

Abstract
Lantana camara L. is an economically important essential oil producing plant belonging to family Verbenaceae. It is used in medication for treating various diseases like cancer, ulcers, tumor, asthma and fever. The plant is a useful source of essential bioactive compounds such as steroids, flavonoids and phenylpropanoid glycosides etc. Nonetheless, very little is known about the genomic or transcriptomic resources of L. camara, and this might be the reason of hindering molecular studies leading to identification of improved lines. Here we used Illumina sequencing platform and performed the L. camara leaf (LCL) and root (LCR) de novo transcriptome analyses. A total of 70,155,594 and 84,263,224 clean reads were obtained and de novo assembly generated 72,877 and 513,985 unigenes from leaf (LCL) and root (LCR) respectively. Furthermore, the pathway analysis revealed the presence of 229 and 943 genes involved in the phenylpropanoid biosynthesis in leaf and root tissues respectively. Similarity search was performed against publically available genome databases and best matches were found with Sesamum indicum (67.5%) that were much higher than that of Arabidopsis thaliana (3.9%). To the best of our knowledge, this is the first comprehensive transcriptomic analysis of leaf and root tissues of this non-model plant from family Verbenaceae and may serve as a baseline for further molecular studies.
Subject terms: RNA sequencing, Plant genetics
Introduction
Lantana camara L. belonging to family Verbenaceae is an evergreen shrub, native to the Neotropics and grown worldwide for its medicinal and ornamental value1. The plant may grow up to 4 m, forming dense thickets2 and is extensively used in traditional herbal medicines of many cultures including Saudi Arabia3. The medicinal properties of L. camara are attributed to the presence of many bioactive compounds with therapeutic potential such as steroids, flavonoids, triterpenoids, oligosaccharides, iridoide glycosides, naphthoquinones and phenylpropanoid glycosides4–6. Furthermore, L. camara is used in medication for treating cancer, ulcers, tumors, tetanus, cuts, eczema, measles, chicken pox, fevers, rheumatism and asthma1,7,8. Several important phytochemicals have been isolated from L. camara including ursolic acid, oleanolic acid, linaroside, lantanoside, verbascoside, camarinic acid, phytol and umuhengerin etc. and their biological activities such as anticancer, antibacterial, antioxidant, antiulcer and nematocidal have been reported9–11. Nonetheless, L. camara is also known as a major source of easily available plant essential oil known as Lantana oil12,13. Literature survey indicates the substantial diversity in the composition of essential oils isolated from L. camara growing in diverse localities14–18.
In spite, much is known about the phytochemistry, toxicology and medicinal properties of L. camara19 very little is known about the genomic architecture of the plant. To date, only 41 sequences including (rps3, atpB, ccsA, rpoC1, rpoC2, FT, GLO1, rpl32 and rbcL) have been deposited to the NCBI Genbank database20. To stop the spread of L. camara as an invasive weed, transcriptomes of young ovaries were recently studied and possible mechanisms of unreduced female gametes formation were elaborated21. Complete genome sequences provide invaluable insights into the biological functions of individual genes and proteins and therefore, hold immense promise for crop improvement and breeding of new cultivars. The genome of L. camara has not been sequenced yet and no details are available on the genomics or transcriptomics of the plant. Therefore, a detailed research strategy is needed to elucidate various aspects of genomics and transcriptomics of this important medicinal plant. Recently we have also reported the complete chloroplast genome of L.camara22. Nowadays, transcriptomics using next generation sequencing (NGS) technology has emerged to be one of the most powerful and cost-effective approaches generating huge data on transcribed sequences that may be implemented in wide research purposes23. The practical applications of transcriptome sequencing have been utilized in various medicinal plants as well as crop plants including rice24, maize25, chickpea26, wheat27, Camelina sativa28, Quercus pubescens29, finger millet30, Stellera chamaejasme31 and Aconitum heterophyllum32. As a step forward, here we report the first de novo transcriptome assembly of L. camara leaf and root tissues that may lay the foundation for rapid identification of functional genes discovery and genomics-assisted breeding for developing more efficient L. camara lines with desired traits.
Materials and methods
Plant material and RNA quantification
Plants were grown in the laboratory of plant physiology King Abdulaziz University, Jeddah, Saudi Arabia under 28 °C/22 °C day and night temperature in a semi-controlled green house. No specific permits were required for the described study. Fresh leaf (LCL) and root (LCR) tissues were harvested from 02 months old plant, washed thoroughly with sterile water, and immediately frozen in liquid nitrogen. RNA isolation was carried out using RNeasy Plus Mini Kit (Qiagen, Cat. No: 74134). RNA degradation and contamination were monitored on 1% agarose gel. Purity of RNA was checked on NanoPhotometer® spectrophotometer (IMPLEN, CA, USA). Quantification and integrity of RNA was assessed using the RNA Nano 6000 Assay Kit (Agilent Technologies, CA, USA).
Library preparation for transcriptome sequencing
NEBNext® Ultra™ RNA Library Prep Kit was used to generate sequencing libraries. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Divalent cations were used to carry out fragmentation under high temperature in NEBNext (FSSR) first strand synthesis reaction Buffer (5×). Random hexamer primers were used to synthesize first strand cDNA using M-MuLV Reverse Transcriptase (RNase H-). Second strand cDNA synthesis was carried out using DNA Polymerase I and RNase H. Exonuclease/polymerase activity was performed to convert remaining overhangs into blunt ends. Ligation of NEBNext adaptor with hairpin loop was performed after adenylation of 3′end of DNA fragments in order to prepare sample for hybridization. The cDNA fragments of 250–300 bp were preferentially selected and purification of the library fragments was carried out with AMPure XP system (Beckman Coulter, Beverly, USA). 3 µl of Enzyme (NEB, USA) was used with size-selected, adaptor-ligated cDNA at 37 °C for 15 min, followed by 5 min at 95 °C. PCR was carried out with Phusion High-Fidelity DNA polymerase, Universal PCR primers and Index (X) Primer. PCR products were purified (AMPure XP system) and library quality was assessed on the Agilent Bioanalyzer 2100 system. Paired-end library of 150 bp (PE150) was prepared following Illumina protocol/instructions.
Clustering, quality control and transcriptome assembly
Cluster generation system (PE Cluster Kit cBot-HS Illumina) was used to perform clustering of the index coded samples following manufacturer’s instructions. The library preparations were sequenced on an Illumina platform after cluster generation and paired-end reads were generated. Transcriptome assembly was performed using Trinity software33 with min_kmer_cov set to 2, and all other parameters with default settings.
Gene functional annotation and biological pathways assignment
Libraries of LCL and LCR tissues were annotated using BLASTX against the NCBI database and all unigenes were applied for homology searches. Gene functional annotation was performed using the following 7 databases: NCBI non-redundant protein sequences (NR), NCBI non-redundant nucleotide sequences (Nt), Protein family (Pfam), Clusters of Orthologous Groups of proteins (KOG/COG), manually annotated and reviewed protein sequence database (Swiss-Prot), KEGG Ortholog database (KO)34 and Gene Ontology (GO) database. Best aligning results were selected to annotate the unigenes; if aligning results of these databases were not similar, NR database results were preferentially selected. For assignment of function to unigenes, Gene Ontology (GO) enrichment analysis was performed using AgriGO (https://bioinfo.cau.edu.cn/agriGO/analysis.php)35 and annotated sequence/s may have more than one GO term. Similarly, to understand the high-level functions and gain an overview of gene pathway networks KEGG36,37 was used. Enzyme commission (EC) numbers were assigned to unique sequences, based on the BLASTx search of protein databases, using a cutoff E value 10 − 5. The statistical enrichment of DEGs in KEGG pathways was carried out using KOBAS38,39.
Quantification and differential expression analysis
The RNA-seq by Expectation Maximization (RSEM) package was used to estimate gene expression levels of both root and leaf tissues40. At first clean data was mapped back onto the assembled transcriptome and then read count for each gene was obtained from the mapping results. Prior to differential gene expression (DEGs) analysis of the sequenced library of root and leaf tissues, read counts were adjusted through one scaling normalized factor and analysis was performed using DEGseq (2010) R package41. P value was adjusted using q value of < 0.005 and |log2(foldchange)| > 1 was set as the threshold for significantly differential expression. To identify DEGs between the LCR and LCL datasets, raw read counts were initially filtered to exclude orphan transcripts. Pairwise comparison was performed using the default parameters of DEGseq (2010) R package and an adaptive t shrinkage estimator was followed for ranking and visualization of log-fold changes (log2FC, LFC) of the DEGs42.
Results and discussion
RNA sequencing and de novo assembly
Advances in genomics have led to the development of NGS based trait mapping approaches that have tremendously increased the efficiency of traits selection and mapping in complex genomes43. Among the high throughput genotyping approaches, Next Generation RNA Sequencing technology has contributed to a more comprehensive understanding of functional genes and is widely used for characterization of transcriptome profiles of various model and non-model plants44. De novo transcriptome analysis is not only providing an excellent platform for finding novel genes, molecular markers development but also cater a base for the construction of networks of gene expressions for various tissues and organs of animals as well as plants. Although, the number of available high-quality reference genomes has been constantly growing still, de novo transcriptome approaches are mainly used for non-model species where whole genomes information are missing. The remarkable advances in RNA sequencing provide a cost-effective way to obtain large amounts of transcriptome data from many organisms and tissue types, especially in the complete absence of a reference genome thereby, allowing us to identify all expressed transcripts33,34. Here we provide, the first report on transcriptome profiling and DEGs of L. camara leaf and root tissues; RNA-Seq library was constructed and RNA samples with RIN value more than 6 were used (Supplementary file 1). A total of 76,315,644 and 87,218,768 raw reads were obtained for LCL and LCR respectively. For the removal of adapters, poly-A tail, primer sequences and short as well as low quality sequences trimming process was performed that resulted in a total of 70,155,594 clean reads from LCL and 84,263,224 from LCR. The total size of clean bases generated was 10.5 GB for LCL sample with percent error (0.01%), Q20 (97.39%), Q30 (93.47%) and GC content (45.61%), whereas for LCR sample 12.6 GB with a percent error (0.03%), Q20 (96.81%), Q30 (92.01%) and GC content (43.73%) (Table 1). The sequences (raw data) generated were deposited to NCBI as PRJNA503321 (for leaf sample) and PRJNA605469 (for root sample). For samples lacking reference genomes, clean reads need to be assembled to get a reference sequence. We used Trinity assembler33 to get the leaf and root transcriptome information.
Table 1.
Quality of reads obtained after RNA-sequencing of leaf and root transcriptomes.
| Samples | Raw reads | Clean reads | Clean bases (GB) | Error (%) | Q20 (%) | Q30 (%) | GC (%) |
|---|---|---|---|---|---|---|---|
| Leaf | 76,315,644 | 70,155,594 | 10.5 | 0.01 | 97.39 | 93.47 | 45.61 |
| Root | 87,218,768 | 84,263,224 | 12.6 | 0.03 | 96.81 | 92.01 | 43.73 |
A total of 72,898 and 516,026 transcripts were obtained from LCL and LCR respectively and length of transcripts varied for leaf (201 to 14,753 bp) and root (201 to 16,796 bp) (Table 2). A total 72,877 unigenes were detected in LCL tissues of which 19,955 were within 200–500 bp; 21,816 within 500–1kbp; 20,062 within 1 k–2 kbp and 11,044 were > 2 kbp, with an N50 of 1,650 bp (Table 2). Relatively high number i.e. 513,985 of unigenes were identified in LCR tissue, of which 320,891 were within 200–500 bp; 107,176 within 500-1kbp; 59,405 within 1 k-2 kbp and 26,513 were within > 2 kbp, with an N50 of 939 bp (Table 2).
Table 2.
Summary of transcripts length distribution and unigenes assembled.
| Length interval | Leaf | Root | ||
|---|---|---|---|---|
| No of transcripts | No of unigenes | No of transcripts | No of unigenes | |
| 200–500 bp | 19,976 | 19,955 | 322,930 | 320,891 |
| 500–1 kbp | 21,816 | 21,816 | 107,178 | 107,176 |
| 1 k–2 kbp | 20,062 | 20,062 | 59,405 | 59,405 |
| > 2 kbp | 11,044 | 11,044 | 26,513 | 26,513 |
| Total | 72,898 | 72,877 | 516,026 | 513,985 |
| Minimum length | 201 | 201 | 201 | 201 |
| Maximum length | 14,753 | 14,753 | 16,796 | 16,796 |
| N50 | 1,650 | 1,650 | 937 | 939 |
| N90 | 541 | 541 | 279 | 280 |
| Total nucleotides | 83,817,239 | 83,812,099 | 336,871,604 | 336,370,588 |
Functional annotation of genes
Unigenes of the LCL and LCR tissues were annotated using BLAST search against the NR, NT, PFAM, GO and KOG databases. Gene Ontology assignments based on the protein match and annotation results are based on Götz et al.45 Of the 72,877 and 513,985 unique transcript sequences of LCL and LCR annotated, a large proportion of the sequences (73.27% and 70.11%) had hits in databases. Of the LCL tissue, a total of 50,540 (69.34%) unigenes matched with known proteins in the NR database, while 38,975 (53.48%) unigenes were annotated with entries in the Swiss-prot database, 36,209 (49.68%) unigenes matched with proteins in Gene Ontology (GO) database, 35,711 (49%) matched with proteins in the PFAM database (Table 3). Similarly, the LCR transcriptome revealed that 253,381 (49.29%) of unigenes matched with proteins in the NR database, 250,499 (48.73%) unigenes matched with entries in the Swiss-prot database, 255,964 (49.79%) unigenes matched with proteins in Gene Ontology (GO) database, and 252,005 (49.02%) were annotated with proteins in the PFAM database (Table 3). Of the assembled unigenes, only 9,849 (13.51%) and 45,051 (8.76%) in LCL and LCR respectively, were successfully annotated in all databases. Reasons for the non-annotated sequences could be, lack of conserved protein domains in short sequences, or in some cases the transcriptome have non-coding genes, UTRs, random transcriptional noise or incomplete spliced introns which are non-homologues to the sequences available in the public databases. This may also be one of the possible reasons that genes have not shown expression at the time of RNA extraction or those genes are expressed at very low levels46.
Table 3.
The ratio of successfully annotated genes.
| Annotated databases | Leaf | Root | ||
|---|---|---|---|---|
| No of unigenes | Percentage (%) | No of unigenes | Percentage (%) | |
| NR | 50,540 | 69.34 | 253,381 | 49.29 |
| NT | 36,890 | 50.61 | 177,562 | 34.54 |
| KO | 20,506 | 28.13 | 124,076 | 24.14 |
| SwissProt | 38,975 | 53.48 | 250,499 | 48.73 |
| PFAM | 35,711 | 49 | 252,005 | 49.02 |
| GO | 36,209 | 49.68 | 255,964 | 49.79 |
| KOG | 19,818 | 27.19 | 155,979 | 27.44 |
| All databases | 9,849 | 13.51 | 45,051 | 8.76 |
| At least one database | 53,397 | 73.27 | 360,397 | 70.11 |
| Total unigenes | 72,877 | 100 | 513,985 | 100 |
The assembled unigenes annotation of LCL and LCR tissues is given as Venn diagram (Fig. 1) against PFAM, GO, KOG, NR and NT databases. For leaf tissues, a total of 13,534 unigenes were annotated in 5 databases; 6,060 unigenes sequences showed homology in 3 databases including PFAM, NR and GO. While 1,167 unigenes were annotated in KOG as well as the above three databases and 2,030 unigenes were annotated in PFAM and GO databases (Fig. 1A). Likewise for root tissues, a total of 57,114 unigenes were annotated in five databases where, 28,396 unigenes were homologous in PFAM, NR and GO databases (Fig. 1B).
Figure 1.
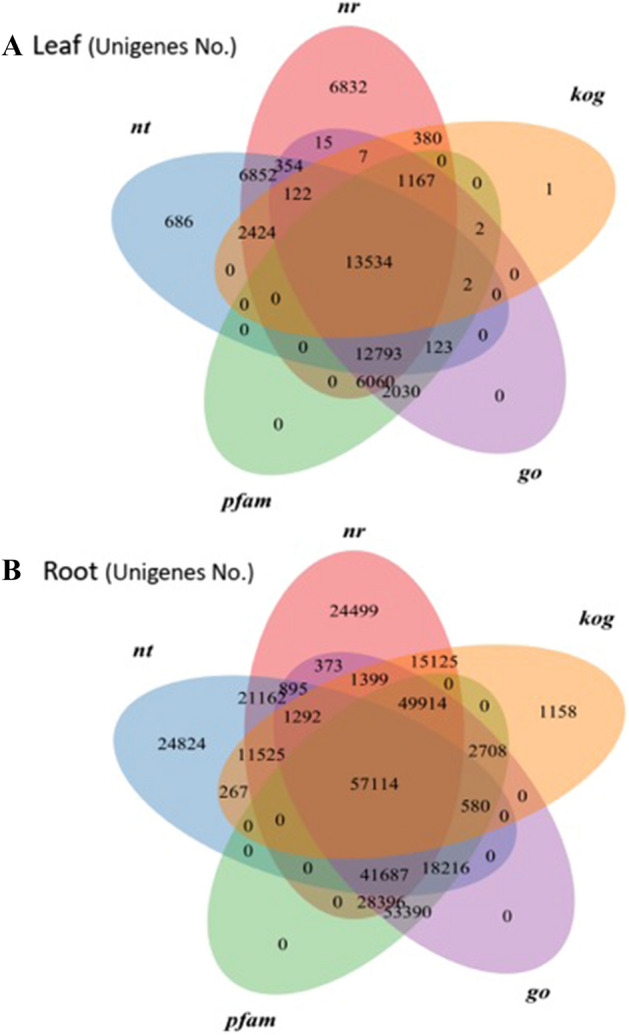
Venn diagram mapping with database annotation.
Annotation in NR database
After the high annotation hit score was ascertained in NR database, we considered identifying homologues features of BlastX hits for the annotated unigenes. The E-value distribution in leaf tissue revealed that 35.6% of the annotated unigenes had E-value of 0-1e-100, followed by 19.9% unigenes with 1e−100 to 1e−60 E-value (Fig. 2A). Similarity distribution showed that 45.2% of the unigenes had 80–90% similarities. Furthermore, 39.1% of the unigenes had 60–80% and 5.2% had 95–100% similarities (Fig. 2B). Additionally, comparison of the unigenes annotated with homologous sequences of other plant species was also performed44. Among the nucleotide sequences, highest similarity was noted for Sesamum indicum (with the highest similarity score of 67.5%). Other species with sequence homology below 13% included Erythranthe guttata (12.3%), Arabidopsis thaliana (3.9%), Coffea canephora (1.4%) and Vitis vinifera (1.0%) (Fig. 2C).
Figure 2.

E-value distribution (A), similarity distribution (B) and species distribution (C).
E-value distributions in root tissue revealed that 25.8% of the annotated unigenes had E-value of 1e−15 to 1e−5, followed by 23.9% unigenes with 1e−30 to 1e−15 E-value (Fig. 2D). Similarity distribution revealed that 2.8% of the unigenes had 95–100% similarities, 26.6% had 80–95% and 42.9% unigenes had 60–80% similarities (Fig. 2E). Comparison of the annotated unigenes with the homologous sequences revealed highest similarity with Sesamum indicum (26.4%) followed by Erythranthe guttata (5%), Ricinus communis (3.1%) and Guillardia theta (2.8%) (Fig. 2F).
Gene ontology (GO) classification
GO assignments were used to classify the functions of the LCL and LCR unigenes, which classified unigenes under the categories of biological process (BP), molecular function (MF) and cellular component (CC). Sum of 36,209 LCL unigenes were classified into three major categories (Fig. 3A). The biological process category, the unique sequences were classified into 25 groups. The most characterized biological processes were ‘cellular process’ (57.59%, GO-ID: 0009987) followed by ‘metabolic process’ (53.72%, GO-ID: 0008152). The cellular components were divided into 21 groups. Interestingly, we found both the represented cellular components ‘Cell’ (31.80%, GO-ID: 0005623) and ‘Cell part’’ (31.80%, GO: 0044464) were similar to previous report45. The molecular functions category, the unique sequences clustered into 10 classes. Where, the highest sub-category was ‘binding’ (58.35%, GO-ID: 0005488) followed by ‘catalytic activity’ (46.37%, GO: 0003824) (Fig. 3A, Supplementary file 2).
Figure 3.

X-axis is the GO term under the three main GO domains; Y-axis is the number and percentage of the annotated genes in the term (sub-term included).
Similarly, 255,964 LCR unigenes were annotated and also likewise classified into three main categories and further divided into 57 sub-groups (Fig. 3B). Among the biological processes classification, the highest was cellular process 142,929 (55.83%), followed by metabolic process 135,458 (52.92%) and single-organism process 107,769 (42.1%). Furthermore, cellular component was classified in 22 groups, among them high amount of unigenes were related to “cell” 75,691 (29.57%) and “cell part” 75,607 (29.53%), followed by “organelle” 50,774 (19.83%) and “membrane” 35,704 (13.94%) (Fig. 3B, Supplementary file 2). Only few unigenes were assigned to extracellular matrix, extracellular region and extra cellular region part. In the molecular function category, the majority of the unigenes were assigned to “binding” 133,482 (52.14%) and “catalytic activity” 114,050 (44.55%). The annotation and sequence information from Gene Ontology results provide important gene sources for future molecular level studies that underline selection and improvements of L. camara.
KOG classification
On the basis of conserved domain alignment, the annotated unigenes were searched against the KOG database to find the functionally classified orthologous gene products. A total of 19,818 and 155,979 identified genes were annotated in LCL and LCR tissues respectively and these were divided into 26 protein group families (Fig. 4). Among these protein families predicted in LCL tissues, general function prediction (2,719 unigenes, 15.41%) was identified as the highest annotated group, followed by post translational modification, protein turnover, chaperones (2,335 unigenes, 13.23%), signal transduction mechanisms (1,557 unigenes, 8.82%), translation, ribosomal structure and biogenesis (1,355 unigenes, 7.68%), RNA processing and modification (1,257 unigenes, 7.12%), intracellular trafficking, secretion, and vesicular transport (1,241, 7.03%), whereas the smallest groups were nuclear structure (89), extracellular structures (26) and cell motility (15) (Supplementary file 3). In LCR tissues, “Translation, ribosomal structure and biogenesis” consisted the largest category with 21,097 genes (14.95%), followed by “Posttranslational modification, protein turnover, chaperones” with 20,612 genes (14.60%) and “General function prediction only” 17,296 genes (12.25%). These results are not in alignment to previous report where the authors found “General function prediction only” as the highest category with 1,006 (13.86%) genes47. Fewer genes were assigned to “cell motility” (92) and unnamed (4) proteins (Fig. 4, Supplementary file 3).
Figure 4.

X-axis: names of the 26 KOG group; Y-axis: percentage of annotated genes under this group in the total annotated genes.
Functional characterization using KEGG
For biological functioning of genes, pathway-based analyses are imperative and this assignment was performed in the KEGG database. In LCL sample, of the 20,506 unigenes, 18,853 were assigned to 5 major groups in KEGG with 26 sub-categories and 131 biochemical pathways (Fig. 5, Supplementary file 4). These major groups were cellular processes (1,004 unigenes), environmental information processing (694 unigenes), genetic information processing (4,285 unigenes), metabolism (8,737 unigenes) and organismal systems (878 unigenes). In these 5 groups, the topmost was the metabolism with 8,737 unigenes, and its further evaluation classified it into 10 subgroups (Fig. 6). Of these, ‘carbohydrate metabolism’ with (1,751) genes involved the highest number of genes, followed by ‘amino acid metabolism’ (1,064) genes and ‘energy metabloism’ with (886) genes (Fig. 6).
Figure 5.

Y-axis: names of KEGG pathways; X-axis is the number of the genes annotated in the pathway and the ratio between the number in this pathway and the total number of annotated genes. The KEGG metabolic pathways gene involved in are divided into five branches; (A) Cellular Processes, (B) Environmental Information Processing, (C) Genetic Information Processing, (D) Metabolism and (E) Organismal Systems.
Figure 6.

Sub categorization of the metabolism group.
Furthermore, 936 genes were identified that coded for important proteins and have matches with 1,113 enzymes. The function of these enzymes was assigned to 21 secondary metabolite pathways (Fig. 7). Among these pathways 546 genes encoded key enzymes involved in terpeniods biosynthesis including monoterpenoid (28 genes), diterpenoids (30 genes), terpenoid backbone (166 genes), sesquiterpenoid and triterpenoids (24 genes). Similarly, 41 genes were related to flavonoid biosynthesis pathway including flavone and flavonol (32 gene). Among all the secondary metabolite pathways, the phenylpropanoid biosynthesis pathway involved the highest numbers (229) of genes (Fig. 7). These results provide valuable insight into the metabolic pathways in leaves of L. camara. These results may provide basis for future studies to identify and characterize genes and transcripts that are involved in important metabolic pathways and may provide insights into the understanding of functions of these genes in the biosynthesis of active compounds in L. camara.
Figure 7.

Assigned enzymes function to secondary metabolite pathways with number of genes.
Of the 124,076 unigenes in LCR transcriptome, 115,383 were assigned into 5 groups in KEGG with 19 sub-categories and 131 biochemical pathways (Fig. 5, Supplementary file 4). The major groups were cellular processes (7,793 unigenes), environmental information processing (3,384 unigenes), genetic information processing (39,630 unigenes), metabolism (50,349 unigenes) and organismal systems (3,111 unigenes). In these 5 groups, the topmost was the metabolism with 50,349 unigenes was further analysed, which we took for further consideration. The metabolism group was further categorized into 10 subgroups (Fig. 6). In all the 10 sub-groups of metabolism ‘carbohydrate metabolism’ was the highest with (11,724) genes, followed by ‘amino acid metabolism’ with (9,120) genes and ‘energy metabolism’ with (6,791) genes (Fig. 6). Furthermore, 4,166 genes were identified that coded for proteins and have important matches to 5,368 enzymes. The function of these enzymes was assigned to 21 KEGG secondary metabolite pathways (Fig. 7). Among these pathways 2,796 genes encoded key enzymes involved in terpeniods biosynthesis including monoterpenoid (92 genes), diterpenoids (75 genes), terpenoid backbone (853 genes), sesquiterpenoid and triterpenoids (103 genes). The results showed 119 genes were related to flavonoid biosynthesis pathway including flavone and flavonol biosynthesis (75 gene), whereas the highest number of genes (943 genes) among all the secondary metabolite were involved in phenylpropanoid biosynthesis pathway (Fig. 7).
Phenylpropanoid biosynthesis
The current study identified the maximum number of genes for phenylpropanoid biosynthesis pathway and this was taken into further consideration. Phenylpropanoids are phyto-based natural compounds that are usually derived from phenylalanine48. Phenylpropanoid plays vital role in plant response to various biotic and abiotic stresses49. The process of phenylpropanoid biosynthesis starts with the formation of cinnamic acid from phenylalanine. This cinnamic acid is then converted into cinnamoyl-CoA, p-Coumaryl-CoA, p-coumaryl quinic acid, caffeoyl quinic acid, caffeoyl-CoA, feruloyl-CoA, and sinapoyl-CoA. caffeoyl quinic acid also known as chlorogenic acid, which is a highly soluble phenylpropanoid in Solanaceae and it has been mentioned to play a vital role as an antioxidant and defense molecule49. Further, we have identified a total of 229 and 943 genes as well as 17 and 19 enzymes in LCL and LCR transcriptomes respectively and these enzymes are required for phenylpropanoid biosynthesis pathway (Table 4). Our results are different from50 where the authors reported only 11 genes from Solanum trilobatum that were involved in the biosynthesis of various compounds of this pathway. The important enzymes identified were cinnamyl-alcohol dehydrogenase [EC:1.1.1.195], followed by caffeoyl-CoA O-methyltransferase [EC:2.1.1.104], trans-cinnamate 4-monooxygenase [EC:1.14.13.11], Cinnamoyl-CoA-reductase [EC: 1.2.1.44], phenylalanine ammonia-lyase [EC:4.3.1.24], 4-Coumarate CoA-ligase [EC: 6.2.1.12], and shikimate O-hydroxycinnamoyl-transferase [EC:2.3.1.133]. Interestingly, in root transcriptome two protein coding genes namely cytochrome P450, family 98, subfamily A, polypeptide 8 [EC:1.14.13.-], and Aromatic-L-amino-acid decarboxylase [EC: 4.1.1.28] were identified, that were not detected in leaf transcriptome for phenylpropanoid biosynthesis pathway. The presence of all these enzymes indicates to the phytotherapeutic potential of L. camara (Fig. 8, Table 4, Supplementary File 5).
Table 4.
Important enzymes identified within phenylpropanoid biosynthesis pathway.
| Enzyme name | EC number | KO ID | Leaf unigenes no | Root unigenes no |
|---|---|---|---|---|
| Cinnamyl-alcohol dehydrogenase | 1.1.1.195 | K00083 | 17 | 177 |
| Peroxidase | 1.11.1.7 | K00430 | 44 | 135 |
| Trans-cinnamate 4-monooxygenase | 1.14.13.11 | K00487 | 3 | 12 |
| Caffeoyl-CoA O-methyltransferase | 2.1.1.104 | K00588 | 9 | 29 |
| Beta-glucosidase | 3.2.1.21 | K01188 | 44 | 141 |
| 4-coumarate-CoA ligase | 6.2.1.12 | K01904 | 14 | 50 |
| Beta-glucosidase | 3.2.1.21 | K05350 | 37 | 99 |
| Cinnamoyl-CoA reductase | 1.2.1.44 | K09753 | 5 | 14 |
| coumaroylquinate(coumaroylshikimate) 3′-monooxygenase | 1.14.13.36 | K09754 | 4 | 16 |
| Ferulate-5-hydroxylase | 1.14.-.- | K09755 | 14 | 29 |
| Phenylalanine ammonia-lyase | 4.3.1.24 | K10775 | 10 | 35 |
| Peroxiredoxin 6, 1-Cys peroxiredoxin | 1.11.1.7 | K11188 | 1 | 86 |
| Coniferyl-aldehyde dehydrogenase | 1.2.1.68 | K12355 | 4 | 76 |
| Coniferyl-alcohol glucosyltransferase | 2.4.1.111 | K12356 | 2 | 4 |
| Shikimate O-hydroxycinnamoyltransferase | 2.3.1.133 | K13065 | 12 | 17 |
| Caffeic acid 3-O -methyltransferase | 2.1.1.68 | K13066 | 8 | 16 |
| Caffeoylshikimate esterase | 3.1.1.- | K18368 | 1 | 5 |
| Cytochrome P450, family 98, subfamily A, polypeptide 8/9 | 1.14.13.- | K15506 | 0 | 1 |
| Aromatic-l-amino-acid decarboxylase | 4.1.1.28 | K01593 | 0 | 1 |
Figure 8.

KEGG analysis showing different enzymes identified (one color for each enzyme code or EC) of the Phenylpropanoid biosynthesis pathway. KEGG Pathway ko00940 is adapted from https://www.kegg.jp/kegg/kegg1.html. The KEGG database has been described previously36,37.
Gene expression analysis
De novo transcriptome filtered by Corset was used as a reference51. RSEM40 that map reads back to transcriptome and quantify their expression level was applied. Total reads in gene expression analysis of LCL tissues were 70,155,594, followed by total mapped 54,875,118 (78.22%); whereas total reads in gene expression analysis of LCR tissues were 84,263,224 followed by total mapped 47,187,580 (56.00%). To calculate the gene expression level, RSEM analyzed the mapping results of Bowtie and read count for each gene was converted into FPKM value. In RNA-seq, it is the most common method of estimating gene expression levels, which takes into account the effects of both sequencing depth and gene length on counting of fragments. These results are summarized in Supplementary File 6. Venn diagram, heat map and volcano plot highlights the DEGs and shows genes that are unique and common to leaf and root samples. The total numbers of expressed genes in LCL and LCR tissues were 67,714 and 359,010 respectively, while a total of 49,072 unigenes were found as commonly expressed in both tissues (Fig. 9A). We used DESeq software, to analyze the expression of unigenes in both LCL and LCR transcriptomes by normalizing the values to Fragments Per Kilobase Million (FPKM). The Benjiamini–Hochberg method was used to verify and revise the p-values. Volcano plot demonstrates the fold changes in the expression and statistical comparison (Fig. 9B). A total of 20,044 differentially expressed genes (DEGs) were identified. Within these DEGs 11,496 genes were up regulated and 8,548 genes were down regulated (Fig. 9B). Further, the hierarchical clustering analysis was used to screen the DEGs and cluster them according to their expression in LCL and LCR samples (Fig. 10A). On the basis of pathway enrichment, 20 metabolic and/or biosynthetic processes were predominantly involved. Of these spliceosome and protein processing in the endoplasmic reticulum was comprised by approximately 300 genes (Fig. 10B). Other important processes identified included; synaptic vesicle cycle, starch and sucrose metabolism, plant hormone signal transduction, photosynthesis, carbon fixation in photosynthetic organisms, amino sugar and nucleotide sugar metabolism and galactose metabolism.
Figure 9.
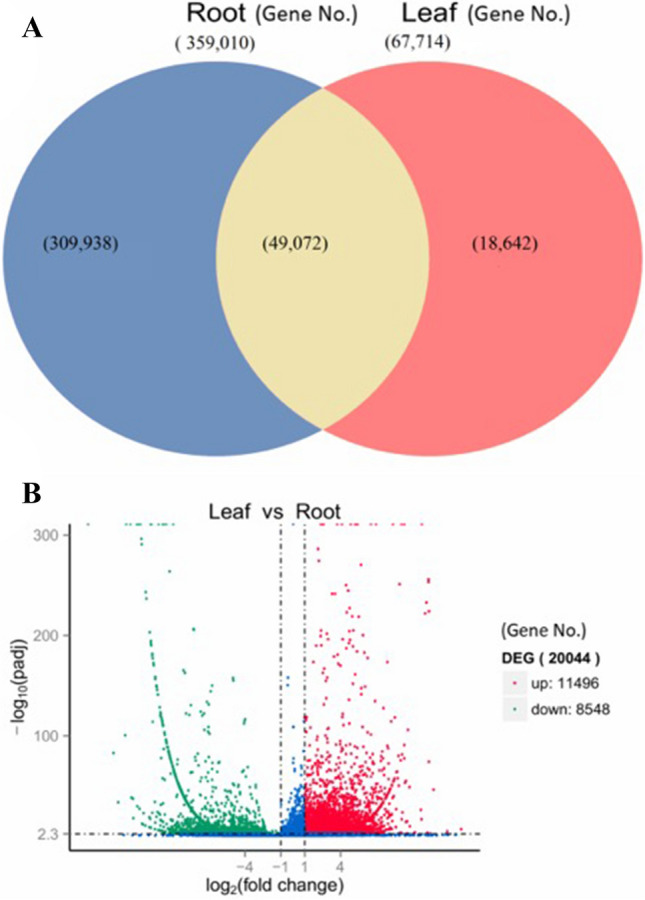
(A) Venn diagram showing differentially expressed genes identified in leaf and root of L.camara, (B) volcano plot showing map of DEGs.
Figure 10.

(A) Heatmap of upregulated and downregulated genes, (B) scatter plot based on number of genes involved in different biological processes.
Conclusion
Molecular studies on L. camara are rare and high-throughput genotyping efforts are almost non-existent. This study investigated the transcriptomes assembly of L. camara leaf and root tissues. A massive data of 70,155,594 and 84,263,224 clean reads were de novo assembled that revealed 72,877 and 513,985 unigenes from leaf and root tissues respectively. Further, the identified unigenes were annotated and functionally characterized in 7 databases. Notably, the numbers of expressed genes in LCL and LCR tissues varied; and 49,072 unigenes were commonly expressed in both tissues. Still, of the 20,044 DEGs 11,496 were up regulated and 8,548 genes were down regulated. Pathway analysis revealed the involvement of 229 and 943 genes (coding for 17 and 19 enzymes) in the biosynthesis of phenylpropanoid pathway in leaf and root tissues respectively. Nonetheless, the genomic resources will not only provide foundation of genomic research in L. camara but it will also provide detailed insight into the expression as well as functional analysis, gene cloning and avenues for genomics-assisted breeding in L. camara. This study will also serve as baseline to understand the regulation and biosynthesis of crucial bioactive compounds and to select superior alleles/haplotypes of L. camara with desired traits in the future.
Supplementary information
Acknowledgments
This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under grant No. (DF-368-130-1441). The authors, therefore, gratefully acknowledge DSR technical and financial support.
Author contributions
M.S. performed the experiments, M.S., M.M. and Y.A. collected the data, M.S. and I.U.R. analyzed the data. M.S. drafted the manuscript, N.A. and I.U.R. helped in interpretation of the results, writing and editing of the manuscript. H.F.A. and K.R.H. supervised the project.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Muzammil Shah, Email: muzammilshah100@outlook.com.
Niaz Ali, Email: niazalitk25@gmail.com.
Inayat Ur Rahman, Email: hajibotanist@outlook.com.
Supplementary information
is available for this paper at 10.1038/s41598-020-70635-5.
References
- 1.Ghisalberti EL. Lantana camara L. (verbenaceae) Fitoterapia. 2000;71:467–486. doi: 10.1016/s0367-326x(00)00202-1. [DOI] [PubMed] [Google Scholar]
- 2.Day, M. D., Wiley, C. J., Playford, J. & Zalucki, M. P. Lantana: Current Management Status and Future Prospects-ACIAR Monograph No. 102. Australian Centre for International Agricultural Research (2003).
- 3.Khan M, Mahmood A, Alkhathlan HZ. Characterization of leaves and flowers volatile constituents of Lantana camara growing in central region of Saudi Arabia. Arab. J. Chem. 2016;9:764–774. [Google Scholar]
- 4.Sharma OP, Sharma S, Pattabhi V, Mahato SB, Sharma PD. A review of the hepatotoxic plant Lantana camara. Crit. Rev. Toxicol. 2007;37:313–352. doi: 10.1080/10408440601177863. [DOI] [PubMed] [Google Scholar]
- 5.Sousa, E. O., Almeida, T. S., Menezes, I. R., Rodrigues, F. F., Campos, A. R., Lima, S. G., & da Costa, J. G. Chemical composition of essential oil of Lantana camara L. (Verbenaceae) and synergistic effect of the aminoglycosides gentamicin and amikacin. Rec. Nat. Prod. (2012).
- 6.Begum S, Ayub A, Qamar Zehra S, Shaheen Siddiqui B, Iqbal Choudhary M. Leishmanicidal triterpenes from Lantana camara. Chem. Biodivers. 2014;11:709–718. doi: 10.1002/cbdv.201300151. [DOI] [PubMed] [Google Scholar]
- 7.Sagar L, Sehgal R, Ojha S. Evaluation of antimotility effect of Lantana camara L. var. acuelata constituents on neostigmine induced gastrointestinal transit in mice. BMC Complement. Altern. Med. 2005;5:18. doi: 10.1186/1472-6882-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sathish R, Vyawahare B, Natarajan K. Antiulcerogenic activity of Lantana camara leaves on gastric and duodenal ulcers in experimental rats. J. Ethnopharmacol. 2011;134:195–197. doi: 10.1016/j.jep.2010.11.049. [DOI] [PubMed] [Google Scholar]
- 9.Herbert JM, et al. Verbascoside isolated from Lantana camara, an inhibitor of protein kinase C. J. Nat. Prod. 1991;54:1595–1600. doi: 10.1021/np50078a016. [DOI] [PubMed] [Google Scholar]
- 10.Qamar F, Begum S, Raza SM, Wahab A, Siddiqui BS. Nematicidal natural products from the aerial parts of Lantana camara Linn. Nat. Prod. Res. 2005;19:609–613. doi: 10.1080/14786410512331330594. [DOI] [PubMed] [Google Scholar]
- 11.Begum S, Wahab A, Siddiqui BS. Antimycobacterial activity of flavonoids from Lantana camara Linn. Nat. Prod. Res. 2008;22:467–470. doi: 10.1080/14786410600898714. [DOI] [PubMed] [Google Scholar]
- 12.Weyerstahl P, Marschall H, Eckhardt A, Christiansen C. Constituents of commercial Brazilian lantana oil. Flavour Fragr. J. 1999;14:15–28. [Google Scholar]
- 13.Randrianalijaona J-A, Ramanoelina PA, Rasoarahona JR, Gaydou EM. Seasonal and chemotype influences on the chemical composition of Lantana camara L.: Essential oils from Madagascar. Anal. Chim. Acta. 2005;545:46–52. [Google Scholar]
- 14.Sefidkon F. Essential oil of Lantana camara L. occurring in Iran. Flavour and Fragr. J. 2002;17:78–80. [Google Scholar]
- 15.Sundufu AJ, Shoushan H. Chemical composition of the essential oils of Lantana camara L. occurring in south China. Flavour Fragr. J. 2004;19:229–232. [Google Scholar]
- 16.Kasali AA, et al. Essential oil of Lantana camara L. var. aculeata from Nigeria. J. Essent. Oil Res. 2004;16:582–584. [Google Scholar]
- 17.Ouamba J-M, et al. Volatile constituents of the essential oil leaf of Lantana salvifolia Jacq. (Verbenaceae) Flavour Fragr. J. 2006;21:158–161. [Google Scholar]
- 18.de Sena Filho JG, et al. Chemical and molecular characterization of fifteen species from the Lantana (Verbenaceae) genus. Biochem. Syst. Ecol. 2012;45:130–137. [Google Scholar]
- 19.Satyal P, et al. The chemical diversity of Lantana camara: Analyses of essential oil samples from Cuba, Nepal, and Yemen. Chem. Biodivers. 2016;13:336–342. doi: 10.1002/cbdv.201500271. [DOI] [PubMed] [Google Scholar]
- 20.Goswami-Giri AS, Oza R. Bioinformatics overview of Lantana camara, an environmental weed. Res. J. Pharm. Biol. Chem. Sci. 2014;5:1712. [Google Scholar]
- 21.Peng Z, Bhattarai K, Parajuli S, Cao Z, Deng Z. Transcriptome analysis of young ovaries reveals candidate genes involved in gamete formation in Lantana camara. Plants. 2019;8:263. doi: 10.3390/plants8080263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yaradua SS, Shah M. The complete chloroplast genome of Lantana camara L. (Verbenaceae) Mitochondrial DNA Part B. 2020;5:918–919. doi: 10.1080/23802359.2020.1719920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z, Gerstein M, Snyder M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang G, et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010;20:646–654. doi: 10.1101/gr.100677.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li P, et al. The developmental dynamics of the maize leaf transcriptome. Nat. Genet. 2010;42:1060–1067. doi: 10.1038/ng.703. [DOI] [PubMed] [Google Scholar]
- 26.Garg R, Patel RK, Tyagi AK, Jain MD. novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 2011;18:53–63. doi: 10.1093/dnares/dsq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duan J, Xia C, Zhao G, Jia J, Kong X. Optimizing de novo common wheat transcriptome assembly using short-read RNA-Seq data. BMC Genomics. 2012;13:392. doi: 10.1186/1471-2164-13-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mudalkar S, Golla R, Ghatty S, De Reddy AR. novo transcriptome analysis of an imminent biofuel crop, Camelina sativa L. using Illumina GAIIX sequencing platform and identification of SSR markers. Plant Mol. Biol. 2014;84:159–171. doi: 10.1007/s11103-013-0125-1. [DOI] [PubMed] [Google Scholar]
- 29.Torre S, et al. RNA-seq analysis of Quercus pubescens leaves: De novo transcriptome assembly, annotation and functional markers development. PLoS ONE. 2014;9:e112487. doi: 10.1371/journal.pone.0112487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar A, Gaur VS, Goel A, De Gupta AK. novo assembly and characterization of developing spikes transcriptome of finger millet (Eleusine coracana): A minor crop having nutraceutical properties. Plant Mol. Biol. Report. 2015;33:905–922. [Google Scholar]
- 31.Zhang Y-H, Zhang S-D, Ling L-Z. De novo transcriptome analysis to identify flavonoid biosynthesis genes in Stellera chamaejasme. Plant Gene. 2015;4:64–68. [Google Scholar]
- 32.Pal T, Malhotra N, Chanumolu SK, Chauhan RS. Next-generation sequencing (NGS) transcriptomes reveal association of multiple genes and pathways contributing to secondary metabolites accumulation in tuberous roots of Aconitum heterophyllum Wall. Planta. 2015;242:239–258. doi: 10.1007/s00425-015-2304-6. [DOI] [PubMed] [Google Scholar]
- 33.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mao X, Cai T, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21:3787–3793. doi: 10.1093/bioinformatics/bti430. [DOI] [PubMed] [Google Scholar]
- 35.Tian T, et al. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017;45:W122–W129. doi: 10.1093/nar/gkx382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu J, Mao X, Cai T, Luo J, Wei L. KOBAS server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006;34:W720–W724. doi: 10.1093/nar/gkl167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie C, et al. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39:W316–W322. doi: 10.1093/nar/gkr483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li B, Dewey CN. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 42.Galachyants YP, Zakharova YR, Volokitina NA, Morozov AA, Likhoshway YV, Grachev MAD. De novo transcriptome assembly and analysis of the freshwater araphid diatom Fragilaria radians, Lake Baikal. Sci. Data. 2019;6:1–11. doi: 10.1038/s41597-019-0191-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Travisany D, et al. RNA-Seq analysis and transcriptome assembly of raspberry fruit (Rubus idaeus “Heritage”) revealed several candidate genes involved in fruit development and ripening. Sci. Hortic. 2019;254:26–34. [Google Scholar]
- 45.Götz S, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kang SW, et al. Sequencing and de novo assembly of visceral mass transcriptome of the critically endangered land snail Satsuma myomphala: Annotation and SSR discovery. Comp. Biochem. Physiol. D Genomics Proteomics. 2017;21:77–89. doi: 10.1016/j.cbd.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 47.Yang Y, Xu M, Luo Q, Wang J, Li HD. novo transcriptome analysis of Liriodendron chinense petals and leaves by Illumina sequencing. Gene. 2014;534:155–162. doi: 10.1016/j.gene.2013.10.073. [DOI] [PubMed] [Google Scholar]
- 48.Michal G, Schomburg D. Biochemical Pathways: An Atlas of Biochemistry and Molecular Biology. New York: Wiley; 2012. [Google Scholar]
- 49.Vogt T. Phenylpropanoid biosynthesis. Mol. Plant. 2010;3:2–20. doi: 10.1093/mp/ssp106. [DOI] [PubMed] [Google Scholar]
- 50.Lateef A, Prabhudas SK, Natarajan P. RNA sequencing and de novo assembly of Solanum trilobatum leaf transcriptome to identify putative transcripts for major metabolic pathways. Sci. Rep. 2018;8:1–13. doi: 10.1038/s41598-018-33693-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davidson NM, Oshlack A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014;15:410. doi: 10.1186/s13059-014-0410-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.