

Abstract
The atmosphere is composed of nitrogen, oxygen and argon, a variety of trace gases, and particles or aerosols from a variety of sources. Reactive, trace gases have short mean residence time in the atmosphere and large spatial and temporal variations in concentration. Many trace gases are removed by reaction with hydroxyl radical and deposition in rainfall or dryfall at the Earth's surface. The upper atmosphere, the stratosphere, contains ozone that screens ultraviolet light from the Earth's surface. Chlorofluorocarbons released by humans lead to the loss of stratospheric ozone, which might eventually render the Earth's land surface uninhabitable. Changes in the composition of the atmosphere, especially rising concentrations of CO2, CH4, and N2O, will lead to climatic changes over much of the Earth's surface.
Keywords: Acid rain, Aerosols, Climate change, Atmospheric deposition, Dryfall, Hydroxyl radical, Oxygen, Ozone, Ozone hole, Troposphere, Stratosphere, Wetfall
Introduction
There are several reasons to begin our treatment of biogeochemistry with a consideration of the atmosphere. The composition of the atmosphere has evolved as a result of the history of life on Earth (Chapter 2), and is now changing rapidly as a result of human activities. The atmosphere controls Earth's climate and ultimately determines the conditions in which we live—our supplies of food and water, our health, and our economy. Further, the atmosphere is relatively well mixed, so changes in its composition can be taken as a first index of changes in biogeochemical processes at the global level. The circulation of the atmosphere transports biogeochemical constituents between the oceans and land, contributing to the global cycles of chemical elements.
We begin our discussion with a brief consideration of the structure, circulation, and composition of the atmosphere. Then we examine reactions that occur among various gases, especially in the lower atmosphere. Many of these reactions remove constituents from the atmosphere, depositing them on the surface of the land and sea. In the face of constant losses, the composition of the atmosphere is maintained by biotic processes that supply gases to the atmosphere. We mention the sources of atmospheric gases here briefly, but they will be treated in more detail in later chapters of this book, especially as we examine the microbial reactions that occur in soils, wetlands, and ocean sediments. Finally, we discuss human impacts on the global atmosphere, as seen in ozone depletion and climate change.
Structure and circulation
The atmosphere is held on Earth's surface by the gravitational attraction of the Earth. At any altitude, the downward force (F) is related to the mass (M) of the atmosphere above that point:
| (3.1) |
where g is the acceleration due to gravity (980 cm/s2 at sea level). Pressure (force per unit area) decreases with increasing altitude because the mass of the overlying atmosphere is smaller (Walker, 1977). Decline in atmospheric pressure (P in bars) with altitude (A in km) is approximated by the logarithmic relation:
| (3.2) |
over the whole atmosphere (Fig. 3.1 ).
Fig. 3.1.
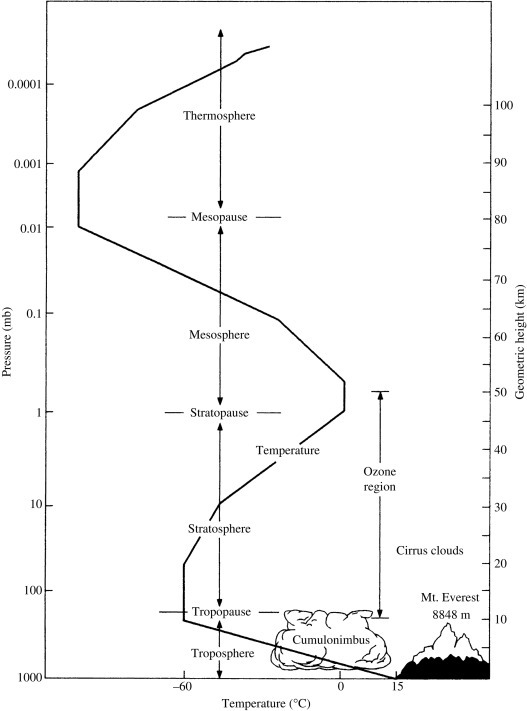
Vertical structure and zonation of the atmosphere, showing the temperature profile to 100-km altitude. Note the logarithmic decline in pressure (left axis) as a function of altitude.
Although the chemical composition of the atmosphere is relatively uniform, when we visit high mountains, we often say that the atmosphere seems “thinner” than at sea level. The composition is the same, but the abundance of molecules in each volume of the atmosphere is greater at sea level, because it is compressed by the pressure of the overlying atmosphere. The lower atmosphere, the troposphere, contains about 80% of the atmospheric mass (Warneck, 2000). The lower density of the upper atmosphere is the reason that jet aircraft flying at high altitudes require cabin pressurization for their passengers.
Certain atmospheric constituents, such as ozone, aerosols, and clouds absorb and reflect portions of the radiation that the Earth receives from the Sun, so only about half of the Sun's radiation penetrates the atmosphere to be absorbed or reflected by the Earth's surface (Fig. 3.2 ). The overall reflectivity or albedo of the Earth, as measured by the CERES satellite (Clouds and the Earth's Radiant Energy System) and by “earthshine” received by the Moon, is about 29% (Goode et al., 2001; Kim and Ramanathan, 2012). Albedo is affected by the reflectivity of the Earth's surface, which differs among sea water, forests and ice cover. Particles in the atmosphere absorb or reflect about 5–10% of the incoming solar radiation. Although some regions with air pollution have greater albedo than pristine areas, the Earth's overall albedo has not changed appreciably during the past couple of decades (Palle et al., 2016). Greater albedo reduces the radiation reaching Earth's surface and leads to “global dimming.”
Fig. 3.2.
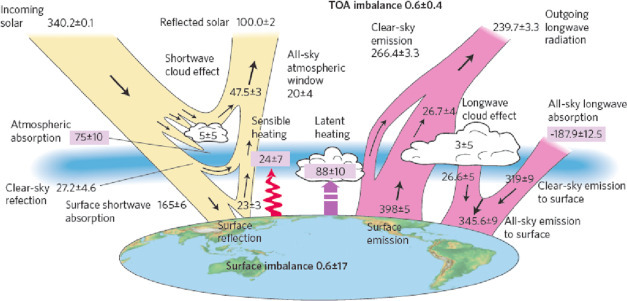
The radiation budget for Earth, showing the proportional fate of the energy that the Earth receives from the Sun, about 340 W/m2 largely in short wavelengths. About one third of this radiation is reflected back to space and the remainder is absorbed by the atmosphere (23%) or the surface (46%). Long-wave radiation (infra-red) is emitted from the Earth's surface, some of which is absorbed by atmospheric gases, warming the atmosphere—the greenhouse effect. The atmosphere emits long-wave radiation, so that the total energy received is balanced by the total energy emitted from the planet.
Source: Stephens et al. (2012).
The land and ocean surfaces reradiate long wave (heat) radiation to the atmosphere, so the atmosphere is heated from the bottom and is warmest at the Earth's surface (Fig. 3.1). Because warm air is less dense and rises, the troposphere is well mixed. The top of the troposphere extends to 8–17 km, varying seasonally and with latitude. The temperature of the upper troposphere is about − 60 °C, which ensures that the atmosphere above 10 km contains only small amounts of water vapor and ice particles, especially over Antarctica.
Above the troposphere, the stratosphere is defined by the zone in which temperatures increase with altitude, extending to about 50 km (Fig. 3.1). The increase is largely due to the absorption of ultraviolet light by ozone. Vertical mixing in the stratosphere is limited, as is exchange across the boundary between the troposphere and the stratosphere, the tropopause. Thus, materials that enter the stratosphere remain there for long periods, allowing for high altitude transport around the globe.
The thermal mixing of the troposphere is largely responsible for the global circulation of the atmosphere, as well as local weather patterns (Fig. 3.3 ). The large annual receipt of solar energy at the equator causes warming of the atmosphere (sensible heat) and the evaporation of large amounts of water, carrying latent heat, from tropical oceans and rainforests. As this warm, moist air rises, it cools, producing a large amount of precipitation in equatorial regions. Having lost its moisture, the rising air masses are deflected towards the poles by the Coriolus forces. In a belt centered on approximately 30°N or S latitude, these dry air masses sink to the Earth's surface, undergoing compressional heating. Most of the world's major deserts are associated with the downward movement of hot, dry air at this latitude. These circulation patterns of rising, cooling air at the equator and sinking, warming air at 30°N and S latitude are known as Hadley Cells. A similar, but much weaker, circulation pattern is found at the poles, where cold air sinks and moves north or south along the Earth's surface to lower latitudes. Frictional drag between the polar cells and Hadley cells drives an indirect circulation in each hemisphere between 40° and 60° latitude, producing regional storm systems and the prevailing west winds that we experience in the temperate zone.a
Fig. 3.3.
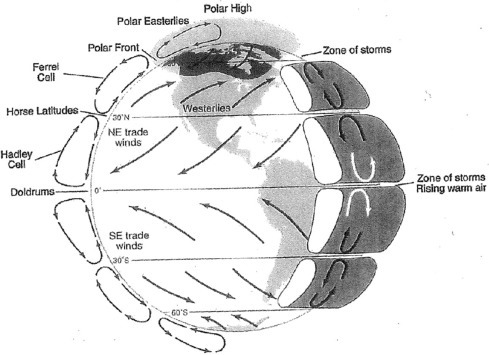
Generalized pattern of global circulation showing surface patters, vertical patterns, and the origin of the Coriolis force. As air masses move across different latitudes, they are deflected by the Coriolis force, which arises because of the different speeds of the Earth's rotation at different latitudes. For instance, if you were riding on an air mass moving at a constant speed south from 30°N latitude, you would begin your journey seeing 1446 km of the Earth's surface pass to the east every hour. By the time your air mass reached the equator, 1670 km would be passing to the east each hour. While moving south at a constant velocity, you would find that you had traveled 214 km west of your expected trajectory. The Coriolis force means that all movements of air in the northern hemisphere are deflected to the right; those in the southern hemisphere are deflected to the left.
Modified from Berner and Berner (2012).
The tropospheric air in each hemisphere mixes on a time scale of a few months (Warneck, 2000), allowing for regional transport of air pollutants that persist for more than a few days. For instance, in 1995 carbon monoxide (CO) from Canadian forest fires contributed to air pollutant loads in the eastern United States (Wotawa and Trainer, 2000). The eruption of the Eyjafjallajökull volcano in Iceland on 13–14 April 2010 produced a cloud of volcanic ash over Poland several days later (Pietruczuk et al., 2010; Langmann et al., 2012) and disrupted airplane travel over much of Europe for several weeks. Vertical mixing in the troposphere is driven by convection, especially in thunderstorms, so that much of the air in the upper troposphere is less than a week old (Brunner et al., 1998; Bertram et al., 2007). Each year, there is also complete mixing of tropospheric air between the Northern and the Southern Hemispheres across the intertropical convergence zone (ITCZ). If a gas shows a higher concentration in one hemisphere, we can infer that a large natural or human source must exist in that hemisphere, overwhelming the tendency for atmospheric mixing to equalize the concentrations (Fig. 3.4 ).
Fig. 3.4.
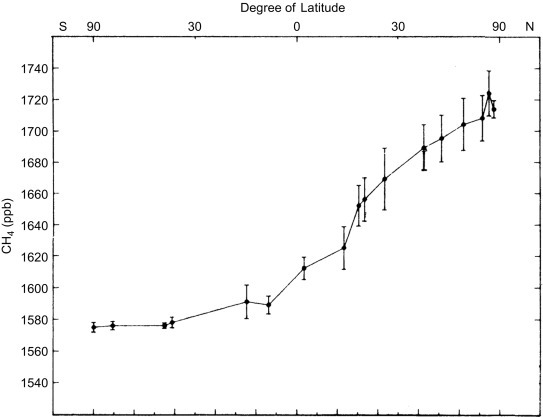
The latitudinal variation in the mean concentration of methane (CH4) in Earth's atmosphere.
From Steele et al. (1987). Used with permission of Reidel Publishing.
Exchange between the troposphere and the stratosphere is driven by several processes (Warneck, 2000). In the tropical Hadley cells, rising air masses carry some tropospheric air to the stratosphere (Holton et al., 1995; Fueglistaler et al., 2004). The strength of the updraft varies seasonally, as a result of variations in the radiation received from the Sun. When the height of the tropopause drops, tropospheric air is trapped in the stratosphere, or vice versa. There is also exchange across the tropopause due to large-scale wind movements, thunderstorms, and eddy diffusion.
Atmospheric scientists have examined the exchange of air mass between the troposphere and the stratosphere by following the fate of industrial pollutants released to the troposphere and radioactive contaminants released to the stratosphere in tests of atomic weapons during the 1950s and early 1960s (Warneck, 2000). In these considerations, the concept of mean residence time is useful. For any reservoir that is in steady state, mean residence time (MRT) is defined as:
| (3.3) |
where flux may be either the input or the loss from the reservoir.b Since the stratosphere is not well mixed vertically, the mean residence time of stratospheric air increases with altitude (Waugh and Hall, 2002). However, the return of stratospheric air to the troposphere, about 4 × 1017 kg/yr (Seo and Bowman, 2002), amounts to about 40% of the stratospheric mass each year, leading to an overall mean residence time of 2.6 years for stratospheric air. Thus, when a large volcano injects sulfur dioxide into the stratosphere, about half of it will remain after 2 years and about 5% will remain after 7.5 years.
Atmospheric composition
Gases
Table 3.1 gives the globally averaged concentration of some important gases in the atmosphere. Three gases—nitrogen, oxygen, and argon—make up 99% of the atmospheric mass of 5.14 × 1021 g (Trenberth and Guillemot, 1994). The mean residence times of these gases are much longer than the rate of atmospheric mixing. Because of their long residence times, the concentrations of N2, O2, and all noble gases (He, Ne, Ar, Kr, and Xe) are globally uniform and time-invariant.
Table 3.1.
Global average concentration of well mixed atmospheric constituents.a
| Compounds | Formula | Concentration | Total mass (g) |
|---|---|---|---|
| Major constituents (%) | |||
| Nitrogen | N2 | 78.084 | 3.87 × 1021 |
| Oxygen | O2 | 20.946 | 1.19 × 1021 |
| Argon | Ar | 0.934 | 6.59 × 1019 |
| Parts-per-million constituents (ppm = 10− 6or μL/L) | |||
| Carbon dioxide | CO2 | 400 | 3.11 × 1018 |
| Neon | Ne | 18.2 | 6.49 × 1016 |
| Helium | He | 5.24 | 3.70 × 1015 |
| Methane | CH4 | 1.83 | 5.19 × 1015 |
| Krypton | Kr | 1.14 | 1.69 × 1016 |
| Parts-per-billion constituents (ppb = 10− 9or nL/L) | |||
| Hydrogen | H2 | 510 | 1.82 × 1014 |
| Nitrous oxide | N2O | 320 | 2.49 × 1013 |
| Xenon | Xe | 87 | 2.02 × 1015 |
| Parts-per-trillion constituents (ppt = 10− 12) | |||
| Carbonyl sulfide | COS | 500 | 5.30 × 1012 |
| Chlorofluorocarbons | |||
| CFC 11 | CCl3F | 280 | 6.79 × 1012 |
| CFC 12 | CCl2F2 | 550 | 3.12 × 1013 |
| Methylchloride | CH3Cl | 620 | 5.53 × 1012 |
| Methylbromide | CH3Br | 11 | 1.84 × 1011 |
Those with a mean residence time > 1 year. Assuming a dry atmosphere with a molecular weight of 28.97, the overall mass of the atmosphere sums to 514 × 1019 g.
Source: Updated from Trenberth and Guillemot (1994).
Several hundred trace gases, including a wide variety of volatile organic compounds (VOCs), are also found in the atmosphere. The most abundant volatile organic compound from vegetation is isoprene, which is commonly emitted from many coniferous forest species (Guenther et al., 2000). For comparison, Table 3.2 shows the volatile emission of organic compounds from representative species in various world biomes. The various volatile organic compounds (VOCs) derived from vegetation include nonmethane hydrocarbons (NMHC) and oxygenated organic molecules, such as methanol (Park et al., 2013). Human activities also add a wide variety of trace organic gases to the atmosphere, including ethane and oxygenated organic gases, such as acetone and alcohols (Piccot et al., 1992; Huang et al., 2015). In urban areas, organic gases are released from a variety of products, including paints, pesticides, cleaning agents, and personal care products (McDonald et al., 2018).
Table 3.2.
Biogenic volatile organic carbon (VOC) emission rates (μg-C g/h) from leaves of woody species, adjusted to standard conditions of temperature and light using algorithms of Guenther et al. (1993).
| Desert species | Isoprene | α-Pinene | β-Pinene | Camphene | d-Limonene | ∑ Monoterpenes | Reference |
|---|---|---|---|---|---|---|---|
| Ambrosia dumosa | < 0.1 | 1.6 | 3.0 | 0.06 | 2.0 | 7.9 | Geron et al., 2006a |
| Chrysothamnus nauseosus | < 0.1 | 0.28 | 0 | 0 | 0.21 | 0.65 | Geron et al., 2006a |
| Hymenoclea salsola | < 0.1 | 1.4 | 0.06 | 0.02 | 0.30 | 2.6 | Geron et al., 2006a |
| Larrea tridentata | < 0.1 | 0.37 | 0.12 | 0.44 | 0.74 | 2.0 | Geron et al., 2006a |
| Boreal | |||||||
| Abies balsamea | < 0.1 | 0.61 | 1.9 | 0.51 | 0 | 3.4 | Ortega et al., 2008 |
| Picea glauca | 14.9 | 0.25 | 0.19 | 0.07 | 0.44 | 1.4 | Kempf et al., 1996 |
| Pinus sylvestris | < 0.1 | 0.34 | 0.02 | 0.02 | 0.03 | 0.8 | Janson, 1993 |
| Temperate | |||||||
| Acer rubrum | < 0.1 | 0.18 | 0.53 | 0.04 | 0.04 | 1.4 | Ortega et al., 2008 |
| Pinus taeda | < 0.1 | 0.08 | 0.02 | 0.01 | 0.01 | 0.14 | Ortega et al., 2008 |
| Pinus ponderosa | < 0.1 | 0.58 | 0.33 | 0.04 | 0.11 | 1.6 | Helmig et al., 2013 |
| Quercus rubra | 67 | 0.28 | 0.10 | 0.05 | 0.20 | 1.7 | Geron et al., 2001; Ortega et al., 2008 |
| Tropical | |||||||
| Apeiba tibourbou | 0 | 1.0 | 0.43 | 0 | 0.14 | 3.6 | Kuhn, 2002 |
| Eucalyptus globules | 56 | 1.0 | 0.4 | 0 | 0.14 | 3.6 | Kuhn, 2002 |
| Hymenaea courbaril | 46 | 0 | 0 | 0 | 0 | 0 | Kuhn, 2002 |
| Cecropia sciadophylla | < 0.1 | 6.5 | 2.2 | 0 | 0 | 155 | Jardine et al., 2015a |
| Hevea brasiliensis | < 0.1 | 2.6 | 2.1 | 0 | 0 | 30 | Geron et al., 2006b |
| Azadirachta indica | < 0.1 | 0 | 0.15 | 0.9 | 0.38 | 2.43 | Singh et al., 2011 |
| Ficus religiosa | 77 | 0 | 0 | 0 | 0 | 0 | Varshney and Singh, 2003 |
| Citrus limon | 0.61 | 0.6 | 1.1 | 0 | 3.8 | 7.9 | Varshney and Singh, 2003 |
Data compiled and provided by Chris Geron (US EPA).
Most trace gases are highly reactive and thus have short mean residence times, so it is not surprising that they are minor constituents in the atmosphere (Atkinson and Arey, 2003). The concentration of such gases varies in space and time. For instance, we expect high concentrations of certain pollutants (O3, CO2 and CO etc.) over cities (e.g., Kort et al., 2012; Pommier et al., 2013), high levels of ammonia (NH3) near areas where cattle and pigs are kept (Leifer et al., 2017), and high concentrations of some reduced gases (methane and hydrogen sulfide) over swamps and other areas of anaerobic decomposition (e.g., Harriss et al., 1982; Steudler and Peterson, 1985). Remote sensing from satellites allows us to measure the spatial variation in the concentration of CO2, CH4, NH3, NO2, and other gases from space and to identify “hot spots” of emission (Hakkarainen et al., 2016; Jacob et al., 2016; Frankenberg et al., 2011; Warner et al., 2017; Van Damme et al., 2018; Griffin et al., 2019). Winds mix the concentrations of these gases to their average tropospheric background concentration within a short distance downwind of local sources. We can best perceive global changes in atmospheric composition, such as the current increase in CH4, by making long-term measurements in remote locations.
Junge (1974) related geographic variations in the atmospheric concentration of various gases to their estimated mean residence time in the atmosphere (Fig. 3.5 ). Gases that have short mean residence times are highly variable from place to place, whereas those that have long mean residence times relative to atmospheric mixing show relatively little spatial variation. For example, the average volume of water in the atmosphere is equivalent to about 13,000 km3 at any time, or 24.6 mm above any point on the Earth's surface (Trenberth, 1998). The average daily precipitation would be about 2.73 mm if it were deposited evenly around the globe. Thus, the mean residence time for water vapor in the atmosphere is:
| (3.4) |
Fig. 3.5.
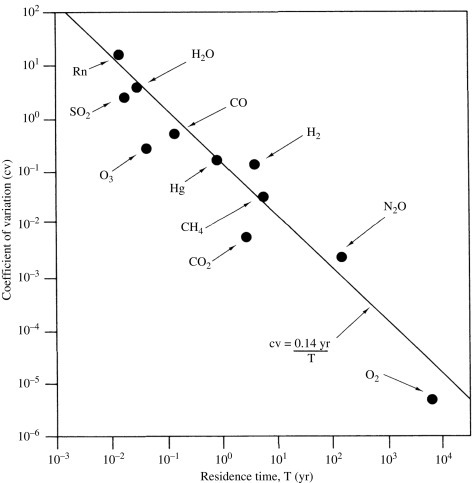
Variability in the concentration of atmospheric gases, expressed as the coefficient of variation among measurements, as a function of their estimated mean residence times in the atmosphere.
Modified from Junge (1974), as updated by Slinn (1988).
This is a short time compared to the circulation of the troposphere, so we should expect water vapor to show highly variable concentrations in space and time (Fig. 3.5). Most of the volatile organic compounds have short residence times in the atmosphere, so they are found at low concentrations in remote regions, such as Antarctica (Beyersdorf et al., 2010). This relationship between variation in concentration and residence time in the atmosphere extends to trace organic species (e.g., propane), which have residence times of a few days (Jobson et al., 1999).
The mean residence time for carbon dioxide in the atmosphere is about 3 years—only slightly longer than the mixing time for the atmosphere.c Owing to the seasonal uptake of CO2 by plants, CO2 shows a minor seasonal and latitudinal variation (± about 1%) in its global concentration of ~ 400 ppm (Figs. 1.1 and 3.6 ). In contrast, painstaking analyses are required to show any variation in the concentration of O2 because the amount in the atmosphere is so large and its mean residence time, ~ 4000 years, is so much longer than the mixing time of the atmosphere (Keeling and Shertz, 1992).
Fig. 3.6.
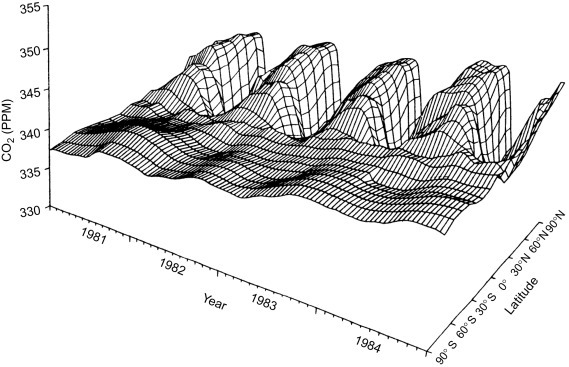
Seasonal fluctuations in the concentration of atmospheric CO2 (1981–1984), shown as a function of 10° latitudinal belts (Conway et al., 1988). Note the smaller amplitude of the fluctuations in the southern hemisphere, reaching peak concentrations during the northern hemisphere's winter.
Gases with mean residence times of < 1 year in the troposphere do not persist long enough for appreciable mixing into the stratosphere. Indeed, one of the most valuable, but dangerous, industrial properties of chlorofluorocarbons (CFCs)d is that they are chemically inert and thus long-lived in the troposphere (Rowland, 1989). This allows chlorofluorocarbons to mix into the stratosphere, where they lead to the destruction of ozone by ultraviolet light (see Eqs. (3.54), (3.55), (3.56), (3.57)).
Aerosols
In addition to gaseous components, the atmosphere contains particles, known as aerosols, that arise from a variety of sources (Table 3.3 ). Increasingly, we recognize that the concentrations of particles in the atmosphere have important impacts on biogeochemistry, human health (Lelieveld et al., 2015), and climate (Mahowald, 2011). The total burden of aerosols in the atmosphere is expressed as Aerosol Optical Depth (AOD), which is now measured from space by satellites (Fig. 3.7 ).e , f
Table 3.3.
The global production and atmospheric burden of aerosols from natural and human-derived sources.
| Mass emission |
Mass burden |
Number produced |
Number |
|
|---|---|---|---|---|
| 1012 g/yr | Tg | per year | Burden | |
| Carbonaceous aerosols | ||||
| Primary organic (0–2 μm) | 95 | 1.2 | – | 310 × 1024 |
| Biomass burning | 54 | – | 7 × 1027 | – |
| Fossil fuel | 4 | – | – | – |
| Biogenic | 35 | 0.2 | – | – |
| Black carbon (0–2 μm) | 10 | 0.1 | – | 270 × 1024 |
| Open burning and biofuel | 6 | – | – | – |
| Fossil fuel | 4.5 | – | – | – |
| Secondary organic | 28 | 0.8 | – | – |
| Biogenic | 25 | 0.7 | – | – |
| Anthropogenic | 3.5 | 0.08 | – | – |
| Sulfates | 200 | 2.8 | 2 × 1028 | – |
| Biogenic | 57 | 1.2 | – | – |
| Volcanic | 21 | 0.2 | – | – |
| Anthropogenic | 122 | 1.4 | – | – |
| Nitrates | 18 | 0.49 | – | – |
| Industrial dust, etc. | 100 | 1.1 | – | – |
| Sea salt | ||||
| d < 1 μm | 180 | 3.5 | 7.4 × 1026 | – |
| d = 1–16 μm | 9940 | 12 | 4.6 × 1026 | – |
| Total | 10,130 | 15 | 1.2 × 1027 | 27 × 1024 |
| Mineral (soil) dust | ||||
| < 1 μm | 165 | 4.7 | 4.1 × 1025 | – |
| 1–2.5 μm | 496 | 12.5 | 9.6 × 1025 | – |
| 2.5–10 μm | 992 | 6 | – | – |
| Total | 1600 | 18 ± 5 | 1.4 × 1026 | 11 × 1024 |
Fig. 3.7.
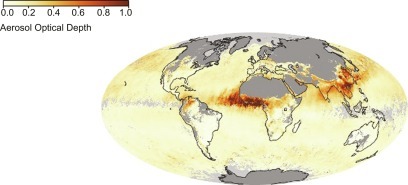
Aerosols in Earth's atmosphere, measured as Aerosol Optical Depth (AOD) by the NASA MODIS satellite during March 2010. See Footnote e, p. 61 for the derivation of AOD. Note high amounts of aerosols exiting the southern Sahel region of Africa, blowing westward to the Amazon, and high concentrations of aerosols emitted from the deserts of China, blowing eastward across the Pacific Ocean.
From http://earthobservatory.nasa.gov/GlobalMaps/view.php?d1=MODAL2_M_AER_OD.
Soil particles are dispersed by wind erosion (also known as deflation weathering or eolian transport) from arid and semiarid regions (Pye, 1987; Ravi et al., 2011). Particles with diameter < 1.0 μm are held aloft by turbulent motion and are subject to long-range transport. Current estimates suggest that up to 2 × 1015 g/yr of soil particles enter the atmosphere from arid and barren agricultural soils (Zender et al., 2004), and about 20% of these particles are involved in long-range transport. While natural sources account for 81% of the global dust emissions (Chen et al., 2018a), the flux of soil dust has increased due to human cultivation, especially in semiarid lands (Tegen et al., 2004; Mulitza et al., 2010; Routson et al., 2019) and increases during droughts. Dust from the deserts of central Asia falls in the Pacific Ocean (Duce et al., 1980), where it contributes much of the iron needed by oceanic phytoplankton (Mahowald et al., 2005b, Chapter 9). Similarly, dust from the Sahara supplies nutrients to phytoplankton in the Atlantic Ocean (Talbot et al., 1986; Jickells et al., 2005) and phosphorus to Amazon rainforests (Swap et al., 1992; Okin et al., 2004; Yu et al., 2015). Some particulate pollution in the western United States is derived from the deserts of China (Yu et al., 2012), and emissions from arid lands in the Colorado Plateau affect the biogeochemistry in Rocky Mountain forests (Field et al., 2010). Dust from desert soils is monitored by several satellites, including NASA's MODIS satellite (Tanre et al., 2001; Kaufman et al., 2002; see Fig. 3.7). Typically, while it is in transit, soil dust warms the atmosphere over land and cools the atmosphere over the oceans, which have lower surface albedo (reflectivity) (Ackerman and Chung, 1992; Kellogg, 1992; Yang et al., 2009).
An enormous quantity of particles enters the atmosphere from the ocean as a result of tiny droplets that become airborne with the bursting of bubbles at the surface (MacIntyre, 1974; Wu, 1981). As the water evaporates from these bubbles, the salts crystallize to form seasalt aerosols, which carry the approximate chemical composition of seawater (Glass and Matteson, 1973; Möller, 1990). As in the case of soil dust, most seasalt aerosols are relatively large and settle from the atmosphere quickly, but a significant proportion remains in the atmosphere for global transport. Lewis and Schwartz (2004) compile global estimates of seasalt production, averaging 5 × 1015 g/yr, whereas Andreae and Rosenfeld, 2008; Table 3.3) estimate a total seasalt production of 10 × 1015 g/yr, which carries about 200 × 1012 g of chloride from sea to land (see Fig. 3.17 later).
Fig. 3.17.
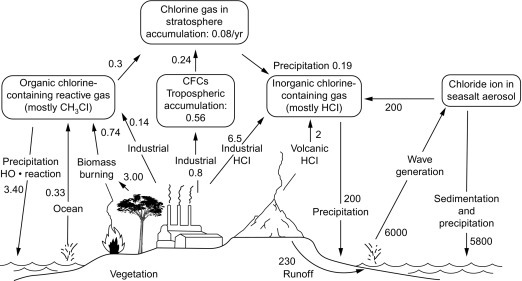
A global budget for Cl in the troposphere and the stratosphere. All data are given in 1012 g Cl/yr.
Modified and updated from Möller (1990), Graedel and Crutzen (1993), Graedel and Keene (1995), with new data from McCulloch et al. (1999) and other sources listed in Table 3.7.
Organic particles are produced from a wide variety of sources, including pollen, plant fragments, and bacteria (Després et al., 2012). Some plant aerosols contain significant quantities of potassium (Crozat, 1979; Pöhlker et al., 2012). Forest fires produce particles of charcoal that are carried throughout the troposphere, and small organic particles (soot) are produced by the condensation of volatile hydrocarbons from the smoke of forest fires (Hahn, 1980; Cachier et al., 1989). Forest fires in the Amazon are thought to release as much as 1 × 1013 g of particulate matter to the atmosphere each year (Kaufman et al., 1990). It is likely that the global production of aerosols from forest fires has increased markedly as a result of higher rates of biomass burning in the tropics (Andreae, 1991; Cahoon et al., 1992). Aerosols from these fires may affect regional patterns of rainfall (Cachier and Ducret, 1991) and global climate (Penner et al., 1992). Recent catastrophic fires in the western United States have contributed to the atmospheric burden of aerosols and to impacts on human health (McClure and Jaffe, 2018; Ford et al., 2018).
Periodically, volcanoes inject finely divided rock material—volcanic ash—into the atmosphere where it is deposited over large areas (Table 3.4 ), contributing to soil development in regions that are downwind from major eruptions (Watkins et al., 1978; Dahlgren et al., 1999; Zobel and Antos, 1991). Volcanic gases and ash that are transported to the stratosphere by violent eruptions undergo global transport, affecting climate for several years (Sigl et al., 2015).g , h During the past several centuries, aerosols from volcanoes have dwarfed those produced by incoming meteors, although the latter have been a potential source of plant nutrients, such as Fe, in the geologic past (Reiners and Turchyn, 2018).
Table 3.4.
Composition of a volcanic ash sample collected during the eruption of Mt. St. Helens (Washington, USA) on 19 May 1980.
| Constituent | Particulate sample | Average ash |
|---|---|---|
| Major elements (%) | ||
| SiO2 | ≡ 65.0 | 65.0 |
| Fe2O3 | 6.7 | 4.81 |
| CaO | 3.0 | 4.94 |
| K2O | 2.0 | 1.47 |
| TiO2 | 0.42 | 0.69 |
| MnO | 0.054 | 0.077 |
| P2O5 | – | 0.17 |
| Trace elements (ppm) | ||
| S | 3220 | 940 |
| CI | 1190 | 660 |
| Cu | 61 | 36 |
| Zn | 34 | 53 |
| Br | < 8 | ~ 1 |
| Rb | < 17 | 32 |
| Sr | 285 | 460 |
| Zr | 142 | 170 |
| Pb | 36 | 8.7 |
Source: From Fruchter et al. (1980) and Hooper et al. (1980).
Small particles, known as secondary aerosols, are also produced by reactions between gases in the atmosphere. For instance, when SO2 is oxidized to sulfuric acid (H2SO4) in the atmosphere, particles rich in (NH4)2SO4 may be produced by a subsequent reaction with atmospheric ammonia (NH3; Behera and Sharma, 2011; Kirkby et al., 2011):
| (3.5) |
(Ammonia is derived from a variety of sources, primarily associated with agricultural activities; Chapter 12.) Sulfate aerosols are also produced during the oxidation of dimethylsulfide released from the ocean (Chapter 9). Sulfate aerosols increase the albedo of the Earth's atmosphere, so estimates of the abundance of sulfate aerosols are an important component of global climate models (Kiehl and Briegleb, 1993; Mitchell et al., 1995).
Secondary aerosols are also produced from volatile organic compounds, such as isoprene, that are released from plants (Kavouras et al., 1998; Jimenez et al., 2009; Zhang et al., 2018b). Sulfuric acid vapor can oxidize volatile organic compounds to produce secondary aerosols that are important to cloud formation (Riccobono et al., 2014).
Finally, a wide variety of particles are produced from human industrial processes, especially the burning of coal (Hulett et al., 1980; Shaw, 1987). Globally, the release of particles during the combustion of fossil fuels rivals the mobilization of elements by rock weathering at the Earth's surface (Bertine and Goldberg, 1971, see Table 1.1). Overall, human activities probably account for about 10% of the burden of aerosols in today's atmosphere (Table 3.3). Fine particulate air pollution has significant human health effects (Samet et al., 2000; Pope et al., 2009; Lelieveld et al., 2015). Nanoparticles (< 0.3 μm), derived from a variety of natural and manufactured sources (Kumar et al., 2010; Hendren et al., 2011), are of particular concern.
Fortunately, the mass of industrial aerosols has declined in many developed countries where pollution controls have been instituted. One of the most widespread anthropogenic aerosols, particles of lead from automobile exhaust, has declined in global abundance over the past 40 years due to a reduction in the use of leaded gasoline (Boutron et al., 1991). In other regions, where air pollution is unregulated, concentrations of aerosols have increased (Streets et al., 2008; Dey and Di Girolamo, 2011), contributing to regional observations of global dimming. The largest changes are observed for East Asia and Africa (Mao et al., 2014; He et al., 2018).
Small particles (< 1.0 μm)i are much more numerous in the atmosphere than large particles, but it is the large particles that contribute the most to the total airborne mass (Warneck, 2000; Raes et al., 2000). The mass of aerosols declines with increasing altitude, from values ranging between 1 and 50 μg/m3 near unpolluted regions of the Earth's surface. Although there is an inverse relation between the size of particles and their persistence in the atmosphere, the overall mean residence time for tropospheric aerosols is about 5 days (Warneck, 2000). Thus, aerosols are not uniform in their distribution in the atmosphere. As a result of their longer mean residence time, small particles have the greatest influence on Earth's climate, and annually they carry the largest mass of material through the atmosphere.
The composition of tropospheric aerosols varies greatly depending on the proximity of continental, maritime, or anthropogenic sources (Heintzenberg, 1989; Murphy et al., 1998). Over land, aerosols are often dominated by soil minerals and human pollutants (Shaw, 1987; Gillette et al., 1992). Over the ocean, the composition of aerosols is a mixture of contributions from silicate minerals of continental origin and seasalt from the ocean (Andreae et al., 1986). Various workers have used ratios among the elemental constituents of aerosols to deduce the relative contribution of different sources (e.g., Moyers et al., 1977; Rahn and Lowenthal, 1984).
Aerosols are important in reactions with atmospheric gases and as nuclei for the condensation of raindrops. The latter are known as cloud condensation nuclei, often abbreviated CCN. Raindrops are formed when water vapor begins to condense on aerosols > 0.1 μm in diameter. As raindrops enlarge and fall to the ground, they collide with other particles and absorb atmospheric gases. Soil dusts often contain a large portion of insoluble material (Reheis and Kihl, 1995), but seasalt aerosols and those derived from pollution sources are readily soluble and contribute to the dissolved chemical content of rainwater. Reactions of atmospheric gases with aerosols or raindrops are known as heterogeneous or multiphase reactions (Ravishankara, 1997). Such reactions are responsible for the ultimate removal of many reactive gases from the atmosphere.
Biogeochemical reactions in the troposphere
Major constituents—Nitrogen
It is perhaps not surprising that the major constituents of the atmosphere, N2, O2, and Ar, have nearly uniform concentrations and long mean residence times in the atmosphere. Argon is inert and has accumulated in the Earth's atmosphere since the earliest degassing of its crust (Chapter 2). From a biogeochemical perspective, N2 is practically inert; reactive N is found only in molecules such as NH3 and NO. Collectively the reactive nitrogen gases are sometimes called “odd” nitrogen, because the molecules have an odd number of N atoms (versus N2 or N2O).j Despite its abundance in the atmosphere, N2 is so inert that the rate of formation of odd, or reactive, nitrogen is the primary factor that limits the growth of plants on land (LeBauer and Treseder, 2008). Among atmospheric gases, only argon and the other noble gases are less reactive.
Conversion of N2 to reactive compounds, N fixation, occurs in lightning bolts, but the estimated global production of NO by lightning (< 9 × 1012 g N/yr; Chapter 12) is too low to account for a significant turnover of N2 in the atmosphere. By far the most important source of fixed nitrogen for the biosphere derives from the bacteria that convert N2 to NH3 in the process of biological nitrogen fixation (Eq. 29.). The global rate of biological N fixation is poorly known because it must be extrapolated from small-scale measurements to the entire surface of the Earth (Chapters 6 and 9). Including human activities, global nitrogen fixation (land + marine) is not likely to exceed 430 × 1012 g N/yr, with the production of synthetic nitrogen fertilizer now accounting for about one-third of the total (Battye et al., 2017, Chapter 12).
The natural rate of nitrogen fixation would remove the pool of N2 from the atmosphere in about 9 million years.k Fortunately, denitrification (Eq. 2.20) returns N2 to the atmosphere. At present, we have little evidence that the rate of either N fixation or denitrification changes significantly in response to changes in the concentration of N2 in the atmosphere. The biosphere is responsible for the maintenance of N2 in Earth's atmosphere over geologic time, but it plays a minor role in stabilizing the concentration of atmospheric N2 over shorter periods, since the pool of N2 in the atmosphere is so large (Walker, 1984).
Oxygen
In Chapter 2 we discussed the accumulation of O2 in the atmosphere during the evolution of life on Earth. The atmosphere now contains only a small portion of the total O2 that has been released by photosynthesis through geologic time (Fig. 2.8). However, the atmosphere contains much more O2 than can be explained by the storage of carbon in land plants today. The instantaneous combustion of all the organic matter now stored on land would reduce the atmospheric oxygen content by only 0.45% (Chapter 5). The accumulation of O2 in the atmosphere is the result of the long-term burial of reduced carbon in ocean sediments (Berner, 1982), which contain nearly all of the reduced, organic carbon on Earth (Table 2.3). The rate of burial is determined by the area and depth of the ocean floor that is subject to anoxic conditions (Walker, 1977; Hartnett et al., 1998). Because the area and depth vary inversely with the concentration of atmospheric O2, the balance between the burial of organic matter and its oxidation maintains O2 at a steady-state concentration of about 21% (see also Chapters 9 and 11).
A large amount of O2 has been consumed in weathering of reduced crustal minerals, especially Fe and S, through geologic time (Fig. 2.8); the current rate of exposure of these minerals would consume all atmospheric oxygen in about 70 million years (Lenton, 2001; see Fig. 11.8). However, the rate of exposure is not likely to vary greatly in response to changes in atmospheric O2, so weathering is not the major factor controlling O2 in the atmosphere (Bolton et al., 2006). In sum, despite the potential reactivity of O2, its rate of reaction with reduced compounds is rather slow, and O2 is a stable component of the atmosphere. Oxygen has declined only 0.7% over the past 800,000 years, presumably due to lower rates of organic burial and weathering (Stolper et al., 2016). The mean residence time of O2 in the atmosphere is on the order of 4000 years, largely determined by exchange with the biosphere (compare Figs. 3.5 and 11.8). As such O2 is well mixed and uniform in the atmosphere. Annual photosynthesis and respiration cause seasonal variations in O2 concentration of about ± 0.002% (Fig. 1.2).
Carbon dioxide
Carbon dioxide is not reactive with other gases in the atmosphere. The concentration of CO2 is affected by interactions with the Earth's surface, including the reactions of the carbonate-silicate cycle (Fig. 1.3), gas exchange with surface seawaters following Henry's Law (Eq. 2.4), consumption and production of CO2 by photosynthesis and respiration by heterotrophs (Figs. 1.2 and 3.6). For the Earth's land surface, our best estimates of plant uptake (120 × 1015 g C/yr; Chapter 5) suggest a mean residence time of about 6 years before a hypothetical molecule of CO2 in the atmosphere is captured by photosynthesis by land plants. The annual exchange of CO2 with seawater, dominated by areas of cold, downwelling water and high productivity (Chapter 9), is about 1.5 × as large as the annual uptake of CO2 by land plants. Both plant and ocean uptake are likely to increase with increasing concentrations of atmospheric CO2, potentially buffering fluctuations in its concentration (Chapters 5, 9, and 11). Following Eq. 3.3, the mean residence time for CO2, determined by the total flux from the atmosphere (the sum of land and ocean uptake), is about 3 years, so CO2 shows small seasonal and latitudinal variation in the atmosphere (Figs. 1.1 and 3.6).
The carbonate-silicate cycle (Fig. 1.3) also buffers the concentration of CO2 in the atmosphere, but does not affect the concentration of atmospheric CO2 significantly in periods of less than ~ 100,000 years (Hilley and Porder, 2008; Colbourn et al., 2015). We will compare the relative importance of these processes in more detail in Chapter 11, which examines the global carbon cycle. The current increase in atmospheric CO2 is a non-steady-state condition, caused by the combustion of fossil fuels and destruction of land vegetation. CO2 is released by these processes faster than it can be taken up by land vegetation and the sea. If these activities were to cease, atmospheric CO2 would return to a steady state, and after several hundred years nearly all of the CO2 released by humans would reside in the oceans (Laurmann, 1979). In the meantime, higher concentrations of CO2 are likely to cause significant atmospheric warming through the “greenhouse effect” (refer to Fig. 3.2).
Trace biogenic gases
Volcanoes are the original source of volatiles in the Earth's atmosphere (Chapter 2) and a small continuing source of some of the reduced gases (H2S, H2, NH3, CH4) that are found in the atmosphere today (Table 2.2). However, in most cases, the concentrations of these gases in today's atmosphere are dominated by supply from the biosphere, particularly by microbial activity (Monson and Holland, 2001). Methane is largely produced by anaerobic decomposition in wetlands (Chapters 7 and 11), nitrogen oxides by fossil fuel combustion and soil microbial transformations (Chapters 6> and 12), carbon monoxide by combustion of biomass and fossil fuels (Chapters 5 and 11), and volatile hydrocarbons, especially isoprene, by vegetation and human industrial activities (Chapter 5). The production of trace gases containing N and S contributes to the global cycling of these elements, which is controlled by the biosphere (Crutzen, 1983). These and other trace gases are found at concentrations well in excess of what is predicted from equilibrium geochemistry in an atmosphere with 21% O2 (Table 3.5 ).
Table 3.5.
Some trace biogenic gases in the atmosphere.
| Compound | Formula | Concentration (ppb) |
Mean residence time | Percentage of sink due to OH | |
|---|---|---|---|---|---|
| Expecteda | Actualb | ||||
| Carbon compounds | |||||
| Methane | CH4 | 10− 148 | 1830 | 9 years | 90 |
| Carbon monoxide | CO | 10− 51 | 45–250 | 60 days | 80 |
| Isoprene | CH2 C(CH3)—CH CH2 | 0.2–10.0 | < 1 day | 100 | |
| Nitrogen compounds | |||||
| Nitrous oxide | N2O | 10− 22 | 320 | 120 years | 0 |
| Nitric oxides | NOx | 10− 13 | 0.02–10.0 | 1 day | 100 |
| Ammonia | NH3 | 10− 63 | 0.08–5.0 | 5 days | < 2 |
| Sulfur compounds | |||||
| Dimethylsulfide | (CH3)2S | 0.004–0.06 | 1 day | 50 | |
| Hydrogen sulfide | H2S | < 0.04 | 4 days | 100 | |
| Carbonyl sulfide | COS | 0 | 0.50 | 5 years | 20 |
| Sulfur dioxide | SO2 | 0 | 0.02–0.10 | 3 days | 50 |
Approximate values in equilibrium with an atmosphere containing 21% O2 (Chameides and Davis, 1982).
For short-lived gases, the value is the range expected in remote, unpolluted atmospheres.
Unlike major atmospheric constituents, many of the trace biogenic gases in the atmosphere are highly reactive, showing short mean residence times and variable concentrations in space and time (refer to Fig. 3.5). Concentrations of these gases in the atmosphere are determined by the balance between local sources and chemical reactions—known as sinks—that remove these gases from the atmosphere. Sinks are largely driven by oxidation reactions and the capture of the reaction products by rainfall. Currently the concentration of nearly all these constituents is increasing as a result of human activities, suggesting that humans are affecting biogeochemistry at the global level (Prinn, 2003).
Oxidation reactions in the atmosphere
Despite its abundance, O2 does not directly oxidize reduced gases in the atmosphere. Instead, a small proportion of the oxygen is converted to the powerful atmospheric oxidants ozone (O3) and hydroxyl radicals (OH) through a series of reactions driven by sunlight (Logan, 1985; Thompson, 1992). Ozone and OH are the primary gases that oxidize many of the trace gases to CO2, HNO3, and H2SO4.
It is important to understand the natural production, occurrence, and reactions of ozone in the atmosphere. Nearly daily we read seemingly contradictory reports of the harmful effects of ozone depletion in the stratosphere and harmful effects of ozone pollution in the troposphere. In each case, human activities are upsetting the natural concentrations of ozone that are critical to atmospheric biogeochemistry.
Ozone is produced by the reaction of sunlight with O2 in the stratosphere, as described in the next section. Some of this ozone is transported to the Earth's surface by the mixing of stratospheric and tropospheric air (e.g., Hocking et al., 2007), where it contributes to the budget of tropospheric ozone. (Table 3.6 ). However, observations of high ozone concentrations in the smog of polluted cities (e.g., Los Angeles) alerted atmospheric chemists to reactions by which ozone is also produced in the troposphere (Warneck, 2000).
Table 3.6.
Tropospheric ozone budget. All values in Tg (1012 g) of O3/yr.
| Sources | |
| Chemical production | 4960 |
| Downward transport from stratosphere | 325 |
| Total | 5290 |
| Sinks | |
| Chemical loss | 4360 |
| Dry deposition | 908 |
| Wet deposition | 19 |
| Total | 5290 |
From Hu et al. (2017).
When NO2 is present in the atmosphere, it is dissociated by sunlight (hv),
| (3.6) |
followed by a reaction producing ozone:
| (3.7) |
This reaction sequence is an example of a homogeneous gas reaction, that is, a reaction between atmospheric constituents that are all in the gaseous phase. The net reaction is:
| (3.8) |
which is an equilibrium reaction, so high concentrations of NO tend to drive the reaction backward. Sunlight is essential to form ozone by these pathways, so they are known as photochemical reactions. At night, ozone is consumed by reactions with NO2 to form nitric acid (Brown et al., 2006b).
Both NO2 and NO, collectively known as NOx, are found in polluted air, in which they are derived from industrial and automobile emissions.l Small concentrations of both of these constituents are also found in the natural atmosphere, where they are derived from forest fires, lightning discharges, and microbial processes in the soil (Chapters 6 and 12). Thus, the production of ozone from NO2 has probably always occurred in the troposphere, and the present-day concentrations of tropospheric ozone have simply increased as industrial emissions have raised the concentration of NO2 and other precursors to O3 formation (Lelieveld et al., 2004; Cooper et al., 2010; Schneider and van der A, 2012). NOx concentrations have increased over much of China (Fig. 3.8 ). As air pollution regulations have reduced emissions of NOx in Europe and the U.S., O3 concentrations have declined (Kim et al., 2006; Butler et al., 2011).m
Fig. 3.8.
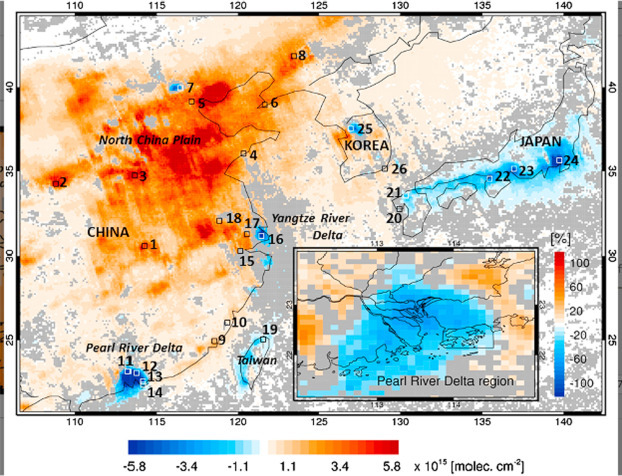
Changes in NOx concentrations over East Asia from 2005 to 2014, as monitored by satellite.
From Duncan et al. (2016).
Ozone is subject to further photochemical reaction in the troposphere,
| (3.9) |
where hv is ultraviolet light with wavelengths < 318 nm and O(1D) is an excited atom of oxygen. Reaction of O(1D) with water yields hydroxyl radicals:
| (3.10) |
The formation of hydroxyl radicals is strongly correlated with the amount of ultraviolet radiation (Rohrer and Berresheim, 2006). Hydroxyl radicals may further react to produce HO2 and H2O2,
| (3.11) |
| (3.12) |
which are other short-lived oxidizing compounds in the atmosphere (Thompson, 1992; Crutzen et al., 1999).
Hydroxyl radicals exist with a mean concentration of about 1 × 106 molecules/cm3 (Prinn et al., 1995; Wolfe et al., 2019). The highest concentrations occur in daylight (Platt et al., 1988; Mount, 1992) and at tropical latitudes, where the concentration of water vapor is greatest (Hewitt and Harrison, 1985). The average OH radical persists for only a few seconds in the atmosphere, so concentrations of OH are highly variable. Local concentrations can be measured using beams of laser-derived light, which is absorbed as a function of the number of OH radicals in its path (Dorn et al., 1988; Mount et al., 1997).
Because of its short mean residence time, the global mean concentration of OH radicals must be estimated indirectly. For this purpose, atmospheric chemists have relied on methylchloroform (trichloroethane), a gas that is known to result only from human activity. Methylchloroform has a mean residence time of about 4.8 years (Prinn et al., 1995), so it is reasonably well mixed in the atmosphere. In the laboratory, it reacts with OH,
| (3.13) |
and the rate constant, K, for the reaction is 0.85 × 10− 14 cm3 molecule− 1 s− 1 at 25 °C (Talukdar et al., 1992). Then, knowing the industrial production of CH3CCl3, its accumulation in the atmosphere and K, one can calculate the concentration of OH that must be present, namely,
| (3.14) |
Hydroxyl radicals are the major source of oxidizing power in the troposphere. For example, in an unpolluted atmosphere, hydroxyl radicals destroy methane in a series of reactions,
| (3.15) |
| (3.16) |
| (3.17) |
| (3.18) |
| (3.19) |
for which the net reaction is:
| (3.20) |
Note that the hydroxyl radical has acted as a catalyst to initiate the oxidation of CH4 and its byproducts by O2. Other volatile organic compounds are also oxidized through this pathway, which yields formaldehyde (CH2O; Atkinson, 2000; Atkinson and Arey, 2003). Changes in the concentration of formaldehyde can be used as an index of the burden of organic gases in the atmosphere from natural sources and air pollution (Zhu et al., 2017).
The formaldehyde that is produced in these reactions is further oxidized to carbon monoxide,
| (3.21) |
and CO is oxidized by OH to produce CO2,
| (3.22) |
| (3.23) |
| (3.24) |
for which the net reaction is
| (3.25) |
Thus, OH acts to scrub the atmosphere of a wide variety of reduced carbon gases, ultimately oxidizing their carbon atoms to carbon dioxide.
Hydroxyl radicals can also react with NO2 and SO2 in homogeneous gas reactions:
| (3.26) |
| (3.27) |
and the latter reaction is followed by a heterogeneous reaction with raindrops:
| (3.28) |
which removes sulfur dioxide from the atmosphere, causing acid rain. Sulfur dioxide is also oxidized by hydrogen peroxide, according to Chandler et al. (1988):
| (3.29) |
The reaction of OH with NO2 is very fast, and it produces nitric acid that is removed from the atmosphere by a heterogeneous interaction with raindrops (Munger et al., 1998). The reactions with SO2 are much slower, accounting for the long-distance transport of SO2 as a pollutant in the atmosphere (Rodhe, 1981). Hydrogen sulfide (H2S) and dimethylsulfide (CH3)2S, released from anaerobic soils (Chapter 7) and the ocean surface (Chapter 9), are also removed by reactions with OH and other oxidizing compounds, leading to the deposition of H2SO4 (Toon et al., 1987). Thus, OH radicals cleanse the atmosphere of trace N and S gases by converting them to acid anions NO3 − and SO4 2 − in the atmosphere.
The vast majority of OH radicals in the atmosphere are consumed in reactions with CO and CH4. Although the concentration of methane is much higher than that of carbon monoxide in unpolluted atmospheres, the reaction of OH with CO is much faster. The speed of reaction of CO with OH accounts for the short mean residence time of CO in the atmosphere (Table 3.5). The mean residence time for methane is much longer, accounting for its more uniform distribution in the atmosphere (Fig. 3.5). One explanation for the current increase in methane in the atmosphere is that the anthropogenic release of CO consumes OH radicals previously available for the oxidation of methane (Khalil and Rasmussen, 1985; Rigby et al., 2017). Consistent with the relative distribution of sinks for OH, the concentration of OH in the atmosphere is slightly lower in the Northern hemisphere (Wolfe et al., 2019), but various global estimates suggest that OH concentrations have declined only slightly (or perhaps not at all) in recent years (Prinn et al., 1995, Prinn et al., 2005; Montzka et al., 2011b). An increasing deposition of formaldehyde in the Greenland snowpack indicates the oxidation of methane has kept up with its increasing concentration in the atmosphere (Eq. 3.20; Staffelbach et al., 1991).
In unpolluted atmospheres, all these reactions consume OH. In “dirty” atmospheres, a different set of reactions pertains, in which there can be a net production of O3, and thus OH, during the oxidation of reduced gases (Jenkin and Clemitshaw, 2000; Sillman, 1999). When the concentration of NO is > 10 ppt, which we will define as a “dirty” atmosphere (Jacob and Wofsy, 1990), the oxidation of carbon monoxide begins by reaction with hydroxyl radical and proceeds as follows (Crutzen and Zimmermann, 1991):
| (3.30) |
| (3.31) |
| (3.32) |
| (3.33) |
| (3.34) |
The net reaction is:
| (3.35) |
Similarly, the oxidation of methane in the presence of high concentrations of NO proceeds through a large number of steps, yielding a net reaction of:
| (3.36) |
In both cases, NO acts as a catalyst leading to the oxidation of reduced gases by oxygen.
Fig. 3.9 shows the contrasting pathways of carbon monoxide oxidation in clean and dirty atmospheres. Crutzen (1988) points out that the oxidation of one molecule of CH4 could consume up to 3.5 molecules of OH and 1.7 molecules of O3 when the NO concentration is low, whereas it would yield a net gain of 0.5 OH and 3.7 O3 in polluted environments (see also Wuebbles and Tamaresis, 1993). Although they were first discovered in urban areas, the reactions of dirty atmospheres are likely to be relatively widespread in nature. NO is produced naturally by soil microbes (Chapter 6) and forest fires, and concentrations of NO > 10 ppt are present over most of the Earth's land surface (Chameides et al., 1992; Levy et al., 1999). In the presence of NO, oxidation of volatile hydrocarbons emitted from vegetation, and CO emitted from both vegetation and forest fires, can account for unexpectedly high concentrations of O3 over rural areas of the southeastern United States (Fig. 3.10 )n (Jacob et al., 1993; Kleinman et al., 1994; Kang et al., 2003) and in remote tropical regions (Crutzen et al., 1985; Zimmerman and Greenberg, 1988; Jacob and Wofsy, 1990; Andreae et al., 1994). In urban areas, where the concentration of NOx is especially high due to industrial pollution, effective control of atmospheric O3 levels may also depend on the regulation of volatile hydrocarbons (Chameides et al., 1988; Seinfeld, 1989). In rural areas, ozone formation is usually limited by the concentration of NOx, especially during the growing season, when vegetation actively emits volatile organic compounds (Fig. 3.11 ; Aneja et al., 1996).
Fig. 3.9.
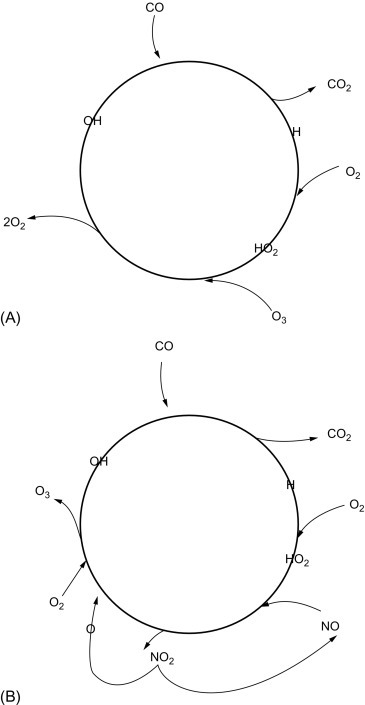
Reaction chain for the oxidation of CO in clean (A) and dirty (B) atmospheric conditions.
Fig. 3.10.
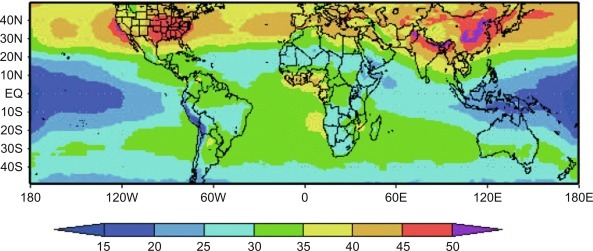
Distribution of ozone in Earth's troposphere, for summer months, averaged over 1979–1991. Data are in Dobson Units, see footnote n, p. 75. Note high ozone concentrations over the eastern United States and China.
From Fishman et al. (2003). Used with permission of European Geosciences Union.
Fig. 3.11.
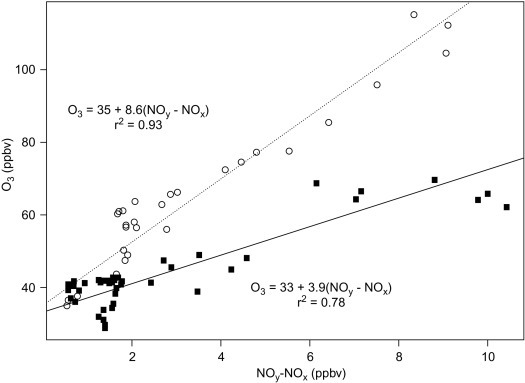
Ambient O3 vs. NOy-NOx concentrations in the atmosphere at Harvard Forest, northern Massachusetts (USA), 6–12 May 1990 (▪) and 24–30 August 1992 (O).
From Hirsch et al. (1996). Used with permission of the American Geophysical Union.
Understanding changes in the concentration of O3 and OH in the atmosphere is critical to predicting future trends in the concentration of trace gases, such as CH4, that can contribute to greenhouse warming. Some models predict an increase in O3 (Isaksen and Hov, 1987; Hough and Derwent, 1990; Thompson, 1992; Prinn, 2003) in the atmosphere as a result of increasing human emissions of NO, creating dirty atmosphere conditions over much of the planet. These predictions are consistent with observations that the ozone concentrations in Europe have risen since the late 1800s (Volz and Kley, 1988; Marenco et al., 1994), though perhaps only 40% or less globally (Yeung et al., 2019). Concentrations of H2O2, derived from OH (Eqs. (3.11), (3.12)), have increased in layers of Greenland ice deposited during the last 200 years, suggesting a greater oxidizing capacity in the northern hemisphere as a result of human activities (Fig. 3.12 ). The models are also consistent with indirect observations that the global concentration of OH has remained fairly stable in recent years, despite increasing emissions of reduced gases that should scrub OH from the atmosphere (Prinn et al., 1995, Prinn et al., 2005; Montzka et al., 2011b). Oxidizing radicals in the atmosphere may increase in future warmer climates (Geng et al., 2017).
Fig. 3.12.
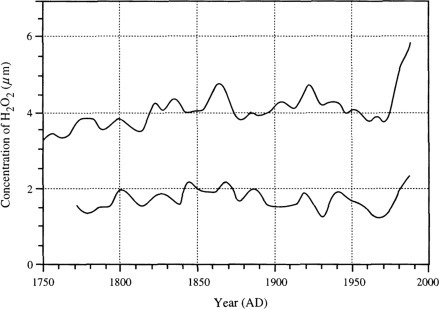
Variation in the mean annual H2O2 concentration during the past 200 years as seen in two cores from the Greenland ice pack.
Modified with permission of Macmillan from Sigg and Neftel (1991).
Several recent papers suggest additional pathways leading to the formation of OH (Li et al., 2008; Hofzumahaus et al., 2009), so that its production may not be restricted to the photochemical reactions outlined in Eqs. (3.8), (3.9), (3.10). Soil microbes that produce nitrite (NO2 −) are a potential source of nitrous acid (HONO) in the atmosphere and OH radicals (Su et al., 2011).
Ozone has a mean lifetime of about 24 days in the troposphere (Hu et al., 2017). Some of the O3 produced over the continents undergoes long-distance transport (Jacob et al., 1993; Parrish et al., 1993; Cooper et al., 2010; Brown-Steiner and Hess, 2011), resulting in the appearance of O3 and its byproducts at considerable distances from their source (Fig. 3.10). In some rural areas, concentrations of O3 from local production and transport from nearby cities inhibit the growth of agricultural crops and trees (Chameides et al., 1994). Other workers disagree, finding that local atmospheric conditions, rather than global changes in transport from polluted areas, determine the oxidizing capacity of the atmosphere over much of the planet (Oltmans and Levy, 1992; Ayers et al., 1992; Kang et al., 2003).
Atmospheric deposition
Elements of biogeochemical interest are deposited on the Earth's surface as a result of rainfall, dry deposition, and the direct absorption of gases from the atmosphere. The importance of each of these processes differs for different regions and for different elements (Gorham, 1961).
In many forests, a large fraction of the annual uptake and circulation of nutrient elements in vegetation may be derived from the atmosphere (Kennedy et al., 2002a; Avila et al., 1998; Miller et al., 1993). The atmosphere accounts for nearly all of the nitrogen and sulfur that circulates in terrestrial ecosystems, with rock weathering providing only smaller amounts (see Table 4.5 and Chapter 6). The chemical composition of rainfall has received great attention, as a result of widespread concern about dissolved constituents that lead to “acid rain.”
Processes
The dissolved constituents in rainfall are often separated into two fractions. The rainout component consists of constituents derived from cloud processes, such as the nucleation of raindrops. The washout component is derived from below cloud level, by scavenging of aerosol particles and the dissolution of gases in raindrops as they fall (Brimblecombe and Dawson, 1984; Shimshock and de Pena, 1989). In one study, rainout contributed about 1/3 of the deposition of NO3 − but 50–80% of the deposition of SO4 2 −, which typically forms cloud condensation nuclei (Aikawa et al., 2014). The dissolved content in both fractions represents the results of heterogeneous reactions between gases and raindrops in the atmosphere.
The relative contribution of rainout and washout varies depending on the length of the rainstorm. As washout cleanses the lower atmosphere, the content of dissolved materials in rainfall declines. Thus, the concentration of dissolved constituents in precipitation is inversely related to the rate of precipitation (Gatz and Dingle, 1971) and to the total volume that has fallen (Likens et al., 1984; Lesack and Melack, 1991; Minoura and Iwasaka, 1996). The concentration of dissolved constituents also varies inversely as a function of mean raindrop size (Georgii and Wötzel, 1970; Bator and Collett, 1997). This inverse relation explains why extremely high concentrations of dissolved constituents are found in fog waters (Weathers et al., 1986; Waldman et al., 1982; Clark et al., 1998; Elbert et al., 2000). Capture of fog and cloud water by vegetation is an important component of the deposition of nutrient elements from the atmosphere in some high-elevation and coastal ecosystems (Lovett et al., 1982; Waldman et al., 1985; Weathers et al., 2000; Templer et al., 2015).
The relative efficiency of scavenging by rainwater is often expressed as the washout ratio:
| (3.37) |
With units of m3/L, this ratio gives an indication of the volume of atmosphere cleansed by each liter of rainfall as it falls. Large ratios are generally found for ions that are derived from relatively large aerosols or from highly water-soluble gases in the atmosphere. Snowfall is generally less efficient at scavenging than rainfall.
Whereas the deposition of nutrients by precipitation is often called wetfall, dryfall is the result of gravitational sedimentation of particles during periods without rain (Hidy, 1970; Wesely and Hicks, 2000). Dryfall of dusts downwind of arid lands is often spectacular; Liu et al. (1981) reported 100/g/m2 /h of dustfall in Beijing, China, as a result of a single dust storm on 18 April 1980. In some regions, enormous deposits of wind-deposited soil, known as loess, were laid down during glacial periods, when large areas of semiarid land were subject to wind erosion (Pye, 1987; Simonson, 1995; Muhs et al., 2001). Today, various elements necessary for plant growth are released by chemical weathering of soil minerals in these deposits (Chapter 4).
The dryfall received in many areas contains a significant fraction that is easily dissolved by soil waters and immediately available for plant uptake. Despite the high rainfall found in the southeastern United States, Swank and Henderson (1976) reported that 19–64% of the total annual atmospheric deposition of ions such as Ca, Na, K, and Mg, and up to 89% of the deposition of P, was derived from dryfall. Dryfall inputs of P may assume special significance to plant growth in areas where the release of P from rock weathering is very small (Newman, 1995; Chadwick et al., 1999; Okin et al., 2004). Dry deposition contributes about 30–60% of the deposition of sulfur in New Hampshire (Likens et al., 1990; cf. Tanaka and Turekian, 1995). Similarly, 34% of the atmospheric inputs of nitrogen to Harvard Forest (Massachusetts) are derived from dry deposition (Munger et al., 1998). Organic nitrogen compounds deposited from the atmosphere are decomposed by soil microbes, providing additional plant nutrients (Neff et al., 2002a; Mace et al., 2003; Zhang et al., 2012c).
Dryfall is often measured in collectors that are designed to close during rainstorms. When open to the atmosphere, these instruments capture particles that are deposited vertically, known as sedimentation. In natural ecosystems, dryfall is also derived by the capture of particles on vegetation surfaces. When vegetation captures particles that are moving horizontally in the airstream, the process is known as impaction (Hidy, 1970). Impaction is a particularly important process in the capture of seasalt aerosols near the ocean (Art et al., 1974; Potts, 1978).
In addition to the uptake of CO2 in photosynthesis, vegetation also absorbs N- and S-containing gases directly from the atmosphere (Hosker and Lindberg, 1982; Lindberg et al., 1986; Sparks et al., 2003; Turnipseed et al., 2006). Uptake of pollutant O3, SO2, and NO2 by vegetation is particularly important in humid regions (McLaughlin and Taylor, 1981; Rondon and Granat, 1994), where plant stomata remain open for long periods. Lovett and Lindberg, 1986, Lovett and Lindberg, 1993 found that uptake of HNO3 vapor accounted for 75% of the annual dry deposition of nitrogen (4.8 kg/ha) in a deciduous forest in Tennessee, where dry deposition was nearly half of the total annual deposition of nitrogen from the atmosphere. Globally, dry deposition of NO2 and SO2 comprises a significant fraction of the total deposition of N and S on land (Nowlan et al., 2014; Jaeglé et al., 2018). Vegetation can also be a source or a sink for atmospheric NH3, depending on the ambient concentration in the atmosphere (Langford and Fehsenfeld, 1992; Sutton et al., 1993; Pryor et al., 2001), and plants can also remove volatile organic compounds from the air (Simonich and Hites, 1994).
The total capture of dry particles and gases by land plants is difficult to measure. When rainfall is collected inside a forest, it contains materials that have been deposited on the plant surfaces, but also large quantities of elements that are derived from the plants themselves (Parker, 1983, Chapter 6). Artificial collectors (surrogate surfaces) are often used to approximate the capture by vegetation (White and Turner, 1970; Vandenberg and Knoerr, 1985; Lindberg and Lovett, 1985). The capture on known surfaces can be compared to the airborne concentrations to calculate a deposition velocity (Sehmel, 1980):
| (3.38) |
In units of cm/s, these velocities can be multiplied by the estimated surface area of vegetation (cm2) and the concentration in the air to calculate total deposition for an ecosystem. For example, Lovett and Lindberg et al. (1986) used a deposition velocity of 2.0 cm/s to calculate a nitrogen deposition of 3.0 kg/ha/yr in a forest with a leaf area indexo of 5.8 m2/m2 and an ambient concentration of 0.82 mg N m3 in the form of nitric acid vapor. It is often unclear if deposition velocities measured using artificial surfaces apply to natural surfaces (e.g., bark), and accurate estimates of the surface area of vegetation are difficult (Whittaker and Woodwell, 1968). Clearly, further work on dry deposition is needed (Lovett, 1994; Petroff et al., 2008).
Atmospheric deposition on the surface of the sea is often estimated from collections of wetfall and dryfall on remote islands (Duce et al., 1991). The surface of the sea can also exchange gases with the atmosphere (Liss and Slater, 1974), often acting as a sink for atmospheric CO2 (Sabine et al., 2004) and SO2 (Beilke and Lamb, 1974) and as a source of NH3 (Paulot et al., 2016) and dimethylsulfide (Chapter 9).
Regional patterns and trends
Regional patterns of rainfall chemistry in the United States reflect the relative importance of different constituent sources and deposition processes in different areas (Munger and Eisenreich, 1983). Coastal areas are dominated by atmospheric inputs from the sea, with large inputs of Na, Mg, Cl, and SO4 that are the major constituents in the seasalt aerosol (Junge and Werby, 1958; Hedin et al., 1995). Areas of arid and semiarid land show high concentrations of soil-derived constituents, such as Ca, in rainfall (Fig. 3.13 ; Young et al., 1988; Sequeira, 1993; Gillette et al., 1992). Areas downwind of regional pollution show exceedingly low pH and high concentrations of SO4 2 − and NO3 − (Schwartz, 1989; Ollinger et al., 1993), whereas agricultural areas have high deposition of NH4 + (Stephen and Aneja, 2008).
Fig. 3.13.
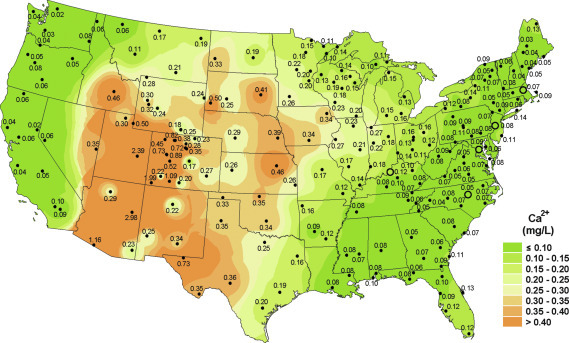
Mean calcium concentration (mg/L) in wetfall precipitation in the United States for 2017.
From the National Atmospheric Deposition Program http://nadp.slh.wisc.edu/maplib/pdf/2017/Ca_conc_2017.pdf.
The ratio among ionic constituents in rainfall can be used to trace their origin. Except in unusual circumstances, nearly all the sodium (Na) in rainfall is derived from the ocean. When magnesium is found in a ratio of 0.12 with respect to Na—the ratio in seawater (refer to Table 9.1)—we may presume that most of the Mg is also of marine origin. In the southeastern United States, however, Mg/Na ratios in wetfall range from 0.29 to 0.76 (Swank and Henderson, 1976). Here the Mg content has increased relative to Na, presumably because the airflow that brings precipitation to this region has crossed the United States, picking up Mg from soil dust and other sources. Schlesinger et al. (1982) used this approach to deduce nonmarine sources of Ca and SO4 in the rainfall in coastal California (Fig. 3.14 ).
Fig. 3.14.
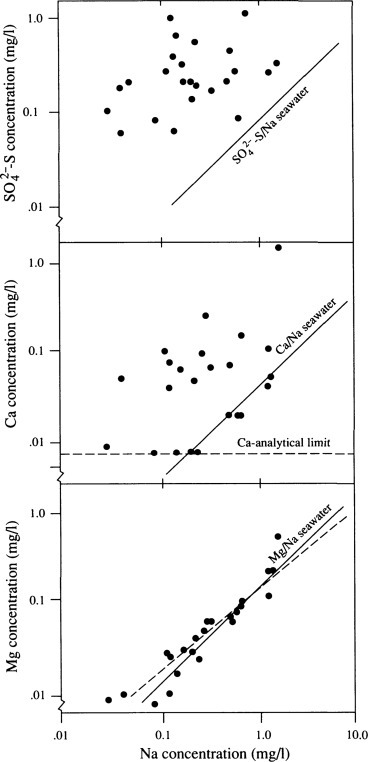
Concentrations of SO4, Ca, and Mg in wetfall precipitation near Santa Barbara, California (USA), plotted as a logarithmic function of Na concentration in the same samples (Schlesinger et al., 1982). The solid line represents the ratio of these ions to Na in seawater. Ca and SO4 are enriched in wetfall relative to seawater, whereas Mg shows a correlation (dashed line) that is not significantly different from the ratio expected in seawater.
Iron (Fe) and aluminum (Al) are largely derived from the soil, and ratios of various ions to these elements in soil can be used to predict their expected concentrations in rainfall when soil dust is a major source (Lawson and Winchester, 1979; Warneck, 2000). High concentrations of Al in dryfall on Hawaii were traced to springtime dust storms on the central plains of China (Parrington et al., 1983). Soil mineralogy in dusts from the 1930s’ Great Plains dust bowl can be identified in layers of the Greenland ice pack (Donarummo et al., 2003). Windborne particles of soil and vegetation contribute significantly to the global transport of trace metals in the atmosphere (Nriagu, 1989, Table 1.1).
In many areas downwind of pollution, a strong correlation between H+ and SO4 2 − is the result of the production of H2SO4 during the oxidation of SO2 and its dissolution in rainfall (Eqs. (3.27), (3.28); Cogbill and Likens, 1974; Irwin and Williams, 1988). Nitrate (NO3 −) also contributes to the strong acid content in rainfall (HNO3). These constituents depress the pH of rainfall below 5.6, which would be expected for water in equilibrium with atmospheric CO2 (Galloway et al., 1976). In contrast, ammonia (NH3) is a net source of alkalinity in rainwater, since its dissolution produces OH:
| (3.39) |
The pH of rainfall is determined by the concentration of strong acid anions that are not balanced by NH4 + and Ca2 + (from CaCO3), namely (from Gorham et al., 1984),
| (3.40) |
In Kanpur, India, ammonia dominated the neutralization of acidity in rainfall during the wet season, while Ca played a similar role in the dry season, when more soil dust was present in the atmosphere (Shukla and Sharma, 2010). Low pH of rainfall in Europe is partially alleviated by Ca from the Sahara desert and NH3 emissions from agriculture (Lajtha and Jones, 2013).
Globally, about 37% of the atmosphere's acidity is neutralized by NH3 (Chapter 13), with a higher proportion in the Southern Hemisphere where there is less industrial pollution (Savoie et al., 1993). Across the United States, airborne concentrations of NH3 and the proportional deposition of NH4 + have increased markedly in recent years, as air pollution regulations have lowered the emissions of NOx (Kharol et al., 2018; Li et al., 2016a). Reduced forms of N (i.e., NH4) are also important in China (Zhan et al., 2015). Monitoring of ammonia in the atmosphere has improved markedly with its monitoring on tall towers (Griffis et al., 2019) and the application of satellite remote sensing (Van Damme et al., 2015, Van Damme et al., 2018; Warner et al., 2017; Kharol et al., 2018).
In the eastern United States, the acidity of rainfall is often directly correlated to the concentration of SO4 2 −, which is related to pollutant emissions of SO2 in areas upwind (Likens et al., 2005). Similar relationships are seen between emissions of NOx and the NO3 − content of rain (Butler et al., 2003, Butler et al., 2005). In the western United States, the relationship between acidity and the acid-forming anions is less clear because they have often reacted with soil aerosols containing CaCO3 (Young et al., 1988).
In the past 100 years, increases in the concentration of NO3 and SO4 in the Greenland and Tibetan snowpacks have reflected the changes in the abundance of anthropogenic pollutants due to industrialization in the Northern Hemisphere (Mayewski et al., 1986, Mayewski et al., 1990; Thompson et al., 2000). There are no apparent changes in the deposition of these ions in the Southern Hemisphere as recorded by Antarctic ice (Langway et al., 1994). Similarly, the uppermost sediments in lakes and peat bogs of the Northern Hemisphere contain higher concentrations of many trace metals, presumably from industrial sources (Galloway and Likens, 1979; Swain et al., 1992; Allan et al., 2013).
Long-term records of precipitation chemistry are rare, but the collections at the Hubbard Brook Ecosystem in central New Hampshire and eastern Tennessee suggest a recent decline in the concentrations of SO4 that may reflect improved control of emissions (Likens et al., 1984, Likens et al., 2002; cf. Kelly et al., 2002; Zbieranowski and Aherne, 2011; Lutz et al., 2012b). Improvements in air quality as a result of the implementation of the Clean Air Act in 1990 have resulted in significant decreases in the acidity of rainfall over the eastern United States and Canada (Figs. 3.15 and 13.2; Hedin et al., 1987; Lajtha and Jones, 2013). Similarly, the uppermost layers of ice in glaciers of the French Alps and Greenland show lower concentrations of SO4 2 − and NO3 −, presumably due to control of pollutant emissions in upwind sources in recent years (Preunkert et al., 2001, Preunkert et al., 2003; Fischer et al., 1998).
Fig. 3.15.
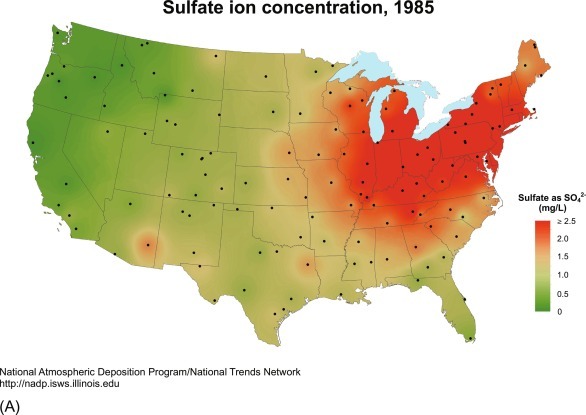
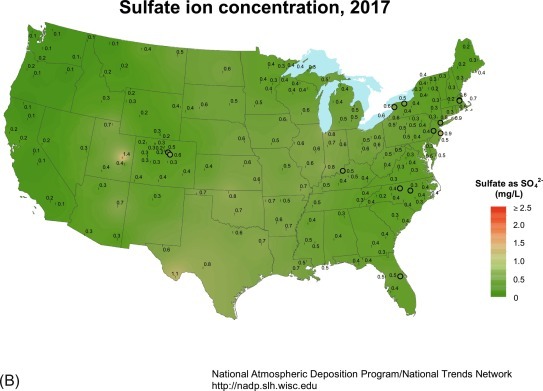
Sulfate (SO4) concentration (mg/L) measured in samples of wetfall precipitation across the United States, showing the effect of the Clean Air Act in reducing SO2 emissions and thus, SO4 deposition between 1985 and 2017.
From the National Atmospheric Deposition Program.
Even with pollutant abatement, long-term records suggest that many natural ecosystems currently receive a greater input of N, S, and other elements of biogeochemical importance than before widespread emissions from human activities. Pollutant emissions have more than doubled the annual input of S-containing gases to the atmosphere globally (Chapter 13). Global deposition of inorganic nitrogen has increased about 8% between 1984 and 2016 (Ackerman et al., 2019), with most of the increase seen for NH4 + (Li et al., 2016a; Kharol et al., 2018). Excess deposition of nitrogen might be expected to enhance the growth of forests, but in combination with acidity, this fertilization effect may lead to deficiencies of P, Mg, and other plant nutrients (Chapters 4 and 6). Atmospheric deposition of nitrogen makes a significant contribution to the nutrient load and eutrophication of lakes (Bergstrom and Jansson, 2006), estuaries (Nixon et al., 1996; Latimer and Charpentier, 2010), and coastal waters (Paerl et al., 1999). The western North Atlantic Ocean receives about 20–40% of the sulfur and nitrogen oxides emitted in eastern North America (Galloway and Whelpdale, 1987; Liang et al., 1998; Jaeglé et al., 2018), and increasing nitrogen deposition is recorded in the North Pacific Ocean (Kim et al., 2014). Although pollutant emissions have declined in North America and Europe, increasing emissions are seen from India and China (Lelieveld et al., 2001; Richter et al., 2005; Stern, 2006). The airborne concentrations of many air pollutants can now be measured using satellite technology (Richter et al., 2005; Clarisse et al., 2009; Martin, 2008; Yang et al., 2013a, Yang et al., 2014; Griffin et al., 2019).
Biogeochemical reactions in the stratosphere
Ozone
Ozone is produced in the stratosphere by the dissociation of oxygen atoms that are exposed to shortwave solar radiation. The reaction accounts for most of the absorption of ultraviolet sunlight (hv) at wavelengths of 180–240 nm and proceeds as follows:
| (3.41) |
| (3.42) |
Some ozone from the stratosphere mixes down into the troposphere, where the production of O3 by these reactions is limited because there is less ultraviolet light. Most of the remaining ozone is destroyed by a variety of reactions in the stratosphere. Absorption of ultraviolet light at wavelengths between 200 and 320 nm destroys ozone:
| (3.43) |
| (3.44) |
This absorption warms the stratosphere (refer to Fig. 3.1) and protects the Earth's surface from the ultraviolet portion of the solar spectrum that is most damaging to living tissue (uvB). Stratospheric ozone is also destroyed by reaction with OH (Wennberg et al., 1994),
| (3.45) |
| (3.46) |
and by reactions stemming from the presence of nitrous oxide (N2O), which mixes up from the troposphere. Tropospheric N2O is produced in a variety of wkays (Chapters 6 and 12), but it is inert in the lower atmosphere. The only significant sink for N2O is photolysis in the stratosphere. About 80% of the N2O reaching the stratosphere is destroyed in a reaction producing N2 (Warneck, 2000),
| (3.47) |
and about 20% in reactions with the O(1D) produced in Eq. 3.47, mainly
| (3.48) |
but with a small amount forming NO:
| (3.49) |
The nitric oxide (NO) produced in reaction 3.49 destroys ozone in a series of reactions,
| (3.50) |
| (3.51) |
| (3.52) |
for which the net reaction is:
| (3.53) |
Note that the mean residence time of NO in the troposphere is too short for an appreciable amount to reach the stratosphere, where it might contribute to the destruction of ozone. Nearly all the NO in the stratosphere is produced in the stratosphere from N2O; only a small amount is contributed by high-altitude aircraft.
Eventually NO2 is removed from the stratosphere by reacting with OH to produce nitric acid (Eq. 3.26), which mixes down to the troposphere and is removed by the heterogeneous reaction with raindrops.p
Finally, stratospheric ozone is destroyed by chlorine, which acts as a catalyst in the reaction:
| (3.54) |
| (3.55) |
| (3.56) |
for a net reaction of:
| (3.57) |
Although each Cl produced may cycle through these reactions and destroy many molecules of O3, Cl is eventually converted to HCl and removed from the stratosphere by downward mixing and heterogeneous interaction with cloud drops in the troposphere (Rowland, 1989; Solomon, 1990).
The balance between ozone production (refer to Eqs. (3.41), (3.42)) and the various reactions that destroy ozone maintains a steady-state concentration of stratospheric O3 with a peak of approximately 7 × 1018 molecules/m3 at 30 km altitude (Warneck, 2000). Although the photochemical production of O3 is greatest at the equator, the density of the ozone layer is normally thickest at the poles.
Since the mid-1980s, field measurements have indicated that the total density of ozone molecules in the atmospheric column has declined significantly over Antarctica, resulting in an ozone “hole” over a large area (Farman et al., 1985; Fig. 3.16 ).q The decline, as much as 0.3%/yr, was unprecedented and represents a perturbation of global biogeochemistry. Destruction of ozone is likely to lead to an increased flux of ultraviolet radiation to the Earth's surface (Correll et al., 1992; Kerr and McElroy, 1993; McKenzie et al., 1999) and increased incidence of skin cancer and cataracts in humans (Norval et al., 2007). Greater uvB radiation at the Earth's surface is likely to reduce marine production in the upper water column of the Southern Ocean around Antarctica (Smith et al., 1992b; Meador et al., 2002; Arrigo et al., 2003). Ultraviolet radiation also causes deleterious effects on land plants (Caldwell and Flint, 1994; Day and Neale, 2002). Because previous, steady-state ozone concentrations were maintained in the face of natural photochemical reactions that produce and consume ozone, attention focused on how this balance might have been disrupted by human activities (Cicerone, 1987; Rowland, 1989; Hegglin et al., 2015).
Fig. 3.16.
The average abundance of ozone in the atmosphere of the Southern Hemisphere during October 2006. The ozone “hole,” seen in blue and purple, is actually the area where the abundance of ozone in the stratosphere is below 220 Dobson units—perhaps better described as a thinning rather than a hole. Data are in Dobson Units for the entire atmosphere, see Footnote n, p. 75.
From http://ozonewatch.gsfc.nasa.gov/monthly/monthly_2006-10.html.
Chlorofluorocarbons (freons), which were produced as aerosol propellants, refrigerants, and solvents, have no known natural source in the atmosphere (Prather, 1985). These compounds are chemically inert in the troposphere, so they eventually mix into the stratosphere where they are decomposed by photochemical reactions producing active chlorine (Molina and Rowland, 1974; Rowland, 1989, Rowland, 1991):
| (3.58) |
which can destroy ozone by the reactions of Eqs. (3.54), (3.55), (3.56). These reactions are greatly enhanced in the presence of ice particles, which accounts for the first observations of the O3 “hole” in the springtime over Antarctica (Farman et al., 1985; Solomon et al., 1986). In the absence of ice particles atmosphere, ClO reacts with NO2 to form ClONO2, an inactive compound that removes both gases from O3 destruction. In the presence of ice clouds, ClONO2 breaks down:
| (3.59) |
| (3.60) |
producing active chlorine for ozone destruction (Molina et al., 1987; Solomon, 1990). Significantly, during the last 40 years, levels of active chlorine over Antarctica have increased in a mirror image to the loss of ozone from the stratosphere (Solomon, 1990). Stratospheric temperatures in the arctic are not as cold as over Antarctica, so there a fewer ice particles and a lower capacity for ozone destruction.
The relative importance of chlorofluorocarbons versus natural sources of chlorine in the stratosphere is apparent in a global budget for atmospheric chlorine (Fig. 3.17 ). Seasalt aerosols are the largest natural source of chlorine in the troposphere, but they have such a short mean residence time that they do not contribute Cl to the stratosphere. There is also no good reason to suspect that seasalt aerosols have increased in abundance in the last few decades. Similarly, industrial emissions of HCl are rapidly removed from the troposphere by rainfall. Especially violent volcanic eruptions can inject gases directly into the stratosphere, sometimes adding to stratospheric Cl (Johnston, 1980; Mankin and Coffey, 1984; Cadoux et al., 2015). However, in most cases only a small amount of Cl reaches the stratosphere, because various processes remove HCl from the rising volcanic plume (Tabazadeh and Turco, 1993; Textor et al., 2003). After the Mount Pinatubo eruption, which released 4.5 × 1012 g of HCl, stratospheric Cl increased by < 1% (Mankin et al., 1992).
The only significant natural source of Cl in the stratosphere stems from the production of methylchlorider by the ocean surface, by plants, especially tropical vegetation, and by forest fires (Table 3.7 ). Methylchloride concentrations have fluctuated during the Holocene (Verhulst et al., 2013), but there is no strong implication of an industrial source that might be indicated by an increase in the concentration of CH3Cl in the Antarctic ice pack during the past 100 years (Butler et al., 1999; Saltzman et al., 2009), or by a significant difference in the concentration of methylchloride between the Northern and Southern Hemispheres (Beyersdorf et al., 2010). The current budget for CH3Cl is slightly imbalanced, with sources exceeding sinks. Methylchloride has a mean residence time of about 1.3 years in the atmosphere, so a small portion mixes into the stratosphere.
Table 3.7.
Budgets of CH3Cl and CH3Br in the atmosphere (Tg/yr).
| CH3Cl | CH3Br | References | |
|---|---|---|---|
| Sources | |||
| Oceans | 0.70 | 0.0015 | Hu et al., 2012, Hu et al., 2013 |
| Coastal vegetation | 0.03–0.17 | 0.001–0.008 | Rhew et al., 2014 |
| Hu et al., 2010 | |||
| Deventer et al., 2018 | |||
| Upland vegetation | 2.20 | Verhulst et al., 2013 | |
| Bahlmann et al., 2019 | |||
| Freshwater wetlands | 0.74 | 0.035 | Hardacre and Heal, 2013 |
| Biomass burning | 0.73 | 0.034 | Andreae, 2019 |
| Verhulst et al., 2013 | |||
| Industrial uses | 0.11–0.16 | 0.05 | McCulloch et al., 1999 |
| Thompson et al., 2002 | |||
| Total of sources (best estimates) |
4.60 | 0.12 | |
| Sinks | |||
| Ocean uptake | 0.37 | 0 | Hu et al., 2013 |
| Soil uptake | 0.25 | 0.022–0.042 | Shorter et al., 1995 |
| Serca et al., 1998 | |||
| Reaction with OH | 3.37 | 0.09 | Thompson et al., 2002 |
| Loss to stratosphere | 0.28 | 0.006 | Thompson et al., 2002 |
| Total of sinks (best estimates) |
4.27 | 0.24 | |
In the global Cl budget, the relatively small industrial production of chlorofluorocarbons, which are inert in the troposphere, is a major source of Cl delivered to the stratosphere (Fig. 3.16; Russell et al., 1996). Increasing concentrations of these compounds have been strongly implicated in ozone destruction (Rowland, 1989; Butler et al., 1999). Happily, with the advent of the Montreal Protocol in 1987, which limits the use of these compounds worldwide,s there is already some evidence that the growth rate of these compounds in the atmosphere is slowing (Elkins et al., 1993; Montzka et al., 1996; Solomon et al., 2006) and that the ozone hole may be starting a slow recovery (Fig. 3.18; Solomon et al., 2016; Chipperfield et al., 2017).t Indeed, the main cause of continued human impacts on stratospheric ozone may stem from our continuing contributions to the rise in N2O in Earth's atmosphere (Ravishankara et al., 2009; Chapter 12).
Fig. 3.18.
Size of the ozone hole, defined as the area over which the ozone content is less than 220 Dobson Units, over Antarctica since the 1980s, showing its stabilization in recent years as a result of the Montreal Protocol, instituted in 1987.
Adapted and updated from Solomon et al. (2016).
Similar reactions are possible with compounds containing bromine; in fact, Br compounds may be even more potent in the destruction of stratospheric O3 than Cl (Wennberg et al., 1994). Industry is a source of methylbromide (CH3Br), which has a long history of use as an agricultural fumigant (Yagi et al., 1995). Methylbromide is also released from the ocean's surface, vegetation, and biomass burning (Table 3.7). Sinks of CH3Br include uptake by the oceans and soils and oxidation by OH radical. After rising throughout the Industrial Revolution (Saltzman et al., 2008; Khalil et al., 1993a), the atmospheric concentration of methylbromide appears to have declined in recent years (Yvon-Lewis et al., 2009). The global budget of CH3Br and its mean residence time (about 0.8 years; Colman et al., 1998) in the atmosphere are poorly constrained—sinks exceed sources (Table 3.7). However, some CH3Br persists long enough to reach the stratosphere, where it can lead to ozone destruction.
Among other halogen-containing gases, the lifetimes of bromoform (CHBr3; Quack and Wallace, 2003; Stemmler et al., 2015) and methyliodide (CH3I; Bell et al., 2002; Stemmler et al., 2014; Yokouchi et al., 2012), both produced by marine phytoplankton, and various inorganic fluoride compounds (e.g., CH3F) are too short for appreciable mixing into the stratosphere. The observed increase of fluoride in the stratosphere appears solely due to the upward transport of chlorofluorocarbons, and it is an independent verification of their destruction in the stratosphere by ultraviolet light (Russell et al., 1996). However, F is ineffective as a catalyst for ozone destruction.
Satellite observations have greatly aided our understanding of changes in stratospheric ozone. The loss of ozone from the atmosphere has been monitored since 1979 when the first Total Ozone Mapping Spectrometer (TOMS) began records of the abundance of O3 in a column extending from the bottom to the top of the atmosphere (Fig. 3.18 ). Both the area and the minimum column abundance of ozone appear to have stabilized in recent years, after strong O3 losses at the beginning of the record.u Similar, though less extensive, ozone losses are reported for the Arctic (Solomon et al., 2007; Manney et al., 2011) resulting in greater exposure to uVB radiation in Europe (Petkov et al., 2014).
Stratospheric sulfur compounds
Sulfate aerosols in the stratosphere are important to the albedo of the Earth (Warneck, 2000). A layer of sulfate aerosols, known as the Junge layer, is found in the stratosphere at about 20 to 25 km altitude. Its origin is twofold. Large volcanic eruptions can inject SO2 into the stratosphere, where it is oxidized to sulfate (Eqs. (3.27), (3.28)). Large eruptions have the potential to increase the abundance of stratospheric sulfate 100-fold (Arnold and Bührke, 1983; Hofmann and Rosen, 1983), and these sulfate aerosols persist in the stratosphere for several years, cooling the planet (McCormick et al., 1995; Briffa et al., 1998; Sigl et al., 2015). Following the eruption of Tambora in Indonesia, the year 1816 was known as the “year without a summer” in New England and much of Europe.
During periods without volcanic activity, the dominant source of stratospheric sulfate derives from carbonyl sulfide (COS)v that mixes up from the troposphere (Sheng et al., 2015). Showing an average concentration of about 500 parts per trillion, COS is the most abundant sulfur gas in the atmosphere (Table 3.1). The pool in the atmosphere contains about 2.8 × 1012 g S (Chin and Davis, 1995). Based on the global budget shown in Table 3.8 , the mean residence time for COS in the atmosphere is ~ 5 years. Whereas most sulfur gases are so reactive that their mean residence time is much less than one year, the mean residence time of COS allows about one-third of the annual production to enter the stratosphere. Additional COS may be lofted to the stratosphere in the smoke plumes of large wildfires (Notholt et al., 2003).
Table 3.8.
Global budget for carbonyl sulfide (COS) in the atmosphere.
| Source or sink | COS (1012 g S /yr) | References |
|---|---|---|
| Sources | ||
| Oceans | 0.04a | Kettle et al., 2002 |
| Anoxic soils | 0.03 | Kettle et al., 2002 |
| Biomass burning | 0.31–0.60 | Andreae, 2019 |
| Stinecipher et al., 2019 | ||
| Industrial | 0.26 | Campbell et al., 2015 |
| Volcanoes | 0.02 | Chin and Davis, 1993 |
| Oxidation of natural CS2 | 0.14b | Kettle et al., 2002 |
| Campbell et al., 2015 | ||
| Oxidation of DMS | 0.15 | Kettle et al., 2002 |
| Total sources | 0.95–1.25 | |
| Sinks | ||
| Vegetation uptake | 0.24–0.74 | Kettle et al., 2002 |
| Berry et al., 2013 | ||
| Soil uptake (oxic) | 0.13 | Kettle et al., 2002 |
| Oxidation by OH | 0.09 | Kettle et al., 2002 |
| Stratospheric photolysis | 0.02 | Kettle et al., 2002 |
| Total sinks | 0.48–0.98 | |
net.
assumes that 44% of the global annual CS2 source is industrial.
The global budget for COS in the atmosphere has been revised repeatedly during the last few decades, and a current budget of COS shows a slight excess of sources over sinks. Several components of the budget, including net ocean uptake, are poorly constrained (Table 3.8). The concentration of COS in the atmosphere has increased during the Industrial Revolution (Aydin et al., 2002; Montzka et al., 2004), but it has declined slightly during the past couple of decades (Sturges et al., 2001; Rinsland et al., 2002), perhaps due to enhanced plant uptake (Campbell et al., 2017).
A large source of COS in the atmosphere stems from the oxidation of carbon disulfide (CS2) emitted from anoxic soils and industrial sources. The oceans are an indirect source of COS, stemming from the oxidation of dimethylsulfide, emitted from phytoplankton (Chapters 9 and 13). In its global budget (Table 3.8), the small direct source of COS from the oceans is a net value, recognizing that COS is also taken up by seawater (Weiss et al., 1995). Wetland soils are a small source, and the global emission of COS from salt marshes is limited by the small extent of salt marsh vegetation (Aneja et al., 1979; Steudler and Peterson, 1985; Carroll et al., 1986). Other sources of COS include biomass burning (Nguyen et al., 1995; Andreae, 2019) and direct industrial emissions, including combustion of coal (Zumkehr et al., 2017; Du et al., 2016). Anthropogenic sources may account for half of the annual sources of COS in the atmosphere (Zumkehr et al., 2018).
Some COS is oxidized in the troposphere via OH radicals, but the major tropospheric sink for COS, first reported by Goldan et al. (1988), appears to be uptake by vegetation and upland soils (Steinbacher et al., 2004; Kuhn et al., 1999; Simmons et al., 1999). COS shows seasonal fluctuations in the atmosphere, parallel to CO2 and likely to reflect plant activity (Montzka et al., 2004, Montzka et al., 2007; Campbell et al., 2008; Geng and Mu, 2006). Kesselmeier and Merk (1993) found that a variety of crop plants take up COS whenever the ambient concentration is greater than 150 ppt. Uptake by vegetation is now believed to account for half of the total annual destruction of COS globally (Table 3.8), and several workers have suggested that measurements of changes in the abundance of COS in the atmosphere may be useful as an indirect measure of changes in global photosynthesis (Blonquist et al., 2011; Chapter 5).
The small amount of COS that mixes into the stratosphere is destroyed by a photochemical reaction involving the OH radicals, producing SO4 and contributing to the Junge layer. In fact, aside from the periodic eruptions of large volcanoes, COS appears to be the main source of SO4 aerosols in the stratosphere (Hofmann and Rosen, 1983; Servant, 1986). Eventually, these aerosols are removed from the stratosphere by downward mixing of stratospheric air.
There is some evidence that these sulfate aerosols have increased in the stratosphere recent years (Hofmann, 1990). These aerosols affect the amount of solar radiation entering the troposphere, and they are an important component of the radiation budget of the Earth (Turco et al., 1980). Through direct and indirect (CS2) sources, humans appear to make large contributions to the budget of COS (Table 3.8), and any increase in COS-derived aerosols in the stratosphere has potential consequences for predictive models of future global warming (Hofmann and Rosen, 1980). In return, global warming and an increasing flux of uvB radiation penetrating to the Earth's surface may enhance the production of COS in ocean waters (Najjar et al., 1995).
The effect of volcanic eruptions that loft SO2 to the stratosphere is so dramatic that some workers have suggested organized programs to seed SO4 aerosols in the stratosphere to combat global climate change (Crutzen, 2006). This form of geoengineering is not without critics, given its potential impacts on other aspects of Earth system function (Robock et al., 2009; Keith et al., 2016).
Models of the atmosphere and global climate
A large number of models have been developed to explain the physical properties and chemical reactions in the atmosphere. When these models attempt to predict the characteristics in a single column of the atmosphere, they are known as one-dimensional (1D) and radiative convective models. For example, Fig. 3.2 is a simple 1D-model for the greenhouse effect, which assumes that the behavior of the Earth's atmosphere can be approximated by average values applied to the entire surface. Two-dimensional (2D) models can be developed using the vertical dimension and a single horizontal dimension (e.g., latitude) to examine the change in atmospheric characteristics across a known distance of the Earth's surface (e.g., Brasseur and Hitchman, 1988; Hough and Derwent, 1990). On a regional scale, these are particularly useful in following the fate of pollution emissions (e.g., Rodhe, 1981; Asman and van Jaarsveld, 1992; Berge and Jakobsen, 1998). Three-dimensional (3D) models attempt to follow the fate of particular parcels of air as they move both horizontally and vertically in the atmosphere. These dynamic 3D models are known as general circulation models (GCMs) for the globe (Fig. 3.19 ).
Fig. 3.19.
Conceptual structure of a dynamic three-dimensional general circulation model for the Earth's atmosphere, indicating the variables that must be included for a global model to function properly.
From Henderson-Sellers and McGuffie (1987). Copyright ©1987. Reprinted by permission of John Wiley and Sons, Ltd.
Many models are constructed to include both chemical reactions and physical phenomena, such as the circulation of the atmosphere due to temperature differences. Chemical transformations are parameterized using the rate and equilibrium coefficients for the reactions that we have examined in this chapter. Because there are a large number of reactions, most of these models are quite complex (e.g., Logan et al., 1981; Isaksen and Hov, 1987; Lelieveld and Crutzen, 1990), but they give useful predictions of future atmospheric composition when the input of several constituents is changing simultaneously.
Nearly all climate models predict that a substantial warming of the atmosphere (2–4.5 °C) will accompany increasing concentrations of CO2, N2O, CH4, and chlorofluorocarbons in the atmosphere (IPCC, 2013).w Largely stemming from fossil fuel combustion, the concentration of CO2 is now higher than at any time in the past 5 million years (Stap et al., 2016). During the past 150 years, the concentrations of CH4, and N2O have risen above levels seen at any time during the past 10,000 years—spanning the entire history of human civilization (Flückiger et al., 2002). Atmospheric warming, resulting from the absorption of infrared (heat) radiation emitted from the Earth's surface, is known as the greenhouse effect or radiative forcing (Fig. 3.2). The predicted warming of future climate is greatest near the poles, where there is normally the greatest net loss of infrared radiation relative to incident sunlight (Manabe and Wetherald, 1980). Dramatic, recent declines in the extent of Arctic sea ice suggest that these predictions are already proving correct (Serreze et al., 2007; Notz and Stroeve, 2016; Chapter 10). For the same reason, future nighttime and wintertime temperatures worldwide are likely to show large changes relative to today's conditions. Presumably the oceans will warm more slowly than the atmosphere, but eventually warmer ocean waters will allow greater rates of evaporation, increasing the circulation of water in the global hydrologic cycle (Chapter 10). Water vapor also absorbs infrared radiation, so it is likely to further accelerate the potential greenhouse effect (Raval and Ramanathan, 1989; Rind et al., 1991; Soden et al., 2005; Willett et al., 2007). Thus, most models predict that higher concentrations of CO2 and other trace gases in the atmosphere will make the Earth a warmer and more humid planet.
Incoming solar radiation delivers about 340 W/m2 to the Earth (Fig. 3.2).x The natural greenhouse effect warms the planet about 33o C by trapping 153 W/m2 of outgoing radiation (Ramanathan, 1988).y For the past 30 years or so, there has been a small increase in the Sun's luminosity (+ 0.12 to + 0.16 W/m2) (IPCC, 2013; Foukal et al., 2006; Pinker et al., 2005), but the human impact on radiative forcing due to increasing concentrations of atmospheric trace gases currently adds about 2.3 W/m2 to the natural greenhouse effect (IPCC, 2013), causing measurable changes in the spectral distribution of radiation leaving the Earth (Harries et al., 2001). Aerosols tend to cool the atmosphere, and increases in aerosols due to human activities are thought to reduce the global radiative forcing by about 1.2 W/m2 (i.e., global dimming; IPCC, 2013; Bellouin et al., 2005; Mahowald, 2011). It is interesting to note that aerosol concentrations were higher (Patterson et al., 1999; Lambert et al., 2008) and CO2 concentrations and temperatures were lower during the last glacial period (Fig. 1.4).
Long-term records from tree rings and ice cores document substantial and continuing warming of the global climate, coincident with rising CO2 (Mann et al., 1999; Thompson et al., 2000). Recent temperatures in Europe are greater than at any point in the past 500 years (Luterbacher et al., 2004). These changes in climate cannot be explained by natural phenomena alone (Crowley, 2000; Stott et al., 2000). How rapidly these changes in climate occur will be moderated by the thermal buffer capacity of the world's oceans, which can absorb enormous quantities of heat. Already, several long-term records suggest increases in the ocean's temperature worldwide (Barnett et al., 2005; Levitus et al., 2001).
Differential warming of the atmosphere and oceans will also change global patterns of precipitation and evapotranspiration (Manabe and Wetherald, 1986; Rind et al., 1990; Zhang et al., 2007), causing substantial changes in soil moisture of most areas outside the tropics. Arid regions, such as the southwestern United States, are especially likely to experience increased drought (Cook et al., 2004; Seager et al., 2007), consistent with recent trends in rainfall in this region (Milly et al., 2005), and discussed further in Chapter 10.
Climate change affects biogeochemistry (and vice versa) in a variety of ways. Land plants and ocean waters take up substantial quantities of CO2 from the atmosphere, potentially slowing the rate of climate change (Chapters 5, 9, and 11). Clearing vegetation alters the albedo of the Earth's land surface, potentially altering radiative forcing (Forzieri et al., 2017). Warmer temperatures may increase the rate of decomposition of soil carbon now frozen in Arctic permafrost (Dorrepaal et al., 2009; Schuur et al., 2009), and trigger the release of methane now frozen in ocean sediments, resulting in positive feedbacks that further exacerbate global warming (Chapter 11). Changes in climate are likely to affect the distribution of many plants and animals, potentially causing extinctions (Thomas et al., 2004); they also impact a wide range of conditions affecting human health and economic activity (Hsiang et al., 2017).
Summary
In this chapter we have examined the physical structure, circulation, and composition of the atmosphere. Major constituents, such as N2, are rather unreactive and have long mean residence times in the atmosphere. CO2 is largely controlled by plant photosynthetic uptake and by its dissolution in waters on the surface of the Earth. The atmosphere contains a variety of minor constituents, many of which are reduced gases. These gases are highly reactive in homogeneous reactions with hydroxyl (OH) radicals and heterogeneous reactions with aerosols and cloud droplets, which scrub them from the atmosphere. Changes in the concentration of many trace gases are indicative of global change, perhaps leading to future climatic warming and higher surface flux of ultraviolet light. The oxidized products of trace gases are deposited in land and ocean ecosystems, resulting in inputs of N, S, and other elements of biogeochemical significance. Pollution of the atmosphere by the release of oxidized gases containing N and S as a result of human activities results in acid deposition in downwind ecosystems. The enhanced deposition of N and S represents altered biogeochemical cycling on a regional and global basis. Changes in stratospheric ozone and global climate are early warnings of the human impact on the atmosphere of our planet.
Footnotes
NASA provides an excellent visualization of wind patterns across the Earth's surface at: https://youtu.be/w3SmRTh5wJ4.
Assuming exponential decay of a tracer from a well-mixed reservoir that is in steady state, the fractional loss per year (− k) is equal to the reciprocal of the mean residence time in years (i.e., 1/MRT). The amount remaining in the reservoir at any time t (in years) as a fraction of the original content is equal to e− kt, the half-life of the reservoir in years is 0.693/k, and 95% will have disappeared from the reservoir after 3/k years.
For visualization of the mixing of CO2 in Earth's atmosphere over an annual cycle, showing emissions from major industrial regions, see https://youtu.be/x1SgmFa0r04.
CFCs are identified using code in which the units digit is the number of fluorine atoms, the tens digit the number of hydrogen atoms plus one, and the hundreds digit the number of carbon atoms minus one (cf. Table 3.1; Rowland (1989).
Aerosol optical depth (AOD) is a dimensionless value that is the natural logarithm of the ratio of incident to transmitted light through the atmosphere.
NASA visualization shows the global emission and circulation of aerosols, with soil dusts (red), sea salt (blue), smoke (green), and sulfate particles (white) from industrial and volcanic sources. See: https://www.nasa.gov/multimedia/imagegallery/image_feature_2393.html.
Aerosols refract sunlight, so sunsets are particularly red in the years following major volcanic eruptions, as recorded inadvertently in landscape paintings in the 1800s (Zerefos et al., 2014).
The U.S. EPA designates small aerosols, those < 2.5 mm, as PM2.5.
The term “odd nitrogen” is not ideal. In practice it refers only to the various oxidized forms of nitrogen in the atmosphere, including N2O5, but not to NH3, which also has an odd number of N atoms.
Mass of N in the atmosphere (Table 3.1) divided by the global rate of biological nitrogen fixation (430 × 1012 g N/yr) gives a mean residence time of ~ 9,000,000 years for N2 in the atmosphere. Thus, k = 1.1 × 10− 7and 3/k = 27 million years.
NOx (pronounced “knocks”) refers to the sum of NO and NO2. NOy is used to refer to the sum of NOx plus all other oxidized forms of nitrogen—for example, HNO3 and CH3C(O)O2NO2 (peroxyacetyl nitrate or PAN).
Reductions in NOx emissions from power plants during the 2003 North American electrical blackout resulted in widespread reductions in O3 levels (Marufu et al., 2004). NOx emissions were also much lower during the economic shutdown in major cities following the coronavirus outbreak (Bauwens et al., 2020).
The customary unit for the total number of ozone molecules in an atmospheric column, the Dobson Unit, is equivalent to 2.69 × 1016 molecules/cm2 of the Earth's surface.
Leaf area index (LAI) is a measure of the total area of leaves (m2) over a certain area of the ground, usually 1 m2.
Atmospheric chemists refer to this reaction as denitrification. It is not to be confused with the denitrification performed by certain bacteria, which remove NO3 and produce N2 in anaerobic soils and sediments (Chapter 7).
The ozone hole is the area in which the abundance of O3 in the atmosphere is less than 220 Dobson Units (see footnote n, p. 75).
Also known as chloromethane.
Other Cl-containing, ozone depleting substances were also banned by the Montreal Protocol, but there is some evidence of continuing clandestine emissions of CCl4 (Liang et al., 2014) and CFC-11 (Montzka et al., 2018; Rigby et al., 2019).
Unfortunately, some of the replacements for CFCs contribute significantly to the greenhouse warming of the Earth (Wuebbles et al., 2013; Rigby et al., 2014) and their use will be curtailed by a recent international agreement.
Also abbreviated OCS.
Through its industrial use as a solvent, nitrogen trifluoride (NF3) is potentially an important contributor to Earth's greenhouse effect, but it is not included in most assessments of climate change (Prather and Hsu, 2008; Weiss et al., 2008).
The Sun's radiation, measured outside the Earth's atmosphere, delivers 1379 W/m2, known as the solar constant (McElroy, 2002). A one-dimensional model for the Earth's radiation budget shows an annual input of ~ 340 W/m2 because only ¼ of the Earth's surface is exposed to sunlight at any given moment.
The Earth's greenhouse effect is dominated by H2O and CO2. O2 and N2 provide only a trivial contribution to Earth's radiation balance, together adding 0.28 W/m2 to the greenhouse effect (Höpfner et al., 2012).
"To view the full reference list for the book, click here"
References☆
- Ackerman S.A., Chung H. Radiative effects of airborne dust on regional energy budgets at the top of the atmosphere. J. Appl. Meteorol. 1992;31:223–233. [Google Scholar]
- Ackerman D., Millet D.B., Chen X. Global estimates of inorganic nitrogen deposition across four decades. Glob. Biogeochem. Cycles. 2019;33(1):100–107. [Google Scholar]
- Aikawa M., Kajino M., Hiraki T., Mukai H. The contribution of site to washout and rainout: precipitation chemistry based on sample analysis from 0.5 Mm precipitation increments and numerical simulation. Atmos. Environ. 2014;95(October):165–174. [Google Scholar]
- Allan M., Le Roux G., De Vleeschouwer F., Bindler R., Blaauw M., Piotrowska N., Sikorski J., Fagel N. High-resolution reconstruction of atmospheric deposition of trace metals and metalloids since AD 1400 recorded by ombrotrophic peat cores in Hautes-Fagnes, Belgium. Environ. Pollut. 2013;178(July):381–394. doi: 10.1016/j.envpol.2013.03.018. [DOI] [PubMed] [Google Scholar]
- Andreae M.O. In: Global Biomass Burning: Atmospheric, Climatic, and Biospheric Implications. Levine J.S., editor. MIT Press; Cambridge, MA: 1991. Biomass burning: its history, use, and distribution and its impact on environmental quality and global climate; pp. 3–21. [Google Scholar]
- Andreae M.O. Emission of trace gases and aerosols from biomass burning—an updated assessment. Atmos. Chem. Phys. 2019;19(13):8523–8546. [Google Scholar]
- Andreae M.O., Rosenfeld D. Aerosol-cloud-precipitation interactions: Part 1. The nature and sources of cloud-active aerosols. Earth Sci. Rev. 2008;89:13–41. [Google Scholar]
- Andreae M.O., Charlson R.J., Bruynseels F., Storms H., Grieken R.V.A.N., Maenhaut W. Internal mixture of sea salt, silicates, and excess sulfate in marine aerosols. Science. 1986;232(4758):1620–1623. doi: 10.1126/science.232.4758.1620. [DOI] [PubMed] [Google Scholar]
- Andreae M.O., Anderson B.E., Blake D.R., Bradshaw J.D., Collins J.E., Gregory G.L., Sachse G.W., Shipham M.C. Influence of plumes from biomass burning on atmospheric chemistry over the equatorial and tropical South Atlantic during CITE-3. J. Geophys. Res. 1994;99:12793–12808. [Google Scholar]
- Aneja V.P., Overton J.H., Cupitt L.T., Durham J.L., Wilson W.E. Carbon disulfide and carbonyl sulfide from biogenic sources and their contributions to the global sulfur cycle. Nature. 1979;282:493–496. [Google Scholar]
- Aneja V.P., Kim D.S., Das M., Hartsell B.E. Measurements and analysis of reactive nitrogen species in the rural troposphere of Southeast United States: southern oxidant study site SONIA. Atmos. Environ. 1996;30:649–659. [Google Scholar]
- Arnold F., Bührke T. New H2SO4 and HSO3 vapour measurements in the stratosphere—evidence for a volcanic influence. Nature. 1983;301(5898):293–295. [Google Scholar]
- Arrigo K.R., Lubin D., van Dijken G.L., Holm-Hansen O., Morrow E. Impact of a deep ozone hole on southern ocean primary production. J. Geophys. Res. 2003;108 [Google Scholar]
- Art H.W., Bormann F.H., Voigt G.K., Woodwell G.M. Barrier island forest ecosystems: the role of meteorological inputs. Science. 1974;184:60–62. doi: 10.1126/science.184.4132.60. [DOI] [PubMed] [Google Scholar]
- Asman W.A.H., van Jaarsveld H.A. A variable-resolution transport model applied for NHχ in Europe. Atmos. Environ. A, Gen. Top. 1992;26(3):445–464. [Google Scholar]
- Atkinson R. Atmospheric chemistry of VOCs and NOx. Atmos. Environ. 2000;34:2063–2101. [Google Scholar]
- Atkinson R., Arey J. Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review. Atmos. Environ. 2003;37:S197–S219. [Google Scholar]
- Avila A., Alarcon M., Queralt I. The chemical composition of dust transported in red rains—its contribution to the biogeochemical cycle of a Holm oak forest in Catalonia (Spain) Atmos. Environ. 1998;32:179–191. [Google Scholar]
- Aydin M., De Bruyn W.J., Saltzman E.S. Preindustrial atmospheric carbonyl sulfide (OCS) from an Antarctic Ice Core. Geophys. Res. Lett. 2002;29 [Google Scholar]
- Ayers G.P., Penkett S.A., Gillett R.W., Bandy B., Galbally I.E., Meyer C.P., Elsworth C.M., Bentley S.T., Forgan B.W. Evidence for photochemical control of ozone concentrations in unpolluted marine air. Nature. 1992;360:446–449. [Google Scholar]
- Bahlmann E., Keppler F., Wittmer J., Greule M., Schöler H.F., Seifert R., Zetzsch C. Evidence for a major missing source in the global chloromethane budget from stable carbon isotopes. Atmos. Chem. Phys. 2019;19(3):1703–1719. [Google Scholar]
- Barnett T.P., Pierce D.W., Achutarao K.M., Gleckler P.J., Santer B.D., Gregory J.M., Washington W.M. Penetration of human-induced warming into the world’s oceans. Science. 2005;309(5732):284–287. doi: 10.1126/science.1112418. [DOI] [PubMed] [Google Scholar]
- Bator A., Collett J.L. Cloud chemistry varies with drop size. J. Geophys. Res. D: Atmos. 1997;102:28071–28078. [Google Scholar]
- Battye W., Aneja V.P., Schlesinger W.H. Is nitrogen the next carbon? Earth’s Future. 2017;5(9):894–904. [Google Scholar]
- Bauwens M., Compernolle S., Stavrakou T., Muller J.-F., van Gent J., Eskes H., Levelt P.F., van der A.R., Veefkind J.P., Vlietinck J., Yu H., Zehner C. Impact of coronavirus outbreak on NO2 pollution assessed using TROPOMI and OMI observations. Geophys. Res. Lett. 2020 doi: 10.1029/2020GL087978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behera S.N., Sharma M. Degradation of SO2, NO2 and NH3 leading to formation of secondary inorganic aerosols: an environmental chamber study. Atmos. Environ. 2011;45:4015–4024. [Google Scholar]
- Beilke S., Lamb D. On the absorption of SO2 in ocean water. Tellus. 1974;26:268–271. [Google Scholar]
- Bell N., Hsu L., Jacob D.J., Schultz M.G., Blake D.R., Butler J.H., King D.B., Lobert J.M., Maier-Reimer E. Methyl iodide: atmospheric budget and use as a tracer of marine convection in global models. J. Geophys. Res. D: Atmos. 2002;107 [Google Scholar]
- Bellouin N., Boucher O., Haywood J., Shekar Reddy M. Global estimate of aerosol direct radiative forcing from satellite measurements. Nature. 2005;438(7071):1138–1141. doi: 10.1038/nature04348. [DOI] [PubMed] [Google Scholar]
- Berge E., Jakobsen H.A. A regional scale multilayer model for the calculation of long-term transport and deposition of air pollution in Europe. Tellus Ser. B Chem. Phys. Meteorol. 1998;50(3):205–223. [Google Scholar]
- Bergstrom A.-K., Jansson M. Atmospheric nitrogen deposition has caused nitrogen enrichment and eutrophication of lakes in the northern hemisphere. Glob. Chang. Biol. 2006;12(4):635–643. [Google Scholar]
- Berner R.A. Burial of organic carbon and pyrite sulfur in the modern ocean: its geochemical and environmental significance. Am. J. Sci. 1982;282:451–473. [Google Scholar]
- Berner E.K., Berner R.A. Princeton University Press; 2012. Global Environment: Water, Air, and Geochemical Cycles. [Google Scholar]
- Berry J., Adam W., Elliott Campbell J., Baker I., Blake N., Don B., Scott Denning A., et al. A coupled model of the global cycles of carbonyl sulfide and CO2: a possible new window on the carbon cycle: carbonyl sulfide as a global carbon cycle tracer. J. Geophys. Res. Biogeosci. 2013;118(2):842–852. [Google Scholar]
- Bertine K.K., Goldberg E.D. Fossil fuel combustion and the major sedimentary cycle. Science. 1971;173(3993):233–235. doi: 10.1126/science.173.3993.233. [DOI] [PubMed] [Google Scholar]
- Bertram T.H., Perring A.E., Wooldridge P.J., Crounse J.D., Kwan A.J., Wennberg P.O., Scheuer E., et al. Direct measurements of the convective recycling of the upper troposphere. Science. 2007;315(5813):816–820. doi: 10.1126/science.1134548. [DOI] [PubMed] [Google Scholar]
- Beyersdorf A.J., Blake D.R., Swanson A., Meinardi S., Rowland F.S., Davis D. Abundances and variability of tropospheric volatile organic compounds at the south pole and other Antarctic locations. Atmos. Environ. 2010;44:4565–4574. [Google Scholar]
- Bolton E.W., Berner R.A., Petsch S.T. The weathering of sedimentary organic matter as a control on atmospheric O2: II. Theoretical modeling. Am. J. Sci. 2006;306(8):575–615. [Google Scholar]
- Boutron C.F., Gorlach U., Candelone J.P., Bolshov M.A., Delmas R.J. Decrease in anthropogenic lead, cadmium and zinc in Greenland snows since the late 1960s. Nature. 1991;353:153–156. [Google Scholar]
- Brasseur G., Hitchman M.H. Stratospheric response to trace gas perturbations: changes in ozone and temperature distributions. Science. 1988;240(4852):634–637. doi: 10.1126/science.240.4852.634. [DOI] [PubMed] [Google Scholar]
- Briffa K.R., Jones P.D., Schweingruber F.H., Osborn T.J. Influence of volcanic eruptions on northern hemisphere summer temperature over the past 600 years. Nature. 1998;393:450–455. [Google Scholar]
- Brimblecombe P., Dawson G.A. Wet removal of highly soluble gases. J. Atmos. Chem. 1984;2:95–107. [Google Scholar]
- Brown S.S., Ryerson T.B., Wollny A.G., Brock C.A., Peltier R., Sullivan A.P., Weber R.J., et al. Variability in nocturnal nitrogen oxide processing and its role in regional air quality. Science. 2006;311(5757):67–70. doi: 10.1126/science.1120120. [DOI] [PubMed] [Google Scholar]
- Brown-Steiner B., Hess P. Asian influence on surface ozone in the United States: a comparison of chemistry, seasonality, and transport mechanisms. J. Geophys. Res. 2011;116 [Google Scholar]
- Brunner D., Staehelin J., Jeker D. Large-scale nitrogen oxide plumes in the tropopause region and implications for ozone. Science. 1998;282(5392):1305–1309. doi: 10.1126/science.282.5392.1305. [DOI] [PubMed] [Google Scholar]
- Butler J.H., Battle M., Bender M.L., Montzka S.A., Clarke A.D., Saltzman E.S., Sucher C.M., Severinghaus J.P., Elkins J.W. A record of atmospheric halocarbons during the twentieth century from polar firn air. Nature. 1999;399:749–755. [Google Scholar]
- Butler T.J., Likens G.E., Vermeylen F.M., Stunder B.J.B. The relation between NOx emissions and precipitation NO3− in the eastern USA. Atmos. Environ. 2003;37:2093–2104. [Google Scholar]
- Butler T.J., Likens G.E., Vermeylen F.M., Stunder B.J.B. The impact of changing nitrogen oxide emissions on wet and dry nitrogen deposition in the northeastern USA. Atmos. Environ. 2005;39:4851–4862. [Google Scholar]
- Butler T.J., Vermeylen F.M., Rury M., Likens G.E., Lee B., Bowker G.E., McCluney L. Response of ozone and nitrate to stationary source NOx emission reductions in the eastern USA. Atmos. Environ. 2011;45(5):1084–1094. [Google Scholar]
- Cachier H., Ducret J. Influence of biomass burning on equatorial African rains. Nature. 1991;352:228–230. [Google Scholar]
- Cachier H., Bremond M.P., Buatmenard P. Carbonaceous aerosols from different tropical biomass burning sources. Nature. 1989;340:371–373. [Google Scholar]
- Cadoux A., Scaillet B., Bekki S., Oppenheimer C., Druitt T.H. Stratospheric ozone destruction by the bronze-age Minoan eruption (Santorini Volcano, Greece) Sci. Rep. 2015;5(1):12243. doi: 10.1038/srep12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoon D.R., Stocks B.J., Levine J.S., Cofer W.R., Oneill K.P. Seasonal distribution of African savanna fires. Nature. 1992;359:812–815. [Google Scholar]
- Caldwell M.M., Flint S.D. Stratospheric ozone reduction, solar UvB radiation and terrestrial ecosystems. Clim. Chang. 1994;28:375–394. [Google Scholar]
- Campbell J.E., Carmichael G.R., Chai T., Mena-Carrasco M., Tang Y., Blake D.R., Blake N.J., et al. Photosynthetic control of atmospheric carbonyl sulfide during the growing season. Science. 2008;322(5904):1085–1088. doi: 10.1126/science.1164015. [DOI] [PubMed] [Google Scholar]
- Campbell J.E., Whelan M.E., Seibt U., Smith S.J., Berry J.A., Hilton T.W. Atmospheric carbonyl sulfide sources from anthropogenic activity: implications for carbon cycle constraints: atmospheric OCS sources. Geophys. Res. Lett. 2015;42(8):3004–3010. [Google Scholar]
- Campbell J.E., Berry J.A., Seibt U., Smith S.J., Montzka S.A., Launois T., Belviso S., Bopp L., Laine M. Large historical growth in global terrestrial gross primary production. Nature. 2017;544(7648):84–87. doi: 10.1038/nature22030. [DOI] [PubMed] [Google Scholar]
- Carroll M.A., Heidt L.E., Cicerone R.J., Prinn R.G. OCS, H2S, and CS2 fluxes from a saltwater marsh. J. Atmos. Chem. 1986;4:375–395. [Google Scholar]
- Chadwick O.A., Derry L.A., Vitousek P.M., Huebert B.J., Hedin L.O. Changing sources of nutrients during four million years of ecosystem development. Nature. 1999;397:491–497. [Google Scholar]
- Chameides W.L., Davis D.D. Chemistry in the troposphere. Chem. Eng. News. 1982;60:38–52. [Google Scholar]
- Chameides W.L., Lindsay R.W., Richardson J., Kiang C.S. The role of biogenic hydrocarbons in urban photochemical smog: Atlanta as a case study. Science. 1988;241(4872):1473–1475. doi: 10.1126/science.3420404. [DOI] [PubMed] [Google Scholar]
- Chameides W.L., Fehsenfeld F., Rodgers M.O., Cardelino C., Martinez J., Parrish D., Lonneman W., et al. Ozone precursor relationships in the ambient atmosphere. J. Geophys. Res. 1992;97:6037–6055. [Google Scholar]
- Chameides W.L., Kasibhatla P.S., Yienger J., Levy H., 2nd. Growth of continental-scale metro-agro-plexes, regional ozone pollution, and world food production. Science. 1994;264(5155):74–77. doi: 10.1126/science.264.5155.74. [DOI] [PubMed] [Google Scholar]
- Chandler A.S., Choularton T.W., Dollard G.J., Eggleton A.E.J., Gay M.J., Hill T.A., Jones B.M.R., Tyler B.J., Bandy B.J., Penkett S.A. Measurements of H2O2 and SO2 in clouds and estimates of their reaction rate. Nature. 1988;336:562–565. [Google Scholar]
- Chen S., Jiang N., Huang J., Xu X., Zhang H., Zang Z., Huang K., et al. Quantifying contributions of natural and anthropogenic dust emission from different climatic regions. Atmos. Environ. 2018;191(October):94–104. [Google Scholar]
- Chin M., Davis D.D. Global sources and sinks of OCS and CS2 and their distributions. Glob. Biogeochem. Cycles. 1993;7:321–337. [Google Scholar]
- Chin M., Davis D.D. A reanalysis of carbonyl sulfide as a source of stratospheric background sulfur aerosol. J. Geophys. Res. 1995;100:8993–9005. [Google Scholar]
- Chipperfield M.P., Bekki S., Dhomse S., Harris N.R.P., Hassler B., Hossaini R., Steinbrecht W., Thiéblemont R., Weber M. Detecting recovery of the stratospheric ozone layer. Nature. 2017;549(7671):211–218. doi: 10.1038/nature23681. [DOI] [PubMed] [Google Scholar]
- Cicerone R.J. Changes in stratospheric ozone. Science. 1987;237(4810):35–42. doi: 10.1126/science.237.4810.35. [DOI] [PubMed] [Google Scholar]
- Clarisse L., Clerbaux C., Dentener F., Hurtmans D., Coheur P.F. Global ammonia distribution derived from infrared satellite observations. Nat. Geosci. 2009;2:479–483. [Google Scholar]
- Clark K.L., Nadkarni N.M., Schaefer D., Gholz H.L. Cloud water and precipitation chemistry in a tropical montane forest, Monteverde, Costa Rica. Atmos. Environ. 1998;32:1595–1603. [Google Scholar]
- Cogbill C.V., Likens G.E. Acid precipitation in the northeastern United States. Water Resour. Res. 1974;10:1133–1137. [Google Scholar]
- Colbourn G., Ridgwell A., Lenton T.M. The time scale of the silicate weathering negative feedback on atmospheric CO2: silicate weathering feedback time scale. Glob. Biogeochem. Cycles. 2015;29(5):583–596. [Google Scholar]
- Colman J.J., Blake D.R., Rowland F.S. Atmospheric residence time of CH3Br estimated from the junge spatial variability relation. Science. 1998;281(5375):392–396. doi: 10.1126/science.281.5375.392. [DOI] [PubMed] [Google Scholar]
- Conway T.J., Tans P., Waterman I.S., Thoning K.W., Masarie K.A., Gammon R.H. Atmospheric carbon dioxide measurements in the remote global atmosphere, 1981–1984. Tellus Ser. B Chem. Phys. Meteorol. 1988;40:81–115. [Google Scholar]
- Cook E.R., Woodhouse C.A., Mark Eakin C., Meko D.M., Stahle D.W. Long-term aridity changes in the Western United States. Science. 2004;306(5698):1015–1018. doi: 10.1126/science.1102586. [DOI] [PubMed] [Google Scholar]
- Cooper O.R., Parrish D.D., Stohl A., Trainer M., Nédélec P., Thouret V., Cammas J.P., et al. Increasing springtime ozone mixing ratios in the free troposphere over Western North America. Nature. 2010;463(7279):344–348. doi: 10.1038/nature08708. [DOI] [PubMed] [Google Scholar]
- Correll D.L., Clark C.O., Goldberg B., Goodrich V.R., Hayes D.R., Klein W.H., Schecher W.D. Spectral ultraviolet-B radiation fluxes at the Earth’s surface—long-term variations at 39°N, 77°W. J. Geophys. Res. 1992;97:7579–7591. [Google Scholar]
- Crowley T.J. Causes of climate change over the past 1000 years. Science. 2000;289(5477):270–277. doi: 10.1126/science.289.5477.270. [DOI] [PubMed] [Google Scholar]
- Crozat G. Emission of potassium aerosols in tropical forest. Tellus. 1979;31:52–57. [Google Scholar]
- Crutzen P.J. In: The Major Biogeochemical Cycles and Their Interactions. Bolin B., Cook R.B., editors. Wiley; 1983. Atmospheric interactions: homogeneous gas reactions of C, N, and S containing compounds; pp. 67–114. [Google Scholar]
- Crutzen P.J. In: Scales and Global Change. Rosswall R.G.W.T., Risser P.G., editors. Wiley; 1988. Variability atmospheric chemical systems; pp. 81–108. [Google Scholar]
- Crutzen P.J. Albedo enhancement by stratospheric sulfur injections: a contribution to resolve a policy dilemma? Clim. Chang. 2006;77:211–219. [Google Scholar]
- Crutzen P.J., Zimmermann P.H. The changing photochemistry of the troposphere. Tellus A Dyn. Meteorol. Oceanogr. 1991;43:136–151. [Google Scholar]
- Crutzen P.J., Delany A.C., Greenberg J., Haagenson P., Heidt L., Lueb R., Pollock W., Seiler W., Wartburg A., Zimmerman P. Tropospheric chemical composition measurements in Brazil during the dry season. J. Atmos. Chem. 1985;2:233–256. [Google Scholar]
- Crutzen P.J., Lawrence M.G., Poschl U. On the background photochemistry of tropospheric ozone. Tellus A Dyn. Meteorol. Oceanogr. 1999;51:123–146. [Google Scholar]
- Dahlgren R.A., Ugolini F.C., Casey W.H. Field weathering rates of Mt St. Helens tephra. Geochim. Cosmochim. Acta. 1999;63:587–598. [Google Scholar]
- Day T.A., Neale P.J. Effects of UV-B radiation on terrestrial and aquatic primary producers. Annu. Rev. Ecol. Syst. 2002;33:371–396. [Google Scholar]
- Després V., Huffman J.A., Burrows S.M., Hoose C., Safatov A., Buryak G., Fröhlich-Nowoisky J., et al. Primary biological aerosol particles in the atmosphere: a review. Tellus Ser. B Chem. Phys. Meteorol. 2012;64(1):15598. [Google Scholar]
- Deventer M.J., Jiao Y., Knox S.H., Anderson F., Ferner M.C., Lewis J.A., Rhew R.C. Ecosystem-scale measurements of methyl halide fluxes from a brackish tidal Marsh invaded with perennial pepperweed (Lepidium latifolium) J. Geophys. Res. Biogeosci. 2018;123(7):2104–2120. [Google Scholar]
- Dey S., Di Girolamo L. A decade of change in aerosol properties over the Indian subcontinent. Geophys. Res. Lett. 2011;38 [Google Scholar]
- Donarummo J., Ram M., Stoermer E.F. Possible deposit of soil dust from the 1930’s US dust bowl identified in Greenland ice. Geophys. Res. Lett. 2003;30 [Google Scholar]
- Dorn H.-P., Callies J., Platt U., Ehhalt D.H. Measurement of tropospheric OH concentration by laser long-path absorption spectroscopy. Tellus Ser. B Chem. Phys. Meteorol. 1988;40:437–445. [Google Scholar]
- Dorrepaal E., Toet S., van Logtestijn R.S.P., Swart E., van de Weg M.J., Callaghan T.V., Aerts R. Carbon respiration from subsurface peat accelerated by climate warming in the subarctic. Nature. 2009;460:616–619. [Google Scholar]
- Du Q., Zhang C., Yujing M., Cheng Y., Zhang Y., Liu C., Song M., et al. An important missing source of atmospheric carbonyl sulfide: domestic coal combustion: COS from domestic coal combustion. Geophys. Res. Lett. 2016;43(16):8720–8727. [Google Scholar]
- Duce R.A., Unni C.K., Ray B.J., Prospero J.M., Merrill J.T. Long-range atmospheric transport of soil dust from Asia to the tropical north Pacific: temporal variability. Science. 1980;209(4464):1522–1524. doi: 10.1126/science.209.4464.1522. [DOI] [PubMed] [Google Scholar]
- Duce R.A., Liss P.S., Merrill J.T., Atlas E.L., Buat-Menard P., Hicks B.B., Miller J.M., et al. The atmospheric input of trace species to the world ocean. Glob. Biogeochem. Cycles. 1991;5:193–259. [Google Scholar]
- Duncan B.N., Lamsal L.N., Thompson A.M., Yoshida Y., Lu Z., Streets D.G., Hurwitz M.M., Pickering K.E. A space-based, high-resolution view of notable changes in urban NOx pollution around the world (2005–2014): notable changes in urban NOx pollution. J. Geophys. Res. D: Atmos. 2016;121(2):976–996. [Google Scholar]
- Elbert W., Hoffmann M.R., Kramer M., Schmitt G., Andreae M.O. Control of solute concentrations in cloud and fog water by liquid water content. Atmos. Environ. 2000;34:1109–1122. [Google Scholar]
- Elkins J.W., Thompson T.M., Swanson T.H., Butler J.H., Hall B.D., Cummings S.O., Fisher D.A., Raffo A.G. Decrease in the growth rates of atmospheric chlorofluorocarbon-11 and chlorofluorocarbon-12. Nature. 1993;364:780–783. [Google Scholar]
- Farman J.C., Gardiner B.G., Shanklin J.D. Large losses of total ozone in antarctica reveal seasonal ClOx/NOx interaction. Nature. 1985;315:207–210. [Google Scholar]
- Field J.P., Belnap J., Breshears D.D., Neff J.C., Okin G.S., Whicker J.J., Painter T.H., Ravi S., Reheis M.C., Reynolds R.L. The ecology of dust. Front. Ecol. Environ. 2010;8(8):423–430. [Google Scholar]
- Fischer H., Wagenbach D., Kipfstuhl J. Sulfate and nitrate firn concentrations on the Greenland ice sheet: 2. Temporal anthropogenic deposition changes. J. Geophys. Res.-Atmos. 1998;103:21935–21942. [Google Scholar]
- Fishman J., Wozniak A.E., Creilson J.K. Global distribution of tropospheric ozone from satellite measurements using the empirically corrected tropospheric ozone residual technique: identification of the regional aspects of air pollution. Atmos. Chem. Phys. 2003;3:893–907. [Google Scholar]
- Flückiger J., Monnin E., Stauffer B., Schwander J., Stocker T.F., Chappellaz J., Raynaud D., Barnola J.-M. High-resolution holocene N2O ice core record and its relationship with CH4 and CO2. Glob. Biogeochem. Cycles. 2002;16(1):10–11. [Google Scholar]
- Ford B., Val Martin M., Zelasky S.E., Fischer E.V., Anenberg S.C., Heald C.L., Pierce J.R. Future fire impacts on smoke concentrations, visibility, and health in the contiguous United States. GeoHealth. 2018;2(8):229–247. doi: 10.1029/2018GH000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forzieri G., Alkama R., Miralles D.G., Cescatti A. Satellites reveal contrasting responses of regional climate to the widespread greening of Earth. Science. 2017;356(6343):1180–1184. doi: 10.1126/science.aal1727. [DOI] [PubMed] [Google Scholar]
- Foukal P., Fröhlich C., Spruit H., Wigley T.M.L. Variations in solar luminosity and their effect on the Earth’s climate. Nature. 2006;443(7108):161–166. doi: 10.1038/nature05072. [DOI] [PubMed] [Google Scholar]
- Frankenberg C., Aben I., Bergamaschi P., Dlugokencky E.J., van Hees R., Houweling S., van der Meer P., Snel R., Tol P. Global column-averaged methane mixing ratios from 2003 to 2009 as derived from SCIAMACHY: trends and variability. J. Geophys. Res. 2011;116(D4) [Google Scholar]
- Fruchter J.S., Robertson D.E., Evans J.C., Olsen K.B., Lepel E.A., Laul J.C., Abel K.H., et al. Mount St. Helens Ash from the 18 May 1980 Eruption: chemical, physical, mineralogical, and biological properties. Science. 1980;209(4461):1116–1125. doi: 10.1126/science.209.4461.1116. [DOI] [PubMed] [Google Scholar]
- Fueglistaler S., Wernli H., Peter T. Tropical troposphere-to-stratosphere transport inferred from trajectory calculations. J. Geophys. Res. 2004;109 [Google Scholar]
- Galloway J.N., Likens G.E. Atmospheric enhancement of metal deposition in Adirondack lake sediments. Limnol. Oceanogr. 1979;24:427–433. [Google Scholar]
- Galloway J.N., Whelpdale D.M. WATOX-86 overview and Western North Atlantic Ocean S and N atmospheric budgets. Glob. Biogeochem. Cycles. 1987;1:261–281. [Google Scholar]
- Galloway J.N., Likens G.E., Edgerton E.S. Acid precipitation in the Northeastern United States: pH and acidity. Science. 1976;194(4266):722–724. doi: 10.1126/science.194.4266.722. [DOI] [PubMed] [Google Scholar]
- Gatz D.F., Dingle A.N. Trace substances in rain water: concentration variations during convective rains, and their interpretation. Tellus. 1971;23:14–27. [Google Scholar]
- Geng C.M., Mu Y.J. Carbonyl sulfide and dimethyl sulfide exchange between trees and the atmosphere. Atmos. Environ. 2006;40:1373–1383. [Google Scholar]
- Geng L., Murray L.T., Mickley L.J., Lin P., Qiang F., Schauer A.J., Alexander B. Isotopic evidence of multiple controls on atmospheric oxidants over climate transitions. Nature. 2017;546(7656):133–136. doi: 10.1038/nature22340. [DOI] [PubMed] [Google Scholar]
- Georgii H.-W., Wötzel D. On the relation between drop size and concentration of trace elements in rainwater. J. Geophys. Res. 1970;75(9):1727–1731. [Google Scholar]
- Geron C., Harley P., Guenther A. Isoprene emission capacity for US tree species. Atmos. Environ. 2001;35(19):3341–3352. [Google Scholar]
- Geron C., Guenther A., Greenberg J., Karl T., Rasmussen R. Biogenic volatile organic compound emissions from desert vegetation of the southwestern US. Atmos. Environ. 2006;40:1645–1660. [Google Scholar]
- Geron C., Owen S., Guenther A., Greenberg J., Rasmussen R., Bai J.H., Li Q.-J., Baker B. Volatile organic compounds from vegetation in southern Yunnan Province, China: emission rates and some potential regional implications. Atmos. Environ. 2006;40(10):1759–1773. [Google Scholar]
- Gillette D.A., Stensland G.J., Williams A.L., Barnard W., Gatz D., Sinclair P.C., Johnson T.C. Emissions of alkaline elements calcium, magnesium, potassium, and sodium from open sources in the contiguous United States. Glob. Biogeochem. Cycles. 1992;6:437–457. [Google Scholar]
- Glass S.J., Matteson M.J. Ion enrichment in aerosols dispersed from bursting bubbles in aqueous salt solutions. Tellus. 1973;25:272–280. [Google Scholar]
- Goldan P.D., Fall R., Kuster W.C., Fehsenfeld F.C. Uptake of COS by growing vegetation: a major tropospheric sink. J. Geophys. Res.-Atmos. 1988;93:14186–14192. [Google Scholar]
- Goode P.R., Qiu J., Yurchyshyn V., Hickey J., Chu M.C., Kolbe E., Brown C.T., Koonin S.E. Earthshine observations of the Earth’s reflectance. Geophys. Res. Lett. 2001;28:1671–1674. [Google Scholar]
- Gorham E. Factors influencing supply of major ions to inland waters, with special reference to the atmosphere. Geol. Soc. Am. Bull. 1961;72:795–840. [Google Scholar]
- Gorham E., Martin F.B., Litzau J.T. Acid rain: ionic correlations in the eastern United States, 1980–1981. Science. 1984;225(4660):407–409. doi: 10.1126/science.225.4660.407. [DOI] [PubMed] [Google Scholar]
- Graedel T.E., Crutzen P.J. W.H. Freeman; 1993. Atmospheric Change. [Google Scholar]
- Graedel T.E., Keene W.C. Tropospheric budget of reactive chlorine. Glob. Biogeochem. Cycles. 1995;9:47–77. [Google Scholar]
- Griffin D., Zhao X., McLinden C.A., Boersma F., Bourassa A., Dammers E., Degenstein D., et al. High-resolution mapping of nitrogen dioxide with TROPOMI: first results and validation over the Canadian oil sands. Geophys. Res. Lett. 2019;46(2):1049–1060. doi: 10.1029/2018GL081095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffis T., Hu C., Baker J., Wood J., Millet D., Erickson M., Yu Z., Deventer M., Winker C., Chen Z. Tall tower ammonia observations and emission estimates in the US midwest. J. Geophys. Res. Biogeosci. 2019;124:3432–3447. [Google Scholar]
- Guenther A.B., Zimmerman P.R., Harley P.C., Monson R.K., Fall R. Isoprene and monoterpene emission rate variability: model evaluations and sensitivity analyses. J. Geophys. Res. Crop Soc. Am. S. 1993;98(D7):12609. [Google Scholar]
- Guenther A., Geron C., Pierce T., Lamb B., Harley P., Fall R. Natural emissions of non-methane volatile organic compounds; carbon monoxide, and oxides of nitrogen from North America. Atmos. Environ. 2000;34:2205–2230. [Google Scholar]
- Hahn J. Organic constituents of natural aerosols. Ann. N. Y. Acad. Sci. 1980;338:359–376. [Google Scholar]
- Hakkarainen J., Ialongo I., Tamminen J. Direct space-based observations of anthropogenic CO2 emission areas from OCO-2: direct observations of anthropogenic CO2. Geophys. Res. Lett. 2016;43(21):11,400–411,406. [Google Scholar]
- Hardacre C.J., Heal M.R. Characterization of methyl bromide and methyl chloride fluxes at temperate freshwater wetlands: methyl halide fluxes at wetlands. J. Geophys. Res. D: Atmos. 2013;118(2):977–991. [Google Scholar]
- Harries J.E., Brindley H.E., Sagoo P.J., Bantges R.J. Increases in greenhouse forcing inferred from the outgoing longwave radiation spectra of the earth in 1970 and 1997. Nature. 2001;410(6826):355–357. doi: 10.1038/35066553. [DOI] [PubMed] [Google Scholar]
- Harriss R.C., Sebacher D.I., Day F.P. Methane flux in the great dismal swamp. Nature. 1982;297(5868):673–674. [Google Scholar]
- Hartnett H.E., Keil R.G., Hedges J.I., Devol A.H. Influence of oxygen exposure time on organic carbon preservation in continental margin sediments. Nature. 1998;391:572–574. [Google Scholar]
- He Y., Wang K., Zhou C., Wild M. A revisit of global dimming and brightening based on the sunshine duration. Geophys. Res. Lett. 2018;45(9):4281–4289. [Google Scholar]
- Hedin L.O., Likens G.E., Bormann F.H. Decrease in precipitation acidity resulting from decreased SO42 concentration. Nature. 1987;325:244–246. [Google Scholar]
- Hedin L.O., Armesto J.J., Johnson A.H. Patterns of nutrient loss from unpolluted old-growth forests: evaluation of biogeochemical theory. Ecology. 1995;76:493–509. [Google Scholar]
- Hegglin M.I., et al. World Meteorological Organization; Geneva, Switzerland: 2015. Twenty Questions and Answers About the Ozone Layer: 2014 Update, Scientific Assessment of Ozone Depletion. [Google Scholar]
- Heintzenberg J. Fine particles in the global troposphere: a review. Tellus B Chem. Phys. Meteorol. 1989;41:149–160. [Google Scholar]
- Helmig D., Daly R.W., Milford J., Guenther A. Seasonal trends of biogenic terpene emissions. Chemosphere. 2013;93(1):35–46. doi: 10.1016/j.chemosphere.2013.04.058. [DOI] [PubMed] [Google Scholar]
- Henderson-Sellers A., McGuffie K. Wiley; 1987. A Climate Modeling Primer. [Google Scholar]
- Hendren C.O., Mesnard X., Dröge J., Wiesner M.R. Estimating production data for five engineered nanomaterials as a basis for exposure assessment. Environ. Sci. Technol. 2011;45(7):2562–2569. doi: 10.1021/es103300g. [DOI] [PubMed] [Google Scholar]
- Hewitt C.N., Harrison R.M. Tropospheric concentrations of the hydroxyl radical—a review. Atmos. Environ. 1985;19:545–554. [Google Scholar]
- Hidy G.M. In: Precipitation Scavenging. Englemann R.J., Slinn W.G.N., editors. U.S. Atomic Energy Commission, Division of Technical Information; Oak Ridge, Tennessee: 1970. Theory of diffusive and impactive scavenging; pp. 355–371. [Google Scholar]
- Hilley G.E., Porder S. A framework for predicting global silicate weathering and CO2 drawdown rates over geologic time-scales. Proc. Natl. Acad. Sci. U. S. A. 2008;105(44):16855–16859. doi: 10.1073/pnas.0801462105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch A.I., Munger J.W., Jacob D.J., Horowitz L.W., Goldstein A.H. Seasonal variation of the ozone production efficiency per unit NOx at Harvard Forest, Massachusetts. J. Geophys. Res. 1996;101:12659–12666. [Google Scholar]
- Hocking W.K., Carey-Smith T., Tarasick D.W., Argall P.S., Strong K., Rochon Y., Zawadzki I., Taylor P.A. Detection of stratospheric ozone intrusions by Windprofiler radars. Nature. 2007;450(7167):281–284. doi: 10.1038/nature06312. [DOI] [PubMed] [Google Scholar]
- Hofmann D.J. Increase in the stratospheric background sulfuric acid aerosol mass in the past 10 years. Science. 1990;248(4958):996–1000. doi: 10.1126/science.248.4958.996. [DOI] [PubMed] [Google Scholar]
- Hofmann D.J., Rosen J.M. Stratospheric sulfuric acid layer: evidence for an anthropogenic component. Science. 1980;208(4450):1368–1370. doi: 10.1126/science.208.4450.1368. [DOI] [PubMed] [Google Scholar]
- Hofmann D.J., Rosen J.M. Sulfuric acid droplet formation and growth in the stratosphere after the 1982 eruption of El Chichon. Science. 1983;222(4621):325–327. doi: 10.1126/science.222.4621.325. [DOI] [PubMed] [Google Scholar]
- Hofzumahaus A., Rohrer F., Lu K., Bohn B., Brauers T., Chang C.-C., Fuchs H., et al. Amplified trace gas removal in the troposphere. Science. 2009;324(5935):1702–1704. doi: 10.1126/science.1164566. [DOI] [PubMed] [Google Scholar]
- Holton J.R., Haynes P.H., McIntyre M.E., Douglass A.R., Rood R.B., Pfister L. Stratosphere-troposphere exchange. Rev. Geophys. 1995;33:403–439. [Google Scholar]
- Hooper P.R., Herrick I.W., Laskowski E.R., Knowles C.R. Composition of the Mount St. Helens Ashfall in the Moscow-Pullman area on 18 May 1980. Science. 1980;209(4461):1125–1126. doi: 10.1126/science.209.4461.1125. [DOI] [PubMed] [Google Scholar]
- Höpfner M., Milz M., Buehler S., Orphal J., Stiller G. The natural greenhouse effect of atmospheric oxygen (O2) and nitrogen (N2): natural greenhouse effect of O2 and N2. Geophys. Res. Lett. 2012;39(10) [Google Scholar]
- Hosker R.P., Lindberg S.E. Atmospheric deposition and plant assimilation of gases and particles. Atmos. Environ. 1982;16:889–910. (Review) [Google Scholar]
- Hough A.M., Derwent R.G. Changes in the global concentration of tropospheric ozone due to human activities. Nature. 1990;344:645–648. [Google Scholar]
- Hsiang S., Kopp R., Jina A., Rising J., Delgado M., Shashank M., Rasmussen D.J., et al. Estimating economic damage from climate change in the United States. Science. 2017;356(6345):1362–1369. doi: 10.1126/science.aal4369. [DOI] [PubMed] [Google Scholar]
- Hu L., Yvon-Lewis S.A., Liu Y., Salisbury J.E., O’Hern J.E. Coastal emissions of methyl bromide and methyl chloride along the Eastern Gulf of Mexico and the East Coast of the United States. Glob. Biogeochem. Cycles. 2010;24 [Google Scholar]
- Hu L., Yvon-Lewis S., Liu Y., Bianchi T.S. The ocean in near equilibrium with atmospheric methyl bromide: oceanic saturation state of CH3 Br. Glob. Biogeochem. Cycles Geophys. Monogr. Ser. 2012;26(3):165. [Google Scholar]
- Hu L., Yvon-Lewis S.A., Butler J.H., Lobert J.M., King D.B. An improved oceanic budget for methyl chloride: oceanic budget for MeCl. J. Geophys. Res. C: Oceans. 2013;118(2):715–725. [Google Scholar]
- Hu L., Jacob D.J., Liu X., Zhang Y., Lin Z., Kim P.S., Sulprizio M.P., Yantosca R.M. Global budget of tropospheric ozone: evaluating recent model advances with satellite (OMI), aircraft (IAGOS), and ozonesonde observations. Atmos. Environ. 2017;167(October):323–334. [Google Scholar]
- Huang Y., Shen H., Chen Y., Zhong Q., Chen H., Wang R., Shen G., Liu J., Li B., Tao S. Global organic carbon emissions from primary sources from 1960 to 2009. Atmos. Environ. 2015;122(December):505–512. [Google Scholar]
- Hulett L.D., Jr., Weinberger A.J., Northcutt K.J., Ferguson M. Chemical species in fly ash from coal-burning power plants. Science. 1980;210(4476):1356–1358. doi: 10.1126/science.210.4476.1356. [DOI] [PubMed] [Google Scholar]
- Irwin J.G., Williams M.L. Acid rain: chemistry and transport. Environ. Pollut. 1988;50(1–2):29–59. doi: 10.1016/0269-7491(88)90184-4. [DOI] [PubMed] [Google Scholar]
- Isaksen I.S.A., Hov O. Calculation of trends in the tropospheric concentrations of O3, OH, CO, CH4, and NOx. Tellus B Chem. Phys. Meteorol. 1987;39:271–285. [Google Scholar]
- Jacob D.J., Wofsy S.C. Budgets of reactive nitrogen, hydrocarbons, and ozone over the Amazon Forest during the wet season. J. Geophys. Res.-Atmos. 1990;95:16737–16754. [Google Scholar]
- Jacob D.J., Logan J.A., Gardner G.M., Yevich R.M., Spivakovsky C.M., Wofsy S.C., Sillman S., Prather M.J. Factors regulating ozone over the United States and its export to the global atmosphere. J. Geophys. Res.—Atmos. 1993;98:14817–14826. [Google Scholar]
- Jacob D.J., Turner A.J., Maasakkers J.D., Sheng J., Sun K., Liu X., Chance K., Aben I., McKeever J., Frankenberg C. Satellite observations of atmospheric methane and their value for quantifying methane emissions. Atmos. Chem. Phys. 2016;16(22):14371–14396. [Google Scholar]
- Jaeglé L., Shah V., Thornton J.A., Lopez-Hilfiker F.D., Lee B.H., McDuffie E.E., Fibiger D., et al. Nitrogen oxides emissions, chemistry, deposition, and export over the Northeast United States during the winter aircraft campaign. J. Geophys. Res. D: Atmos. 2018;123(21):12368–12393. [Google Scholar]
- Janson R.W. Monoterpene emissions from scots pine and Norwegian Spruce. J. Geophys. Res. SCOPE Rep. 1993;98(D2):2839–2850. [Google Scholar]
- Jardine A.B., Jardine K.J., Fuentes J.D., Martin S.T., Martins G., Durgante F., Carneiro V., Higuchi N., Manzi A.O., Chambers J.Q. Highly reactive light-dependent monoterpenes in the Amazon. Geophys. Res. Lett. 2015;42(5):1576–1583. [Google Scholar]
- Jenkin M.E., Clemitshaw K.C. Ozone and other secondary photochemical pollutants: chemical processes governing their formation in the planetary boundary layer. Atmos. Environ. 2000;34:2499–2527. [Google Scholar]
- Jickells T.D., An Z.S., Andersen K.K., Baker A.R., Bergametti G., Brooks N., Cao J.J., et al. Global iron connections between desert dust, ocean biogeochemistry, and climate. Science. 2005;308(5718):67–71. doi: 10.1126/science.1105959. [DOI] [PubMed] [Google Scholar]
- Jimenez J.L., Canagaratna M.R., Donahue N.M., Prevot A.S.H., Zhang Q., Kroll J.H., DeCarlo P.F., et al. Evolution of organic aerosols in the atmosphere. Science. 2009;326(5959):1525–1529. doi: 10.1126/science.1180353. [DOI] [PubMed] [Google Scholar]
- Jobson B.T., McKeen S.A., Parrish D.D., Fehsenfeld F.C., Blake D.R., Goldstein A.H., Schauffler S.M., Elkins J.C. Trace gas mixing ratio variability versus lifetime in the troposphere and stratosphere: observations. J. Geophys. Res.-Atmos. 1999;104:16091–16113. [Google Scholar]
- Johnston D.A. Volcanic contribution of chlorine to the stratosphere: more significant to ozone than previously estimated? Science. 1980;209(4455):491–493. doi: 10.1126/science.209.4455.491. [DOI] [PubMed] [Google Scholar]
- Junge C.E. Residence time and variability of tropospheric trace gases. Tellus. 1974;26:477–488. [Google Scholar]
- Junge C.E., Werby R.T. The concentration of chloride, sodium, potassium, calcium, and sulfate in rain water over the United States. J. Meteorol. 1958;15:417–425. [Google Scholar]
- Kang D.W., Aneja V.P., Mathur R., Ray J.D. Nonmethane hydrocarbons and ozone in three rural Southeast United States National Parks: a model sensitivity analysis and comparison to measurements. J. Geophys. Res. Atmos. 2003;108 [Google Scholar]
- Kaufman Y.J., Tucker C.J., Fung I. Remote sensing of biomass burning in the tropics. J. Geophys. Res.-Atmos. 1990;95:9927–9939. [Google Scholar]
- Kaufman Y.J., Tanré D., Boucher O. A satellite view of aerosols in the climate system. Nature. 2002;419(6903):215–223. doi: 10.1038/nature01091. [DOI] [PubMed] [Google Scholar]
- Kavouras I.G., Mihalopoulos N., Stephanou E.G. Formation of atmospheric particles from organic acids produced by forests. Nature. 1998;395:683–686. [Google Scholar]
- Keeling R.F., Shertz S.R. Seasonal and interannual variations in atmospheric oxygen and implications for the global carbon cycle. Nature. 1992;358:723–727. [Google Scholar]
- Keith D.W., Weisenstein D.K., Dykema J.A., Keutsch F.N. Stratospheric solar geoengineering without ozone loss. Proc. Natl. Acad. Sci. U. S. A. 2016;113(52):14910–14914. doi: 10.1073/pnas.1615572113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg W.W. Aerosols and global warming. Science. 1992;256(5057):598. doi: 10.1126/science.256.5057.598. [DOI] [PubMed] [Google Scholar]
- Kelly V.R., Lovett G.M., Weathers K.C., Likens G.E. Trends in atmospheric concentration and deposition compared to regional and local pollutant emissions at a rural site in southeastern New York. Atmos. Environ. 2002;36:1569–1575. [Google Scholar]
- Kempf K., Allwine E., Westberg H., Claiborn C., Lamb B. Hydrocarbon emissions from spruce species using environmental chamber and branch enclosure methods. Atmos. Environ. 1996;30(9):1381–1389. [Google Scholar]
- Kennedy M.J., Hedin L.O., Derry L.A. Decoupling of unpolluted temperate forests from rock nutrient sources revealed by natural 87Sr/86Sr and 84Sr tracer addition. Proc. Natl. Acad. Sci. 2002;99:9639–9644. doi: 10.1073/pnas.152045499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr J.B., McElroy C.T. Evidence for large upward trends of ultraviolet-B radiation linked to ozone depletion. Science. 1993;262(5136):1032–1034. doi: 10.1126/science.262.5136.1032. [DOI] [PubMed] [Google Scholar]
- Kesselmeier J., Merk L. Exchange of carbonyl sulfide (COS) between agricultural plants and the atmosphere—studies on the deposition of COS to peas, corn and rapeseed. Biogeochemistry. 1993;23:47–59. [Google Scholar]
- Kettle A.J., Kuhn U., von Hobe M., Kesselmeier J., Andreae M.O. Global budget of atmospheric carbonyl sulfide: temporal and spatial variations of the dominant sources and sinks. J. Geophys. Res. 2002;107 [Google Scholar]
- Khalil M.A.K., Rasmussen R.A. Causes of increasing atmospheric methane: depletion of hydroxyl radicals and the rise of emissions. Atmos. Environ. 1985;19:397–407. [Google Scholar]
- Khalil M.A.K., Rasmussen R.A., Gunawardena R. Atmospheric methyl bromide: trends and global mass balance. J. Geophys. Res. 1993;98:2887–2896. [Google Scholar]
- Kharol S.K., Shephard M.W., McLinden C.A., Zhang L., Sioris C.E., O’Brien J.M., Vet R., et al. Dry deposition of reactive nitrogen from satellite observations of ammonia and nitrogen dioxide over North America. Geophys. Res. Lett. 2018;45(2):1157–1166. [Google Scholar]
- Kiehl J.T., Briegleb B.P. The relative roles of sulfate aerosols and greenhouse gases in climate forcing. Science. 1993;260(5106):311–314. doi: 10.1126/science.260.5106.311. [DOI] [PubMed] [Google Scholar]
- Kim D., Ramanathan V. Improved estimates and understanding of global albedo and atmospheric solar absorption. Geophys. Res. Lett. 2012;39(24):38. [Google Scholar]
- Kim S.W., Heckel A., McKeen S.A., Frost G.J., Hsie E.Y., Trainer M.K., Richter A., Burrows J.P., Peckham S.E., Grell G.A. Satellite-observed US power plant NOx emission reductions and their impact on air quality. Geophys. Res. Lett. 2006;33 [Google Scholar]
- Kirkby J., Curtius J., Almeida J., Dunne E., Duplissy J., Ehrhart S., Franchin A., et al. Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation. Nature. 2011;476(7361):429–433. doi: 10.1038/nature10343. [DOI] [PubMed] [Google Scholar]
- Kleinman L., Lee Y.N., Springston S.R., Nunnermacker L., Zhou X.L., Brown R., Hallock K., et al. Ozone formation at a rural site in the southeastern United States. J. Geophys. Res. 1994;99:3469–3482. [Google Scholar]
- Kort E.A., Frankenberg C., Miller C.E., Oda T. Space-based observations of megacity carbon dioxide: space-based observations of megacity CO2. Geophys. Res. Lett. 2012;39(17) [Google Scholar]
- Kuhn U. Isoprene and monoterpene emissions of Amazônian tree species during the wet season: direct and indirect investigations on controlling environmental functions. J. Geophys. Res. 2002;107(D20):1616. [Google Scholar]
- Kuhn U., Ammann C., Wolf A., Meixner F.X., Andreae M.O., Kesselmeier J. Carbonyl sulfide exchange on an ecosystem scale: soil represents a dominant sink for atmospheric COS. Atmos. Environ. 1999;33:995–1008. [Google Scholar]
- Kumar P., Robins A., Vardoulakis S., Britter R. A review of the characteristics of nanoparticles in the urban atmosphere and the prospects for developing regulatory controls. Atmos. Environ. 2010;44:5035–5052. [Google Scholar]
- Lajtha K., Jones J. Trends in cation, nitrogen, sulfate and hydrogen ion concentrations in precipitation in the United States and Europe from 1978 to 2010: a new look at an old problem. Biogeochemistry. 2013;116(1):303–334. [Google Scholar]
- Lambert F., Delmonte B., Petit J.R., Bigler M., Kaufmann P.R., Hutterli M.A., Stocker T.F., Ruth U., Steffensen J.P., Maggi V. Dust-climate couplings over the past 800,000 years from the EPICA dome C ice core. Nature. 2008;452(7187):616–619. doi: 10.1038/nature06763. [DOI] [PubMed] [Google Scholar]
- Langford A.O., Fehsenfeld F.C. Natural vegetation as a source or sink for atmospheric ammonia: a case study. Science. 1992;255(5044):581–583. doi: 10.1126/science.255.5044.581. [DOI] [PubMed] [Google Scholar]
- Langmann B., Folch A., Hensch M., Matthias V. Volcanic ash over Europe during the eruption of Eyjafjallajokull on Iceland, April-May 2010. Atmos. Environ. 2012;48:1–8. [Google Scholar]
- Langway C.C., Osada K., Clausen H.B., Hammer C.U., Shoji H., Mitani A. New chemical stratigraphy over the last millennium for Byrd Station, Antarctica. Tellus B—Chem. Phys. Meteorol. 1994;46:40–51. [Google Scholar]
- Latimer J.S., Charpentier M.A. Nitrogen inputs to seventy-four southern New England estuaries: application of a watershed nitrogen loading model. Estuar. Coast. Shelf Sci. 2010;89:125–136. [Google Scholar]
- Laurmann J.A. Market penetration characteristics for energy production and atmospheric carbon dioxide growth. Science. 1979;205(4409):896–898. doi: 10.1126/science.205.4409.896. [DOI] [PubMed] [Google Scholar]
- Lawson D.R., Winchester J.W. Standard crustal aerosol as a reference for elemental enrichment factors. Atmos. Environ. 1979;13:925–930. [Google Scholar]
- LeBauer D.S., Treseder K.K. Nitrogen limitation of net primary productivity in terrestrial ecosystems is globally distributed. Ecology. 2008;89(2):371–379. doi: 10.1890/06-2057.1. [DOI] [PubMed] [Google Scholar]
- Leifer I., Melton C., Tratt D.M., Buckland K.N., Clarisse L., Coheur P., Frash J., et al. Remote sensing and in situ measurements of methane and ammonia emissions from a megacity dairy complex: Chino, CA. Environ. Pollut. 2017;221(February):37–51. doi: 10.1016/j.envpol.2016.09.083. [DOI] [PubMed] [Google Scholar]
- Lelieveld J., Crutzen P.J. Influences of cloud photochemical processes on tropospheric ozone. Nature. 1990;343:227–233. [Google Scholar]
- Lelieveld J., Crutzen P.J., Ramanathan V., Andreae M.O., Brenninkmeijer C.M., Campos T., Cass G.R., et al. The Indian Ocean experiment: widespread air pollution from south and Southeast Asia. Science. 2001;291(5506):1031–1036. doi: 10.1126/science.1057103. [DOI] [PubMed] [Google Scholar]
- Lelieveld J., van Aardenne J., Fischer H., de Reus M., Williams J., Winkler P. Increasing ozone over the Atlantic Ocean. Science. 2004;304(5676):1483–1487. doi: 10.1126/science.1096777. [DOI] [PubMed] [Google Scholar]
- Lelieveld J., Evans J.S., Fnais M., Giannadaki D., Pozzer A. The contribution of outdoor air pollution sources to premature mortality on a global scale. Nature. 2015;525(7569):367–371. doi: 10.1038/nature15371. [DOI] [PubMed] [Google Scholar]
- Lenton T.M. The role of land plants, phosphorus weathering and fire in the rise and regulation of atmospheric oxygen. Glob. Chang. Biol. 2001;7:613–629. [Google Scholar]
- Lesack L.F.W., Melack J.M. The deposition, composition, and potential sources of major ionic solutes in rain of the Central Amazon Basin. Water Resour. Res. 1991;27:2953–2977. [Google Scholar]
- Levitus S., Antonov J.I., Wang J., Delworth T.L., Dixon K.W., Broccoli A.J. Anthropogenic warming of Earth’s climate system. Science. 2001;292(5515):267–270. doi: 10.1126/science.1058154. [DOI] [PubMed] [Google Scholar]
- Levy H., Moxim W., Klonecki A., Kasibhatla P.S. Simulated tropospheric NOx: its evaluation, global distribution and individual source contributions. J. Geophys. Res. 1999;104:26279–26306. [Google Scholar]
- Lewis R., Schwartz E. vol. 152. American Geophysical Union; Washington, DC: 2004. Sea Salt Aerosol Production: Mechanisms, Methods, Measurements and Models—A Critical Review. Geophysical Monograph Series. [Google Scholar]
- Lewis S.L., Lopez-Gonzalez G., Sonké B., Affum-Baffoe K., Baker T.R., Ojo L.O., Phillips O.L., et al. Increasing carbon storage in intact African tropical forests. Nature. 2009;457(7232):1003–1006. doi: 10.1038/nature07771. [DOI] [PubMed] [Google Scholar]
- Li S., Matthews J., Sinha A. Atmospheric hydroxyl radical production from electronically excited NO2 and H2O. Science. 2008;319(5870):1657–1660. doi: 10.1126/science.1151443. [DOI] [PubMed] [Google Scholar]
- Li Y., Schichtel B.A., Walker J.T., Schwede D.B., Chen X., Lehmann C.M.B., Puchalski M.A., Gay D.A., Collett J.L., Jr. Increasing importance of deposition of reduced nitrogen in the United States. Proc. Natl. Acad. Sci. U. S. A. 2016;113(21):5874–5879. doi: 10.1073/pnas.1525736113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J.Y., Horowitz L.W., Jacob D.J., Wang Y.H., Fiore A.M., Logan J.A., Gardner G.M., Munger J.W. Seasonal budgets of reactive nitrogen species and ozone over the United States, and export fluxes to the global atmosphere. J. Geophys. Res. 1998;103:13435–13450. [Google Scholar]
- Liang Q., Newman P.A., Daniel J.S., Reimann S., Hall B.D., Dutton G., Kuijpers L.J.M. Constraining the carbon tetrachloride (CCl4) budget using its global trend and inter-hemispheric gradient: the carbon tetrachloride budget. Geophys. Res. Lett. 2014;41(14):5307–5315. [Google Scholar]
- Likens G.E., Bormann F.H., Pierce R.S., Eaton J.S., Munn R.E. Long-term trends in precipitation chemistry at Hubbard Brook, New Hampshire. Atmos. Environ. 1984;18:2641–2647. [Google Scholar]
- Likens G.E., Bormann F.H., Hedin L.O., Driscoll C.T., Eaton J.S. Dry deposition of sulfur: a 23 year record for the Hubbard Brook Forest ecosystem. Tellus B Chem. Phys. Meteorol. 1990;42:319–329. [Google Scholar]
- Likens G.E., Driscoll C.T., Buso D.C., Mitchell M.J., Lovett G.M., Bailey S.W., Siccama T.G., Reiners W.A., Alewell C. The biogeochemistry of sulfur at Hubbard Brook. Biogeochemistry. 2002;60:235–316. [Google Scholar]
- Likens G.E., Buso D.C., Butler T.J. Long-term relationships between SO2 and NOx emissions and SO42 and NO3 concentration in bulk deposition at the Hubbard Brook experimental forest, NH. J. Environ. Monitor. 2005;7:964–968. doi: 10.1039/b506370a. [DOI] [PubMed] [Google Scholar]
- Lindberg S.E., Lovett G.M. Field measurements of particle dry deposition rates to foliage and inert surfaces in a forest canopy. Environ. Sci. Technol. 1985;19(3):238–244. doi: 10.1021/es00133a003. [DOI] [PubMed] [Google Scholar]
- Lindberg S.E., Lovett G.M., Richter D.D., Johnson D.W. Atmospheric deposition and canopy interactions of major ions in a forest. Science. 1986;231(4734):141–145. doi: 10.1126/science.231.4734.141. [DOI] [PubMed] [Google Scholar]
- Liss P.S., Slater P.G. Flux of gases across the air-sea interface. Nature. 1974;247:181–184. [Google Scholar]
- Liu T.S., Gu X.F., An Z.S., Fan Y.X. In: Desert Dust: Origin, Characteristics, and Effects on Man. Pewe T.L., editor. Geological Society of America; Boulder, CO: 1981. The dust fall in Beijing, China on April 18, 1980; pp. 149–157. [Google Scholar]
- Logan J.A. Tropospheric ozone—seasonal behavior, trends, and anthropogenic influence. J. Geophys. Res. 1985;90:10463–10482. [Google Scholar]
- Logan J.A., Prather M.J., Wofsy S.C., McElroy M.B. Tropospheric chemistry: a global perspective. J. Geophys. Res. D: Atmos. 1981;86:7210–7254. [Google Scholar]
- Lovett G.M. Atmospheric deposition of Nutrients and pollutants in North America: An ecological perspective. Ecol. Appl. 1994;4:629–650. [Google Scholar]
- Lovett G.M., Lindberg S.E. Dry deposition of nitrate to a deciduous forest. Biogeochemistry. 1986;2:137–148. [Google Scholar]
- Lovett G.M., Lindberg S.E. Atmospheric deposition and canopy interactions of nitrogen in forests. Can. J. Forest Res. 1993;23:1603–1616. [Google Scholar]
- Lovett G.M., Reiners W.A., Olson R.K. Cloud droplet deposition in subalpine balsam fir forests: hydrological and chemical inputs. Science. 1982;218(4579):1303–1304. doi: 10.1126/science.218.4579.1303. [DOI] [PubMed] [Google Scholar]
- Luterbacher J., Dietrich D., Xoplaki E., Grosjean M., Wanner H. European seasonal and annual temperature variability, trends, and extremes since 1500. Science. 2004;303(5663):1499–1503. doi: 10.1126/science.1093877. [DOI] [PubMed] [Google Scholar]
- Lutz B.D., Mulholland P.J., Bernhardt E.S. Long-term data reveal patterns and controls on stream water chemistry in a forested stream: walker branch Tennessee. Ecol. Monogr. 2012;82:367–387. [Google Scholar]
- Mace K.A., Duce R.A., Tindale N.W. Organic nitrogen in rain and aerosol at cape grim, Tasmania, Australia. J. Geophys. Res.-Atmos. 2003;108 [Google Scholar]
- MacIntyre F. The top millimeter of the ocean. Sci. Am. 1974;230:62–77. [Google Scholar]
- Mahowald N. Aerosol indirect effect on biogeochemical cycles and climate. Science. 2011;334(6057):794–796. doi: 10.1126/science.1207374. [DOI] [PubMed] [Google Scholar]
- Mahowald N.M., Baker A.R., Bergametti G., Brooks N., Duce R.A., Jickells T.D., Kubilay N., Prospero J.M., Tegen I. Atmospheric global dust cycle and iron inputs to the ocean. Glob. Biogeochem. Cycles. 2005;19 [Google Scholar]
- Manabe S., Wetherald R.T. Distribution of climate change resulting from an increase in the CO2 content of the atmosphere. J. Atmos. Sci. 1980;37:99–118. [Google Scholar]
- Manabe S., Wetherald R.T. Reduction in summer soil wetness induced by an increase in atmospheric carbon dioxide. Science. 1986;232(4750):626–628. doi: 10.1126/science.232.4750.626. [DOI] [PubMed] [Google Scholar]
- Mankin W.G., Coffey M.T. Increased stratospheric hydrogen chloride in the El Chichon cloud. Science. 1984;226(4671):170–172. doi: 10.1126/science.226.4671.170. [DOI] [PubMed] [Google Scholar]
- Mankin W.G., Coffey M.T., Goldman A. Airborne observations of SO2, HCl, and O3 in the stratospheric plume of the Pinatubo volcano in July 1991. Geophys. Res. Lett. 1992;19:179–182. [Google Scholar]
- Mann M.E., Bradley R.S., Hughes M.K. Northern hemisphere temperatures during the past millennium: inferences, uncertainties, and lim itations. Geophys. Res. Lett. 1999;26:759–762. [Google Scholar]
- Manney G.L., Santee M.L., Rex M., Livesey N.J., Pitts M.C., Veefkind P., Nash E.R., et al. Unprecedented Arctic ozone loss in 2011. Nature. 2011;478(7370):469–475. doi: 10.1038/nature10556. [DOI] [PubMed] [Google Scholar]
- Mao K.B., Ma Y., Xia L., Chen W.Y., Shen X.Y., He T.J., Xu T.R. Global aerosol change in the last decade: an analysis based on MODIS data. Atmos. Environ. 2014;94(September):680–686. [Google Scholar]
- Marenco A., Gouget H., Nedelec P., Pages J.P., Karcher F. Evidence of a long-term increase in tropospheric ozone from PIC DU MIDI data series: consequences: positive radiative forcing. J. Geophys. Res. 1994;99:16617–16632. [Google Scholar]
- Martin R.V. Satellite remote sensing of surface air quality. Atmos. Environ. 2008;42(March):7823–7843. [Google Scholar]
- Marufu L.T., Taubman B.F., Bloomer B., Piety C.A., Doddridge B.G., Stehr J.W., Dickerson R.R. The 2003 North American electrical blackout: an accidental experiment in atmospheric chemistry. J. Geophys. Res. 2004;31 [Google Scholar]
- Mayewski P.A., Lyons W.B., Spencer M.J., Twickler M., Dansgaard W., Koci B., Davidson C.I., Honrath R.E. Sulfate and nitrate concentrations from a South Greenland ice core. Science. 1986;232(4753):975–977. doi: 10.1126/science.232.4753.975. [DOI] [PubMed] [Google Scholar]
- Mayewski P.A., Lyons W.B., Spencer M.J., Twickler M.S., Buck C.F., Whitlow S. An ice-core record of atmospheric response to anthropogenic sulfate and nitrate. Nature. 1990;346:554–556. [Google Scholar]
- McClure C.D., Jaffe D.A. US particulate matter air quality improves except in wildfire-prone areas. Proc. Natl. Acad. Sci. U. S. A. 2018;115(31):7901–7906. doi: 10.1073/pnas.1804353115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick M.P., Thomason L.W., Trepte C.R. Atmospheric effects of the Mt. Pinatubo eruption. Nature. 1995;373:399–404. [Google Scholar]
- McCulloch A., Aucott M.L., Benkovitz C.M., Graedel T.E., Kleiman G., Midgley P.M., Li Y.F. Global emissions of hydrogen chloride and chloromethane from coal combustion, incineration and industrial activities: reactive chlorine emissions inventory. J. Geophys. Res. 1999;104:8391–8403. [Google Scholar]
- McDonald B.C., de Gouw J.A., Gilman J.B., Jathar S.H., Akherati A., Cappa C.D., Jimenez J.L., et al. Volatile chemical products emerging as largest petrochemical source of urban organic emissions. Science. 2018;359(6377):760–764. doi: 10.1126/science.aaq0524. [DOI] [PubMed] [Google Scholar]
- McElroy M.B. Princeton University Press; 2002. The Atmospheric Environment. [Google Scholar]
- McKenzie R., Connor B., Bodeker G. Increased summertime UV radiation in New Zealand in response to ozone loss. Science. 1999;285(5434):1709–1711. doi: 10.1126/science.285.5434.1709. [DOI] [PubMed] [Google Scholar]
- McLaughlin S.B., Taylor G.E. Relative humidity: important modifier of pollutant uptake by plants. Science. 1981;211(4478):167–169. doi: 10.1126/science.211.4478.167. [DOI] [PubMed] [Google Scholar]
- Meador J., Jeffrey W.H., Kase J.P., Dean Pakulski J., Chiarello S., Mitchell D.L. Seasonal fluctuation of DNA photodamage in marine plankton assemblages at Palmer Station, Antarctica. Photochem. Photobiol. 2002;75(3):266–271. doi: 10.1562/0031-8655(2002)075<0266:sfodpi>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Miller E.K., Blum J.D., Friedland A.J. Determination of soil exchangeable cation loss and weathering rates using Sr isotopes. Nature. 1993;362:438–441. [Google Scholar]
- Milly P.C.D., Dunne K.A., Vecchia A.V. Global pattern of trends in streamflow and water availability in a changing climate. Nature. 2005;438(7066):347–350. doi: 10.1038/nature04312. [DOI] [PubMed] [Google Scholar]
- Minoura H., Iwasaka Y. Rapid change in nitrate and sulfate concentrations observed in early stage of precipitation and their deposition processes. J. Atmos. Chem. 1996;24:39–55. [Google Scholar]
- Mitchell J.F.B., Johns T.C., Gregory J.M., Tett S.F.B. Climate response to increasing levels of greenhouse gases and sulfate aerosols. Nature. 1995;376:501–504. [Google Scholar]
- Molina M.J., Rowland F.S. Stratospheric sink for chlorofluoromethanes: chlorine atom-catalyzed destruction of ozone. Nature. 1974;249:810–812. [Google Scholar]
- Molina M.J., Tso T.L., Molina L.T., Wang F.C. Antarctic stratospheric chemistry of chlorine nitrate, hydrogen chloride, and ice: release of active chlorine. Science. 1987;238(4831):1253–1257. doi: 10.1126/science.238.4831.1253. [DOI] [PubMed] [Google Scholar]
- Möller D. The Na/Cl ratio in rainwater and the seasalt chloride cycle. Tellus B Chem. Phys. Meteorol. 1990;42(3):254–262. [Google Scholar]
- Monson R.K., Holland E.A. Biospheric trace gas fluxes and their control over tropospheric chemistry. Annu. Rev. Ecol. Syst. 2001;32:547–576. [Google Scholar]
- Montzka S.A., Butler J.H., Myers R.C., Thompson T.M., Swanson T.H., Clarke A.D., Lock L.T., Elkins J.W. Decline in the tropospheric abundance of halogen from halocarbons: implications for stratospheric ozone depletion. Science. 1996;272(5266):1318–1322. doi: 10.1126/science.272.5266.1318. [DOI] [PubMed] [Google Scholar]
- Montzka S.A., Aydin M., Battle M., Butler J.H., Saltzman E.S., Hall B.D., Clarke A.D., Mondeel D., Elkins J.W. A 350-year atmospheric history for carbonyl sulfide inferred from Antarctic firn air and air trapped in ice. J. Geophys. Res. 2004;109 [Google Scholar]
- Montzka S.A., Calvert P., Hall B.D., Elkins J.W., Conway T.J., Tans P.P., Sweeney C. On the global distribution, seasonality, and budget of atmospheric carbonyl sulfide (COS) and some similarities to CO2. J. Geophys. Res. D: Atmos. 2007;112 [Google Scholar]
- Montzka S.A., Krol M., Dlugokencky E., Hall B., Jöckel P., Lelieveld J. Small interannual variability of global atmospheric hydroxyl. Science. 2011;331(6013):67–69. doi: 10.1126/science.1197640. [DOI] [PubMed] [Google Scholar]
- Montzka S.A., Dutton G.S., Yu P., Ray E., Portmann R.W., Daniel J.S., Kuijpers L., et al. An unexpected and persistent increase in global emissions of ozone-depleting CFC-11. Nature. 2018;557(7705):413–417. doi: 10.1038/s41586-018-0106-2. [DOI] [PubMed] [Google Scholar]
- Mount G.H. The measurement of tropospheric OH by long-path absorption. 1. Instrumentation. J. Geophys. Res. 1992;97:2427–2444. [Google Scholar]
- Mount G.H., Brault J.W., Johnston P.V., Marovich E., Jakoubek R.O., Volpe C.J., Harder J., Olson J. Measurement of tropospheric OH by long-path laser absorption at Fritz peak observatory, Colorado, during the OH photochemistry experiment, fall 1993. J. Geophys. Res. 1997;102:6393–6413. [Google Scholar]
- Moyers J.L., Ranweiler L.E., Hopf S.B., Korte N.E. Evaluation of particulate trace species in the Southwest Desert atmosphere. Environ. Sci. Technol. 1977;11:789–795. [Google Scholar]
- Muhs D.R., Bettis E.A., Been J., McGeehin J.P. Impact of climate and parent material on chemical weathering in loess-derived soils of the Mississippi river valley. Soil Sci. Soc. Am. J. 2001;65:1761–1777. [Google Scholar]
- Mulitza S., Heslop D., Pittauerova D., Fischer H.W., Meyer I., Stuut J.-B., Zabel M., et al. Increase in African dust flux at the onset of commercial agriculture in the Sahel region. Nature. 2010;466(7303):226–228. doi: 10.1038/nature09213. [DOI] [PubMed] [Google Scholar]
- Munger J.W., Eisenreich S.J. Continental-scale variations in precipitation chemistry. Environ. Sci. Technol. 1983;17:32–42. [Google Scholar]
- Munger J.W., Fan S.M., Bakwin P.S., Goulden M.L., Goldstein A.H., Colman A.S., Wofsy S.C. Regional budgets for nitrogen oxides from continental sources: variations of rates for oxidation and deposition with season and distance from source regions. J. Geophys. Res. D: Atmos. 1998;103:8355–8368. [Google Scholar]
- Murphy D.M., Thomson D.S., Mahoney M.J. In situ measurements of organics, meteoritic material, mercury, and other elements in aerosols at 5 to 19 kilometers. Science. 1998;282(5394):1664–1669. doi: 10.1126/science.282.5394.1664. [DOI] [PubMed] [Google Scholar]
- Najjar R.G., Erickson D.J., Madronich S. In: The Role of Nonliving Organic Matter in the Earth’s Carbon Cycle. Zepp R.G., Sonntag C., editors. Wiley; 1995. Modeling the air-sea fluxes of gases formed from the decomposition of dissolved organic matter: carbonyl sulfide and carbon monoxide; pp. 107–132. [Google Scholar]
- Neff J.C., Holland E.A., Dentener F.J., McDowell W.H., Russell K.M. The origin, composition and rates of organic nitrogen deposition: a missing piece of the nitrogen cycle? Biogeochemistry. 2002;57:99–136. [Google Scholar]
- Newman E.I. Phosphorus inputs to terrestrial ecosystems. J. Ecol. 1995;83:713–726. [Google Scholar]
- Nguyen B.C., Mihalopoulos N., Putaud J.P., Bonsang B. Carbonyl sulfide emissions from biomass burning in the tropics. J. Atmos. Chem. 1995;22:55–65. [Google Scholar]
- Nixon S.W., Ammerman J.W., Atkinson L.P., Berounsky V.M., Billen G., Boicourt W.C., Boynton W.R., et al. The fate of nitrogen and phosphorus at the land sea margin of the North Atlantic Ocean. Biogeochemistry. 1996;35:141–180. [Google Scholar]
- Norval M., Cullen A.P., de Gruijl F.R., Longstreth J., Takizawa Y., Lucas R.M., Noonan F.P., van der Leun J.C. The effects on human health from stratospheric ozone depletion and its interactions with climate change. Photochem. Photobiol. Sci.: Off. J. Eur. Photochem. Assoc. Eur. Soc. Photobiol. 2007;6(3):232–251. doi: 10.1039/b700018a. [DOI] [PubMed] [Google Scholar]
- Notholt J., Kuang Z., Rinsland C.P., Toon G.C., Rex M., Jones N., Albrecht T., et al. Enhanced upper tropical tropospheric COS: impact on the stratospheric aerosol layer. Science. 2003;300(5617):307–310. doi: 10.1126/science.1080320. [DOI] [PubMed] [Google Scholar]
- Notz D., Stroeve J. Observed Arctic sea-ice loss directly follows anthropogenic CO2 emission. Science. 2016;354(6313):747–750. doi: 10.1126/science.aag2345. [DOI] [PubMed] [Google Scholar]
- Nowlan C.R., Martin R.V., Philip S., Lamsal L.N., Krotkov N.A., Marais E.A., Wang S., Zhang Q. Global dry deposition of nitrogen dioxide and sulfur dioxide inferred from space-based measurements. Glob. Biogeochem. Cycles. 2014;28(10):1025–1043. [Google Scholar]
- Nriagu J.O. A global assessment of natural sources of atmospheric trace metals. Nature. 1989;338:47–49. [Google Scholar]
- Okin G.S., Mahowald N., Chadwick O.A., Artaxo P. Impact of desert dust on the biogeochemistry of phosphorus in terrestrial ecosystems. Glob. Biogeochem. Cycles. 2004;18 [Google Scholar]
- Ollinger S.V., Aber J.D., Lovett G.M., Millham S.E., Lathrop R.G., Ellis J.M. A spatial model of atmospheric deposition for the northeastern United States. Ecol. Appl. 1993;3:459–472. doi: 10.2307/1941915. [DOI] [PubMed] [Google Scholar]
- Oltmans S.J., Levy H. Seasonal cycle of surface ozone over the Western North Atlantic. Nature. 1992;358:392–394. [Google Scholar]
- Ortega J., Helmig D., Daly R.W., Tanner D.M., Guenther A.B., Herrick J.D. Approaches for quantifying reactive and low-volatility biogenic organic compound emissions by vegetation enclosure techniques—part B: applications. Chemosphere. 2008;72(3):365–380. doi: 10.1016/j.chemosphere.2008.02.054. [DOI] [PubMed] [Google Scholar]
- Paerl H.W., Willey J.D., Go M., Peierls B.L., Pinckney J.L., Fogel M.L. Rainfall stimulation of primary production in Western Atlantic ocean waters: Roles of different nitrogen sources and co-limiting nutrients. Mar. Ecol.—Prog. Ser. 1999;176:205–214. [Google Scholar]
- Palle E., Goode P.R., Montañés-Rodríguez P., Shumko A., Gonzalez-Merino B., Martinez Lombilla C., Jimenez-Ibarra F., et al. Earth’s albedo variations 1998–2014 as measured from ground-based earthshine observations. Geophys. Res. Lett. 2016;43(9):4531–4538. [Google Scholar]
- Park J.-H., Goldstein A.H., Timkovsky J., Fares S., Weber R., Karlik J., Holzinger R. Active atmosphere-ecosystem exchange of the vast majority of detected volatile organic compounds. Science. 2013;341(6146):643–647. doi: 10.1126/science.1235053. [DOI] [PubMed] [Google Scholar]
- Parker G.G. Throughfall and stemflow in the forest nutrient cycle. Adv. Ecol. Res. 1983;13:57–133. [Google Scholar]
- Parrington J.R., Zoller W.H., Aras N.K. Asian dust: seasonal transport to the Hawaiian islands. Science. 1983;220(4593):195–197. doi: 10.1126/science.220.4593.195. [DOI] [PubMed] [Google Scholar]
- Parrish D.D., Holloway J.S., Trainer M., Murphy P.C., Fehsenfeld F.C., Forbes G.L. Export of North American ozone pollution to the North Atlantic Ocean. Science. 1993;259(5100):1436–1439. doi: 10.1126/science.259.5100.1436. [DOI] [PubMed] [Google Scholar]
- Patterson D.B., Farley K.A., Norman M.D. 4He as a tracer of continental dust: a 1.9-million-year record of Aeolian flux to the West equatorial Pacific Ocean. Geochim. Cosmochim. Acta. 1999;63:615–625. [Google Scholar]
- Paulot F., Ginoux P., Cooke W.F., Donner L.J., Fan S., Lin M.-Y., Mao J., Naik V., Horowitz L.W. Sensitivity of nitrate aerosols to ammonia emissions and to nitrate chemistry: implications for present and future nitrate optical depth. Atmos. Chem. Phys. 2016;16(3):1459–1477. [Google Scholar]
- Penner J.E., Dickinson R.E., Oneill C.A. Effects of aerosols from biomass burning on the global radiation budget. Science. 1992;256:1432–1434. doi: 10.1126/science.256.5062.1432. [DOI] [PubMed] [Google Scholar]
- Petkov B.H., Vitale V., Tomasi C., Siani A.M., Seckmeyer G., Webb A.R., Smedley A.R.D., et al. Response of the ozone column over Europe to the 2011 Arctic ozone depletion event according to ground-based observations and assessment of the consequent variations in surface UV irradiance. Atmos. Environ. 2014;85(March):169–178. [Google Scholar]
- Petroff A., Mailliat A., Amielh M., Anselmet F. Aerosol dry deposition on vegetative canopies. Part I: Review of present knowledge. Atmos. Environ. 2008;42:3625–3653. [Google Scholar]
- Piccot S.D., Watson J.J., Jones J.W. A global inventory of volatile organic compound emissions from anthropogenic sources. J. Geophys. Res. 1992;97:9897–9912. [Google Scholar]
- Pietruczuk A., Krzyscin J.W., Jaroslawski J., Podgorski J., Sobolewski P., Wink J. Eyjafjallajokull volcano ash observed over Belsk (52°N, 21°E), Poland, in April 2010. Int. J. Remote Sens. 2010;31:3981–3986. [Google Scholar]
- Pinker R.T., Zhang B., Dutton E.G. Do satellites detect trends in surface solar radiation? Science. 2005;308(5723):850–854. doi: 10.1126/science.1103159. [DOI] [PubMed] [Google Scholar]
- Platt U., Rateike M., Junkermann W., Rudolph J., Ehhalt D.H. New tropospheric OH measurements. J. Geophys. Res. D: Atmos. 1988;93:5159–5166. [Google Scholar]
- Pöhlker C., Wiedemann K.T., Sinha B., Shiraiwa M., Gunthe S.S., Smith M., Hang S., et al. Biogenic potassium salt particles as seeds for secondary organic aerosol in the Amazon. Science. 2012;337(6098):1075–1078. doi: 10.1126/science.1223264. [DOI] [PubMed] [Google Scholar]
- Pommier M., McLinden C.A., Deeter M. Relative changes in CO emissions over megacities based on observations from space. Geophys. Res. Lett. 2013;40(14):3766–3771. [Google Scholar]
- Potts M.J. Deposition of air-borne salt on Pinus radiata and underlying soil. J. Appl. Ecol. 1978;15:543–550. [Google Scholar]
- Prather M.J. Continental sources of halocarbons and nitrous oxide. Nature. 1985;317:221–225. [Google Scholar]
- Prather M.J., Hsu J. NF3, the greenhouse gas missing from Kyoto. Geophys. Res. Lett. 2008;35 [Google Scholar]
- Preunkert S., Legrand M., Wagenbach D. Sulfate trends in a col Du dome (French Alps) ice core: a record of anthropogenic sulfate levels in the European midtroposphere over the twentieth century. J. Geophys. Res. 2001;106:31991–31994. [Google Scholar]
- Preunkert S., Wagenbach D., Legrand M. A seasonally resolved alpine ice core record of nitrate: comparison with anthropogenic inventories and estimation of preindustrial emissions of NO in Europe. J. Geophys. Res. 2003;108 [Google Scholar]
- Prinn R.G. The cleansing capacity of the atmosphere. Annu. Rev. Environ. Resour. 2003;28:29–57. [Google Scholar]
- Prinn R.G., Weiss R.F., Miller B.R., Huang J., Alyea F.N., Cunnold D.M., Fraser P.J., Hartley D.E., Simmonds P.G. Atmospheric trends and lifetime of CH3CCl3 and global OH concentrations. Science. 1995;269:187–192. doi: 10.1126/science.269.5221.187. [DOI] [PubMed] [Google Scholar]
- Prinn R.G., Huang J., Weiss R.F., Cunnold D.M., Fraser P.J., Simmonds P.G., McCulloch A., et al. Evidence for variability of atmospheric hydroxyl radicals over the past quarter century. Geophys. Res. Lett. 2005;32 [Google Scholar]
- Pryor S.C., Barthelmie R.J., Sorensen L.L., Jensen B. Ammonia concentrations and fluxes over a forest in the Midwestern USA. Atmos. Environ. 2001;35:5645–5656. [Google Scholar]
- Pye K. Academic Press; London: 1987. Eolian Dust and Dust Deposits. [Google Scholar]
- Quack B., Wallace D.W.R. Air-sea flux of bromoform: controls, rates, and implications. Glob. Biogeochem. Cycles. 2003;17 [Google Scholar]
- Raes F., Van Dingenen R., Vignati E., Wilson J., Putaud J.P., Seinfeld J.H., Adams P. Formation and cycling of aerosols in the Global troposphere. Atmos. Environ. 2000;34:4215–4240. [Google Scholar]
- Rahn K.A., Lowenthal D.H. Elemental tracers of distant regional pollution aerosols. Science. 1984;223(4632):132–139. doi: 10.1126/science.223.4632.132. [DOI] [PubMed] [Google Scholar]
- Ramanathan V. The greenhouse theory of climate change: a test by an inadvertent global experiment. Science. 1988;240(4850):293–299. doi: 10.1126/science.240.4850.293. [DOI] [PubMed] [Google Scholar]
- Raval A., Ramanathan V. Observational determination of the greenhouse effect. Nature. 1989;342:758–761. [Google Scholar]
- Ravi S., D’Odorico P., Breshears D.D., Field J.P., Goudie A.S., Huxman T.E., Li J., et al. Aeolian processes and the biosphere. Rev. Geophys. 2011;49 [Google Scholar]
- Ravishankara A.R. Heterogeneous and multiphase chemistry in the troposphere. Science. 1997;276:1058–1065. [Google Scholar]
- Ravishankara A.R., Daniel J.S., Portmann R.W. Nitrous oxide (N2O): the dominant ozone-depleting substance emitted in the 21st century. Science. 2009;326(5949):123–125. doi: 10.1126/science.1176985. [DOI] [PubMed] [Google Scholar]
- Reheis M.C., Kihl R. Dust deposition in southern Nevada and California, 1984–1989: relations to climate, source area, and source lithology. J. Geophys. Res.-Atmos. 1995;100:8893–8918. [Google Scholar]
- Reiners P.W., Turchyn A.V. Extraterrestrial dust, the marine lithologic record, and global biogeochemical cycles. Geology. 2018;46(10):863–866. [Google Scholar]
- Rhew R.C., Whelan M.E., Min D.-H. Large methyl halide emissions from South Texas salt marshes. Biogeosciences. 2014;11(22):6427–6434. [Google Scholar]
- Riccobono F., Schobesberger S., Scott C.E., Dommen J., Ortega I.K., Rondo L., Almeida J., et al. Oxidation products of biogenic emissions contribute to nucleation of atmospheric particles. Science. 2014;344(6185):717–721. doi: 10.1126/science.1243527. [DOI] [PubMed] [Google Scholar]
- Richter A., Burrows J.P., Nüss H., Granier C., Niemeier U. Increase in tropospheric nitrogen dioxide over China observed from space. Nature. 2005;437(7055):129–132. doi: 10.1038/nature04092. [DOI] [PubMed] [Google Scholar]
- Rigby M., Prinn R.G., O’Doherty S., Miller B.R., Ivy D., Mühle J., Harth C.M., et al. Recent and future trends in synthetic greenhouse gas radiative forcing: synthetic greenhouse gas trends. Geophys. Res. Lett. 2014;41(7):2623–2630. [Google Scholar]
- Rigby M., Montzka S.A., Prinn R.G., White J.W.C., Young D., O’Doherty S., Lunt M.F., et al. Role of atmospheric oxidation in recent methane growth. Proc. Natl. Acad. Sci. U. S. A. 2017;114(21):5373–5377. doi: 10.1073/pnas.1616426114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigby M., Park S., Saito T., Western L.M., Redington A.L., Fang X., Henne S., et al. Increase in CFC-11 emissions from eastern China based on atmospheric observations. Nature. 2019;569(7757):546–550. doi: 10.1038/s41586-019-1193-4. [DOI] [PubMed] [Google Scholar]
- Rind D., Goldberg R., Hansen J., Rosenzweig C., Ruedy R. Potential evapotranspiration and the likelihood of future drought. J. Geophys. Res.-Atmos. 1990;95:9983–10004. [Google Scholar]
- Rind D., Chiou E.W., Chu W., Larsen J., Oltmans S., Lerner J., McCormick M.P., McMaster L. Positive water-vapor feedback in climate models confirmed by satellite data. Nature. 1991;349:500–503. [Google Scholar]
- Rinsland C.P., Goldman A., Mahieu E., Zander R., Notholt J., Jones N.B., Griffith D.W.T., Stephen T.M., Chiou L.S. Ground-based infrared spectroscopic measurements of carbonyl sulfide: free tropospheric trends from a 24-year time series of solar absorption measurements. J. Geophys. Res.—Atmos. 2002;107 [Google Scholar]
- Robock A., Marquardt A., Kravitz B., Stenchikov G. Benefits, risks, and costs of stratospheric geoengineering. Geophysical Research Letters. 2009;36 [Google Scholar]
- Rodhe H. Formation of sulfuric and nitric acid in the atmosphere during long-range transport. Tell’Us. 1981;33:132–141. [Google Scholar]
- Rohrer F., Berresheim H. Strong correlation between levels of tropospheric hydroxyl radicals and solar ultraviolet radiation. Nature. 2006;442(7099):184–187. doi: 10.1038/nature04924. [DOI] [PubMed] [Google Scholar]
- Rondon A., Granat L. Studies on the dry deposition of NO2 to Coniferous species at low NO2 concentrations. Tellus B Chem. Phys. Meteorol. 1994;46(5):339–352. [Google Scholar]
- Routson C.C., Arcusa S.H., McKay N.P., Overpeck J.T. A 4,500-year-long record of southern rocky mountain dust deposition. Geophys. Res. Lett. 2019;46(14):8281–8288. [Google Scholar]
- Rowland F.S. Chlorofluorocarbons and the depletion of stratospheric ozone. Am. Sci. 1989;77:36–45. [Google Scholar]
- Rowland F.S. Stratospheric ozone depletion. Ann. Rev. Phys. Chem. 1991;42:731–768. [Google Scholar]
- Russell J.M., Luo M.Z., Cicerone R.J., Deaver L.E. Satellite confirmation of the dominance of chlorofluorocarbons in the global stratospheric chlorine budget. Nature. 1996;379:526–529. [Google Scholar]
- Sabine C.L., Feely R.A., Gruber N., Key R.M., Lee K., Bullister J.L., Wanninkhof R., et al. The oceanic sink for anthropogenic CO2. Science. 2004;305(5682):367–371. doi: 10.1126/science.1097403. [DOI] [PubMed] [Google Scholar]
- Saltzman E.S., Aydin M., Tatum C., Williams M.B. 2,000-Year record of atmospheric methyl bromide from a south pole ice core. J. Geophys. Res.—Atmos. 2008;113 [Google Scholar]
- Saltzman E.S., Aydin M., Williams M.B., Verhulst K.R., Gun B. Methyl chloride in a deep ice core from Siple Dome, Antarctica. Geophys. Res. Lett. 2009;36 [Google Scholar]
- Samet J.M., Dominici F., Curriero F.C., Coursac I., Zeger S.L. Fine particulate air pollution and mortality in 20 U.S. cities, 1987–1994. N. Engl. J. Med. 2000;343(24):1742–1749. doi: 10.1056/NEJM200012143432401. [DOI] [PubMed] [Google Scholar]
- Savoie D.I., Prospero J.M., Larsen R.J., Huang F., Izaguirre M.A., Huang T., Snowdon T.H., Custals L., Sanderson C.G. Nitrogen and sulfur species in Antarctic aerosols at Mawson, Palmer Station, and Marsh (King George Island) J. Atmos. Chem. 1993;17:95–122. [Google Scholar]
- Schlesinger W.H., Gray J.T., Gilliam F.S. Atmospheric deposition processes and their importance as sources of nutrients in a chaparral ecosystem of Southern California. Water Resour. Res. 1982;18:623–629. [Google Scholar]
- Schneider P., R. J. van der A. A global single-sensor analysis of 2002–2011 tropospheric nitrogen dioxide trends observed from space: global No 2 trends observed from space. J. Geophys. Res. 2012;117(D16) [Google Scholar]
- Schuur E.A.G., Vogel J.G., Crummer K.G., Lee H., Sickman J.O., Osterkamp T.E. The effect of permafrost thaw on old carbon release and net carbon exchange from tundra. Nature. 2009;459(7246):556–559. doi: 10.1038/nature08031. [DOI] [PubMed] [Google Scholar]
- Schwartz S.E. Acid deposition: unraveling a regional phenomenon. Science. 1989;243(4892):753–763. doi: 10.1126/science.243.4892.753. [DOI] [PubMed] [Google Scholar]
- Seager R., Ting M., Held I., Kushnir Y., Lu J., Vecchi G., Huang H.-P., et al. Model projections of an imminent transition to a more arid climate in southwestern North America. Science. 2007;316(5828):1181–1184. doi: 10.1126/science.1139601. [DOI] [PubMed] [Google Scholar]
- Sehmel G.A. Particle and gas dry deposition: a review. Atmos. Environ. 1980;14:983–1011. [Google Scholar]
- Seinfeld J.H. Urban air pollution: state of the science. Science. 1989;243(4892):745–752. doi: 10.1126/science.243.4892.745. [DOI] [PubMed] [Google Scholar]
- Seo K.H., Bowman K.P. Lagrangian estimate of global stratosphere-troposphere mass exchange. J. Geophys. Res. 2002;107 [Google Scholar]
- Sequeira R. On the large-scale impact of arid dust on precipitation chemistry of the continental northern hemisphere. Atmos. Environ. A—Gen. Top. 1993;27:1553–1565. [Google Scholar]
- Serca D., Guenther A., Klinger L., Helmig D., Hereid D., Zimmerman P. Methyl bromide deposition to soils. Atmos. Environ. 1998;32:1581–1586. [Google Scholar]
- Serreze M.C., Holland M.M., Stroeve J. Perspectives on the Arctic’s shrinking sea-ice cover. Science. 2007;315(5818):1533–1536. doi: 10.1126/science.1139426. [DOI] [PubMed] [Google Scholar]
- Servant J. The burden of the sulphate layer of the stratosphere during volcanic “quiescent” periods. Tellus B Chem. Phys. Meteorol. 1986;38:74–79. [Google Scholar]
- Shaw R.W. Air pollution by particles. Sci. Am. 1987;257:96–103. [Google Scholar]
- Sheng J.-X., Weisenstein D.K., Luo B.-P., Rozanov E., Stenke A., Anet J., Bingemer H., Peter T. Global atmospheric sulfur budget under volcanically quiescent conditions: aerosol-chemistry-climate model predictions and validation. J. Geophys. Res. D: Atmos. 2015;120(1):256–276. [Google Scholar]
- Shimshock J.P., de Pena R.G. Below-cloud scavenging of tropospheric ammonia. Tellus B Chem. Phys. Meteorol. 1989;41:296–304. [Google Scholar]
- Shorter J.H., Kolb C.E., Crill P.M., Kerwin R.A., Talbot R.W., Hines M.E., Harriss R.C. Rapid degradation of atmospheric methyl bromide in soils. Nature. 1995;377:717–719. [Google Scholar]
- Shukla S.P., Sharma M. Neutralization of rainwater acidity at Kanpur, India. Tellus B—Chem. Phys. Meteorol. 2010;62:172–180. [Google Scholar]
- Sigg A., Neftel A. Evidence for a 50-percent increase in H2O2 over the past 200 years from a Greenland ice Core. Nature. 1991;351:557–559. [Google Scholar]
- Sigl M., Winstrup M., McConnell J.R., Welten K.C., Plunkett G., Ludlow F., Büntgen U., et al. Timing and climate forcing of volcanic eruptions for the past 2,500 years. Nature. 2015;523(7562):543–549. doi: 10.1038/nature14565. [DOI] [PubMed] [Google Scholar]
- Sillman S. The relation between ozone, NOx and hydrocarbons in Urban and polluted rural environments. Atmos. Environ. 1999;33:1821–1845. [Google Scholar]
- Simmons J.S., Klemedtsson L., Hultberg H., Hines M.E. Consumption of atmospheric carbonyl sulfide by coniferous boreal forest soils. J. Geophys. Res. 1999;104:11569–11576. [Google Scholar]
- Simonich S.L., Hites R.A. Importance of vegetation in removing polycyclic aromatic hydrocarbons from the atmosphere. Nature. 1994;370:49–51. [Google Scholar]
- Simonson R.W. Airborne dust and its significance to soils. Geoderma. 1995;65:1–43. [Google Scholar]
- Singh A.P., Singh R., Mina U., Singh M.P., Varshney C.K. Emissions of monoterpene from tropical Indian plant species and assessment of VOC emission from the forest of Haryana state. Atmos. Pollut. Res. 2011;2(1):72–79. [Google Scholar]
- Slinn W.G.N. A simple model for Junge’s relationship between concentration fluctuations and residence times for tropospheric trace gases. Tellus B Chem. Phys. Meteorol. 1988;40:229–232. [Google Scholar]
- Smith T.M., Leemans R., Shugart H.H. Sensitivity of terrestrial carbon storage to CO2-induced climate change: comparison of four scenarios based on general-circulation models. Clim. Chang. 1992;21:367–384. [Google Scholar]
- Soden B.J., Jackson D.L., Ramaswamy V., Schwarzkopf M.D., Huang X. The radiative signature of upper tropospheric moistening. Science. 2005;310(5749):841–844. doi: 10.1126/science.1115602. [DOI] [PubMed] [Google Scholar]
- Solomon S. Progress towards a quantitative understanding of Antarctic ozone depletion. Nature. 1990;347:347–354. [Google Scholar]
- Solomon S., Garcia R.R., Rowland F.S., Wuebbles D.J. On the depletion of Antarctic ozone. Nature. 1986;321:755–758. [Google Scholar]
- Solomon P., Barrett J., Mooney T., Connor B., Parrish A., Siskind D.E. Rise and decline of active chlorine in the stratosphere. Geophys. Res. Lett. 2006;33 [Google Scholar]
- Solomon S., Portmann R.W., Thompson D.W.J. Contrasts between Antarctic and Arctic ozone depletion. Proc. Natl. Acad. Sci. U. S. A. 2007;104(2):445–449. doi: 10.1073/pnas.0604895104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon S., Ivy D.J., Kinnison D., Mills M.J., Neely R.R., 3rd, Schmidt A. Emergence of healing in the Antarctic ozone layer. Science. 2016;353(6296):269–274. doi: 10.1126/science.aae0061. [DOI] [PubMed] [Google Scholar]
- Sparks J.P., Roberts J.M., Monson R.K. The uptake of gaseous organic nitrogen by leaves: a significant global nitrogen transfer process. Geophys. Res. Lett. 2003;30 [Google Scholar]
- Staffelbach T., Neftel A., Stauffer B., Jacob D. A record of the atmospheric methane sink from formaldehyde in polar ice cores. Nature. 1991;349:603–605. [Google Scholar]
- Stap L.B., de Boer B., Ziegler M., Bintanja R., Lourens L.J., van de Wal R.S.W. CO2 over the past 5 million years: continuous simulation and new δ11B-based proxy data. Earth Planet. Sci. Lett. 2016;439(April):1–10. [Google Scholar]
- Steele L.P., Fraser P.J., Rasmussen R.A., Khalil M.A.K., Conway T.J., Crawford A.J., Gammon R.H., Masarie K.A., Thoning K.W. The global distribution of methane in the troposphere. J. Atmos. Chem. 1987;5:125–171. [Google Scholar]
- Steinbacher M., Bingemer H.G., Schmidt U. Measurements of the exchange of carbonyl sulfide (OCS) and carbon disulfide (CS2) between soil and atmosphere in a spruce Forest in Central Germany. Atmos. Environ. 2004;38:6043–6052. [Google Scholar]
- Stemmler I., Hense I., Quack B., Maier-Reimer E. Methyl iodide production in the open ocean. Biogeosciences. 2014;11(16):4459–4476. [Google Scholar]
- Stemmler I., Hense I., Quack B. Marine sources of bromoform in the global open ocean—global patterns and emissions. Biogeosciences. 2015;12(6):1967–1981. [Google Scholar]
- Stephen K., Aneja V.P. Trends in agricultural ammonia emissions and ammonium concentrations in precipitation over the southeast and midwest United States. Atmos. Environ. 2008;42(14):3238–3252. [Google Scholar]
- Stephens G.L., Li J., Wild M., Clayson C.A., Loeb N., Kato S., L’Ecuyer T., Stackhouse P.W., Lebsock M., Andrews T. An update on Earth’s energy balance in light of the latest global observations. Nat. Geosci. 2012;5(10):691–696. [Google Scholar]
- Stern D.I. Reversal of the trend in global anthropogenic sulfur emissions. Glob. Environ. Change-Hum. Policy Dimens. 2006;16:207–220. [Google Scholar]
- Steudler P.A., Peterson B.J. Annual cycle of gaseous sulfur emissions from a New England Spartina Alterniflora Marsh. Atmos. Environ. 1985;19(9):1411–1416. [Google Scholar]
- Stinecipher J.R., Cameron-Smith P.J., Blake N.J., Kuai L., Lejeune B., Mahieu E., Simpson I.J., Campbell J.E. Biomass burning unlikely to account for missing source of carbonyl sulfide. Geophys. Res. Lett. 2019;46:14912–14920. [Google Scholar]
- Stolper D.A., Bender M.L., Dreyfus G.B., Yan Y., Higgins J.A. A pleistocene ice core record of atmospheric O2 concentrations. Science. 2016;353(6306):1427–1430. doi: 10.1126/science.aaf5445. [DOI] [PubMed] [Google Scholar]
- Stott P.A., Tett S.F., Jones G.S., Allen M.R., Mitchell J.F., Jenkins G.J. External control of 20th century temperature by natural and anthropogenic forcings. Science. 2000;290(5499):2133–2137. doi: 10.1126/science.290.5499.2133. [DOI] [PubMed] [Google Scholar]
- Streets D.G., Yu C., Wu Y., Chin M., Zhao Z., Hayasaka T., Shi G. Aerosol trends over China, 1980–2000. Atmos. Res. 2008;88:174–182. [Google Scholar]
- Sturges W.T., Penkett S.A., Barnola J.M., Chappellaz J., Atlas E., Stroud V. A long-term record of carbonyl sulfide (COS) in two hemispheres from firn air measurements. Geophys. Res. Lett. 2001;28:4095–4098. [Google Scholar]
- Su H., Cheng Y., Oswald R., Behrendt T., Trebs I., Meixner F.X., Andreae M.O., Cheng P., Zhang Y., Pöschl U. Soil nitrite as a source of atmospheric HONO and OH radicals. Science. 2011;333(6049):1616–1618. doi: 10.1126/science.1207687. [DOI] [PubMed] [Google Scholar]
- Sutton M.A., Pitcairn C.E.R., Fowler D. The exchange of ammonia between the atmosphere and plant communities. Adv. Ecol. Res. 1993;24:301–393. [Google Scholar]
- Swain E.B., Engstrom D.R., Brigham M.E., Henning T.A., Brezonik P.L. Increasing rates of atmospheric mercury deposition in Midcontinental North America. Science. 1992;257(5071):784–787. doi: 10.1126/science.257.5071.784. [DOI] [PubMed] [Google Scholar]
- Swank W.T., Henderson G.S. Atmospheric input of some cations and anions to forest ecosystems in North Carolina and Tennessee. Water Resour. Res. 1976;12:541–546. [Google Scholar]
- Swap R., Garstang M., Greco S., Talbot R., Kallberg P. Saharan dust in the Amazon Basin. Tellus B Chem. Phys. Meteorol. 1992;44:133–149. [Google Scholar]
- Tabazadeh A., Turco R.P. Stratospheric chlorine injection by volcanic eruptions: HCl scavenging and implications for ozone. Science. 1993;260:1082–1086. doi: 10.1126/science.260.5111.1082. [DOI] [PubMed] [Google Scholar]
- Talbot R.W., Harriss R.C., Browell E.V., Gregory G.L., Sebacher D.I., Beck S.M. Distribution and geochemistry of aerosols in the tropical North Atlantic troposphere: relationship to Saharan dust. J. Geophys. Res.-Atmos. 1986;91:5173–5182. [Google Scholar]
- Talukdar R.K., Mellouki A., Schmoltner A.M., Watson T., Montzka S., Ravishankara A.R. Kinetics of the OH reaction with methyl chloroform and its atmospheric implications. Science. 1992;257(5067):227–230. doi: 10.1126/science.257.5067.227. [DOI] [PubMed] [Google Scholar]
- Tanaka N., Turekian K.K. Determination of the dry deposition flux of SO2 using cosmogenic 35S and 7Be measurements. J. Geophys. Res.-Atmos. 1995;100:2841–2848. [Google Scholar]
- Tanre D., Breon F.M., Deuze J.L., Herman M., Goloub P., Nadal F., Marchand A. Global observation of anthropogenic aerosols from satellite. Geophys. Res. Lett. 2001;28:4555–4558. [Google Scholar]
- Tegen I., Werner M., Harrison S.P., Kohfeld K.E. Relative importance of climate and land use in determining present and future global soil dust emission. Geophys. Res. Lett. 2004;31 [Google Scholar]
- Templer P.H., Weathers K.C., Ewing H.A., Dawson T.E., Mambelli S., Lindsey A.M., Webb J., Boukili V.K., Firestone M.K. Fog as a source of nitrogen for redwood trees: evidence from fluxes and stable isotopes. J. Ecol. 2015;103(6):1397–1407. Austin, A. (Ed.) [Google Scholar]
- Textor C., Graf H.F., Herzog M., Oberhuber J.M. Injection of gases into the stratosphere by explosive volcanic eruptions. J. Geophys. Res. 2003;108 [Google Scholar]
- Thompson A.M. The oxidizing capacity of the Earth’s atmosphere: probable past and future changes. Science. 1992;256(5060):1157–1165. doi: 10.1126/science.256.5060.1157. [DOI] [PubMed] [Google Scholar]
- Thompson L.G., Yao T., Mosley-Thompson E., Davis M.E., Henderson K.A., Lin P. A high-resolution millennial record of the South Asian monsoon from Himalayan ice cores. Science. 2000;289(5486):1916–1920. doi: 10.1126/science.289.5486.1916. [DOI] [PubMed] [Google Scholar]
- Thompson A.E., Anderson R.S., Rudolph J., Huang L. Stable carbon isotope signatures of background tropospheric chloromethane and CFC113. Biogeochemistry. 2002;60:191–211. [Google Scholar]
- Toon O.B., Kasting J.F., Turco R.P., Liu M.S. The sulfur cycle in the marine atmosphere. J. Geophys. Res. 1987;92:943–963. [Google Scholar]
- Trenberth K.E. Atmospheric moisture residence times and cycling implications from rainfall rate and climate change. Clim. Chang. 1998;39:667–694. [Google Scholar]
- Trenberth K.E., Guillemot C.J. The total mass of the atmosphere. J. Geophys. Res. 1994;99:23079–23088. [Google Scholar]
- Turco R.P., Whitten R.C., Toon O.B., Pollack J.B., Hamill P. OCS, stratospheric aerosols and climate. Nature. 1980;283:283–286. [Google Scholar]
- Turnipseed A.A., Huey L.G., Nemitz E., Stickel R., Higgs J., Tanner D.J., Slusher D.L., Sparks J.P., Flocke F., Guenther A. Eddy covariance fluxes of peroxyacetyl nitrates (PANs) and NOy to a coniferous Forest. J. Geophys. Res. 2006;111 [Google Scholar]
- Van Damme M., Erisman J.W., Clarisse L., Dammers E., Whitburn S., Clerbaux C., Dolman A.J., Coheur P.-F. Worldwide spatiotemporal atmospheric ammonia (NH3) columns variability revealed by satellite: NH3 spatiotemporal variability. Geophys. Res. Lett. 2015;42(20):8660–8668. [Google Scholar]
- Van Damme M., Clarisse L., Whitburn S., Hadji-Lazaro J., Hurtmans D., Clerbaux C., Coheur P.-F. Industrial and agricultural ammonia point sources exposed. Nature. 2018;564(7734):99–103. doi: 10.1038/s41586-018-0747-1. [DOI] [PubMed] [Google Scholar]
- Vandenberg J.J., Knoerr K.R. Comparison of surrogate surface techniques for estimation of sulfate dry deposition. Atmos. Environ. 1985;19:627–635. [Google Scholar]
- Varshney C.K., Singh A.P. Isoprene emission from Indian trees. J. Geophys. Res. 2003;108(D24) [Google Scholar]
- Verhulst K.R., Aydin M., Saltzman E.S. Methyl chloride variability in the Taylor Dome ice core during the Holocene: Taylor Dome methyl chloride. J. Geophys. Res. D: Atmos. 2013;118(21):12218–12228. [Google Scholar]
- Volz A., Kley D. Evaluation of the Montsouris series of ozone measurements made in the 19th-century. Nature. 1988;332:240–242. [Google Scholar]
- Waldman J.M., Munger J.W., Jacob D.J., Flagan R.C., Morgan J.J., Hoffmann M.R. Chemical composition of acid fog. Science. 1982;218(4573):677–680. doi: 10.1126/science.218.4573.677. [DOI] [PubMed] [Google Scholar]
- Waldman J.M., Munger J.W., Jacob D.J., Hoffmann M.R. Chemical characterization of stratus cloudwater and its role as a vector for pollutant deposition in a Los Angeles pine forest. Tellus B—Chem. Phys. Meteorol. 1985;37:91–108. [Google Scholar]
- Walker J.C.G. Macmillan; 1977. Evolution of the Atmosphere. [Google Scholar]
- Walker J.C. How life affects the atmosphere. Bioscience. 1984;34(8):486–491. [PubMed] [Google Scholar]
- Warneck P. second ed. Academic Press; 2000. Chemistry of the Natural Atmosphere. [Google Scholar]
- Warner J.X., Dickerson R.R., Wei Z., Strow L.L., Wang Y., Liang Q. Increased atmospheric ammonia over the world’s major agricultural areas detected from space. Geophys. Res. Lett. 2017;44(6):2875–2884. doi: 10.1002/2016GL072305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins N.D., Sparks R.S.J., Sigurdsson H., Huang T.C., Federman A., Carey S., Ninkovich D. Volume and extent of Minoan tephra from Santorini volcano: new evidence from deep sea sediment cores. Nature. 1978;271:122–126. [Google Scholar]
- Waugh D.W., Hall T.M. Age of stratospheric air: theory, observations, and models. Rev. Geophys. 2002;40 [Google Scholar]
- Weathers K.C., Likens G.E., Bormann F.H., Eaton J.S., Bowden W.B., Andersen J.L., Cass D.A., et al. A regional acidic cloud fog water event in the eastern United States. Nature. 1986;319:657–658. [Google Scholar]
- Weathers K.C., Lovett G.M., Likens G.E., Lathrop R. The effect of landscape features on deposition to Hunter Mountain, Catskill Mountains, New York. Ecol. Appl. 2000;10:528–540. [Google Scholar]
- Weiss P.S., Johnson J.E., Gammon R.H., Bates T.S. Reevaluation of the open ocean source of carbonyl sulfide to the atmosphere. J. Geophys. Res. 1995;100:23083–23092. [Google Scholar]
- Weiss R.F., Muehle J., Salameh P.K., Harth C.M. Nitrogen trifluoride in the global atmosphere. Geophys. Res. Lett. 2008;35 [Google Scholar]
- Wennberg P.O., Cohen R.C., Stimpfle R.M., Koplow J.P., Anderson J.G., Salawitch R.J., Fahey D.W., et al. Removal of stratospheric O3 by radicals: in situ measurements of OH, HO2, NO, NO2, ClO, and BrO. Science. 1994;266(5184):398–404. doi: 10.1126/science.266.5184.398. [DOI] [PubMed] [Google Scholar]
- Wesely M.L., Hicks B.B. A review of the current status of knowledge on dry deposition. Atmos. Environ. 2000;34:2261–2282. [Google Scholar]
- White E.J., Turner F. A method of estimating income of nutrients in catch of airborne particles by a woodland canopy. J. Appl. Ecol. 1970;7:441–461. [Google Scholar]
- Whittaker R.H., Woodwell G.M. Dimension and production relations of trees and shrubs in the Brookhaven Forest. J. Ecol. 1968;56:1–25. [Google Scholar]
- Willett K.M., Gillett N.P., Jones P.D., Thorne P.W. Attribution of observed surface humidity changes to human influence. Nature. 2007;449(7163):710–712. doi: 10.1038/nature06207. [DOI] [PubMed] [Google Scholar]
- Wolfe G.M., Nicely J.M., St Clair J.M., Hanisco T.F., Liao J., Oman L.D., Brune W.B., et al. Mapping hydroxyl variability throughout the global remote troposphere via synthesis of airborne and satellite formaldehyde observations. Proc. Natl. Acad. Sci. U. S. A. 2019;116(23):11171–11180. doi: 10.1073/pnas.1821661116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotawa G., Trainer M. The influence of Canadian forest fires on pollutant concentrations in the United States. Science. 2000;288(5464):324–328. doi: 10.1126/science.288.5464.324. [DOI] [PubMed] [Google Scholar]
- Wu J. Evidence of sea spray produced by bursting bubbles. Science. 1981;212(4492):324–326. doi: 10.1126/science.212.4492.324. [DOI] [PubMed] [Google Scholar]
- Wuebbles D.J., Tamaresis J.S. In: Atmospheric Methane: Sources, Sinks, and Role in Global Climate. Khalil M.A.K., editor. Springer; 1993. The role of methane in the global environment; pp. 469–513. [Google Scholar]
- Wuebbles D.J., Dong W., Patten K.O., Olsen S.C. Analyses of new short-lived replacements for HFCs with large GWPs. Geophys. Res. Lett. 2013;40(17):4767–4771. [Google Scholar]
- Yagi K., Williams J., Wang N.Y., Cicerone R.J. Atmospheric methyl bromide (CH3Br) from agricultural soil fumigations. Science. 1995;267(5206):1979–1981. doi: 10.1126/science.267.5206.1979. [DOI] [PubMed] [Google Scholar]
- Yang E.S., Gupta P., Christopher S.A. Net radiative effect of dust aerosols from satellite measurements over Sahara. Geophys. Res. Lett. 2009;36 [Google Scholar]
- Yang K., Dickerson R.R., Carn S.A., Ge C., Wang J. First observations of SO2 from the satellite Suomi NPP OMPS: widespread air pollution events over China. Geophys. Res. Lett. 2013;40(18):4957–4962. [Google Scholar]
- Yang K., Carn S.A., Ge C., Wang J., Dickerson R.R. Advancing measurements of tropospheric NO2 from space: new algorithm and first global results from OMPS. Geophys. Res. Lett. 2014;41(13):4777–4786. [Google Scholar]
- Yeung L.Y., Murray L.T., Martinerie P., Witrant E., Hu H., Banerjee A., Orsi A., Chappellaz J. Isotopic constraint on the twentieth-century increase in tropospheric ozone. Nature. 2019;570(7760):224–227. doi: 10.1038/s41586-019-1277-1. [DOI] [PubMed] [Google Scholar]
- Yokouchi Y., Nojiri Y., Toom-Sauntry D., Fraser P., Inuzuka Y., Tanimoto H., Nara H., Murakami R., Mukai H. Long-term variation of atmospheric methyl iodide and its link to global environmental change: long-term change of methyl iodide. Geophys. Res. Lett. 2012;39(23) [Google Scholar]
- Young J.R., Ellis E.C., Hidy G.M. Deposition of air-borne acidifiers in the Western environment. J. Environ. Qual. 1988;17:1–26. [Google Scholar]
- Yu H., Remer L.A., Chin M., Bian H., Tan Q., Yuan T., Zhang Y. Aerosols from overseas rival domestic emissions over North America. Science. 2012;337(6094):566–569. doi: 10.1126/science.1217576. [DOI] [PubMed] [Google Scholar]
- Yu H., Chin M., Yuan T., Bian H., Remer L.A., Prospero J.M., Omar A., et al. The fertilizing role of African dust in the Amazon rainforest: a first multiyear assessment based on data from cloud-aerosol Lidar and infrared pathfinder satellite observations: dust deposition in the Amazon rainforest. Geophys. Res. Lett. 2015;42(6):1984–1991. [Google Scholar]
- Zbieranowski A.L., Aherne J. Long-term trends in atmospheric reactive nitrogen across Canada: 1988–2007. Atmos. Environ. 2011;45:5853–5862. [Google Scholar]
- Zender C.S., Miller R.L., Tegen I. Supplemental material to “quantifying mineral dust mass budgets: systematic terminology, constraints, and current estimates”. Eos Trans. Aguiaine/Societe D’etudes Folkloriques Du Centre-Ouest. 2004;85:509. [Google Scholar]
- Zerefos C.S., Tetsis P., Kazantzidis A., Amiridis V., Zerefos S.C., Luterbacher J., Eleftheratos K., Gerasopoulos E., Kazadzis S., Papayannis A. Further evidence of important environmental information content in red-to-green ratios as depicted in paintings by great masters. Atmos. Chem. Phys. 2014;14(6):2987–3015. [Google Scholar]
- Zhan X., Yu G., He N., Jia B., Zhou M., Wang C., Zhang J., et al. Inorganic nitrogen wet deposition: evidence from the north-south transect of eastern China. Environ. Pollut. 2015;204(September):1–8. doi: 10.1016/j.envpol.2015.03.016. [DOI] [PubMed] [Google Scholar]
- Zhang X., Zwiers F.W., Hegerl G.C., Hugo Lambert F., Gillett N.P., Solomon S., Stott P.A., Nozawa T. Detection of human influence on twentieth-century precipitation trends. Nature. 2007;448(7152):461–465. doi: 10.1038/nature06025. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Song L., Liu X.J., Li W.Q., Lu S.H., Zheng L.X., Bai Z.C., Cai G.Y., Zhang F.S. Atmospheric organic nitrogen deposition in China. Atmos. Environ. 2012;46:195–204. [Google Scholar]
- Zhang H., Yee L.D., Lee B.H., Curtis M.P., Worton D.R., Isaacman-VanWertz G., Offenberg J.H., et al. Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States. Proc. Natl. Acad. Sci. U. S. A. 2018;115(9):2038–2043. doi: 10.1073/pnas.1717513115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L., Mickley L.J., Jacob D.J., Marais E.A., Jianxiong Sheng L.H., Abad G.G., Chance K. Long-term (2005–2014) trends in formaldehyde (HCHO) columns across North America as seen by the OMI satellite instrument: evidence of changing emissions of volatile organic compounds: HCHO trend across North America. Geophys. Res. Lett. 2017;44(13):7079–7086. [Google Scholar]
- Zimmerman P.R., Greenberg J.P. Measurements of atmospheric hydrocarbons and biogenic emission fluxes in the Amazon boundary layer. J. Geophys. Res. 1988;93:1407–1416. [Google Scholar]
- Zobel D.B., Antos J.A. 1980 tephra from Mount St. Helens: spatial and temporal variation beneath forest canopies. Biol. Fertil. Soils. 1991;12(1):60–66. [Google Scholar]
- Zumkehr A., Hilton T.W., Whelan M., Smith S., Elliott Campbell J. Gridded anthropogenic emissions inventory and atmospheric transport of carbonyl sulfide in the U.S. J. Geophys. Res. D: Atmos. 2017;122(4):2169–2178. [Google Scholar]
- Zumkehr A., Hilton T.W., Whelan M., Smith S., Kuai L., Worden J., Elliott Campbell J. Global gridded anthropogenic emissions inventory of carbonyl sulfide. Atmos. Environ. 2018;183(June):11–19. [Google Scholar]