

Key Points
Question
What are the safety and immunogenicity of an inactivated vaccine against coronavirus disease 2019 (COVID-19)?
Findings
This was an interim analysis of 2 randomized placebo-controlled trials. In 96 healthy adults in a phase 1 trial of patients randomized to aluminum hydroxide (alum) only and low, medium, and high vaccine doses on days 0, 28, and 56, 7-day adverse reactions occurred in 12.5%, 20.8%, 16.7%, and 25.0%, respectively; geometric mean titers of neutralizing antibodies at day 14 after the third injection were 316, 206 and 297 in the low-, medium-, and high-dose groups, respectively. In 224 healthy adults randomized to the medium dose, 7-day adverse reactions occurred in 6.0% and 14.3% of the participants who received injections on days 0 and 14 vs alum only, and 19.0% and 17.9% who received injections on days 0 and 21 vs alum only, respectively; geometric mean titers of neutralizing antibodies in the vaccine groups at day 14 after the second injection were 121 vs 247, respectively.
Meaning
This inactivated COVID-19 vaccine had a low rate of adverse reactions and demonstrated immunogenicity, but longer-term assessment of safety and efficacy will require phase 3 trials.
Abstract
Importance
A vaccine against coronavirus disease 2019 (COVID-19) is urgently needed.
Objective
To evaluate the safety and immunogenicity of an investigational inactivated whole-virus COVID-19 vaccine in China.
Interventions
In the phase 1 trial, 96 participants were assigned to 1 of the 3 dose groups (2.5, 5, and 10 μg/dose) and an aluminum hydroxide (alum) adjuvant–only group (n = 24 in each group), and received 3 intramuscular injections at days 0, 28, and 56. In the phase 2 trial, 224 adults were randomized to 5 μg/dose in 2 schedule groups (injections on days 0 and 14 [n = 84] vs alum only [n = 28], and days 0 and 21 [n = 84] vs alum only [n = 28]).
Design, Setting, and Participants
Interim analysis of ongoing randomized, double-blind, placebo-controlled, phase 1 and 2 clinical trials to assess an inactivated COVID-19 vaccine. The trials were conducted in Henan Province, China, among 96 (phase 1) and 224 (phase 2) healthy adults aged between 18 and 59 years. Study enrollment began on April 12, 2020. The interim analysis was conducted on June 16, 2020, and updated on July 27, 2020.
Main Outcomes and Measures
The primary safety outcome was the combined adverse reactions 7 days after each injection, and the primary immunogenicity outcome was neutralizing antibody response 14 days after the whole-course vaccination, which was measured by a 50% plaque reduction neutralization test against live severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
Results
Among 320 patients who were randomized (mean age, 42.8 years; 200 women [62.5%]), all completed the trial up to 28 days after the whole-course vaccination. The 7-day adverse reactions occurred in 3 (12.5%), 5 (20.8%), 4 (16.7%), and 6 (25.0%) patients in the alum only, low-dose, medium-dose, and high-dose groups, respectively, in the phase 1 trial; and in 5 (6.0%) and 4 (14.3%) patients who received injections on days 0 and 14 for vaccine and alum only, and 16 (19.0%) and 5 (17.9%) patients who received injections on days 0 and 21 for vaccine and alum only, respectively, in the phase 2 trial. The most common adverse reaction was injection site pain, followed by fever, which were mild and self-limiting; no serious adverse reactions were noted. The geometric mean titers of neutralizing antibodies in the low-, medium-, and high-dose groups at day 14 after 3 injections were 316 (95% CI, 218-457), 206 (95% CI, 123-343), and 297 (95% CI, 208-424), respectively, in the phase 1 trial, and were 121 (95% CI, 95-154) and 247 (95% CI, 176-345) at day 14 after 2 injections in participants receiving vaccine on days 0 and 14 and on days 0 and 21, respectively, in the phase 2 trial. There were no detectable antibody responses in all alum-only groups.
Conclusions and Relevance
In this interim report of the phase 1 and phase 2 trials of an inactivated COVID-19 vaccine, patients had a low rate of adverse reactions and demonstrated immunogenicity; the study is ongoing. Efficacy and longer-term adverse event assessment will require phase 3 trials.
Trial Registration
Chinese Clinical Trial Registry Identifier: ChiCTR2000031809
This interim analysis of 2 randomized trials compares adverse reactions and neutralizing antibody responses to inactivated coronavirus disease 2019 (COVID-19) vs adjuvant-only control vaccination, and compares the outcomes at varying vaccine doses among healthy adults in China.
Introduction
Coronavirus disease 2019 (COVID-19) is an emerging respiratory infectious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that had infected more than 16 million individuals and caused more than 656 000 deaths worldwide by July 29, 2020.1 A safe and effective vaccine against COVID-19 is urgently needed.
There are currently more than 160 COVID-19 candidate vaccines in development worldwide, and 25 are in different phases of clinical trials using different platforms.2 The results of the phase 1 and 2 trials of several vaccines, such as a recombinant adenovirus type-5 (Ad5)–vectored vaccine, a chimpanzee adenovirus-vectored vaccine (ChAdOx1 nCoV-19), and 2 mRNA vaccines, have been published or made available on preprint servers.3,4,5,6,7,8 Inactivated vaccines have been widely used for the prevention of emerging respiratory diseases for decades,9 and results from preclinical studies of 2 inactivated COVID-19 vaccines have shown that the vaccines could protect rhesus macaques against SARS-CoV-2 with varying efficacy.10,11 However, results from clinical trials in humans have not been reported for the inactivated vaccines.
This report is the preliminary assessment of the safety outcomes 28 days and immunogenicity outcomes 14 days after 3 doses in a phase 1 trial and 2 doses in a phase 2 trial of an inactivated COVID-19 vaccine candidate in healthy adults in China.
Methods
Study Design and Participants
These double-blind, randomized, placebo-controlled phase 1 and 2 trials were designed by the Wuhan Institute of Biological Products Co Ltd and Henan Provincial Center for Disease Control and Prevention (CDC). The study protocol, available in Supplement 1, was approved by the institutional review board of Henan Provincial CDC. The statistical analysis plan is available in Supplement 2. Written informed consents were obtained from all participants before enrollment. The ongoing trials are being performed and data collected by the investigators at the CDC of Wuzhi Country, Henan Province, beginning on April 12, 2020. An independent data and safety monitoring board is monitoring the safety data and evaluating the risks among the participants during the trial. An interim analysis was conducted on June 16, 2020, and updated on July 27, 2020.
Healthy adults, aged 18 to 59 years, without history of SARS-CoV (via on-site inquiry) or SARS-CoV-2 infection (via serological and nucleic acid test) were eligible for enrollment in the study, and details of the inclusion and exclusion criteria are provided in the eMethods in Supplement 3. The participants were sequentially assigned a computer-generated randomization number, and stratified block randomization (block size, 8) by subgroups was adopted. Within each randomization block, the ratio of vaccine vs placebo was 3:1. All the vaccines and placebos were supplied in coded, identical-appearing, single-dose vials. Participants received 200 renminbi (US $29) for each blood sample donation.
In the phase 1 trial, participants were randomly and equally assigned to 3 vaccine dose groups (low, medium, and high doses with 2.5-, 5-, and 10-μg antigen protein content per dose, respectively, corresponding to 100, 200, and 400 Wuhan units/dose in the protocol) and a control group of aluminum hydroxide (alum) adjuvant only, and received intramuscular injections on days 0, 28, and 56 (Figure 1). In the phase 2 trial, participants were randomly divided into 2 schedule groups (days 0 and 14, and days 0 and 21) using the medium (5-μg) dose. Within each schedule group, the ratio of receiving vaccine and alum only was 3:1 (Figure 1).
Figure 1. Screening, Randomization, and Inclusion in Safety and Immunogenicity Analyses of Inactivated Vaccine for SARS-CoV-2.
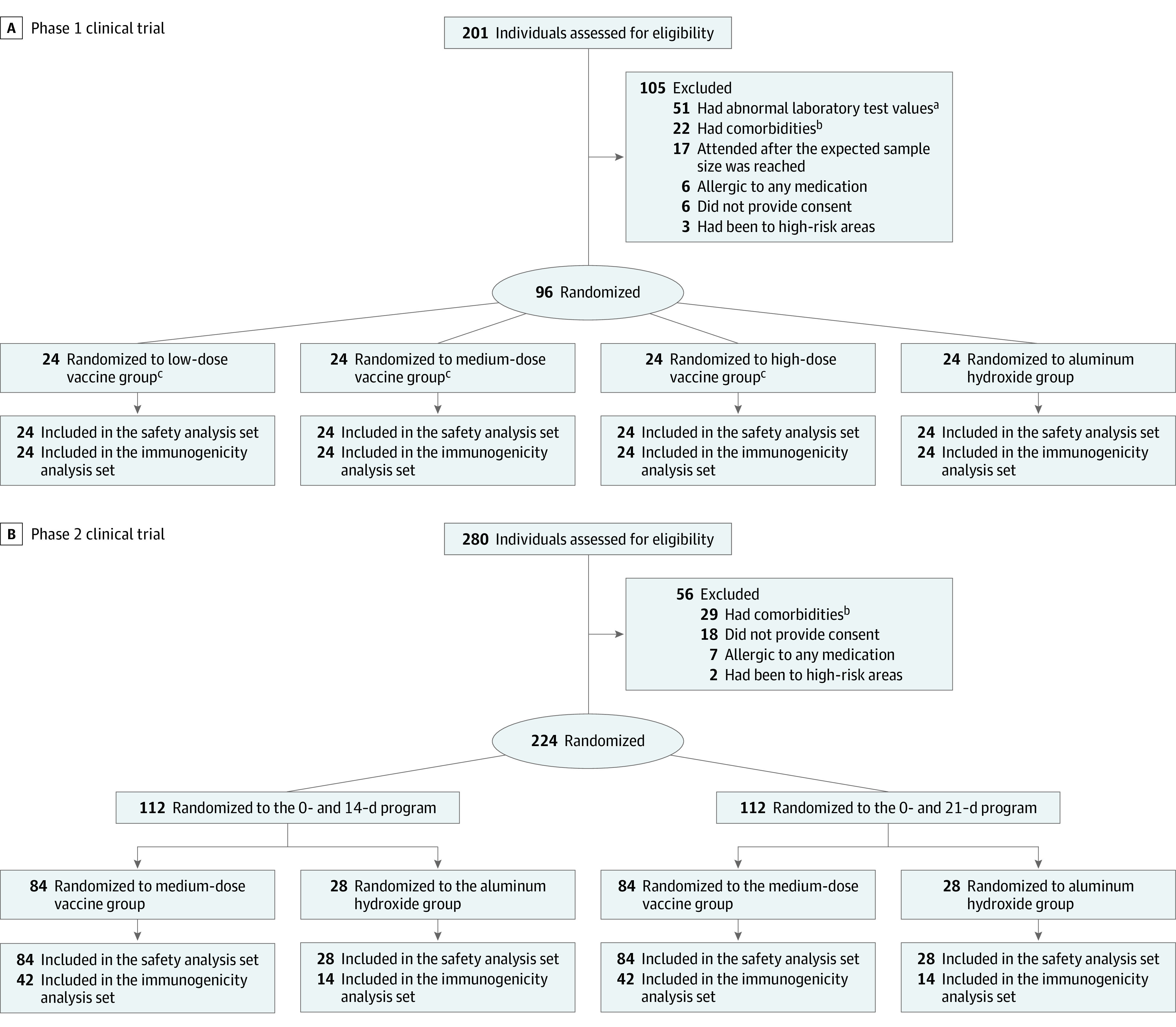
aLaboratory tests included routine blood tests, liver enzymes, total bilirubin, creatinine, urea nitrogen, urine protein, urine sugar, and urinary occult blood. Details of the list and definition of abnormal values are provided in the protocol in Supplement 1.
bThe comorbidities in the exclusion criteria included cardiovascular disease, cancer, respiratory disease, autoimmune disease, tuberculosis, severe liver disease, congenital malformation, mental illness, nervous system diseases, uncontrolled hypertension and diabetes, severe malnutrition, fever within 14 days, and other diseases that could affect participation and compliance in the trial as judged by the investigators.
cThe low, medium, and high doses represent 2.5, 5, and 10 μg/dose, respectively.
Participants were sequentially enrolled and received injections based on sentinel data. Participants in the low-dose group in the phase 1 trial received injections first, and adverse events were monitored for 7 days. If no vaccine-related severe adverse events were observed, participants in the medium-dose group in phase 1 received injections on day 8. Subsequently, participants in the high-dose group in phase 1 and participants in the medium-dose group in phase 2 received injections 8 days later if no vaccine-related severe adverse events were observed in the medium-dose group in phase 1.
Vaccine
A SARS-CoV-2 strain (WIV04 strain, National Genomic Data Center of the Chinese Academy of Science accession No. SAMC133237, and GenBank accession number MN996528) was isolated from a patient in the Jinyintan Hospital, Wuhan. The virus was cultivated in a qualified Vero cell line for propagation, and the supernatant of the infected cells was inactivated with β-propiolactone (1:4000 vol/vol at 2 to 8 °C for 48 hours. Following clarification of cell debris and ultrafiltration, the second β-propiolactone inactivation was performed in the same conditions as the first inactivation. The vaccine was adsorbed to 0.5-mg alum and packed into prefilled syringes in 0.5-mL sterile phosphate-buffered saline without preservative. The placebo group contained only sterile phosphate-buffered saline and alum adjuvant. All the vaccines and placebos were approved by the National Institutes for Food and Drug Control of China.
Outcomes
The participants were asked to record any injection site–specific adverse reactions (eg, pain, redness, and swelling) and systemic adverse reactions (eg, fever, headache, and fatigue) on diary cards within 7 days of each injection, which were summed and considered as the primary safety outcome in both phases. Any other unsolicited symptoms were also recorded during a 28-day follow-up period after each injection, and were considered as the secondary safety outcome. In the phase 1 trial, laboratory safety tests (including routine blood tests, liver enzymes, total bilirubin, creatinine, urea nitrogen, urine protein, urine sugar, and urinary occult blood) were performed before and 4 days after each injection to assess any toxic effects after vaccination. Blood lymphocyte subset distribution (eg, CD3+CD4+ and CD3+CD8+ T cells; natural killer cells; and B cells) and key cytokines (eg, tumor necrosis factor, IFN-γ, IL-2, IL-4, IL-5, and IL-6) were measured via flow cytometry before and 14 days after each injection. The schedule of biospecimen sample collections and related tests are shown in eFigure 1 in Supplement 3. The hematologic and biochemical markers were considered secondary safety measures in the phase 1 trial, and detailed methods for the laboratory measures are described in the eMethods in Supplement 3. The grading criteria of adverse reactions or events and the relationship with receiving injections were decided by the investigators before unblinding according to the standard guidelines issued by the National Medical Products Administration of China, and details are shown in eTable 1 in Supplement 3.
The primary humoral immunogenicity outcomes included the neutralizing antibody titers and the specific IgG-binding antibody titers. In the phase 1 trial, blood samples were collected before and at days 4 and 14 after each injection, as well as day 21 after the first dose. In the phase 2 trial, blood samples were collected before the first dose among all participants and at day 14 after the second dose among half of the participants whose randomization numbers were in the first half of the group list, which was considered as the immunogenicity subset in the analysis. The neutralization capacity induced by vaccine against live SARS-CoV-2 (BetaCoV/Wuhan/AMMS01/2020 activated) was analyzed in triplicate by plaque reduction neutralization test (PRNT), and the PRNT50 values were reported as a measure to determine the extent to which serum can be diluted and still reduce SARS-CoV-2 plaque formation by 50%. The total specific IgG antibody responses were measured with an in-house–developed enzyme-linked immunosorbent assay kit, which used the inactivated whole SARS-CoV-2 as coating antigen. Details of the immunogenicity assays have been previously reported12 and are provided in the eMethods in Supplement 3. The lower limit of detection was 5 for the neutralizing antibody test and 10 for the specific IgG antibody test, and those below the detection limit (eg, all baseline values) were assigned to 5 or 10, respectively, for further analysis. Seroconversion rate, as a secondary immunogenicity outcome, was defined as at least a 4-fold increase of antibody titers over baseline, thus the threshold was 20 for the neutralizing antibody titers and 40 for the specific IgG antibody titers.
Statistical Analysis
The statistical analysis plan is included in Supplement 2. The sample size was not determined based on the statistical power calculation; however, a minimum sample size of 20 to 30 participants was recommended for the phase 1 vaccine trial by the National Medical Products Administration of China. Both phases were designed at the same time, thus the sample size in the phase 2 trial was not calculated based on results from the phase 1 trial, but determined based on expert opinion of previous research experiences to be at least 3-fold that of the phase 1 trial.
The safety analysis was performed on data from all participants who received at least 1 dose. The number and proportion of participants with adverse reactions or events and the detailed safety profiles were compared across groups. The immunogenicity analysis was performed on data from the full analysis set of participants who received at least 1 dose and had results of any blood biomarker measurements before or after injections, and missing values were planned to be imputed by the last observation carried forward method; however, there were no missing data. The χ2 test or Fisher exact test (when data were sparse) was used to analyze categorical data, and the t test or the Mann-Whitney U test (for nonnormally distributed data) was used to analyze log-transformed antibody titers between vaccine and alum-only groups. Differences across groups at different time points were analyzed by analysis of variance. Analyses were conducted using SPSS software, version 25.0 (IBM SPSS Inc). Hypothesis testing was 2-sided with an α value of .05. Because of the potential type I error due to multiple comparisons, findings for analyses of secondary end points should be interpreted as exploratory.
To accelerate the vaccine development procedure and provide evidence for the phase 3 trial, an interim analysis was added to the protocol on June 14, 2020, to partially unblind some groups while the trial was ongoing. The groups that were unblinded in the current analysis were chosen for 2 major reasons. First, data were available for up to 14 days after 2 injections in those groups in both phases. Second, those groups had results for 3 different doses (low, medium, and high doses) and 3 injection procedures (2 injections on days 0 and 14, on days 0 and 21, or on days 0 and 28), which provided evidence for the design of the next phase. The data and safety monitoring board reviewed the data and results on June 16, 2020, and recommended that the preliminary analysis report be provided to the principal investigator and sponsor. The summary-level results of safety and humoral immunogenicity for these groups were released to the public; however, the individual data were not made public. The field investigators and participants remained blinded and the planned assessments were continued as specified in the protocol. On July 27, 2020, the results were further updated for the phase 1 trial with data available up to 28 days for safety assessment and 14 days for humoral immunogenicity assessment after the third injection. Data of extended follow-up visits (days 28, 90, 180, and 360 after the whole-course vaccination in both phases) were not yet available and are not reported in the current analysis. The other groups in the phase 2 trial (low-, medium-, and high-dose groups with 3 injections [days 0, 28, and 56; n = 80 in each group], a medium-dose group with 2 injections [days 0 and 28; n = 112], and a 1-injection group with the high dose [n = 112]) remain blinded as specified in the protocol, and results from those groups are not yet available.
Results
Study Participants
Between April 12 and May 2, 2020, a total of 481 volunteers were screened, and 96 were included in the phase 1 trial and 224 in the phase 2 trial (Figure 1). All participants completed immunizations and scheduled visits within the prescribed time. The baseline characteristics of the participants are shown in Table 1. The mean (SD) age was 41.2 (9.6) years and 43.5 (9.1) years in the phase 1 and 2 trials, respectively; there were 58 women (60.4%) in phase 1 and 142 women (63.4%) in phase 2.
Table 1. Baseline Characteristics of the Study Participantsa.
| Characteristic | No. (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| Phase 1 clinical trial; 0, 28, and 56–d group |
Phase 2 clinical trial | |||||||
| 0 and 14–d Group | 0 and 21–d Group | |||||||
| Low dose | Medium dose | High dose | Alum only | Medium dose | Alum only | Medium dose | Alum only | |
| No. of participants | 24 | 24 | 24 | 24 | 84 | 28 | 84 | 28 |
| Age, mean (SD), y | 36.0 (8.5) | 43.5 (8.9) | 43.1 (10.5) | 42.4 (8.7) | 43.8 (8.8) | 45.9 (8.7) | 41.4 (9.3) | 46.2 (8.9) |
| Age groups, y | ||||||||
| 18-29 | 6 (25.0) | 1 (4.2) | 5 (20.8) | 3 (12.5) | 5 (6.0) | 2 (7.1) | 13 (15.5) | 1 (3.6) |
| 30-44 | 13 (54.2) | 9 (37.5) | 7 (29.2) | 12 (50.0) | 36 (42.9) | 8 (28.6) | 35 (41.7) | 11 (39.3) |
| 45-59 | 5 (20.8) | 14 (58.3) | 12 (50.0) | 9 (37.5) | 43 (51.2) | 18 (64.3) | 36 (42.9) | 16 (57.1) |
| Sex | ||||||||
| Women | 13 (54.2) | 13 (54.2) | 13 (54.2) | 19 (79.2) | 52 (61.9) | 19 (67.9) | 52 (61.9) | 19 (67.9) |
| Men | 11 (45.8) | 11 (45.8) | 11 (45.8) | 5 (20.8) | 32 (38.1) | 9 (32.1) | 32 (38.1) | 9 (32.1) |
Abbreviation: alum, aluminum hydroxide.
The low, medium, and high doses represent 2.5, 5, and 10 μg/dose, respectively.
Safety Outcomes
The combined injection site and systemic reaction symptoms (according to solicited reports) are shown in Table 2. Within 7 days after injection, adverse reactions were reported by 48 (15.0%) of 320 participants in the trials. Specifically, the number of participants reporting adverse reactions was 5 (20.8%), 4 (16.7%), 6 (25.0%), and 3 (12.5%) in the low-dose, medium-dose, high-dose, and alum-only groups, respectively, in the phase 1 trial; and 5 (6.0%), 4 (14.3%), 16 (19.0%) and 5 (17.9%) in the groups who received the medium dose at days 0 and 14, alum only at days 0 and 14, the medium dose at days 0 and 21, and alum only at days 0 and 21, respectively, in the phase 2 trial. The most common adverse reaction was injection site pain (14 in phase 1 and 21 in phase 2), followed by fever (2 in phase 1 and 8 in phase 2). All adverse reactions were mild (grade 1 or 2), transient, and self-limiting, and did not require any treatment. No other adverse reactions were reported between days 8 and 28 after injection. There were no major differences across 3 injections in phase 1 or 2 injections in phase 2 (eTable 2 in Supplement 3). Unsolicited adverse events (regardless of relations with the immunization) are shown in eTable 3 in Supplement 3. Four severe adverse events (grade 3) occurred during the follow-up but all were unrelated to the immunization.
Table 2. Adverse Reactions After 3 Doses in the Phase 1 Trial and 2 Doses in the Phase 2 Trial in the Safety Seta.
| Adverse reaction | Phase 1 clinical trial; 0, 28, and 56–d group |
Phase 2 clinical trial | ||||||
|---|---|---|---|---|---|---|---|---|
| 0 and 14–d Group | 0 and 21–d Group | |||||||
| Low dose (n = 24) | Medium dose (n = 24) | High dose (n = 24) | Alum only (n = 24) | Medium dose (n = 84) | Alum only (n = 28) | Medium dose (n = 84) | Alum only (n = 28) | |
| 0-7 d | ||||||||
| Total adverse reactions | 5 (20.8) | 4 (16.7) | 6 (25.0) | 3 (12.5) | 5 (6.0) | 4 (14.3) | 16 (19.0) | 5 (17.9) |
| Systemic reactions | 0 | 3 (12.5) | 1 (4.2) | 1 (4.2) | 4 (4.8) | 2 (7.1) | 4 (4.8) | 2 (7.1) |
| Coughing | 0 | 0 | 0 | 0 | 1 (1.2) | 0 | 0 | 0 |
| Diarrhea | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.2) | 0 |
| Fatigue | 0 | 1 (4.2) | 0 | 0 | 1 (1.2) | 0 | 0 | 0 |
| Fever | 0 | 1 (4.2) | 1 (4.2) | 0 | 4 (4.8) | 1 (3.6) | 2 (2.4) | 1 (3.6) |
| Headache | 0 | 0 | 0 | 0 | 1 (1.2) | 1 (3.6) | 0 | 1 (3.6) |
| Nausea and vomiting | 0 | 1 (4.2) | 0 | 1 (4.2) | 0 | 0 | 1 (1.2) | 1 (3.6) |
| Pruritus (noninoculated site) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (3.6) |
| Local reactions | 5 (20.8) | 1 (4.2) | 6 (25.0) | 2 (8.3) | 2 (2.4) | 3 (10.7) | 13 (15.5) | 4 (14.3) |
| Itching | 0 | 0 | 0 | 0 | 0 | 0 | 1 (1.2) | 1 (3.6) |
| Pain | 5 (20.8) | 1 (4.2) | 6 (25.0) | 2 (8.3) | 2 (2.4) | 3 (10.7) | 12 (14.3) | 4 (14.3) |
| Redness | 0 | 0 | 1 (4.2) | 0 | 0 | 0 | 0 | 1 (3.6) |
| Swelling | 1 (4.2) | 0 | 1 (4.2) | 0 | 0 | 0 | 1 (1.2) | 1 (3.6) |
| Other reactions | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 0-28 d | ||||||||
| Total adverse reactions | 5 (20.8) | 4 (16.7) | 6 (25.0) | 3 (12.5) | 5 (6.0) | 4 (14.3) | 16 (19.0) | 5 (17.9) |
Abbreviation: alum, aluminum hydroxide.
The safety set included all participants who received at least 1 dose. The low, medium, and high doses represent 2.5, 5, and 10 μg/dose, respectively. Data are shown as No. of participants with event (%). A participant was only counted once in the specific reaction category even though a participant could have more than 1 adverse reaction. For example, a participant who had the same symptom (eg, injection site pain) after each dose was counted once in the symptom category. Similarly, if a participant had more than 1 symptom in the reaction class (total, systemic, and local), they were only counted once in that adverse reaction class. Detailed adverse reactions after each dose are shown in eTable 2 in Supplement 3.
In the phase 1 trial, the laboratory safety tests before and on day 4 after each injection revealed very few transient abnormalities in blood biochemical tests or urinalysis tests without special treatment (eTable 4 in Supplement 3). The blood lymphocyte subset and cytokine analysis showed no notable changes over time in different groups or substantial differences across groups at a certain time point (eFigures 2, 3, 4, 5, and 6 in Supplement 3).
Immunogenicity Outcomes
None of the participants had any detectable neutralizing antibody response against live SARS-CoV-2 at baseline or in the alum groups during the follow-up. In the phase 1 trial, the geometric mean titer (GMT) of neutralizing antibody at day 14 after the third injection was 316 (95% CI, 218-457) in the low-dose group, 206 (95% CI, 123-343) in the medium-dose group, and 297 (95% CI, 208-424) in the high-dose group (Table 3). In the phase 2 trial using the medium dose, the GMT was 121 (95% CI, 95-154) in the group who received injections on days 0 and 14 and 247 (95% CI, 176-345) in the group who received injections on days 0 and 21. Seroconversion was noted in all participants (100%) receiving vaccines in the low- and high-dose groups in phase 1, 23 of 24 participants (95.8%) in the medium-dose group in phase 1, and 41 of 42 participants (97.6%) in the 2 groups in phase 2, but none in the alum-only group.
Table 3. Antibody Responses 14 Days After 3 Doses in the Phase 1 Trial and 2 Doses in the Phase 2 Triala.
| Phase 1 clinical trial; 0, 28, and 56–d group |
Phase 2 clinical trial | |||||||
|---|---|---|---|---|---|---|---|---|
| 0 and 14–d Group | 0 and 21–d Group | |||||||
| Low dose (n = 24) | Medium dose (n = 24) | High dose (n = 24) | Alum only (n = 24) | Medium dose (n = 42) | Alum only (n = 14) | Medium dose (n = 42) | Alum only (n = 14) | |
| Neutralizing antibodies to live SARS-CoV-2 | ||||||||
| Geometric mean titer (95% CI) | 316 (218-457) | 206 (123-343) | 297 (208-424) | 5 (5-5) | 121 (95-154) | 5 (5-5) | 247 (176-345) | 5 (5-5) |
| Geometric mean ratio (95% CI)b | 63.1 (43.6-91.5) | 41.1 (24.6-68.7) | 59.4 (41.5-84.9) | NA | 24.1 (19.0-30.7) | NA | 49.3 (35.2-69.0) | NA |
| Seroconversion rate, % (95% CI)b | 100.0 (60.0-100.0) | 95.8 (56.7-100.0) | 100.0 (60.0-100.0) | 0 | 97.6 (67.7-100.0) | 0 | 97.6 (67.7-100.0) | 0 |
| Specific IgG-binding antibody responses to whole SARS-CoV-2 antigen | ||||||||
| Geometric mean titer (95% CI) | 415 (288-597) | 349 (258-472) | 311 (229-422) | 10 (10-10) | 74 (56-97) | 10 (10-10) | 215 (157-296) | 10 (10-10) |
| Geometric mean ratio (95% CI)b | 41.5 (28.8-59.7) | 34.9 (25.8-47.2) | 31.1 (22.9-42.2) | NA | 7.4 (5.6-9.7) | NA | 21.5 (15.7-29.6) | NA |
| Seroconversion rate, % (95% CI)b | 100.0 (60.0-100.0) | 100.0 (60.0-100.0) | 100.0 (60.0-100.0) | 0 | 85.7 (57.7-100.0) | 0 | 100.0 (60.0-100.0) | 0 |
Abbreviations: alum, aluminum hydroxide; NA, not applicable; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Immunogenicity population is defined as randomized participants who received at least 1 dose injection with nonmissing immunogenicity data before or after injections. All participants in the phase 1 trial and the first half of the participants in the phase 2 trial were scheduled for the hormoral immunogenicity measurement, and there were no missing data. The low, medium, and high doses represent 2.5, 5, and 10 μg/dose, respectively.
The geometric mean ratio was calculated as the geometric mean titer at day 14 after the whole-course injection over the geometric mean titer at baseline. Seroconversion was defined by at least a 4-fold increase in postinjection titer from baseline. The baseline values were imputed by the lower limit of detection of the assays, which was 5 for the neutralizing antibody measurement and 10 for the specific IgG-binding antibody measurement.
The GMTs of the specific IgG antibody were 415 (95% CI, 288-597), 349 (95% CI, 258-472), and 311 (95% CI, 229-422) in the low-dose, medium-dose, and high-dose groups in the phase 1 trial, as well as 74 (95% CI, 56-97) in the medium-dose group who received injections on days 0 and 14 and 215 (95% CI, 157-296) in the group who received injections on days 0 and 21 in the phase 2 trial (Table 3). Seroconversion was noted in all participants (100%) in phase 1 and those who received injections on days 0 and 21 in phase 2, while it was 85.7% (36/42) in the group who received injections on days 0 and 14 in phase 2 and none in the alum-only group.
The dynamic changes of the antibody responses in the phase 1 trial were further examined (Figure 2; eTable 5 in Supplement 3). Most participants started to generate antibody responses after the second injection, and remained at high levels 14 days after the third injection. The antibody responses 14 days after the second injection in the phase 2 trial are also shown in Figure 3, and results in more time points are currently not available.
Figure 2. Antibody Responses at Different Time Points in the Phase 1 Trial.

The dots represent individual participant values. Boxplots show the 25th, 50th (median), and 75th percentiles. Whiskers extend to the upper and lower adjacent values, the farthest values within 1.5 × the interquartile range beyond the 25th and 75th percentiles. The numbers below the boxes indicate the number of participants at the lowest measurable value. Alum indicates aluminum hydroxide; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Figure 3. Antibody Responses 14 Days After the Second Dose in the Phase 2 Trial.

Prevaccination is not shown; there were 14 participants in both aluminum hydroxide (alum)–only groups and 42 in both of the medium-dose vaccine groups. No measurable antibody responses could be detected at baseline, and thus, the baseline values were all imputed by the lower limit of detection of the assays, which was 5 for the neutralizing antibody measurement and 10 for the specific IgG-binding antibody measurement. The dots represent individual participant values. Boxplots show the 25th, 50th (median), and 75th percentiles. Whiskers extend to the upper and lower adjacent values, the farthest values within 1.5 × the interquartile range beyond the 25th and 75th percentiles. The numbers below the boxes indicate the number of participants at the lowest measurable value. Only half of the participants in the phase 2 trial were scheduled for the hormoral immunogenicity measurement at day 14 after the second injection. SARS-CoV-2 indicates severe acute respiratory syndrome coronavirus 2.
Discussion
To our knowledge, this is the first report of phase 1 and 2 clinical trials of a whole-virus inactivated COVID-19 vaccine among healthy adults. The inactivated vaccine was well tolerated in all dose groups under different injection procedures with no vaccine-related serious adverse events. The most common adverse reaction was injection site pain, which was mild and self-limiting. The incidence rate of adverse reactions in the current study (15.0% among all participants) was lower compared with results of other candidate vaccines.3,4,5,6,7,8 Although all these studies reported that the adverse reactions were mostly mild and moderate in severity and self-limiting, the incidence rates in the vaccine groups (mostly >60% and in some studies 100%) were higher compared with the control group. Therefore, the inactivated vaccine in the current study suggests a relatively better safety profile compared with vaccines using other platforms. However, these comparisons should be cautiously interpreted given the small sample size in the studies, and that few serious adverse events occurred.
The neutralizing antibody response was monitored over 14 days after injections in the current preliminary report, and the results suggested that the inactivated vaccine may effectively induce antibody production. The results in both phases indicated that a longer interval (21 days and 28 days) between the first and second injections produced higher antibody responses compared with a shorter interval schedule (14-day group). The antibody titers started to increase after the second injection and further increased after the third injection, suggesting the need for a booster injection. Nevertheless, the optimal interval between injections and times of booster injections of the inactivated vaccine remain unclear, and the full analysis of the trial data with extended follow-up and other intervention groups is needed.
The observed humoral immune response was likely generated by the vaccine, not through natural infections, because the screening procedure (including serological screening and nucleic acid testing) was conducted to try to ensure that none of the participants was infected with SARS-CoV-2 before enrollment. Furthermore, no new COVID-19 cases were reported at the study area and no participant developed any symptoms of SARS-CoV-2 infection during the trial.
Convalescent serum samples in a comparison group were unavailable to provide a benchmark to interpret the magnitude of the antibody responses. However, when compared with published results from other COVID-19 vaccine trials, the neutralizing antibody titers (measured by the PRNT assay in the current study) were comparable with the levels in other studies using a similar method, such as the mRNA-1273 vaccine,7 the BNT162b1 RNA vaccine,5,6 and the ChAdOx1 nCoV-19 vaccine,8 and higher than the Ad5-vectored vaccine.3,4 However, such direct comparisons should be interpreted cautiously because of different vaccine doses in those trials and different assay methods. Comparison of the specific IgG antibody titers across studies cannot be done because of varied assay methods.
Epidemiological studies have reported that neutralizing antibody titers vary widely in convalescent serum samples and may be related to several factors (eg, age, sex, disease severity, and days since infection),13,14,15,16,17,18 but none of those studies used the PRNT method. In addition, some studies have indicated that the antibody titers may decline over time in patients recovered from COVID-19,19,20,21 particularly in those who were asymptomatic. A study of 56 patients recovered from SARS found that the neutralizing and IgG antibodies quickly declined after 16 months and continued to decline further to a very low level or even became undetectable after 3 years.22 Trials of SARS vaccines also suggest that the neutralizing antibody responses may decline over time.23,24 However, a recent study suggested that patients who had recovered from SARS (n = 23) still possessed long-lasting memory T cells reactive to SARS nucleocapsid protein 17 years after infection.25 Thus, it is unclear whether vaccine-induced antibody levels could persist and, if not, whether the long-lasting memory T cells could affect susceptibility and pathogenesis of SARS-CoV-2 infection. The participants in the current study are scheduled to be followed up to 1 year. A phase 3 trial has been initiated (Chinese Clinical Trial Registry No. ChiCTR2000034780), which will provide information on immune persistence and efficacy.
One concern about COVID-19 vaccines is the antibody-dependent enhancement (ADE) phenomenon that vaccine could make the subsequent SARS-CoV-2 infection more severe.26,27 The ADE phenomenon has been reported in studies of Middle East respiratory syndrome–CoV and SARS-CoV vaccines in animal challenge models.26,27 However, this was not observed in the preclinical study in an immunization-challenge model of rhesus macaques using the same vaccines in the current study or in reports from preclinical studies of other COVID-19 vaccine candidates, including 2 other inactivated COVID-19 vaccines.10,11,28 Studies have shown that previous infection of SARS-CoV-2 could protect against rechallenge in rhesus macaques.29,30
Another concern with whole-inactivated virus, particularly with alum adjuvant that can induce T helper 2 cell–biased responses, was vaccine-associated enhanced respiratory disease (VAERD). VAERD was reported in young children in the 1960s when whole-inactivated virus vaccine with alum adjuvant was tested for measles and respiratory syncytial virus.31,32 However, most of the inactivated vaccines against COVID-19 under development used alum adjuvant,10,11 and no evidence of VAERD has been seen. Instead, alum may reduce immunopathology compared with unadjuvanted coronavirus vaccines.33 In the current study, notable changes in the lymphocyte subset distribution or various cytokines (including T helper 2 cell–related cytokines IL-4, IL-5, and IL-10) in various vaccine groups or alum-only group were not observed. However, T-cell—mediated immune responses on stimulation were not measured in the current study. Furthermore, alum is the most widely tested adjuvant component and has been commonly used in many types of vaccines on the market.34 Nevertheless, safety, including the potential possibility of ADE and VAERD, will be closely monitored in the extended follow-up visits as well as in the phase 3 trial.
Limitations
This study has several limitations. First, the interim analysis was not prespecified in the original protocol; it was added during the study to provide necessary information for the design of a phase 3 trial. Therefore, results from the unplanned interim analysis should be cautiously interpreted.
Second, the current analysis reported results from only some groups, and the study was likely underpowered for comparisons of adverse events. Therefore, the full analysis of the entire trial population (extended follow-up and other injection schedule groups as specified in the protocol) is needed to provide a comprehensive profile of the inactivated vaccine in terms of tolerability, immunogenicity, and immune persistence.
Third, although the inactivated vaccine elicited robust antibody responses, whether it could protect individuals against COVID-19 remains unknown.
Conclusions
In this interim report of the phase 1 and phase 2 trials of an inactivated COVID-19 vaccine, patients had a low rate of adverse reactions and demonstrated immunogenicity; the study is ongoing. Efficacy and longer-term adverse event assessment will require phase 3 trials.
Trial Protocol
Statistical Analysis Plan
eMethods
eTable 1. Grading Scales for Systemic and Local Adverse Events
eTable 2. Total Adverse Reactions After Each Dose in the Phase 1 and 2 Trials
eTable 3. Total Adverse Events 28 Days after Three Doses in the Phase 1 Trial and Two Doses in the Phase 2 Trial
eTable 4. Abnormal Laboratory Parameters after three Vaccinations in the Phase 1 Clinical Trial
eTable 5. Geometric Mean Titer (95% Confidence Interval) of Antibodies in Different Time Points in the Two Trials
eFigure 1. Schedule of Biospecimen Collection and Related Tests
eFigure 2. Lymphocyte Subset Distribution Analysis in the Phase 1 Trial
eFigure 3. Changes in T Helper 1 Cell Related Cytokines in the Phase 1 Trial
eFigure 4. Changes in T Helper 2 Cell Related Cytokines in the Phase 1 Trial
eFigure 5. Changes in T Helper 17 Cell Related Cytokines in the Phase 1 Trial
eFigure 6. Changes in Other Cytokines in the Phase 1 Trial
Data Sharing Statement
References
- 1.World Health Organization WHO coronavirus disease (COVID-19) dashboard. Accessed July 30, 2020. https://covid19.who.int/
- 2.World Health Organization Draft landscape of COVID-19 candidate vaccines. Accessed July 30, 2020. https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines
- 3.Zhu FC, Li YH, Guan XH, et al. Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: a dose-escalation, open-label, non-randomised, first-in-human trial. Lancet. 2020;395(10240):1845-1854. doi: 10.1016/S0140-6736(20)31208-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu FC, Guan XH, Li YH, et al. Immunogenicity and safety of a recombinant adenovirus type-5-vectored COVID-19 vaccine in healthy adults aged 18 years or older: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. Published online July 20, 2020. doi: 10.1016/S0140-6736(20)31605-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sahin U, Muik A, Derhovanessian E, et al. Concurrent human antibody and TH1 type T-cell responses elicited by a COVID-19 RNA vaccine. medRxiv Preprint posted July 20, 2020. doi: 10.1101/2020.07.17.20140533 [DOI]
- 6.Mulligan MJ, Lyke KE, Kitchin N, et al. Phase 1/2 study to describe the safety and immunogenicity of a COVID-19 RNA vaccine candidate (BNT162b1) in adults 18 to 55 years of age: interim report. medRxiv Preprint posted July 1, 2020. doi: 10.1101/2020.06.30.20142570 [DOI]
- 7.Jackson LA, Anderson EJ, Rouphael NG, et al. ; mRNA-1273 Study Group . An mRNA vaccine against SARS-CoV-2 - preliminary report. N Engl J Med. Published online July 14, 2020. doi: 10.1056/NEJMoa2022483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Folegatti PM, Ewer KJ, Aley PK, et al. ; Oxford COVID Vaccine Trial Group . Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet. Published online July 20, 2020. doi: 10.1016/S0140-6736(20)31604-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stern PL. Key steps in vaccine development. Ann Allergy Asthma Immunol. 2020;125(1):17-27. doi: 10.1016/j.anai.2020.01.025 [DOI] [PubMed] [Google Scholar]
- 10.Gao Q, Bao L, Mao H, et al. Development of an inactivated vaccine candidate for SARS-CoV-2. Science. 2020;369(6499):77-81. doi: 10.1126/science.abc1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H, Zhang Y, Huang B, et al. Development of an inactivated vaccine candidate, BBIBP-CorV, with potent protection against SARS-CoV-2. Cell. Published online June 6, 2020. doi: 10.1016/j.cell.2020.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duan K, Liu B, Li C, et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc Natl Acad Sci U S A. 2020;117(17):9490-9496. doi: 10.1073/pnas.2004168117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Long QX, Liu BZ, Deng HJ, et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat Med. 2020;26(6):845-848. doi: 10.1038/s41591-020-0897-1 [DOI] [PubMed] [Google Scholar]
- 14.Luchsinger LL, Ransegnola B, Jin D, et al. Serological analysis of New York City COVID19 convalescent plasma donors. medRxiv Preprint posted June 9, 2020. doi: 10.1101/2020.06.08.20124792 [DOI]
- 15.Robbiani DF, Gaebler C, Muecksch F, et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature. Published online June 18, 2020. doi: 10.1038/s41586-020-2456-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Guo X, Xin Q, et al. Neutralizing antibodies responses to SARS-CoV-2 in COVID-19 inpatients and convalescent patients. Clin Infect Dis. Published online June 4, 2020. doi: 10.1093/cid/ciaa721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu F, Wang A, Liu M, et al. Neutralizing antibody responses to SARS-CoV-2 in a COVID-19 recovered patient cohort and their implications. medRxiv Preprint posted April 20, 2020. doi: 10.1101/2020.03.30.20047365 [DOI]
- 18.Zhao J, Yuan Q, Wang H, et al. Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. Clin Infect Dis. Published online March 28, 2020. doi: 10.1093/cid/ciaa344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu T, Wu S, Tao H, et al. Prevalence of IgG antibodies to SARS-CoV-2 in Wuhan: implications for the ability to produce long-lasting protective antibodies against SARS-CoV-2. medRxiv Preprint posted June 16, 2020. doi: 10.1101/2020.06.13.20130252 [DOI]
- 20.Long QX, Tang XJ, Shi QL, et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat Med. Published online June 18, 2020. doi: 10.1038/s41591-020-0965-6 [DOI] [PubMed] [Google Scholar]
- 21.Ibarrondo FJ, Fulcher JA, Goodman-Meza D, et al. Rapid decay of anti–SARS-CoV-2 antibodies in persons with mild Covid-19. N Engl J Med. Published online July 21, 2020. doi: 10.1056/NEJMc2025179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao WC, Liu W, Zhang PH, Zhang F, Richardus JH. Disappearance of antibodies to SARS-associated coronavirus after recovery. N Engl J Med. 2007;357(11):1162-1163. doi: 10.1056/NEJMc070348 [DOI] [PubMed] [Google Scholar]
- 23.Lin JT, Zhang JS, Su N, et al. Safety and immunogenicity from a phase I trial of inactivated severe acute respiratory syndrome coronavirus vaccine. Antivir Ther. 2007;12(7):1107-1113. [PubMed] [Google Scholar]
- 24.Martin JE, Louder MK, Holman LA, et al. ; VRC 301 Study Team . A SARS DNA vaccine induces neutralizing antibody and cellular immune responses in healthy adults in a phase I clinical trial. Vaccine. 2008;26(50):6338-6343. doi: 10.1016/j.vaccine.2008.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le Bert N, Tan AT, Kunasegaran K, et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. Published online July 15, 2020. doi: 10.1038/s41586-020-2550-z [DOI] [PubMed] [Google Scholar]
- 26.Eroshenko N, Gill T, Keaveney MK, Church GM, Trevejo JM, Rajaniemi H. Implications of antibody-dependent enhancement of infection for SARS-CoV-2 countermeasures. Nat Biotechnol. 2020;38(7):789-791. doi: 10.1038/s41587-020-0577-1 [DOI] [PubMed] [Google Scholar]
- 27.Graham BS. Rapid COVID-19 vaccine development. Science. 2020;368(6494):945-946. doi: 10.1126/science.abb8923 [DOI] [PubMed] [Google Scholar]
- 28.Corbett KS, Flynn B, Foulds KE, et al. Evaluation of the mRNA-1273 vaccine against SARS-CoV-2 in nonhuman primates. N Engl J Med. Published online July 28, 2020. doi: 10.1056/NEJMoa2024671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chandrashekar A, Liu J, Martinot AJ, et al. SARS-CoV-2 infection protects against rechallenge in rhesus macaques. Science. Published online May 20, 2020. doi: 10.1126/science.abc4776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng W, Bao L, Liu J, et al. Primary exposure to SARS-CoV-2 protects against reinfection in rhesus macaques. Science. Published online July 2, 2020. doi: 10.1126/science.abc5343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fulginiti VA, Eller JJ, Downie AW, Kempe CH. Altered reactivity to measles virus: atypical measles in children previously immunized with inactivated measles virus vaccines. JAMA. 1967;202(12):1075-1080. doi: 10.1001/jama.1967.03130250057008 [DOI] [PubMed] [Google Scholar]
- 32.Kim HW, Canchola JG, Brandt CD, et al. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am J Epidemiol. 1969;89(4):422-434. doi: 10.1093/oxfordjournals.aje.a120955 [DOI] [PubMed] [Google Scholar]
- 33.Hotez PJ, Corry DB, Bottazzi ME. COVID-19 vaccine design: the Janus face of immune enhancement. Nat Rev Immunol. 2020;20(6):347-348. doi: 10.1038/s41577-020-0323-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hotez PJ, Corry DB, Strych U, Bottazzi ME. COVID-19 vaccines: neutralizing antibodies and the alum advantage. Nat Rev Immunol. 2020;20(7):399-400. doi: 10.1038/s41577-020-0358-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial Protocol
Statistical Analysis Plan
eMethods
eTable 1. Grading Scales for Systemic and Local Adverse Events
eTable 2. Total Adverse Reactions After Each Dose in the Phase 1 and 2 Trials
eTable 3. Total Adverse Events 28 Days after Three Doses in the Phase 1 Trial and Two Doses in the Phase 2 Trial
eTable 4. Abnormal Laboratory Parameters after three Vaccinations in the Phase 1 Clinical Trial
eTable 5. Geometric Mean Titer (95% Confidence Interval) of Antibodies in Different Time Points in the Two Trials
eFigure 1. Schedule of Biospecimen Collection and Related Tests
eFigure 2. Lymphocyte Subset Distribution Analysis in the Phase 1 Trial
eFigure 3. Changes in T Helper 1 Cell Related Cytokines in the Phase 1 Trial
eFigure 4. Changes in T Helper 2 Cell Related Cytokines in the Phase 1 Trial
eFigure 5. Changes in T Helper 17 Cell Related Cytokines in the Phase 1 Trial
eFigure 6. Changes in Other Cytokines in the Phase 1 Trial
Data Sharing Statement