

Abstract
Glioblastoma multiforme (GBM) is a highly aggressive and malignant brain tumor that is refractory to existing therapeutic regimens, which reflects the presence of stem-like cells, termed glioma-initiating cells (GICs). The complex interactions between different signaling pathways and epigenetic regulation of key genes may be critical in the maintaining GICs in their stem-like state. Although several signaling pathways have been identified as being dysregulated in GBM, the prognosis of GBM patients remains miserable despite improvements in targeted therapies. In this report, we identified that BRG1, the catalytic subunit of the SWI/SNF chromatin remodeling complex, plays a fundamental role in maintaining GICs in their stem-like state. In addition, we identified a novel mechanism by which BRG1 regulates glycolysis genes critical for GICs. BRG1 downregulates the expression of TXNIP, a negative regulator of glycolysis. BRG1 knockdown also triggered the STAT3 pathway, which led to TXNIP activation. We further identified that TXNIP is an STAT3-regulated gene. Moreover, BRG1 suppressed the expression of interferon-stimulated genes, which are negatively regulated by STAT3 and regulate tumorigenesis. We further demonstrate that BRG1 plays a critical role in the drug resistance of GICs and in GIC-induced tumorigenesis. By genetic and pharmacological means, we found that inhibiting BRG1 can sensitize GICs to chemotherapeutic drugs, temozolomide and carmustine. Our studies suggest that BRG1 may be a novel therapeutic target in GBM. The identification of the critical role that BRG1 plays in GIC stemness and chemosensitivity will inform the development of better targeted therapies in GBM and possibly other cancers.
Keywords: BRG1, STAT3, Glioblastoma, Gene expression, Tumorigenesis
Introduction
Glioblastoma multiforme (GBM) is a highly aggressive and malignant brain tumor that is refractory to current therapeutic regimens. Despite advanced treatment strategies and improved molecular characterization, the prognosis for GBM patients continues to be dismal and the mean patient survival remains <2 years. A population of glioma-initiating cells (GICs) has been identified in GBM tumors, which can self-renew and give rise to the heterogeneous mixture of cells in the developing tumor. GICs possess potent tumor-initiating capacity and are considered to be responsible for the drug resistance associated with GBM [1]. The maintenance of GICs in a stem-like state requires the precise interplay of signaling pathways and transcriptional networks, which underscores the possible involvement of epigenetic factors in maintaining stem cell-like identity of GICs. Here we describe the role of Brahma-related gene 1 (BRG1), one of the catalytic subunits of the SWI/SNF chromatin remodeling complex, in the maintenance of stem cell identity and tumorigenic potential of GICs.
The SWI/SNF complex is an evolutionary conserved multi-subunit complex that is critical for gene regulation, differentiation, DNA repair, and development. The two catalytic subunits, BRM (also called BRAMA, SMARCA2) and BRG1 (also called BRAHMA RELATED GENE 1, SMARCA4), provide energy from ATP hydrolysis required to reposition and/or remodel nucleosomes at targeted loci to open or close chromatin and regulate transcription [2, 3]. The importance of BRG1 in embryonic development was demonstrated by the finding that BRG1 knockout in mice was embryonically lethal at the peri-implantation stage [4], and BRG1-negative embryonic stem (ES) cells have not yet been isolated. BRG1 promotes pluripotency of both mouse and human ES cells, and targeted knockdown of BRG1 induces cell differentiation [4,5]. In ES cells, the SWI/SNF or BAF complex has a unique composition (esBAF), and expression of the esBAF components in fibroblasts causes a reversal to a pluripotent state [5, 6]. Furthermore, the SWI/SNF complex not only enhances the efficiency of ESC pluripotency induced by the forced expression of Oct4, Sox2, KLF4, and c-Myc [7], but also can replace c-Myc in this pathway [7].
Interestingly, BRG1 is the sole catalytic subunit in the esBAF complex [8–10]. BRG1 binds to the promoters of various pluripotency specific genes such as Oct4, Nanog, and the polycomb group (PcG) of genes [5]. BRG1 has been found to have tumor suppressing [11,12] and tumor promoting activity [13,14] in a highly cancer-context-specific manner. BRG1 has been identified as a major tumor suppressor with silencing or loss-of-function mutations enriched in cancers such as lung, ovaries, skin, thoracic sarcoma, and lymphoma [12,15–18]. However, BRG1 mutations are relatively rare in GBM [19], and BRG1 appears to be overexpressed in glioma as compared to adjacent normal brain tissue [20]. Interestingly, high BRG1 expression was found to be associated with poor prognosis in many types of tumors and more aggressive tumors in several types of cancer using data from The Cancer Genome Atlas (TCGA) and other databases [21]. Although other BAF components have been found to regulate GIC stemness [22], the precise role of BRG1 in GICs remains poorly understood. In this article, we demonstrate that the BRG1 subunit of the human SWI/SNF complex is essential for maintaining the stem-like identity of GICs. Through targeted microarrays, we uncovered a novel mechanism by which BRG1 regulates the expression of glycolysis and glucose transport genes in GICs by attenuating the expression of TXNIP, a negative regulator of glycolysis [23,24]. We also determined that BRG1 knockdown led to upregulation of the STAT3 signaling pathway and the subsequent downregulation of interferon-stimulated genes (ISGs). Moreover, we demonstrate that TXNIP is an STAT3-regulated gene. Inhibiting BRG1 by genetic and pharmacologic means promotes GIC differentiation and chemosensitizes GICs to temozolomide (TMZ) and carmustine. Our studies suggest that BRG1 may be a novel, druggable therapeutic target in GBM.
Materials and Methods
Cell Culture
GBM6, GBMX10, and GBMX16 patient-derived xenolines (PDXs) were maintained as xenografts in immunocompromised mice [25]. GICs were isolated and maintained in flasks precoated with poly-d-lysine and laminin, and grown in NeuroBasal-A medium (Invitrogen, Carlsbad, CA) containing 2% B27 supplement, 2 mM l-glutamine, 100 units/ml penicillin, 100 g/ml streptomycin, EGF (20 ng/ml), and basic FGF (40 ng/ml).
Knockdown of BRG1 and TXNIP
pLKO.1 plasmids containing gene-specific or scrambled shRNA (control) were used to knockdown (KD) BRG1 or TXNIP expression (Open Biosystems, Thermo Scientific, Lafayette, CO). Lentivirus was produced by packaging in 293FT cells [26]. BRG1-KD or TXNIP-KD GICs were generated by transduction with shRNA-encoding lentivirus, selected with 5 μg/ml puromycin, and stable pools maintained without puromycin.
Inducible STAT3 Knockdown (iSTAT3-KD) and Restoration of STAT3 Expression
The doxycycline-inducible STAT3 knockdown system was used as described previously [27].
Cell Proliferation Assays
GICs were seeded in 96-well plates (4,000 cells/well) and the number of viable GICs was measured using the CellTiter-Glo luminescence viability assay (Promega, Madison, WI). TMZ was purchased from Sigma (St Louis, MO), carmustine from Toronto Research Chemicals (Toronto, Canada), and PFI-3 from Tocris (Minneapolis, MN).
Gene Expression Analysis
Total RNA was extracted using the QIAshredder and RNeasy mini kits (Qiagen Inc., Frederick, MD). Gene expression was determined by quantitative real-time PCR (qPCR) using gene-specific primers (Supporting Information, Table 1) and an iScript one-step RT-PCR kit with SYBR Green (Bio-Rad). Reaction parameters were as follows: cDNA synthesis at 50°C for 20 minutes, transcriptase inactivation at 95°C for 5 minutes, and PCR cycling at 95°C for 10 seconds and 60 °C for 30 seconds for 40 cycles.
Immunoblotting
Protein extracts (50 μg) were prepared from GICs in urea-based lysis buffer as previously described [28,29], separated by SDS-PAGE, transferred to polyvinylidene difluoride membranes (Millipore, Burlington, MA), and immunoblotted with the following antibodies: BRG1 and BRM (Proteintech, Rosemont, IL), TXNIP (Abcam, Cambridge, MA), STAT3 (BD Biosciences, San Jose, CA), pY705-STAT3 and GFAP (Abcam), CDK1, CDK2, Rb, Rb 107, Rb 110 (Cell Signaling, Danvers, MA), and Actin (Santa Cruz, Dallas, TX). Following addition of IRDye800CW goat anti-mouse IgG or IRDye680 goat anti-rabbit IgG, blots were visualized on an Odyssey infrared imaging system (LICOR Biosciences, Lincoln, NE).
Immunohistochemistry
Cells were grown in 8-well chamber slides (Millipore, Burlington, MA) precoated with poly-d-lysine and laminin to ~50% confluency and fixed with 4% paraformaldehyde and permeabilized with Triton X-100. After blocking with 5% goat serum, cells were incubated with antibodies against GFAP (Molecular Probes, Carlsbad, CA), S100B (Abcam, Cambridge, MA), and Olig2 (Abcam, Cambridge, MA) and subsequently stained with Alexa Fluor 488 or 594 (Molecular Probes, Carlsbad, CA). Images were captured on a laser-scanning confocal microscope (Zeiss model LSM700, San Diego, CA).
Tumor Xenografts in Mice
All animal experiments were performed with at least 5 mice in each experimental group in accordance with a protocol approved by the Institutional Animal Care and Use Committee of the University of Tennessee Health Science Center. GICs were dissociated with HyQTase, resuspended in PBS, and enumerated on a cellometer (Nexelcom, Lawrence, MA). Heterotopic tumor xenografts were established in 5-week-old male NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratory, Bar Harbor, ME) by injecting control and BRG1-KD GICs expressing luciferase (106 cells/100 μl PBS) into the flanks. Tumor burden was assessed weekly by palpation with slide caliper measurement, and live animal imaging following luciferin injection and bioluminescence was analyzed with Living Image software (IVIS, Perkin Elmer, Waltham, MA) [30]. After confirming tumor initiation, TMZ was administered at 10 mg/kg of mice on alternate days by intraperitoneal (IP) administration. For orthotopic xenograft studies, luciferase-expressing GICs (5 × 105 cells in 100 μl PBS) were injected stereotactically into the superficial brain parenchyma through a burr hole in the skull, and tumor burden was assessed as previously described [31]. Tumor growth was analyzed using Graphpad Prism 7 software (La Jolla, CA). At 3 weeks following injection, animals were sacrificed.
Chromatin Immunoprecipitation (ChIP)
ChIP analysis was carried out using the ChIP-IT™ Express Enzymatic kit (Active Motif, Carlsbad, CA) according to the manufacturer’s instructions. In brief, chromatin was cross-linked with 1% formaldehyde (10 minutes at 22°C), sheared to an average size of ~200 bp by sonication (Bioruptor Pico, Diagenode, Denville, NJ), and immunoprecipitated with anti-STAT3 (Santa Cruz Biotechnology). The ChIP-PCR primers were designed to amplify a proximal promoter region containing putative binding sites on the TXNIP promoter using ExPASy and the JASPAR database.
Nanostring Gene Expression Analysis
Total RNA from X16 and the X16 BRG1-KD cells was extracted using the QIAshredder and RNeasy mini kits (Qiagen Inc., Frederick, MD). Nanostring arrays were conducted with the PanCancer Immune Panel on the nCounter Analysis system (NanoString, Seattle, WA), and the data (GEO accession number GSE116545) were analyzed with nSolver software using a twofold cutoff value as previously described [32].
Data Analysis
At least three independent experiments were performed in triplicate, and data are presented as mean ± SEM. Analysis of variance or Student’s t tests were performed and p ≤ 0.05 was considered to be statistically significant.
Results
BRG1 Subunit of the SWI/SNF Complex is Expressed in GICs
We assessed the expression of the catalytic subunits of the SWI/SNF complex in a panel of GICs obtained from independent GBM tumors (GBM6, X10, and X16), which all have high tumor-initiating activity [27]. Immunoblotting with a BRG1-specific antibody showed that BRG1 was robustly expressed in all three GICs (Fig. 1A). In contrast, BRM was detected in both GBM6 and X16 GICs, but not in X10 GICs (Fig. 1A). The expression of BRG1 in all the three GICs indicates that BRG1, and not BRM, may be an essential component for GIC function. This finding is consistent with BRG1 but not BRM being essential for the maintenance of pluripotency in hESCs and mESCs [4,5], and that BRG1 is the only catalytic component found in the embryonic stem cells (esBAF) SWI/SNF complex [4,5,9].
Figure 1.
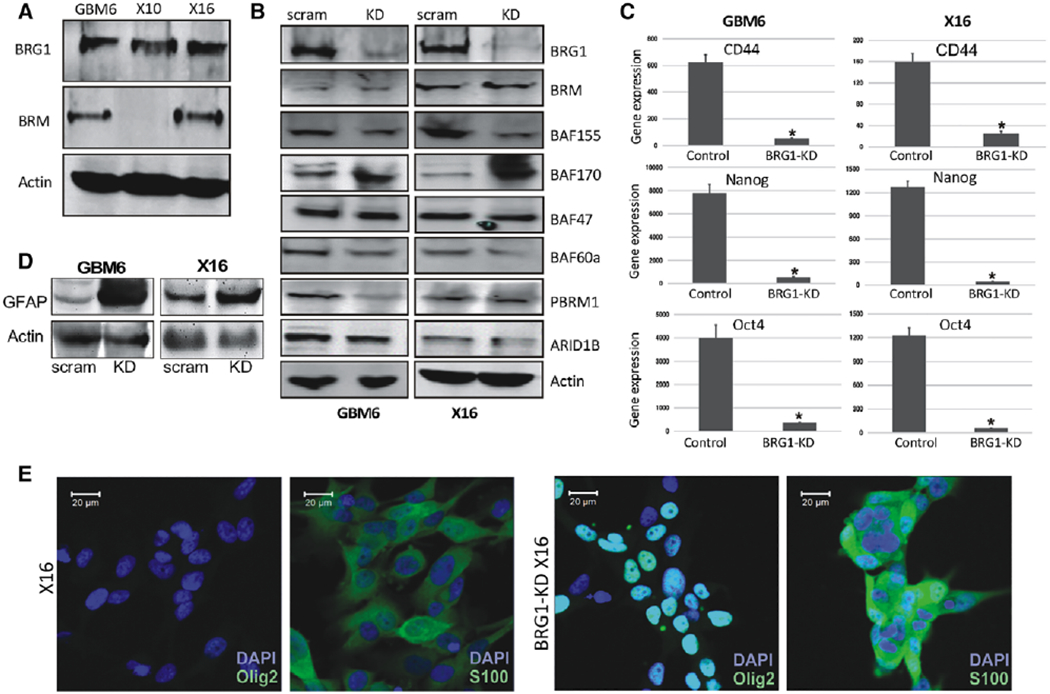
Effect of BRG1-KD in GICs on BAF components and stem cell markers. (A): Protein lysates from GBM6, X10, and X16 GICs were immunoblotted for BRG1 and BRM. (B): Protein lysates from BRG1-KD GBM6 and X16 GICs were immunoblotted for core and accessory subunits of the BAF complex. The quantitation of the signals in GBM6 and X16 GICs versus BRG1-KD cells relative to actin, respectively, were BRG1 (0.1, 0.1); BAF155 (0.6, 0.5); BAF170 (1.7, 2.2); BAF47 (1.1, 1); BAF60A (0.9, 0.8); PBRM1 (0.6, 1); ARID1B (0.9, 1). (C): RNA was prepared from control and BRG1-KD GBM6 and X16 GICs, and CD44, Nanog and Oct4 expression was determined by qPCR. (D): Protein lysates from GBM6 and X16 GICs were immunoblotted for GFAP. (E): Immunohistochemistry for Olig2, S100B in X16 and X16 BRG1-KD GICs. A scale bar of 20 pm is provided for reference. *p ≤ .05 was considered to be statistically significant.
BRG1 is Critical for Maintaining GIC Sternness
To elucidate the role of BRG1 in GBM6 and X16 GICs, we then generated BRG1-KD GICs by transducing GICs with lentivirus encoding scrambled or BRG1-shRNA RNAs. After puromycin selection, stable pools of GICs were maintained without puromycin. Immunoblotting showed decreased BRG1 expression in BRG1-KD cells, but BRM levels were unaffected (Fig. 1B). In contrast, BRG1 and BRM levels were not reduced in scrambled-shRNA treated cells (Fig. 1B). We then examined the effect of BRG1-KD on expression of the esBAF components in GICs, and found that BRG1-KD had subtle effects on some components of the esBAF complex in a GIC-specific manner (Fig. 1B), such as attenuating PBRM1 levels in GBM6 (40% decrease) but not in X16 GICs, and attenuating BAF155 levels in both GBM6 and X16 GICs (40%–50% decrease). Since GICs express a number of stem cell markers (CD44, OCT4, and Nanog), we also determined the effect of BRG1-KD on the expression of these marker genes by qPCR and found that BRG1-KD significantly attenuated their expression in both GBM6 and X16 GICs (Fig. 1C). Since we found that GBM6 and X16 GICs can be induced to differentiate into cells of the astrocytic lineage [33], we evaluated the expression of astrocytic marker, Glial fibrillary acidic protein (GFAP), following BRG1-KD by immunoblotting. BRG1-KD in both GBM6 and X16 GICs significantly upregulated GFAP levels (Fig. 1D). In addition, BRG1-KD in GICs also increased expression of astrocytic marker S100B and oligodendroglial marker Olig2, as determined by immunohistochemistry (Fig. 1E). Taken together, these data strongly suggest BRG1 is critical for maintaining GICs in a stem-like, undifferentiated state.
BRG1-KD GICs Show Accelerated Cellular Proliferation in vitro and Produce Larger Intracranial Tumors
BRG1 plays an important role in cell cycle regulation and proliferation of hESCs [4,6]. We next examined the role of BRG1 on GIC proliferation by CellTiter-Glo assay, which quantifies the number of viable cells. BRG1-KD GBM6 and X16 GICs showed a marked increase in cell proliferation (more than twofold increase in cell number after 7 days) when compared to control GICs (Fig. 2A). The increased proliferation of BRG1-KD GICs was consistent with BRG1 maintaining GICs in a more quiescent dormant state; hence, BRG1-KD cells were more actively dividing. Previous studies show that BRG1 binds directly to the retinoblastoma (RB) protein to inhibit cell proliferation [34], and that BRG1 loss is associated with the inactivation of the function of the RB family members, p107 and p130, through their hyperphosphorylation. To elucidate the underlying mechanism by which BRG1 regulates GIC growth, we next analyzed the phosphorylation state of Rb1, p107, and p130. We observed increased p107 and p130 phosphorylation in the BRG1-KD GICs (Fig. 2B), but no change in Rb1 phosphorylation (data not shown). Additionally, Cyclin A2 expression increased in BRG1-KD GBM6 and X16 GICs, while CDK1 levels remained unchanged (Fig. 2C). Taken together these results suggest that BRG1 restrained GIC proliferation.
Figure 2.
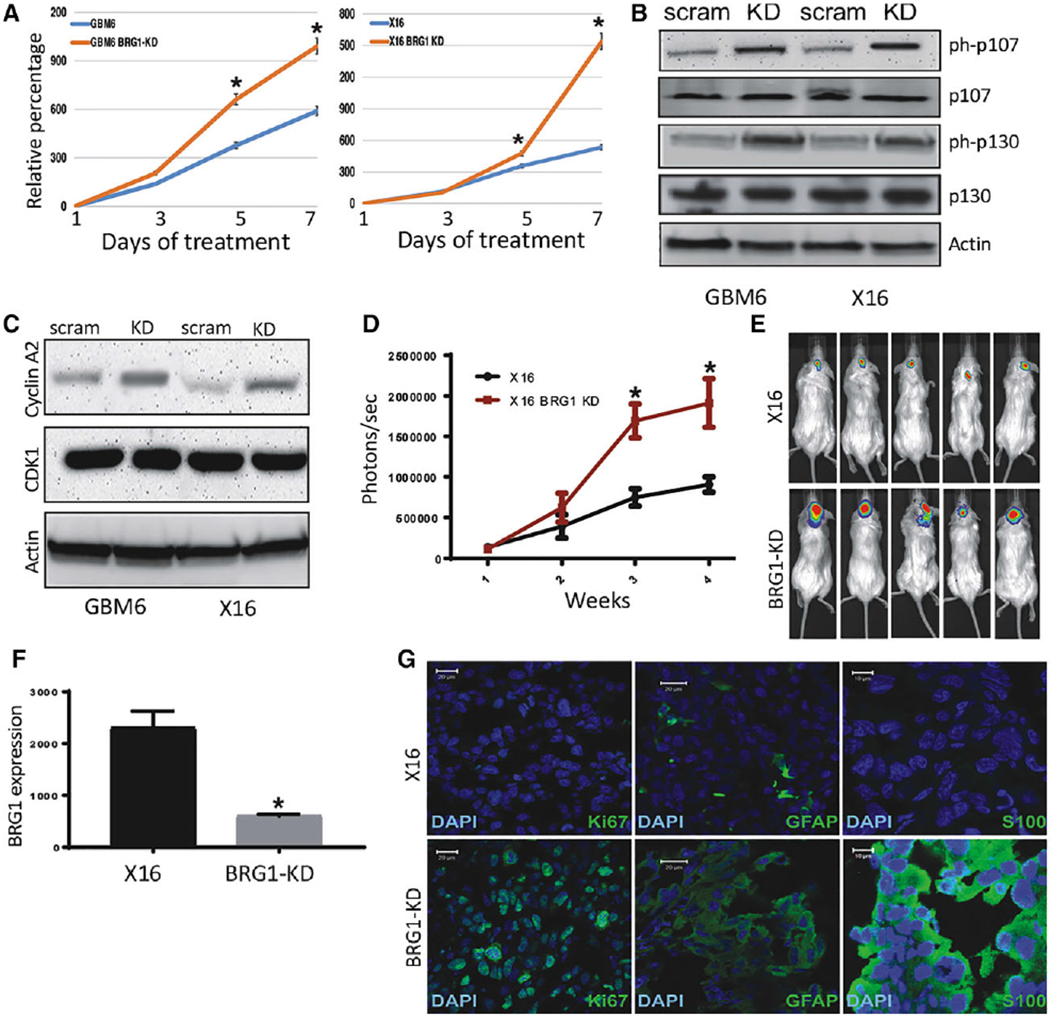
Effect of BRG1-KD on proliferation in vitro and tumorigenicity of GBM6 and X16 GICs. (A): Proliferation of BRG1-KD GBM6 and X16 GICs was determined by CellTiter-Glo assays. (B,C): Protein lysates from control and BRG1-KD X16 GICs were immunoblotted for (B) phosphorylated and total Rb protein family members, and (C) Cyclin A2 and CDK1. (D-G): Tumorigenicity was assessed by injection of 106 tumor cells into the brains of NSG mice and live animal imaging was performed at weekly intervals. (D): Quantification of the bioluminescence signal detected at 1, 2, and 3 weeks postinjection. (E): Representative bioluminescent images of mice at 21 days postinjection. (F): RNA was prepared from control and BRG1-KD X16 GIC-induced tumors and BRG1 gene expression was determined by qPCR. (G): Immunohistochemistry for GFAP, S100B, and Ki67 in X16 and X16-BRG1 KD tumor tissue. A scale bar of 20 μm is provided for reference. *p ≤ .05 was considered to be statistically significant.
We next determined the biological role of BRG1 in vivo by examining the tumorigenic potential of BRG1-KD and control GICs by intracranial injection of luciferase-labeled cells into immunocompromised NSG mice. Tumor progression was determined at weekly intervals by live animal imaging after luciferin injection. BRG1-KD GICs produced tumors more rapidly (Fig. 2D) and the tumors were larger at the end-point of the study (Fig. 2E) as determined by bioluminescence. In addition, at necropsy BRG1 expression in BRG1-KD tumors was verified to be reduced in the tumor tissue by qPCR (Fig. 2F). Since BRG1-KD resulted in GFAP and S100B upregulation in vitro (Fig. 1D, 1E), we also evaluated GFAP and S100B expression in tumor tissue by immunohistochemistry. BRG1-KD X16 GIC-induced tumors had markedly higher GFAP and S100B protein expression as compared to the EV tumors, and Ki67 (a marker for proliferation) staining was also much higher in BRG1-KD tumors (Fig. 2G). These results suggest that BRG1 restrains GIC differentiation and proliferation in vivo and in vitro.
BRG1 Regulates an STAT3/TXNIP Pathway
To assess the role of BRG1 on cancer-related gene pathways in GICs, we conducted targeted microarrays on X16 BRG1-KD GICs and scrambled-shRNA transduced GICs serving as a control. Interestingly, as illustrated in the heat maps (Fig. 3A), multiple IFN-simulated genes (ISGs) were downregulated in BRG1-KD GICs, including CXCL11, IRF7, and IL6, whose differential expression were validated by qPCR (Supporting Information, Fig. S1). We recently demonstrated that these ISGs were negatively regulated by STAT3 in GICs [27], suggesting that BRG1 might regulate a STAT3-dependent pathway. A functional link between STAT3 signaling and BRG1/SMARCA4 is further supported by protein–protein interaction (PPI) analysis of the BRG1-regulated genes (Supporting Information, Fig. S2), as well as by previous studies [35–37]. BRG1-KD resulted in increased expression of LIF (STAT3 pathway activator [42]) and decreased SOCS1 (STAT3 pathway repressor [38,39]) in GICs, which was validated by qPCR (Fig. 3A and Supporting Information, Fig. S1). Thus, these results provide further evidence that BRG1 regulates a STAT3-dependent pathway. In addition, TXNIP (Thioredoxin-interacting protein) expression was markedly upregulated in BRG1-KD GICs (Fig. 3A and Supporting Information, Fig. S1). TXNIP is involved in redox regulation and acts as a negative regulator of glycolysis [23,40–43]. To assess whether BRG1 regulated a STAT3 pathway leading to TXNIP upregulation, lysates from control (scrambled shRNA), and BRG1-KD GBM6 and X16 GICs were prepared and immunoblotted. As shown in Figure 3B, BRG1 levels were markedly reduced in BRG1-KD GBM6 and X16 GICs, while levels of pY705-STAT3 and TXNIP were increased. Since our data indicate that BRG1 alters TXNIP expression which regulates cellular glucose uptake [44–46], we then examined the effect of BRG1-KD on several genes encoding enzymes involved in glucose transport and glycolysis (GLUT1, FBP2, and PKM) and found BRG1-KD resulted in a significant downregulation of these genes (Fig. 3C). To determine whether TXNIP directly regulated the expression of these genes in GICs, we knocked down TXNIP expression in the BRG1-KD X16 GICs (Fig. 3D), and found that it restored their expression (Fig. 3E). Taken together, our data suggest that BRG1 may maintain glucose availability in GICs by downregulating TXNIP expression.
Figure 3.
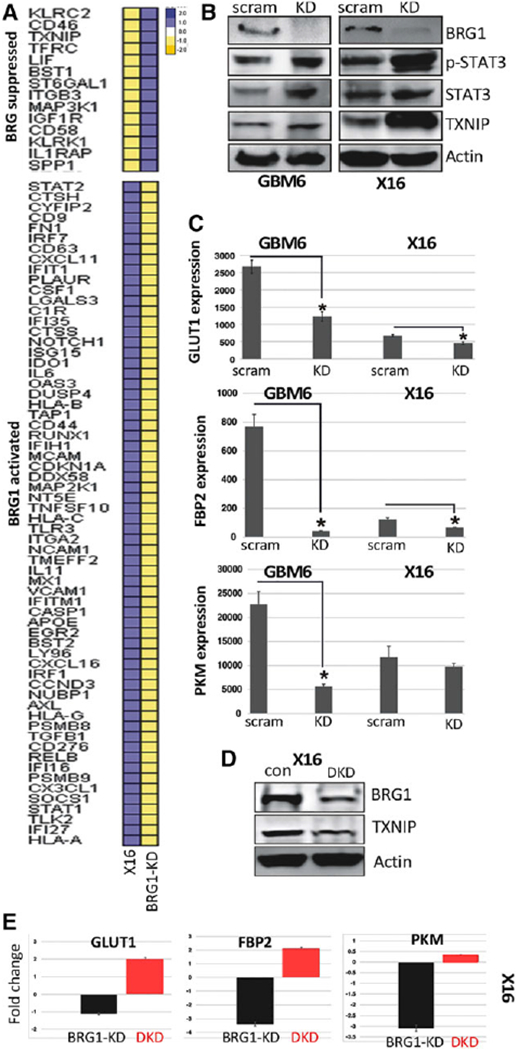
Effect of BRG1-KD on gene and protein expression. (A): Total RNA was prepared from X16 and BRG1-KD GICs, and gene expression profiling was conducted on the nCounter Analysis System using the PanCancer Immune Panels, and heat maps of BRG1 suppressed and activated genes are shown. The scale reflects the levels of relative gene expression. (B): Protein lysates from control and BRG1-KD X16 GICs were immunoblotted for BRG1, STAT3, pTyr705-STAT3, and TXNIP. (C): RNA was prepared from control and BRG1-KD GICs, and expression of GLUT1, FBP2, and PKM was determined by qPCR. (D): Protein lysates from control and BRG1-KD X16 GICs were immunoblotted for BRG1, TXNIP, and actin. (E): RNA was prepared from control X16 GICs, BRG1-KD X16 GICs, and X16 GICs with both BRG1 and TXNIP (DKD) knocked down, and GLUT1, FBP2, and PKM was determined by qPCR. Data were expressed relative to gene expression in control X16 GICs. *p ≤ .05 was considered to be statistically significant.
To directly determine the role of STAT3 on TXNIP expression in GICs, X16 and STAT3-KD GICs were examined for TXNIP expression. STAT3-KD reduced TXNIP gene (Fig. 4A) and protein expression (Fig. 4B) in the STAT3-KD GICs as compared to control GICs. Most importantly, rescue of STAT3 expression in STAT3-KD GICs restored TXNIP expression, indicating that STAT3 regulates TXNIP expression in GICs (Fig. 4A, 4B). Similar results were also observed in the X10 GICs (Supporting Information, Fig. S3). We then identified three putative STAT3 binding sites on the TXNIP promoter, and by chromatin immunoprecipitation (ChIP) assays with the GBM6 and X16 GICs found that STAT3 binds to the TXNIP promoter at these sites in the BRG1-KD GICs (Fig. 4C, 4D). BRG1 was previously reported to be essential in STAT3-mediated gene transcription [5,35,47], and our results suggest TXNIP expression in GICs is STAT3-dependent and lies downstream of BRG1, providing a mechanism of how BRG1 negatively regulates TXNIP expression in GICs.
Figure 4.
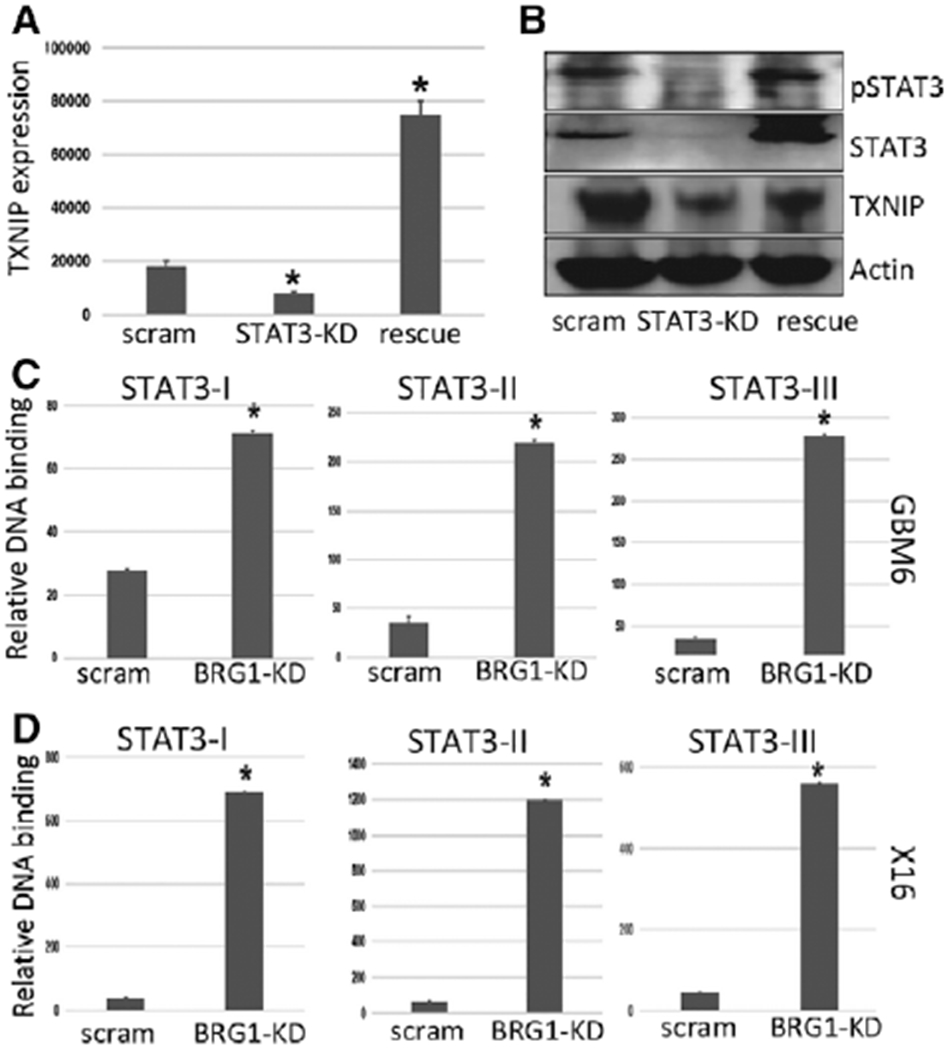
TXNIP is an STAT3-regulated gene. GBMX16 GICs were transduced with a lentiviral vector containing a Dox-inducible shRNA against STAT3, and then transduced with wild-type (WT) STAT3 construct to restore STAT3 expression. (A): Total RNA was prepared from X16 GICs, and STAT3-KD GICs with or without STAT3 rescue, and TXNIP expression was determined by qPCR. (B): Protein lysates from control, STAT3-KD, and STAT3 rescued KD GICs were immunoblotted for pTyr-STAT3, total STAT3, TXNIP, and actin. (C,D): ChIP analyses of STAT3 binding to the STAT3-I, -II, and -III sites within the TXNIP promoter in control and BRG1-KD (C) GBM6 GICs, and (D) X16 GICs. The ChIP-enriched DNA levels analyzed by qPCR were normalized to input DNA, followed by subtraction of nonspecific binding determined by control IgG. *p ≤ .05 was considered to be statistically significant.
We then related our findings on BRG1-regulated gene expression in GICs to gene expression patterns of GBM patient tumor specimens in the TCGA database. Among the molecular features defined by the PanCancer Atlas molecular classification [48,49], we found that BRG1 gene expression levels were positively correlated to the stemness index defined by mRNA expression patterns (mRNAsi) and to the IFN-score (Fig. 5), which assess the degree of oncogenic dedifferentiation [48] and activation of IFN response pathway, respectively [49]. These observations provide further evidence in supporting for a role of BRG1 in the regulating of genes involved in maintaining cancer sternness and the IFN response of GBM.
Figure 5.
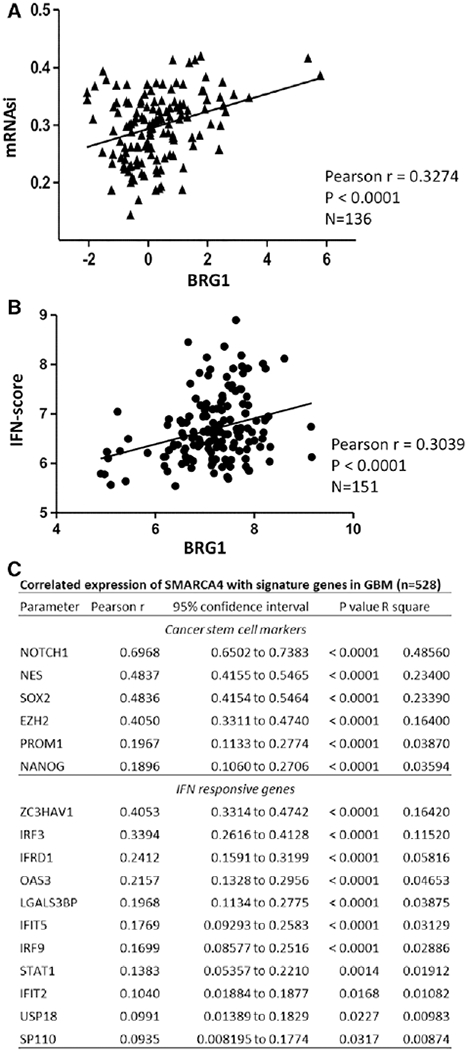
BRG1 regulates sternness and IFN response genes in GBM patient samples. SMARCA4/BRG1 expression in GBM patient samples in the TCGA database was correlated with the cancer sternness indices (mRNAsi) [48] (A) and the IFN-score [49] (B). Signature genes of cancer stem cells and IFN-score that are significantly correlated with BRG1/SMARCA4 is listed in the table (C).
BRG1-KD Sensitizes X16 GICs to TMZ in vitro and In Vivo
Although the alkylating agent TMZ is front-line chemotherapy in the treatment of GBM [50], a significant clinical problem is intrinsic or adaptive resistance to TMZ [50]. As such, we next sought to determine if GICs are resistant to TMZ. To evaluate for TMZ sensitivity of GBM6 or X16 GICs, cells were treated with 300 and 500 pM TMZ, and cell proliferation was determined in Cell Titer-Glo assays. GBM6 GICs were highly sensitive to both TMZ concentrations and TMZ resulted in a marked reduction in cell viability (Fig. 6A). In contrast, X16 GICs were highly resistant to the effects of TMZ (Fig. 6A). TMZ resistance in GBM is linked to O6-methylguanine methyltransferase (MGMT) expression, which is a DNA repair protein [50,51]. To assess the involvement of MGMT in the TMZ sensitivity of GICs, we next determined MGMT mRNA and protein expression and found significantly higher MGMT expression in X16 GICs as compared to GBM6 GICs (Fig. 6B, 6C). The SWI/SNF complex plays an important role in facilitating DNA repair [52–55], and BRG1 has been implicated in DNA double-strand break repair [53–55]. Thus, we hypothesized that BRG1-KD in GICs would prevent the repair of TMZ-induced DNA damage and sensitize GICs to TMZ. The X16 BRG1-KD GICs were treated with 500 pM TMZ and displayed significantly higher sensitivity to TMZ (Fig. 6D). Moreover, BRG1-KD in X16 GICs resulted in a reduction in MGMT expression consistent with their sensitization to TMZ (Fig. 6E).
Figure 6.
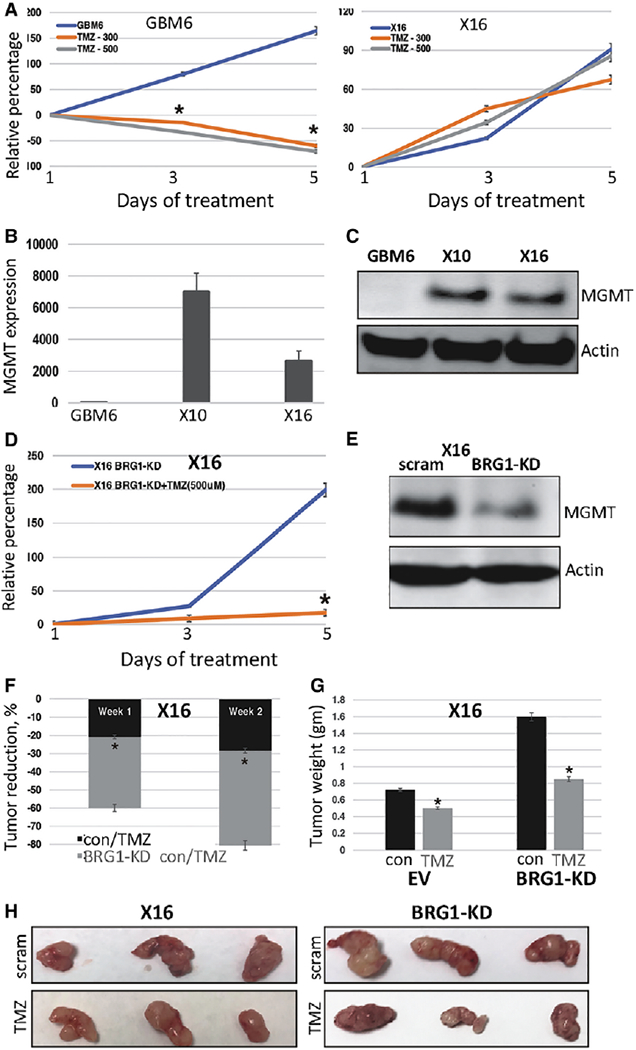
BRG1 regulates GIC sensitivity to TMZ. (A): GBM6 and X16 GICs were treated with 300 or 500 μM of TMZ, and cell proliferation was determined by CellTiter-Glo assays. (B): Total RNA was prepared from GBM6, X10 and X16 GICs, and MGMT expression was determined by qPCR. (C): Protein lysates from GBM6, X10 and X16 GICs were immunoblotted for MGMT and actin. (D): Control and BRG1-KD X16 GICs were treated with or without 500 μM of TMZ, and cell proliferation was determined by CellTiter-Glo assays. (E): Protein lysates from control (scrambled shRNA) and BRG1-KD GICs were immunoblotted for MGMT and actin. (F): The effect of TMZ on the tumorigenicity of control and BRG-KD GICs was assessed by injection of 106 tumor cells into the flanks of NSG mice and live animal imaging was performed at weekly intervals. After confirming tumor initiation by live animal imaging, mice were treated with TMZ by IP injection (10 mg/kg on alternate days), and the reduction in tumor volume was calculated and 1 and 2 weeks post-treatment. (G) Tumor weights and (H) photographs of tumors at necropsy. *p ≤ .05 was considered to be statistically significant.
To determine whether BRG1-KD sensitized GICs to TMZ in vivo, we conducted animal studies with control (scrambled shRNA) and BRG1-KD X16 GICs injected into the flanks of NSG mice. Following confirmation of tumor initiation by live animal imaging, mice were treated with TMZ. At 1 and 2 weeks posttreatment, TMZ markedly reduced tumor growth in BRG1-KD GICs (Fig. 6F), and at necropsy, the tumors were markedly smaller (Fig. 6G, 6H). Thus, despite X16 BRG1-KD GICs forming larger tumors, the tumors had a more differentiated phenotype and were sensitized to TMZ therapy.
Pharmacologic Inhibition of BRG1 Sensitizes X16 GICs to TMZ
PFI-3 is a competitive inhibitor of the SWI/SNF catalytic components with higher affinity for BRG1 than BRM [56]. We examined the effect of PFI-3 on TMZ sensitivity and found that the combination of TMZ and PFI-3 had a markedly greater effect on X16 GIC proliferation than either agent alone (Fig. 7A). Since BRG1-KD upregulated GFAP expression, we also examined the effect of PFI-3 on GFAP expression in X16 GICs and found that PFI-3 upregulated GFAP protein expression (Fig. 7B). The X10 GICs have been isolated from another PDX which was TMZ-resistant. Both X10 and X16 GICs have high MGMT expression compared to the GBM6 GICs (Fig. 6B, 6C). Most importantly, although X10 GICs were highly resistant to TMZ, the combination of PFI-3 and TMZ had a marked effect on cell proliferation (Fig. 7C). Similar to the findings with TMZ, although X10 and X16 GICs were resistant to carmustine (another DNA alkylating agent used to treat GBM patients [57]), a combination treatment of carmustine and PFI-3 rendered the X10 and X16 GICs sensitive to the drugs (Fig. 7D).
Figure 7.
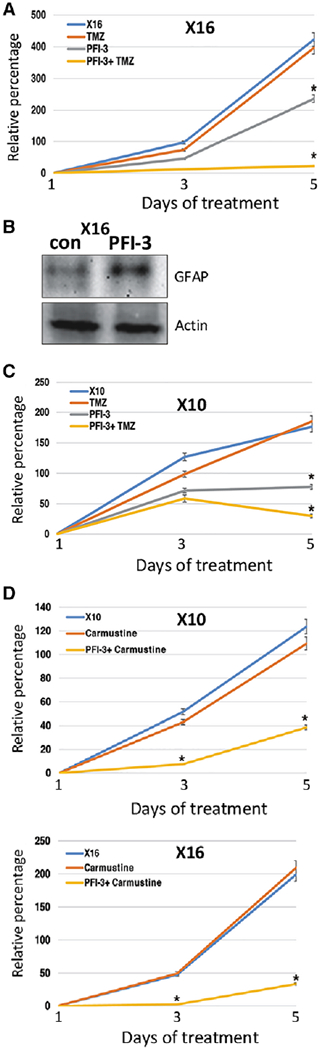
The BRG1 inhibitor PFI-3 sensitizes GICs to chemotherapy. (A): X16 GICs were treated with TMZ (500 μM) and PFI-3 (1 μM), and cell proliferation was determined by CellTiter-Glo assays. (B): Protein lysates from X16 GICs treated with PFI-3 were immunoblotted for GFAP and actin. (C): X10 GICs were treated with TMZ (500 μM) and PFI-3 (1 μM), and cell proliferation was determined by CellTiter-Glo assays. (D): X10 and X16 GICs were treated with carmustine (10 μM) and PFI-3 (1 μM), and CellTiter-Glo assays performed. *p ≤ .05 was considered to be statistically significant.
Discussion
In GBM, the subpopulation of neural stem-like GICs is considered responsible for tumor initiation and progression, inherent and acquired resistance to therapy, and intratumoral heterogeneity. Therefore, it is imperative to identify and elucidate the mechanisms deregulated in GICs. The SWI/SNF complex of chromatin remodelers are global regulators of gene expression, which are recruited to the promoters and enhancers of genes across the genome [58–61]. This process alters the positioning of histones and enables the recruitment of transcription factors that activate gene transcription [62,63]. The SWI/SNF complex plays critical roles during embryonic development, and regulates stem cell renewal and differentiation [9,64]. Of the two catalytic components, BRG1 is the sole catalytic component found to be expressed in the ESCs [5,8–10]. In the present study, we investigated the role of BRG1 in the maintenance and function of GICs. We found that the BRG1 protein was broadly expressed in GICs isolated from multiple GBM patients. In contrast, BRM was expressed in only some GICs. This observation suggests that BRG1 is a critical component in GICs and that BRM maybe dispensable, since all GICs studied display characteristics of stem cells and have high tumor-initiating capability.
Loss of the different components of the SWI/SNF complex has been implicated in several cancers but its role appears to be highly context dependent. In some cancers, loss of SWI/SNF complex components leads to tumor progression, while in others, it leads to tumor suppression [65–69]. To determine the role of BRG1 in GICs, BRG1 expression was knocked down by an shRNA-based approach in BRG1-expressing GICs. Similar to the findings described in ESCs [4,6], BRG1 loss in GICs significantly reduced stem cell markers (CD44, Oct4, and Nanog) and induced differentiation markers (GFAP, S100, and Olig2). CD44 expression has been previously shown to be BRG1-regulated [70,71]. These findings indicated that BRG1 is involved in maintaining GIC stemness; hence, BRG1 loss leads to a more differentiated state. We next examined whether BRG1 loss alters other components of the BAF complex. We observed subtle modulation of several members of the BAF complex, including BAF155 and PBRM1, in a GIC-specific manner. Thus, BRG1 loss has effects on the BAF complex, which may regulate stem cell identity in GICs due to destabilization of the BAF complex or aberrant activity of the residual complex [61,72,73].
Although BRG1-KD in ESCs and in GICs has been reported to reduce proliferation [4,6,22], BRG1 loss also has been shown to increase the proliferation of some cell lines [67,74–77]. Consistent with the latter reports, we found that BRG1-KD enhanced proliferation of both GBM6 and X16 GICs. Moreover, BRG1 loss resulted in hyperphosphorylation of the p107 and p130 members of RB-like proteins, which is consistent with reports that BRG1 mediates cell growth inhibition through the Rb pathway [34,76]. Since p107 controls G1/S phase transition, while p130 controls G1/G0 progression [78,79], BRG1 loss in association with p107 and p130 inactivation may promote proliferation of BRG1-KD GICs. Furthermore, BRG1 loss also led to increased expression of Cyclin A2, which interacts with CDK1/2 and promotes G1/S and G2/M cell cycle transition [80]. Taken together our findings strongly suggest that BRG1 plays an important role in restraining GIC proliferation, which may also contribute to GIC drug resistance since chemotherapeutic drugs target proliferating cancer cells [1,81]. Consistent with the finding that BRG1 loss increased GIC proliferation in vitro, BRG1-KD GICs injected orthotopically into the brains of mice showed accelerated tumor growth. Although BRG1-KD tumors were larger and more proliferative (as indicated by Ki67 staining), the tumors were more differentiated since they had high GFAP, S100B, and Olig2 expression, which is consistent with the finding that BRG1 loss led to an increased ESC differentiation [5].
To elucidate the molecular pathways that BRG1 regulates in GICs, we conducted targeted microarrays with cancer-related gene panels on RNA prepared from X16 and the X16 BRG1-KD cells, and identified several BRG1-regulated genes, whose altered expression was validated by qPCR and immunoblotting. An interesting finding was that BRG1 loss upregulated TXNIP expression in GBM6 and X16 GICs. TXNIP is a major redox regulator that negatively regulates thioredoxin, an ROS scavenger [42,82]. TXNIP also blocks glucose uptake in cells by targeting the glycolytic pathway [42,82]. TXNIP has been implicated as a tumor suppressor, and TXNIP downregulation is associated with tumor aggressiveness [43,82,83]. Cancer stem cells (CSCs) have been found to have lower mitochondrial respiration and higher glucose requirements [81]. By modulating mitochondrial respiration, CSCs regulate ROS levels which are important in resistance to DNA damage [84]. Studies have further demonstrated that high glucose levels are critical for CSC survival and maintenance [85]. This led us to hypothesize that BRG1 regulates the expression of genes implicated in the glucose requirement of GICs, which was substantiated by the observation that BRG1-KD resulted in the downregulation of glucose transporter GLUT1, and FBP2 and PKM of the glycolytic pathway [86–88]. We found that silencing of TXNIP in BRG1-KD GICs restored GLUT1, FBP2, and PKM expression, indicating that BRG1 promotes the expression of these genes in GICs through the TXNIP-regulated pathway. Interestingly, BRG1 has been linked to cancer cell metabolism through reprogramming lipid synthesis in breast cancer cells [89]. In addition, BRG1 regulates the glycolytic pathway in cardiomyocytes, providing another link of BRG1 to cell metabolism [90].
BRG1 loss in GICs also downregulated ISG expression, and we recently showed that ISG expression was negatively regulated by a STAT3 pathway in GICs [27]. These results suggest an interaction of the STAT3 pathway with BRG1. PPI hub analysis of our microarray data provided additional evidence that BRG1 loss leads to activation of the STAT3 pathway. Furthermore, BRG1-KD in GICs increased LIF expression but decreased SOCS1 expression, which together would consequently result in STAT3 pathway activation. We also confirmed that BRG1-KD resulted in increased tyrosine-phosphorylated and total STAT3 protein levels in both the GBM6 and X16 GICs. The BRG1 subunit of the esBAF complex employs STAT3 in driving pluripotency genes such as Sox2, Oct4, and Nanog and is associated with the PRC2 complex to inhibit the expression of the Hox gene clusters that are essential for differentiation [5]. However, loss of BRG1 allows the PRC2 complex to silence the BRG1-mediated transcription of the stem cell markers [5]. Our findings suggest that increased STAT3 activity in BRG1-KD GICs contributed toward their differentiation, which is consistent with the findings in ESCs where BRG1 drives the transcription of the pluripotent genes through STAT3, and BRG1 loss allows STAT3 to induce transcription of genes involved in differentiation [5]. Furthermore, our findings also implicate STAT3 in regulating TXNIP expression. STAT3-KD in GICs reduced TXNIP expression, while rescue with STAT3 restored TXNIP expression. ChIP analysis showed increased STAT3 binding to several sites on the TXNIP promoter in BRG1-KD GICs. These observations provide further insights into how BRG1 promotes GIC stem cell maintenance and inhibits differentiation. We found that BRG1-KD in GICs increased the STAT3 signaling pathway, which mediates the transcription of the genes promoting stem cell differentiation and proliferation. In addition, STAT3 induces the transcription of TXNIP, which then blocks increased glucose uptake essential for GIC maintenance. Interestingly, we have also observed an increase in TXNIP expression in xenograft tumors of BRG1-KD cells (Supporting Information, Fig. S4).
The SWI/SNF complex regulates various DNA repair pathways, including DNA double-strand breaks by enabling homologous recombination repair, and BRG1 loss sensitizes cancer cells to cisplatin and UV radiation [53,55]. The DNA alkylating agent TMZ is front-line chemotherapy in GBM patients following tumor resection. However, a significant clinical problem is inherent and acquired resistance of GBM patients to TMZ therapy. TMZ resistance is in part due to the expression of the DNA repair protein MGMT [91,92]. We found that while GBM6 GICs were sensitive to TMZ, X16 GICs were relatively TMZ-resistant and that MGMT expression in the GBM6 and X16 GICs correlated with TMZ resistance. Most importantly, BRG1 loss resulted in decreased MGMT expression and sensitized X16 GICs to TMZ treatment. Furthermore, although tumors produced by X16 BRG1-KD cells were larger, the tumors were more sensitive to TMZ treatment than the tumors produced by control GICs. Additionally, although BRG1-KD in vitro and in vivo promotes GIC proliferation and differentiation, BRG1-KD GICs are sensitized to chemotherapy, which is consistent with previous findings that BRG1 loss sensitizes cancer cell lines to chemotherapy [67,93]. GICs are considered responsible for chemoresistance and radioresistance in GBM, and increased DNA repair capacity [1]. We also used the small molecule inhibitor of the SWI/SNF catalytic subunits PFI-3 to pharmacologically inhibit BRG1 function. Interestingly, PFI-3 treatment previously was not found to phenocopy the effect that BRG1/BRM knockdown has on growth inhibition of various tumor cell lines [94]. We treated TMZ-resistant X16 GICs with PFI-3 and TMZ, and found significant cell growth inhibition compared to TMZ treatment alone. Furthermore, PFI-3 treatment attenuated MGMT expression. X10 GICs, which are BRM-deficient and MGMT-positive, were also TMZ resistant, but were sensitized by a combination of PFI-3 with TMZ. Similar to our findings with TMZ, PFI-3 also sensitized GICs to carmustine, another DNA alkylating agent. Our findings provide strong evidence that BRG1 plays critical roles in the drug-resistance of GICs. Through a process, yet unknown, BRG1 regulates MGMT expression in GICs, which in turn promotes drug resistance. Future studies will be directed toward understanding the BRG1/ MGMT-mediated drug resistance pathway in GBM which will be critical for developing improved therapeutic interventions.
Supplementary Material
Significance Statement.
Glioma-initiating cells (GICs) are implicated in tumor heterogeneity and therapeutic resistance of GBM. Results of this study show that the BRG1 subunit of the SWI/SNF chromatin remodeling complex plays critical roles in maintaining GICs in a stem-like state. Furthermore, the authors identified a novel mechanism by which BRG1 regulates expression of TXNIP, which is a redox regulator and also negatively regulates glycolysis. It was found that BRG1 regulates TXNIP through a STAT3-dependent pathway and that BRG1 plays a critical role in the drug resistance of GICs and in GIC-induced tumorigenesis. Thus, the BRG1-STAT3-TXNIP axis is a potential therapeutic target in GBM.
Acknowledgments
This work was supported in part by the Department of Defense W81XWH-11-1-0533 (LMP), National Institutes of Health CA197206 (MF), West Cancer Center grant (LMP), and the Muir-head Chair Endowment (LMP). We thank Bhaskar Kahali, St. Jude Children’s Research Hospital, for his valuable comments.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
See www.StemCells.com for supporting information available online.
References
- 1.Bao S, Wu Q, McLendon RE et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444:756–760. [DOI] [PubMed] [Google Scholar]
- 2.Trotter KW, Archer TK. The BRG1 transcriptional coregulator. Nucl Recept Signal 2008;6: e004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tolstorukov MY, Sansam CG, Lu P et al. Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc Natl Acad Sci U S A 2013;110:10165–10170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X, Li B, Li W et al. Transcriptional repression by the BRG1-SWI/SNF complex affects the pluripotency of human embryonic stem cells. Stem Cell Rep 2014;3:460–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho L, Miller EL, Ronan JL et al. esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nat Cell Biol 2011;13:903–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kidder BL, Palmer S, Knott JG. SWI/SNF-Brg1 regulates self-renewal and occupies core pluripotency-related genes in embryonic stem cells. Stem Cells 2009;27:317–328. [DOI] [PubMed] [Google Scholar]
- 7.Singhal N, Graumann J, Wu G et al. Chromatin-remodeling components of the BAF complex facilitate reprogramming. Cell 2010;141: 943–955. [DOI] [PubMed] [Google Scholar]
- 8.Gao X, Tate P, Hu P et al. ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc Natl Acad Sci U S A 2008;105: 6656–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho L, Ronan JL, Wu J et al. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc Natl Acad Sci U S A 2009; 106:5181–5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaeser MD, Aslanian A, Dong MQ et al. BRD7, a novel PBAF-specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. J Biol Chem 2008; 283:32254–32263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kadoch C, Hargreaves DC, Hodges C et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 2013;45: 592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One 2013;8:e55119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sentani K, Oue N, Kondo H et al. Increased expression but not genetic alteration of BRG1, a component of the SWI/SNF complex, is associated with the advanced stage of human gastric carcinomas. Pathobiology 2001;69:315–320. [DOI] [PubMed] [Google Scholar]
- 14.Sun A, Tawfik O, Gayed B et al. Aberrant expression of SWI/SNF catalytic subunits BRG1/BRM is associated with tumor development and increased invasiveness in prostate cancers. Prostate 2007;67:203–213. [DOI] [PubMed] [Google Scholar]
- 15.Imielinski M, Berger AH, Hammerman PS et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012;150:1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Love C, Sun Z, Jima D et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet 2012;44:1321–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Witkowski L, Carrot-Zhang J, Albrecht S et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet 2014;46: 438–443. [DOI] [PubMed] [Google Scholar]
- 18.Le Loarer F, Watson S, Pierron G et al. SMARCA4 inactivation defines a group of undifferentiated thoracic malignancies transcriptionally related to BAF-deficient sarcomas. Nat Genet 2015;47:1200–1205. [DOI] [PubMed] [Google Scholar]
- 19.Hodges HC, Stanton BZ, Cermakova K et al. Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nat Struct Mol Biol 2018;25:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bai J, Mei PJ, Liu H et al. BRG1 expression is increased in human glioma and controls glioma cell proliferation, migration and invasion in vitro. J Cancer Res Clin Oncol 2012;138: 991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guerrero-Martinez JA, Reyes JC. High expression of SMARCA4 or SMARCA2 is frequently associated with an opposite prognosis in cancer. Sci Rep 2018;8:2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hiramatsu H, Kobayashi K, Kobayashi K et al. The role of the SWI/SNF chromatin remodeling complex in maintaining the stemness of glioma initiating cells. Sci Rep 2017;7:889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen L, O’Shea JM, Kaadige MR et al. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc Natl Acad Sci U S A 2015;112:5425–5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu N, Zheng B, Shaywitz A et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell 2013;49:1167–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garner JM, Ellison DW, Finkelstein D et al. Molecular heterogeneity in a patient-derived glioblastoma xenoline is regulated by different cancer stem cell populations. PLoS One 2015;10: e0125838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang CH, Yue J, Fan M et al. IFN induces miR-21 through a signal transducer and activator of transcription 3-dependent pathway as a suppressive negative feedback on IFN-induced apoptosis. Cancer Res 2010;70:8108–8116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ganguly D, Fan M, Yang CH et al. The critical role that STAT3 plays in glioma-initiating cells: STAT3 addiction in glioma. Oncotarget 2018;9:22095–22112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glaros S, Cirrincione GM, Muchardt C et al. The reversible epigenetic silencing of BRM: Implications for clinical targeted therapy. Oncogene 2007;26:7058–7066. [DOI] [PubMed] [Google Scholar]
- 29.Kahali B, Gramling SJ, Marquez SB et al. Identifying targets for the restoration and reactivation of BRM. Oncogene 2014;33:653–664. [DOI] [PubMed] [Google Scholar]
- 30.Yang CH, Yue J, Pfeffer SR et al. MicroRNA miR-21 regulates the metastatic behavior of B16 melanoma cells. J Biol Chem 2011;286:39172–39178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang CH, Wang Y, Sims M et al. MiRNA203 suppresses the expression of protumorigenic STAT1 in glioblastoma to inhibit tumorigenesis. Oncotarget 2016;7:84017–84029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang CH, Yue J, Sims M et al. The curcumin analog EF24 targets NF-kappaB and miRNA-21, and has potent anticancer activity in vitro and in vivo. PLoS One 2013;8:e71130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du Z, Cai C, Sims M et al. The effects of type I interferon on glioblastoma cancer stem cells. Biochem Biophys Res Commun 2017;491: 343–348. [DOI] [PubMed] [Google Scholar]
- 34.Strobeck MW, Knudsen KE, Fribourg AF et al. BRG-1 is required for RB-mediated cell cycle arrest. Proc Natl Acad Sci U S A 2000;97: 7748–7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ni Z, Bremner R. Brahma-related gene 1-dependent STAT3 recruitment at IL-6-inducible genes. J Immunol 2007;178:345–351. [DOI] [PubMed] [Google Scholar]
- 36.Ito K, Noguchi A, Uosaki Y et al. Gfap and Osmr regulation by BRG1 and STAT3 via inter-chromosomal gene clustering in astrocytes. Mol Biol Cell 2018;29:209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giraud S, Hurlstone A, Avril S et al. Implication of BRG1 and cdk9 in the STAT3-mediated activation of the p21waf1 gene. Oncogene 2004;23:7391–7398. [DOI] [PubMed] [Google Scholar]
- 38.Souma Y, Nishida T, Serada S et al. Antiproliferative effect of SOCS-1 through the suppression of STAT3 and p38 MAPK activation in gastric cancer cells. Int J Cancer 2012;131:1287–1296. [DOI] [PubMed] [Google Scholar]
- 39.Megeney LA, Perry RLS, Lecouter JE et al. bFGF and LIF signaling activates STAT3 in proliferating myoblasts. Dev Genet 1996;19:139–145. [DOI] [PubMed] [Google Scholar]
- 40.Alhawiti NM, Al Mahri S, Aziz MA et al. TXNIP in metabolic regulation: Physiological role and therapeutic outlook. Curr Drug Targets 2017;18:1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, Yue Z, Xiong W et al. TXNIP overexpression suppresses proliferation and induces apoptosis in SMMC7221 cells through ROS generation and MAPK pathway activation. Oncol Rep 2017;37:3369–3376. [DOI] [PubMed] [Google Scholar]
- 42.Malone CF, Emerson C, Ingraham R et al. mTOR and HDAC inhibitors converge on the TXNIP/thioredoxin pathway to cause catastrophic oxidative stress and regression of RAS-driven tumors. Cancer Discov 2017;7:1450–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagaraj K, Lapkina-Gendler L, Sarfstein R et al. Identification of thioredoxin-interacting protein (TXNIP) as a downstream target for IGF1 action. Proc Natl Acad Sci U S A 2018;115:1045–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elgort MG, O’Shea JM, Jiang Y et al. Transcriptional and translational downregulation of thioredoxin interacting protein is required for metabolic reprogramming during G(1). Genes Cancer 2010;1:893–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Labak CM, Wang PY, Arora R et al. Glucose transport: Meeting the metabolic demands of cancer, and applications in glioblastoma treatment. Am J Cancer Res 2016;6:1599–1608. [PMC free article] [PubMed] [Google Scholar]
- 46.DeBalsi KL, Wong KE, Koves TR et al. Targeted metabolomics connects thioredoxin-interacting protein (TXNIP) to mitochondrial fuel selection and regulation of specific oxidoreductase enzymes in skeletal muscle. J Biol Chem 2014;289:8106–8120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang M, Qian F, Hu Y et al. Chromatinremodelling factor BRG1 selectively activates a subset of interferon-alpha-inducible genes. Nat Cell Biol 2002;4:774–781. [DOI] [PubMed] [Google Scholar]
- 48.Malta TM, Sokolov A, Gentles AJ et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 2018;173:338–354.e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thorsson V, Gibbs DL, Brown SD et al. The immune landscape of cancer. Immunity 2018; 48:812–830.e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sang YL. Temozolomide resistance in glioblastoma multiforme. Genes Dis 2016;3: 198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gutenberg A, Bock HC, Bruck W et al. MGMT promoter methylation status and prognosis of patients with primary or recurrent glioblastoma treated with carmustine wafers. Br J Neurosurg 2013;27:772–778. [DOI] [PubMed] [Google Scholar]
- 52.Smith-Roe SL, Nakamura J, Holley D et al. SWI/SNF complexes are required for full activation of the DNA-damage response. Oncotarget 2015;6:732–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kothandapani A, Gopalakrishnan K, Kahali B et al. Downregulation of SWI/SNF chromatin remodeling factor subunits modulates cisplatin cytotoxicity. Exp Cell Res 2012;318:1973–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Park JH, Park EJ, Lee HS et al. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting gamma-H2AX induction. EMBO J 2006;25:3986–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qi W, Wang R, Chen H et al. BRG1 promotes the repair of DNA double-strand breaks by facilitating the replacement of RPA with RAD51. J Cell Sci 2015;128:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fedorov O, Castex J, Tallant C et al. Selective targeting of the BRG/PB1 bromodomains impairs embryonic and trophoblast stem cell maintenance. Sci Adv 2015;1:e1500723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Attenello F, Raza SM, Dimeco F et al. Chemotherapy for brain tumors with polymer drug delivery. Handb Clin Neurol 2012;104:339–353. [DOI] [PubMed] [Google Scholar]
- 58.Bourgo RJ, Siddiqui H, Fox S et al. SWI/SNF deficiency results in aberrant chromatin organization, mitotic failure, and diminished proliferative capacity. Mol Biol Cell 2009;20:3192–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Flanagan JF, Peterson CL. A role for the yeast SWI/SNF complex in DNA replication. Nucleic Acids Res 1999;27:2022–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muchardt C, Yaniv M. ATP-dependent chromatin remodelling: SWI/SNF and Co. are on the job. J Mol Biol 1999;293:187–198. [DOI] [PubMed] [Google Scholar]
- 61.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 2011;11:481–492. [DOI] [PubMed] [Google Scholar]
- 62.Alexander JM, Hota SK, He D et al. Brg1 modulates enhancer activation in mesoderm lineage commitment. Development 2015;142: 1418–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim SI, Bultman SJ, Kiefer CM et al. BRG1 requirement for long-range interaction of a locus control region with a downstream promoter. Proc Natl Acad Sci U S A 2009;106:2259–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bultman SJ, Gebuhr TC, Magnuson T. A Brg1 mutation that uncouples ATPase activity from chromatin remodeling reveals an essential role for SWI/SNF-related complexes in beta-globin expression and erythroid development. Genes Dev 2005;19:2849–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Buscarlet M, Krasteva V, Ho L et al. Essential role of BRG, the ATPase subunit of BAF chromatin remodeling complexes, in leukemia maintenance. Blood 2014;123:1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lan J, Li H, Luo X et al. BRG1 promotes VEGF-A expression and angiogenesis in human colorectal cancer cells. Exp Cell Res 2017;360: 236–242. [DOI] [PubMed] [Google Scholar]
- 67.Liu X, Tian X, Wang F et al. BRG1 promotes chemoresistance of pancreatic cancer cells through crosstalking with Akt signalling. Eur J Cancer 2014;50:2251–2262. [DOI] [PubMed] [Google Scholar]
- 68.Roy N, Malik S, Villanueva KE et al. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev 2015;29:658–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi J, Whyte WA, Zepeda-Mendoza CJ et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev 2013;27:2648–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Strobeck MW, DeCristofaro MF, Banine F et al. The BRG-1 subunit of the SWI/SNF complex regulates CD44 expression. J Biol Chem 2001;276:9273–9278. [DOI] [PubMed] [Google Scholar]
- 71.Reisman DN, Strobeck MW, Betz BL et al. Concomitant down-regulation of BRM and BRG1 in human tumor cell lines: Differential effects on RB-mediated growth arrest vs CD44 expression. Oncogene 2002;21:1196–1207. [DOI] [PubMed] [Google Scholar]
- 72.Nakayama RT, Pulice JL, Valencia AM et al. SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat Genet 2017;49:1613–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hohmann AF, Vakoc CR. A rationale to target the SWI/SNF complex for cancer therapy. Trends Genet 2014;30:356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marquez-Vilendrer SB, Rai SK, Gramling SJ et al. Loss of the SWI/SNF ATPase subunits BRM and BRG1 drives lung cancer development. Oncoscience 2016;3:322–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hendricks KB, Shanahan F, Lees E. Role for BRG1 in cell cycle control and tumor suppression. Mol Cell Biol 2004;24:362–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dunaief JL, Strober BE, Guha S et al. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell 1994;79:119–130. [DOI] [PubMed] [Google Scholar]
- 77.Shanahan F, Seghezzi W, Parry D et al. Cyclin E associates with BAF155 and BRG1, components of the mammalian SWI-SNF complex, and alters the ability of BRG1 to induce growth arrest. Mol Cell Biol 1999;19:1460–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mayol X, Garriga J, Grana X. G1 cyclin/CDK-independent phosphorylation and accumulation of p130 during the transition from G1 to G0 lead to its association with E2F-4. Oncogene 1996; 13:237–246. [PubMed] [Google Scholar]
- 79.Rodier G, Makris C, Coulombe P et al. p107 inhibits G1 to S phase progression by downregulating expression of the F-box protein Skp2. J Cell Biol 2005;168:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 2013;14:518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Deshmukh A, Deshpande K, Arfuso F et al. Cancer stem cell metabolism: A potential target for cancer therapy. Mol Cancer 2016;15:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang P, Gao J, Wang X et al. A novel indication of thioredoxin-interacting protein as a tumor suppressor gene in malignant glioma. Oncol Lett 2017;14:2053–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morrison JA, Pike LA, Sams SB et al. Thioredoxin interacting protein (TXNIP) is a novel tumor suppressor in thyroid cancer. Mol Cancer 2014;13:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu M, Neilson A, Swift AL et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol 2007;292:C125–C136. [DOI] [PubMed] [Google Scholar]
- 85.Palorini R, Votta G, Balestrieri C et al. Energy metabolism characterization of a novel cancer stem cell-like line 3AB-OS. J Cell Biochem 2014;115:368–379. [DOI] [PubMed] [Google Scholar]
- 86.Shibuya K, Okada M, Suzuki S et al. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 2015; 6:651–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vaupel P, Harrison L. Tumor hypoxia: Causative factors, compensatory mechanisms, and cellular response. Oncologist 2004;9(Suppl 5): 4–9. [DOI] [PubMed] [Google Scholar]
- 88.Wu C, Khan SA, Peng LJ et al. Roles for fructose-2,6-bisphosphate in the control of fuel metabolism: Beyond its allosteric effects on glycolytic and gluconeogenic enzymes. Adv Enzyme Regul 2006;46:72–88. [DOI] [PubMed] [Google Scholar]
- 89.Wu Q, Madany P, Dobson JR et al. The BRG1 chromatin remodeling enzyme links cancer cell metabolism and proliferation. Oncotarget 2016;7:38270–38281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Banerjee R, Bultman SJ, Holley D et al. Non-targeted metabolomics of Brg1/Brm double-mutant cardiomyocytes reveals a novel role for SWI/SNF complexes in metabolic homeostasis. Metabolomics 2015;11:1287–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fan CH, Liu WL, Cao H et al. O6-methylguanine DNA methyltransferase as a promising target for the treatment of temozolomide-resistant gliomas. Cell Death Dis 2013;4:e876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kitange GJ, Carlson BL, Schroeder MA et al. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro Oncol 2009;11:281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wu Q, Sharma S, Cui H et al. Targeting the chromatin remodeling enzyme BRG1 increases the efficacy of chemotherapy drugs in breast cancer cells. Oncotarget 2016;7:27158–27175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vangamudi B, Paul TA, Shah PK et al. The SMARCA2/4 ATPase domain surpasses the bromodomain as a drug target in SWI/SNF-mutant cancers: Insights from cDNA rescue and PFI-3 inhibitor studies. Cancer Res 2015;75:3865–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.