

Abstract
Among glycan-binding proteins, galectins, β-galactoside-binding lectins, exhibit relevant biological roles and are implicated in many diseases, such as cancer and inflammation. Their involvement in crucial pathologies makes them interesting targets for drug discovery. In this review, we gather the last approaches toward the specific design of glycomimetics as potential drugs against galectins. Different approaches, either using specific glycomimetic molecules decorated with key functional groups or employing multivalent presentations of lactose and N-acetyl lactosamine analogs, have provided promising results for binding and modulating different galectins. The review highlights the results obtained with these approximations, from the employment of S-glycosyl compounds to peptidomimetics and multivalent glycopolymers, mostly employed to recognize and/or detect hGal-1 and hGal-3.
Keywords: galectins, glycomimetics, glycans, molecular recogntion, drug design
Introduction
Galectins are a large family of glycan-binding proteins with preference for β-galactoside- (β-Gal) containing structures. In mammals, 16 different members have been described, which can be found both in the intra- and extracellular environments. On the basis of the supramolecular organization of their carbohydrate-recognition domains (CRDs), they are classified (Figure 1) as prototype galectins, when a single CRD spontaneously forms homodimers (human galectins-1, -2, -5, -7, -10, -11, -13, -14, -15, -16), as tandem-repeat galectins, when two homologous CRDs are connected by a short aminoacidic linker (human galectin-4, -6, -8, -9, -12), or as chimera-type galectins (human galectin-3), when the CRD is connected to an N-terminal aminoacidic non-lectin region (Leffler et al., 2002; Yang et al., 2008; Dings et al., 2018). The sequence identity between galectins is moderately high, and the CRD's structure of six (from S1 to S6) and five (from F1 to F5) antiparallel beta strands is always conserved (Figures 1, 2). The β-Gal-binding site is located in the S-face, centered on subsite C and extended away, occupying subsites A, B, D, and E, which display variations between the galectin members (Figure 1).
Figure 1.

Above: representation of the three group of galectins. On the bottom: representation of the CRD of hGal-1 in complex with N-acetyllactosamine (PDB ID: 1W6P) with subsites (A, B, C, D, and E) of the binding site and architecture of the beta strands (S1, S2, S3, S4, S5, S6, F1, F2, F3, F4, F5, F6) highlighted.
Figure 2.

On the top: sequence alignment of the galectins discussed in this review. The residues conserved among galectins are highlighted in bold; the blue ones are those mainly involved in the interaction with lactose. On the bottom: galectin's binding sites of complexes (A) hGal-1/lactose (PDB ID: 1GZW), (B) hGal-3/lactose (PDB ID: 2NN8), and (C) hGal-8-Nterminal/lactose (PDB ID: 5T7S). Residues involved in the interaction are displayed as sticks; H bonds are displayed as yellow dots.
The canonical binding site of galectins is shallow and solvent exposed, and the interaction with β-galactosides occurs through several hydrogen bonds between the hydroxyl groups of the β-Gal and specific histidine, arginine, and asparagine galectin residues and through the CH-π stacking interaction between the non-polar face of the β-Gal moiety and a conserved tryptophan residue (Figure 2). Even though these residues in the binding site are highly conserved, the divergence in few neighboring sites may modulate the glycan-binding preferences among galectins (Johannes et al., 2018).
Galectins are involved in a wide range of biological activities, such as homeostasis, apoptosis, and vascular embryogenesis (Chan et al., 2018). Their involvement in pathological contexts has been described for inflammation (Brinchmann et al., 2018), host-pathogen interaction (Vasta, 2009), antibacterial autophagy (Weng et al., 2018), and cancer (Liu and Rabinovich, 2005; Girotti et al., 2020). In fact, many members of the family are associated with the phenomena of carcinogenesis being involved in processes such as apoptosis, adhesion, migration, cell transformation, invasion, metastasis, immune escape, and angiogenesis (Dings et al., 2018). The heterogeneous roles of galectins in these physiological and pathophysiological processes have motivated the rational design of fine-tuned inhibitors with precise selectivity and specificity. The characterization at the molecular level of the interaction of galectins with their natural binders has provided the structural basis for the design of potent antagonists (Ardá and Jiménez-Barbero, 2018; Valverde et al., 2019a).
Notwithstanding carbohydrates are the natural ligands of galectins, they are not the best antagonist candidates because of their inherent properties that make them not good enough as pharmacological agents. The main disadvantages are the low metabolic stability and high hydrophilicity. Moreover, it is well-known that the affinities that characterize single carbohydrate-lectin interactions are usually rather weak, in the μM-mM range, far from the nM scale required for a good drug candidate. This evidence derives from many factors intrinsically connected to the nature of carbohydrates, such as the absence of hydrophobic groups that can interact with the protein surface, the hydrogen bond-based interplay with the binding site that suffers from the competition with the bulk water, and the large enthalpic penalties for desolvation of the shallow binding site (Hevey, 2019).
Thus, in the last two decades, molecular scaffolds differently derivatized have been employed as mimetics capable of recognizing and blocking galectins, especially human galectins 1 and 3 (hGal-1 and hGal-3) (Tellez-Sanz et al., 2013; Blanchard et al., 2014, 2016; Girard and Magnani, 2018). These glycomimetics are molecules that mimic carbohydrates structurally and functionally but display improved pharmacological properties (Magnani and Ernst, 2009).
The majority of the molecules synthesized for this purpose are based on sugar scaffolds, although some non-saccharide-based inhibitors, as peptide-based mimetics, have also been reported. The chemical modifications of those based on sugar scaffolds mainly concern the replacement of the endocyclic oxygen of the pyranose ring (carbasugars, iminosugars, thiosugars, and phosphasugars) or the exocyclic oxygen (C-glycosyl, N-glycosyl, S-glycosyl). Indeed, the substitution of the bridging exocyclic oxygen by other atoms has been proved to be effective in increasing the resistance of carbohydrates to hydrolysis (Tamburrini et al., 2019).
The workflow for the development of new glycomimetics aims at finely modulate affinity and stability, as well as selectivity. Since, as discussed, the CRD of mammalian galectins is highly conserved, with minor amino acid sequence variation between members of the family, achieving selective glycomimetics is somewhat difficult and challenging (Figure 2) (Compain, 2018; Tamburrini et al., 2019).
In addition to the modifications of the endocyclic or exocyclic oxygen of the sugar scaffold, which modulate mostly stability and conformation, the introduction of new functional groups as substituents enables reaching new binding pockets and establishes additional contacts, in order to increase affinity and selectivity. Moreover, the development of potent inhibitors usually resorts to the employment of multivalent ligands, which exploits lectin avidity, and allows reaching increased affinities. We have arbitrarily divided the glycomimetics design in this area into two categories: strategies that exploit the presentation of a single strong inhibitor (monovalent carbohydrate-based inhibitors) and those that use multimerized ligands (multivalent carbohydrate-based inhibitors). Below, the most recent and promising examples for both types of inhibitors are presented and contextualized.
Single Presentation Strategies
As canonical natural ligands of galectins, lactose or N-acetyllactosamine have been the most-used scaffolds to build synthetic glycomimetics, although monosaccharides and other disaccharides have also been taken into consideration and have led to the synthesis of mimetics with promising characteristics (Dings et al., 2018). All the monovalent carbohydrate-based inhibitors can be defined as modified Gal-containing mono- or disaccharides decorated with diverse molecular fragments.
On the molecular basis, the interaction of galectins with β-galactoside sugars, besides the stacking interaction with the conserved Trp moiety, mainly involves hydroxyl O4 and O6 of galactose (Figure 2). Therefore, the other positions are free to accept modifications that can enhance the effectiveness of the mimetic.
In fact, lactosamine derivates with modifications at Gal-C3 (4-phenoxyaryl, via ether or triazole linkages) and Glc-N2 (attachment of an aromatic phthaloyl or benzoyl) have been synthetized, and their affinities have been measured using fluorescence polarization assays (Sörme et al., 2004). These molecules reached dissociation constants in the low μM range for hGal-3 and hGal-1 (Van Hattum et al., 2013). Especially, compound 1 (KD 1.2 μM) was far more selective toward hGal-3 over hGal-1 (230-fold) (Figure 5). The larger aromatic substitution at Glc-N2 is better accommodated in the binding site of hGal-3. Nevertheless, analogous modifications on a non-hydrolysable thio-digalactoside (TDG) scaffold led to the synthesis of glycomimetics showing high affinity (Van Hattum et al., 2013).
The TDG scaffold (Figure 3) has proved to be an effective platform for designing glycomimetics. These molecules reproduce the interactions of lactosides at subsites C and D of galectins with the advantage of being hydrolytically stable (Figures 3, 4).
Figure 3.

Binding site of the hGal-3/LacNAc (PDB ID: IKJL) and hGal-3/TDG (PDB ID: 4JC1) complexes. Key residues for binding are reported as sticks.
Figure 4.
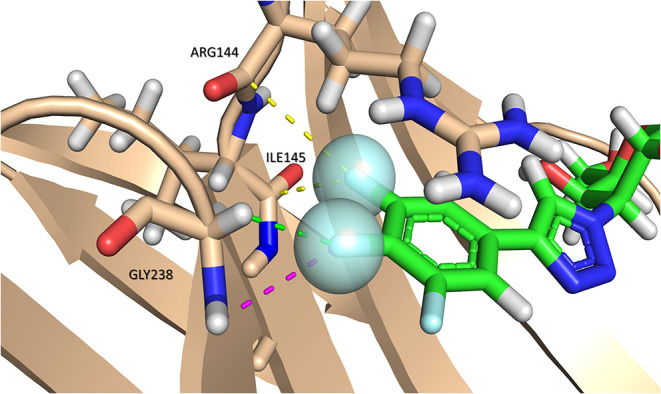
Multipolar interactions between the fluorine atoms (blue sphere) of compound 13 and hGal-3. C-F···H-N interactions are represented with purple dashes. C-F···H-Cα interactions with green dashes. The C-F···C=O interaction with the backbone amides of Arg144 and Ile145 is represented with yellow dashes. PDB ID: 6QLP. The interactions are represented with analog 13 instead of TD139 since it lacks C-F···H-N and C-F···H-Cα interactions.
In particular, TD139, a symmetric TDG substituted with aromatic moieties at each C3 position, has proved to be a very good antagonist of hGal-1 and hGal-3 (MacKinnon et al., 2012; Figure 5). Furthermore, the compound includes fluorinated substituents, which can have an enormous impact in protein-ligand interactions. Besides decreasing hydrophobicity, the presence of fluorine atoms provides multiple ways to interact with protein moieties, such as multipolar interactions with carbonyl groups, amides, or dipole-dipole interactions (Figure 4).
Figure 5.

X-ray structures of hGal-3/TDG (PDB ID: 4JC1), hGal-3/TAZTDG (PDB ID: 5H9R), hGal-3/TD139 (PDB ID: 5H9P), and hGal-7/TAZTDG (PDB ID: 5H9S) complexes. Galectin's subsites and the key residues for the interaction are highlighted.
TD139 is one of the few examples of galectin's antagonist in advanced clinical trials (Galecto Biotech). In particular, it has been employed as a therapeutic agent against hGal-3 for the treatment of Idiopathic Pulmonary Fibrosis. Since the molecule is not well-absorbed orally and must be given parenterally, the method of administration is inhalation of dry powder.
Hsieh et al. (2016) have taken advantage of this scaffold to target and analyze the interaction with hGal-1, -3, and -7. In order to distinguish the implication of each aromatic group of TD139 in the binding, TAZTDG, a mimetic with only one aromatic substitution, was studied (Figure 5). 15N-1H HSQC NMR titrations, 19F relaxation NMR experiments, Isothermal Titration Calorimetry (ITC), and X-ray have unearthed that for hGal-1 and -7, the aromatic ring contributes more to the binding at the subsite E, whereas for hGal-3, the fluorophenyl group is mainly interacting with subsite B (Figure 5). This kind of information can be employed for the rational design of new inhibitors, since different substituents at C3 can determine the selectivity for a single galectin (Hsieh et al., 2016).
Delaine et al. (2016) have presented the synthesis of a battery of doubly C-3 substituted derivatives at C3 (compounds 2-7, Figure 6). The generated 4-aryl-1,2,3-triazolyl thiodigalactoside-based derivatives were tested as antagonists of galectin-1, -2, -3, -4 (N- and C-terminal domain), -7, -8 (N-terminal domain), domain), and 9 (N- and C-terminal domain) (Delaine et al., 2016). The molecules displayed affinities considerably larger than those measured for the unsubstituted thiodigalactosides, especially against hGal-1 and hGal-3. Regarding hGal-1, this lectin bound the phenyl-triazole unsubstituted 2 with higher affinity (4 nM) than that for mimetics displaying larger substitutions (3, 84 μM). Furthermore, only the mimetic substituted with 3- or 4-fluorophenyl moieties (4, 12 nM; 5, 27 nM, respectively) were more akin than unsubstituted 2, strongly suggesting that the galectin subsites display rather limited possibilities for substitution, as previously described (Van Hattum et al., 2013). Nevertheless, the thienyl substituted compound 6 provided a very good inhibition of hGal-1 (KD < 10 nM, 2,400-fold stronger than TDG). Moreover, its selectivity for hGal-1 over hGal-3 (65 nM) was remarkable. Concerning hGal-3, the selectivity profile was similar to that achieved for hGal-1, with the exception of the 4-phenoxy-substituted phenyl 3 that displayed a KD for hGal-3 of 0.36 μM, with a considerably enhanced selectivity over hGal-1 (KD of 84 μM). It is evident that the right choice of the substituent on the triazole ring of this type of compounds guide selectivity for either hGal-1 or hGal-3.
Figure 6.

Structure of mimetics 1 (Van Hattum et al., 2013), TAZTDG (Hsieh et al., 2016), TD139 (MacKinnon et al., 2012), 2–6 (Delaine et al., 2016), 8–13 (Peterson et al., 2018), 14 (Nilsson et al., 2013), 15 (Salameh et al., 2010), and 16–19 (Peterson et al., 2018).
Regarding hGal-4, the aryltriazoles carrying no substituents or holding small appendages at the aryl moiety (2, 4, 5, 6, 7) were the best inhibitors, especially for the C-terminal domain (mid-nM affinities), in line with the observations for hGal-1. In contrast, the N-terminal domain recognized all the molecules of the study with low-medium μM affinities, with some notable exceptions (4, 0.17 μM). The best inhibitor for the C-terminal domain was the fluorophenyl-derivative 5 (73 nM). However, this molecule is not specific, since it also recognized hGal-1 and hGal-3 within the same affinity range. hGal-7 and hGal-9 (both C- and N-terminal domains) did not efficiently bind the larger compounds of the library, while they showed sub- to low-μM affinities for those carrying small substituents. In contrast with the data for hGal-4, no binding preferences were detected when compared the C- and N-terminal domains of hGal-9.
X-ray crystallography structures of hGal-3 complexed with the three best antagonists (4, 5, and 6), together with mutation analysis of the lectin, showed that the aryl-triazol moieties display guanidine-arene interactions involving R144 and R186, as well as additional fluorine-amide orthogonal multipolar interactions, which are obviously absent in the binding with the natural ligands. The inhibition action of 4 against hGal-3 was also corroborated with in vitro and in vivo strategies. The results showed that 4 has intracellular availability and activity blocking the intracellular accumulation of the lectin around damaged vesicles in breast carcinoma MCF-7 cells and also attenuates lung fibrosis in a dose-dependent manner, using a murine model (Delaine et al., 2016).
On the basis of this success, a systematic tuning of the aryl substituents was performed: a series of fluorinated C3 aryltriazolyl-substituted thiodigalactosides, including symmetrical and asymmetrical compounds, were synthesized to optimize the interaction with R144 of hGal-3 (compounds 8-13, Figure 6). As a first step, the interaction of monogalactosides carrying phenyl-triazole moieties with fluorine, chlorine, or bromine atoms was first evaluated, and the three best inhibitors in terms of both affinity and selectivity for hGal-3 over hGal-1 were selected. They include the di- or trifluorophenyltriazol thiogalactosides (8, 9, and 10), with relative affinities of 8.8, 15, and 5.2 μM, respectively. Then, the corresponding symmetrical thiodigalactosides (11, 12, 13) were prepared, which displayed improved affinity and selectivity compared to 4 and 5, as well as higher affinities over the thiogalactosides 8, 9, and 10. However, the selectivity was lower (Peterson et al., 2018).
In contrast, asymmetrically substituted thiodigalactosides showed improved binding affinities and selectivity. The design of these new compounds was based on the good selectivity described for the analogous symmetrical molecules 14 (Nilsson et al., 2013) and 15 (Salameh et al., 2010) and they combined the monofluoro- and the trifluorophenyl moieties present in 4 and 13 with the coumaryl and triazolyl groups in 14 and 15 (Figure 6). This new set of compounds (16, 17, 18, and 19) improved hGal-3 over hGal-1 selectivity maintaining the low nM affinity (Figure 6). In particular, 17 (7.5 nM) was 46-fold more affine for hGal-3. Structural analysis and ITC measurements revealed that increased fluorination of a phenyl triazole group favors the interaction with R144. In conclusion, the combination of C3-aryltriazolyl groups with O3-coumaryl in a thiodigalactoside generates a first-class glycomimetic with affinity and selectivity for hGal-3 without equals (Peterson et al., 2018).
Additional improvements based on the monogalactoside scaffolds 8, 9, and 10 have also been achieved. Zetterberg et al. (2018) used a different approach, derivatizing the Gal moiety at position 1 with an unnatural substituent, thus favoring non-natural lectin-ligand interactions. They include orthogonal multipolar fluorine-amide, phenyl-arginine, sulfur-π, and halogen bond interactions. All the resulting compounds [series of α- and β-thio-D-galactopyranosides with 4-(3,4,5-trifluorophenyl)-1H-triazole at C3] bind to hGal-3 with very good affinities. In particular, the S-glycoside 20, substituted with a 3,4-dichlorophenylthio moiety (Figure 7) presents a KD of 37 nM, and it is fairly selective, with 100-fold lower affinity for most of the other galectins (1, 4N, 7, 8C, 8N, 9C, 9N) and 20- and 4-fold for hGal-2 and hGal-4 C-terms, respectively (Zetterberg et al., 2018).
Figure 7.

Structure of mimetics 20 (Zetterberg et al., 2018), 21 (Santarsia et al., 2018), and 25–29 (Dahlqvist et al., 2019). Structure of TF antigen and histo-blood group antigens H, B, and A of type II (22–24).
The cellular uptake of some of these mimetics (4 with KD of 12 nM, 20 with KD of 37 nM, and 15 with KD of 99 nM) and their hGal-3 inhibition ability have been evaluated in intracellular and extracellular environments (Stegmayr et al., 2019). The permeability assays with monolayers of Caco-2 cells determined that 20 had a significantly higher value of membrane permeability (with passive transport) than 4 and 15. Despite the relatively similar affinities, their polar surface area is different, being significantly lower for 20, in good correlation with the observed results. The extent of non-polar surface areas is relevant for the passive cell membrane permeability and oral bioavailability. Additionally, the extracellular activity was tested using an assay based on the use of a fluorescently-labeled hGal-3 coupled to the cell surface of CHO cells line, revealing that the inhibition potency correlated with the previously measured relative affinities. The determination of the intracellular inhibition was based on the induction of the accumulation of hGal-3 around damaged intracellular vesicles, followed by the subsequent administration of the inhibitors. The results followed the trend of the permeability assay, with 20 showing IC50 values considerably lower than those for 4 and 15 (Stegmayr et al., 2019).
Another promising disaccharide mimetic has been presented by Marcelo and co-workers. The authors focused on the interplay between hGal-3 and the Thomsen-Friedenreich (TF) antigen to develop a mimetic capable of binding hGal-3 and compete with the natural antigen (Santarsia et al., 2018). The TF antigen (20), also known as pancarcinoma-related antigen, is a disaccharide (Galβ1 → 3GalNAc) that decorates glycoproteins located on the cell-surface through O-Ser/Thr linkages (Figure 7). Under healthy conditions, this fragment is not exposed since it is covered by additional glycosylation, and therefore hidden. In contrast, the epitope is exposed and overexpressed in the tumor microenvironment, particularly in adenocarcinomas (Springer, 1997; Glinsky et al., 2001). Now, the TF antigen is accessible and, among other interactors, extracellular hGal-3, also overexpressed in various tumor lines, is capable of recognizing and binding to it, so mediating the adhesion of tumor cells to the endothelium leading to metastasis progression (Jeschke et al., 2006; Fortuna-Costa et al., 2014; Mori et al., 2015). MUC1 is a tumor-related glycoprotein decorated with the TF antigen, and it has been proposed that the aberrant interaction between MUC1 presenting TF disaccharide epitopes, and hGal-3 promotes cancer progression (Glinsky et al., 2001; Goletz et al., 2002; Takenaka et al., 2002). The authors exploited a previously proposed α-O-Gal scaffold (Jiménez-Barbero et al., 2009) to generate the disaccharide TF mimetic (21, Figure 7). 15N-1H HSQC-based titrations provided the chemical shift perturbations of the hGal-3 signals, which revealed that the more affected protein residues belong to the canonical binding site already described for the natural TF antigen, both by NMR and X-ray crystallography (Bian et al., 2011; Yongye et al., 2012). Linewidth 1H NMR and STD-NMR experiments allowed to obtain the ligand-binding epitope (Valverde et al., 2019b; Gimeno et al., 2020). Both experimental procedures pointed out that the disaccharidic moiety is the key epitope for hGal-3, while the unnatural scaffold is exposed to the solvent. Similar observations have been reported for the natural ligand TF, where the attached Thr does not participate in the binding event (Santarsia et al., 2018). The thermodynamic profile of the system indicate that there is a slight gain in terms of KD, which interestingly arises from a favorable entropic contribution (−0.9 kcal mol−1), while there is less enthalpy gain (−4.3 kcal mol−1) compared to the natural TF (–TΔS 7.75 kcal mol−1; ΔH − 12.6 kcal mol−1) (Yongye et al., 2012). The analysis of the X-ray crystallographic structure obtained for the hGal-3/21 complex allowed noticing the absence of four hydrogen bonds involving the GalNAc residue that were present in the complex with the TF antigen, due to the chemical nature of the lactam ring and the removal of the NHAc group. These evidences might explain the loss of enthalpy gain; however, they did not provide a completely definite explanation for the observed positive entropic term. The authors suggested that the amphiphilic character of the lactam ring and the associated changes in the solvation features could be beyond the favorable entropic contribution. This work is an exquisite example of how a simple modification of the chemical nature of the ligand can lead to a big change in the entropy-enthalpy compensation phenomenon without changing the binding epitope. Overall, mimetic 21 might be considered as a putative lead compound to interfere with the aberrant interaction of hGal-3 and the TF antigen (Santarsia et al., 2018).
In this context, and concerning the role of the entropy, a multidisciplinary study of the interaction between the histo-blood group antigens (Figure 7, hBGAs type II 22, 23, 24) and hGal-3, revealed that the typical entropy penalty that occurs upon galectin binding may be modulated by chemical modifications at the ligand. For the hBGA tetrasaccharides, which are fucosylated molecules, it was demonstrated that the fucose moiety rigidifies the motion around the glycosidic linkages of the whole molecule, thus providing a rather stiff and preorganized conformation. This feature is reflected in the existence of faster association rates of these ligands to hGal-3, which also exhibit a less unfavorable binding entropy. Even if the fucose is not directly involved in the binding event, it is capable of guiding the interaction and improving the binding affinity. This evidence may be considered to explore novel avenues for the development of more potent ligands (Gimeno et al., 2019).
Simple C-glycosyl compounds have also been employed as scaffolds for developing selective small-molecule inhibitors, mainly involving C1- and C3-substitutions at the Gal moiety. Nilsson and collaborators have proposed novel C1-galactopyranosyl heterocycle mimetics that were fine-tuned against galectins (Dahlqvist et al., 2019). Different triazoles, oxazoles, isoxazoles, and pyrazoles (25–29, Figure 7) were used as substituents at Gal C1 to generate a library that was tested with galectins 1, 3, 4, 7, 8 (C- and N-terminal), and 9 (C- and N-terminal). Competitive fluorescence polarization assays (Sörme et al., 2004) allowed discriminating the more active compounds. The triazole-substituted 25 and 26 mimetics were fairly selective for hGal-1 (KD 750 μM and 290 μM, respectively) while the oxazole-substituted 27 and 28 were selective for hGal-3 (KD 90 μM and 230 μM, respectively). These compounds presented affinities only in the millimolar range with all the other galectins. On the other hand, the isoxazoles displayed good affinity but poor selectivity, while the pyrazoles were poorly active and selective. Consequently, further design was accomplished using the triazoles and the oxazoles as leads. Better affinity for hGal-1 was found for triazole 29 (KD 170 μM), where the aryl moiety is fluorinated at position 2, keeping the specificity for this galectin. Molecular Dynamics (MD) simulations performed for the hGal-1/29 complex revealed a stacking interaction between the triazole ring of 29 and the His52 side chain of hGal-1. This His52 is unique for hGal-1 and differentiates it from the other galectins. The specificity of 27 for hGal-3 was accounted for by the occupation of the ligand of a unique subsite of the lectin above the β-face of the bound Gal ring. The aforementioned workflow can be followed to develop more high-affinity inhibitors by exploiting the reported heteroaryl-selectivity (Dahlqvist et al., 2019).
Besides sugar-based mimetics, non-carbohydrate inhibitors have also been studied. In particular, carbohydrate-mimetic peptides have been developed as effective inhibitors, taking advantage of the role that peptides may play as immunogens and interfere with protein–sugar interactions (Kieber-Emmons et al., 2014). Using high-throughput phage display analysis, a variety of synthetic peptides capable of binding to the CRD of hGal-3 with high affinity, were identified as well as their inhibition ability to affect metastasis-associated cancer cell adhesion (Zou et al., 2005). The most active peptides shared the common motif [P-(C)-G-P-X-X-X-D-(C)-P]. Notably, G3-C12 (ANTPCGPYTHDCPVKR) recognized hGal-3 with a KD of 70 nM, and its administration significantly reduced metastatic cell deposition in a mice model, consequently decreasing the outgrowth within vasculature (Newton-Northup et al., 2013). Using N-(2-hydroxypropyl)methacrylamide (HPMA) as a carrier molecule, the drug delivery process was further enhanced (Yang et al., 2014).
Anginex is an amphipathic synthetic peptide directed against hGal-1, designed through basic folding principles, which incorporates short sequences of interleukin-8, platelet factor-4 and the bactericidal/permeability increasing protein (Figure 8), (Mayo et al., 2001; Griffioen, 2006). It is able to act as tumor growth inhibitor and to block angiogenesis and migration of cancer cells (Griffioen et al., 2001; Wang et al., 2012). It adopts a major β-sheet topology, as deduced by NMR and CD experiments, and displays a net positive charge as well as a hydrophobic face that are essential for its function (Dings et al., 2003; Mayo et al., 2003). Concerning the specificity, anginex preferentially targets hGal-1, although it is also capable of binding to hGal-2, -7, -8N, and -9N with low affinities. In contrast, it does not show any activity against hGal-3, -4N, -4C, and 9-C 9-C (Salomonsson et al., 2011). Starting from anginex's sequence, other antagonist peptides of hGal-1 with improved properties have been developed. Mayo et al. (2003, 2008) synthetized 6DBF7, in which keeping the β-sheet structure presents a central dibenzofuran (DBF) scaffold that connects six amino acid residues (at the N-term) to other seven amino acids (at the C-term) (Figure 8). 6DBF7, which also targets hGal-1, is more effective in vivo in terms of anti-angiogenic and anti-cancer activity than parent anginex (Mayo et al., 2003, 2008). A soluble analog, DB16, was employed to deduce the binding site by 15N-1H HSQC NMR experiments. The interaction primarily takes place at one edge of the β-sandwich of the lectin and reduces the affinity of hGal-1 to lactose, suggesting the presence of an allosteric interplay (Dings et al., 2013). The DB21 analog, which shows a branched alkyl side chain (Figure 8) shows even higher capacity to inhibit tumor angiogenesis and tumor growth in vivo.
Figure 8.
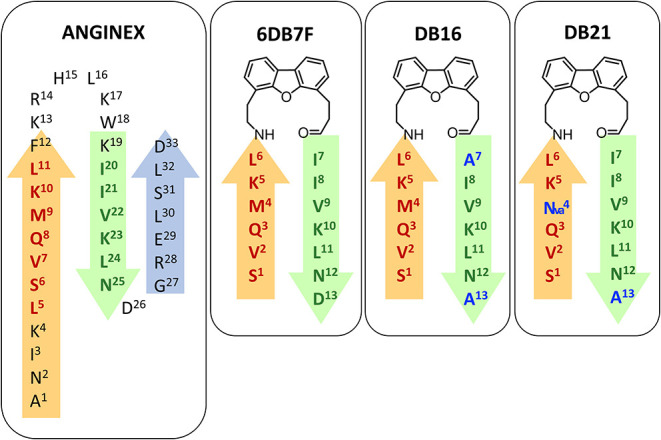
Amino acidic sequence of different mimetics: anginex (Mayo et al., 2001; Griffioen, 2006), 6DB7F (Mayo et al., 2003, 2008), DB16, and DB21. β-sheets structures are reported as arrows.
A series of non-peptidic topomimetics of anginex based on a calix[4]arene scaffold substituted with hydrophobic and hydrophilic moieties have also been employed (Dings et al., 2006). Compound PTX008 (also known as OTX008) (Figure 9) interacts with the lectin at an allosteric site (Dings et al., 2008) and shows potent anti-angiogenic and anti-proliferation activity in vitro and in vivo. In fact, it was approved for Phase I clinical trials for advanced solid tumors (NCT number: NCT01724320) (Thijssen et al., 2006; Mayo, 2014; Koonce et al., 2017).
Figure 9.
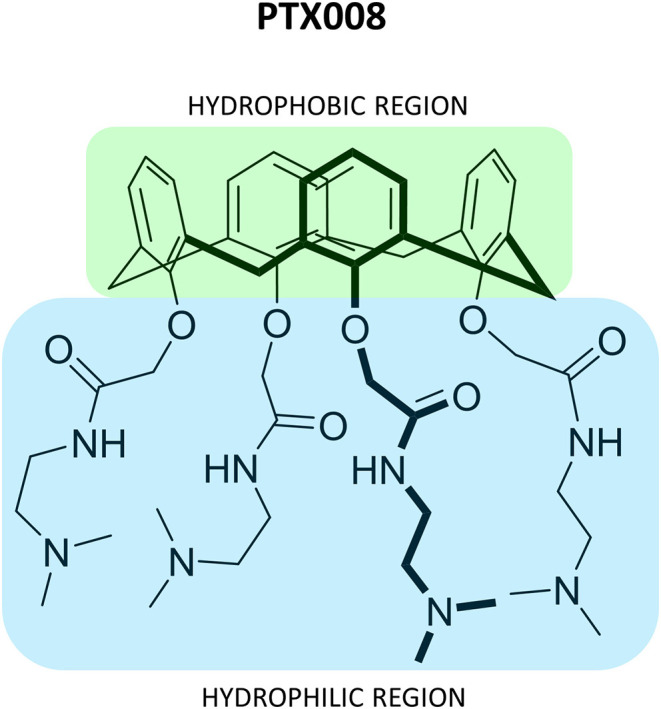
Representation of compound PTX008 (Dings et al., 2006; Mayo, 2014).
A series of peptides and phosphopeptides able to interact with the hGal-3 CRD were also identified using fragments from the N-terminal tail of hGal-3. HSQC experiments using 15N-labeled hGal-3 CRD demonstrated the effective interaction of the peptides with the back-face of the CRD of hGal-3, also showing that phosphorylation of serines 6 and 12 enhances the binding, while phosphorylation of tyrosines 107 and 118 in specific N-peptides attenuates the interaction. These data also suggested a fine modulation of hGal-3 activity through the phosphorylation of the tail (Berbís et al., 2014).
Multivalent Presentation Strategies
In biological systems, low-affinity binding is usually overcome through the engagement of simultaneous interactions that the receptor and ligand can establish a phenomenon known as multivalency (Chittasupho, 2012). Thus, mimicking nature and developing multivalent ligands has been a frequent approach to surmount the low affinity of single galectin-carbohydrate interactions.
There are diverse ways in which multivalent presentations can enhance affinity: chelation, subsite binding, statistical rebinding, steric stabilization, and clustering effects (Cecioni et al., 2015; Zhang et al., 2018; Bücher et al., 2019). However, in the development of multivalent ligands, there are various factors that should be taken into consideration. First, the nature of the scaffold. Rigidity is a crucial feature to consider, since it is directly related to entropy. Flexible linkers may display a large entropic penalty upon binding. Second, flexibility can also be advantageous because the linker might adopt the proper conformation for favorable interactions to take place (Shewmake et al., 2008; Hevey, 2019). Third, the chemical nature of the linker is also relevant, since it might establish additional interactions with secondary binding sites and therefore improve the affinity (Boden et al., 2019). Undoubtedly, the choice of the ligand is a key factor in the outcome as well as its actual concentration within the scaffold. Usually, the higher the concentration of the ligand is, the higher the affinity. However, at a high concentration of the ligand, steric clashes may take place and the effectivity of the approach decreases. Thus, the content of the specific ligand should be carefully chosen (Karaman, 2010).
Despite that most of the described antagonists targeting galectins are monovalent compounds, diverse multivalent glycomimetics have been developed over the last decade. One of the first designs of multivalent ligands targeting galectins was proposed by Gouin et al. (2010). Multivalent ligands with a glucose-based backbone and a lactose epitope targeting hGal-1 and hGal-3 were designed (Gouin et al., 2010; Figure 10). In order to address the relevance of valence, different multimeric compounds were synthesized, varying the number of glucose units and therefore the number of lactose moieties available in the construct. The pendant chains also present ethylene glycol spacers with different lengths, which affect the 3D structure and flexibility of the cluster. Moreover, they were attached to the β-1,4-linked glucose backbone using positions 1 or 6, thus, leading to a variety of compounds that enabled the independent analysis of the effect of epitope-units and the spacer length (Rostovtsev et al., 2002; Figure 13). This strategy has previously been applied for the synthesis and binding studies of multivalent mannosides with ConA (Gouin et al., 2007).
Figure 10.

The multimeric lactosides employed by Gouin et al. (2010).
Competitive fluorescence polarization assays demonstrated that the spacer length of the multimeric lactosides has no relevant effect, since negligible differences in affinity toward hGal-1 and hGal-3 were observed (Rostovtsev et al., 2002). In fact, MD calculations suggested that the lactoside moieties display a random localization, and the spacer length does not alter the availability of the binding epitope. Two-site ELLA experiments were employed to test the crosslinking ability of the multilactosides, first with peanut agglutinin. In this case, the relative potency per lactoside ranged from 2.1 to 3.2 (McCoy et al., 1984). However, the crosslinking ability was not reproduced for galectins, for which the affinity increased in proportion to the lactose content. The best glycomimetic was the trivalent compound, which yielded a KD of 16 μM toward hGal-3, a <5-fold affinity increase in comparison with the monovalent ligand.
A different glucose-based scaffold to bear disaccharide epitopes has been proposed by Rahkila et al. (2019), consisting of a dextran skeleton decorated with LacNAc moieties. The idea behind this architecture was to mimic natural polysaccharides (Rahkila et al., 2018). In particular, the multivalent antagonists used a ca. 70 kDa dextran, which was propargylated and bioconjugated with lactose, as well as with mannobiose and maltose disaccharides (Grischenko et al., 2013) as blank molecules. Interestingly, upon disaccharide coupling through CuAAC, fractionation of the dextran occurred, yielding multivalent ligands of ca. 40–50 kDa. Binding studies of the multivalent conjugates were performed through 15N-1H HSQC NMR titrations with the carbohydrate recognition domain of hGal-3 and dextrans decorated with different lactose contents were evaluated. Surprisingly, dissociation constants ranging from 0.25 to 0.45 mM were recorded, very similar to those reported for free lactose (Diehl et al., 2010; Miller et al., 2016) The lack of affinity enhancement suggests that the multiple interactions do not cooperatively provide additional value to the interaction, which might arise from unfavorable entropic costs (Bernardi et al., 2014). Strikingly, although the titration with the mannobiose multivalent ligand produced small chemical-shift perturbations, a considerable decrease in the signal intensities was observed in ca. 15 peaks, not located at the canonical binding site of the lectin. Interestingly, these residues are located in the opposite face of the binding site of the lectin and belong to the region that was previously reported to interact with β-mannans (Miller et al., 2016). Although the obtained affinities did not provide positive multivalence effects, probably due to the entropic costs caused by the flexible linker, and the dextran backbone constitutes an easy to modify, well-defined, and homogeneous scaffold to mimic natural-like polysaccharides with possible applications as biomaterials.
Alternatively, Böcker et al. (2015) have employed neo-glycoproteins based on bovine serum albumin (BSA) to target human hGal-3. BSA is a highly abundant protein rich in lysine residues, which makes it fairly appropriate for bioconjugation (Blackmore and Eisoldt, 1999; Huang et al., 2004; Luyai et al., 2009). Two different tetratrasaccharides (30, 31) were synthesized in a chemo-enzymatically manner and coupled to squaric acid diethyl ester to provide 32 and 33, which were conjugated to BSA to provide two neo-glycoproteins series (34, 35) with specific glycans attached at different (between 0 and 29) lysine residues (Figure 11).
Figure 11.

General scheme of the synthesis of the neo-glycoproteins. For neo-glycoproteins series 35 and 36 the surface of BSA is depicted in red, except for lysine residues (blue), the attached carbohydrates are drawn with their symbolic representation. For each series of neo-glycoprotein, the number of lysines glycosylated ranged from 0 to 29.
The selective binding of the multivalent compounds to hGal-1 and hGal-3 was tested through ELISA-type assays (Kupper et al., 2013). The BSA conjugated with the LacNAc-LacNAc motif (34) displayed preference for hGal-3, as a result of the capability of this lectin of recognizing the internal LacNAc motif, which is rather weak for hGal-1 (Stowell et al., 2004). Interestingly, the selectivity for hGal-3 decreased when the carbohydrate content increases, even if the affinity was better. For BSA conjugated with the LacDiNAc-LacNAc tetrasaccharide (35), the affinity also increased with a higher carbohydrate content. However, the selectivity between the lectins decreased. The selectivity of ligands 35 toward hGal-3 was always higher, given the low capability of hGal-1 to recognize LacDiNAc moieties (Van den Berg et al., 2004; Rapoport et al., 2008). Therefore, the interaction relies on the weak recognition of the internal LacNAc. The affinity increase per glycan unit was then analyzed at different lectin concentrations. The LacNAc-LacNAc series 34 did not show significant affinity enhancement. In contrast, the LacDiNAc analogs 35, with 14 to 27 glycosylated lysines, displayed multivalent effects with a 100-fold increase in the relative potency per glycan. Overall, the best neo-glycoproteins showed very high affinity (in the nM range) and selectivity toward hGal-3.
As previously described, the addition of hydrophobic residues to the Gal moieties at proper positions enhances the affinity toward galectins. However, the corresponding synthetic approach may involve laborious chemical procedures (Sörme et al., 2003). In order to overcome this issue, Böcker and Elling (2017) biotinylated the glycan epitopes attached to BSA at position 6 of the non-reducing end through enzymatic synthesis (Böcker and Elling, 2017). Fittingly, the presence of biotin increased the affinity of the ligands toward hGal-1 and hGal-3, maintaining the selectivity for hGal-3. In fact, comparing biotinylated and non-biotinylated neo-glycoproteins, the latter required higher glycosylation densities to reach similar multivalent effects. In the presence of biotin, six glycosylations were enough to obtain a KD of 100 nM, whereas, in the absence of biotinylation, only the analog with 14 glycosylations displayed a similar binding affinity.
The LacDiNAc epitope has been widely employed for decorating scaffolds in the seeking of selectivity. Bojarová et al. (2018) have selectively targeted hGal-3 by employing a series of HPMA [N-2(2-hydroxypropyl)methacrylamide]-based nanocarriers (Figure 12) bearing this disaccharide motif (Bojarová et al., 2018). HPMA, a biocompatible nanocarrier, soluble in water and non-toxic (Ulbrich and Šubr, 2010), was conveniently functionalized with alkyne and easily conjugated to the desired sugar epitope. The neighborhood and content of the alkyne-bearing groups was varied to provide multimeric ligands with different carbohydrate contents (3 to 29 mol-%), providing a platform to evaluate structure-activity relationships (Figure 12). Additionally, the effect of the presence of a short linker between the disaccharide and the triazole moieties was also investigated.
Figure 12.

Different scaffolds (A–D) used for targeting hGal-3. R = LacDiNAc. For each scaffold, polymers with different carbohydrate content were synthesized.
Competitive inhibition ELISA-based assays of hGal-3 binding to asialofetuin determined that a sugar content between 8 and 12 mol-% was optimal for providing the best relative potency (rp/n) compared to the single LacDiNAc disaccharide. Direct attachment of the sugar to the triazole moiety resulted in an improvement in affinity compared with the more flexible O-linked 2-ethylazido linker. Additional contacts between the electron-rich ring with Arg186 at subsite E of hGal-3 are probably at the origin of this effect. The presence of a phenyl group attached to the triazole moiety (scaffold C) did not improve the affinity when compared to scaffold A, whereas the presence of the large DBCO linker (scaffold B) was detrimental for binding. Overall, the best results were obtained for scaffold A, with the LacDiNAc disaccharide (8.1 mol-% content) directly attached to the triazole moiety, although a small rp/n value of 2.3 was achieved.
A series of HPMA-based glycopolymers (Figure 13) bearing different LacNAc contents (3.8 to 22.0 in mol-%) have been designed (Tavares et al., 2020) and tested through an ELISA-type assay.
Figure 13.

Glycopolymers designed for targeting hGal-1 and hGal-3. For each scaffold, five polymers were synthesized with different carbohydrate contents.
The selectivity of the designed multimeric compounds for hGal-1 was remarkable. The ligands displayed IC50 values in the μM range vs. hGal-3, whereas nM affinities were achieved for hGal-1. The best results were obtained for the glycopolymers carrying monovalent scaffolds, with 19.9 and 20.6 mol-% of glycan content, which displayed IC50 values in the low nM scale, and for which each LacNAc unit was 166 times more potent than that in the single presentation and 283 times more selective for hGal-1 than for hGal-3.
In the lectin detection side, Martos-Maldonado et al. (2020) have developed a series of lactose-ferrocene conjugates to improve the sensitivity and reproducibility of hGal-3 detection. This design was based in initial works that employed ferrocene-sugar dendrimers and improved the sensing ability toward a given lectin by combining the multivalent presentation features with the multielectron exchange capability due to the presence of multiple ferrocene units (Martos-Maldonado et al., 2013a,b). In particular, four different ligands were designed targeting hGal-3, three dendrimers with different lactose content: 36 (4 lactose units), 37 (8), and 38 (16) and a monovalent ligand attached to gold nanoparticles, with more than 1,500 ligands per nanoparticle (Figure 14).
Figure 14.

The lactose-ferrocene conjugates targeting hGal-3.
ITC data demonstrated that the affinity was higher for the multivalent dendrimers and improved for that with the highest valency values (38). Intriguingly, although the gold nanoparticle analog 39 displayed smaller affinity toward hGal-3, its sensing capability was extraordinary, obtaining a detection limit of 160 nM. Overall, the three dendrimers displayed affinity values in the range of the most potent inhibitors previously developed, whereas the gold nanoparticle is a promising electrochemical probe that may enable the detection of hGal-3 at biologically relevant concentrations.
Conclusions
High affinity and selective antagonists have been designed, especially for hGal-1 and hGal-3, including various ligands that are in clinical trials, although none of them are in the market yet. The success of the mimetics designed on the basis of single presentation strategies relies on the fact that not only the canonical binding site of galectins has been targeted, but also interactions with neighboring regions have been exploited through the wise decoration of the sugar scaffold with specifically designed functional groups, using a canonical medicinal chemistry approach based on the continuous improvement of the binding enthalpy factor. The use of symmetric molecules, such as the TD139 and its pseudo-symmetric relatives, have demonstrated to be in the good pathway. Nevertheless, the control of the entropy factor, with the concomitant enthalpy-entropy compensation phenomenon, remains challenging and the next frontier, not only for the single presentation strategies but especially for the design of glycoconjugates with multiple epitope presentations. Except the chimera-type hGal-3, the other galectins are dimers whose sugar-binding sites are located at opposite spatial directions. Therefore, there is no obvious strategy for tackling both binding sites in a simultaneous manner. The proposed scheme for the best molecule should keep enough flexibility to reach the target binding sites and thus enhance avidity, but without paying a high entropic penalty. Usually, typical lactose or N-acetyllactosamine molecules have been used to decorate the multivalent scaffold, what implies that single-binding events are of moderate affinity. There is room for improvement at this point, using specific functional groups and probably also using strategies that minimize the entropy loss at the nanocarrier and/or at the interaction site. The approaches employed so far have proved to be satisfactory and are good starting points for further efforts. New biological roles for galectins are continuously being shown. Thus, the design, synthesis, and applications of galectin binders and sensors will continue being an active field of research for the years to come.
Author Contributions
AA and JJ-B developed the idea. SB, JQ, and AG wrote the review. AA, AG, and JJ-B revised the manuscript and gave it the final form. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. We thank the European Research Council (RECGLYCANMR, Advanced Grant no. 788143), ISCIII (Grant PRB3 IPT17/0019 to AG), and the Agencia Estatal de Investigación (Spain) for Grants RTI2018-094751-B-C21, Ramón y Cajal contract to AA and the Severo Ochoa Excellence Accreditation (SEV-2016-0644).
References
- Ardá A., Jiménez-Barbero J. (2018). The recognition of glycans by protein receptors. Insights from NMR spectroscopy. Chem. Commun. 54, 4761–4769. 10.1039/C8CC01444B [DOI] [PubMed] [Google Scholar]
- Berbís M. Á., André S., Cañada F. J., Pipkorn R., Ippel H., Mayo K. H., et al. (2014). Peptides derived from human galectin-3 N-terminal tail interact with its carbohydrate recognition domain in a phosphorylation-dependent manner. Biochem. Biophys. Res. Commun. 443, 126–131. 10.1016/j.bbrc.2013.11.063 [DOI] [PubMed] [Google Scholar]
- Bernardi S., Fezzardi P., Rispoli G., Sestito S. E., Peri F., Sansone F., et al. (2014). Clicked and long spaced galactosyl- and lactosylcalix[4]arenes: new multivalent galectin-3 ligands. Beilstein J. Org. Chem. 10, 1672–1680. 10.3762/bjoc.10.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian C. F., Zhang Y., Sun H., Li D. F., Wang D. C. (2011). Structural basis for distinct binding properties of the human galectins to Thomsen-Friedenreich antigen. PLoS ONE 6:e25007. 10.1371/journal.pone.0025007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmore P. F., Eisoldt S. (1999). The neoglycoprotein mannose-bovine serum albumin, but not progesterone, activates T-type calcium channels in human spermatozoa. Mol. Hum. Reprod. 5, 498–506. 10.1093/molehr/5.6.498 [DOI] [PubMed] [Google Scholar]
- Blanchard H., Bum-Erdene K., Bohari M. H., Yu X. (2016). Galectin-1 inhibitors and their potential therapeutic applications: a patent review. Expert Opin. Ther. Pat. 26, 537–554. 10.1517/13543776.2016.1163338 [DOI] [PubMed] [Google Scholar]
- Blanchard H., Yu X., Collins P. M., Bum-Erdene K. (2014). Galectin-3 inhibitors: a patent review (2008-present). Expert Opin. Ther. Pat. 24, 1053–1065. 10.1517/13543776.2014.947961 [DOI] [PubMed] [Google Scholar]
- Böcker S., Elling L. (2017). Biotinylated N-acetyllactosamine- and N,N-diacetyllactosamine-based oligosaccharides as novel ligands for human galectin-3. Bioengineering 4:31. 10.3390/bioengineering4020031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böcker S., Laaf D., Elling L. (2015). Galectin binding to neo-glycoproteins: LacDiNAc conjugated BSA as ligand for human galectin-3. Biomolecules 5, 1671–1696. 10.3390/biom5031671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden S., Reise F., Kania J., Lindhorst T. K., Hartmann L. (2019). Sequence-defined introduction of hydrophobic motifs and effects in lectin binding of precision glycomacromolecules. Macromol. Biosci. 19:e1800425. 10.1002/mabi.201800425 [DOI] [PubMed] [Google Scholar]
- Bojarová P., Tavares M. R., Laaf D., Bumba L., Petrásková L., Konefał R., et al. (2018). Biocompatible glyconanomaterials based on HPMA-copolymer for specific targeting of galectin-3. J. Nanobiotechnol. 16:73. 10.1186/s12951-018-0399-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinchmann M. F., Patel D. M., Iversen M. H. (2018). The role of galectins as modulators of metabolism and inflammation. Mediators Inflammation 2018, 1–11. 10.1155/2018/9186940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bücher K. S., Konietzny P. B., Snyder N. L., Hartmann L. (2019). Heteromultivalent glycooligomers as mimetics of blood group antigens. Chem. Eur. J. 25, 3301–3330. 10.1002/chem.201804505 [DOI] [PubMed] [Google Scholar]
- Cecioni S., Imberty A., Vidal S. (2015). Glycomimetics versus multivalent glycoconjugates for the design of high affinity lectin ligands. Chem. Rev. 115, 525–561. 10.1021/cr500303t [DOI] [PubMed] [Google Scholar]
- Chan Y. C., Lin H. Y., Tu Z., Kuo Y. H., Hsu S. T. D., Lin C. H. (2018). Dissecting the structure–activity relationship of galectin–ligand interactions. Int. J. Mol. Sci. 19:392. 10.3390/ijms19020392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittasupho C. (2012). Multivalent ligand: design principle for targeted therapeutic delivery approach. Ther. Deliv. 3, 1171–1187. 10.4155/tde.12.99 [DOI] [PubMed] [Google Scholar]
- Compain P. (2018). Glycomimetics: design, synthesis, and therapeutic applications. Molecules 23:1658. 10.3390/molecules23071658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlqvist A., Leffler H., Nilsson U. J. (2019). C1-galactopyranosyl heterocycle structure guides selectivity: triazoles prefer galectin-1 and oxazoles prefer galectin-3. ACS Omega 4, 7047–7053. 10.1021/acsomega.9b00320 [DOI] [Google Scholar]
- Delaine T., Collins P., MacKinnon A., Sharma G., Stegmayr J., Rajput V. K., et al. (2016). Galectin-3-binding glycomimetics that strongly reduce bleomycin-induced lung fibrosis and modulate intracellular glycan recognition. ChemBioChem 17, 1759–1770. 10.1002/cbic.201600285 [DOI] [PubMed] [Google Scholar]
- Diehl C., Engström O., Delaine T., Håkansson M., Genheden S., Modig K., et al. (2010). Protein flexibility and conformational entropy in ligand design targeting the carbohydrate recognition domain of galectin-3. J. Am. Chem. Soc. 132, 14577–14589. 10.1021/ja105852y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dings R. P. M., Arroyo M. M., Lockwood N. A., Van Eijk L. I., Haseman J. R., Griffioen A. W., et al. (2003). β-sheet is the bioactive conformation of the anti-angiogenic anginex peptide. Biochem. J. 373, 281–288. 10.1042/bj20030295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dings R. P. M., Chen X., Hellebrekers D. M. E. I., Van Eijk L. I., Zhang Y., Hoye T. R., et al. (2006). Design of nonpeptidic topomimetics of antiangiogenic proteins with antitumor activities. J. Natl. Cancer Inst. 98, 932–936. 10.1093/jnci/djj247 [DOI] [PubMed] [Google Scholar]
- Dings R. P. M., Kumar N., Miller M. C., Loren M., Rangwala H., Hoye T. R., et al. (2013). Structure-based optimization of angiostatic agent 6DBF7, an allosteric antagonist of galectin-1. J. Pharmacol. Exp. Therap. 344, 589–599. 10.1124/jpet.112.199646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dings R. P. M., Laar E. S. V., Webber J., Zhang Y., Griffin R. J., Waters S. J., et al. (2008). Ovarian tumor growth regression using a combination of vascular targeting agents anginex or topomimetic 0118 and the chemotherapeutic irofulven. Cancer Lett. 265, 270–280. 10.1016/j.canlet.2008.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dings R. P. M., Miller M. C., Griffin R. J., Mayo K. H. (2018). Galectins as molecular targets for therapeutic intervention. Int. J. Mol. Sci. 19:905. 10.3390/ijms19030905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortuna-Costa A., Gomes A. M., Kozlowski E. O., Stelling M. P., Pavão M. S. G. (2014). Extracellular galectin-3 in tumor progression and metastasis. Front. Oncol. 4:138. 10.3389/fonc.2014.00138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno A., Delgado S., Valverde P., Bertuzzi S., Berbís M. A., Echavarren J., et al. (2019). Minimizing the entropy penalty for ligand binding: lessons from the molecular recognition of the histo blood-group antigens by human galectin-3. Angew. Chem. Int. Ed. 8, 7268–7272. 10.1002/anie.201900723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno A., Valverde P., Ardá A., Jiménez-Barbero J. (2020). Glycan structures and their interactions with proteins. A NMR view. Curr. Opin. Struct. Biol. 62, 22–20. 10.1016/j.sbi.2019.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard A., Magnani J. L. (2018). Clinical trials and applications of galectin antagonists. Trends Glycosci. Glycotechnol. 30, SE211–SE220. 10.4052/tigg.1744.1SE [DOI] [Google Scholar]
- Girotti M. R., Salatino M., Dalotto-Moreno T., Rabinovich G. A. (2020). Sweetening the hallmarks of cancer: galectins as multifunctional mediators of tumor progression. J. Exp. Med. 217, 2018–2041. 10.1084/jem.20182041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinsky V. V., Glinsky G. V., Rittenhouse-Olson K., Huflejt M. E., Glinskii O. V., Deutscher S. L., et al. (2001). The role of Thomsen-Friedenreich antigen in adhesion of human breast and prostate cancer cells to the endothelium. Cancer Res. 61, 4851–4857. [PubMed] [Google Scholar]
- Goletz S., Cao Y., Danielczyk A., Ravn P., Schoeber U., Karsten U. (2002). Thomsen-friedenreich antigen: the hidden tumor antigen. Adv. Exp. Med. Biol. 535, 147–162. 10.1007/978-1-4615-0065-0_10 [DOI] [PubMed] [Google Scholar]
- Gouin S. G., García-Fernández J. M., Vanquelef E., Dupradeau F. Y., Salomonsson E., Leffer H., et al. (2010). Multimeric lactoside “click clusters” as tools to investigate the effect of linker length in specific interactions with peanut lectin, galectin-1, and−3. ChemBioChem 11, 1430–1442. 10.1002/cbic.201000167 [DOI] [PubMed] [Google Scholar]
- Gouin S. G., Vanquelef E., García-Fernández J. M., Ortiz-Mellet C., Dupradeau F. Y., Kovensky J. (2007). Multi-mannosides based on a carbohydrate scaffold: synthesis, force field development, molecular dynamics studies, and binding affinities for lectin con A. J. Org. Chem. 72, 9032–9045. 10.1021/jo071248a [DOI] [PubMed] [Google Scholar]
- Griffioen A. W. (2006). Functionally Active Recombinant Peptides, Methods for Producing Same and Interactions With Other Peptides. WO Patent No 2006128027A1. Saint Paul, MN: Peptx Inc. [Google Scholar]
- Griffioen A. W., Van der Schaft D. W. J., Barendsz-Janson A. F., Cox A., Struijker Boudier H. A. J., Hillen H. F. P., et al. (2001). Anginex, a designed peptide that inhibits angiogenesis. Biochem. J. 354, 233–242. 10.1042/bj3540233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grischenko L. A., Parshina L. N., Kanitskaya L. V., Larina L. I., Novikova L. N., Trofimov B. A. (2013). Propargylation of arabinogalactan with propargyl halides–a facile route to new functionalized biopolymers. Carbohydr. Chem. 376, 7–14. 10.1016/j.carres.2013.04.031 [DOI] [PubMed] [Google Scholar]
- Hevey R. (2019). Strategies for the development of glycomimetic drug candidates. Pharmaceuticals 12:55. 10.3390/ph12020055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh T. J., Lin H. Y., Tu Z., Lin T. C., Wu S. C., Tseng Y. Y., et al. (2016). Dual thio-digalactoside-binding modes of human galectins as the structural basis for the design of potent and selective inhibitors. Sci. Rep. 6:29457. 10.1038/srep29457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B. X., Kim H. Y., Dass C. (2004). Probing three-dimensional structure of bovine serum albumin by chemical cross-linking and mass spectrometry. J. Am. Soc. Mass Spectrom. 15, 1237–1247. 10.1016/j.jasms.2004.05.004 [DOI] [PubMed] [Google Scholar]
- Jeschke U., Karsten U., Wiest I., Schulze S., Kuhn C., Friese K., et al. (2006). Binding of galectin-1 (gal-1) to the Thomsen-Friedenreich (TF) antigen on trophoblast cells and inhibition of proliferation of trophoblast tumor cells in vitro by gal-1 or an anti-TF antibody. Histochem. Cell Biol. 126, 437–444. 10.1007/s00418-006-0178-1 [DOI] [PubMed] [Google Scholar]
- Jiménez-Barbero J., Dragoni E., Venturis C., Nannucci F., Ardá A., Fontanella M., et al. (2009). α-O-linked glycopeptide mimetics: synthesis, conformation analysis, and interactions with viscumin, a galactoside-binding model lectin. Chem. Eur. J. 15, 10423–10431. 10.1002/chem.200901077 [DOI] [PubMed] [Google Scholar]
- Johannes L., Jacob R., Leffler H. (2018). Galectins at a glance. J. Cell Sci. 131:jcs208884. 10.1242/jcs.208884 [DOI] [PubMed] [Google Scholar]
- Karaman R. (2010). The effective molarity (EM)–a computational approach. Bioorg. Chem. 38, 165–172. 10.1016/j.bioorg.2010.04.002 [DOI] [PubMed] [Google Scholar]
- Kieber-Emmons T., Saha S., Pashov A., Monzavi-Karbassi B., Murali R. (2014). Carbohydrate-mimetic peptides for pan anti-tumor responses. Front. Immunol. 5:308. 10.3389/fimmu.2014.00308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonce N. A., Griffin R. J., Dings R. P. M. (2017). Galectin-1 inhibitor OTX008 induces tumor vessel normalization and tumor growth inhibition in human head and neck squamous cell carcinoma models. Int. J. Mol. Sci. 18:2671. 10.3390/ijms18122671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupper C. E., Böcker S., Liu H. L., Adamzyk C., Van de Kamp J., Recker T., et al. (2013). Fluorescent SNAP-Tag galectin fusion proteins as novel tools in glycobiology. Curr. Pharm. Des. 19, 5457–5467. 10.2174/1381612811319300017 [DOI] [PubMed] [Google Scholar]
- Leffler H., Carlsson S., Hedlund M., Qian Y., Poirier F. (2002). Introduction to galectins. Glycoconj. J. 19, 433–440. 10.1023/B:GLYC.0000014072.34840.04 [DOI] [PubMed] [Google Scholar]
- Liu F. T., Rabinovich G. A. (2005). Galectins as modulators of tumour progression. Nat. Rev. Cancer 5, 29–41. 10.1038/nrc1527 [DOI] [PubMed] [Google Scholar]
- Luyai A., Lasanajak Y., Smith D. F., Cummings R. D., Song X. (2009). Facile preparation of fluorescent neoglycoproteins using p-nitrophenyl anthranilate as a heterobifunctional linker. Bioconjug. Chem. 20, 1618–1624. 10.1021/bc900189h [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon A. C., Gibbons M. A., Farnworth S. L., Leffler H., Nilsson U. J., Delaine T., et al. (2012). Regulation of transforming growth factor-β1-driven lung fibrosis by galectin-3. Am. J. Respir. Crit. Care Med. 185, 537–546. 10.1164/rccm.201106-0965OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani J. L., Ernst B. (2009). Glycomimetic drugs - a new source of therapeutic opportunities. Discov. Med. 8, 247–252. [PubMed] [Google Scholar]
- Martos-Maldonado M. C., Casas-Solvas J. M., Quesada-Soriano I., García-Fuentes L., Vargas-Berenguel A. (2013b). Poly (amido amine)-based mannose-glycodendrimers as multielectron redox probes for improving lectin sensing. Langmuir 29, 1318–1326. 10.1021/la304107a [DOI] [PubMed] [Google Scholar]
- Martos-Maldonado M. C., Quesada-Soriano I., García-Fuentes L., Vargas-Berenguel A. (2020). Multivalent lactose-ferrocene conjugates base don poly (amido amine) dendrimers and gold nanoparticles as electrochemical probes for sensing galectin-3. Nanomaterials 10:203 10.3390/nano10020203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martos-Maldonado M. C., Thygesen M. B., Jensen K. J., Vargas-Berenguel A. (2013a). Glyco-nanoparticles for high-sensitivity electrochemical detection of carbohydrate-lectin interactions. Eur. J. Org. Chem. 2013, 7256–7260. 10.1002/ejoc.201390036 [DOI] [Google Scholar]
- Mayo K. H. (2014). Anti-Tumor Agent otx-008 Targets Human Galectin-1. WO Patent No 2014070214A1. Minneapolis, MN: University of Minnesota. [Google Scholar]
- Mayo K. H., Dings R. P. M., Flader C., Nesmelova I., Hargittai B., Van Der Schaft D. W. J., et al. (2003). Design of a partial peptide mimetic of anginex with antiangiogenic and anticancer activity. J. Bio. Chem. 278, 45746–45752. 10.1074/jbc.M308608200 [DOI] [PubMed] [Google Scholar]
- Mayo K. H., Hoye T. F., Flader-Lavey C. (2008). Partial Peptide Mimetics and Methods. US Patent No 8716343B2. Minneapolis, MN: Reagents of the University of Minnesota. [Google Scholar]
- Mayo K. H., Van Der Schaft D. W. J., Griffioen A. W. (2001). Designed β-sheet peptides that inhibit proliferation and induce apoptosis in endothelial cells. Angiogenesis 4, 45–51. 10.1023/A:1016672117477 [DOI] [PubMed] [Google Scholar]
- McCoy J. P., Varani J., Goldstein I. J. (1984). Enzyme-linked lectin assay (ELLA): II. Detection of carbohydrate groups on the surface of unfixed cells. Exp. Cell Res. 151, 96–103. 10.1016/0014-4827(84)90359-8 [DOI] [PubMed] [Google Scholar]
- Miller M. C., Ippel H., Suylen D., Klyosov A. A., Traber P. G., Hackeng T., et al. (2016). Binding of polysaccharides to human galectin-3 at a noncanonical site in its carbohydrate recognition domain. Glycobiology 26, 88–99. 10.1093/glycob/cwv073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y., Akita K., Yashiro M., Sawada T., Hirakawa K., Murata T., et al. (2015). Binding of galectin-3, a β-galactoside-binding lectin, to MUC1 protein enhances phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2) and akt, promoting tumor cell malignancy. J. Bio. Chem. 290, 26125–26140. 10.1074/jbc.M115.651489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton-Northup J. R., Dickerson M. T., Ma L., Besch-Williford C. L., Deutscher S. L. (2013). Inhibition of metastatic tumor formation in vivo by a bacteriophage display-derived galectin-3 targeting peptide. Clin. Exp. Metastasis. 30, 119–132. 10.1007/s10585-012-9516-y [DOI] [PubMed] [Google Scholar]
- Nilsson U., Leffler H., Mukhopadhyay B., Rajput V. (2013). Novel Galactoside Inhibitors of Galectins. WO Patent No 2013110704. Copenhagen, DK: Galecto Biotech Ab. [Google Scholar]
- Peterson K., Kumar R., Stenström O., Verma P., Verma P. R., Håkansson M., et al. (2018). Systematic tuning of fluoro-galectin-3 interactions provides thiodigalactoside derivatives with single-digit nM affinity and high selectivity. J. Med. Chem. 61, 1164–1175. 10.1021/acs.jmedchem.7b01626 [DOI] [PubMed] [Google Scholar]
- Rahkila J., Ekholm F. S., Ardá A., Delgado S., Savolainen J., Jiménez-Barbero J., et al. (2019). Novel dextran-supported biological probes decorated with disaccharide entities for investigating the carbohydrate-protein interactions of gal-3. ChemBioChem 20, 203–209. 10.1002/cbic.201800423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahkila J., Ekholm F. S., Leino R. (2018). CuI-mediated degradation of polysaccharides leads to fragments with narrow polydispersities. Eur. J. Org. Chem. 20, 1449–1454. 10.1002/ejoc.201800033 [DOI] [Google Scholar]
- Rapoport E. M., Andre S., Kurmyshkina O. V., Pochechueva T. V., Severov V. V., Pazynina G. V., et al. (2008). Galectin-loaded cells as a platform for the profiling of lectin specificity by fluorescent neoglycoconjugates: a case study on galectins-1 and-3 and the impact of assay setting. Glycobiology 18, 315–324. 10.1093/glycob/cwn009 [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V., Green L. G., Fokin V. V., Sharpless K. B. (2002). A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “Ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 41:2596. 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4 [DOI] [PubMed] [Google Scholar]
- Salameh B. A., Cumpstey I., Sundin A., Leffler H., Nilsson U. J. (2010). 1H-1,2,3-triazol-1-yl thiodigalactoside derivatives as high affinity galectin-3 inhibitors. Bioorg. Med. Chem. 8, 5367–5378. 10.1016/j.bmc.2010.05.040 [DOI] [PubMed] [Google Scholar]
- Salomonsson E., Thijssen V. L., Griffioen A. W., Nilsson U. J., Leffler H. (2011). The anti-angiogenic peptide anginex greatly enhances galectin-1 binding affinity for glycoproteins. J. Bio. Chem. 286, 13801–13804. 10.1074/jbc.C111.229096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarsia S., Grosso A. S., Trovão F., Jiménez-Barbero J., Carvalho A. L., Nativi C., et al. (2018). Molecular recognition of a thomsen–friedenreich antigen mimetic targeting human galectin-3. ChemMedChem 13, 2030–2036. 10.1002/cmdc.201800525 [DOI] [PubMed] [Google Scholar]
- Shewmake T. A., Solis F. J., Gillies R. J., Caplan M. R. (2008). Effects of linker length and flexibility on multivalent targeting. Biomacromolecules 9, 3057–3064. 10.1021/bm800529b [DOI] [PubMed] [Google Scholar]
- Sörme P., Kahl-Knutsson B., Huflejt M., Nilsson U. J., Leffler H. (2004). Fluorescence polarization as an analytical tool to evaluate galectin-ligand interactions. Anal. Biochem. 334, 36–47. 10.1016/j.ab.2004.06.042 [DOI] [PubMed] [Google Scholar]
- Sörme P., Kahl-Knutsson B., Wellmar U., Magnusson B. G., Leffler H., Nilsson U. J. (2003). Design and synthesis of galectin inhibitors. Meth. Enzymol. 363, 157–169. 10.1016/S0076-6879(03)01050-4 [DOI] [PubMed] [Google Scholar]
- Springer G. F. (1997). Immunoreactive T and Tn epitopes in cancer diagnosis, prognosis, and immunotherapy. J. Mol. Med. 75, 594–602. 10.1007/s001090050144 [DOI] [PubMed] [Google Scholar]
- Stegmayr J., Zetterberg F., Carlsson M. C., Huang X., Sharma G., Kahl-Knutson B., et al. (2019). Extracellular and intracellular small-molecule galectin-3 inhibitors. Sci. Rep. 9:2186. 10.1038/s41598-019-38497-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowell S. R., Dias-Baruffi M., Penttila L., Renkonen O., Nyame A. K., Cummings R. D. (2004). Human galectin-1 recognition of poly-N-acetyllactosamine and chimeric polysaccharides. Glycobiology 14, 157–167. 10.1093/glycob/cwh018 [DOI] [PubMed] [Google Scholar]
- Takenaka Y., Fukumori T., Raz A. (2002). Galectin-3 and metastasis. Glycoconj. J. 19, 543–549. 10.1023/B:GLYC.0000014084.01324.15 [DOI] [PubMed] [Google Scholar]
- Tamburrini A., Colombo C., Bernardi A. (2019). Design and synthesis of glycomimetics: recent advances. Med. Res. Rev. 40, 495–531. 10.1002/med.21625 [DOI] [PubMed] [Google Scholar]
- Tavares M. R., Bláhová M., Sedláková L., Elling L., Pelantová H., Konefal R., et al. (2020). High-affinity N-(2-hydroxylpropyl)methacrylamide copolymers with tailored N-acetyllactosamine presentation discriminate berween galectins. Biomacromolecules 21, 641–652. 10.1021/acs.biomac.9b01370 [DOI] [PubMed] [Google Scholar]
- Tellez-Sanz R., Garcia-Fuentes L., Vargas-Berenguel A. (2013). Human galectin-3 selective and high affinity inhibitors. Present state and future perspectives. Curr. Med. Chem. 20, 2979–2990. 10.2174/09298673113209990163 [DOI] [PubMed] [Google Scholar]
- Thijssen V. L. J. L., Postel R., Brandwijk R. J. M. G. E., Dings R. P. M., Nesmelova I., Satijn S., et al. (2006). Galectin-1 is essential in tumor angiogenesis and is a target for antiangiogenesis therapy. Proc. Natl. Acad. Sci. U.S.A. 103, 15975–15980. 10.1073/pnas.0603883103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrich K., Šubr V. (2010). Structural and chemical aspects of HPMA copolymers as drug carriers. Adv. Drug Deliv. Rev. 62, 150–166. 10.1016/j.addr.2009.10.007 [DOI] [PubMed] [Google Scholar]
- Valverde P., Ardá A., Reichardt N. C., Jiménez-Barbero J., Gimeno A. (2019a). Glycans in drug discovery. MedChemComm 10, 1678–1691. 10.1039/C9MD00292H [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde P., Quintana J. I., Santos J. I., Ardá A., Jiménez-Barbero J. (2019b). Novel NMR avanues to explore the conformation and interactions of glycans. ACS Omega 4, 13618–13630. 10.1021/acsomega.9b01901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berg T. K., Honing H., Franke N., Van Remoortere A., Schiphorst W., Liu F. T., et al. (2004). Lacdinac-glycans constitute a parasite pattern for galectin-3-mediated immune recognition. J. Immunol. 173, 1902–1907. 10.4049/jimmunol.173.3.1902 [DOI] [PubMed] [Google Scholar]
- Van Hattum H., Branderhorst H. M., Moret E. E., Nilsson U. J., Leffler H., Pieters R. J. (2013). Tuning the preference of thiodigalactoside- and lactosamine-based ligands to galectin-3 over galectin-1. J. Med. Chem. 56, 1350–1354. 10.1021/jm301677r [DOI] [PubMed] [Google Scholar]
- Vasta G. R. (2009). Roles of galectins in infection. Nat. Rev. Microbiol. 7, 424–438. 10.1038/nrmicro2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. B., De Wang M., Li E. X., Dong D. F. (2012). Advances and prospects of anginex as a promising anti-angiogenesis and anti-tumor agent. Peptides 38, 457–462. 10.1016/j.peptides.2012.09.007 [DOI] [PubMed] [Google Scholar]
- Weng I. C., Chen H. L., Lo T. H., Lin W. H., Chen H. Y., Hsu D. K., et al. (2018). Cytosolic galectin-3 and−8 regulate antibacterial autophagy through differential recognition of host glycans on damaged phagosomes. Glycobiology 28, 392–405. 10.1093/glycob/cwy017 [DOI] [PubMed] [Google Scholar]
- Yang R. Y., Rabinovich G. A., Liu F. T. (2008). Galectins: structure, function and therapeutic potential. Expert Rev. Mol. Med. 13, 10–17. 10.1017/S1462399408000719 [DOI] [PubMed] [Google Scholar]
- Yang Y., Li L., Zhou Z., Yang Q., Liu C., Huang Y. (2014). Targeting prostate carcinoma by G3-C12 peptide conjugated N-(2-hydroxypropyl)methacrylamide copolymers. Mol. Pharm. 11, 3251–3260. 10.1021/mp500083u [DOI] [PubMed] [Google Scholar]
- Yongye A. B., Calle L., Ardá A., Jiménez-Barbero J., André S., Gabius H. J., et al. (2012). Molecular recognition of the Thomsen-Friedenreich antigen-threonine conjugate by adhesion/growth regulatory galectin-3: nuclear magnetic resonance studies and molecular dynamics simulations. Biochemistry 51, 7278–7289. 10.1021/bi300761s [DOI] [PubMed] [Google Scholar]
- Zetterberg F. R., Peterson K., Johnsson R. E., Brimert T., Håkansson M., Logan D. T., et al. (2018). Monosaccharide derivatives with low-nanomolar lectin affinity and high selectivity based on combined fluorine–amide, phenyl–arginine, sulfur–π, and halogen bond interactions. ChemMedChem. 3, 133–137. 10.1002/cmdc.201700744 [DOI] [PubMed] [Google Scholar]
- Zhang X., Yao W., Xu X., Sun H., Zhao J., Meng X., et al. (2018). Synthesis of fucosylated chondroitin sulfate glycoclusters: a robust route to new anticoagulant agents. Chem. Eur. J. 24, 1694–1700. 10.1002/chem.201705177 [DOI] [PubMed] [Google Scholar]
- Zou J., Glinsky V. V., Landon L. A., Matthews L., Deutscher S. L. (2005). Peptides specific to the galectin-3 carbohydrate recognition domain inhibit metastasis-associated cancer cell adhesion. Carcinogenesis 26, 309–318. 10.1093/carcin/bgh329 [DOI] [PubMed] [Google Scholar]