

Abstract
Iron- and α-ketoglutarate-dependent dioxygenases (Fe/αKGs) represent a versatile and intriguing enzyme family by virtue of their ability to directly functionalize unactivated C–H bonds at the cost of αKG and O2. Fe/αKGs play an important role in the biosynthesis of natural products, valuable biologically active secondary metabolites frequently pursued as drug leads. The field of natural product total synthesis seeks to contruct these molecules as effeciently as possible, although natural products continue to challenge chemists due to their intricate structural complexity. Chemoenzymatic approaches seek to remedy the shortcomings of traditional synthetic methodology by combining Nature’s biosynthetic machinery with traditional chemical methods to efficiently construct natural products. Although other oxygenase families have been widely employed for this purpose, Fe/αKGs remain underutilized. The following review will cover recent chemoenzymatic total syntheses involving Fe/αKG enzymes. Additionally, related information involving natural product biosynthesis, methods development, and non-chemoenzymatic total syntheses will be discussed to inform retrosynthetic logic and synthetic design.
1. Introduction
The iron- and α-ketoglutarate-dependent dioxygenases (Fe/αKGs) are an intriguing family of enzymes that catalyze diverse and powerful reactions.1–6 Fe/αKGs were first discovered in the 1960s and shown to catalyze collagen, thymine, and γ-butyrobetaine hydroxylation.1,2 Structurally, Fe/αKGs contain a conserved double-stranded β-helix (DSBH or cupin) fold that uses a His1-X-Asp/Glu-Xn-His2 iron-binding motif.1,2 During catalysis, oxidative decomposition of αKG yields a high energy Fe(IV)-oxo intermediate that is capable of performing homolysis of unactivated C–H bonds (Figure 1), yielding a hydroxylated product following radical recombination.1,2 In mammals, Fe/αKGs take part in a variety of important biological processes, such as collagen biosynthesis/regulation, L-carnitine biosynthesis, post-translational protein modification, epigenetic regulation via histone/DNA demethylation, and sensors in energy metabolism.3 Within plants, bacteria, and fungi, Fe/αKGs play an important role in secondary metabolite (natural product) biosynthesis.5–7 Due to their unique biological properties, a large number of natural products and their derivatives have been developed as drugs.8,9 For this reason, the field of natural product total synthesis has grown and evolved over the years to produce these important molecules as efficiently as possible. Despite rapid advances in synthetic methodology, natural products continue to challenge chemists due to their intricate structural complexity.10 Chemoenzymatic approaches have been used to remedy shortcomings in existing synthetic methods by leveraging Nature’s biosynthetic machinery as an alternative.9 Although Fe/αKG enzymes have been known for decades, their development for biocatalysis and chemoenzymatic total synthesis has only begun recently. When compared to more extensively developed oxygenase families, such as cytochrome P450s or flavin-dependent monoxygenases (FMOs), Fe/αKGs can be considered cost effective, since the reaction requires cheap and abundant O2 and αKG compared to NAD(P)H. Furthermore, in contrast to the P450s, Fe/αKGs are self-sufficient enzymes and do not require a dedicated reductase partner.9
Figure 1.
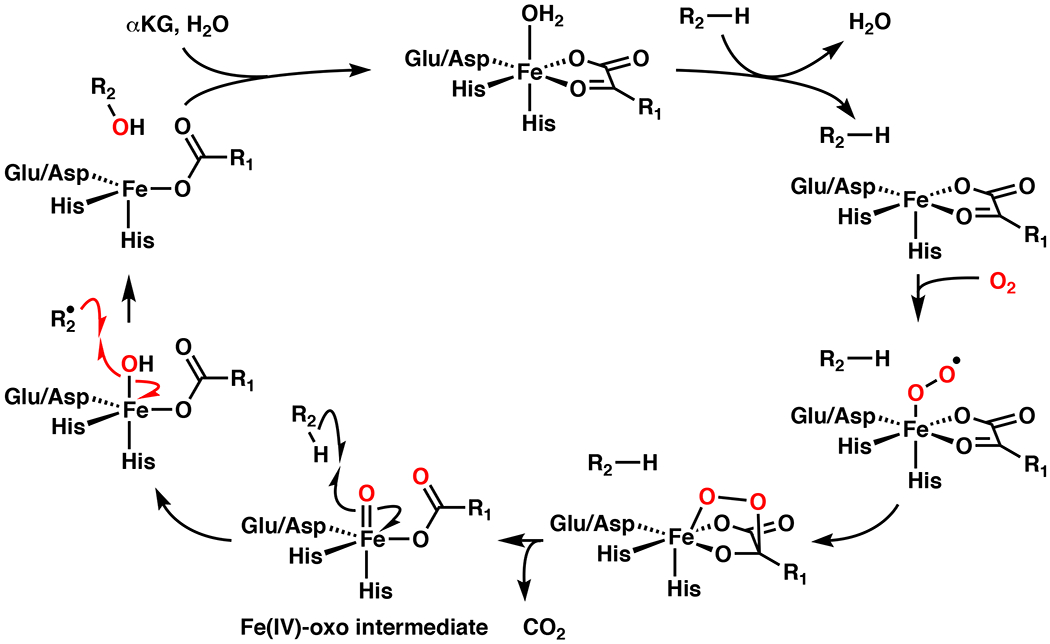
Catalytic cycle for hydroxylation with iron- and α-ketoglutarate-dependent dioxygenase (Fe/αKG)1,2 involving the intermediacy of a high energy Fe(IV)-oxo intermediate for C–H abstraction.
Direct C–H functionalization is often considered the “holy grail” of synthetic chemistry, due to its potential for streamlining synthetic strategies by bypassing prefunctionalization step(s).11–15 Although this field has experienced explosive growth in recent years, we are far away from being able to functionalize any C–H bond of our choosing. C–H functionalization reactions are difficult to develop due to the inherently high strength of most “unactivated” C–H bonds (bond-dissociation energy (BDE)≈96–101 kcal/mol and pKa≈50).15 Additionally, the prevalence of C–H bonds in most organic molecules, paired with subtle differences in their BDEs, typically manifests in poor chemo-, regio-, and stereoselectivity for a given reaction.13 Common methods for chemocatalytic C–H activation typically rely on pathways involving nucleophilic metal complexes that select for electronically or sterically distinct 1° over 2° or 3° sites.11,14 Conversely, radical C–H functionalization methods typically prefer 3°, benzylic, or allylic C–H bonds based on their lower BDEs.15 In contrast, Fe/αKG enzymes are able to perform C–H functionalization reactions with exquisite chemo-, regio-, and stereocontrol at 1°, 2°, and 3° sites depending on the enzyme/substrate pair.5,6 For the reasons listed above, Fe/αKG enzymes can be viewed as powerful biocatalysts with untapped synthetic potential.
The following sections will give a comprehensive overview of all known chemoenzymatic total syntheses that involve Fe/αKG enzymes. Each section will be divided based on the type of reaction catalyzed by the Fe/αKG and then further classified based on the type of substrate(s). When applicable, methods development will also be discussed. Relevant background information will be detailed at the beginning of each section, in addition to the retrosynthetic logic involved in the design of the synthesis. When relevant, chemoenzymatic syntheses will be benchmarked to non-enzymatic approaches for comparison.
2. Hydroxylation
The hydroxyl group represents one of the most commonly encountered functionality among natural products and medicines, and can have a profound impact on molecular properties, such as solubility, polarity, and target interaction.15 Alcohols can also serve as versatile handles or directing group in complex molecule synthesis. For these reasons, many existing methods and those in development for C–H functionalization are focused on alcohol synthesis. Traditional redox reactions require the use of pre-functionalized or activated substrates for alcohol synthesis, whereas newer methods perform redox neutral16,17,18/direct C–H hydroxylation transformations.11,13,15 As mentioned above, Fe/αKG enzymes show untapped synthetic potential in the realm of C–H functionalization. In fact, variants that perform C–H hydroxylation represent the most abundant and well-studied enzymes in this family.1 In turn, a steady increase in the utilization of these enzymes for synthetic application has been observed over the years. The following sections will highlight biocatalytic application of Fe/αKG hydroxylase enzymes for natural product total synthesis and methods development.
2.1. Amino Acid Hydroxylation
The 20 canonical amino acids (AA) represent key building blocks in the biosynthesis of complex primary and secondary metabolites.12 Although the vast majority of these metabolites consist of peptides derived exclusively from canonical amino acids (cAA), a great number of increasingly more complex secondary metabolites/natural products are found to contain non-canonical amino acids (ncAAs) that arises from enzymatic modification(s) of their canonical variant(s).5,6 Additionally, several ncAA intermediates may exist in a biosynthetic pathway en route to the final ncAA or natural product. In recent years, a growing number of Fe/αKG hydroxylase enzymes that operate on free AAs have been discovered and characterized from natural product biosynthetic pathways.5,6 Catalytic remote C–H hydroxylation on free AAs remains an unsolved problem in organic synthesis.12 This methodological gap paired with the ubiquity of Fe/αKG AA hydroxylases, chemical versatility of the newly-introduced hydroxyl group, and the low cost of AAs, makes direct AA hydroxylation particularly attractive for synthetic development. As such, Fe/αKG AA hydroxylases have been utilized more than any other Fe/αKG family members for natural product total synthesis to date.
2.1.1. Aliphatic Amino Acid Hydroxylation
Fe/αKG enzymes serve important roles in producing proline-based ncAAs in proteins and peptide based secondary metabolites (Figure 2). Hydroxylation of Gly-Pro-Pro repeats by C-P4H and C-P3H in collagen is important for regulating its structure.3 Among biosynthetic Fe/αKG enzymes that operate on free amino acids, Pro hydroxylases (PHs) are by far the most well characterized/established for biocatalysis.6 Every enzyme necessary for generating each possible OH-Pro variant has been identified (cis-3-PH, cis-4-PH, trans-3-PH, and trans-4-PH).19 Additionally, a subset of these enzymes are pipecolic acid hydroxylases (PiHs), which are known to act on pipecolic acid (Pip) and Me-Pro variants without any cross-reactivity with Pro.20 A couple examples include fungal enzyme FoPip4H and GetF from the biosynthesis of GE81112.20,21 Fe/αKG enzymes are also important in the production of methylated variants of Pro. These compounds typically arise from δ-oxidation, cyclization, and reduction from L-Leu or L-Ile.22–24 This strategy is used in Nature in the biosynthesis of (2S,4R)-4-methylproline (4-Me-Pro) in the antituberculosis natural product griselimycin.22 In this pathway, 4-Me-Pro is produced from L-Leu following a three-step sequence involving δ-selective C–H hydroxylation by Fe/αKG GriE, alcohol to aldehyde oxidation by zinc-dependent dehydrogenase GriF (leading to spontaneous imine formation), and finally, imine to amine reduction with oxidoreductase GriH. The catalytic activity of GriE is particularly intriguing as distal C–H functionalization constitutes a particularly challenging transformation with only few precedents and δ-OH-L-Leu could serve as a valuable starting point in the synthesis of proline analogues and various natural products.25–27
Figure 2.
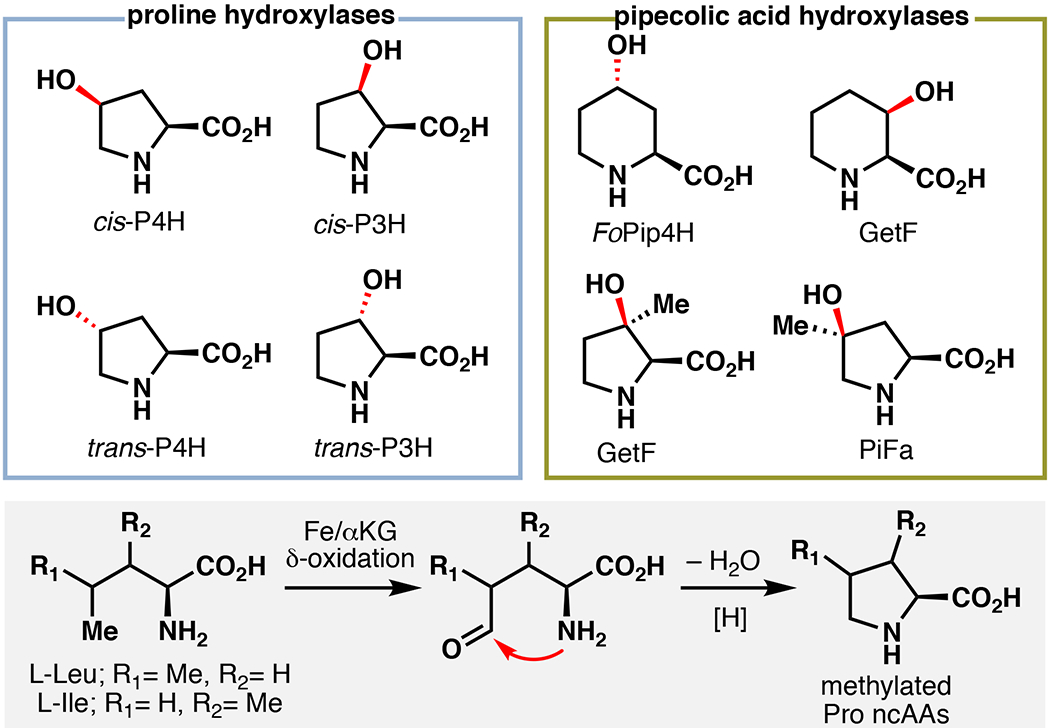
Involvement of Fe/αKG enzymes in proline and pipecolic acid hydroxylation19–21 and the general utility of Fe/αKG enzymes in the biosynthesis of methylated proline derivatives.22–24
Total Synthesis of Manzacidin C
In 2000, Ohfune reported the first total synthesis of manzacidin A and C, bromopyrrole alkaloids originally isolated from the Okinawan sponge Hymeniacidon sp.28 This report proposed that the core structure of the manzacidins may arise biosynthetically from a Leu-derived intermediate (1) containing an isonitrile group and a primary alcohol at the γ-position and the δ-position, respectively (Figure 3A). In their approach, a functionally analogous lactone intermediate (2) was constructed in 13 steps and used to prepare both natural products following a concise two-step protocol. Owing to the brevity of the protocol, lactone 2 has become the target of numerous formal total syntheses of manzacidin A and C.29–36 Nevertheless, 2 remains challenging to access, typically requiring 13–15 step synthesis. Our laboratory envisioned a double C–H functionalization approach to 2 from L-Leu involving C5 hydroxylation with GriE, and chemocatalytic C–H amination at C4 to complete a formal total synthesis of manzacidin C with significantly reduced step count.33,34 Towards this objective, biochemical characterization and substrate scope profiling of GriE were undertaken, revealing its ability to hydroxylate a broad array of AAs with high total turnover numbers (TTNs) and modest to excellent yields (18–92%). Based on the observed reactivity patterns, a substrate structure-activity relationship could be developed to establish structural features within the substrates that improve, diminish, or abolish hydroxylation. Gram-scale hydroxylation on L-Leu could be performed using clarified lysate, affording 4 in 68% two-step yield following Boc protection and lactonization (Figure 3B). While attempts at intermolecular C–H amination/azidation on 4 proved unsuccessful, 37–40 intramolecular C–N bond formation proceeded in 65% yield (77% brsm), following lactone opening and carbamate installation.41–42 Boc protection of the resulting oxazolidinone (6) followed by basic methanolysis and lactonization cleanly afforded 2. This approach constitutes a 9-step formal synthesis of manzacidin C with an 11% overall yield from L-Leu. While this represented the most expedient synthesis of 2 to date (7 steps), formation of the critical C4–N bond through intramolecular C–H insertion was a step-inefficient process. We rationalized that early installation of the C4–N bond would avoid undesirable concession steps and allow for a more direct approach to manzacidin C (Figure 3C). As prior structure-activity relationship studies suggest that additional substitution at the γ-position of the substrate is well tolerated by GriE, we hypothesized that a route towards lactone 2 that features C4–N bond formation prior to C5 enzymatic hydroxylation would be feasible. This reaction sequence also avoided the requirement for asymmetric C–H amination since the γ-position remains prochiral prior to GriE hydroxylation. Reports by Britton on photochemical C–H fluorination of L-Leu inspired the development of a similar C–H azidation protocol with 2,5-di-trifluoromethylsulfonylazide (7).43,44 Using this method, γ-azidated Leu derivative 8 could be obtained in 49% isolated yield following preparative HPLC purification. This represents the first example of a tetrabutylammonium decatungstate (TBADT) mediated azidation, as well as the first direct C–H azidation on an unprotected AA. Furthermore, 8 was readily hydroxylated with >95% conversion using purified GriE. Following hydroxylation, a one-pot three-transformation sequence was developed, whereby the azide was first hydrogenated, the free amines Boc protected, and the remaining free acid/alcohol lactonized to access 2 in 41% yield over two steps. Our second generation approach constitutes a five-step formal synthesis of manzacidin C (11% overall yield). This approach represents the first example of an Fe/αKG AA hydroxylase to be used in total synthesis.
Figure 3.
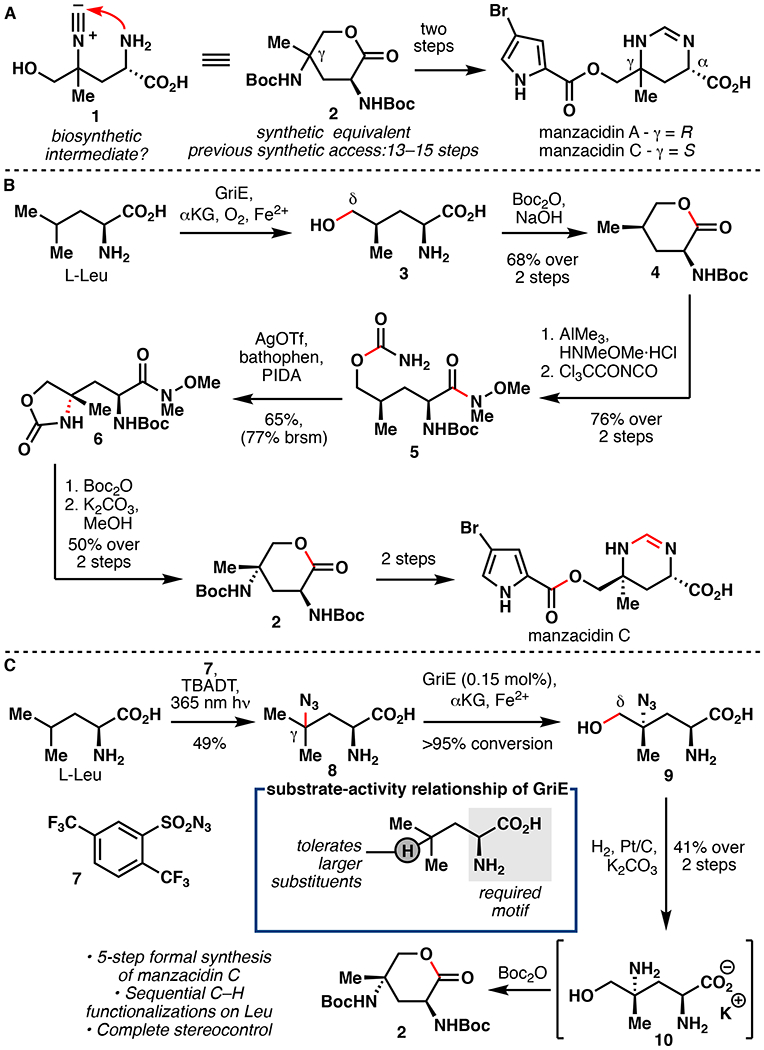
A. Ohfune’s hypothesis for the biosynthesis of the manzacidins.28 B. Renata’s first generation chemoenzymatic formal synthesis of manzacidin C.34 C. Renata’s second generation chemoenzymatic formal synthesis of manzacidin C.33,34 TBADT = tetrabutylammonium decatungstate.
Total Synthesis of Cavinafungin B
During the course of our studies, we observed minor amounts of cyclic imine 11 formation while incubating L-Leu with GriE. This result inspired the development of a chemoenzymatic cascade to access substituted prolines by mimicking 4-Me-Pro biogenesis (Figure 4A).22 Optimization studies showed that increasing the enzyme loading (0.01 – 0.12 mol%) could drive the formation of imine 11 from L-Leu to completion. Subsequent addition of ammonia borane (NH3•BH3) in a one-pot fashion led to the formation of 4-Me-Pro in 88% yield.45 When utilizing cell lysate, we were able to achieve 57% yield of 12 over two steps following Fmoc protection. Extension of this methodology produced a range of 4,4-disubstituted proline derivatives (75–88% yield) in a protecting group free manner with complete stereocontrol. The previous state-of-the-art synthesis of 4-Me-Pro proceeds from 4-OH-Pro in more than 10 steps, and necessitates the use of directed hydrogenation to impart diastereocontrol.46 In an analogous fashion, a two-step protocol was developed to synthesize (2S,3R)-3-hydroxyl-3-methylproline (3-OH-3-Me-Pro), a key structural element in biologically relevant natural products polyoxypeptin A and FR225659.47 Based on the postulated biogenesis of 3-OH-3-Me-Pro, we first utilized the ability of Fe/αKG hydroxylase UcsF to catalyze iterative C5 oxidation of isoleucine in the construction of 3-Me-Pro (13).48 The final β-hydroxylation was then conducted by using GetF, which has previously been shown by Huttel and co-workers to perform this function.20 Finally, efficient access to 4-Me-Pro has also enabled the first total synthesis of the antiviral lipopeptide aldehyde cavinafungin B (Figure 4B).49 Efficient access to this natural product is of particular value, as cavinafugin A (acetylated cavinafungin B) was previously shown to potently inhibit cells infected by each dengue virus serotype (IC50=1–5 nM) or the Zika virus (IC50=150 nM) with 30–100 fold selectivity over uninfected cells.50 To complete the synthesis of cavinafungin B, we utilized solid-phase peptide synthesis (SPPS) starting from a modified Rink oxazolidine tether known to release the mature peptide as a C-terminal aldehyde following resin cleavage.51 Following standard Fmoc-based SPPS peptide assembly and resin cleavage, cavinafungin B could be assembled in 37% yield over 10 steps.
Figure 4.
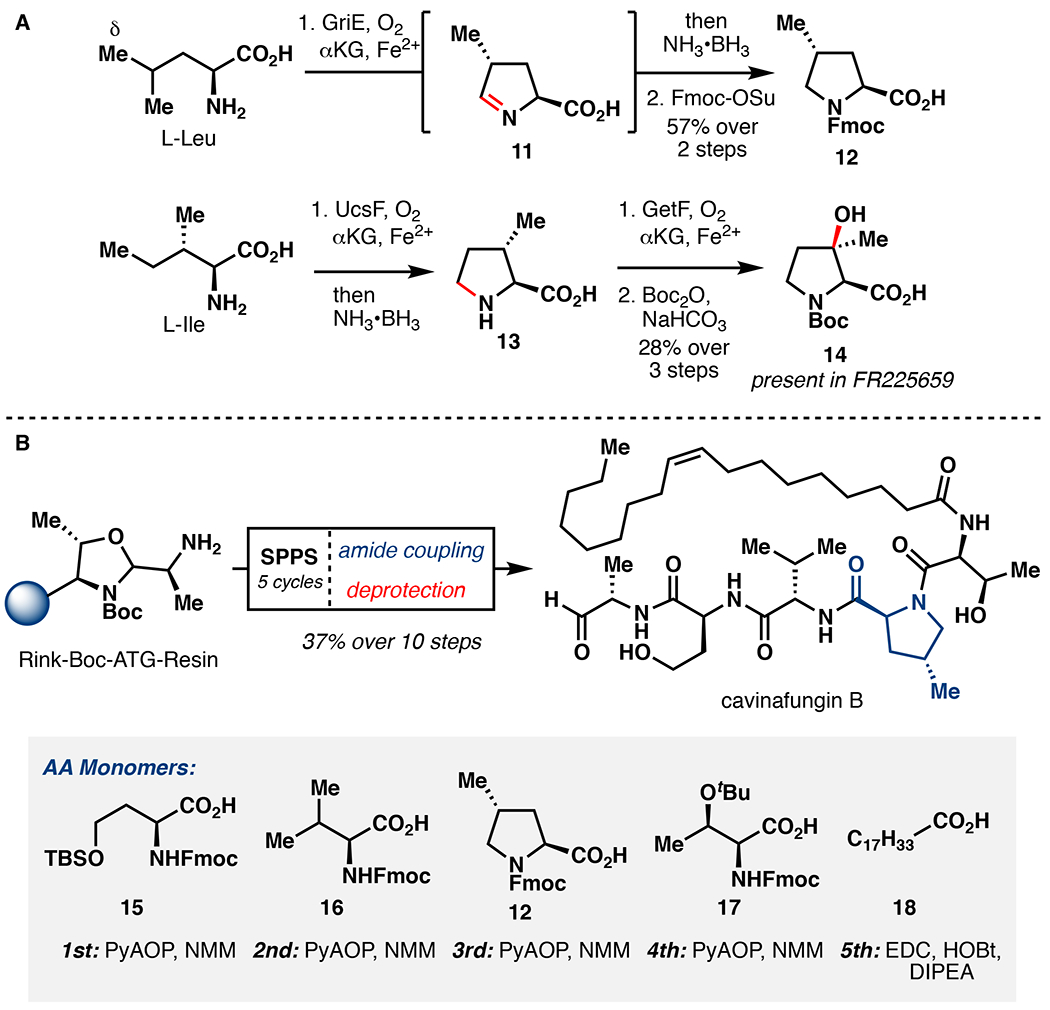
A. One-pot chemoenzymatic synthesis of 4-Me-Pro and 3-OH-3-Me-Pro.33,48 B. Chemoenzymatic synthesis of cavinafungin B.49
2.1.2. L-Lysine Hydroxylation
Fe/αKG enzymes that perform hydroxylation of Lys residues in proteins are important for diverse biological functions. Histones release DNA following Lys demethylation, mediated by Fe/αKG lysine demethylases (JHDMs).52 Lys hydroxylation in procollagen by procollagen-lysine 5-dioxygenases (PLODs), is important for fibrillogenesis, cross-linking, and matrix mineralization to generate collagen.53 Numerous Fe/αKG hydroxylases that act of free Lys or Orn are known, such as the KDOs, K3H/K4Hs, and ODO, and are capable of performing stereoselective β or γ-hydroxylation (Figure 5).54,55 By comparison, contemporary non-enzymatic methods for β or γ C–H functionalization typically require directing groups and often suffer from a lack of stereocontrol.12,26
Figure 5.
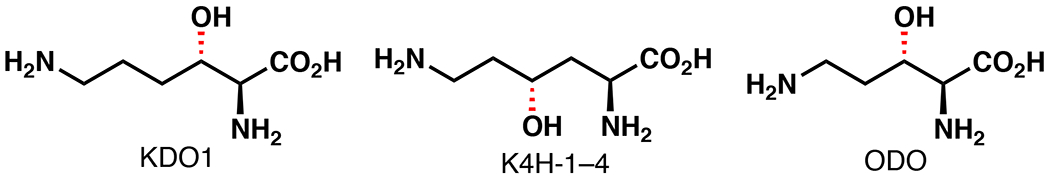
Involvement of Fe/αKG enzymes in lysine and ornithine hydroxylation.54,55
Total synthesis of Tambromycin
In 2016, a new approach to natural product discovery was reported wherein genomic and metabolomic information was correlated to identify structurally novel metabolites and their biosynthetic gene clusters (BGCs).56 This method, termed metabologenomics, resulted in the identification of tambromycin, a non-ribosomal peptide (NRP) peptide containing a rare ncAA tambroline (L-Tam). Based on isotopic feeding experiments with Lys-d9, it was determined that L-Tam biosynthetically originates from L-Lys. Due to missing deuteration at the α-position, these experiments also suggest that this ncAA arises via dehydrogenation of Lys at its C2–C3 bond (19), followed by an intramolecular 5-exo-trig cyclization of the ε-amino group to form the pyrrolidine core (Figure 6). This mechanism is reminiscent of an analogous cyclization in the biosynthesis of ncAA (2S,3R)-capreomycidine (L-Cap) wherein the Fe/αKG enzyme VioC first hydroxylates the β-position of L-Arg, followed by dehydration and a 6-exo-trig cyclization mediated by PLP dependent enzyme VioD.57 While L-Tam’s biosynthesis does not involve a hydroxylated L-Lys intermediate analogous to hydroxylated L-Arg for L-Cap, our laboratory hypothesized that such an intermediate may serve as a valuable precursor for a chemoenzymatic synthesis of L-Tam, and subsequently tambromycin.58
Figure 6.
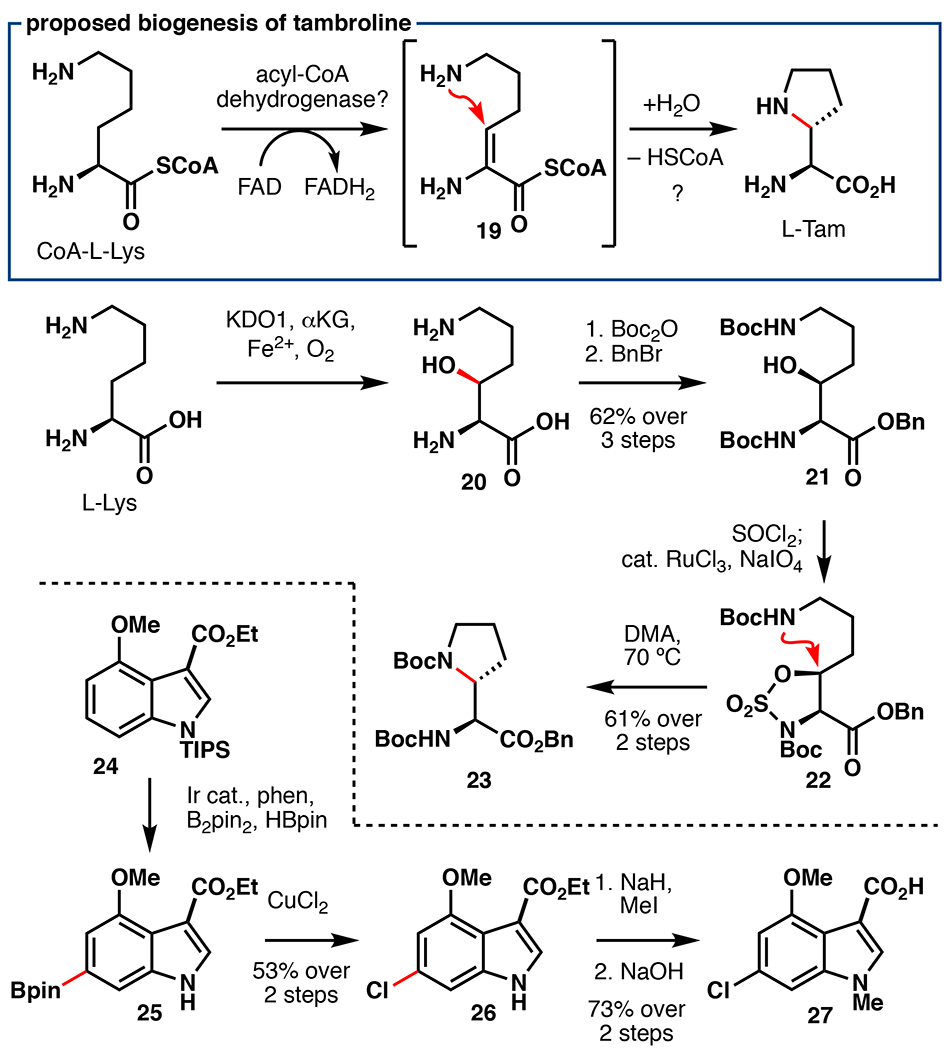
Proposed biogenesis of tambroline (L-Tam) and preparation of key fragments 23 and 27 via Fe/αKG hydroxylation and Ir-catalyzed borylation en route to tambromycin.58
Towards this objective, clarified lysate from E. coli BL21(DE3) cells overexpressing Fe/αKG hydroxylase KDO1 alongside chaperones GroES/EL was used for gram-scale hydroxylation of L-Lys (Figure 6).54 Global Boc protection and benzyl ester formation afforded 21 in 62% over three steps. While our efforts to affect cyclization via Appel or Mitsunobu conditions gave only β-elimination, sulfamidate formation, followed by simple heating in DMF cleanly afforded protected L-Tam (23) in 61% yield over two steps.59 Attempts to target shorter approaches to L-Tam, including photochemical C–H amination and VioD mediated cyclization of 20 showed no reaction.26 Prior to our studies, two routes for the synthesis of 3,4,6-trisubstituted indoles had been described.60,61 As both approaches required relatively high step count (8–9 steps), an alternative strategy was sought. In recent years, iridium-catalyzed borylation has proven to be a powerful method for the direct functionalization of arene C–H bonds.62 Regioselectivity for this reaction is determined based on the steric profile of the substrate, and a previous report published by the Baran lab established conditions for selective borylation at C6 vs C5 on indoles.63 The use of this reaction on a 3,4-disubstituted indole afforded the correct borylation regioselectivity and the desired C6 chlorinated product (26) could be obtained following heating with CuCl2 (52% yield over 2 steps). Fragment assembly was performed in a convergent fashion (Figure 7) by coupling 28 with 23 using hexafluorophosphate azabentotriazole tetramethyluronium (HATU) and 28 with 27 using 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride (DMTMM).64 Although dipeptide 29 contains two free amines which could potentially present regioselectivity issues during peptide coupling, we rationalized that the α-amine of L-Tam would be more sterically accessible and thereby allow for some degree of regiocontrol. To our delight, coupling with HATU gave complete regiocontrol for the α-amine over the β, yielding the first total synthesis of tambromycin in 76% yield over two steps following methyl ester hydrolysis. This total synthesis highlights the power of taking cues from effective biosynthetic steps, as well as combining chemocatalytic and biocatalytic C–H functionalization logic in NRP total synthesis.
Figure 7.

Completion of Renata’s chemoenzymatic total synthesis of tambromycin.58
Glidobactin A Fragment Synthesis
Glidobactin A and cepafungin I are hybrid peptide-polyketide natural products known to potently inhibit the eukaryotic 20S proteasome core particle (IC50<20 nM).65,66 Proteasome inhibitors have recently garnered attention as potential anticancer therapeutics due to their ability to dysregulate degradation of pro-apoptotic factors in malignant cells.67 γ-OH-L-Lys (31) represents a key structural motif in glidobactin A and cepafungin I, although its biogenesis was hitherto uncharacterized. Due to the ubiquity of γ-OH-L-Lys among this important family, our laboratory set out to determine its biogenesis.68 We postulated that Fe/αKG enzyme GlbB from the glidobactin A biosynthetic gene cluster may be responsible for L-Lys hydroxylation (Figure 8). Uniprot annotation suggests that GlbB belongs to the Pfam PF10014, members of which have previously been shown to be amino acid hydroxylases.69,70 Indeed, isolated GlbB, recombinantly produced from E. coli BL21(DE3) cells, demonstrated L-Lys hydroxylation when combined with αKG and FeSO4. Towards synthetic application, we found that 0.02 mol% GlbB was sufficient to achieve >95% conversion to 31. 31 was then forwarded to 32 in 60% yield over three steps following double Boc protection and lactonization. An advanced intermediate en route to glidobactin A was then constructed by simply combining lactone 32 with L-alaninol (33), affording 34 in 92% yield. This report lays the groundwork for total synthetic access to glidobactin A as well as related syrbactins and analogs.
Figure 8.
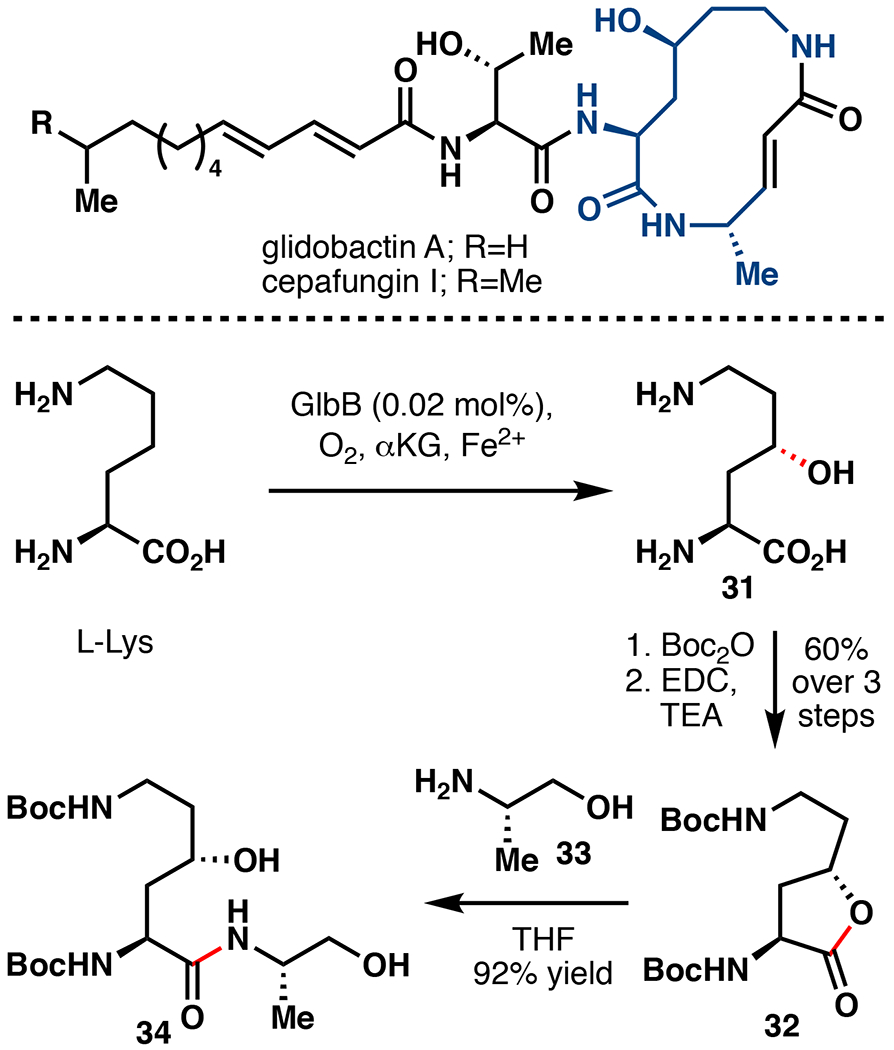
Application of Fe/αKG hydroxylation in the preparation of a key dipeptide fragment of glidobactin A and cepafungin I.68 EDC = 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide.
2.1.3. L-Arginine Hydroxylation
L-Arg hydroxylation represents another prominent reaction performed by Fe/αKG enzymes present in the biosynthesis of natural products (Figure 9). In the biosynthesis of capreomycidine (L-Cap), a key motif present in viomycin and related antibiotics, β-hydroxylation on free Arg by the Fe/αKG VioC is followed by dehydration and cyclization by the PLP-dependent enzyme VioD.57 A similar strategy was also used by Nature in the biosynthesis of the unusual bicycle streptolidine found within the broad-spectrum antibiotic streptothricin F.71 Another noteworthy L-Arg derived ncAA is enduracididine (L-End), present in an array of peptide antibiotics such as teixobactin, mannopeptimycin, and enduracidin.72 The biosynthesis of L-End similarly proceeds via a dehydration and cyclization pathway, although the hydroxylated intermediate is 2-oxo-4-hydroxy-5-guanidinovaleric acid, a deaminated analog of γ-OH-L-Arg.73 While Fe/αKG enzyme OrfP has been shown to catalyze β,γ-dihydroxylation on free Arg, an enzyme that selectively catalyzes γ-hydroxylation has yet to be discovered in Nature. δ-hydroxylation of Arg is also known (Fe/αKG enzyme EFE), although the resulting hydroxylated intermediate rapidly degrades to guanidine and L-Δ−1-pyrroline-5-carboxylate.5
Figure 9.
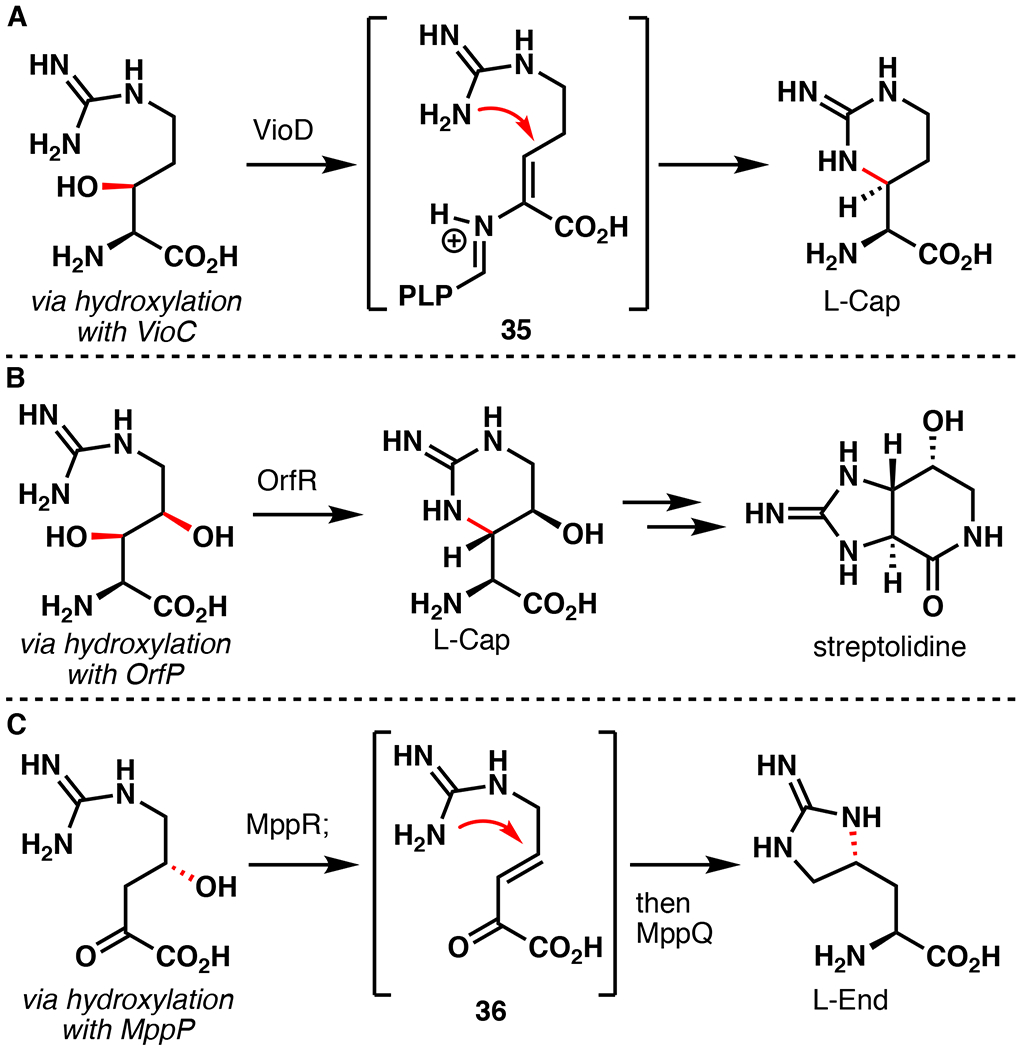
Involvement of Fe/αKG hydroxylation of L-arginine en route to the biosynthesis of (A) capreomycidine (L-Cap), (B) streptolidine, and (C) enduracididine (L-End).57,72,74
Engineering of GetI for Enduracidin Fragment Synthesis
Towards establishing an efficient chemoenzymatic route to L-End, our laboratory has developed a rational engineering approach to convert GetI to an L-Arg C4 hydroxylase.74 Examination of solved crystal structures of VioC and OrfP, sharing 45% and 51% sequence identity with GetI respectively, suggests the presence of a key loop region in their active sites that are vital for recognition of the guanidine moiety of their L-Arg substrate. Based on this finding, a “loop grafting” approach was used to alter the substrate specificity of GetI from L-Cit to L-Arg (Figure 10A). This approach entails sequential site-directed mutagenesis to convert GetI to a VioC-like or OrfP-like enzyme. The VioC-like mutant, GetI DGDF (A257G/T258D/H259F), showed no L-Cit hydroxylation activity and was capable of hydroxylating Arg with 87 TTN. Unfortunately, reaction with GetI DGDF also led to considerable formation of β,γ-di-OH-L-Arg (1:4 ratio of β,γ-di-OH-L-Arg : γ-OH-Arg). The OrfP-like mutant, GetI QDPYF (L143Q/A257P/T258Y/H259F), showed a very similar reactivity profile (94 TTN), but exclusively produced γ-OH-Arg. This example is remarkable because only four mutations are required to completely switch the substrate preference of GetI from L-Cit to L-Arg.
Figure 10.

A. Engineering of GetI for L-Arg hydroxylation via site-directed mutagenesis; TTN = total turnover number.74 B. Synthetic utility of variant GetI QDPYF in the preparation of a dipeptide fragment of enduracidin. DIAD = diisopropyl azodicarboxylate.
With the optimized enzyme variant in hand, we next targeted a chemoenzymatic synthesis of an L-End containing dipeptide fragment from enduracidin (Figure 10B). The γ-OH-L-Arg product from hydroxylation with GetI QDPYF was subjected to Boc protection followed by guanidine hydrazinolysis to afford the free δ-amine (38), which was then converted to lactone 40 in two steps featuring re-introduction of the guanidine side-chain in protected form with reagent 39.75 This sequence proved necessary to avoid N-Boc regioisomerism of γ-OH-L-Arg observed during global Boc protection conditions. This approach can also be adapted for the introduction of methylated guanidine motif, which has been found naturally in the N-Me-γ-OH-L-Arg substructure of the argimicin peptide antibiotics.76 Lactone opening of 40 using a combination of AlMe3 and H-Ala-OtBu (41), followed by Mitsunobu reaction formed the desired L-End containing dipeptide 43.77 Our chemoenzymatic approach compares favorably in step-count (6) and overall yield (6%) to previously reported syntheses of L-End, but also remedies the poor C4 stereocontrol typically observed.72,78 This approach delineates how Fe/αKG engineering can be paired with traditional synthetic methods to efficiently construct ncAAs.
2.2. Alkaloid Hydroxylation
Fe/αKG hydroxylation represents a common means by which nature builds and modifies alkaloid natural products (Figure 11). In the biosyntheses of commercial β-lactamase inhibitor clavulanic acid and antibiotic bicyclomycin, hydroxylation serves as a starting point for complexity building oxidative cyclization reactions.79,80 Late-stage hydroxylation is a common structural feature among β-lactam antibiotics sulfazecin and cephamycin C.79 Vinca alkaloids represent important anticancer therapeutics that share hydroxylation at C17’, installed by the Fe/αKG DV4H.81 Demethylation mediated by Fe/αKG hydroxylases represents a common reaction in the biosynthesis of morphine and related opioids.82 Given their prevalence and potential synthetic utility, it comes as a surprise that an Fe/αKG alkaloid hydroxylase has only been leveraged once for chemoenzymatic total synthesis.
Figure 11.
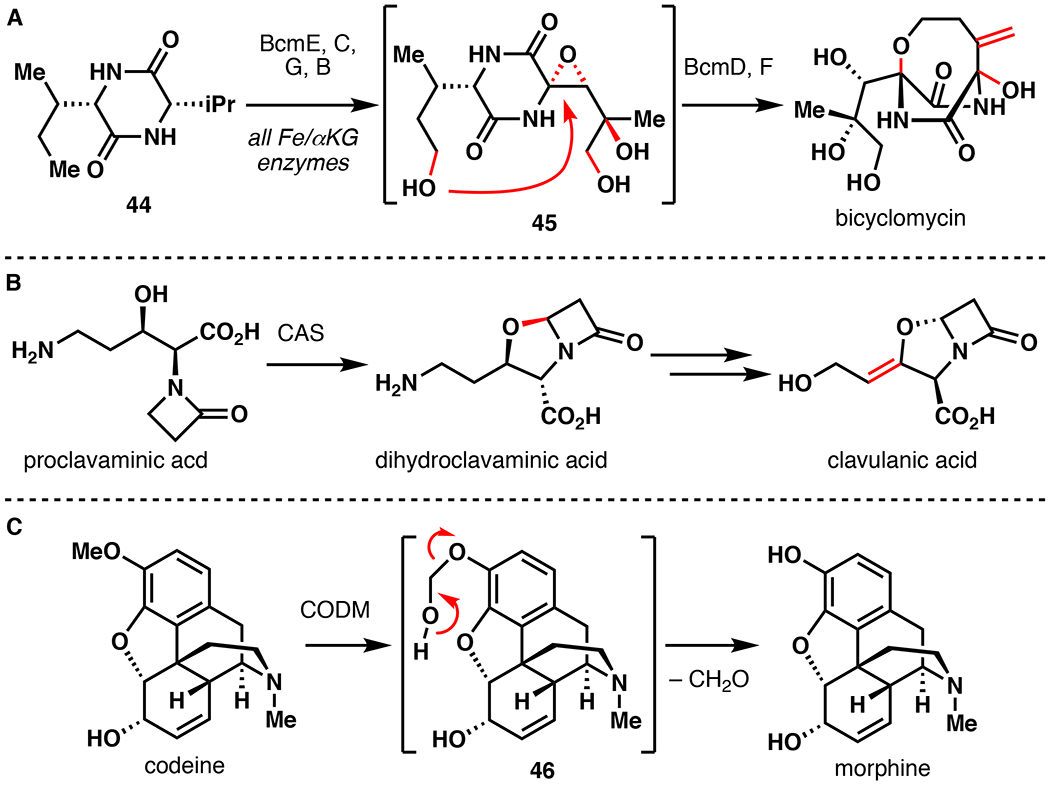
Involvement of Fe/αKG hydroxylation in the biosynthesis of (A) bicyclomycin, (B) clavulanic acid, and (C) morphine.79–82
Total Synthesis of [15N5]-Cylindrospermopsin
Cylindrospermopsin represents a biotoxin that is partially responsible for the health risks of cyanobacterial algal blooms.83,84 Alongside acute toxicity, exposure to cylindrospermopsin or its derivatives is also linked to hepatotoxicity, gastrointestinal inflammation, hemorrhages, anorexia, and liver failure.85 As a means for improved detection of cylindrospermopsin in environmental samples, the use of isotope-dilution mass spectrometry (IDMS) is desired. To enable this strategy, Mailyan et al. completed a chemoenzymatic total synthesis of [15N5]-cylindrospermopsin featuring a late-stage hydroxylation with CyrI, an Fe/αKG from cylindrospermopsin biosynthesis, to serve as an essential internal standard for IDMS.86 Their retrosynthetic analysis targeted convergent assembly using 15N labelled guanidine and pyrimidine 52, recognizing that availability of 15N starting materials such as 15NH4Cl and 15NH3 was a key constraint. Synthesis of 52 commenced from 15NH4Cl using a solid phase reaction with NaOH to generate 15NH3, which was then condensed with diphenyl carbonate to afford [15N2]-urea. Condensation with diethyl malonate to [15N2]-barbituric acid, followed by bromination and methanolysis afforded 52 in good yield. Towards [15N3]-guanidine, [15N2]-thiourea was formed from the reaction of 15NH3 with diphenyl thiocarbonate followed by pyrolysis. Methylation and Boc protection afforded 48. The methanethiolate was then displaced by 15NH3 and the Boc groups were removed in refluxing 12N HCl to afford [15N3]-guanidinium•HCl (49).
With both 15N sources in hand, the total synthesis of [15N5]-cylindrospermopsin commenced (Figure 13). Sonogashira coupling of 52 with 5-hexyn-1-ol afforded 53 in 98% yield. Swern oxidation, followed by crotylation using Leighton’s reagent (54) afforded alcohol 55 in 99% yield and 10:1 dr.87 An efficient guanidine carbamoylation then followed to install the [15N3]-guanidine motif. Electrophilic cycloguanidylation using N-iodosuccinimide (NIS) followed by Mbs protection afforded cyclic guanidine 57 in 71% yield and 2.5:1 dr favoring the desired diastereomer. The alkyne was then converted to 1,3-diene (58) as a 6:1 E/Z mixture using [Pd(PPh3)4] and benzoic acid in refluxing dioxanes (75% yield). Cylindrospermopsin’s guanidine tricycle was constructed following a remarkable one-pot, three-reaction sequence. First, the oxazolidinone was opened using basic methanolysis. This reaction freed the guanidine to perform a spontaneous 1,6/1,4-addition across the alkenes. Finally, concentration, followed by addition of methanol and NaOMe cleaved the remaining methyl carbonate to afford 59 in >93% yield and 3:2 dr (C8). Refluxing in 12 N HCl deprotected the Mbs and hydrolyzed the pyrimidine to the pyrimidine-dione. The use of SO3•pyridine then effected an O-sulfonation, resulting in the formation of 60. To address the final hydroxylation, CyrI was recombinantly expressed and isolated from E. coli cells.88 2.5 mol% CyrI was sufficient to convert 60 to [15N5]-cylindrospermopsin in 95% yield on ~9 mg scale. This approach produced [15N5]-cylindrospermopsin in a 10-step LLS with 15% overall yield, and demonstrates the power of combining traditional chemical steps with late-stage biocatalytic hydroxylation. One important drawback involves scalability due to the need for a large amount of purified CyrI (2.5 mol%) in the reaction and poor water solubility of the substrate. To address this issue, the authors also demonstrated chemical approaches to [15N5]-cylindrospermopsin and [15N5]-7-epi-cylindrospermopsin utilizing a modified sequence. Trimethylsilyl (TMS) protection of 59 was followed by hydroxylation using lithium diisopropylamide (LDA) and camphor derived oxaziridine (61), providing 51% yield (84% brsm) and >20:1 dr. This optimized hydroxylation could be scaled to 1.5 grams, resulting in 46% product along with 41% starting material. With a scalable route to [15N5]-cylindrospermopsin in hand, an assay was developed using LC/ESI-MS/MS that is able to detect cylindrospermopsin with a 2.5 and 8.25 ng/L limit of detection (LOD) and limit of quantification (LOQ), respectively, the highest sensitivity reported to date.89 This approach delineates an effective strategy for the detection of toxic natural products present in low abundance.
Figure 13.
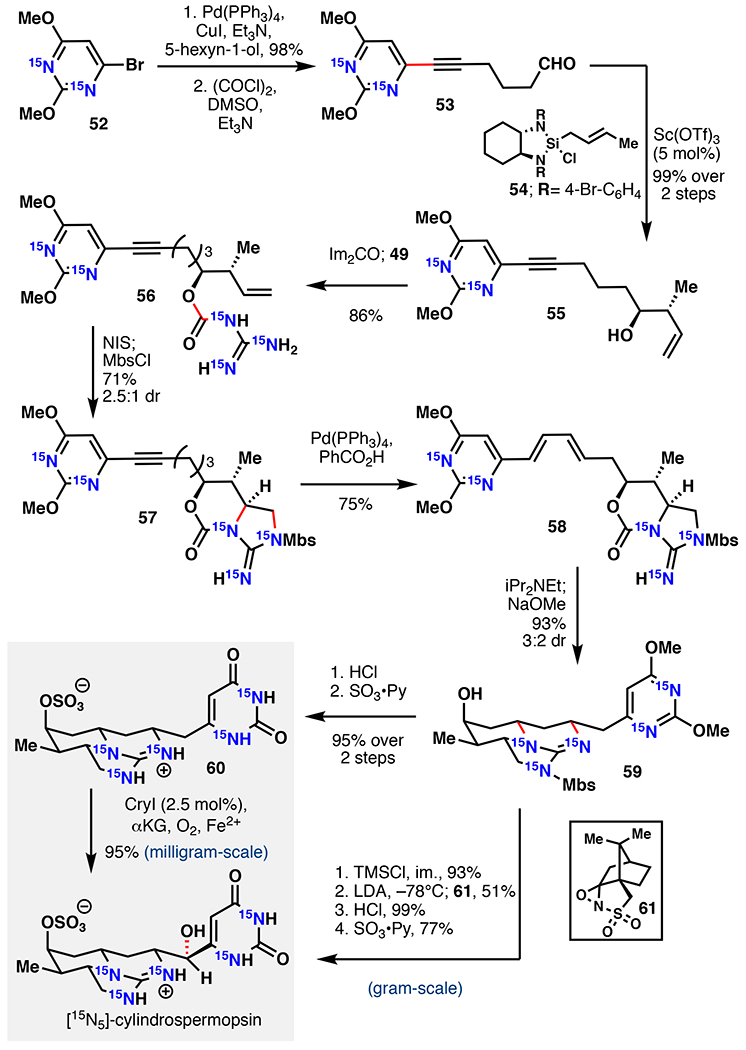
Zakarian’s chemoenzymatic synthesis [15N5]-cylindrospermopsin via either enzymatic hydroxylation with CryI or chemical oxidation.86 NIS = N-iodosuccinimide, LDA = lithium diisopropylamide.
2.3. Aromatic Polyketide Hydroxylation
Fungal polyketides represent a structurally diverse and interesting family of natural products with a wide assortment of bioactivities.90 These molecules are produced biosynthetically from acetyl-CoA and malonyl-CoA by multi-domain polyketide synthases (PKSs), which are classified as highly reducing (HR), partially reducing (PR), or non-reducing (NR) based on their products.90,91,92 The NR-PKS family produces many families of aromatic polyketides such as the azaphilones, sorbicillinoids, tropolones, and citirinin.92 Structural divergence is mediated by enzymatic hydroxylation reactions that serve as a gateway for further oxidation, condensation, annulation, or ring expansion reactions (Figure 14). Based on synthetic novelty and target activity, efforts have been made to develop these hydroxylation reactions as valuable biocatalytic methods with total synthetic applications.
Figure 14.
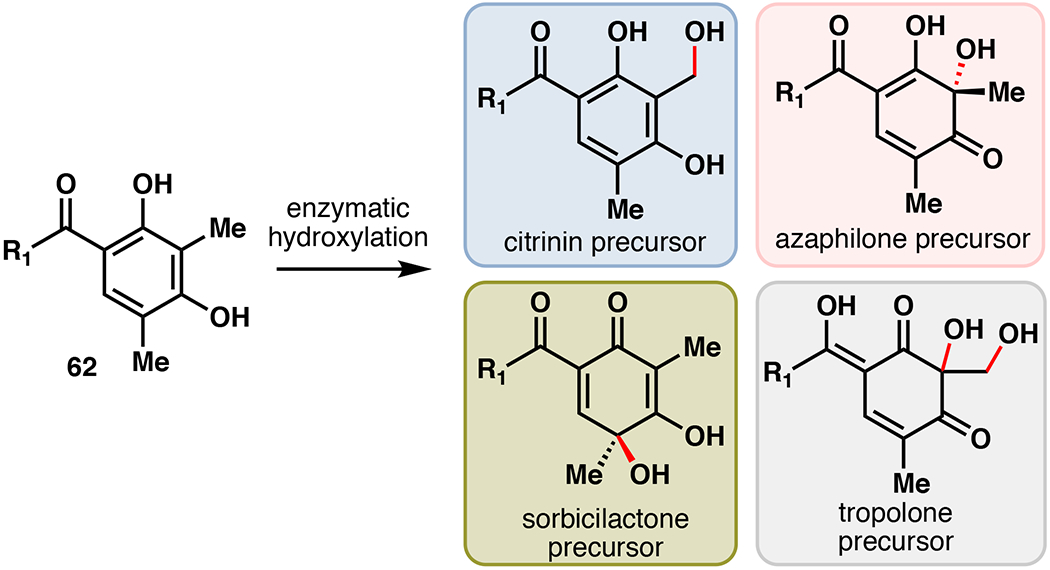
Structural diversity of fungal aromatic polyketides arising from divergent enzymatic oxidation.
Total Synthesis of Stipitatic Aldehyde
In 1942, stipitatic acid became the first tropolone ever discovered. Its discovery contributed greatly to our understanding of aromaticity and bonding in organic chemistry.93 To date, approximately 200 naturally occurring tropolones have been identified, and display a range of biological functions such as anti-bacterial, -fungal, -tumor, and -viral. Although numerous synthetic methods have been developed for the synthesis of tropones and tropolones, efficient construction of non-benzannulated, highly substituted tropolones has remained particularly challenging.94 During the development of flavin-dependent monooxygenases (FMOs) for oxidative dearomatization of phenols, Dockery et al. reported the total synthesis of stipitatic aldehyde via a combined use of the FMO TropB and Fe/αKG hydroxylase TropC.95 TropB and TropC originate from the biosynthesis of stipitatic acid, and have previously been functionally characterized and expressed in E. coli.93 3-methylorcinaldehyde (63) was first synthesized from 2,5-dimethylbenzene-1,3-diol via a Vilsmeier-Haack reaction using POCl3/DMF in 66% yield. 63 was then submitted to a one-pot dearomatization with TropB, and hydroxylation/ring-expansion by TropC to stipitatic aldehyde in 31% isolated yield (54% conversion). This 2-step approach highlights the power of leveraging biosynthetic enzymes in the total synthesis of tropolone natural products.
Total Synthesis of (–)-Xyloketal D
Ortho-quinone methide ylides (o-QM) represent commonly encountered reactive intermediates used in the total synthesis of natural products.96 These versatile intermediates participate in a range of reactions, such as the inverse electron-demand Diels-Alder (IEDDA) or 1,4-additions. Unfortunately, their generation often requires pre-functionalization followed by high heating, or the use of stoichiometric oxidants that are only applicable to electron rich substrates.97,98 To alleviate these shortcomings, Doyon et al. leveraged the Fe/αKG hydroxylase enzymes CitB and ClaD to generate o-QMs under mild conditions (Figure 16A).99 The o-QM intermediates were then submitted to a variety of 1,4-addition or IEDDA reactions. CitB and ClaD share 54% sequence identity, and are involved in the biosyntheses of citrinin as well as the peniphenones and penilactones, respectively.92,100 CitB was initially tested with 3-methylorcinaldehyde as a model substrate, resulting in benzylic oxidation in 82% isolated yield. By comparison, attempts to produce the same compound via chemical means (DDQ, MnO2, Ag2O, K2S2O8, (NH4)2Ce(NO3)6) gave either no reaction, over-oxidation, or degradation. Gentle heating of the hydroxylated product in the presence of thiophenol afforded the thioether in 98% yield, presumably through the o-QM intermediate. Next, substrate profiling of CitB and ClaD was performed with a suite of ortho-cresol derivatives. Both enzymes were capable of hydroxylating a range of arenes with different steric/electronic features. Introduction of additional groups at C5/6 proved problematic with ClaD, but did not prevent productive reactions with CitB. By contrast, modification of the C4 aldehyde to larger electron withdrawing groups (EWGs) was tolerated by ClaD, but not by CitB. Overall, both enzymes require resorcinol substrates that contain an EWG at C4. Preparative scale reactions with CitB or ClaD produced >20 thioether or alcohol products in 7–98% yield (~30–50% average). Among alternative nucleophiles, linear/branched alcohols, morpholine, and thiophenol additionally provided the desired 1,4-adducts in 41–71% yield. A variety of mixed chromane products could also be obtained via IEDDA reactions of o-QMs with electron rich alkenes under mild heating (33–56% isolated yield, 1:2–1:4 dr). Enzymatic o-QM formation could also be forwarded to cysteine-selective bioconjugation of a synthetic peptide in 61% conversion. Finally, this approach was used to complete a total synthesis of (–)-xyloketal D (Figure 16B). Towards this end, crude E. coli lysate overexpressing ClaD gave complete conversion of 67 to the benzylic alcohol, at which point benzene and dienophile (69) were added directly to the mixture and heated to 80°C. (–)-xyloketal D was isolated in 64% yield as a 2:1 ratio along with its diastereomer 70 (28% yield). This one-pot sequence displays improved yields and milder conditions compared to a previous o-QM mediated approaches to this molecule.97 The developed method leverages Fe/αKG hydroxylase enzymes for a more mild and efficient way to access o-QMs.
Figure 16.
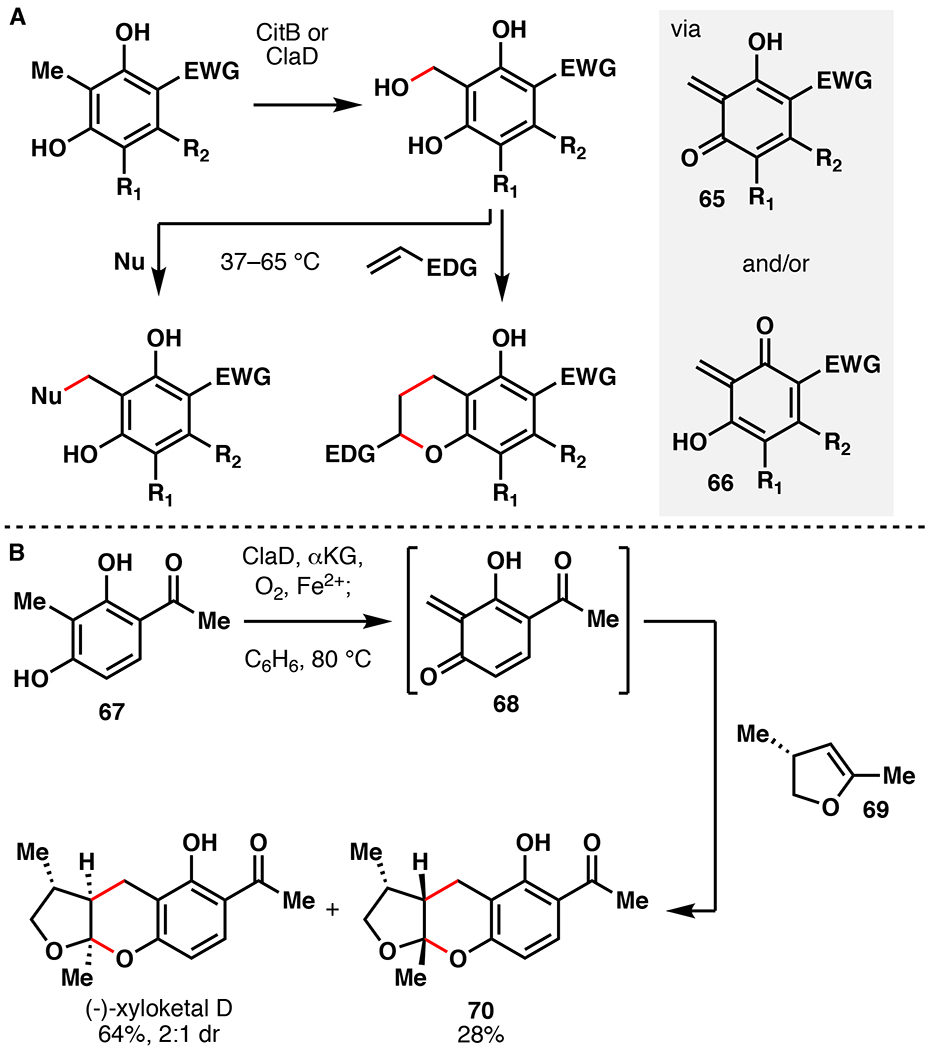
A. Synthetic utility of CitB or ClaD via the intermediacy of a quinone methide species. B. Chemoenzymatic total synthesis of (–)-xyloketal D featuring hydroxylation with ClaD.99
3. Oxidative Cyclization
Oxidative cyclization reactions are prevalent in the biosynthesis of natural products, and can be defined as a reaction that forms a new ring from an oxidative or redox modification.24 These reactions introduce some of the most impressive structural features present among natural products (Figure 17), such the phenol-phenol cross-links in vancomycin and the penam core of the β-lactam antibiotic isopenicillin N.79,101 The majority of these reactions are limited to the realm of biocatalysis, with no existing analogous reactions in traditional chemical methodology. Fe/αKG enzymes mediate many of these transformations, following a similar mechanism to hydroxylation. In fact, many of these reactions have been proposed to proceed through a hydroxylated intermediate prior en route to the end product. The following section summarizes recent applications of Fe/αKG oxidative cyclization enzymes to simplify chemoenzymatic access to bioactive natural products.
Figure 17.
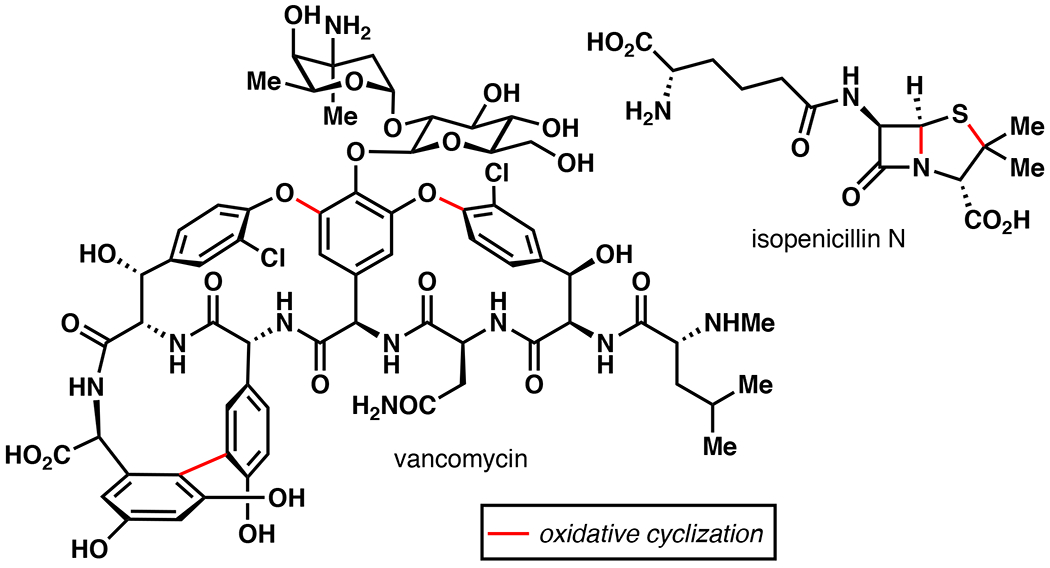
Examples of the use of oxidative cyclization in the biosynthesis of vancomycin101 and isopenicillin N.79
Total Synthesis of Kainic Acid
Kainic acid comprises the active component of Digenea simplex, a tropical seaweed used for centuries as an anthelmintic agent in Asia.102 Although its exact mechanism of action is unknown, kainic acid functions as an ionotropic glutamate receptor (iGluR) agonist that stimulates cation influx, resulting in cell death.103 Kainic acid represents a highly modified L-Pro derivative as well as an L-Glu structural mimic. This natural product has been important in the discovery and characterization of several classes of iGluRs and has been used to create mouse model systems to study neurological disease.104,105 Due to its importance and structural novelty, >70 total synthetic endeavors towards kainic acid have been reported since its discovery in the 1950s.106 Unfortunately, these approaches often suffer from low yields and high step count (at least six steps with less than 40% overall yield). By comparison, kainic acid is biosynthesized in two steps from L-Glu through the action of the N-prenyltransferase KabA and the Fe/αKG enzyme KabC (Figure 18A). Following initial biosynthetic studies, Chekan et al. established a chemoenzymatic total synthesis of kainic acid (Figure 18B).107 In the biosynthesis of kainic acid, KabC is responsible for performing a synthetically enabling oxidative cyclization from prekainic acid to kainic acid. In turn, prekainic acid was synthesized in one-step from L-Glu via a reductive amination with 3-methyl-2-butenal in 56% isolated yield. Prekainic acid was then submitted to a small-scale reaction with purified KabC, affording kainic acid in 46% isolated yield. To improve scalability, crude prekainic acid was fed directly to 1L of E. coli cells overexpressing KabC, resulting in 1.1 grams kainic acid in 57% isolated yield over two steps. This chemoenzymatic approach demonstrates the efficiency of combining biosynthetic and traditional chemical steps to approach the most efficient strategy possible.
Figure 18.
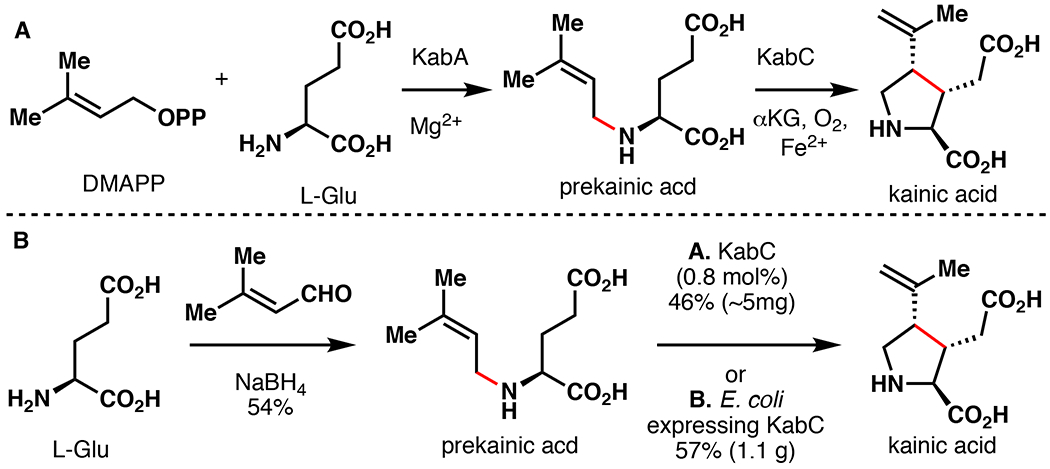
A. Biosynthesis of kainic acid in D. simplex.107 B. Moore’s chemoenzymatic total synthesis of kainic acid via late-stage enzymatic cyclization with KabC.107
Total Syntheses of Podophyllotoxin
Aryltetralin lignans represent an important family of natural products with a range of biological activities and diverse structural features.108 Among them, (–)-podophyllotoxin (71) is undoubtedly the most well-known, due to its ability to potently depolymerize microtubules at the colchicine site of tubulin, resulting in cell death.109 Etoposide and teniposide represent (–)-podophyllotoxin analogs currently used in the treatment of several types of cancer.110 Due to their importance, numerous synthetic approaches have been previously reported, but an efficient asymmetric approach to aryltetralin lignans is still lacking.111 Recently, the Fe/αKG enzyme 2-ODD-PH was shown to construct (–)-deoxypodophyllotoxin from (–)-yatein via an oxidative cyclization.112 This reaction assembles the C-ring by coupling of C2 and C7’ with exquisite regio- and stereocontrol. While several of the biosynthetic steps approaching (–)-yatein can be considered non-strategic (oxidation state adjustments), the reaction catalyzed by 2-ODD-PH shows great synthetic promise.113 For these reasons, our laboratory targeted the application of 2-ODD-PH for the asymmetric synthesis of 71 and related aryltetralin lignans.114 Concurrent to our studies, Lazzarotto et al. reported a similar approach to 71 utilizing 2-ODD-PH in the kinetic resolution of dibenzylbutyrolactones (Figure 19).115 Towards this objective, Lazzarotto et al. first synthesized (±)-yatein following an efficient 3-step procedure starting from piperonal (82% overall yield). During substrate screening of 2-ODD-PH, it was determined that C7-hydroxyl derivative of yatein (75) could be accepted as substrate, but only when the C7 -OH is present in the (S)-stereoconfiguration. In turn, this information was used to perform a kinetic resolution on (±)-7-OH-yatein to selectively form epi-(–)-podophyllotoxin (76) in 39% yield and 95% ee on 2-gram scale. During these studies, 2-ODD-PH was also used to generate a range of epi-(–)-podophyllotoxin analogs with varying structural features in the B-ring. Stereoinversion of the secondary alcohol of 76 could be achieved via Dess-Martin oxidation and reduction by L-selectride to afford (–)-podophyllotoxin in 17% overall yield over five steps.
Figure 19.
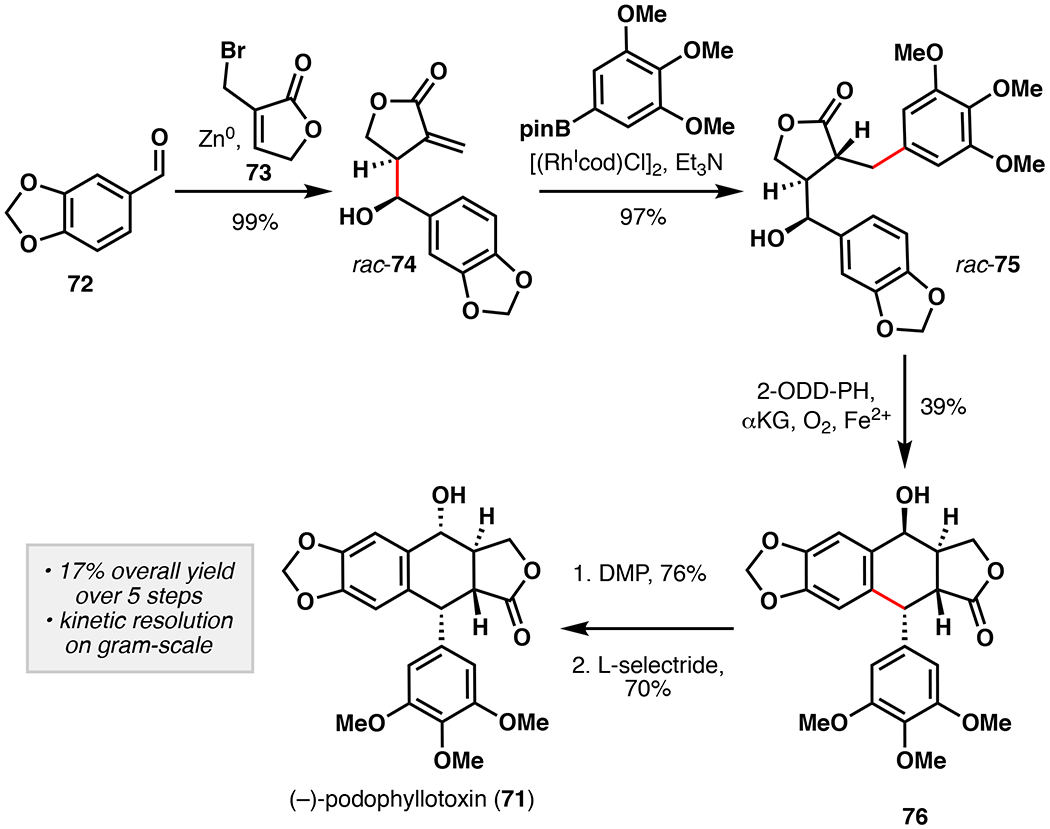
Fuch’s chemoenzymatic total synthesis of (–)-podophyllotoxin featuring the use of 2-ODD-PH for oxidative kinetic resolution of rac-74.115 DMP = Dess-Martin periodinane.
Our approach began by synthesizing (–)-yatein via oxidative enolate coupling methodology developed by Baran and co-workers (Figure 20).116 A one-pot coupling of oxazolidinone 77 and ester 78 followed by DBU mediated epimerization afforded (–)-yatein in 51% yield as a single diastereomer. Submitting (–)-yatein to clarified E. coli cell lysate overexpressing 2-ODD-PH and GroES/EL afforded (–)-deoxypodophyllotoxin in 95% isolated yield on 1.6 gram scale, which translates to a titer yield of more than 3 gram L–1.117 Benzylic oxidation with CrO3 and reduction by L-selectride then afforded (–)-podophyllotoxin in a 58% two step yield. Our approach constitutes a five-step sequence with a 28% overall yield, and compares favorably to previously reported approaches. Substrate profiling was also performed in order to establish structural features important for reaction with 2-ODD-PH. By comparison to the previously reported study, the southern aromatic portion could be modified as long as C7-OH was not present. Additionally, four aryltetralin lignan natural products, (–)-polygamain, (–)-morelensin, (–)-austrobilignan, and (–)-hernandin, could be synthesized using an analogous approach, demonstrating the utility and versatility of our strategy. These contemporary approaches highlight alternative strategic benefits of pairing biocatalysis with traditional chemical methodology for natural product total synthesis.
Figure 20.
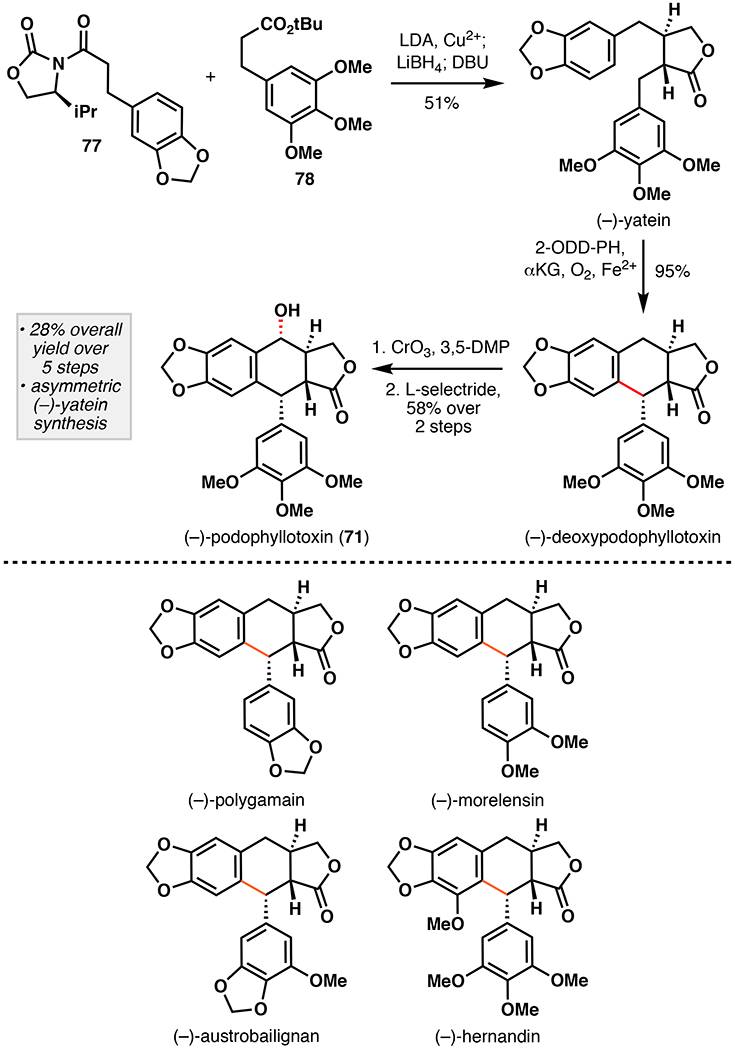
Renata’s chemoenzymatic total synthesis of (–)-podophyllotoxin (71) featuring oxidative enolate coupling and oxidative cyclization of enantiopure (–)-yatein with 2-ODD-PH and substrate scope examination of oxidative cyclization with 2-ODD-PH.114 DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, 3,5-DMP = 3,5-dimethylpyrazole.
4. Conclusions
The examples outlined in this review are powerful case studies on how one can harness Nature’s toolbox to construct molecules of value. By leveraging Fe/αKG enzymes that perform hydroxylation and oxidative cyclization reactions, we have just begun to scratch the “biocatalytic potential” of this amazing enzyme superfamily.1–6 In addition to hydroxylation and oxidative cyclization, Fe/αKG enzymes that perform epoxidation, halogenation, desaturation, endoperoxidation, carbon migration, azetidination, spirocyclization, and epimerization have also been identified.5,6 Furthermore, these functionalities can serve as useful functional handles for alkynylation, cyclopropanation, decarboxylation, dealkylation, ring expansion, ring contraction, and complex rearrangement reactions.5,6,118 Preliminary efforts describing engineering of Fe/αKG enzymes for non-native reactivity, such as halogenation, azidation, nitration, aziridination, and nitrene insertion, promise to broaden the reaction repertoire of this enzyme superfamily even further.119–122 We also expect efforts to broaden the range of natural product scaffolds that are accessible via Fe/αKG biocatalysis to be highly fruitful endeavors. For example, many Fe/αKG enzymes are known to contribute to the breadth of structural diversity present in the terpenoid class of natural products. Astaxanthin, a carotenoid pigment found among many marine animals, is synthesized in Nature from β-carotene following three oxidations mediated by CrtW and CrtZ.5 The gibberellins are a large family of plant-derived terpenoids that play key roles in plant growth and sporulation, and require Fe/αKG hydroxylases GA2βox, GA3βox, GA13ox, and GAC20ox for maturation.1 The fungal meroterpenoid biosyntheses are also known to involve many Fe/αKG-catalyzed transformations, including some unprecedented oxidation/rearrangement cascades.123 These enzymes represent untapped opportunities for use in chemoenzymatic total synthesis of highly modified terpenoids. Finally, as genome mining techniques continue to improve and evolve, so will our ability to identify Fe/αKG enzymes with novel functions directly from sequence.124 Thus, Fe/αKG enzymes hold tremendous potential as useful and highly enabling biocatalysts for chemoenzymatic total synthesis of bioactive natural products.
Figure 12.
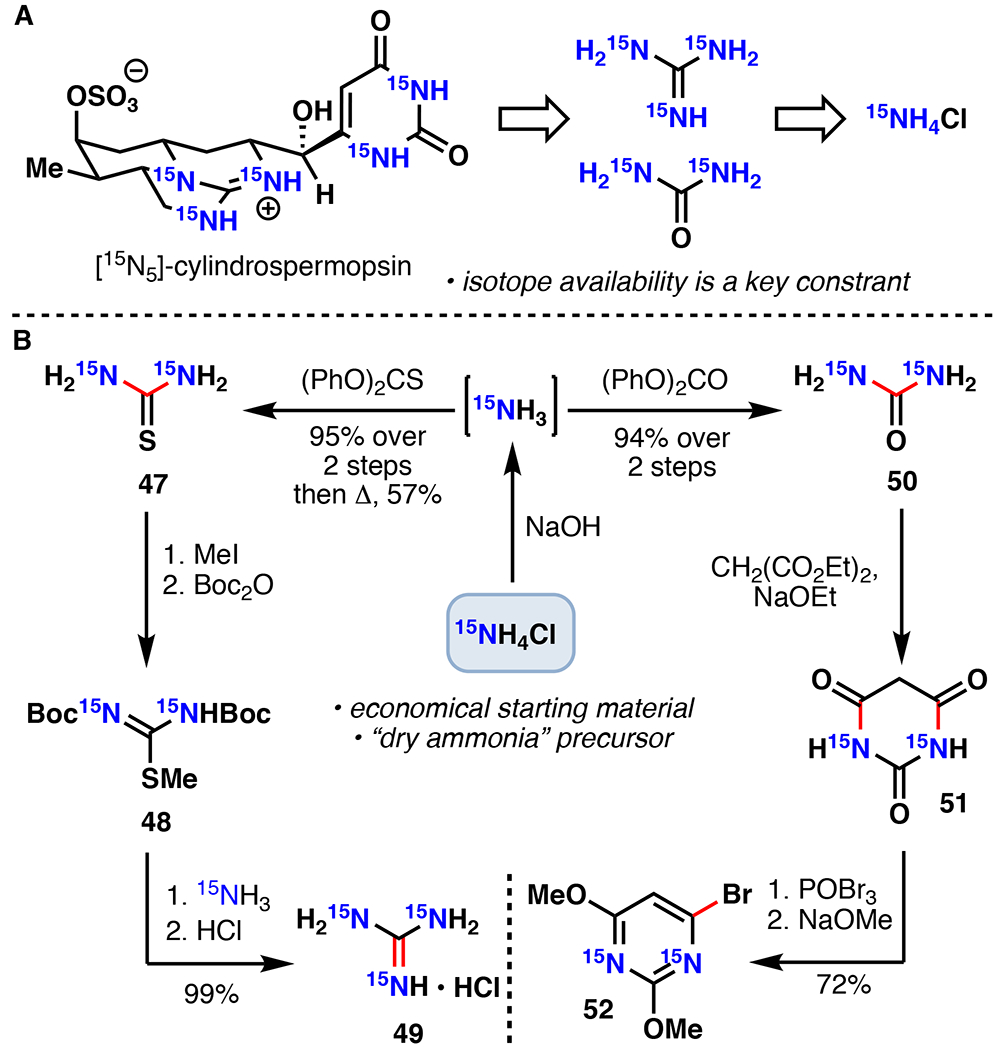
A. Zakarian’s retrosynthetic analysis of [15N5]-cylindrospermopsin. B. Synthesis of key radiolabelled precursors 49 and 52.86
Figure 15.
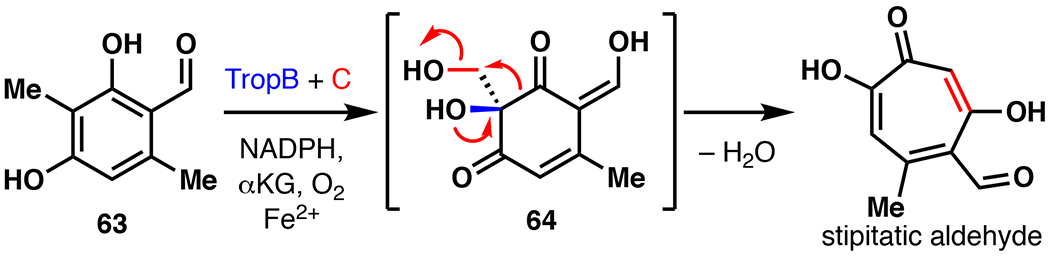
Narayan’s chemoenzymatic synthesis of stipitatic aldehyde.95
Figure 21.
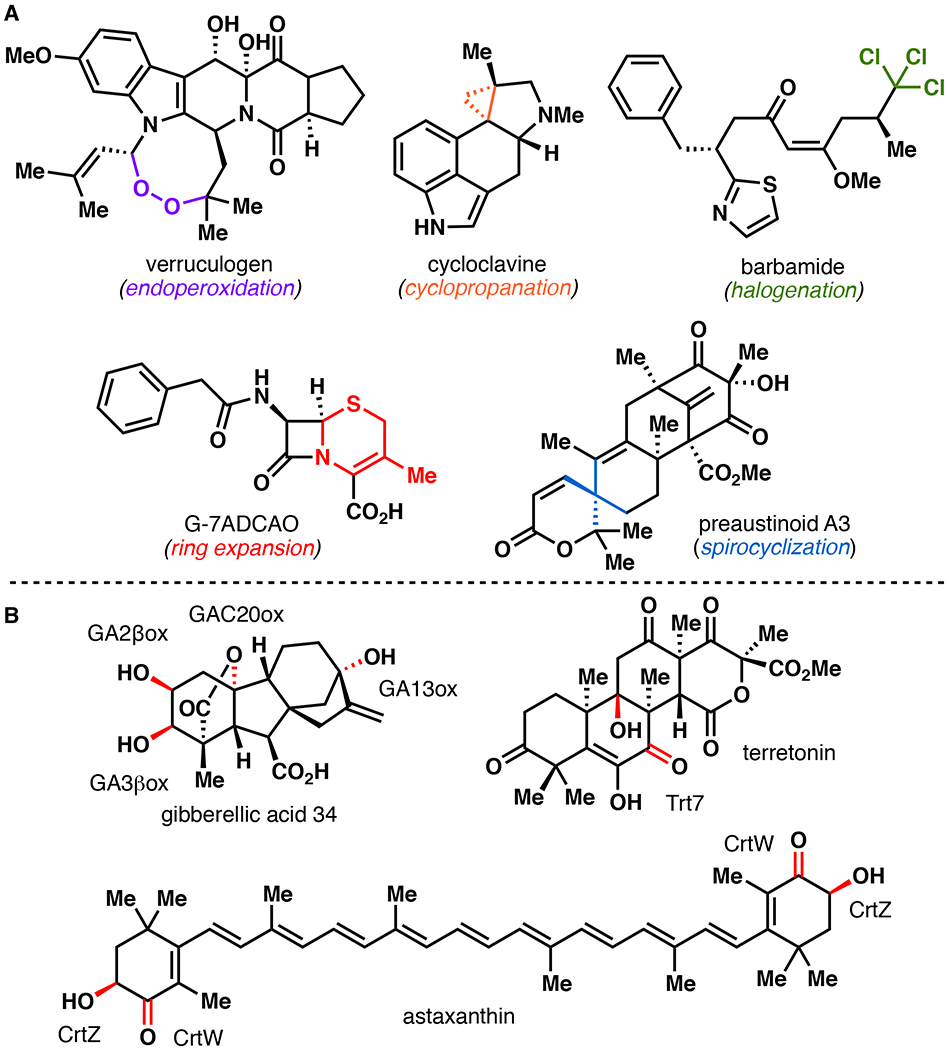
A. Reaction diversity of Fe/αKG enzymes. B. Chemo- and regioselective terpene hydroxylation mediated by Fe/αKG enzymes.
Acknowledgements
The authors acknowledge the support of the National Institute of Health (1R35GM128895) and The Scripps Research Institute. The content is solely the responsibility of the authors and does not represent the official views of any of the funding agencies.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Hausinger RP, in 2-Oxoglutarate-Dependent Oxygenases, ed. Hausinger Robert P., Schofield Christopher J., The Royal Society of Chemistry, Thomas Graham House, Science Park, Milton Road, Cambridge CB4 0WF, UK, 2015, 1, 1–58. [Google Scholar]
- 2.Hausinger RP, Crit. Rev. Biochem. Mol. Bio, 2004, 39, 21. [DOI] [PubMed] [Google Scholar]
- 3.Loenarz C and Schofield CJ, Trends Biochem. Sci, 2011, 36, 7. [DOI] [PubMed] [Google Scholar]
- 4.Hibi M and Ogawa Jun, Appl. Microb. Biotechnol, 2014, 98, 3869. [DOI] [PubMed] [Google Scholar]
- 5.Gao S-S, Naowarojna N, Cheng R, Liu X and Liu P, Nat. Prod. Rep, 2018, 35, 792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu L-F, Meng S and Tang G-L, Biochim. Biophys. Acta, 2016, 1864, 453. [DOI] [PubMed] [Google Scholar]
- 7.Mitchell AJ and Weng J-K, Plant Physiol, 2019, 179, 813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dias DA, Urban S and Roessner U, Metabolites, 2012, 2, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.King-Smith E, Zwick III CR and Renata H, Biochemistry, 2018, 57, 403. [DOI] [PubMed] [Google Scholar]
- 10.Huffman BJ and Shenvi RA, J. Am. Chem. Soc, 2019, 141, 3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newhouse T and Baran PS, Angew. Chem. Int. Ed, 2011, 50, 3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noisier AFM and Brimble MA, Chem. Rev, 2014, 114, 8775. [DOI] [PubMed] [Google Scholar]
- 13.Giri R, Shi B-F, Engle KM, Maugel N and Yu J-Q, Chem. Soc. Rev, 2009, 38, 3242. [DOI] [PubMed] [Google Scholar]
- 14.Hartwig JF and Larsen MA, ACS Cent. Sci, 2016, 2, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White MC and Zhao J, J. Am. Chem. Soc, 2018, 140, 13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leung JC, Geary LM, Chen T-Y, Zbieg JR and Krische MJ, J. Am. Chem. Soc, 2012, 134, 15700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie J, Jin H and Hashmi ASK, Chem. Soc. Rev, 2017, 46, 5193. [DOI] [PubMed] [Google Scholar]
- 18.Cai Y, Zhang J-W, Li F, Liu J-M and Shi S-L, ACS Catal, 2019, 9, 1. [Google Scholar]
- 19.Houwaart S, Youssar L and Hüttel W, ChemBioChem, 2014, 15, 2365. [DOI] [PubMed] [Google Scholar]
- 20.Mattay J and Hüttel W, ChemBioChem, 2017, 18, 1523. [DOI] [PubMed] [Google Scholar]
- 21.Hibi M, Mori R, Miyake R, Kawabata H, Kozono S, Takahashi S and Ogawa J, Appl Environ Microbiol, 2016, 82, 2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lukat P, Katsuyama Y, Wenzel S, Binz T, König C, Blankenfeldt W, Brönstrup M and Müller R, Chem. Sci, 2017, 8, 7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Umezawa K, Ikeda Y, Kawase O, Naganawa H and Kondo S, J. Chem. Soc., Perkin Trans. 1, 2001, 1550. [Google Scholar]
- 24.Tang M-C, Zou Y, Watanabe K, Walsh CT and Tang Y, Chem. Rev, 2017, 117, 5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reddy LR, Subba Reddy BV and Corey EJ, Org. Lett, 2006, 8, 2819. [DOI] [PubMed] [Google Scholar]
- 26.Wappes EA, Fosu SC, Chopko TC and Nagib DA, Angew. Chem. Int. Ed, 2016, 55, 9974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu T, Myers MC and Yu J-Q, Angew. Chem. Int. Ed, 2016, 56, 306. [DOI] [PubMed] [Google Scholar]
- 28.Namba K, Shinada T, Teramoto T and Ohfune Y, J. Am. Chem. Soc, 2000, 122, 10708. [Google Scholar]
- 29.Hashimoto T and Maruoka K, Org. Biomol. Chem, 2008, 6, 829. [DOI] [PubMed] [Google Scholar]
- 30.Ohfune Y, Oe K, Namba K and Shinada T, Heterocycles, 2012, 85, 2617. [Google Scholar]
- 31.Nagatomo M, Nishiyama H, Fujino H and Inoue M, Angew. Chem. Int. Ed, 2014, 54, 1537. [DOI] [PubMed] [Google Scholar]
- 32.Tong TMT, Soeta T, Suga T, Kawamoto K, Hayashi Y and Ukaji Y, J. Org. Chem, 2017, 82, 1969. [DOI] [PubMed] [Google Scholar]
- 33.Zwick III CR and Renata H, J. Am. Chem. Soc, 2018, 140, 1165. [DOI] [PubMed] [Google Scholar]
- 34.Zwick III CR and Renata H, J. Org. Chem, 2018, 83, 7407. [DOI] [PubMed] [Google Scholar]
- 35.Meng S-S, Tang W-B and Zheng W-H, Org. Lett, 2018, 20, 518. [DOI] [PubMed] [Google Scholar]
- 36.Liu Y, Ruan Z, Wang Y, Huang S-H and Hong R, Tetrahedron, 2019, 75, 1767. [Google Scholar]
- 37.Fiori KW and Du Bois J, J. Am. Chem. Soc, 2017, 129, 562. [DOI] [PubMed] [Google Scholar]
- 38.Zhang X, Yang H and Tang P, Org. Lett, 2015, 17, 5828. [DOI] [PubMed] [Google Scholar]
- 39.Karimov RR, Sharma A and Hartwig JF, ACS Cent. Sci, 2016, 2, 715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prier CK, Zhang RK, Buller AR, Brinkmann-Chen S and Arnold FH, Nature Chemistry, 2017, 9, 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wehn PM and Du Bois J, J. Am. Chem. Soc, 2002, 124, 12950. [DOI] [PubMed] [Google Scholar]
- 42.Quasdorf KW, Huters AD, Lodewyk MW, Tantillo DJ and Garg NK, J. Am. Chem. Soc, 2012, 134, 1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Halperin SD, Kwon D, Holmes M, Regalado EL, Campeau L-C, DiRocco DA and Britton R, Org. Lett, 2015, 17, 5200. [DOI] [PubMed] [Google Scholar]
- 44.Nodwell MB, Yang H, Čolović M, Yuan Z, Merkens H, Martin RE, Bénard F, Schaffer P and Britton R, J. Am. Chem. Soc, 2017, 139, 3595. [DOI] [PubMed] [Google Scholar]
- 45.Ghislieri D, Green AP, Pontini M, Willies SC, Rowles I, Frank A, Grogen G and Turner NJ, J. Am. Chem. Soc, 2013, 135, 10863. [DOI] [PubMed] [Google Scholar]
- 46.Del Valle JR and Goodman M, J. Org. Chem, 2003, 68, 3923. [DOI] [PubMed] [Google Scholar]
- 47.Zhang X and Renata H, Tetrahedron, 2019, 75, 3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li L, Cheng Tang M, Tang S, Gao S, Soliman S, Hang L, Xu W, Ye T, Watanabe K and Tang Y, J. Am. Chem. Soc, 2018, 140, 2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zwick CR III and Renata H, Tetrahedron, 2018, 74, 6469. [Google Scholar]
- 50.Estoppey D, Lee CM, Janoschke M, Lee BH, Wan KF, Dong H, Mathys P, Filipuzzi I, Schuhmann T, Riedl R, Aust T, Galuba O, McAllister G, Russ C, Spiess M, Bouwmeester T, Bonamy GMC and Hoepfner D, Cell Rep., 2017, 19, 451. [DOI] [PubMed] [Google Scholar]
- 51.Malins LR, deGruyter JN, Robbins KJ, Scola PM, Eastgate MD, Ghadiri MR and Baran PS, J. Am. Chem. Soc, 2017, 139, 5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Unoki M, Masuda A, Dohmae N, Arita K, Yoshimatsu M, Iwai Y, Fukui Y, Ueda K, Hamamoto R, Shirakawa M, Sasaki H and Nakamura Y, J. Biol. Chem, 2013, 288, 6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamauchi M and Shiiba M, in Post-translational Modifications of Proteins, ed. Kannicht Christoph, Humana Press, 999 Riverview Drive, Suite 208, Totowa, NJ 07512 USA, 2008, 2, 95–108. [Google Scholar]
- 54.Baud D, Saaidi P-L, Monfleur A, Harari M, Cuccaro J, Fossey A, Besnard M, Debard A, Mariage A, Pellouin V, Petit J-L, Salanoubat M, Weissenbach J, de Berardinis V and Zaparucha A, ChemCatChem., 2014, 6, 3012. [Google Scholar]
- 55.Bastard K, Isabet T, Stura EA, Legrand P and Zaparucha A, Sci. Rep, 2018, 8, 16587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goering AW, McClure RA, Doroghazi JR, Albright JC, Haverland NA, Zhang Y, Ju K-S, Thomson RJ, Metcalf WW and Kelleher NL, ACS Cent. Sci, 2016, 2, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ju J, Ozanick SG, Shen B and Thomas MG, ChemBioChem., 2004, 5, 1281. [DOI] [PubMed] [Google Scholar]
- 58.Zhang X, King-Smith E and Renata H, Angew. Chem. Int. Ed, 2018, 57, 5037. [DOI] [PubMed] [Google Scholar]
- 59.Bower JF, Rujirawanich J and Gallagher T, Org. Biomol. Chem, 2010, 8, 1505. [DOI] [PubMed] [Google Scholar]
- 60.Pandey SK, Guttormsen Y, Haug BE, Hedberg C and Bayer A, Org. Lett, 2015, 17, 122. [DOI] [PubMed] [Google Scholar]
- 61.Khan AH and Chen JS, Org. Lett, 2015, 17, 3718. [DOI] [PubMed] [Google Scholar]
- 62.Hartwig JF, Chem. Soc. Rev, 2011, 40, 1992. [DOI] [PubMed] [Google Scholar]
- 63.Feng Y, Holte D, Zoller J, Umemiya S, Simke LR and Baran PS, J. Am. Chem. Soc, 2015, 137, 10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kamiński ZJ, Kolesińska B, Kolesińska J, Sabatino G, Chelli M, Rovero P, Błaszczyk M, Główka ML and Papini A. Maria, J. Am. Chem. Soc, 2005, 127, 16912. [DOI] [PubMed] [Google Scholar]
- 65.Terui Y, Nishikawa J, Hinoo H, Kato T and Shoji J, J. Antibiot, 1990, 43, 788. [DOI] [PubMed] [Google Scholar]
- 66.Groll M, Schellenberg B, Bachmann AS, Archer CR, Huber R, Powell TK, Lindow S, Kaiser M and Dudler R, Nature, 2008, 452, 755. [DOI] [PubMed] [Google Scholar]
- 67.Cromm PM and Crews CM, ACS Cent. Sci, 2017, 3, 830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Amatuni A and Renata H, Org. Biomol. Chem, 2019, 17, 1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dudnik A, Bigler L and Dudler R, Microbiol. Res, 2013, 168, 73. [DOI] [PubMed] [Google Scholar]
- 70.Kodera T, Smirnov SV, Samsonova NN, Kozlov YI, Koyama R, Hibi M, Ogawa J, Yokozeki K and Shimizu S, Biochem. Biophys. Res. Commun, 2009, 390, 506. [DOI] [PubMed] [Google Scholar]
- 71.Chang C-Y, Lyu S-Y, Liu Y-C, Hsu N-S, Wu C-C, Tang C-F, Lin K-H, Ho J-Y, Wu C-J, Tsai M-D and Li T-L, Angew. Chem. Int. Ed, 2014, 53, 1943. [DOI] [PubMed] [Google Scholar]
- 72.Atkinson DJ, Naysmith BJ, Furkert DP and Brimble MA, Beilstein J Org Chem, 2016, 12, 2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Burroughs AM, Hoppe RW, Goebel NC, Sayyed BH, Voegtline TJ, Schwabacher AW, Zabriskie TM and Silvaggi NR, Biochemistry, 2013, 52, 4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zwick III CR, Sosa MB and Renata H, Angew. Chem. Int. Ed, 2019, 58, 18854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kawauchi H, Tohno M, Tsuchiya Y, Hayashida M, Adachi Y, Mukai T, Hayashi I, Kimura S and Kondo S, Int. J. Peptide Protein Res, 1983, 21, 546. [DOI] [PubMed] [Google Scholar]
- 76.Yamaguichi T, Kobayashi Y, Adachi K and Imamura N, J. Antibiot, 2003, 56, 655. [DOI] [PubMed] [Google Scholar]
- 77.Martin SF, Dwyer MP and Lynch CL, Tetrahedron Letters, 1998, 39, 1517. [Google Scholar]
- 78.Goa B, Chen S, Hou YN, Zhao YJ, Ye T and Xu Z, Org. Biomol. Chem, 2019, 17, 1141. [DOI] [PubMed] [Google Scholar]
- 79.Rabe P, Kamps JJAG, Schofield CJ and Lohans CT, Nat. Prod. Rep, 2018, 35, 735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Meng S, Han W, Zhao J, Jian X-H, Pan H-X and Tang G-L, Angew. Chem. Int. Ed, 2017, 57, 719. [DOI] [PubMed] [Google Scholar]
- 81.De Carolis E and De Luca V, J. Biol. Chem, 1993, 268, 5504. [PubMed] [Google Scholar]
- 82.Hagel JM and Facchini PJ, Nat. Chem. Biol, 2010, 6, 273. [DOI] [PubMed] [Google Scholar]
- 83.Cheung MY, Liang S and Lee J, J. Microbiol, 2013, 51, 1. [DOI] [PubMed] [Google Scholar]
- 84.de la Cruz AA, Hiskia A, Kaloudis T, Chernoff N, Hill D, Antoniou MG, He X, Loftin K, O’Shea K, Zhao C, Pelaez M, Han C, Lynch TJ and Dionysiou DD, Environ. Sci.: Processes Impacts, 2013, 15, 1979. [DOI] [PubMed] [Google Scholar]
- 85.Falconer IR and Humpage AR, Environ. Toxicol, 2006, 21, 299. [DOI] [PubMed] [Google Scholar]
- 86.Mailyan AK, Chen JL, Li W, Keller AA, Sternisha SM, Miller BG and Zakarian A, J. Am. Chem. Soc, 2018, 140, 6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim H, Ho S and Leighton JL, J. Am. Chem. Soc, 2011, 133, 6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mazmouz R, Chapuis-Hugon F, Pichon V, Méjean A and Ploux O, ChemBioChem, 2011, 12, 858. [DOI] [PubMed] [Google Scholar]
- 89.Guzmán-Guillén R, Prieto AI, González AG, Soria-Díaz ME and Cameán AM, Environ. Toxicol. Chem, 2012, 31, 2233. [DOI] [PubMed] [Google Scholar]
- 90.Chooi Y-H and Tang Y, J. Org. Chem, 2012, 77, 9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schor R and Cox R, Nat. Prod. Rep, 2018, 35, 230. [DOI] [PubMed] [Google Scholar]
- 92.He Y and Cox RJ, Chem. Sci, 2016, 7, 2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Davison J, al Fahad A, Cai M, Song Z, Yehia SY, Lazarus CM, Bailey AM, Simpson TJ and Cox RJ, Proc. Nat. Acad. Sci. USA, 2012, 109, 7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu N, Song W, Schienebeck CM, Zhang M and Tang W, Tetrahedron, 2014, 70, 9281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Baker Dockrey SA, Lukowski AL, Becker MR and Narayan ARH, Nat. Chem, 2018, 10, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bai W-J, David JG, Feng Z-G, Weaver MG, Wu K-L and Pettus TRR, Acc. Chem. Res, 2014, 47, 3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pettigrew JD, Freeman RP and Wilson PD, Can. J. Chem, 2004, 82, 1640–1648. [Google Scholar]
- 98.Spence JTJ and George JH, Org. Lett, 2013, 15, 3891. [DOI] [PubMed] [Google Scholar]
- 99.Doyon TJ, Perkins JC, Baker Dockrey SA, Romero EO, Skinner KC, Zimmerman PM and Narayan ARH, J. Am. Chem. Soc, 2019, 141, 20269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fan J, Liao G, Kindinger F, Ludwig-Radtke L, Yin W-B and Li S-M, J. Am. Chem. Soc., 2019, 141, 4225. [DOI] [PubMed] [Google Scholar]
- 101.Gibson MI, Forneris CC and Seyedsayamdost MR, in Cyclic Peptides: From Bioorganic Synthesis to Applications, ed Koehnke Jesko, Naismith James and van der Donk Wilfred A, The Royal Society of Chemistry, Thomas Graham House, Science Park, Milton Road, Cambridge CB4 0WF, UK, 2018, 7, 141–163. [Google Scholar]
- 102.Higa T and Kuniyoshi M, J. Toxicol. Toxin Rev, 2000, 19, 119. [Google Scholar]
- 103.Werner P, Voigt M, Keinänen K, Wisden W and Seeburg PH, Nature, 1991, 351, 742. [DOI] [PubMed] [Google Scholar]
- 104.Lodge D, Neuropharmacology, 2009, 56, 6. [DOI] [PubMed] [Google Scholar]
- 105.Yu Zheng X, Zhang H-L, Luo Q and Zhu J, J. Biomed. Biotechnol, 2011, 2011, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stathakis Christos I., Yoiti Efthymia G. and Gallos John K., Eur. J. Org. Chem, 2012, 2012, 4661. [Google Scholar]
- 107.Chekan JR, McKinnie SMK, Moore ML, Poplawksi SG, Michael TP and Moore BS, Angew. Chem. Int. Ed, 2019, 58, 8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Botta B, Monache GD, Misiti D, Vitali A and Zappia G, Curr. Med. Chem, 2001, 8, 1363. [DOI] [PubMed] [Google Scholar]
- 109.Wolff J, Knipling L, Cahnmann HJ and Palumbo G, Proc. Nat. Acad. Sci. USA, 1991, 88, 2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Clark PI and Slevin ML, Clin. Pharmacokinet, 1987, 12, 223. [DOI] [PubMed] [Google Scholar]
- 111.Yu X, Che Z and Xu H, Chem. Eur. J, 2017, 23, 4467. [DOI] [PubMed] [Google Scholar]
- 112.Chang W-C, Yang Z-J, Tu Y-H and Chien T-C, Org. Lett, 2019, 21, 228. [DOI] [PubMed] [Google Scholar]
- 113.Lau W and Sattely ES, Science, 2015, 349, 1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li J, Zhang X and Renata Hans, Angew. Chem. Int. Ed, 2019, 58, 11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lazzarotto M, Hammerer L, Hetmann M, Borg A, Schmermund L, Steiner L, Hartmann P, Belaj F, Kroutil W, Gruber K and Fuchs M, Angew. Chem. Int. Ed, 2019, 58, 8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.DeMartino MP, Chen K and Baran PS, J. Am. Chem. Soc, 2008, 130, 11546. [DOI] [PubMed] [Google Scholar]
- 117.Thomas JG, Ayling A and Baneyx F, Appl. Biochem. Biotechnol, 1997, 66, 197. [DOI] [PubMed] [Google Scholar]
- 118.Marchand JA, Neugebauer ME, Ing MC, Lin C-I, Pelton JG and Chang MCY, Nature, 2019, 567, 420. [DOI] [PubMed] [Google Scholar]
- 119.Matthews ML, Chang W-C, Layne AP, Miles LA, Krebs C and Bollinger JM Jr, Nat. Chem. Biol, 2014, 10, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mitchell AJ, Dunham NP, Bergman JA, Wang B, Zhu Q, Chang W-C, Liu X and Boal AK, Biochemistry, 2017, 56, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Goldberg NW, Knight AM, Zhang RK and Arnold FH, J. Am. Chem. Soc, 2019, 141, 19585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Duewel S, Schmermund L, Faber T, Harms K, Srinivasan V, Meggers E and Hoebenreich S, ACS Catal, 2020, 10, 1272. [Google Scholar]
- 123.Matsuda Y, Iwabuchi T, Wakimoto T, Awakawa T and Abe I, J. Am. Chem. Soc, 2015, 137, 3393. [DOI] [PubMed] [Google Scholar]
- 124.Ziemert N, Alanjery M and Weber T, Nat. Prod. Rep, 2016, 33, 988. [DOI] [PubMed] [Google Scholar]