

Abstract
Synaptotagmin-7 (Syt-7) is one of two major calcium sensors for exocytosis in adrenal chromaffin cells, the other being synaptotagmin-1 (Syt-1). Despite a broad appreciation for the importance of Syt-7, questions remain as to its localization, function in mediating discharge of dense core granule cargos, and role in triggering release in response to physiological stimulation. These questions were addressed using two distinct experimental preparations – mouse chromaffin cells lacking endogenous Syt-7 (KO cells) and a reconstituted system employing cell-derived granules expressing either Syt-7 or Syt-1. First, using immunofluorescence imaging and subcellular fractionation, it is shown that Syt-7 is widely distributed in organelles, including dense core granules. Total internal reflection fluorescence (TIRF) imaging demonstrates that the kinetics and probability of granule fusion in Syt-7 KO cells stimulated by a native secretagogue, acetylcholine, are markedly lower than in WT cells. When fusion is observed, fluorescent cargo proteins are discharged more rapidly when only Syt-1 is available to facilitate release. To determine the extent to which the aforementioned results are attributable purely to Syt-7, granules expressing only Syt-7 or Syt-1 were triggered to fuse on planar supported bilayers bearing plasma membrane SNARE proteins. Here, as in cells, Syt-7 confers substantially greater calcium sensitivity to granule fusion than Syt-1 and slows the rate at which cargos are released. Overall, this study demonstrates that by virtue of its high affinity for calcium and effects on fusion pore expansion, Syt-7 plays a central role in regulating secretory output from adrenal chromaffin cells.
Graphical Abstract
Syt-7 is a high-affinity calcium sensor expressed on chromaffin cell dense core granules. The purpose of this study was to assess the role of Syt-7 in regulating the secretory response to cholinergic stimulation. Acetylcholine elicits secretion by elevating cytosolic calcium. Syt-7 is required for a normal secretory response to acetylcholine. Cells that lack Syt-7 also release peptide hormones at faster rates, implicating a role for Syt-7 in regulating the exocytotic fusion pore. These data demonstrate that Syt-7 has an important role in regulating calcium-triggered exocytosis in cells and is likely to play a role in regulating hormone output, in situ.
Introduction
Adrenomedullary chromaffin cells serve as a key effector arm of the sympathetic nervous system. In response to stimulation by preganglionic sympathetic nerves, they secrete a number of important hormones, including epinephrine, norepinephrine, neuropeptide Y (NPY), and tissue plasminogen activator (tPA) directly into the circulation (Guerineau 2019; Carmichael & Winkler 1985). The trigger for stimulus-evoked exocytosis in adrenal chromaffin cells is a rise in intracellular Ca2+. The level to which intracellular Ca2+ is elevated varies with the stimulus intensity and secretagogue (Augustine & Neher 1992; Fulop et al. 2005; Fulop & Smith 2007). Ca2+ regulates release by acting on the Ca2+-binding Syt-protein family (Chapman 2008; Geppert et al. 1994; Sudhof 2013) to drive its penetration into membranes that harbor anionic lipids (Bai et al. 2000; Bai et al. 2004). These actions result in subsequent zippering of the SNARE complex and opening of a fusion pore to permit diffusion or discharge of granule lumenal contents into the extracellular space (Murthy & De Camilli 2003).
Two Syts in particular – Syt-1 and Syt-7 –account for the vast majority of Ca2+-triggered release from chromaffin cells (Schonn et al. 2008). Although these proteins share a similar topology, they exhibit a number of biochemical differences. For example, in vitro, Syt-7 has a much higher affinity for Ca2+ in the presence of phospholipid membranes than Syt-1 (Bhalla et al. 2005; Sugita et al. 2002). In fact, Syt-7 stimulation of SNARE-mediated liposome fusion occurs with a 400-fold higher sensitivity to Ca2+ than Syt-1 (Bhalla et al. 2005). Once bound to Ca2+, Syt-7 also releases membranes at rates that are orders of magnitude faster than Syt-1(Bhalla et al. 2005; Sugita et al. 2002; Hui et al. 2005; Bendahmane et al. 2018). The biochemical distinctions between Syt-1 and Syt-7 have a strong bearing on their actions during chromaffin cell exocytosis. The much lower affinity of Syt-1 for Ca2+ compared to Syt-7 explains why small elevations in cytosolic Ca2+ levels from baseline are generally ineffective in driving fusion of granules bearing Syt-1, but much more effective at driving fusion of granules bearing Syt-7 (Rao et al. 2017a). Differences in the Ca2+ sensitivities of Syt isoforms, as well as their membrane association and dissociation rates, may also underlie the fast and slow kinetic components of the secretory response (Schonn et al. 2008). These components, revealed as a result of Ca2+ uncaging in combination with capacitance measurements, are differentially reliant on Syt-1 and Syt-7. The rapid phase of release is eliminated in the absence of Syt-1 (Schonn et al. 2008). The remaining delayed component exhibits a Ca2+-threshold that closely mirrors the low, micromolar-range sensitivity of Syt-7 for binding to phospholipids in the presence of Ca2+. Furthermore, deletion of Syt-7 in a Syt-1 knockout (KO) background, almost completely eliminates the slow phase of release (Schonn et al. 2008).
Thus, stimulation paradigms that elevate intracellular Ca2+ to varying degrees allow the impact of biochemical differences in the function of Syt-1 and Syt-7 on exocytosis to be readily appreciated. The purpose of this study is to delineate how such differences impinge on the release kinetics of cargos stored within dense core granules harboring Syt-7 and affect the secretory response to native, cholinergic stimulation. In order to define the localization and function of Syt isoforms – especially Syt-7 – in chromaffin cell exocytosis, multiple experimental preparations were employed. This includes primary chromaffin cells and an assay in which fusion of dense core granules with a planar membrane has been reconstituted (Kreutzberger et al. 2017b; Kreutzberger et al. 2017a; Kreutzberger et al. 2019). Immunolabeling of endogenous Syt proteins in mouse chromaffin cells as well as subcellular fractionation demonstrate that both Syt-1 and Syt-7 are principally sorted to intracellular organelles (including dense core granules). The role of Syt-7 in regulating the characteristics of exocytosis was ascertained by monitoring the discharge of fluorescently labeled lumenal cargo proteins in cells lacking endogenous Syt-7 (Syt-7 KO cells) using TIRF microscopy. These experiments revealed that cargos are discharged from Syt-7 KO cells at significantly faster rates than they are from WT cells, consistent with the idea that endogenous Syt-7 constrains fusion pore expansion (Jaiswal et al. 2004; Brahmachari et al. 1998; Rao et al. 2014; Rao et al. 2017a; Zhang et al. 2011).
The absence of Syt-7 has a pronounced detrimental effect on the secretory response to prolonged cholinergic stimulation. In fact, the likelihood of observing exocytosis at all declines sharply in KO cells after the first few seconds of perfusion with acetylcholine (ACh). In contrast, exocytotic events are evident throughout the stimulation period in WT cells, albeit with a lessening frequency as stimulation proceeds. Exocytosis is sustained in WT cells because of the high affinity of Syt-7 for Ca2+. This idea is underscored by reconstitution studies in which dense core granules consisting of only Syt-1 or Syt-7 were triggered to fuse with synthetic bilayers. Even in this setting, Syt-7-bearing granules fuse at significantly lower Ca2+ concentrations than Syt-1-bearing granules, with fusion pores that dilate at slower rates. Taken together, the data highlight clear functional distinctions in the roles of synaptotagmins. These properties have a direct bearing on the regulation of Ca2+-triggered exocytosis in cells and are likely to play a role in modulating adrenomedullary output, in situ.
Material and Methods for Chromaffin Cell Studies
Animals
Litters of adult male and female Syt-7 −/− (Catalogue # 004950, Jackson labs) (gift of Dr. Joel Swanson; (Chakrabarti et al. 2003) and Syt-7 +/+ (from a C57BL/6J background and obtained from Jackson Labs, Bar Harbor, ME, Catalogue # 000664) were used in these studies. Animals were group housed (2 to 5 per ventilated cage) with 24 hour (12/12 dark/light cycle) access to food and water. All animal procedures and experiments were conducted in accordance with the University of Michigan Institutional Animal Care and Use Committee protocol (PRO00007247). No randomization was performed to allocate subjects in the study.
Mouse chromaffin cells preparation and transfection
Below, we describe a novel method for the isolation and culture of adult mouse chromaffin cells from the adrenal medulla. Although the protocol was adapted from previous studies (Kolski-Andreaco et al. 2007), it is different enough to warrant a more detailed description. Catalogue numbers and vendors of materials used can be found in Table 1. 6 to 16 week-old animals (male and female; 18 to 25 g) were gas anesthetized using an isoflurane drop jar technique and sacrificed by guillotine decapitation. Chromaffin cells are responsible for releasing catecholamines in response to stress, including hypoxia. Isoflurane is used to induce a faster loss of consciousness compared to CO2 euthanasia (30 s to 1 minute versus several minutes) and reduce animal stress. 3–4 mice are euthanized per plate to ensure proper cell density and health. Adrenal glands per condition were rapidly isolated and moved to dishes containing ice cold dissection buffer (148 mM NaCl, 2.57 mM KCl, 2.2 mM K2HPO4•3H2O, 6.5 mM KH2PO4, 10 mM glucose, 5 mM HEPES free acid, 14.2 mM mannitol). Under a dissection microscope, the cortex was rapidly and carefully removed using thin forceps (Dumont Swissmade, Switzerland Cat. # 72891-Bx) and thin micro scissors (World Precision Instruments, 14124-G). The isolated medullas were washed three times in 150 μL drops of enzyme solution containing (450 units/ml Papain (Worthington Biochemical. #LS003126), 250 μg/ml BSA, and 75 μg/ml dithiothreitol). The medullas were then digested for 15 minutes in 0.5 ml of the enzyme solution at 37°C. After 15 minutes, the digesting solution was carefully removed and replaced by 0.5 ml of fresh enzyme solution and left for a maximum of 15 extra minutes at 37°C. The digestion was stopped by transferring the glands into an antibiotic-free culture medium (Dulbecco’s Modified Eagle’s Medium (DMEM) (ThermoFisher Scientific) supplemented with 10% Fetal Bovine Serum (FBS) (ThermoFisher Scientific)). The digested glands were then triturated by a push-pull movement through a 1 ml pipette tip (10 to 12 times). The suspension of digested and broken glands was spun at 1300 x g for 2.5 minutes. The supernatant was discarded, and the pellet was re-suspended in antibiotic-free medium and triturated again in a 200 μl pipette tip for a better cell dissociation (10 to 12 times). The suspension was spun again at 1300 x g for 2.5 minutes. After discarding the supernatant, the pellet was re-suspended in resuspension buffer (Invitrogen, Thermofisher Scientific) for transfection. The cells were rapidly counted and the desired plasmid was added (15 ng/106 cells). The suspended cells were then transiently transfected by electroporation with a single pulse (1050 mV, 40 ms) using the Neon transfection system (Invitrogen, Thermofisher Scientific). In parallel, 35 mm diameter dishes with 14 mm diameter glass-bottom dishes (MatTek Corporation, Ashland, MA. #P35G-1.5–14-C) were pre-coated with Matrigel (Corning, NY, Cat. #356230) diluted in DMEM (1:7) for two hours after which the dishes were washed with DMEM and let to dry. After electroporation, an antibiotic-free medium was added to cells to obtain a final concentration of 1 million cells per ml. Three hundred microliters of the final solution containing the electroporated cells were then deposited in each dish. The cells were stored in an incubator (37°C, 5% CO2) for three to five hours. Culture medium with antibiotics was then added to a final volume of 2 ml (DMEM supplemented with 10% FBS, 9.52 unit/ml Penicillin, 9.52 μg/ml Streptomycin and 238 μg/ml Gentamicin (ThermoFisher Scientific). The media was changed daily, and cells were used within 48 hours after plating. The method we describe here provides consistently healthy cells (Figure S1) which exhibit a high probability of secretion upon stimulation. As a matter of course, cell preps and experiments were usually performed during normal working hours (9 am – 5 pm).
Table 1.
List of Materials
| Chemical/materials | Vendor | Catalog number | notes |
|---|---|---|---|
| Fluriso (Isofluorane) | Vetone | H03A18B | |
| Sodium Chloride | Fisher BioReagents | BP-358–212 | |
| Potassium Chloride | EMD Chemicals | PX1405–1 | |
| Glucose | Sigma | G8270 | |
| Potassium Phosphate monobasic | EMD Chemicals | PX1565–1 | |
| Potassium Phosphate Dibasic | Sigma | P966 | |
| HEPES | Sigma | H3375 | |
| D-Mannitol | Sigma | M4125 | |
| Micro scissors | World Precision Instruments | 14124-G | |
| Papain | Worthington Biochemical | LS003126 | |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | A7906 | |
| Dulbecco’s Modified Eagle’s Medium | ThermoFisher Scientific | F12 [+] L-glutamine [+] 15mM HEPES |
|
| Fetal Bovine Serum (FBS) | Gibco by Life Technologies | 10437–028 | |
| Calcium Chloride | Sigma | C1016 | |
| Magnesium Chloride | Fisher BioReagents | BP214 | |
| Acetylcholine | Sigma | A6625 | |
| Paraformaldehyde | Polysciences | 18814 | |
| Ammonium Chloride | Sigma | A9434 | |
| Gelatin | Sigma | G9391 | |
| MES | Sigma | M3671 | |
| Rabbit polyclonal Anti-syt-7 antibody | Synaptic Systems | 105 173 RRID: AB_887838 |
1:1200 |
| Polyclonal anti-PAI-1 | Abcam | 66705 RRID:AB_1310540 |
1:1000 |
| Uncongugated F(ab’)2-Goat anti-Rabbit IgG | Invitrogen, Thermofisher | A24539 RRID:AB_2536007 |
1:300 |
| Normal Rabbit Serum | Invitrogen, Thermofisher | 016101 RRID:AB_2532937 |
|
| Mouse anti-alpha tubulin | Cedarlane | CLT 9002 RRID:AB_10060319 |
1:10000 |
| Mouse monoclonal anti-LAMP-1 | Abcam | Ab25630 RRID:AB_470708 |
1:500 |
| Rabbit polyclonal anti-LAMP-1 | Abcam | Ab24170 RRID:AB_775978 |
1:1000 |
| Mouse Monoclonal Anti-Syt-1 |
Synaptic Systems | 105 011 RRID:AB_887832 |
1:1200 |
| Licor fluorescent secondaries | IRDye 800CW Donkey Anti-rabbit |
C9012905 | 1:10000 |
| Licor fluorescent secondaries | IRDye 680CW Donkey Anti Rabbit | C92568073 | 1:10000 |
| anti-mouse HRP | GE healthcare | NXA931V RRID: AB_2721110 |
1:5000 |
| anti-rabbit HRP | GE healthcare | NA934V RRID: AB_772206 |
1:5000 |
| SDS | Sigma Life Sciences | L3771 | |
| Glass Bottom Dishes | MatTek Corporation, Ashland MA | P35G-1.5-14-C | |
| Matrigel | Corning, NY | 356230 | |
| Sucrose | Sigma | 84100 | |
| Tris HCl | Roche Diagnostics | 10-708-976-001 | |
| EDTA | Fisher Chemical | S311–100 | |
| mycSyt-7 plasmid | Gifted by Dr. Thomas Sudhof | ||
| NPY-pHluorin | Gifted from Dr. Ronald Holz | Vector: pEGFP-N1 | Kanamycin resistance |
| NPY-GFP | Gifted from Dr. Ronald Holz | Vector: pEGFP-N1 | Ampicillin resistance |
| NPY-mRuby | Gifted from Ronald Holz | Vector: pEGFP-N1 | Ampicillin resistance |
| tPA-pHl | Gifted from Ronald Holz | Vector: pEGFP-N1 | Ampicillin resistance |
| Sylgard elastomer | EMS, Hatfield, PA | 24236–10 |
Chromaffin cells were transfected with rat myc-Syt-7 or cargo proteins, including human Neuropeptide Y (NPY) and human tissue plasminogen activator (tPA). The mycSyt-7 plasmid was a gift from Dr. Thomas Sudhof. The fluorescent tag was located following the C-terminal region of the cargo proteins. NPY and tPA constructs (originally in pEGFP-N1 vectors) were provided by Dr. Ronald W. Holz.
Approximate numbers of animals used per experimental group are listed below:
Immunocytochemistry (Syt-7 + Syt-1): 4 KO and 4 WT
Immunocytochemistry (Syt-7 + PAI-1): 8 KO and 8 WT
Secretion (KCl): 46 KO and 46 WT
Secretion (ACh): 32 KO and 32 WT
Ca2+ imaging: 12 KO and 12 WT
Electrophysiology: 12 KO and 12 WT
Total: 114 KO and 114 WT (males and females, mixed)
Western blotting
Catalogue numbers, vendors of materials used and appropriate dilutions can be found in Table 1. Lysis buffer containing 8 M urea, 5% SDS, and 5 mM N-ethylmaleimide in water was heated to 65°C. 6 to 16 week-old animals (male and female; 18 to 25 g) were gas anesthetized using an isoflurane drop jar technique and sacrificed by guillotine decapitation. Adrenal glands were dissected from five months-old mice and immediately frozen in liquid nitrogen, then handed over to a colleague blinded to the genotype of the mice to complete the western blot. Four total adrenal glands from two mice of each genotype were dissolved into 200 μL of warm lysis buffer, and homogenized using a handheld, battery-operated homogenizer. The homogenate was incubated at 65°C for 20 minutes and mixed 1:1 with 5x PAGE buffer (5% (wt/vol) SDS, 25% (wt/vol) sucrose, 50 mM Tris pH 8, 5 mM EDTA, & bromophenol blue). The lysates were stored at −20°C until use. Samples (10 μL/well) were separated on a 4–12% NuPAGE Bis-Tris Gel in 1x NuPAGE MOPS SDS Running Buffer, for 1 hour at 175 mV. Transfer to nitrocellulose membrane occurred at 120 mV for 1.5 hours, on ice, in 1x NuPAGE transfer buffer. The membrane was blocked with blocking buffer containing 5% Bovine Serum Albumin (BSA) and 0.1% tween in TBS (TBS-T) for 1 hour, before incubation with primary antibodies (Rabbit anti Synaptotagmin-7 1:1000, Synaptic Systems, and Mouse anti alpha-tubulin, 1:10,000, Cedarlane; RRID:AB_10060319) at 4°C, overnight. The membrane was washed 3× 15 minutes with TBS-T and incubated for 1 hour with LiCor fluorescent secondaries (1:10,000) in blocking buffer (multiplexed). After washing 3× 15 minutes in TBS-T, the membrane was imaged using LiCor Odyssey Clx imager.
Chromaffin cells are isolated from bovine adrenal glands as previously described (Holz et al. 1982). Briefly, adrenal glands were obtained from a slaughterhouse and delivered to the lab. Surrounding fat and connective tissues were removed carefully and adrenal glands were perfused with physiological saline solution (PSS) through adreno-lumbar vein to remove residual blood. Glands were then digested at 37°C by perfusing with enzymatic solution A (Liberase TH) for 30 – 40 minutes. Digested medullas were collected and digested again at 37°C in enzymatic solution B (Liberase TH and TL mixture) for 30 minutes under constant agitation. This is then passed through 400 μm filter to obtain chromaffin cells. Subcellular fractionation of chromaffin cells was performed following a published protocol (Kreutzberger et al. 2019) with slight modifications. The fractions (400 μl) were collected for western blotting. Blots containing the transferred proteins were blocked in 5% milk and incubated overnight at 4°C with the following primary antibodies: Rabbit anti Synaptotagmin-7 (1:1000, Synaptic Systems, Cat.no. 105173; RRID: AB_887838), Mouse anti Synaptotagmin-1 (1:1000, Synaptic Systems,Cat # 105 011, 1:1200; RRID: AB_887832), Rabbit anti PAI-1 (1:500, Abcam, Cat. no. ab66705; RRID: AB_1310540), Rabbit anti LAMP-1 (1:1000, Abcam, Cat. no. ab24170; RRID: AB_775978). Following primary antibody incubation, blots were washed with TBS-T and incubated with HRP conjugated secondary antibodies (anti-mouse HRP 1:5000, Cat.no. NXA931V; RRID: AB_2721110 or anti-rabbit HRP 1:5000, Cat.no NA934V; RRID: AB_772206) diluted in 5% milk. Chemiluminescent substrate (Thermofisher) was used to visualize blots on iBright Imager (Invitrogen). Protein band intensity was quantified using Fiji software.
Reverse transcription and quantitative PCR
Reverse transcription was performed on mouse adrenal medullas dissected from adrenal glands and homogenized. Adrenal medullas from two animals in each WT and Syt-7 KO group are considered as one experiment. Specifically, four adrenal medullas from two animals were homogenized in one 1.5-ml Eppendorf tube on ice for ~45 seconds with motorized pestle mixer (Argos Technologies, Inc, Vernon Hills, IL). More than three experiments were performed for each target. RNeasy Mini (Qiagen, Valencia, CA) was used to isolate the RNAs. The first strand cDNA synthesis was performed with 400 ng of RNAs using the qScript cDNA SuperMix kit (Quanta Biosciences, Beverly, MA). The reverse transcription product was kept at −20°C until qPCR was performed. qPCR primers for the target genes were designed with online tools (GenScript PCR Primer Design and NCBI primer designing tool). The forward (fw) and reverse (rv) primer sequences are as follows:
| GAPDH fw | CTGACGTGCCGCCTGGAGAA |
| GAPDH rv | CCCGGCATCGAAGGTGGAAGA |
| Syt-1 fw | GGCGCGATCTCCAGAGTGCT |
| Syt-1 rv | GCCGGCAGTAGGGACGTAGC |
| Syt-7 fw | CCAGACGCCACACGATGAGTC |
| Syt-7 rv | CCTTCCAGAAGGTCTGCATCTGG |
| NPY fw | GTGTGTTTGGGCATTCTGGC |
| NPY rv | TGTCTCAGGGCTGGATCTCT |
| tPA fw | CTCGGCCTGGGCAGACACAA |
| tPA rv | AGGCCACAGGTGGAGCATGG |
| TH fw | GCGCCGGAAGCTGATTGCAG |
| TH rv | CCGGCAGGCATGGGTAGCAT |
Glyceraldehyde 3-phosphatedehydrogenase (GAPDH) was used as an endogenous control run in parallel with target genes. Each assay was performed in triplicate. For the qPCR, we used the PerfeCTa SYBR Green SuperMix (Quanta Biosciences, Beverly, MA). Ten microliters of reverse transcription product was added to the master mix with 10 μM of each primer. The directions for the PCR protocol were followed per the manufacturer’s instructions. The qPCR was performed using the CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad, Hercules, CA). Melting curves were analyzed to verify that no primer dimers were produced.
TIRF microscopy for observation of exocytosis
TIRF imaging was performed using an Olympus cellTIRF-4Line microscope (Olympus, USA) equipped with a TIRF oil-immersion objective (NA 1.49) and an additional 2x lens in the emission path between the microscope and the cooled electron-multiplying charge-coupled device Camera (iXon 897; Andor Technology). The final pixel size of the images was 80 nm. Series of images were acquired at ~ 20 Hz using CellSense software with an exposure time of 30 ms and an EM gain of 100. pHl and GFP were excited using a 488 nm laser.
Cell stimulation
All TIRF experiments were performed at room temperature ~ 24° C. The culture medium was replaced by pre-warmed (37°C) physiological salt solution (PSS) (145 mM NaCl, 5.6 mM KCl, 2.2 mM CaCl2, 0.5 mM MgCl2, 5.6 mM glucose, and 15 mM HEPES, pH 7.4). Chromaffin cells were individually stimulated using a needle (100-μm inner diameter) connected to a perfusion system under positive pressure ALA-VM4 (ALA Scientific Instruments, Westbury, NY). To trigger exocytosis, cells were first perfused with PSS for 5 – 10 s and then stimulated with high potassium containing solution (100 mM KCl) for 70–75 s. For the acetylcholine experiment, 100 μM acetylcholine (Sigma-Aldrich) diluted in PSS was perfused for 120 s.
Image analysis
Fusion sites of granules containing GFP or pHl-tagged proteins undergoing exocytosis were identified. The abruptness with which NPY-GFP puncta disappear is taken as indication that fusion and release has occurred. In the event of slower cargo release, an increase in fluorescence often precedes the disappearance of GFP puncta as the granule moves closer to the brightest part of the evanescent field and the lumenal pH of the granule becomes more neutral (Taraska et al. 2003). Regions of interest (ROIs) measuring 0.8 μm diameter were manually selected at fusion sites and image sequences were analyzed using the Time Series Analyzer v3.0 plugin on Fiji software. For each ROI, the fluorescence intensity was measured for each frame. A nearby ROI of the same size where no fusion events were observed was selected for background subtraction. Using a custom program written in Interactive Data Language (IDL; ITT, Broomfield, CO) by Dr. Daniel Axelrod (University of Michigan), background subtracted intensity versus time curves were plotted and the duration of cargo release was calculated (Bohannon et al. 2017; Abbineni et al. 2019). Briefly, after a user selects fusion events from blinded files, the program generates fluorescence-versus-time curves for each one. A user selects a start time, tstart, just before fluorescence begins and an end time, tend, when fluorescence returns to baseline. The program determines the time of the maximum fluorescence tmax within this time window. The intervals (tstart, tmax) and (tmax, tend) are defined as the rise phase and fall phase, respectively. Each phase is fit with a fifth-degree polynomial function, and a weighted average slope is calculated (upward for the rising phase, and downward for the falling phase). The time period between the baseline intercept of the rising phase straight line and the falling phase straight line is considered to be the duration of the event (Bohannon et al. 2017). Each fusion event was further analyzed by the user to confirm that only events in which cargos were completely released were used in the analysis. Illustrations of the output of the curve-fitting procedure and examples of release durations calculated by the program, are reported in Bohannon et al. (2017) and in Bendahmane et al. (2018).
Cells transfected with GCaMP5G (Akerboom et al. 2012) were analyzed to determine the relative amount of calcium influx into the cell. Three ROIs measuring 1.68 μm diameter were manually selected at different points within each cell and fluorescence was measured for each ROI for each frame. The equation ΔF/F was applied to each ROI for each frame, then the values were averaged between the three ROIs to determine the overall ΔF/F for the whole cell. The values determined were plotted versus time. The exclusion criteria used to select which cells to analyze were based on proper fixation to the dish, no response to the control stimulation, and single response tracings that indicated calcium influx in response to the stimulus trigger.
As a matter of routine, imaging and analysis was performed by an experimenter who was blinded to the genotype of the cell. The individual who euthanized the mice assigned numbers to each groups and then handed over said groups to the experimenter who continued with the preparation. Whether the cells were harvested from WT or KO mice was revealed after analysis was completed. Chromaffin cells that appeared healthy in brightfield (Figure S1) with at least 10 docked granules, and responded to KCl or ACh stimulation with at least 1 fusion event, were included in data sets. No sample calculation was used.
Immunocytochemistry
Immunofluorescence imaging was performed to assess the distribution of endogenous Syt-1, Syt-7, LAMP-1 (Abcam cat# 25630; RRID: AB_470708), and PAI-1 in chromaffin cells. Mouse chromaffin cells were plated on the same Matrigel-precoated 14 mm glass-bottom dishes used for TIRF imaging. All incubations and washing steps were performed on ice unless otherwise stated. Twenty-four hours after plating, the cells were fixed with 4% paraformaldehyde in phosphate buffered solution (PBS) for 30 min. The fixed cells were quickly rinsed with PBS and quenched with 50 mM NH4Cl solution in PBS for 30 min. After a brief wash from the NH4Cl solution with PBS, the cells were permeabilized with methanol for 7 min at −20°C. Following the permeabilization, the cells were washed in Tris-buffered saline (TBS) and blocked in 0.01% gelatin solution for 30 minutes followed by another 30 min incubation in 4% donkey serum and 0.2% bovine serum albumin (BSA) prepared in TBS. Primary and secondary antibodies were diluted in TBS at 0.2%. Cells were incubated for two hours with a combination of polyclonal rabbit anti-Syt-7 antibody (Synaptic Systems, Göttingen, Germany. Cat # 105 173, 1:1200; RRID: AB_887838), and monoclonal mouse anti-Syt-1 (Synaptic Systems, Göttingen, Germany. Cat # 105 011, 1:1200; RRID: AB_887832). The cells were then washed and incubated for 70 minutes at room temperature with Alexa 488/561-conjugated anti-rabbit and anti-mouse secondary antibodies (Molecular Probes, Invitrogen). The double labeled cells were then washed and kept a 4°C until confocal imaging.
For co-labeling of endogenous PAI-1 and Syt-7, both the polyclonal anti-Syt-7 and polyclonal anti-PAI-1 (Abcam cat# 66705; RRID: AB_1310540) antibodies were made in rabbit. In this experiment, to avoid cross-labeling, the cells were first labeled with anti-Syt-7 following the protocol described above. Cells labeled for Syt-7 were then incubated for 60 minutes with polyclonal cross-adsorbed unconjugated F(ab’)2-Goat anti-Rabbit IgG (Invitrogen, ThermoFisher, Cat. # A24539, 1:300; RRID: AB_2536007) diluted in TBS-0.2% BSA solution to bind the possibly remaining free primary antibody sites (anti-Syt-7) not bound by the Alexa 488conjugated donkey anti-rabbit.
Labeling for PAI-1 was performed separately. Briefly, the primary and secondary antibodies (anti-PAI-1, 1:600 – Alexa 561-conjugated anti rabbit, 1:600) were incubated for 70 minutes in reaction tube pre-adsorbed in dry milk 1% diluted in TBS (Kroeber et al. 1998). After incubation, normal rabbit serum (Invitrogen, ThermoFisher Scientific, Cat. # 016101; RRID: AB_2532937) was added to the tube (10% volume/volume dilution) to bind the free secondary antibody for 60 minutes. The mix was then added in the Syt-7 labeled dishes for two hours. In control dishes, either the Syt-7 or PAI-1 antibodies were omitted (Figure S2) to verify the absence of cross-labeling. Cells were subsequently imaged using a Zeiss 880 confocal microscope (Zeiss, Oberkochen, Germany) with a 63x oil immersion objective in airyscan mode. Images were analyzed using Imaris Software (Bitplane, Zurich, Switzerland) using the “spot” function module for puncta detection and colocalization analysis (Rao et al., 2017). Catalogue numbers, vendors of materials used and appropriate dilutions can be found in Table 1.
Electrophysiological recordings
Primary cultures of mouse chromaffin cells were maintained on glass bottom dishes and mounted onto the stage of a Nikon Eclipse TE2000, as previously described (Rao et al. 2017a). A micro-manifold and polyamide-coated capillary was positioned in the field of view and bath solutions were exchanged through using a pressure-driven reservoir system. Standard whole-cell patch clamp methods were used record currents evoked by acetylcholine or by step depolarizations using an Axopatch 200B amplifier and Pulse Control/Igor Pro software. Patch pipettes were constructed out of 1.5 mm o.d. borosilicate glass (#TW150F-4; WPI, Sarasota, FL), coated with Sylgard elastomer (#24236–10, EMS, Hatfield, PA) and fire polished to resistances of 2.5–7 MΩ. The standard intracellular recording solution contained (in mM): 128 N-methyl-d-glucamine-Cl, 40 HEPES, 10 NaCl, 4 MgATP, 0.2 GTP, 0.1 Tris-EGTA, and pH adjusted to 7.2. IACh were induced by 10 s to 300 s applications of 100 μM ACh and recorded in physiological saline (125 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 25 mM glucose, pH 7.4). For recording of ICa, the superfusion solution was changed to a solution containing (in mM): 137 tetraethylammonium chloride, 5 CaCl2, 2 MgCl2, 10 HEPES, and 19 glucose, and pH adjusted to 7.2 with Tris. ICa current-voltage relationship was obtained in response to step depolarizations (30 ms) from a holding membrane potential of −90 mV. Pulses were applied in a randomized series to membrane potentials between −80 and +60 mV. All recordings were performed at room temperature. Cells with leak current greater than −80 pA or access resistance greater than 45 MΩ were excluded from our analysis.
Materials and Methods for Single granule/supported membrane fusion assay
The following materials were purchased and used without further purification: porcine brain L-α-phosphatidylcholine (bPC), porcine brain L- α-phosphatidylethanolamine (bPE), porcine brain L- α-phosphatidylserine (bPS), and L- α-phosphatidylinositol (liver, bovine) (PI), and porcine brain phosphatidylinositol 4,5bisphosphate (bPIP2) were from Avanti Polar Lipids; cholesterol, sodium cholate, EDTA, calcium, Opti-Prep Density Gradient Medium, sucrose, MOPS, glutamic acid potassium salt monohydrate, potassium acetate, and glycerol were from Sigma; CHAPS and DPC were from Anatrace; HEPES was from Research Products International; chloroform, ethanol, Contrad detergent, all inorganic acids and bases, and hydrogen peroxide were from Fisher Scientific. Water was purified first with deionizing and organic-free 3 filters (Virginia Water Systems) and then with a NANOpure system from Barnstead to achieve a resistivity of 18.2 MΩ/cm. Antibodies for synaptotagmin-1 (mouse monoclonal), synaptotagmin-7 (rabbit polyclonal) are from Synaptic Systems.
PC12 Cell Culture
Pheochromocytoma cells (PC12), gifted from Tom Martin, with endogenous synaptotagmin-1 and −9 knocked down, described previously (Kreutzberger et al. 2017a), were cultured on 10 cm plastic cell culture plates at 37°C in 10% CO2 in 1x Dulbecco’s modified Eagle’s medium, high glucose (Gibco) supplemented with 10% horse serum (CellGro), 10% calf serum (Fe+) (HyClone), 1% penicillin/streptomycin mix, and 2 μg/ml of puromycin. Medium was changed every 2 to 3 days and cells were passed after reaching 90% confluency by incubating for 5 minutes in Hank’s balanced salt solution and replating in fresh medium. Cells were passed no more than 30 times total. Cells were transfected by electroporation using ECM 830 Electro Square Porator (BTX). After harvesting and sedimentation, cells were suspended in a small volume of sterile cytomix electroporation buffer (120 mM KCl, 10 mM KH2PO4, 0.15 mM CaCl2, 2 mM EGTA, 20 mM HEPES-KOH, 5 mM MgCl2, 2 mM adenosine triphosphate, and 5 mM glutathione (pH 7.6), then diluted to ~14 × 106 cells/ml. Cell suspensions (~10 × 106 cells in ~700 μl volume) and 30 μg of NPY-mRuby DNA and 30 μg of synaptotagmin-1 or −7 DNA added and placed in an electroporator cuvette with a 4-mm gap. Then two 255-V, 8-ms electroporation pulses were applied. Cells were immediately transferred to a 10-cm cell culture dish with 10 ml of normal growth medium. Cells were cultured under normal conditions for 3 days prior to fractionation.
Dense core granule purification
Dense core granules from PC12 cells were purified as described previously (Kreutzberger et al. 2017a). PC12 cells with shRNA mediated knockdowns of endogenous synaptotagmin-1 and −9 were transfected with NPY-mRuby (~20 10-cm plates) and plasmids for synaptotagmin-1 or −7 (Kreutzberger et al. 2017a; Kreutzberger et al. 2019). Cells were scraped into PBS and pelleted by centrifugation and then suspended and washed in homogenization medium (0.26 M sucrose, 5 mM MOPS, 0.2 mM EDTA) by pelleting and re-suspending. Following re-suspension in 3 ml of medium containing protease inhibitor (Roche Diagnostics), the cells were cracked open using a ball bearing homogenizer with a 0.2507-inch bore and 0.2496-inch diameter ball. The homogenate was spun at 4,000 rpm for 10 minutes at 4° C in fixedangle micro-centrifuge to pellet nuclei and larger debris. The post-nuclear supernatant was collected and spun at 11,000 rpm (8000 x g) for 15 min at 4° C to pellet mitochondria. The post-mitochondrial supernatant was then collected, adjusted to a final concentration of 5 mM EDTA, and incubated 10 min on ice. A working solution of 50% Optiprep (iodixanol) (5 vol 60% Optiprep: 1 vol 0.26M sucrose, 30 mM MOPS, 1 mM EDTA) and homogenization medium were mixed to prepare solutions for discontinuous gradients in Beckman SW55 tubes: 0.5 ml of 30% iodixanol on the bottom and 3.8 ml of 14.5% iodixanol, above which 1.2 ml EDTA-adjusted supernatant was layered. Samples were spun at 45,000 rpm (190,000 x g) for 5 hrs. A clear white band at the interface between the 30% iodixanol and the 14.5% iodixanol was collected as the dense core granule sample. The dense core granule sample was then extensively dialyzed in a cassette with 10,000 kDa molecular weight cutoff (24–48 hrs, 3 × 5L) into the fusion assay buffer (120 mM potassium glutamate, 20 mM potassium acetate, 20 mM HEPES, pH 7.4).
Protein purification
Syntaxin-1a (constructs of residues 1–288), SNAP-25, Munc18, and Munc13 (construct of residues 529–1407 containing the C1C2MUN region), and complexin-1 from Rattus norvegicus were expressed in Escherichia coli strain BL21(DE3) cells described previously (Zdanowicz et al. 2017; Kreutzberger et al. 2017b; Kreutzberger et al. 2017a).
Formation of planar supported bilayers with reconstituted plasma membrane SNAREs
Planar supported bilayers with reconstituted plasma membrane SNAREs were prepared by Langmuir-Blodgett/vesicle fusion technique as described in previous studies (Domanska et al. 2009; Kalb et al. 1992; Wagner & Tamm 2001). Quartz slides were cleaned by dipping in 3:1 sulfuric acid:hydrogen peroxide for 15 minutes using a Teflon holder. Slides were then rinsed in milli-Q water. The first leaflet of the bilayer was prepared by Langmuir-Blodgett transfer onto the quartz slide using a Nima 611 Langmuir-Blodgett trough (Nima, Conventry, UK) by applying the lipid mixture of 70:30:3 bPC:Chol:DPS from a chloroform solution. Solvent was allowed to evaporate for 10 minutes, the monolayer was compressed at a rate of 10 cm2/minute to reach a surface pressure of 32 mN/m. After equilibration for 5 minutes, a clean quartz slide was rapidly (68 mm/minute) dipped into the trough and slowly (5 mm/minute) withdrawn, while a computer maintained a constant surface pressure and monitored the transfer of lipids with head groups down onto the hydrophilic substrate.
Plasma membrane SNARE containing proteoliposomes with a lipid composition of bPC:bPE:bPS:Chol:PI:PI(4,5)P2 (25:25:15:30:4:1) were prepared by mixing the lipids and evaporating the organic solvents under a stream of N2 gas followed by vacuum desiccation for at least 1 hour. The dried lipid films were dissolved in 25 mM sodium cholate in a buffer (20 mM HEPES, 150 mM KCl, pH 7.4) followed by the addition of an appropriate volume of synatxin-1a and SNAP-25 in their respective detergents to reach a final lipid/protein ratio of 3,000 for each protein. After 1 hour of equilibration at room temperature, the mixture was diluted below the critical micellar concentration by the addition of more buffer to the desired final volume. The sample was then dialyzed overnight against 1 L of buffer, with one buffer change after ~4 hour with Biobeads included in the dialysis buffer. To complete formation of the SNARE containing supported bilayers, proteoliposomes were incubated with the Langmuir-Blodgett monolayer with the proteoliposome lipids forming the outer leaflet of the planar supported membrane and most SNAREs oriented with their cytoplasmic domains away from the substrate and facing the bulk aqueous region. A concentration of ~77 mM total lipid in 1.2 ml total volume was used. Proteoliposomes were incubated for 1 hour and excess proteoliposomes were removed by perfusion with 5 ml of buffer (120 mM potassium glutamate, 20 mM potassium acetate (20 mM potassium sulfate was used in buffers with acridine orange labeled granules), 20 mM HEPES, 100 mM EDTA, pH 7.4).
TIRF microscopy for single granule/supported membrane fusion assay
Dense core granule to planar supported bilayer fusion assay experiments were performed on a Zeiss Axio Observer 7 fluorescence microscope (Carl Zeiss), with a 63x water immersion objective (Zeiss, N.A. 0.95) and a prism-based TIRF illumination. Laser light at 514 nm from an argon ion laser (Innova 90C, Coherent), controlled through an acousto-optic modulator (Isomet), and at 640 nm from a diode laser (Cube 640, Coherent) were used as excitation sources. The characteristic penetration depths were between 90 and 130 nm. An OptoSplit (Andor Technology) was used to separate two spectral bands (540 nm – 610 nm, and 655 nm – 725 nm). Fluorescence signals were recorded by an EMCCD (iXon DV887ESC-BV, Andor Technology).
Calcium-triggered single granule – planar supported membrane fusion assay
As previously described (Kreutzberger et al., 2017a), planar supported bilayers containing syntaxin-1a (1–288): dodecylated (d) SNAP-25 (bulk phase-facing leaflet lipid composition of 25:25:15:30:4:1 bPC:bPE:bPS:Chol:PI:bPIP2) were incubated with 0.5 μM Munc18 and 2 μM complexin-1. Secretory granules were then injected while keeping the concentrations of Munc18 and complexin-1 constant. Dense core granule docking was allowed to occur for ~20 minutes before the chamber was placed on the TIRF microscope and the microscope was focused on the planar supported membrane. Fluorescent images were recorded every 200 ms while buffer containing 100 μM calcium and a soluble Alexa647 dye to monitor the arrival of calcium at the observation site was injected.
Movies were analyzed as previously described (Domanska et al. 2010; Kreutzberger et al. 2017b; Kreutzberger et al. 2017a). Fusion efficiencies are reported as the percentage of granules in the field of view that fuse within 15 s. Fluorescent line shapes are presented as average from 20 single events as described previously (Kreutzberger et al. 2017b).
Statistical analysis
Graphpad Prism 7 was used for analysis of data collected from TIRF studies, immunocytochemistry, electrophysiological data and qPCR. Igor was used to fit and analyze data collected from single granule /supported membrane fusion assays. Data collected for studies utilizing primary chromaffin cell culture were collecting using at least 3 different cell preparations. Specifically, co-localization studies include the average and standard error of the mean (SEM). Imaging studies on the release of cargos were analyzed using non-linear regressions and Tukey’s multiple comparisons tests (p<0.05) to determine if there were significant differences between groups. Fusion probability and release data were statistically analyzed using a nonparametric Mann-Whitney test, p<0.05. The choice of using a parametric or nonparametric test was determined by an F-test or a Brown-Forsythe test which determines if variances are significantly different; values are reported in results section if nonparametric tests were used. No sample calculation was performed to determine appropriate sample sizes. The ROUT method was used to identify outliers (Graphpad Prism).
Results
The localization of Syt-7 in mouse chromaffin cells
Fluorescent labeling of endogenous Synaptotagmin-1 and −7 in bovine adrenal chromaffin cells revealed these proteins to exhibit a punctate intracellular distribution, with a low degree of co-localization (Rao et al. 2014). A similar pattern is observed in chromaffin cells harvested from the mouse adrenal medulla. As shown in Figure 1, endogenous Syt-1 and Syt-7 fluorescence is largely non-overlapping (in the range of 3 – 5%), punctate, and intracellular (Figure 1 A, B). It was previously reported that Syt-7 is sorted to the plasma membrane in PC12 cells and rat chromaffin cells (Sugita et al. 2001). However, the punctate pattern of staining observed here is most consistent with the protein being sorted to organelles. The co-localization of Syt-7 with plasminogen activator inhibitor-1 (PAI-1) (Figure 1 C, D) – a ubiquitous dense core marker in chromaffin cells (Bohannon et al. 2017; Abbineni et al. 2019) – shows that Syt-7 is present in secretory granules. Confocal imaging of immunolabeled Syt-7 demonstrates that it is co-localized with lysosome associated membrane protein-1 (LAMP-1), in the range of 14 – 18% (Figure 1 E, F). LAMP-1 and PAI-1 exhibited a roughly 10 – 11% co-localization frequency (Figure 1 G, H). LAMP-1 has been previously shown to label both granules and lysosomes in chromaffin cells (Hao et al. 2015).
Figure 1. Endogenous Syt-7 is co-localized with PAI-1, a protein of the granule dense core.
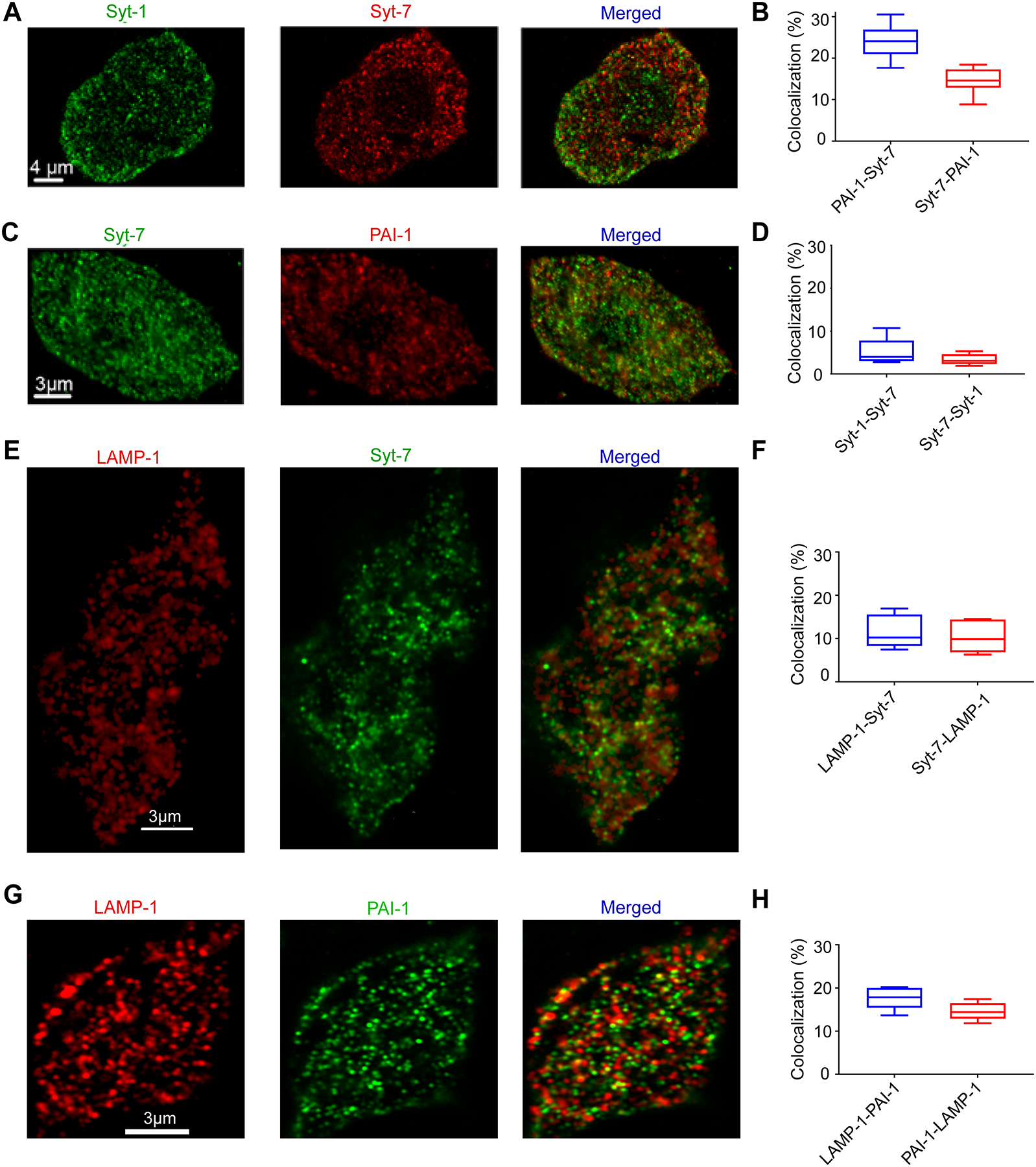
A. Immunolabeling was performed in WT cells for endogenous Syt-7 (red) and Syt-1 (green). Images from a representative cell showing Syt-7 fluorescence, Syt-1 fluorescence, and fluorescence of the merged channels. B. Box plot indicates the percentage of Syt-1 puncta that harbor Syt-7 (5.18 ± 1.03 %; n = 8 cells; 3 primary cell preparations) and Syt-7 puncta that harbor Syt-1 (3.36 ± 0.43 %; n = 8 cells; 3 primary cell preparations). C. Immunolabeling was performed in WT cells for Syt-7 (green) and PAI-1, a dense core granule marker (red). D. Box plot showing the percentage of PAI-1 puncta that are co-localized with Syt-7 (24.06 ± 1.41 %; n = 8 cells; 3 primary cell preparations) and the percentage of Syt-7 puncta that are co-localized with PAI-1 (14.65 ± 1.075 %; n = 8 cells; 3 primary cell preparations). E. Immunolabeling was performed in WT cells for endogenous LAMP-1 (red) and Syt-7 (green). Images from two representative cells showing LAMP-1 fluorescence, Syt-7 fluorescence, and fluorescence of the merged channels. F. Box plot shows the percentage of LAMP-1 staining that harbor Syt-7 puncta (17.59 ± 2.28 %; n= 6 cells; 2 primary cell preparations) and Syt-7 puncta that harbor LAMP-1 staining (14.58 ± 1.83 %; n= 6 cells: 2 primary cell preparations). G. Immunolabeling was performed in WT cells for endogenous LAMP-1 (red), a lysosomal marker, and PAI-1 a dense core granule marker (green). H. Box plot shows the percentage of LAMP-1 staining that are co-localized with PAI-1 puncta (11.42 ± 3.44%; n= 6 cells; 2 primary cell preparations) and the percentage of PAI-1 puncta that are co-localized with LAMP-1 (10.31 ± 3.47%; n= 6 cells; 2 primary cell preparations). For all box-plots, box dimensions extend from the 25th to the 75th percentiles; whiskers represent the minimum and maximum values; line represents median.
To further characterize the subcellular distribution of Syt-7, bovine chromaffin cell lysates were layered on an iodixanol gradient and centrifuged to separate organelles by density (see Methods). Sequential fractions of 400 μl were collected and run on a polyacrylamide gel for western blotting. Immunoreactive bands corresponding to PAI-1, Syt-7, Syt-1, and LAMP-1 are evident in Figure S3. Given the intensity of the PAI-1 reactivity, fraction 13 likely contains dense core granule constituents. Less dense fractions (4–6) presumably contain lysosomes or endosomes. Syt-7 and Syt-1 also appear in these lighter fractions, in addition to those that contain PAI-1.
Endogenous Syt-7 slows dense core granule cargo release
A major goal of this study was to delineate the ways in which the chromaffin cell secretory system depends on the presence of Syt-7. To avoid the potential ambiguities associated with overexpression of synaptotagmins in cells already expressing a full complement of endogenous proteins, most of the experiments in this study instead rely on cells harvested from Syt-7 KO mice. Figure S4A shows that compared to WT cells, expression of Syt-1 transcript remains unchanged in KO cells, while Syt-7 transcript is almost undetectable. Western Blot was also performed on adrenal gland lysates to verify the loss of Syt-7 protein in the KO. The alpha variant of Syt-7 (403 amino acids), targeted by Andrews and colleagues when generating the original Syt-7 KO mouse, has been reported to run at approximately 45 kDa (marked by arrow) (Martinez et al., 2000). That band is absent in the KO (Figure S4B).
Our first objective was to determine the release rate of NPY-pHl in Syt-7 KO cells compared to WT cells. Cells expressing fluorescent protein were identified by a brief exposure to 10 mM NH4Cl (Anantharam et al., 2010). Single cells were then depolarized by local perfusion of 100 mM KCl while exocytosis was imaged with a TIRF microscope. Figures 2A, B, and D show that the time necessary for NPY to be completely released from fused chromaffin granules is broadly distributed in WT cells (times ranged from 0.049 s to 8.10 s; the data are not normally distributed). In Syt-7 KO cells, NPY release durations were shifted to shorter times (ranging from 0.084 s to 1.17 s) (Figure 2C and D). A cumulative frequency distributions representing the time at which granules fused with respect to the start of stimulation, are plotted in Figure S5.
Figure 2. Syt-7 slows release of lumenal cargos.
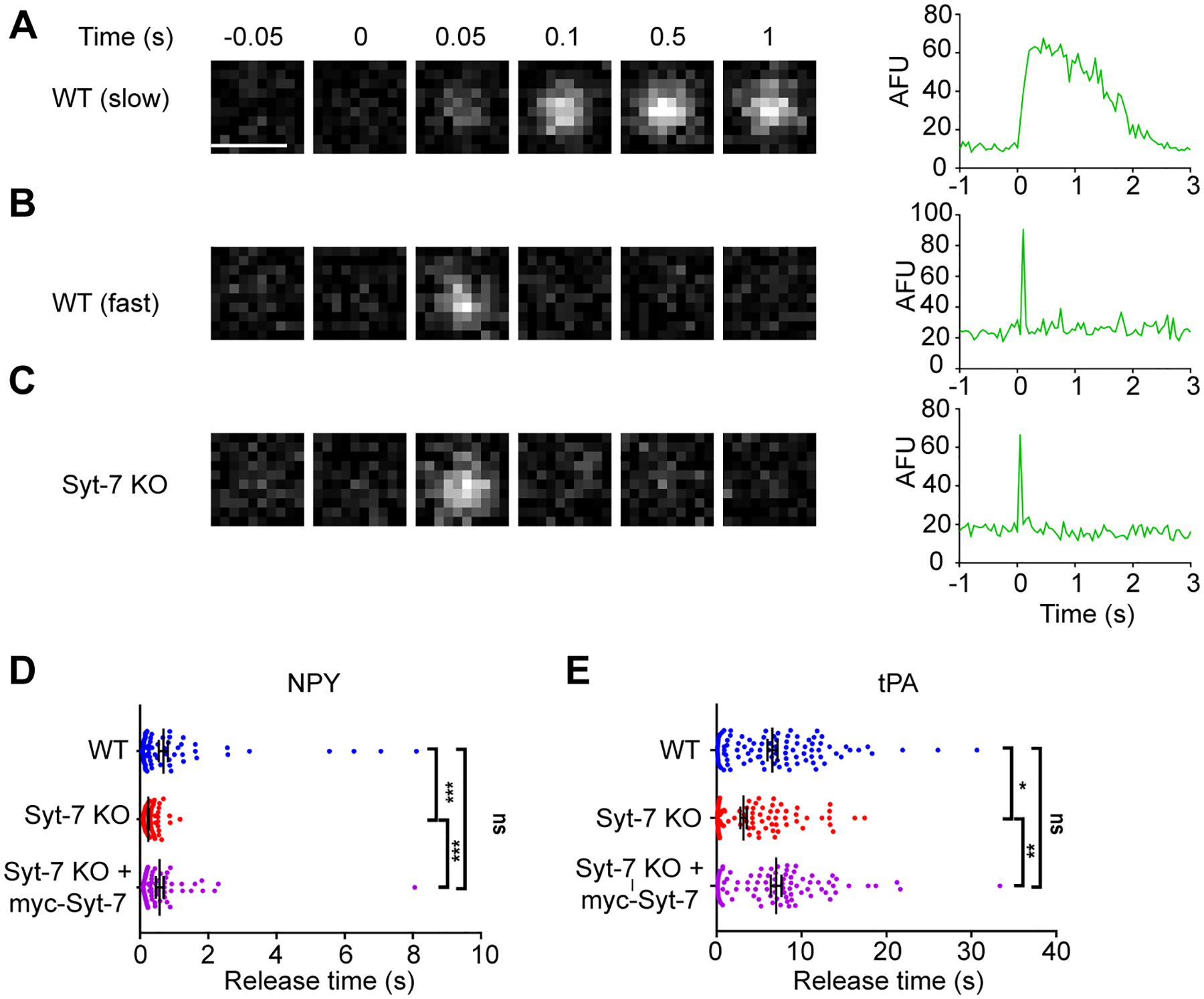
WT and Syt-7 KO cells overexpressing pHl-tagged NPY and tPA were stimulated with 100 mM KCl. Fusion events were observed using TIRFM and cargo release times were measured. A-C. Representative images showing fusion events of NPY-pHl expressing granules from WT and Syt-7 KO chromaffin cells alongside intensity vs. time curves. Release times in WT cells ranged from fast to slow (A). However, release in Syt-7 KO cells was shifted to faster times (C). D. Individual times for NPY release are shown in a scatter plot. In WT cells, these ranged from 49 ms to 8.10 s (average time = 0.67 ± 0.13 s; n = 100 events; 3 primary cell preparations). In Syt-7 KO cells, the distribution of the release times was narrower, and ranged from 84 ms to 1.17 s (average time = 0.25 ± 0.17 s; n = 73 events; 3 primary cell preparations). The overexpression of myc-Syt-7 in Syt-7 KO cells restored the broader distribution of release times, which now ranged from 76 ms to 8.05 s (average time = 0.57 ± 0.11 s; n = 77 events; 3 primary cell preparations). E. Release times for individual tPA events shown in a scatter plot. Secretion events from WT cells show a large distribution of release times ranging from 68 ms to 30.67 s (average time = 6.57 ± 0.59 s; n = 103 events; 3 primary cell preparations). In Syt-7 KO cells, the distribution of the release time was somewhat narrower, and ranged from 84 ms to 17.44 s (average time = 3.16 ± 0.38 s; n = 120 events; 3 primary cell preparations). The overexpression of myc-Syt-7 in the Syt-7 KO cells again restored the broader distribution of release times, which now ranged from 100 ms to 33.4 s (average time = 7.00 ± 0.62 s; n = 89 events; 3 primary cell preparations). * p<0.05, ** p<0.005 Tukey’s multiple comparison test.
To assess if Syt-7 is responsible for slower rates of NPY release, myc-tagged Syt-7 and NPY-pHl (myc expression was verified post hoc) were co-expressed Syt-7 KO cells. Cells were stimulated with KCl and the rate of NPY release determined, as above. In cells expressing myc-Syt-7, a slow population of NPY release times was again observed (times ranged from 0.076 s to 8.05 s) (Figure 2D).
Certain lumenal cargo proteins, such as tissue Plasminogen Activator (tPA), exhibit intrinsically slower release or “discharge” times during exocytosis (Perrais et al., 2004; Weiss et al., 2014). In the case of tPA, this has been attributed to the protein’s ability to stabilize the curvature of the fusion pore and thereby limit its rate of expansion (Bohannon et al., 2017). Here, the goal was to address whether tPA release from fused granules may be hastened by the abrogation of Syt-7 expression – a protein which imposes its own constraints on fusion pore expansion (Rao et al., 2014; Rao et al., 2017). As before, the rate of release of tPA-pHl was measured in both WT and Syt-7 KO cells depolarized with KCl. The average rate at which tPA is released is considerably slower than it is for NPY (tPA: 6.57 ± 0.59 s, NPY: 0.67 ± 0.13 s, Mann Whitney test, p<0.0001, Brown-Forsythe test, p<0.05) (Figure 2E). Transfection with tPA yielded secretion events from WT cells with a large distribution of release times ranging from 68 ms to 30.67 s (average time = 6.57 ± 0.59 s; n = 103 events). In Syt-7 KO cells, the distribution of the release time was somewhat narrower, and ranged from 84 ms to 17.44 s (average time = 3.16 ± 0.38 s; n = 120 events). The overexpression of myc-Syt-7 in the Syt-7 KO cells again restored the broader distribution of release times, which ranged from 100 ms to 33.4 s (average time = 7.00 ± 0.62 s; n = 89 events). * p<0.05, ** p<0.005 Tukey’s multiple comparison test.
Granule fusion probability and mobility in WT and Syt-7 KO cells
The number of NPY-GFP-labeled, docked granules that fused in response to 100 mM KCl depolarization in WT and KO cells was determined and used to calculate a granule fusion probability (Figure 3A; see also Supplementary Movie 1). The criterion for “docked” in this case, is that the granule is resident in the evanescent field at the start of KCl stimulation. The absence of Syt-7 severely disrupts the secretory response to KCl-based depolarization. This is evidenced by the fact that fusion probability of granules in KO cells is approximately 5-fold lower than that of granules in WT cells (Figure 3B).
Figure 3. Syt-7 endows granules with an increased fusion probability.
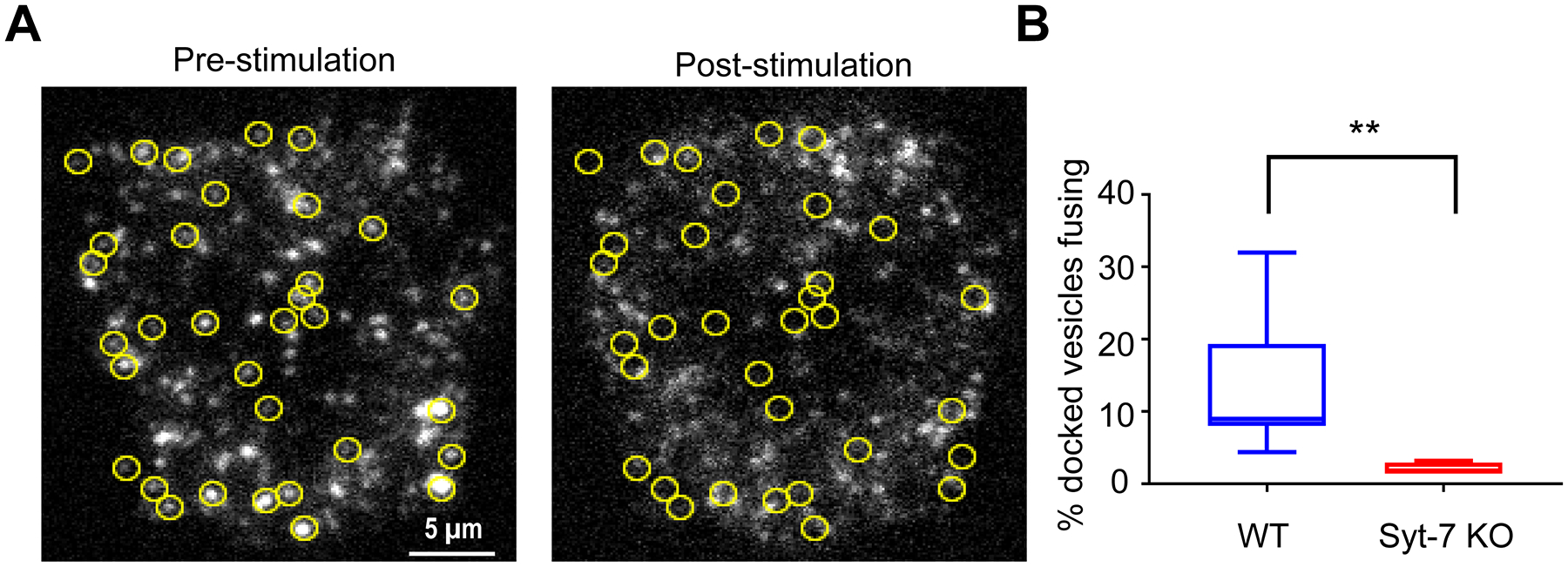
WT and Syt-7 KO cells overexpressing GFP-tagged NPY were stimulated with 100 mM KCl. Fusion events were observed using TIRFM. A. Pre- and post-stimulation images of fluorescent NPY puncta in a WT cell. Yellow circles indicate the position of granules that fused during the stimulation period. B. Box plot showing the percentage of docked granules that fused in WT (13.49 ± 3.23 %; n = 8 cells; 4 primary cell preparations) and Syt-7 KO cells (2.14 ± 0.36 s; n = 5 cells; 4 primary cell preparations). Box dimensions extend from the 25th to the 75th percentiles; whiskers represent the minimum and maximum values; line represents median. ** p<0.005, Mann-Whitney test, Brown-Forsythe, p< 0.05)
In a previous study, we had reported that the frame-to-frame displacement (ΔR) of granules in bovine chromaffin cells harboring GFP-Syt-7 was, on average, lower than that of granules harboring GFP-Syt-1 (Rao et al., 2017). Thus, we also set out to determine whether granule mobility varies as a function of Syt-7 expression. NPY-GFP-labeled granules in WT and KO cells were tracked over a minimum of 200 frames (10 s in duration) (Figure S6A and B). From those tracks, ΔR of individual granules was calculated (Figure S6C) (Rao et al. 2017a). The distribution of ΔRs for granules from both cell-types, WT and KO, were best fit to a sum of two gaussians. The average ΔR from the slower WT population was 29.32 +/− 1.41 nm, and 48.39 +/− 1.13 nm for the faster population, with 53.58 +/− 30.67% of granules residing in the slower population. The differences between the slower and faster populations of KO granules were less pronounced, with a slow population ΔR of 27.82 +/− 0.59, a fast population ΔR of 39.97 +/− 3.76 nm, and 47.84 +/− 59.77% of granules residing in the slower population. Thus, the mobility of chromaffin granules labeled with NPY-GFP was not significantly different between WT and Syt-7 KO groups.
Stimulation of secretion in WT and Syt-7 KO cells with a physiological agonist
Chromaffin cell secretion, in situ, is triggered by activation of nicotinic receptors and subsequent Ca2+ influx (Douglas & Rubin 1963; Douglas 1968). Therefore, to understand how the lack of Syt-7 might disrupt secretion in a physiological setting, WT and KO cells were stimulated with ACh delivered locally via a perfusion pipet. ACh-triggered release kinetics of NPY-pHl was measured first (Figure 4A–D). As with elevated KCl stimulation, the range of NPY release times in response to ACh stimulation was broader in WT cells (0.06 s to 24.05 s) than in Syt-7 KO cells (0.07 s to 2.93 s). On average, the rate at which NPY is released was slower in WT cells compared to Syt-7 KO cells (WT: 1.47 ± 0.34 s, KO: 0.44 ± 0.1 s, Mann Whitney test, p < 0.05, Brown-Forsythe, p<0.05) (Figure 4D). Moreover, the overall fusion probability of NPY-GFP-labeled granules in Syt-7 KO cells was substantially lower than WT cells (Supplementary Movie 2) following cholinergic stimulation (3.2 ± 0.7% in KO versus 19.1 ± 3.4% in WT) (Figure 5A, B).
Figure 4. Syt-7 slows release of overexpressed NPY in cells stimulated with ACh.
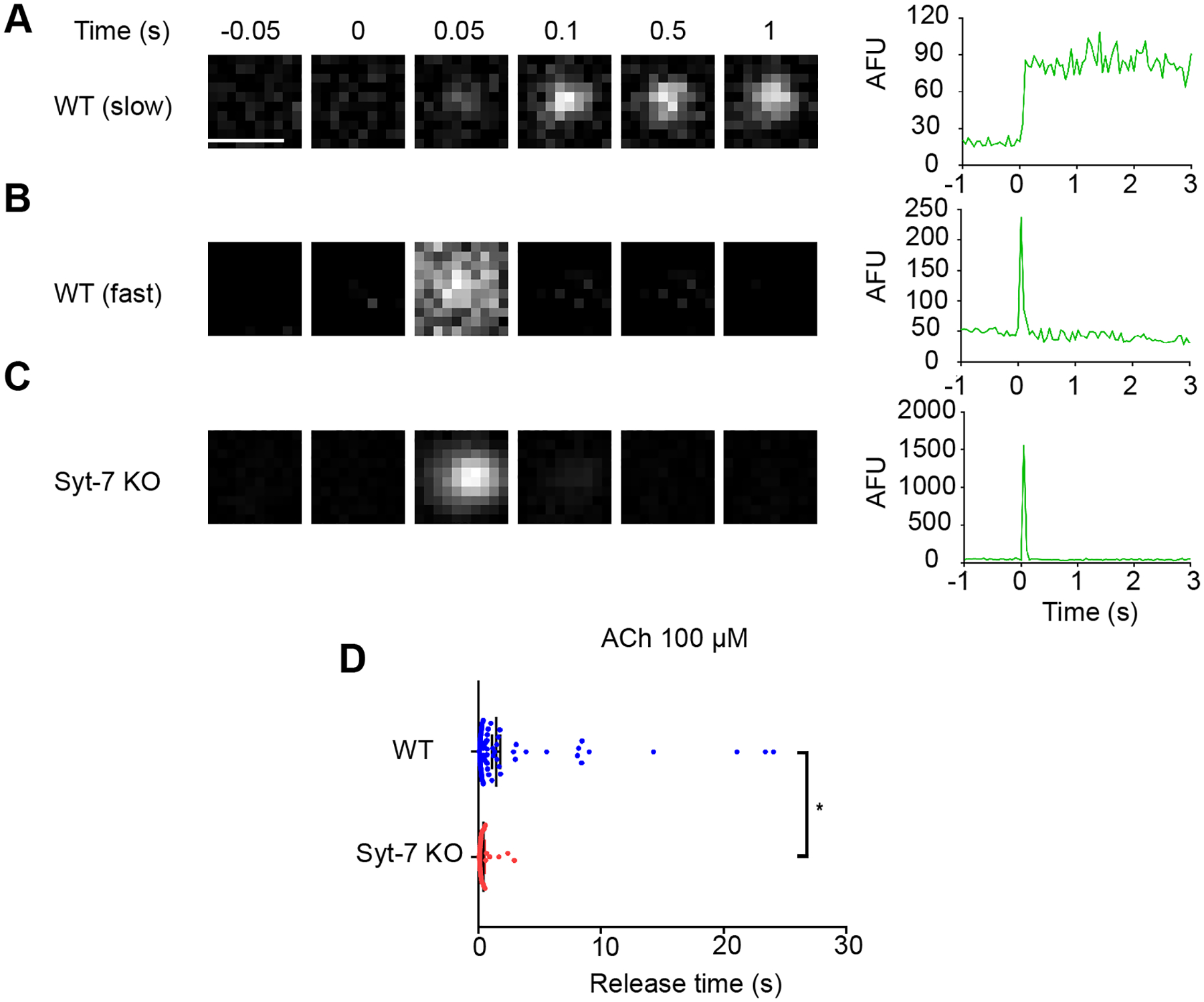
WT and Syt-7 KO cells overexpressing NPY were stimulated with 100 μM ACh for two minutes. Fusion events were monitored using TIRFM and NPY release times were measured. A-C. Representative images showing fusion events from WT and Syt-7 KO chromaffin cells alongside intensity vs. time curves. Release times in WT cells ranged from fast to slow (A). Release times in Syt-7 KO cells were typically faster (C). D. Release times for individual NPY fusion events shown in a scatter plot. Release times in WT cells ranged from 67 ms to 24.05 s (average time = 1.47 ± 0.34 s; n = 130 events, 7 cells; 3 primary cell preparations). In Syt-7 KO cells, the distribution of the release times was narrower and ranged from 70 ms to 2.93 s (average time = 0.44 ± 0.10 s (n = 38 events, 9 cells; 3 primary cell preparations).
Figure 5. Granules in Syt-7 KO cells stimulated by ACh evince a lower fusion probability than those in WT cells.
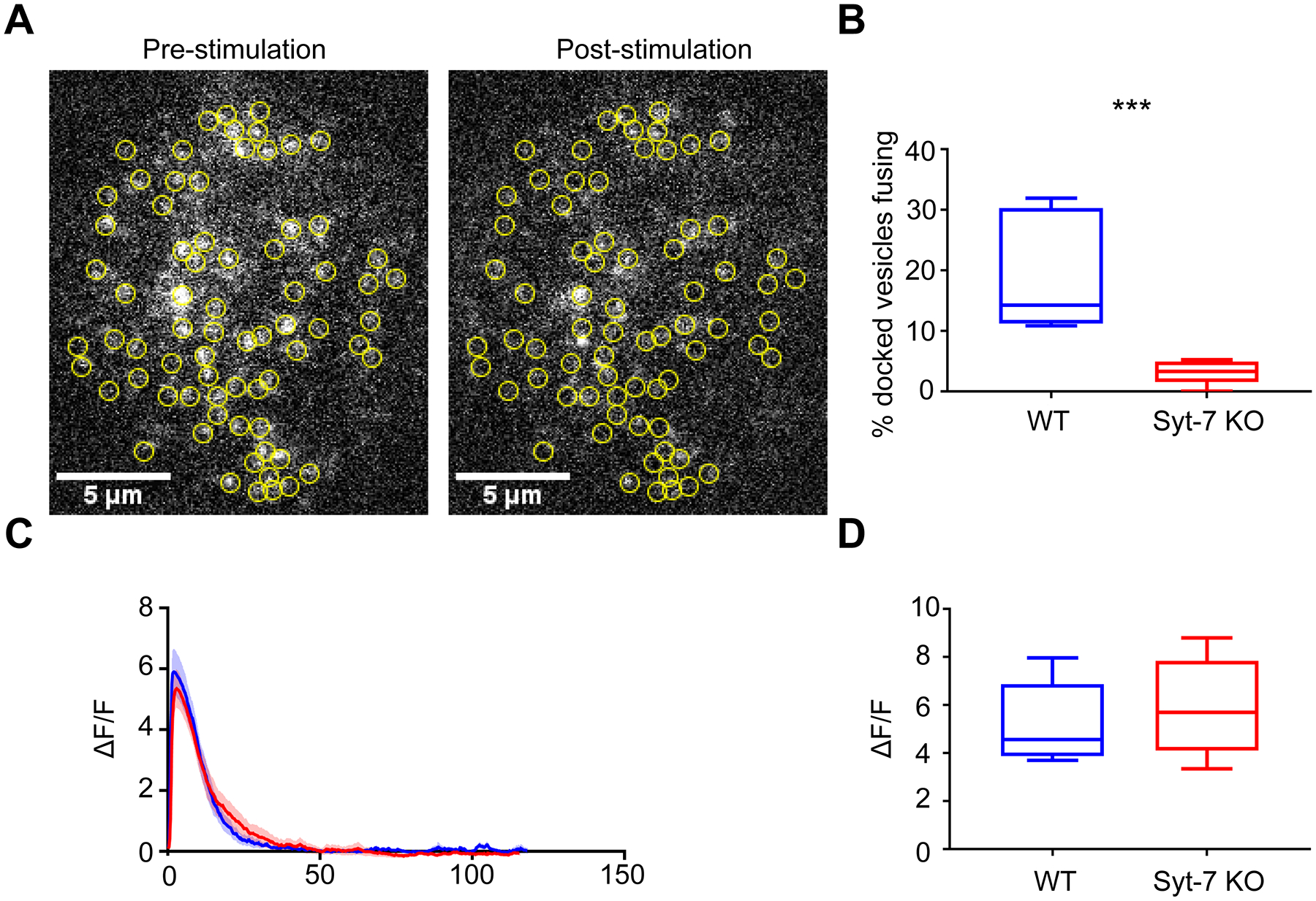
WT and Syt-7 KO cells overexpressing GFP-tagged NPY were stimulated with 100 μM ACh for two minutes. Fusion events were observed using TIRFM. A. Pre- and post-stimulation images of fluorescent NPY puncta in a WT cell. Yellow circles indicate the position of granules that fused during the stimulation period. B. Box plot showing the percentage of docked granules that fused in WT (19.1 ± 3.4 %; n = 8 cells; 4 primary cell preparations) and Syt-7 KO cells (3.2 ± 0.7 %; n = 7 cells; 4 primary cell preparations). *** p<0.005, Mann-Whitney test, Brown-Forsythe, p<0.05). C-D. WT and Syt-7 KO cells transfected with GCaMP5G were stimulated with 100 μM ACh for two minutes. Fluorescence was monitored using TIRFM, with ΔF/F was calculated for each cell. C. Average ΔF/F vs. time traces in response to ACh stimulation for WT (n = 7 cells, blue; 5 primary cell preparations) and Syt-7 KO (n= 7 cells, red; 5 primary cell preparations) cells. D. Box plot showing the average value of the peak ΔF/F for WT (5.41 ± 0.62) and Syt-7 KO (5.94 ± 0.76) cells. The average values were not statistically significant (Student’s t-test, p > 0.05). For all box plots, box dimensions extend from the 25th to the 75th percentiles; whiskers represent the minimum and maximum values; line represents median.
One potential explanation for the difference in granule fusion probability observed between the two cell types is that Ca2+ signaling is compromised in cells lacking Syt-7. Therefore, qualitative changes in intracellular Ca2+ in response to ACh stimulation were monitored using the fluorescent Ca2+ indicator, GCaMP5G (Akerboom et al. 2012). Neither the kinetics nor the peak amplitude of the GCaMP5G signal differed between WT and Syt-7 KO cells (Figure 5C, D). The possibility that differences in the magnitude of cholinergic or Ca2+ currents might underlie differences in the secretory phenotype of WT and KO cells was also tested. However, these were not different between the two groups (Figures S7A–D).
Sustained chromaffin cell secretory output relies on the presence of Syt-7
During the course of the experiments described in Figure 4, it became evident that the times at which NPY-pHl-labeled granules fused with respect to the start of stimulation varied depending on the genotype of the cell. To examine this issue more closely, WT and KO cells expressing NPY-pHl were stimulated for 2 minutes with 100 μM ACh while images were continuously acquired. The secretory response of a single WT and Syt-7 KO cell to prolonged ACh stimulation (beginning at 5 s after imaging begins) is shown in Figures 6A and B. Each of the vertical lines (blue for WT and red for KO) corresponds to the time of an individual NPY fusion event during the period of stimulation. In contrast to WT cells, the secretory response of KO cells rapidly wanes after an initial burst of fusion events.
Figure 6. Syt-7 is necessary to sustain secretion during prolonged ACh stimulation.
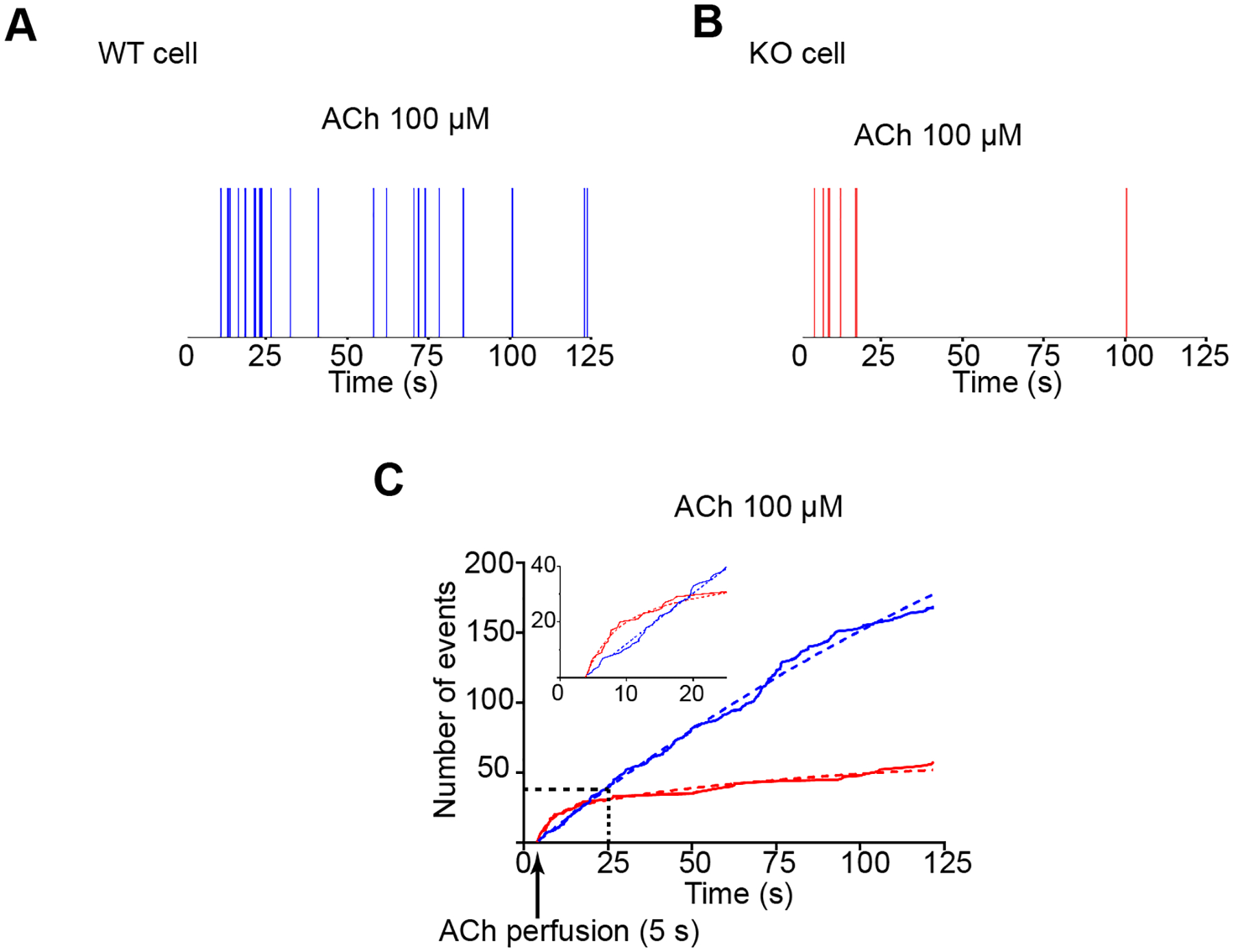
NPY-pHl was overexpressed in WT and Syt-7 KO chromaffin cells. Secretion was triggered by local perfusion of 100 μM ACh. A-B. Time distribution of fusion events in a WT (A) and Syt-7 KO (B) cell. Each vertical line corresponds to the occurrence of a single fusion event during the stimulation period. C. Cumulative frequency distribution showing the time at which fusion events occurred in WT (blue) and Syt-7 KO (red) cells (n=7 cells; 4 primary cell preparations for each group). WT cells continue to secrete throughout the course of the stimulation period; secretory activity in Syt-7 KO cells largely ceases after 25 sec. Note that in the 5 s - 25 s window (inset), the rate at which fusion events occur is actually higher in the Syt-7 KO cell. Curves were fit by a two phase association function in Prism 7 with R2 ≥ 0.99; WT: kfast = 1.3 s−1; kslow = 4.3 × 10−3 s−1; KO: kfast = 0.2 s−1; kslow = 1.3 × 10–2s−1.
The cumulative distribution of all fusion events in WT and Syt-7 KO cells (n = 7 cells for each group) occurring after ACh stimulation is plotted in Figure 6C. A distinctive feature of the cumulative time course for Syt-7 KO events is that the curve plateaus. Such a result would be expected if the absence of Syt-7 prevents a sustained response to cholinergic stimulation. The cumulative time course for WT cells stimulated with ACh does not plateau; secretion is observed for as long as the cell is stimulated although the frequency with which events occur does decline at later times. The curves were fit by the sum of two exponential (Figure 6C). Interestingly, over the first approximately 20 s of ACh stimulation, there is more secretory activity in the Syt-7 KO than in the WT cell (see inset, Figure 6C).
Syt-7 endows dense core granules with distinct Ca2+-sensing and fusion properties
To better assign mechanistic roles to Syt-1 and Syt-7 in exocytosis, we utilized a previously characterized purified secretory granule to planar supported bilayer fusion assay (Figure 7A). PC12 cells lacking endogenous synaptotagmin (Kreutzberger et al. 2017a) were transfected to overexpress either Syt-1 or Syt-7 protein ((Kreutzberger et al. 2019) and Figure 7B). In the presence of SNARE regulatory proteins Munc18 and complexin-1, these granules will bind in an arrested state to planar supported bilayers (lipid composition of 70:30 bPC:Chol in the extracellular mimicking leaflet and 25:25:15:30:4:1 bPC:bPE:bPS:Chol:PI:PIP2) containing the plasma membrane SNARE proteins (syntaxin-1a and dSNAP-25). Injection of Ca2+ into this system readily stimulates fusion of dense core granules with the planar bilayer. Increasing the level of Ca2+ in the chamber containing docked dense core granules, triggers fusion with different efficiencies depending on the sensitivity to Ca2+ of the expressed synaptotagmin isoform (Figure 7C). Syt-7 containing dense core granules consistently fused at lower Ca2+ concentrations than those bearing Syt-1. The higher sensitivity of Syt-7 bearing granules to Ca2+ is consistent with the observation in cells that even mild depolarization selectively activates Syt-7 containing granules (Rao et al. 2014; Rao et al. 2017b; Bendahmane et al. 2018).
Figure 7. In vitro fusion of Syt-7 and Syt-1 granules on planar supported bilayers.
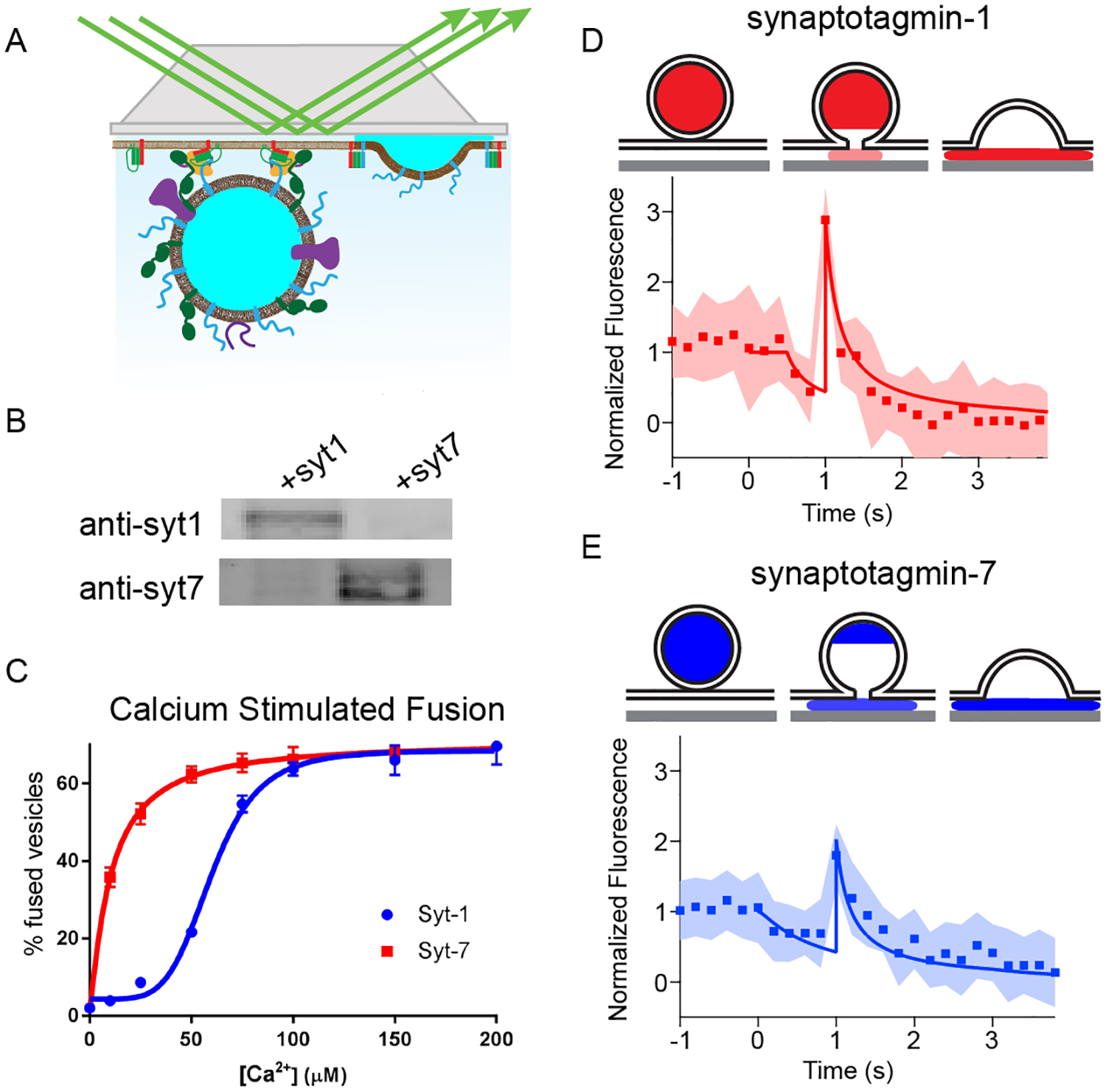
A, B. Fusion of dense core granules containing either Syt-1 or Syt-7 with planar supported bilayers containing syntaxin-1a, dSNAP-25, Munc18, and complexin-1 were examined. As previously described, dense core granules labeled with NPY-mRuby stably dock to the supported bilayer, where fusion is triggered by injection of calcium. C. The calcium sensitivity of fusion is dependent on the Syt-isoform expressed on purified dense core granules (K1/2 = 63 ± 3 μM for Syt-1 granules and 10 ± 1 μM for Syt-7 granules). Example of a characteristic NPY-mRuby signal (D, E) is observed for fusion. There is an initial decrease in NPY-mRuby fluorescence caused by diffusion of the protein away from the site of fusion under the cleft of the supported bilayer. This is followed by an increase in fluorescence caused by a collapse of the granule into the supported bilayer. This action pulls the remaining lumenal content towards the brightest portion of the evanescent field. Eventually, the fluorescence diminishes as NPY-mRuby diffuses away from the site of fusion. Averaging more than 20 individual traces (D, E) for Syt-1 fusion events, revealed a short delay (D) between the initial decrease in fluorescence and the subsequent increase in fluorescence during granule collapse. The delay time between these phases of the fluorescent signal was prolonged for Syt-7 granule fusion. The average tracings of Syt-7 and Syt-1 events are shown as an overlay (F). The cartoons illustrate the mathematical model (solid line in C and D) used to fit the data.
The intensity time course of NPY-mRuby fluorescence during release has noteworthy characteristics. Initially, a decrease in fluorescence is observed as NPY-mRuby begins to diffuse out of the early fusion pore and away from the fusion site (note dip in fluorescence in Figures 7D and 7E). After a brief delay, a sharp increase in fluorescence intensity is observed as the fusion pore expands and the granule membrane collapses (Kreutzberger et al. 2017b).
Previously, the delay time from the onset of NPY-mRuby release was shown to be sensitive to the presence of lipids with geometries that promote or inhibit fusion pore stability (Kreutzberger et al. 2017b). Here, we show this feature is also sensitive to the synaptotagmin isoform expressed. A close examination of the fluorescence intensity profile of Syt-1 granules shows that there is approximately a 0.4 s delay time from the initial decrease in fluorescence until the collapse of the granule into the supported bilayer (Figure 7D). In Syt-7 granules, the delay is lengthened to approximately 1 s (Figure 7E). An overlay of averaged release profiles of NPY from Syt-1 and Syt-7 granules is shown in Figure 7F. Images of individual NPY-mRuby release events from Syt-1 and Syt-7-bearing granules are shown in Figure S8.
Discussion
Despite the broad interest in Syt-7 as a regulator of exocytosis (MacDougall et al. 2018), surprisingly basic aspects of its function remain unresolved, including its intracellular localization and role in regulating fusion pore expansion. Using a combination of subcellular fractionation and immunolabeling methods, we report that endogenous Syt-7 is co-localized with markers of the lysosome and dense core granule (LAMP-1 and PAI-1, respectively). The marker used in this study to identify granules, PAI-1, is ubiquitously expressed in chromaffin cells (Bohannon et al. 2017) and exhibits a high degree of co-localization with other markers of the dense core, tPA and dopamine β-hydroxylase (Bohannon et al. 2017; Abbineni et al. 2019). The conclusion that fraction 13 (Fr-13, Figure S3) contained the majority of dense core granule constituents is based on the strong PAI-1 signal. Syt-7 was most abundant in fractions 13 and 4–6. Approximately 20% of the total amount of Syt-7 in the cell can be found in fraction 13 (i.e., granules) – a number that is also consistent with the immunocytochemical data (Figure 1). Fractions 4–6 represent less dense organelles, including lysosomes. Immunoreactivity for LAMP-1 is greatest in fractions 4–6, but is also detected in fraction 13. This suggests that dense core granules house LAMP1 in addition to Syts 1 and 7 – a conclusion which is supported by immuncytochemical data (Figure 1) and published reports (Hao et al. 2015).
We presume that membrane proteins, such as Syt-7, are turned over more slowly than secreted proteins (Winkler 1971). The recycling of synaptotagmin isoforms (e.g., after fusion) would account for their higher abundance in lighter organelles which may form part of the endolysosomal system. Interestingly, Syt-1 is more abundant in fraction 13, as a percentage of total protein, than is Syt-7. This may reflect differences in the steady-state turnover of granules to which Syt-1 and Syt-7 are sorted.
The basic finding that Syt-7 is sorted to chromaffin granules corroborates what has been previously published in PC12 cells and chromaffin cells (Fukuda et al. 2004; Tsuboi & Fukuda 2007; Zhang et al. 2011; Rao et al. 2014). In contrast, neuronal Syt-7 has been reported to exist primarily as a plasma membrane bound protein (Weber et al. 2014; Jackman et al. 2016; Sugita et al. 2001) (Jackman et al., 2016; Sugita et al., 2001; Weber et al., 2014). The reason for differences in Syt-7 localization between neurons and neuroendocrine cells is unclear at this time.
A second key unresolved issue concerning the function of Syt-7 in chromaffin cells is whether it constrains (Rao et al. 2014), or alternatively, promotes (Zhang et al. 2019) fusion pore expansion. Here, two distinct experimental preparations were used to address this issue: 1) primary mouse chromaffin cells lacking Syt-7; and, 2) a reconstituted single granule fusion assay employing PC12 granules only expressing either Syt-1 or Syt-7. Data gathered from both experimental preparations provide support for the principle that Syt-7 imposes limits on the rate at which cargos are discharged during exocytosis. In the case of cargos that exhibit an intrinsically slow release profile (e.g., tPA), the effect of Syt-7 is additive. The findings are also consistent with TIRF-based measurements of secretion in other systems, including PC12 cells (Zhang et al. 2011) and mouse embryonic fibroblasts (Jaiswal et al. 2004), where it has been shown that Syt-7 restricts post-fusion soluble content release and diffusion of granule membrane proteins into the plasma membrane.
Although the mechanism by which Syt-7 acts at the pore to slow discharge is not resolved, the protein’s high affinity for anionic phospholipids likely plays a key role. According to this model, the tight binding of the Syt-7 C2 domains to lipids constituting the fusion pore is a determining factor of pore stabilization (Bendahmane et al. 2018; Voleti et al. 2017; Tran et al. 2019). In support of this hypothesis, a mutant of Syt-1, whose C2B domain Ca2+/phospholipid-binding loops were exchanged for those of Syt-7, exhibited a higher intrinsic affinity for phospholipids and a slower dissociation from phospholipids in the presence of Ca2+, than the WT Syt-1 protein (Bendahmane et al. 2018). When expressed in cells, this Syt-1 mutant also stabilized membrane curvature associated with the fused granule/plasma membrane domain, thereby slowing the discharge of NPY-pHl (Bendahmane et al. 2018). It should be noted here that fluorescent peptide hormone release observed by TIRF microscopy is likely to represent a slower process than the transition from pre-spike foot to burst of neurotransmitter release captured by amperometry recordings, which occurs in a matter of milliseconds (Albillos et al. 1997; Lindau & Alvarez de Toledo 2003; Ales et al. 1999; Chow et al. 1992; Wightman et al. 1991; Segovia et al. 2010). Although both phenomena have been interpreted as expansion of the fusion pore, they are likely to represent temporally distinct kinetic steps in the process of exocytosis. Therefore, a clear distinction between fusion pore dynamics as described by imaging and electrochemical methods should be made.
This study shows that Syt-7 and Syt-1 are both necessary for a robust secretory response to native stimuli that elevate intracellular Ca2+ level (Figure 6). The number of fusion events, and the rate at which those fusion events occur, is significantly lower in Syt-7 KO cells than in WT cells. The decreased likelihood of observing fusion events in cells that do not express Syt-7 is not due to impaired Ca2+ “handling” or excitability. There is also no difference in the magnitude of Ca2+ or cholinergic current in Syt-7 KO cells compared to WT cells (Figure S7). Based on the data shown in Figures 6 and 7, a mechanism is proposed to explain why the secretory response to prolonged cholinergic stimulation is disrupted in Syt-7 KO cells. Over the course of a two-minute exposure to ACh, nicotinic receptors (nAChRs) undergo desensitization (Figure S7A). Free cytosolic Ca2+, initially elevated as a result of Ca2+ influx through nAChRs, and also possibly Cavs, is rapidly sequestered, buffered, or extruded (Neher & Augustine 1992). Despite the collapse of the Ca2+ gradient, fusion persists in WT cells throughout the period of ACh perfusion (Figure 8B, C). On the other hand, in Syt-7 KO cells, secretory activity quickly ceases after an initial burst of fusion. Thus, expression of Syt-7 is necessary for sustained activity in the setting of rapidly declining levels of intracellular free Ca2+ following nAChR desensitization. Presumably, the residual Ca2+ that remains is effective at triggering fusion only when Syt-7 is present (Figure 8C). The stark differences in Ca2+ affinities between Syt-1 and Syt-7 are likely sufficient to account for this phenomenon (Bhalla et al. 2005; Sugita et al. 2002). Dose-response curves (Figure 7C) show that there is at least a 6-fold difference in the [Ca2+]1/2 for fusion of purified dense core granules bearing Syt-1 (63 μM) versus Syt-7 (10 μM). In fact, at 10 μM Ca2+, there is no appreciable fusion of Syt-1 granules.
Figure 8. Chromaffin cells require Syt-7 for sustained secretory activity during cholinergic stimulation.
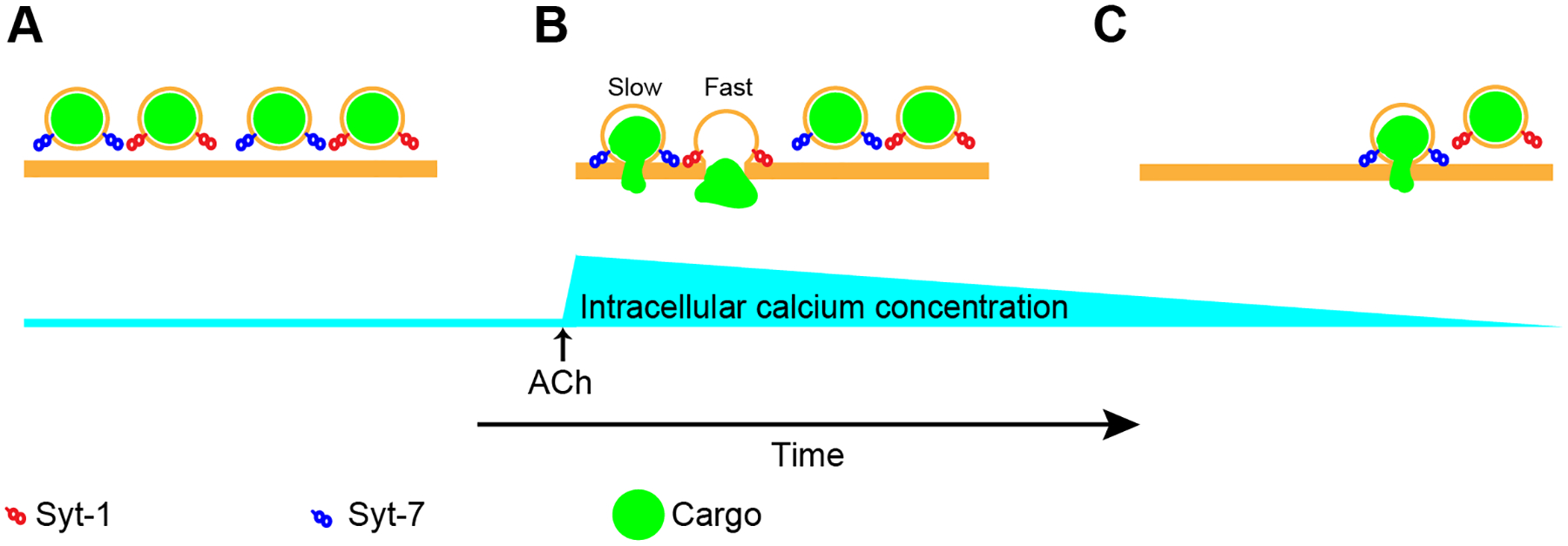
A. Chromaffin cell granules harbor Syt-1, Syt-7, or both (not shown). When stimulated with ACh, intracellular calcium concentrations increase rapidly (Figure 5C), leading to fusion of Syt-1 and Syt-7 granule populations (B). C. Prolonged stimulation with ACh causes nicotinic receptor desensitization (Figure S7A). Associated with desensitization is a corresponding decline in intracellular Ca2+ which is buffered, sequestered, or otherwise removed from the cytosol. Because Syt-1 has a lower affinity for Ca2+ than Syt-7, granules bearing this isoform are less likely to fuse as intracellular Ca2+ levels decline. On the other hand, Syt-7-bearing granules continue to fuse in the setting of near-baseline cytosolic Ca2++.
Interestingly, the time course of the cumulative frequency distribution of fusion events in Syt-7 KO cells stimulated by ACh and KCl is different (Figure S5 compare to Figure 6). This can be explained by the fact that Ca2+ elevations evoked by ACh stimulation (Figure 5) decay more rapidly in the subplasmalemma than those evoked by KCl secretion (Rao et al. 2014; Fulop & Smith 2007) which, in the absence of Syt-7, results in a more rapid inhibition of the secretory response. The rate at which NPY was released as a result of fusion in WT cells differed depending on whether KCl or ACh was used to trigger release (compare Figure 2 and Figure 4). It may be that ACh-based stimulation relies to a greater extent on activation of granules bearing Syt-7 than does KCl-based stimulation, or such variability may simply reflect the fact that experiments were performed in different groups of cells at different times. Irrespective of the mode of stimulation, the absence of Syt-7 has the consistent effect of speeding NPY cargo release, irrespective of the stimulus.
We note that no significant difference in the mobility of NPY-GFP-labeled chromaffin granules in WT and Syt-7 KO cells was observed in this study. A previous study, in which fluorescently-tagged Syts were overexpressed in cells, revealed that the frame-to-frame movement of granules harboring Syt-1 was greater than those harboring Syt-7 (Rao et al. 2017a). Therefore, the degree to which the molecular identity of a chromaffin granule regulates its mobility and whether this depends at all on the expression of a particular Syt isoform, all remain open questions for future work. We also note that the absence of Syt-7 inhibits the secretory response to a greater extent in these TIRF-based studies than what had been previously reported in studies that measured secretion by capacitance or amperometry (Schonn et al. 2008; Segovia et al. 2010). One can speculate as to the reasons why. What is reported here is the probability of fusion, which requires overexpression of NPY-GFP in order to identify docked granules. It is only the fusion of these labeled granules, which likely constitute a minority of the total fusion-competent population of granules within the cell, which is documented. The manner in which secretion was elicited in these studies, and previous ones in which capacitance was measured, is also different. Here, cells were stimulated via local perfusion of elevated KCl or ACh. In studies employing capacitance measurements, cells were stimulated by either directly depolarizing the membrane potential via the patch pipet, or by uncaging Ca2+ to concentrations in the range of tens of micromolar (Schonn et al. 2008). Secretion elicited by a strong stimulus, and associated with a greater increase in intracellular Ca2+, may be less likely to rely on fusion mediated by Syt-7.
To conclude, the experiments in this study demonstrate that the absence of Syt-7 has a profound impact on Ca2+-triggering of exocytosis and the discharge rate of peptide hormones. A key result is that the delayed kinetic component of the secretory response to cholinergic stimulation, during which subplasmalemmal Ca2+ elevations have collapsed to near baseline levels, is impaired in Syt-7 KO cells. This is shown in cumulative frequency histograms of the time course of fusion events, which can be fit by the sum of two exponentials representing the fast and slow/sustained phases of secretion. The fast phase, in WT cells, is a minor component of the curve (0.66%), whereas the slow phase dominates. In the Syt-7 KO, the fast phase of release constitutes a greater proportion of the span of the curve (58.08%) than the slow phase. The idea that the initial and later phases of exocytosis have different Ca2+ sensitivities is supported by previous work performed in permeabilized bovine chromaffin cells (Bittner & Holz 1992). By measuring [3H] norepinephrine in the medium after cells were exposed to different levels of Ca2+, Bittner and Holz (1992) concluded that an early phase of release (i.e., occurring with 5 s of stimulation) must correspond to a lower affinity process ([Ca2+]1/2 ≈ 100 μM) than a later phase of release (i.e., occurring between 5 −10 s ([Ca2+]1/2 ≈ 10 μM)). In this study, we propose a molecular mechanism to account for these observations – a mechanism that relies on the sequential activation of specialized low and high affinity Ca2+ sensors.
Supplementary Material
Acknowledgements
The authors thank Drs. J. David Castle and Ronald W. Holz for critical reading of the manuscript. This study was supported by NIH grants R01GM111997 to A.A. and P01GM072694 to L.K.T. A.C-M. and J.M.P. are supported by a fellowship from the Pharmacological Sciences Training Program T32GM007767 to the University of Michigan. The unrevised version of this manuscript has been pre-printed in bioRxiv 704205; doi: https://doi.org/10.1101/704205. Authors declare no conflict of interest.
Abbreviations:
- Syt
Synaptotagmin
- SNARE
Soluble NSF (N-Ethylmaleimide-Sensitive Factor) Attachment Protein Receptor
- GFP
Green fluorescent protein
- pHl
pHluorin
- PAI-1
Plasminogen Activator inhibitor 1
- ROI
Region of Interest
- BSA
Bovine Serum Albumin
- PC12 cells
Pheochromocytoma cells
- TIRFM
Total internal reflection fluorescence microscopy
Footnotes
--Human subjects --
Involves human subjects:
If yes: Informed consent & ethics approval achieved:
=> if yes, please ensure that the info “Informed consent was achieved for all subjects, and the experiments were approved by the local ethics committee.” is included in the Methods.
ARRIVE guidelines have been followed:
Yes
=> if it is a Review or Editorial, skip complete sentence => if No, include a statement in the “Conflict of interest disclosure” section: “ARRIVE guidelines were not followed for the following reason:” (edit phrasing to form a complete sentence as necessary).
=> if Yes, insert in the “Conflict of interest disclosure” section:
“All experiments were conducted in compliance with the ARRIVE guidelines.” unless it is a Review or Editorial
Conflicts of interest: none
=> if ‘none’, insert “The authors have no conflict of interest to declare.”
=> otherwise insert info unless it is already included
References
- Abbineni PS, Bittner MA, Axelrod D and Holz RW (2019) Chromogranin A, the major lumenal protein in chromaffin granules, controls fusion pore expansion. J Gen Physiol 151, 118–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerboom J, Chen TW, Wardill TJ et al. (2012) Optimization of a GCaMP calcium indicator for neural activity imaging. The Journal of neuroscience : the official journal of the Society for Neuroscience 32, 13819–13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albillos A, Dernick G, Horstmann H, Almers W, Alvarez de Toledo G and Lindau M (1997) The exocytotic event in chromaffin cells revealed by patch amperometry. Nature 389(6650), 509–512. [DOI] [PubMed] [Google Scholar]
- Ales E, Tabares L, Poyato JM, Valero V, Lindau M and Alvarez d. (1999) High calcium concentrations shift the mode of exocytosis to the kiss-and-run mechanism.[comment]. Nature Cell Biology. 1, 40–44. [DOI] [PubMed] [Google Scholar]
- Augustine GJ and Neher E (1992) Calcium requirements for secretion in bovine chromaffin cells. The Journal of physiology 450, 247–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Earles CA, Lewis JL and Chapman ER (2000) Membrane-embedded synaptotagmin penetrates cis or trans target membranes and clusters via a novel mechanism. Journal of Biological Chemistry 275, 25427–25435. [DOI] [PubMed] [Google Scholar]
- Bai J, Tucker WC and Chapman ER (2004) PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nature Structural & Molecular Biology. 11, 36–44. [DOI] [PubMed] [Google Scholar]
- Bendahmane M, Bohannon KP, Bradberry MM et al. (2018) The synaptotagmin C2B domain calcium-binding loops modulate the rate of fusion pore expansion. Mol Biol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla A, Tucker WC and Chapman ER (2005) Synaptotagmin isoforms couple distinct ranges of Ca2+, Ba2+, and Sr2+ concentration to SNARE-mediated membrane fusion. Mol Biol Cell 16, 4755–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner MA and Holz RW (1992) Kinetic analysis of secretion from permeabilized adrenal chromaffin cells reveals distinct components. Journal of Biological Chemistry 267, 16219–16225. [PubMed] [Google Scholar]
- Bohannon KP, Bittner MA, Lawrence DA, Axelrod D and Holz RW (2017) Slow fusion pore expansion creates a unique reaction chamber for co-packaged cargo. J Gen Physiol 149, 921–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmachari NS, Anantharaman DS, Rao BR, Gupte MD, Rao SK and Mahalingam VN (1998) Underutilization of the available services by the needy disabled leprosy patients in Government Leprosy Control Unit Puttoor, Chittoor district, Andhra Pradesh, South India. Indian J Lepr 70 Suppl, 47S–61S. [PubMed] [Google Scholar]
- Carmichael SW and Winkler H (1985) The adrenal chromaffin cell. Scientific American 253, 40–49. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Kobayashi KS, Flavell RA, Marks CB, Miyake K, Liston DR, Fowler KT, Gorelick FS and Andrews NW (2003) Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. J Cell Biol 162, 543–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman ER (2008) How does synaptotagmin trigger neurotransmitter release? Annu Rev Biochem 77, 615–641. [DOI] [PubMed] [Google Scholar]
- Chow RH, von Ruden L and Neher E (1992) Delay in vesicle fusion revealed by electrochemical monitoring of single secretory events in adrenal chromaffin cells. Nature 356, 60–63. [DOI] [PubMed] [Google Scholar]
- Domanska MK, Kiessling V, Stein A, Fasshauer D and Tamm LK (2009) Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J Biol Chem 284, 32158–32166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domanska MK, Kiessling V and Tamm LK (2010) Docking and fast fusion of synaptobrevin vesicles depends on the lipid compositions of the vesicle and the acceptor SNARE complex-containing target membrane. Biophysical journal 99, 2936–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas WW (1968) Stimulus-secretion coupling: the concept and clues from chromaffin and other cells. Br J Pharmacol 34, 451–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas WW and Rubin RP (1963) The mechanism of catecholamine release from the adrenal medulla and the role of calcium in stimulus-secretion coupling. The Journal of physiology 167, 288–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Kanno E, Satoh M, Saegusa C and Yamamoto A (2004) Synaptotagmin VII is targeted to dense-core vesicles and regulates their Ca2+ -dependent exocytosis in PC12 cells. J Biol Chem 279, 52677–52684. [DOI] [PubMed] [Google Scholar]
- Fulop T, Radabaugh S and Smith C (2005) Activity-Dependent Differential Transmitter Release in Mouse Adrenal Chromaffin Cells. Journal of Neuroscience 25, 7324–7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop T and Smith C (2007) Matching native electrical stimulation by graded chemical stimulation in isolated mouse adrenal chromaffin cells. Journal of neuroscience methods 166, 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF and Sudhof TC (1994) Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79, 717–727. [DOI] [PubMed] [Google Scholar]
- Guerineau NC (2019) Cholinergic and peptidergic neurotransmission in the adrenal medulla: A dynamic control of stimulus-secretion coupling. IUBMB Life. [DOI] [PubMed] [Google Scholar]
- Hao Z, Wei L, Feng Y, Chen X, Du W, Ma J, Zhou Z, Chen L and Li W (2015) Impaired maturation of large dense-core vesicles in muted-deficient adrenal chromaffin cells. J Cell Sci 128, 1365–1374. [DOI] [PubMed] [Google Scholar]
- Holz RW, Senter RA and Frye RA (1982) Relationship between Ca2+ uptake and catecholamine secretion in primary dissociated cultures of adrenal medulla. Journal of Neurochemistry 39, 635–646. [DOI] [PubMed] [Google Scholar]
- Hui E, Bai J, Wang P, Sugimori M, Llinas RR and Chapman ER (2005) Three distinct kinetic groupings of the synaptotagmin family: Candidate sensors for rapid and delayed exocytosis. Proceedings of the National Academy of Sciences 102, 5210–5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman SL, Turecek J, Belinsky JE and Regehr WG (2016) The calcium sensor synaptotagmin 7 is required for synaptic facilitation. Nature 529, 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal JK, Chakrabarti S, Andrews NW and Simon SM (2004) Synaptotagmin VII Restricts Fusion Pore Expansion during Lysosomal Exocytosis. PLoS Biology 2, e233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalb E, Frey S and Tamm LK (1992) Formation of supported planar bilayers by fusion of vesicles to supported phospholipid monolayers. Biochim Biophys Acta 1103, 307–316. [DOI] [PubMed] [Google Scholar]
- Kolski-Andreaco A, Cai H, Currle DS, Chandy KG and Chow RH (2007) Mouse adrenal chromaffin cell isolation. Journal of visualized experiments : JoVE, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzberger AJB, Kiessling V, Liang B, Seelheim P, Jakhanwal S, Jahn R, Castle JD and Tamm LK (2017a) Reconstitution of calcium-mediated exocytosis of dense-core vesicles. Sci Adv 3, e1603208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzberger AJB, Kiessling V, Liang B, Yang ST, Castle JD and Tamm LK (2017b) Asymmetric Phosphatidylethanolamine Distribution Controls Fusion Pore Lifetime and Probability. Biophysical journal 113, 1912–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzberger AJB, Kiessling V, Stroupe C et al. (2019) In vitro fusion of single synaptic and dense core vesicles reproduces key physiological properties. Nat Commun 10, 3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeber S, Schomerus C and Korf HW (1998) A specific and sensitive double-immunofluorescence method for the demonstration of S-antigen and serotonin in trout and rat pinealocytes by means of primary antibodies from the same donor species. Histochem Cell Biol 109, 309–317. [DOI] [PubMed] [Google Scholar]
- Lindau M and Alvarez de Toledo G (2003) The fusion pore. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1641, 167–173. [DOI] [PubMed] [Google Scholar]
- MacDougall DD, Lin Z, Chon NL, Jackman SL, Lin H, Knight JD and Anantharam A (2018) The high-affinity calcium sensor synaptotagmin-7 serves multiple roles in regulated exocytosis. J Gen Physiol 150, 783–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN and De Camilli P (2003) Cell biology of the presynaptic terminal. Annu Rev Neurosci 26, 701–728. [DOI] [PubMed] [Google Scholar]
- Neher E and Augustine GJ (1992) Calcium gradients and buffers in bovine chromaffin cells. J.Physiol.(Lond.) 450, 273–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao TC, Passmore DR, Peleman AR, Das M, Chapman ER and Anantharam A (2014) Distinct fusion properties of synaptotagmin-1 and synaptotagmin-7 bearing dense core granules. Mol Biol Cell 25, 2416–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao TC, Rodriguez ZS, Bradberry MM et al. (2017a) Synaptotagmin isoforms confer distinct activation kinetics and dynamics to chromaffin cell granules. Journal of General Physiology 149, 763–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao TC, Santana Rodriguez Z, Bradberry MM et al. (2017b) Synaptotagmin isoforms confer distinct activation kinetics and dynamics to chromaffin cell granules. J Gen Physiol 149, 763–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonn JS, Maximov A, Lao Y, Sudhof TC and Sorensen JB (2008) Synaptotagmin-1 and −7 are functionally overlapping Ca2+ sensors for exocytosis in adrenal chromaffin cells. Proc Natl Acad Sci U S A 105, 3998–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segovia M, Ales E, Montes MA, Bonifas I, Jemal I, Lindau M, Maximov A, Sudhof TC and Alvarez de Toledo G (2010) Push-and-pull regulation of the fusion pore by synaptotagmin-7. Proc Natl Acad Sci U S A 107, 19032–19037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC (2013) A molecular machine for neurotransmitter release: synaptotagmin and beyond. Nat Med 19, 1227–1231. [DOI] [PubMed] [Google Scholar]
- Sugita S, Han W, Butz S, Liu X, Fernandez-Chacon R, Lao Y and Sudhof TC (2001) Synaptotagmin VII as a plasma membrane Ca(2+) sensor in exocytosis. Neuron 30, 459–473. [DOI] [PubMed] [Google Scholar]
- Sugita S, Shin OH, Han W, Lao Y and Sudhof TC (2002) Synaptotagmins form a hierarchy of exocytotic Ca(2+) sensors with distinct Ca(2+) affinities. EMBO J 21, 270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraska JW, Perrais D, Ohara-Imaizumi M, Nagamatsu S and Almers W (2003) Secretory granules are recaptured largely intact after stimulated exocytosis in cultured endocrine cells. Proceedings of the National Academy of Sciences of the United States of America 100, 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, Anderson LH and Knight JD (2019) Membrane-Binding Cooperativity and Coinsertion by C2AB Tandem Domains of Synaptotagmins 1 and 7. Biophysical journal 116, 1025–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi T and Fukuda M (2007) Synaptotagmin VII modulates the kinetics of dense-core vesicle exocytosis in PC12 cells. Genes Cells 12, 511–519. [DOI] [PubMed] [Google Scholar]
- Voleti R, Tomchick DR, Sudhof TC and Rizo J (2017) Exceptionally tight membrane-binding may explain the key role of the synaptotagmin-7 C2A domain in asynchronous neurotransmitter release. Proc Natl Acad Sci U S A 114, E8518–E8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner ML and Tamm LK (2001) Reconstituted Syntaxin1A/SNAP25 Interacts with Negatively Charged Lipids as Measured by Lateral Diffusion in Planar Supported Bilayers. Biophysical Journal 81, 266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JP, Toft-Bertelsen TL, Mohrmann R, Delgado-Martinez I and Sorensen JB (2014) Synaptotagmin-7 is an asynchronous calcium sensor for synaptic transmission in neurons expressing SNAP-23. PLoS One 9, e114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman RM, Jankowski JA, Kennedy RT, Kawagoe KT, Schroeder TJ, Leszczyszyn DJ, Near JA, Diliberto EJ Jr. and Viveros OH (1991) Temporally resolved catecholamine spikes correspond to single vesicle release from individual chromaffin cells. Proceedings of the National Academy of Sciences of the United States of America 88, 10754–10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler H (1971) The membrane of the chromaffin granule. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 261, 293–303. [DOI] [PubMed] [Google Scholar]
- Zdanowicz R, Kreutzberger A, Liang B, Kiessling V, Tamm LK and Cafiso DS (2017) Complexin Binding to Membranes and Acceptor t-SNAREs Explains Its Clamping Effect on Fusion. Biophysical journal 113, 1235–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Liu B, Wu Q et al. (2019) Differential Co-release of Two Neurotransmitters from a Vesicle Fusion Pore in Mammalian Adrenal Chromaffin Cells. Neuron 102, 173–183 e174. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wu Y, Wang Z, Dunning FM, Rehfuss J, Ramanan D, Chapman ER and Jackson MB (2011) Release mode of large and small dense-core vesicles specified by different synaptotagmin isoforms in PC12 cells. Mol Biol Cell 22, 2324–2336.21551071 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.