014;170(1):13–18. doi: 10.1016/j.neurol.2013.07.029. [DOI] [PubMed] [Google Scholar]
- 69.Vanlandewijck M, He L, Mäe MA, et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature. 2018;554(7693):475–480. doi: 10.1038/nature25739. [DOI] [PubMed] [Google Scholar]
- 70.Hainsworth AH, Brittain JF, Khatun H. Pre-clinical models of human cerebral small vessel disease: Utility for clinical application. J Neurol Sci. 2012;322(1–2):237–240. doi: 10.1016/j.jns.2012.05.046. [DOI] [PubMed] [Google Scholar]
- 71.Regenhardt RW, Mecca AP, Desland F, et al. Centrally administered angiotensin-(1–7) increases the survival of stroke-prone spontaneously hypertensive rats. Exp Physiol. 2014;99(2):442–453. doi: 10.1113/expphysiol.2013.075242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hattori Y, Enmi J-i., Kitamura A, et al. A Novel Mouse Model of Subcortical Infarcts with Dementia. J Neurosci. 2015;35(9):3915–3928. doi: 10.1523/JNEUROSCI.3970-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Regenhardt RW, Desland F, Mecca AP, et al. Anti-inflammatory effects of angiotensin-(1–7) in ischemic stroke. Neuropharmacology. 2013;71:154–163. doi: 10.1016/j.neuropharm.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rosenzweig S, Carmichael ST. Age-dependent exacerbation of white matter stroke outcomes: a role for oxidative damage and inflammatory mediators. Stroke. 2013;44(9):2579–2586. doi: 10.1161/STROKEAHA.113.001796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Naik P, Cucullo L. In vitro blood-brain barrier models: current and perspective technologies. J Pharm Sci. 2012;101(4):1337–1354. doi: 10.1002/jps.23022. [DOI] [PMC free article] [PubMed] [Google Scholar]