

Abstract
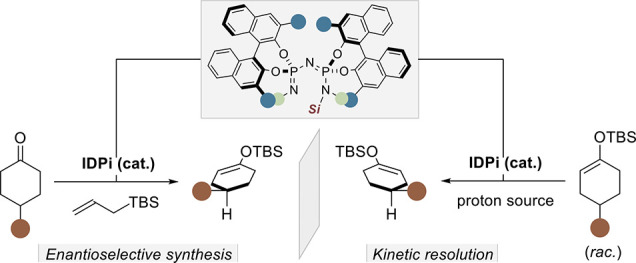
The use of chiral enol silanes in fundamental transformations such as Mukaiyama aldol, Michael, and Mannich reactions as well as Saegusa–Ito dehydrogenations has enabled the chemical synthesis of enantiopure natural products and valuable pharmaceuticals. However, accessing these intermediates in high enantiopurity has generally required the use of either stoichiometric chiral precursors or stoichiometric chiral reagents. We now describe a catalytic approach in which strongly acidic and confined imidodiphosphorimidates (IDPi) catalyze highly enantioselective interconversions of ketones and enol silanes. These “silicon–hydrogen exchange reactions” enable access to enantiopure enol silanes via tautomerizing σ-bond metatheses, either in a deprotosilylative desymmetrization of ketones with allyl silanes as the silicon source or in a protodesilylative kinetic resolution of racemic enol silanes with a carboxylic acid as the silyl acceptor.
Silicon–hydrogen exchange reactions can be described as formal σ-bond metatheses, interconverting silylated (C–SiR3 or O–SiR3) and hydrogenated (C–H or O–H) compounds (Scheme 1A). These reactions are used to install protecting groups,1 synthesize reagents such as trimethylsilyl cyanide2 or enol silanes,3−6 and also increase the volatility of compounds for analytical applications.7 Despite the value of silicon–hydrogen exchange reactions and their potential to generate enantiopure silylated compounds that are otherwise challenging to obtain, their use in asymmetric catalysis has been rare, and applications toward chiral enol silanes are currently unknown.8−12 Access to enantiopure enol silanes traditionally requires stoichiometric amounts of strong chiral bases such as Simpkins’ lithium amides (Scheme 1B).13 Alternatively, silicon–hydrogen exchange reactions have recently been used by Takasu et al.14 to prepare achiral and racemic enol silanes from ketones with silylated triflimide as a catalyst. Independently, we found that, under certain conditions, enol silanes can also form as side products in Mukaiyama aldol and Hosomi–Sakurai reactions via silylium asymmetric counteranion-directed catalysis (Si-ACDC).15−17 Inspired by these observations, we became intrigued by the opportunity to develop asymmetric silicon–hydrogen exchange reactions toward enantiopure enol silanes. We envisioned a catalytic cycle in which, upon the formation of a chiral silylium ion-equivalent (R3SiX*) and propene, via protodesilylation of an allylsilane by the strong acid HX*, a siloxocarbenium ion intermediate could be reversibly generated from the starting ketone (Scheme 1C). Its enantioselective deprotonation by the chiral counteranion X*– would then produce the enantioenriched enol silane and simultaneously regenerate the Brønsted acid precatalyst HX*.
Scheme 1. Design of a Catalytic Asymmetric Silicon–Hydrogen Exchange Reaction.

While the overall transformation is generally disfavored thermodynamically, here it would presumably be driven by the release of volatile propene. In contrast, the corresponding reverse reaction, the protodesilylation of an enol silane, is thermodynamically favored and indeed well-known.18−22 Such reactions are readily catalyzed by acids, and an asymmetric protodesilylation of racemic silyl enol ethers should be attainable by applying the same catalysis concept outlined above. Only now, instead of using an allylsilane as the silylating reagent, the reaction would be mediated by a suitable proton source (Scheme 1D). We speculated that, if the identical chiral catalyst would be used in the protodesilylation and if the mechanism of the protonation step would be the exact reverse of or very similar to the above oxocarbenium deprotonation, then, in line with the principle of microscopic reversibility,23 the same enantiomer that is created in the forward reaction should be selectively protodesilylated in the reverse. As a result, the opposite enantiomer would be obtained in a kinetic resolution.24
Indeed, when we reacted 4-phenylcyclohexanone (1a) with commercially available allyl silane 2a in the presence of IDPi catalyst 4a, enol silane product 3a was obtained in 95% yield with a promising e.r. of 82:18 (Table 1, entry 1). Other commonly used Brønsted acid catalysts such as phosphoric acids, imidodiphosphates, and disulfonimides did not catalyze the reaction under these conditions (Supporting Information, Table S1).
Table 1. Optimization of the Catalytic Asymmetric Synthesis of Enol Silanea.

| entry | catalyst | solvent | T (°C) | yieldb (%) | e.r.c |
|---|---|---|---|---|---|
| 1 | 4a | toluene | 25 | 95 | 82:18 |
| 2 | 4b | toluene | 25 | 99 | 85.5:14.5 |
| 3 | 4c | toluene | 25 | 99 | 88:12 |
| 4 | 4d | toluene | 25 | 99 | 80:20 |
| 5 | 4e | toluene | 25 | 80 | 80:20 |
| 6 | 4f | toluene | 25 | 99 | 75:25 |
| 7 | 4c | toluene:dioxane (2:1) | 0 | 99 | 94:6 |
| 8 | 4c | toluene:dioxane (2:1) | –20 | 99 | 97:3 |
Reactions were performed with 1a (0.025 mmol), 2a (2.0 equiv), and 4a–4f (1 mol %).
1H NMR yield.
e.r. determined by HPLC.
With this encouraging result, we began optimizing the reaction. IDPi catalysts with different steric environments were designed by further modifying the sulfonamide core and the substitution pattern of the 3,3′-arenes of the chiral 1,1′-bi-2-naphthol (BINOL) scaffold. With the newly synthesized catalysts 4b–4f, the e.r. was further improved (entries 2–6). Gratifyingly, using a mixed solvent system at a lower temperature significantly improved the enantioselectivity with catalyst 4c, ultimately giving the product with an e.r. of 97:3 in quantitative yield (entries 7 and 8).
Using the developed conditions, a variety of 4-aryl-substituted enol silanes were prepared and isolated with generally excellent enantioselectivities (up to 98:2 e.r.) and yields (up to 99%) (Table 2, products 3a–3o). In addition, 4-alkyl substituted cyclohexanones 1p–1r were also tolerated, giving the desired products 3p–3r with enantioselectivities in the range of 92:8 to 97:3. 4,4-Disubstituted cyclohexanone 1s provided product 3s, featuring a quaternary stereogenic carbon center, in 94:6 e.r. Satisfyingly, product 3t bearing an ester group was generated smoothly in 92% yield with an excellent e.r. of 98:2. Additionally, we explored a bicyclic cyclopentanone (1u) and a 3-substituted cyclobutanone (1v) to probe the generality of the methodology. To our delight, the corresponding enol silanes 3u and 3v were obtained with 95:5 e.r. (98% yield) and 89:11 e.r. (96% yield), respectively.
Table 2. Substrate Scope of the Catalytic Asymmetric Deprotosilylation of Ketonesa.


Performed with 1 (0.2 mmol), 2a (2.0 equiv), and 4c (1 mol %) in 2.4 mL of toluene/dioxane. Isolated yields. e.r. determined by HPLC.
With 4d.
With 4a.
With 4b;
With 4e.
With triethyl(2-methylallyl)silane (2b).
To illustrate the synthetic utility of the method, the reaction between ketone 1a and allylsilane 2a was conducted on a gram scale and furnished the product without erosion of enantioselectivity (Scheme 2). Meanwhile, IDPi catalyst 4c could be recovered (93%) and remained active in further transformations, demonstrating the practicality of the process. The produced enol silane 3a could be readily transformed into different structural motifs via α-bromination,25 α-fluorination,26 Saegusa–Ito oxidation,27 [2 + 2] cycloaddition,28 and α-benzylation reactions.29 All of these transformations proceeded smoothly to give products 5–9, with complete conservation of the enantiomeric purity. Importantly, our method could successfully be applied to furnish enol silane 3w, which is a known intermediate toward the synthesis of the prostacyclin analogue iloprost, a commercial drug used for the treatment of pulmonary arterial hypertension.30
Scheme 2. Synthetic Applications.

As proposed above, our catalytic asymmetric ketone silylation reaction is likely driven by the irreversible protodesilylation of the allylsilane. We therefore hypothesized that, if a suitable proton source could be identified, a kinetic resolution of racemic enolsilanes may also be achievable with IDPi catalysts, in what is effectively the reverse of our enol silane synthesis. Importantly, such a kinetic resolution would not only be expected to be applicable to racemic enol silanes derived from symmetric ketones, such as 4-substituted cyclohexanones, but possibly also to enol silanes derived from unsymmetrical ketones. To explore this possibility, we investigated the protodesilylation of racemic enol silane 3x as a model substrate and isopropanol as the initial proton source. Indeed, the reaction proceeded smoothly and provided moderate enantioselectivity at 68% conversion when we used catalyst 4c (Table 3, entry 1). Interestingly, sulfonamide-modified catalyst 4d gave significantly higher selectivity and was therefore further investigated (entry 2). Lowering the temperature to −30 °C increased the s factor to a promising 12 (entry 3).31,32 We also explored different proton sources using IDPi catalyst 4d at lower temperatures (entries 4–6). While water and TMP proved to be unsuitable (entry 4), 2-biphenyl carboxylic acid (BCA) 10 turned out to be an optimal reagent (entry 6). Finally, recovered silyl enol ether 3x was obtained in 97:3 e.r. at 51% conversion, corresponding to a selectivity of 70, when the reaction was conducted at −60 °C for 24 h (entry 7). As expected, even though this protonation presumably occurs via a slightly different mechanism than the enol silane synthesis described above, the opposite enantiomer of product 3x was indeed obtained, confirming the possibility to produce both enantiomers of the enol silane using the same catalyst enantiomer.
Table 3. Optimization of the Protodesilylative Kinetic Resolution of rac-3xa.

| entry | catalyst | proton source | conversionb (%) | e.r.c | sd |
|---|---|---|---|---|---|
| 1 | 4c | i-PrOH | 68 | 64:36 | 2 |
| 2 | 4d | i-PrOH | 66 | 87:13 | 5 |
| 3e | 4d | i-PrOH | 55 | 91:9 | 12 |
| 4e | 4d | H2O | 49 | 66:34 | 3 |
| 5e | 4d | TMP | 62 | 85:15 | 5 |
| 6e | 4d | BCA | 52 | 91:9 | 18 |
| 7f | 4d | BCA | 51 | 97:3 | 70 |
Performed with 3x (0.1 mmol), proton sources (0.25–0.5 equiv), and catalyst (1 mol %) in 0.2 mL of toluene.
Conversion determined by GC analysis.
e.r. determined by HPLC.
s = ln[(1 – C)(1 – ee)]/ln[(1 – C)(1 + ee)].
–30 °C.
–60 °C.
Under the optimized reaction conditions, the kinetic resolution of a broad range of enol silanes was realized (Table 4). For example, enol silane 3a was recovered in 50% yield after 11 h with an e.r. of 94:6 corresponding to an s-factor of 122. Similarly, the enantioenriched 4-alkyl enol silanes 3x–3z and 4-ester group substituted (S)-3t were recovered in high yields and enantioselectivities. Moreover, 2-substituted enol silanes 3aa and 3ab also underwent an efficient kinetic resolution and were recovered in remarkably high selectivities, with both an aryl and an alkyl substituent. However, the ketone byproducts 1aa and 1ab did undergo partial racemization (see the Supporting Information, Figures S7 and S8). Importantly, 3-phenyl cyclopentanone derived enol silane 3ac was also recovered with synthetically useful selectivity.
Table 4. Kinetic Resolution of the Racemic Silyl Enol Ethersa.


Performed with 3 (0.2 mmol), 10 (0.5 equiv), and 4d (1 mol %) in 0.4 mL of toluene.
Conversions and yields determined by GC analysis or isolation.
e.r. determined by HPLC.
s = ln[(1 – C)(1 – ee)]/ln[(1 – C)(1 + ee)].
–60 °C.
To gain insight into the reaction mechanism, we first compared the reactions using either triflimide (Tf2NH) or IDPi catalyst 4c in the reaction of 4-phenylcyclohexanone 1a with allyl silane 2a. Remarkably, when we used triflimide as the catalyst, only homoaldol product 11 was obtained in quantitative yield as a mixture of diastereomers.33 Even when we lowered the temperature to −20 °C, only ∼5% of the enol silane was observed and the homoaldol adducts remained the major products. In contrast, IDPi 4c exclusively led to the formation of product 3a in 99% isolated yield and 88:12 e.r. (Scheme 3A). In light of our recent studies,34 we explain this remarkable chemoselectivity by invoking confinement effects: the competing self-aldolization is sterically challenged, as its relatively larger cationic transition state cannot be accommodated by the confined active site of our IDPi catalyst anion. The acidity difference may also play a role.35
Scheme 3. Mechanistic Studies.
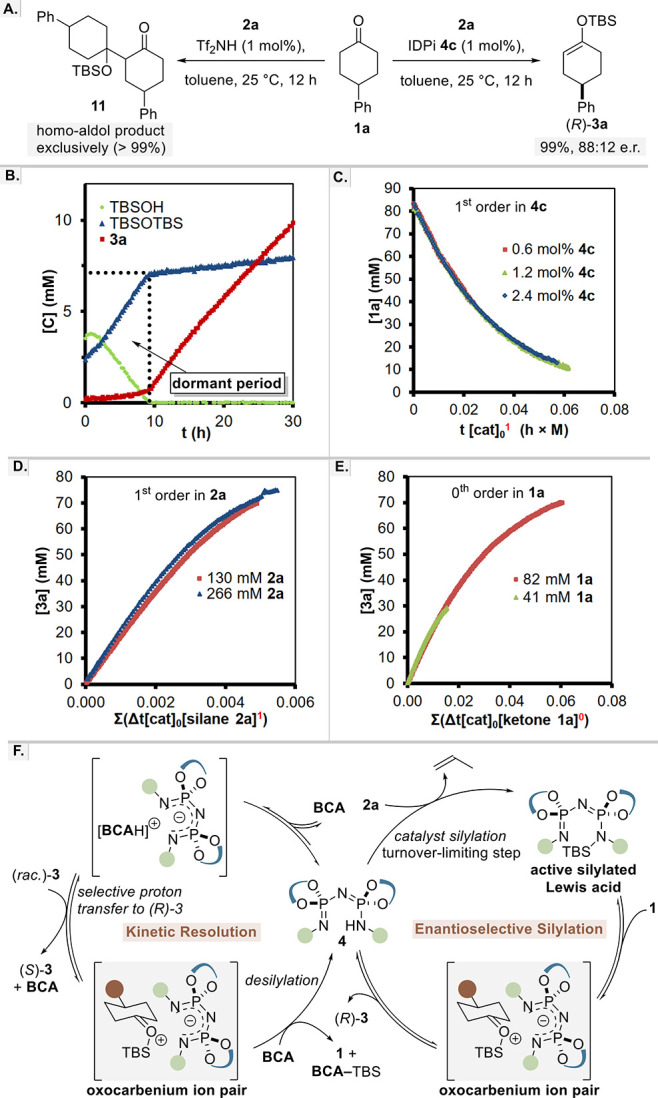
We furthermore investigated the reaction progression of cyclohexanone 1a with 2a by 1H NMR and 31P NMR spectroscopy. When following the reaction under standard conditions in toluene-d8:dioxane-d8 mixture, a significant dormant period, in which adventitious water and TBSOH were first converted to TBS2O, was observed before the enol silane formation began (Scheme 3B). Such dormant periods are quite characteristic for silylium–ACDC reactions and have been described before in detail.36 A shorter dormant period was observed with only toluene as solvent; hence, toluene-d8 was used for all further NMR experiments.
Toward a deeper understanding of the mechanism, we analyzed the reaction by variable time normalization analysis with kinetic data obtained from 1H NMR (Scheme 3C–E). When following the procedures described by the Burés group,37 we found that the overall reaction is first order in catalyst and allyl silane but zeroth order in ketone.38 Alternatively, 31P NMR data acquired at the beginning of the reaction shows the presence of catalyst 4c as a sharp singlet at −16.9 ppm. After approximately 12 h, small amounts of silylated catalyst 4c can be detected. The two doublets at −11.3 and −20.8 ppm with J = 132 Hz indicate that the TBS group is bound preferentially to one site of the activated catalyst. After 24 h, the reaction is complete and the catalyst is almost exclusively present in its silylated form (see the Supporting Information, Figure S9). On the basis of the data obtained, we propose that the initial silylation of the catalyst with silane is the turnover-limiting step of the reaction (Scheme 3F, right). Only if all easily exchangeable protons are consumed, the silylated IDPi catalyst can engage in the reversible activation of the ketone. Interestingly, it is possible that the ketone silylation could already initiate the desymmetrization, by furnishing two different diastereomeric silyloxocarbenium–IDPi ion pairs. Ultimately, it is the final deprotonation step at the α-position of the silyloxocarbenium ion which establishes the enantiopurity of the produced enol silane, while regenerating the free catalyst 4c.
Based on previously reported asymmetric protonations of enol silanes with phenols,22 we speculate that the protodesilylative kinetic resolution is initiated via protonation of the achiral BCA reagent by the IDPi catalyst 4d to form an ion pair [BCAH]+ X*–. In fact, the formation of this species was supported by ESI-MS (see the Supporting Information, Figure S10). As the enantioselectivity depends on the proton source, we propose that it is this complex and not the free IDPi catalyst that engages in the enantioselective protonation of the (R)-enol silane, leaving the corresponding (S)-enantiomer untouched. This step furnishes the same silyloxocarbenium–IDPi ion pair intermediate that is also generated in the corresponding forward reaction. Finally, the catalytic cycle is completed upon desilylation of the oxocarbenium ion by the carboxylic acid BCA, liberating ketone 1 and ester BCA–TBS, while regenerating the IDPi catalyst (Scheme 3F, left).
We report a general asymmetric catalytic methodology for tautomeric σ-bond metathesis reactions between ketones and enol silanes. Our reactions are catalyzed by strong and confined acids and enable both desymmetrizations of achiral cyclic ketones and kinetic resolutions of racemic cyclic enol silanes. Our findings suggest further utility in selective reactions of carbonyl compounds and enol silanes.
Acknowledgments
Generous support from the Max Planck Society, the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), Leibniz Award to B.L. and under Germany’s Excellence Strategy–EXC 2033–390677874–RESOLV, and the European Research Council (ERC, European Union’s Horizon 2020 research and innovation program “C–H Acids for Organic Synthesis, CHAOS” Advanced Grant Agreement No. 694228) is gratefully acknowledged. The authors thank Benjamin Mitschke for his help during the preparation of this manuscript and several members of the group for crowd reviewing. We also thank the technicians of our group and the members of our NMR, MS, and chromatography groups for their excellent service.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c06677.
Experimental details and analytical data for all new compounds (PDF)
The authors declare no competing financial interest.
This paper published ASAP on August 3, 2020 with errors in the TOC graphic and Scheme 3. The errors were corrected and the paper was reposted when the issue published on August 12, 2020.
Supplementary Material
References
- Lalonde M.; Chan T. Use of Organosilicon Reagents as Protective Groups in Organic Synthesis. Synthesis 1985, 1985, 817–845. 10.1055/s-1985-31361. [DOI] [Google Scholar]
- Weber W. P.Chemistry of Trimethylsilyl Cyanide. Silicon Reagents for Organic Synthesis, 1st ed.; Springer: Berlin, Heidelberg, 1983; pp 6–20. [Google Scholar]
- Brownbridge P. Silyl Enol Ethers in Synthesis-Part I. Synthesis 1983, 1983, 1–28. 10.1055/s-1983-30204. [DOI] [Google Scholar]
- Brownbridge P. Silyl Enol Ethers in Synthesis-Part II. Synthesis 1983, 1983, 85–104. 10.1055/s-1983-30234. [DOI] [Google Scholar]
- Smietana M.; Mioskowski C. Preparation of Silyl Enol Ethers Using (bistrimethylsilyl) Acetamide in Ionic Liquids. Org. Lett. 2001, 3, 1037–1039. 10.1021/ol015602h. [DOI] [PubMed] [Google Scholar]
- Song J. J.; Tan Z.; Reeves J. T.; Fandrick D. R.; Yee N. K.; Senanayake C. H. N-Heterocyclic Carbene-Catalyzed Silyl Enol Ether Formation. Org. Lett. 2008, 10, 877–880. 10.1021/ol703025b. [DOI] [PubMed] [Google Scholar]
- Villas-Bôas S. G.; Smart K. F.; Sivakumaran S.; Lane G. A. Alkylation or Silylation for Analysis of Amino and Non-Amino Organic Acids by GC-MS. Metabolites 2011, 1, 3–20. 10.3390/metabo1010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Rodrigo J.; Hoveyda A. H.; Snapper M. L. Enantioselective Silyl Protection of Alcohols Catalysed by an Amino-Acid-Based Small Molecule. Nature 2006, 443, 67–70. 10.1038/nature05102. [DOI] [PubMed] [Google Scholar]
- Hyodo K.; Gandhi S.; van Gemmeren M.; List B. Brønsted Acid Catalyzed Asymmetric Silylation of Alcohols. Synlett 2015, 26, 1093–1095. 10.1055/s-0034-1380409. [DOI] [Google Scholar]
- Park S. Y.; Lee J.-W.; Song C. E. Parts-Per-Million Level Loading Organocatalysed Enantioselective Silylation of Alcohols. Nat. Commun. 2015, 6, 7512. 10.1038/ncomms8512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T.; Sato D.; Kiyoto T.; Kumar A.; Koga K. Stereoselective Reactions. 29. Lithium-Hydrogen Interchange between Achiral Tridentate Lithium Amides and Chiral Bidentate Amines. An Approach to Catalytic Enantioselective Deprotonation. Tetrahedron 1997, 53, 16987–16998. 10.1016/S0040-4020(97)10144-2. [DOI] [Google Scholar]
- Claraz A.; Oudeyer S.; Levacher V. J. T. A. Enantioselective Desymmetrization of Prochiral Ketones via an Organocatalytic Deprotonation Process. Tetrahedron: Asymmetry 2013, 24, 764–768. 10.1016/j.tetasy.2013.05.011. [DOI] [Google Scholar]
- Simpkins N. S. Recent Advances in Asymmetric Synthesis Using Chiral Lithium Amide Bases. Pure Appl. Chem. 1996, 68, 691–694. 10.1351/pac199668030691. [DOI] [Google Scholar]
- Kurahashi K.; Yamaoka Y.; Takemoto Y.; Takasu K. Silyl Enol Etherification by a Tf2NH/Amine Co-Catalytic System for Minimizing Hazardous Waste Generation. React. Chem. Eng. 2018, 3, 626–630. 10.1039/C8RE00039E. [DOI] [Google Scholar]
- Bae H. Y.; Höfler D.; Kaib P. S.; Kasaplar P.; De C. K.; Döhring A.; Lee S.; Kaupmees K.; Leito I.; List B. Approaching Sub-PPM-Level Asymmetric Organocatalysis of a Highly Challenging and Scalable Carbon-Carbon Bond Forming Reaction. Nat. Chem. 2018, 10, 888–894. 10.1038/s41557-018-0065-0. [DOI] [PubMed] [Google Scholar]
- Kaib P. S.; Schreyer L.; Lee S.; Properzi R.; List B. Extremely Active Organocatalysts Enable a Highly Enantioselective Addition of Allyltrimethylsilane to Aldehydes. Angew. Chem., Int. Ed. 2016, 55, 13200–13203. 10.1002/anie.201607828. [DOI] [PubMed] [Google Scholar]
- Schreyer L.; Properzi R.; List B. IDPi Catalysis. Angew. Chem., Int. Ed. 2019, 58, 12761–12777. 10.1002/anie.201900932. [DOI] [PubMed] [Google Scholar]
- Ishihara K.; Nakamura S.; Kaneeda M.; Yamamoto H. First Example of a Highly Enantioselective Catalytic Protonation of Silyl Enol Ethers Using a Novel Lewis Acid-Assisted Brønsted Acid System. J. Am. Chem. Soc. 1996, 118, 12854–12855. 10.1021/ja962414r. [DOI] [Google Scholar]
- Cheon C. H.; Kanno O.; Toste F. D. Chiral Brønsted Acid from a Cationic Gold (I) Complex: Catalytic Enantioselective Protonation of Silyl Enol Ethers of Ketones. J. Am. Chem. Soc. 2011, 133, 13248–13251. 10.1021/ja204331w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa A.; Touge T.; Arai T. Enantioselective Protonation of Silyl Enolates Catalyzed by a Binap AgF Complex. Angew. Chem., Int. Ed. 2005, 44, 1546–1548. 10.1002/anie.200462325. [DOI] [PubMed] [Google Scholar]
- Beck E. M.; Hyde A. M.; Jacobsen E. N. Chiral Sulfinamide/Achiral Sulfonic Acid Cocatalyzed Enantioselective Protonation of Enol Silanes. Org. Lett. 2011, 13, 4260–4263. 10.1021/ol201608a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheon C. H.; Yamamoto H. A Brønsted Acid Catalyst for the Enantioselective Protonation Reaction. J. Am. Chem. Soc. 2008, 130, 9246–9247. 10.1021/ja8041542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolman R. C. The Principle of Microscopic Reversibility. Proc. Natl. Acad. Sci. U. S. A. 1925, 11, 436. 10.1073/pnas.11.7.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G.; Shabat D.; List B.; Anderson J.; Sinha S. C.; Lerner R. A.; Barbas C. F. III Catalytic Enantioselective Retro-Aldol Reactions: Kinetic Resolution of β-Hydroxyketones with Aldolase Antibodies. Angew. Chem., Int. Ed. 1998, 37, 2481–2484. . [DOI] [PubMed] [Google Scholar]
- Mitachi K.; Shimizu T.; Miyashita M.; Tanino K. Cyclooctanone Synthesis via a Formal [6 + 2] Cycloaddition Reaction of a Dicobalt Acetylene Complex. Tetrahedron Lett. 2010, 51, 3983–3986. 10.1016/j.tetlet.2010.05.120. [DOI] [Google Scholar]
- Gao R.; Yi C. S. Catalytic Formation of Silyl Enol Ethers and Its Applications for Aldol-Type Condensation and Aminomethylation Reactions. ACS Catal. 2011, 1, 544–547. 10.1021/cs200087c. [DOI] [Google Scholar]
- Kato M.; Watanabe M.; Tooyama Y.; Vogler B.; Yoshikoshi A. Efficient and Convenient Syntheses of (R)-(−)-Cryptone and (S)-(−)-4-Isopropenyl-2-cyclo. Synthesis 1992, 1992, 1055–1057. 10.1055/s-1992-26297. [DOI] [Google Scholar]
- Inanaga K.; Ogawa Y.; Nagamoto Y.; Daigaku A.; Tokuyama H.; Takemoto Y.; Takasu K. Facile Isomerization of Silyl Enol Ethers Catalyzed by Triflic Imide and Its Application to One-Pot Isomerization-(2+ 2) Cycloaddition. Beilstein J. Org. Chem. 2012, 8, 658–661. 10.3762/bjoc.8.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kad G.; Singh V.; Khurana A.; Chaudhary S.; Singh J. Regioselective α-Alkylation of Silyl Enolates Using a Mild Catalyst-ZnCl2 Doped on Acidic Alumina. Synth. Commun. 1999, 29, 3439–3442. 10.1080/00397919908085973. [DOI] [Google Scholar]
- Chandrasekhar S.; Sridhar C.; Srihari P. J. T. A. A Carbohydrate Approach for the Formal Total Synthesis of the Prostacyclin Analogue (16S)-Iloprost. Tetrahedron: Asymmetry 2012, 23, 388–394. 10.1016/j.tetasy.2012.03.005. [DOI] [Google Scholar]
- Kagan H.; Fiaud J.. Topics in Stereochemistry, 1st ed.; Wiley: New York, 1988. [Google Scholar]
- Larrow J. F.; Schaus S. E.; Jacobsen E. N. Kinetic Resolution of Terminal Epoxides via Highly Regioselective and Enantioselective Ring Opening with TMSN3. An Efficient, Catalytic Route to 1, 2-Amino Alcohols. J. Am. Chem. Soc. 1996, 118, 7420–7421. 10.1021/ja961708+. [DOI] [Google Scholar]
- Bae H. Y.; List B. Triflimide: An Overlooked High-Performance Catalyst of the Mukaiyama Aldol Reaction of Silyl Ketene Acetals with Ketones. Chem. - Eur. J. 2018, 24, 13767–13772. 10.1002/chem.201803142. [DOI] [PubMed] [Google Scholar]
- Schreyer L.; Kaib P. S.; Wakchaure V. N.; Obradors C.; Properzi R.; Lee S.; List B. Confined Acids Catalyze Asymmetric Single Aldolizations of Acetaldehyde Enolates. Science 2018, 362, 216–219. 10.1126/science.aau0817. [DOI] [PubMed] [Google Scholar]
- Höfler D.; Goddard R.; Nöthling N.; List B. A Dendralenic C-H Acid. Synlett 2019, 30, 433–436. 10.1055/s-0037-1612246. [DOI] [Google Scholar]
- Zhang Z.; Bae H. Y.; Guin J.; Rabalakos C.; van Gemmeren M.; Leutzsch M.; Klussmann M.; List B. Asymmetric Counteranion-Directed Lewis Acid Organocatalysis for the Scalable Cyanosilylation of Aldehydes. Nat. Commun. 2016, 7, 12478. 10.1038/ncomms12478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burés J. Variable time normalization analysis: General Graphical Elucidation of Reaction Orders from Concentration Profiles. Angew. Chem., Int. Ed. 2016, 55, 16084–16087. 10.1002/anie.201609757. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Klussmann M.; List B.. Kinetic Study of Disulfonimide Catalyzed Cyanosilylation of Aldehyde Using a Method of Progress Rates. Synlett [Online early access]. 10.1055/s-0040-1707129. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.