

Abstract
Leptin plays a role in CNS developmental programs and intercurrent physiological processes related to body fat regulation. The timing and neuro-molecular mechanisms for these effects are relevant to the prevention and treatment of obesity. Factors implicated in a body weight “set point” such as dietary fat, circulating leptin, and other adipokines tend to covary with adiposity and are difficult to disarticulate experimentally. To dissociate the effects of leptin from adiposity and diet, we created a transgenic mouse in which leptin expression is regulated by doxycycline exposure in a dose-responsive manner. Using this system, we investigated the physiological consequences of developmentally timed transient hyperleptinemia on subsequent adiposity. Specifically, we evaluated the physiological effects of leptin elevation during adulthood (9–29 weeks old), “adolescence” (3–8 weeks), and the immediate postnatal period (postnatal days 0–22 [P0-P22]) on both long-term adiposity and susceptibility to gain weight when fed a high fat diet (HFD) ad libitum. We found that inducing chronic hyperleptinemia in adult or “adolescent” mice did not alter body weight when excess leptin was discontinued and, when later exposed to a HFD, their weight gain did not differ from controls. However, transient elevation of circulating leptin from P0-P22 increased weight and fat gain in response to HFD, indicating greater susceptibility to obesity as adults. Thus, transient elevations of circulating leptin—mimicking one aspect of transient adiposity—increased susceptibility of mice to gain weight on a HFD, although these effects were restricted to a critical developmental (P0-P22) time window. These findings may have clinical implications for weight management in infancy.
One Sentence Summary:
Developmentally timed hyperleptinemia without weight gain during the immediate postnatal period exacerbates later diet-induced obesity in male and female mice.
Introduction
Childhood obesity is highly correlated with subsequent adult obesity (1). 90% of children who are obese at 3 years of age are overweight or obese as adolescents, and infants born large for their gestational age have a 1.5-fold higher risk of being obese as adolescents compared to babies normal or small for their gestational age (2). These correlations of childhood obesity with obesity later in life likely reflect interactions of genetic predisposition (3, 4) with developmental (5) and environmental (3, 6) factors. The increasing prevalence of childhood and adult obesity during the past 30 years (7, 8) suggests a substantial impact of non-genetic factors. Accordingly, close attention is being paid to the gestational and early childhood influences mediated by maternal adiposity (9) and early feeding practices associated with adiposity (10–12). The mechanistic bases for such effects (including genetic susceptibility) are difficult to disarticulate experimentally, but these distinctions are critical to developing preventive measures for obesity (13, 14). The studies reported here are based on the premise that the hormone leptin, by virtue of its responsiveness to systemic nutritional status and adiposity (15, 16) and importance in the development of CNS circuits that subserve energy homeostasis (17, 18), could mediate some of the long-term effects of transient perturbations of adiposity.
Dietary fat and adiposity alter leptin signaling in the CNS (19). The extent to which molecular (leptin homeostasis) and neuroanatomic (neuronal circuit alteration) processes involved in the establishment of the neuroregulatory mechanisms of adiposity can be influenced by environmental factors such as adequacy and quality of intrauterine metabolic fuels and endocrine processes, postnatal diet, and intercurrent obesity is unclear. Specifically, can the long-term regulation of adiposity be altered by perinatal factors, and what are the mechanisms underlying such alterations? Dietary fat content (19), adiposity (19), overfeeding (20), and leptin (21) have been implicated in such effects, but because these factors are experimentally correlated, isolation of individual factors during development has not been possible. The underlying cellular and molecular mechanisms for maternal programming of adult obesity remain unclear (22–28).
Leptin is a neurotrophic factor that plays a critical role in the development of hypothalamic feeding circuits (17). Leptin deficiency in mice during the first 12 days of life impairs the formation of projections from the arcuate nucleus of hypothalamus (ARH) to other brain regions involved in energy homeostasis [the paraventricular nucleus (PVH), the dorsomedial hypothalamic nucleus (DMH), and the lateral hypothalamic area (LHA)] (17). Other experiments have shown that maternal high fat feeding or hyperinsulinemia can impair the formation of these circuits (20, 29). We hypothesized that excess leptin during this time period could also influence circuit formation.
Here we investigated the physiological consequences of developmentally timed transient hyperleptinemia on subsequent adiposity. Our hypothesis was that periods of transiently increased leptin lead to higher subsequent body weight by virtue of effects on neural structures that regulate adiposity, and that the timing of this exposure would be an important factor in such responses. We generated a doxycycline (dox)-responsive leptin transgenic mouse in which non-invasive induction of hyperleptinemia can be isolated from diet composition, obesity and its resulting metabolic consequences such as elevated FFA and glucose, insulin insensitivity, and fatty liver, etc. A tetracycline-controlled Tet-On gene expression system (Tet-On) enabled transgenic leptin expression regulated by exposure to doxycycline (dox) in a dose-responsive manner that can be rapidly turned on and off. We evaluated the physiological effects of transient elevation of leptin in ad libitum chow-fed mice during (1) “adulthood” (9 to 29 weeks old, postnatal day 63 to 203; P63–203), (2) “adolescence” (3 to 8 weeks old, P22–56) and (3) “postnatal period” (P0–22) on subsequent adiposity and susceptibility to gain weight when offered a highly palatable diet ad libitum.
Results
Generation and validation of leptin-overexpressing mice
We generated leptin-overexpressing mouse embryonic stem cells (KH2-Lep) (fig. S1 A–D) and subsequently used to create transgenic mice via tetraploid blastocyst injection (31). The transgenic mouse generated for the experiments reported in this manuscript requires two transgenes for leptin expression in response to dox: (1) the reverse tet-transactivator (M2rtTA) at the Rosa26 locus (R26-rtTA), and the leptin transgene in Col1A1 locus controlled by tetracycline responsive elements (TREs) and the CMV promoter (TRE-Lep). Dox-induced leptin overexpressing mice (“2TG”) respond to dox exposure by producing leptin, whereas control mice (“1TG”) contain only the TRE-Lep transgene and do not respond to dox (fig. S1B).
Induction of leptin in 2TG mice is proportional to dox concentration (Fig. 1A). Circulating leptin in 2TG mice increased at the lowest dox dose of 10 μg/ml (1.9 ±0.4 ng/ml to 30.6 ±5.4 ng/ml) whereas 1TG controls maintained proportionality of leptin to fat mass (Fig. 1A). The induction of leptin in 2TG mice ceased rapidly upon withdrawal of dox (Fig. 1B). 2TG mice were exposed to successive intervals of 24-hours of ad libitum access to 200 μg/ml of dox immediately followed by 24 hours of dox withdrawal. Within 24h of dox exposure, circulating leptin in 2TG mice increased from 18.1 ±5.8 ng/ml to 98.6 ±11.5 ng/ml and returned to baseline within 24h of dox removal (16.2 ±6.1 ng/ml; Fig. 1B). After two cycles of 24h dox exposure, circulating leptin in 2TG mice returned to concentrations seen in animals provided with dox-free water, but these concentrations were significantly below the baseline values (13.1 ±5.5 ng/ml vs 18.1 ±5.8 ng/ml, P < 0.01; Fig. 1B) suggesting that overexpression of leptin for 48 hours reduced endogenous leptin production, possibly due to fat loss. To assess the kinetics of leptin overexpression, 2TG mice were gavaged with 400 μg of dox. Plasma leptin concentrations were induced within 4h and peaked after 6–8 hours (83.62 ±18.68 ng/ml; Fig. 1C). Within 24 hours after the single dox gavage, plasma leptin concentrations returned to baseline with no residual effects on plasma leptin (Fig. 1C).
Fig. 1. Validation of leptin-overexpressing mice.
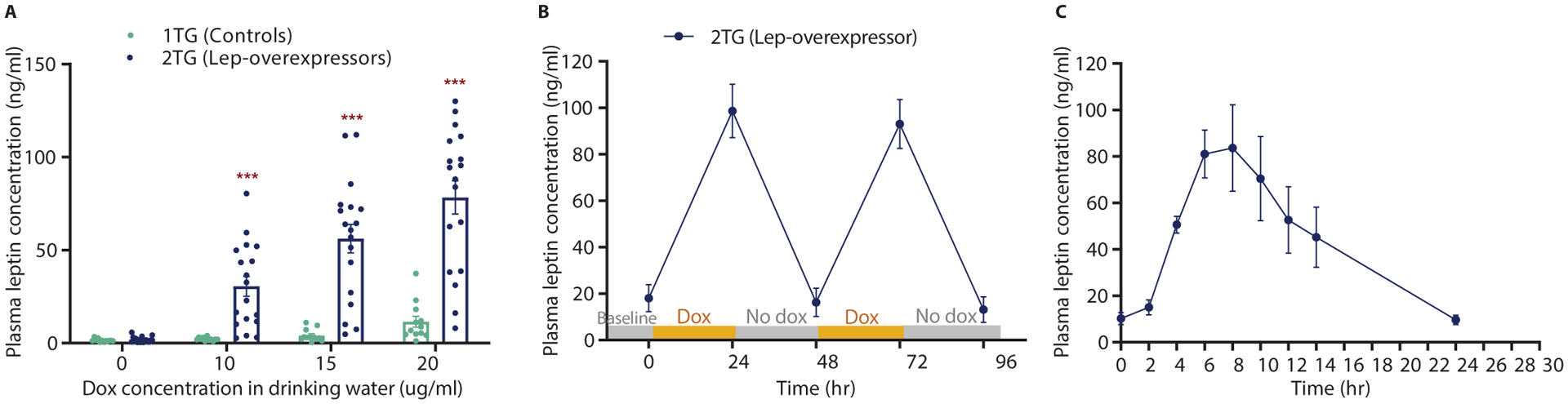
(A) Dose response of 1TG control and 2TG dox-induced leptin-overexpressing mice to increasing amounts of dox in drinking water in. (B) Rapid control of leptin expression with two 24h dox exposures in 2TG leptin-overexpressing mice. (C) Dynamic induction of circulating leptin after dox gavage 2TG leptin-overexpressing mice. All values are means ±SEM. Student’s t-test, ***p<0.001.
Because rtTA was inserted in the Rosa26 locus, its expression was ubiquitous. The primary source of the increased plasma leptin in the dox-induced 2TG mice was the gastrointestinal tract (stomach, duodenum, jejunum, and ileum) which was likely due to both the oral route of dox administration and the capacity of the gut to secrete peptides (fig. S1, E–I). Other tissues expressing transgenic leptin included adipose tissue (perigonadal, subcutaneous, and brown), liver, kidney, lung, spleen, and the hypothalamus (fig. S1, E–I). Exposing adult 2TG mice to a dox concentration of 200 μg/ml in drinking water for two weeks induced circulating leptin concentrations that were at the high end of the physiological range in obese animals (70–227 ng/ml) and resulted in reduced body fat (fig. S2), indicating that dox-induced leptin was bioactive and capable of causing physiological responses. When 1TG nursing dams were exposed to dox at parturition, the dams remained euleptinemic, but the 2TG pups had significantly higher circulating leptin concentrations than 1TG littermates (P < 0.001, fig. S3). Thus, bioactive dox is transferred in breast milk.
Hyperleptinemia in adult male mice (P63-P203)
To investigate whether transient hyperleptinemia affects long term body weight in adult animals, we exposed 9-week-old 1TG (controls) and 2TG (lep-overexpressors) mice fed ad libitum chow to increasing concentrations of dox in drinking water containing 5% sucrose (2TG +Dox group; Fig. 2A). Dox exposure was continued for 20 weeks. The dox dose was escalated on a bi-weekly basis to mimic in the 2TG mice the leptin concentration profile seen with HFD-induced weight gain (34). Circulating leptin concentrations in the 2TG +dox group were significantly higher than the control group starting at the 12.5 μg/ml dox dose (P < 0.05) and remained significantly elevated for the remaining 16 weeks until the cessation of dox (P < 0.01, Fig. 2B).
Fig. 2. Dox-induced chronic hyperleptinemia in adult mice (P63-P203).
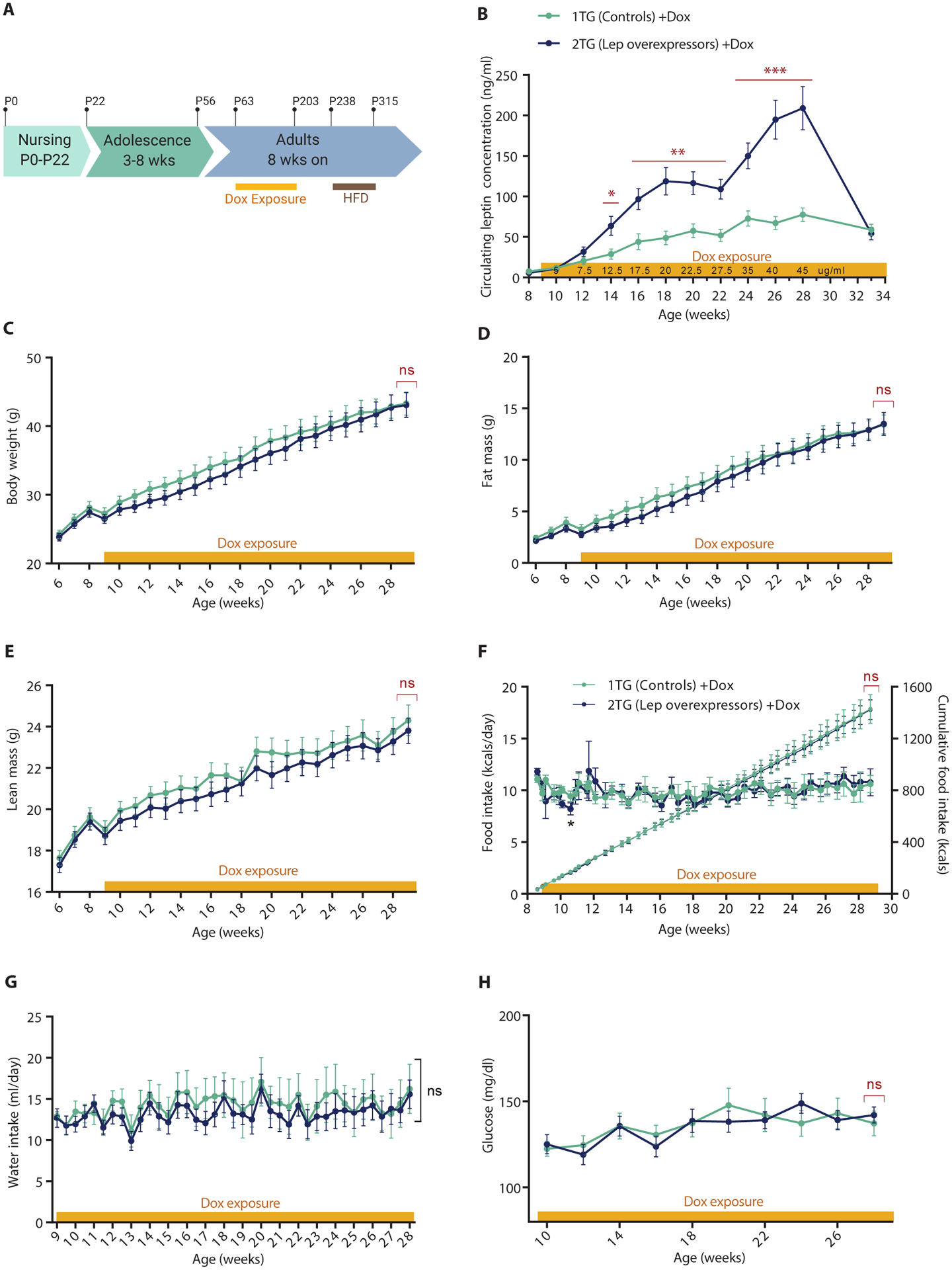
(A) Schematic of the study timeline. P, postnatal day. (B) Plasma leptin concentrations (Student’s t-test at each dox dose), (C) Body weight (two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,32) = 0.34, p = 0.56), (D) Fat mass (two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,32) = 0.24, p = 0.63), (E) Lean mass (two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,32) = 0.44, p = 0.51, (F) Daily caloric food intake per mouse (left axis; two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,8) = 0.0036, p = 0.53) and cumulative food intake (right axis), (G) Daily water intake per mouse (two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,8) = 0.18, p = 0.68), and (H) Venous whole blood glucose concentration (two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,32) = 0.016, p = 0.90) in 1TG controls and 2TG dox-induced leptin overexpressing mice given dox in 5% sucrose water during 20 weeks of escalating dox exposure. All values are means ± SEM. Red brackets indicate a comparison by Student’s t-test of the final datapoint. *P < 0.05, **P < 0.01, ***P < 0.001 between 1TG and 2TG groups by Student’s t-test (in red) or Fisher’s LSD (in black).
Body weight, fat mass, lean mass, food intake, water intake, and plasma glucose were not different between the groups during the 20 weeks of hyperleptinemia (Fig. 2C–H).
Following a 20-week period of gradual elevation of circulating leptin concentrations, dox was eliminated from the drinking water, and leptin in 2TG mice declined to concentrations appropriately proportional to body fat (Fig. 2B). Leptin concentrations measured in 1TG and 2TG mice 5 weeks post-dox cessation were indistinguishable (Fig. 2B). Body weight, composition and food intake for 5 weeks after the cessation of hyperleptinemia were the same in both groups (Fig. 3). Mice were then fed a 60% HFD ad libitum to determine if formerly hyperleptinemic mice were more responsive to highly palatable food. Mice were monitored for an additional 11 weeks but did not show any consistently significant differences in body weight, composition, or food intake (Fig. 3).
Fig. 3. Release of adult mice from dox-induced chronic (P63-P203) hyperleptinemia.
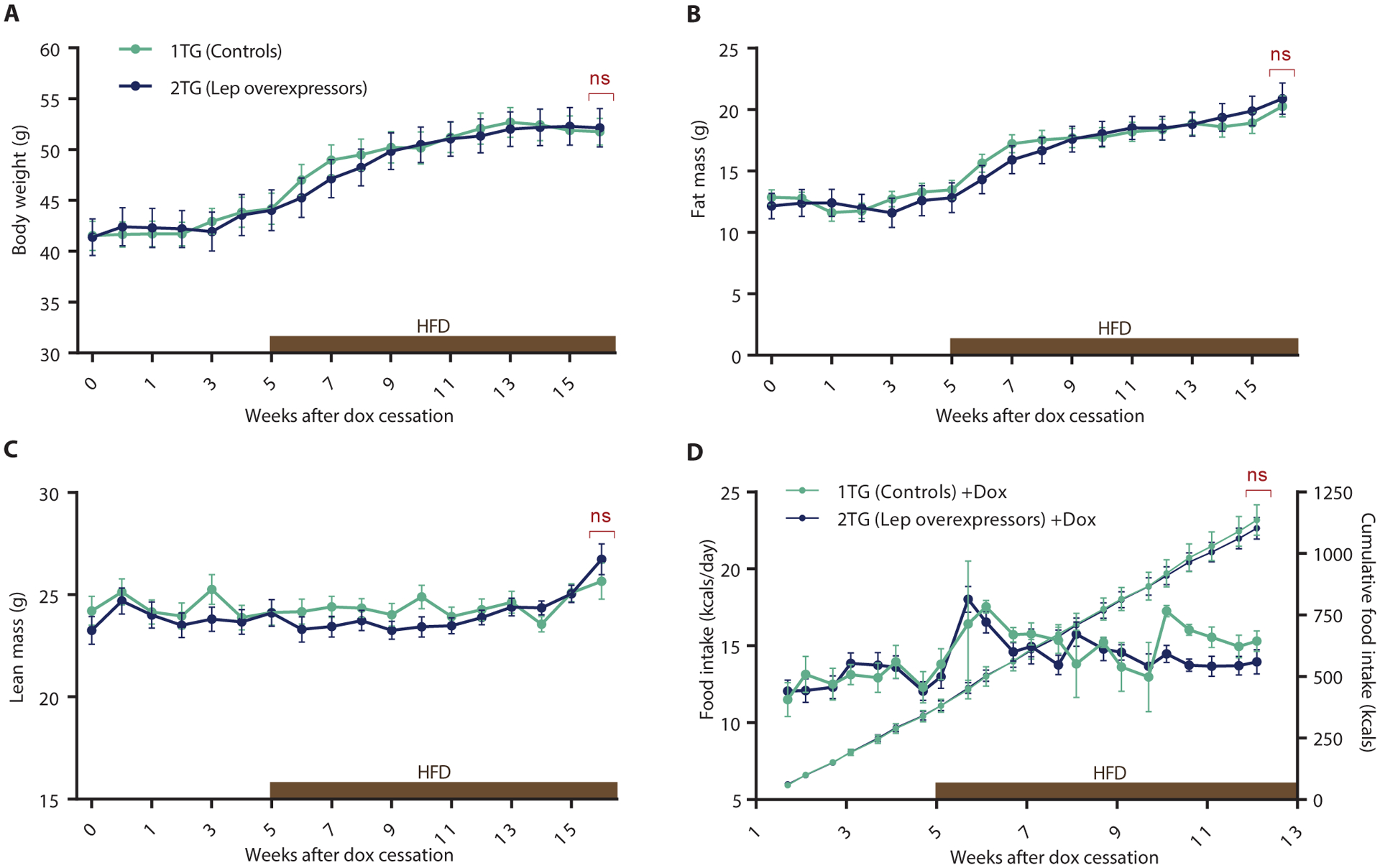
(A) Body weight (Mixed effect model with Fisher’s LSD post hoc analysis, genotype: F(1,32) = 0.22, p = 0.88), (B) Fat mass (Mixed effect model with Fisher’s LSD post hoc analysis, genotype: F(1,32) = 0.012, p = 0.91), (C) Lean mass (Mixed effect model with Fisher’s LSD post hoc analysis, genotype: F(1,32) = 0.37, p = 0.55), (D) Daily caloric food intake (left axis; two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,8) = 0.24, p = 0.63) and cumulative food intake (right axis) per mouse after dox was discontinued in 1TG controls and 2TG dox-induced leptin-overexpressing mice. Five weeks after dox cessation mice were switch from chow to 60% HFD (indicated by brown bar). All values are means ± SEM. Red brackets indicate a comparison by Student’s t-test of the final datapoint. *P < 0.01 between 1TG and 2TG groups by Student’s t-test (in red) or Fisher’s LSD (in black).
We repeated the experiment described above using a slightly different paradigm. Similarly to the previous experiment, adult male 1TG controls and 2TG lep-overexpressing mice were exposed to increasing concentrations of dox in water every 2 weeks for 20 weeks (fig. S4A). By the second dose of dox, the 2TG mice had gained less weight than 1TG mice and had significantly lower body weight (P < 0.01), fat (P < 0.01) and lean mass (P < 0.05); weight of 2TG mice recovered to the levels of controls within 4 weeks while still maintaining elevated circulating leptin concentrations (fig. S4, B–D). Both groups were then (7 weeks after the initiation of dox exposure) given ad libitum access to 60% HFD concurrently with dox exposure. Both groups increased their caloric intake when exposed to HFD and gained a similar amount of weight (~18 g over 12 weeks; fig. S4, B and E). Body weight and composition of the HFD 1TG and 2TG animals did not differ for the remainder of the dox/HFD exposure period or after their cessation (fig. S4 and S5).
Hyperleptinemia in “adolescent ” (P22-P56) male mice
At weaning (P22), mice were separated by genotype and 3 mice per cage were placedwith ad libitum access to chow, and given access to 50 |ig/ml of dox in drinking water for 5 weeks (Fig. 4A). In response to dox, circulating leptin concentrations increased in 2TG mice by about 25fold compared to 1TG controls. Leptin concentrations remained significantly elevated throughout the duration of the exposure (P < 0.001, Fig. 4B). Food intake was significantly reduced in 2TG mice compared to 1TG controls for 2 weeks after the switch to dox water (P < 0.01, Fig. 4C). Body weight, fat and lean mass of 2TG mice were significantly lower than 1TG mice during dox exposure (P < 0.001, Fig. 4, D–F).
Fig. 4. Dox-induced hyperleptinemia during “adolescent” period (P22-P56), followed by 60% HFD ad libitum at 14 weeks.
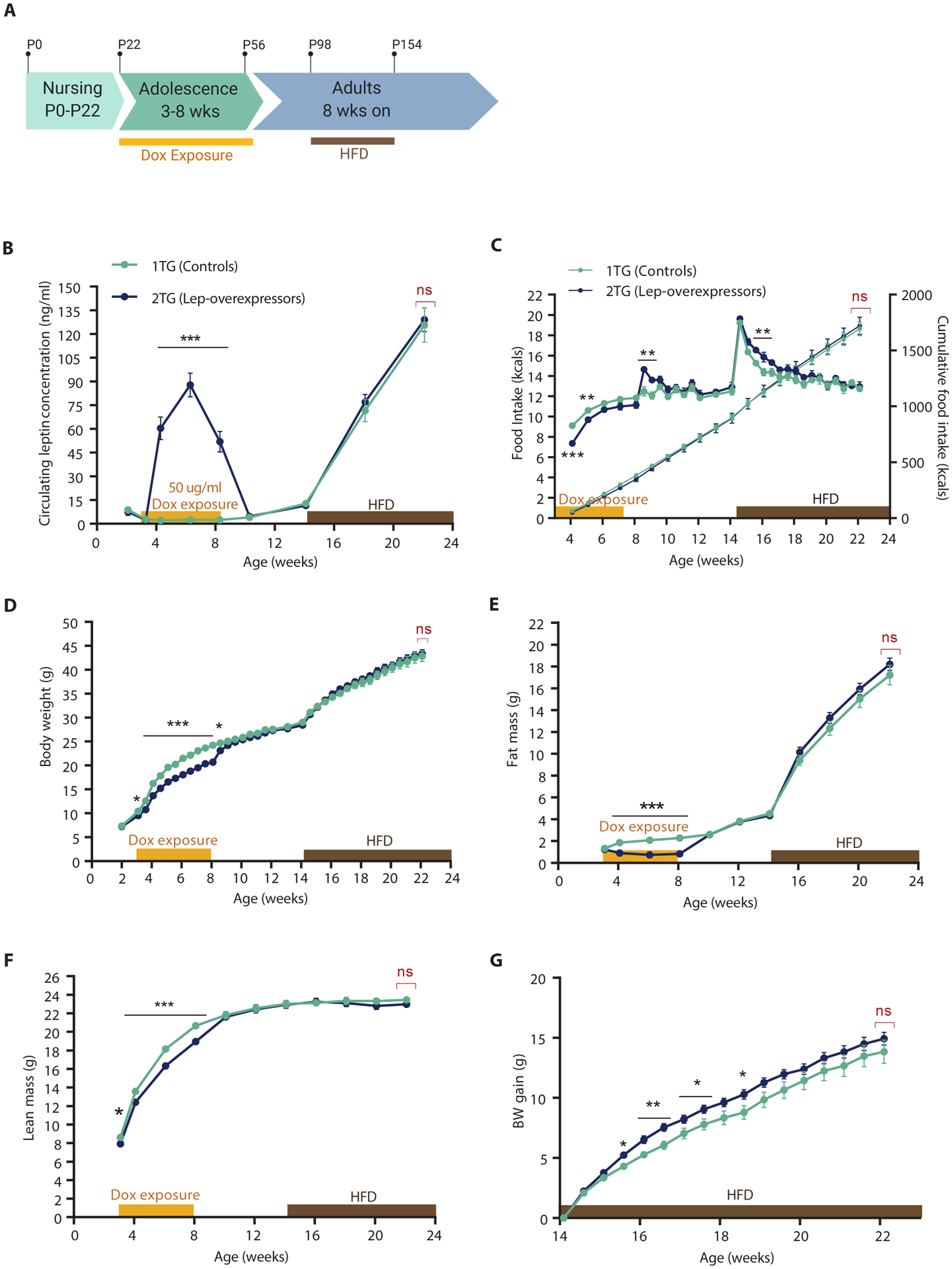
(A) Schematic of the study timeline. P, postnatal day. (B) Circulating leptin concentrations (Mixed effect model with Fisher’s LSD post hoc analysis, genotype: F(1,34) = 38.6, p < 0.0001), (C) Daily caloric food intake (left axis; two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,10) = 1.27, p = 0.29) and cumulative food intake (right axis), (D) Body weight (two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,34) = 1.56, p = 0.22), (E) Body weight gain (two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,34) = 3.01, p = 0.092), (F) Fat mass (two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,34) = 0.016, p = 0.90), and (G) Lean mass (two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,34) = 2.67, p = 0.11) of 1TG controls and 2TG dox-induced leptin-overexpressing mice throughout the study. All values are means ± SEM. Red brackets indicate a comparison by Student’s t-test of the final datapoint. Significance of differences between 1TG and 2TG groups calculated with Student’s t-test (in red) or post hoc Fisher’s LSD (in black), *p < 0.05, **p < 0.01, ***p<0.001.
At 8 weeks of ages dox was discontinued, and plasma leptin concentrations measured 1 week after dox withdrawal had returned to fat mass-proportional concentrations in the 2TG mice (Fig. 4B). After the sharp drop in circulating leptin, 2TG mice increased their food intake significantly for 1 week (P < 0.01, Fig. 4C) until their body weights increased to that of 1TG controls. Similarly, fat mass and lean mass were restored to those seen in 1TG mice within 1 week (Fig. 4, E and F). Once the 2TG mice caught up with controls, body weight, body composition, and food intake were not different from 1TG controls for the following 6 weeks while mice were maintained on chow (Fig. 4, C–F).
Both 1TG and 2TG mice were switched from chow to 60% (calories as fat) HFD at 14 weeks (6 weeks post dox exposure). During the first 2 weeks, hyperphagia in response to the HFD was significantly greater in 2TG compared to 1TG mice (P < 0.01, Fig. 4C). During that time, 2TG mice gained significantly more weight than the 1TG controls (P < 0.05, Fig. 4G). After 4 weeks of HFD exposure, caloric intake remained identical in both groups until the end of the study. No statistically significant difference was detected in body weight between 1TG and 2TG mice after 8-week exposure to HFD (P = 0.72, Fig. 4D).
Hyperleptinemia in postnatal (P0-P22) male and female mice
1TG dams were given dox in drinking water immediately following parturition until weaning of their progeny at P22 (Fig. 5A). Plasma leptin concentrations in 2TG lep-overexpressing pups at P15 and P22 were elevated compared to 1TG control littermates (Fig. 5B) and were higher on P22 than on P15, presumably as a result of mice ingesting dox through drinking water directly at P22 as opposed to having dox transferred primarily through mother’s milk at P15.
Fig. 5. Dox-induced hyperleptinemia in male and female mice during postnatal period (P0-P22) followed by switch from ad libitum chow to 60% HFD at 10 weeks.
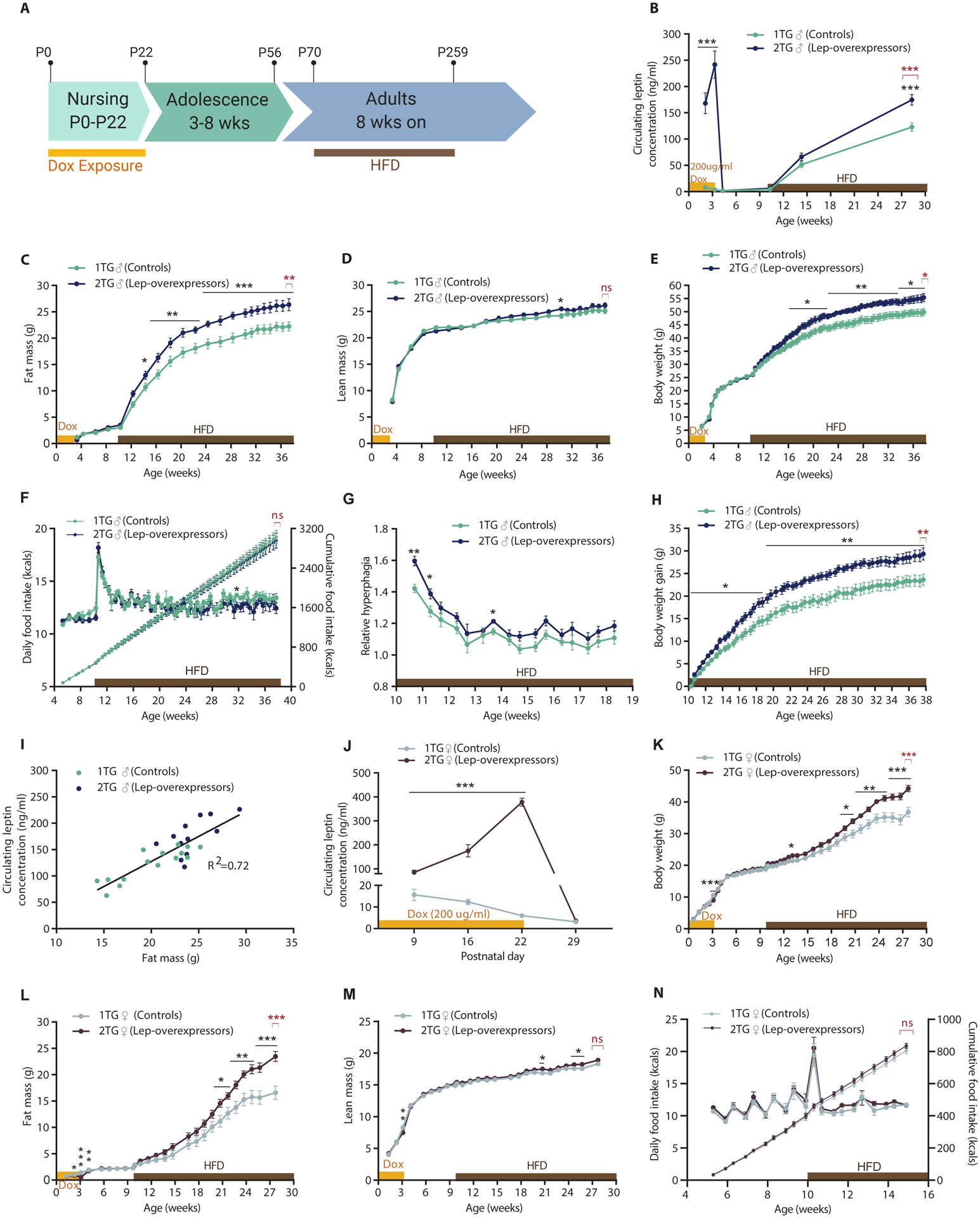
(A) Schematic of the study timeline. P, postnatal day. (B) Circulating leptin concentrations (Mixed effect model with Fisher’s LSD post hoc analysis, genotype: F(1,25) = 67, p < 0.0001), (C) Fat mass (Mixed effect model with Fisher’s LSD post hoc analysis, genotype: F(1,25) = 9.4, p < 0.01), (D) Lean mass (Mixed effect model with Fisher’s LSD post hoc analysis, genotype: F(1,25) = 0.062, p = 0.44), (E) Body weight (two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,25) = 7.0, p < 0.05), (F) Daily caloric food intake (left axis; two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,8) = 0.62, p = 0.45) and cumulative food intake (right axis) per mouse, (G) Fold increase in caloric intake after the initiation of HFD feeding (two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,8) = 5.0, p = 0.56), and (H) Body weight gain (two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,25) = 11.27, p < 0.01) of 1TG controls and 2TG dox-induced leptin-overexpressing mice throughout the study, and (I) Regression of fat mass vs. circulating leptin concentrations of male (♂) 1TG and 2TG mice at 28 weeks of age, after 18 weeks of HFD feeding. (J) Plasma leptin concentrations (Mixed effect model with Fisher’s LSD post hoc analysis, genotype: F(1,39) = 296.4, p < 0.0001), (K) Body weight (two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,39) = 6.74, p < 0.013), (L) Fat mass (Mixed effect model with Fisher’s LSD post hoc analysis, genotype: F(1,39) = 6.90, p < 0.05), (M) Lean mass (Mixed effect model with Fisher’s LSD post hoc analysis, genotype: F(1,39) = 2.23, p = 0.14), (N) Estimated daily caloric food intake (left axis; two-way RM ANOVA with Fisher’s LSD post hoc analysis, genotype: F(1,13) = 1.55, p = 0.24) and cumulative food intake (right axis) per mouse, of female (♀) 1TG controls and 2TG dox-induced leptin-overexpressing mice throughout the study. All values are means ± SEM. Red brackets indicate a comparison by Student’s t-test of the final datapoint. Significance of differences between 1TG and 2TG groups assessed with Student’s t-test (in red) or post hoc Fisher’s LSD (in black), *p < 0.05, **p < 0.01, ***p<0.001.
Body weight and composition were not significantly different between the 2TG lep-overexpressing and 1TG control male mice during the period of hyperleptinemia (P0-P22; Fig. 5C–E). At P22, pups were weaned and dox exposure was stopped (Fig. 5A). One week after dox exposure, plasma leptin concentrations, body weight, and composition did not differ between 2TG and 1TG mice (Fig. 5B).
Until 10 weeks of age mice were fed ad libitum chow, which resulted in no genotype-related differences in body weight, body composition, or food intake. Mice were then switched to 60% HFD. Both groups of mice increased food intake by about 50% in the first 3 days. There was no significant difference in caloric intake between the two groups (Fig. 5F). However, the relative hyperphagia of 2TG mice (defined as caloric intake after the switch to HFD divided by caloric intake on the last week of regular chow before the switch) was significantly higher in the first 7 days of HFD feeding (P < 0.05, Fig. 5G). Within the first 3 days of HFD feeding, 2TG mice gained more weight than controls (2TG: 2.6 ±0.2g, 1TG: 1.6 ±0.2g, P < 0.01). 2TG mice continued to gain body weight at a greater rate than 1TG, and at the end of the study (38 weeks of age) the difference in body weight gain by genotype was 5.7g (P < 0.01, Fig. 5H). The difference in absolute body weights reached significance 6 weeks after the start of HFD feeding (P < 0.05; Fig. 5E) and remained significant until the end of the study at 38 weeks of age (1TG: 49.9 ±1.3 g; 2TG: 55.4 ±1.5 g; P < 0.05; Fig. 5E). Throughout the study, lean mass did not differ by genotype (Fig. 5D). Fat mass was significantly higher in 2TG mice 4 weeks after the diet switch (P < 0.05) and remained higher until the end of the study (p < 0.001; Fig. 5C). Circulating leptin concentrations were measured 4 and 18 weeks after the start of HFD exposure and were significantly higher in 2TG mice after 18 weeks of HFD feeding (1TG: 122.9 ±8.0 ng/ml, 2TG: 172.6 ±9.5 ng/ml; P < 0.001; Fig. 5B). Circulating leptin concentrations were proportional to fat mass in animals of both genotypes (Fig. 5I).
We repeated the experiment described above in female mice under similar conditions, withxposure to 200 μg/ml of dox in drinking water during the first three weeks of life, followed by HFD feeding at 10 weeks of age (Fig. 5A). The first measurements of plasma leptin concentrations were made at P9 (a week earlier than in the male cohort), and then at P16 and P22. Plasma leptin concentrations were significantly elevated throughout the dox exposure (P < 0.001, Fig. 5J).
Fat mass was lower in 2TG lep-overexpressing females compared to 1TG controls between P16 and P28. At this age mice have very little body fat, nonetheless, the 2TG females had less fat than 1TG controls (Fig. 5L). In addition, lean mass and body weight were decreased at P22 in the 2TG vs 1TG group (Fig. 5, K and M). In males, we did not detect any differences in body weight or composition at this time.
Similar to the male cohort, female 2TG mice did not differ from the 1TG group in body weight, body composition, or food intake while maintained on ad libitum chow. Once exposed to HFD, 1TG and 2TG female mice gained weight at a slower rate than males, and hyperphagia (relative to chow) in both 1TG and 2TG females was detected only during the first 3 days after initiation of HFD exposure (Fig. 5N). However in males, HFD feeding induced hyperphagia for the duration of HFD exposure (Fig. 5F). There was no detectable difference in mean daily or cumulative food intake between the female 1TG and 2TG animals during the first 5 weeks of the HFD (Fig. 5N). After approximately 6 weeks of ad libitum HFD feeding, the females began to gain weight at a faster rate than the males, and 2TG females started to increase in body weight and fat mass compared to 1TG females, with genotype-related differences in body weight and fat mass reaching statistical significance at 10 and 11 weeks of HFD feeding, respectively (Fig. 5, J and K). In females, the final differences in body weight and fat mass at 28 weeks of age, by genotype, were 7.5g (P < 0.001) and 6.9g (P < 0.001), respectively (Fig. 5, J and K). Lean mass was not different by genotype at the end of the study (Fig. 5M).
After 16 weeks of HFD feeding, a subset of 16 females (n=8 for each genotype) were housed in metabolic cages (TSE Systems) to assess energy expenditure (fig. S6). Absolute energy expenditure and energy expenditure per unit of lean mass were not significantly different between 1TG and 2TG female mice (fig. S6A–B). Neither cumulative food intake over the 6-day measurement (fig. S6C) nor the respiratory exchange ratio (fig. S6D) differed significantly between the 1TG and 2TG groups.
Discussion
Genetic changes cannot account for the rapid increase in obesity prevalence in the past 4 decades, and the relevant environmental factors are challenging to dissociate experimentally. Among potential factors of interest is exposure to hypernutrition in early infancy (10, 11). We used transient elevation in circulating leptin as a surrogate for transient increases in adiposity, in order to avoid the confounding effects of hyperphagia (35), diet composition (34, 36), and weight change (34, 37). Obese or HFD-fed animals have complex metabolic phenotypes that include increased circulating free fatty acids, elevated glucose, decreased insulin sensitivity, increased circulating insulin, and fatty liver. However, the transgenic mouse model used in this study isolates hyperleptinemia from confounders of HFD feeding and maternal obesity. Additionally, mice in the experimental and control groups share dams and are therefore developed under shared embryonic and postnatal conditions with leptin concentrations being the only factor differing between the groups.
In the leptin/rtTA double transgenic mouse, leptin produced by the transgene is bioactive and emanates primarily from secretory cells in the gut. The transient weight loss of the animals in response to dox is proportional to circulating concentrations of leptin, consistent with intact conventional leptin physiology in these animals.
We evaluated the effects of transient hyperleptinemia at three distinct developmental periods on body weight and subsequent responses to a highly palatable food. We found that inducing chronic hyperleptinemia in adult mice does not increase long term body weight after hyperleptinemia is discontinued and does not affect hyperphagia in response to a HFD. We then showed that hyperleptinemia during “adolescence” does not alter body weight after the cessation of dox exposure, but does transiently increase the intake of a HFD, without affecting weight gain or body composition after 8 weeks of being fed a HFD. Lastly we found that transient elevation of circulating leptin in the immediate postnatal period increases the hyperphagic response to a HFD diet and renders animals more susceptible to obesity as adults, 7 weeks after their transient hyperleptinemia. These results emphasize the importance of the timing of exposure to hyperleptinemia on the phenotypes of progeny. We identified the immediate postnatal period (P0-P22) as a critical time window during which exposure to elevated leptin increases the body weight in adult offspring under conditions of ad libitum access to highly palatable food.
We found no effect on subsequent body weight of transient hyperleptinemia in adult mice. This result is consistent with an earlier study by our group (21). The absence of leptin’s effect on body weight is consistent with leptin not providing a physiological potent defense against fat gain (38). It was shown that chronic (18 weeks) elevation of circulating leptin by administration of exogenous leptin via osmotic pumps did not trigger the physiological or behavioral mechanism in mice to defend a higher body weight (21). One of the limitations of this study is that the amount of exogenous leptin delivered via a mini-pump was limited due to low leptin solubility at high concentrations. Additionally, delivery efficiency of exogenous leptin via the mini-pump was highly variable in individual mice, and the repeated surgeries necessary to replace minipumps caused weight loss. These issues were avoided in the present study.
Inducing hyperleptinemia for 5 weeks in “adolescent” mice did not affect long-term body weight in response to HFD feeding ad libitum. Transgenic leptin expression induced by dox administration resulted in transiently reduced chow intake and slowed weight gain in 2TG versus 1TG controls only during active leptin overexpression. When challenged with HFD, 2TG mice increased food intake transiently and gained more weight than 1TG controls within the first 4 weeks of HFD exposure but, ultimately (at 8 weeks of HFD), there were no differences in body weight or body fat between 1TG and 2TG groups. Greater hyperphagia in response to highly palatable diet in the 2TG mice suggests that exposure to hyperleptinemia during P22-P56 may have altered leptin-responsive neural circuitry.
In mice, a physiologic leptin surge (5–10 fold increase in circulating leptin concentrations) occurs between P8-P12, independent of pup fat mass (39). This surge—the mechanism of which is unclear—is a major developmental signal that affects the outgrowth of neuronal projections from the ARH to PVH, DMH, and LHA involved in feeding circuits (17). Maternal HFD feeding during nursing causes the leptin surge in pups to initiate earlier with higher intensity and to last for a longer period of time; the pups have a higher body weight compared to the offspring of chow-fed dams (29). This surge is not due to leptin secreted in breast milk (29, 40–42).
We found that isolated hyperleptinemia during the immediate postnatal period (P0-P22) predisposed mice (both males and females) to hyperphagia and increased weight gain when exposed to highly palatable HFD at 10 weeks of age. At 3 weeks of age, mice have little body fat. Nonetheless, hyperleptinemic 2TG female mice had reduced body weight due to decreased somatic fat compared to 1TG mice. Exogenous leptin in mice does not suppress food intake during the first 2 weeks of life (43, 44), but is an important neurotrophic factor affecting the development of ARH projections to downstream hypothalamic targets (17). It is possible that decreased fat mass in the hyperleptinemic 2TG female mice was caused by decreased energy intake of the 2TG pups, and that effect may constitute a confounding factor with regard to the interpretation of possible leptin-specific effects in the CNS, especially as postnatal undernutrition in rodents can cause growth retardation and decreases body weight throughout life even when animals are subsequently exposed to high fat diet (45, 46). The effect of this potential confounder would presumably be to reduce the magnitude of the increases in body weight and fat that we observed in adult HFD-fed animals following transient hyperleptinemia at P0-P22. That is, the bias would be against our inference of a genotype-mediated difference in body weight.
One to two weeks after release from hyperleptinemia and ad libitum chow feeding, male and female 1TG and 2TG mice were indistinguishable in body weight, composition, and circulating leptin concentrations until 10 weeks of age when they were switched to 60% HFD. Immediately following HFD exposure, 2TG males and females gained more weight than 1TG littermates. In male mice the difference in fat mass was approximately 4 grams per mouse after 14 weeks of HFD, representing an increase of only 38 kcals out of approximately 1300 kcals ingested over this period (47), or a positive energy balance of 3%. This difference is not detectable with current instrumentation for energy intake or expenditure. It should be noted that we estimated energy intake on a per-cage basis, whereas all body weights, composition and energy expenditure measurements were determined in individual animals. Based upon these considerations, it appears most likely that the genotype-related differences in weight gain of the P0-P22 animals fed a HFD were due primarily to subtle effects on food intake.
Transgenic mice that congenitally overexpress leptin have been described previously, and their body weight phenotypes are consistent with our results (48, 49). These congenital leptin-overexpressing mice initially display reduced body weight and fat stores, but by 33 weeks of age are indistinguishable in body mass and composition from wild type littermates. Mice congenitally overexpressing leptin (off the adipocyte-specific aP2 promoter) are more prone to high fat diet-induced obesity when exposed to 20 weeks of high fat diet initiated at 9 weeks of age (50). The congenital leptin-overexpressing mouse models suggests that the phenotype we report in mice that are postnatally hyperleptinemic (P0-P22) is specifically a result of elevated leptin exposure as opposed to the withdrawal of transgenic leptin at the time of weaning. Although transgenic mice that constitutively overexpress leptin are useful in assessing the consequences of life-long hyperleptinemia, congenital models do not permit assessment of the effects of transient hyperleptinemia at specified developmental time points.
Three other groups have studied the physiological consequences for body weight of postnatal exogenous leptin supplementation in rats; these studies varied in the timing and route of leptin administration and have somewhat conflicting results. One study administered leptin orally to rats from P0 to P20. At weaning rats were fed ad libitum HFD, and at 6 months of age, the leptin-fed rats had ~7% lower body weight than the controls (51). Plasma leptin concentrations in leptin-supplemented pups were not reported; therefore, it is unclear whether oral leptin elevated circulating leptin concentrations. Another study administered leptin to neonatal rats via IP injections either during the first or last 10 days of nursing and showed that when fed chow ad libitum, by 16 weeks of age, leptin-injected rats had 11% and 10% higher body weights, respectively, than controls (52). When suckling rats were injected with IP leptin from P3 to P13, the leptin-supplemented rats gained 10% more weight than controls after they were switched to HFD at weaning (53). The latter two studies are consistent with results reported in this manuscript. In both studies, neonatal leptin treatment lead to transient decreases in weight gain of the pups (52, 53).
Under normal physiological conditions, leptin is secreted in a diurnal pattern with highest plasma concentrations at night, and lowest in the morning (32). The half-life of leptin is about 40 minutes with a sharp peak 30 min after intraperitoneal (IP) injection and full clearance within 4h (33). Thus, in studies using daily injections to manipulate circulating leptin concentrations, large transient spikes of non-physiological leptin concentrations are created, followed by a 20-hour period during which plasma concentrations are not elevated. Although the leptin-overexpressing mouse reported here does not fully recapitulate normal circadian physiology either, the mouse has ad libitum access to dox-supplemented water throughout the 24h period.
The mechanisms that might mediate the effects of transiently elevated circulating leptin concentrations during the postnatal period on subsequent body weight are unclear. Leptin has been shown to play a neurotrophic role in the development of feeding circuitry (17). In Lepob/ob mice, ARH projections to hypothalamic regions (the PVH, DMH, and LHA) are greatly reduced, and the formation of these pathways is temporally delayed. For example, in WT mice ARH neurons innervate the PVH on P12, whereas in Lepob/ob littermates no axons from the ARH are detected at this time (17). Administration of leptin to P4-P12 Lepob/ob mice restores the density of the innervation from the ARH to the PVH to the levels of a WT control. However, administering leptin to adult Lepob/ob mice does not rescue the density of AgRP and α-MSH immunoreactive innervation in the PVH to levels seen in WT mice, indicating that there is a restricted neonatal time window in which ambient leptin impacts the formation of ARH connections (17). It has been shown that the offspring of dams exposed to HFD during gestation and lactation gained more weight than offspring of dams fed regular chow, and displayed decreased hypothalamic leptin sensitivity (reduced leptin-induced pSTAT3 in the ARH and VMH) at P30 and P90 (29). The density of AgRP immunoreactive fibers in the PVH was decreased in the offspring of HFD fed dams compared to chow-fed dams (29). The authors hypothesized that early hyperleptinemia induces leptin resistance, thereby attenuating leptin signaling and impairing the development of hypothalamic projections (29). The most critical time for the effects of maternal HFD feeding on offspring body weight is during the immediate postnatal period (birth to weaning) and the fiber density of AgRP and α-MSH from ARH to three areas downstream of the hypothalamus (PVH, DMH and LHA) is reduced in progeny of HFD-fed dams (20). Similar alterations in hypothalamic circuits are reported in Lepob/ob mice (17). We found the pre-weaning period to be critical for the effects of hyperleptinemia on body weight and response to a HFD later in life. It is plausible that the neurobiological changes seen in the offspring of HFD-fed dams result, at least in part, from the hyperleptinemia associated with increased body fat of the pups. The normal plasticity of hypothalamic development during the 2nd postnatal week renders this an optimal time window for permanent structural impact of altered humoral signals such as leptin.
Other potential mechanisms linking hyperleptinemia during the postnatal period to developmental programming include epigenetic modifications, hypothalamic inflammation, and neuroanatomic changes in hedonic or reward circuits. Over-nutrition of weanling rats (by decreasing litter size) during the perinatal period results in hypermethylation of the hypothalamic POMC promoter which is negatively correlated with hypothalamic POMC expression adjusted for circulating leptin concentrations (54).
Exposure to highly palatable food results in 1–2 weeks of hyperphagia in adult mice (55). This response is mediated, at least in part, by hedonic, reward-based mechanisms that can override homeostatic regulation during abundant food availability and drive consumption of highly palatable foods in the absence of hunger (56). The neuroanatomy of the hedonic circuits may also be affected by hypernutrition in the postnatal period. Because the increased weight gain in mice that were postnatally hyperleptinemic is evident only after exposure to a highly palatable diet, it is likely that hedonic pathways are affected in these mice (57).
Finally, studies implicate hypothalamic inflammation as a mediator of DIO. It has been demonstrated that within 1–3 days of HFD feeding, prior to substantial weight gain, mice develop hypothalamic inflammation characterized by activation of local microglia and astrocytes (gliosis) (58). In rodents, microglia cell number in the brain markedly increases in the first two weeks of postnatal life (59), coinciding with the leptin surge, then starts declining in the 3rd postnatal week until it reaches mature levels by 6 weeks of age (59). The proportion of microglia is lower in Lepob/ob mice fed chow or HFD (60), suggesting that leptin is a partial mediator of microglial activation. Administration of leptin between P8-P12 increases proliferation of astrocytes in the hypothalamus, whereas deletion of LepRb from astrocytes decreases astrogenesis (61). Additionally, leptin-induced suppression of food intake is reduced in astrocyte-specific LepRb adult knockout mice (62). These data suggest that high ambient hypothalamic leptin during development may influence the susceptibility to HFD later in life via effects on proliferation and activation of astrocytes.
In humans, maternal circulating leptin is elevated throughout gestation, peaks at ~ 1.5–2 fold above the concentration accounted for by adipose tissue mass in the late second/early third trimester of pregnancy (63–66), decreases within 3 days postpartum to concentrations below those present pregravid (67, 68), and then gradually increases to concentrations appropriate to fat mass by 6 months postpartum (67). In humans, umbilical vein leptin concentrations are positively correlated with neonatal fat mass and gestational size, suggesting that in humans, fetal adipose tissue is the primary source of circulating leptin (69, 70).
The developmental timing of brain maturation differs greatly between mice and humans. In this regard, the immediate postnatal period in mice is analogous to the 3rd trimester of human (or primate) gestation (71). In rodents the hypothalamic circuitry is immature until the 3rd week of postnatal life, whereas in primates this circuitry matures functionally in utero (17, 72). From this perspective, studies investigating the effects of HFD feeding and obesity in postnatal rodents may be more relevant to the neuro-developmental impact of maternal obesity and metabolic status during human late gestation.
Nutritional changes during critical developmental periods can have lasting effects on energy homeostasis. The hypothalamus is particularly sensitive to changes in the hormonal milieu. In mice during the perinatal period, changes in circulating leptin (17), insulin (20, 28) and ghrelin alter the architecture of key hypothalamic centers involved in energy and glucose homeostasis, however the mechanisms of these developmental effects remain unclear. Many physiological and molecular changes are triggered by obesity and HFD feeding; however, the direct mediators of phenotypes observed in offspring are largely unknown. In this study we identified the immediate postnatal period in mice as a critical time window in which exposure to hyperleptinemia is associated with alterations in subsequent responses to highly palatable food. Further studies are needed to define the cell-molecular mechanism(s) by which hyperleptinemia during the postnatal period in mice mediates the developmental programming of body weight regulation. In humans, the development of energy homeostasis neurocircuits may be influenced by the environmental cues—including the leptin concentrations. The critical time window in humans that is analogous to the suckling period in mice (P0-P22) likely occurs from the 3rd trimester into early infancy (71).
There are several limitations of this work. In our study, hyperleptinemia in the immediate postnatal period transiently decreased fat mass, possibly as a consequence of reduced caloric intake. The difference in fat mass and body weight between 1TG and 2TG mice could potentially be larger than observed if the 2TG mice maintained the same fat mass as 1TG mice during the immediate postnatal period. There are also some limitations of the leptin transgenic mouse model. The 2TG mice had a wide range of responses of circulating leptin concentrations to the same concentrations of dox in the drinking water. Much of this variability was probably due to differences in water intake. Bacterial DNA within transgenes has been reported to cause stochastic silencing of transgenes in mice (73), however, and it is possible that some of the variability in transgenic leptin expression seen in our LepTg/rtTA double transgenic mice was due to mosaic expression. At very high doses of dox, ectopic leptin production in the hypothalamus could contribute to the functional consequences of dox exposure. In some experimental circumstances this effect could confound mechanistic inferences regarding effects of transient leptin overexpression on subsequent ingestive behaviors. At doses of dox that produce circulating leptin concentrations that are within the physiological range, this potential hypothalamic effect is likely not substantial as the dose administered to the adolescent mice and the maximal dose administered to the adult mice was 6-fold lower than the dose which induced low amounts of leptin secretion in the hypothalamic incubates. Production of leptin in the gastrointestinal tract and the liver of dox-treated animals could also possibly influence vagal signaling by paracrine effects. Our dox-treated animals showed the anticipated behavioral responses to plasma concentrations of leptin that are comparable to those in animals administered exogenous leptin (74). Hence, we do not believe that local effects of ectopic leptin production are physiologically meaningful.
Materials and Methods
Study Design
The objective of the study was to determine the effects of elevated leptin in mice at different stages of life (nursing period, adolescence, and adulthood) on body weight later in life and the subsequent susceptibility of mice to gain weight when exposed to high fat diet. The study included a series of controlled laboratory experiments carried out in a transgenic mouse model. The transgenic mice used in the experiments were generated on a mixed genetic background of C57BL/6 and 129/Sv and back-crossed to C57BL/6J for at least 4 generations. All experiments were performed in mice from P0 up to 37 weeks of age. The transgenic experimental and control mice used in these experiments were born in the same litters at the approximate ratio of 1:1 and all of them were exposed to doxycycline; therefore, no additional randomization was necessary. The investigators were not blinded while collecting and analyzing the data because the mice had to be assigned to appropriate cages per genotype. Sample sizes were based on previous experience with similar types of experiments. All mice in each cohort were born at the same time. Data collection was stopped early for mice with skin lesions (common side effect of extended high fat diet feeding). The data for these mice were excluded from the analysis. Experiments investigating adult and postnatal (P0-P22) hyperleptinemia were replicated in independent cohorts of mice.
Animals
Throughout the study, animals were maintained at room ambient 22–24°C with a 12-h dark-light cycle (lights on at 0700h) in a pathogen-free barrier facility. Mice were maintained on 9% fat chow (Purina LabDiet, 22% calories from fat) or fed high fat diet (Research Diets, Inc.; 60% calories from fat; HFD) as indicated. The protocol was approved by the Columbia University Institutional Animal Care and Use Committee.
All experiments were carried out with dox-inducible Leptin/rtTA double transgenic mice. Leptin/rtTA double transgenic mice were generated using a commercially available KH2 embryonic stem (ES) cell line (30) (Mirimus, Inc; details on the transgenic mouse are provided in supplementary methods).
Hyperleptinemia in adult (P63-P203) male mice
Lep-overexpressing (2TG, n=21) and control (1TG, n=15) mice shared dams and were born and raised in the same litters. After weaning, mice were group-housed 3 per cage by genotype with ad libitum access to chow and water. Dox exposure began at 9 weeks of age and all mice were fed chow ad libitum during this period. Mice were exposed to increasing concentrations of dox in 5% sucrose water (for palatability) every two weeks. Dox water was changed twice weekly. Baseline blood was collected 1 week prior to dox exposure. Dox concentrations in water were 5, 7.5, 12.5, 17.5, 20, 22.5, 27.5, 35, 40, and 45 μg/ml and were increased stepwise every two weeks. Circulating leptin concentration was measured from plasma isolated from submandibular blood every two weeks, one week after each dox dose escalation. Glucose was measured in submandibular whole blood. Dox degrades over time, as was observed on week 22 when circulating leptin concentrations were lower than the previous period despite higher nominal concentration of dox in the drinking water. A new batch of dox was purchased for the 24-week exposure.
Body weight and composition were measured weekly throughout the experiment. Food was placed on the wire cage tops and food intake was measured twice a week for all mice (on a per cage basis) throughout the study. During dox exposure, water intake was measured twice a week to ensure that there was no dox (taste) effect on the amount of water consumed.
Dox-free water was provided after 20-weeks of dox exposure. Monitoring of body weight, composition and food intake continued weekly after dox exposure was discontinued. Five weeks after release from dox exposure, mice were switched from chow to 60% HFD. Mice were sacrificed 16 weeks after release from hyperleptinemia.
Two mice from were excluded from the analysis: one died during MRI assessment (1TG group, at 29 weeks of age, fed chow diet); the other animal (2TG group, at 22 weeks of age, fed chow diet) was not gaining weight due to skin lesions. Exclusion of the first mouse decreased the mean body weight of 1TG group by 0.19% at 29 weeks of age and exclusion of the second mouse increased the mean body weight of 2TG group by 0.9% at 22 weeks of age.
Hyperleptinemia in “adolescent ” (P22-P56) male mice
1TG (n=18) and 2TG (n=18) littermates were weaned on P22. At weaning, pups were separated by genotype and group-housed 3 per cage with ad libitum access to chow. At the same time mice began a 5-week exposure to 50 μg/ml of dox in drinking water. Blood was collected at P15 and P22 for baseline plasma leptin measurement and at weeks 4, 6, and 8 during dox exposure. At 8 weeks of age, mice were released from dox exposure and continued ad libitum access to chow. At 14 weeks (6 weeks post release from dox) both groups were switched to ad libitum 60% HFD. Throughout the experiment, body weight and food intake were recorded twice per week on a per cage basis. Body composition was measured biweekly. Plasma leptin was measured every 4 weeks during the post-dox period.
Hyperleptinemia in postnatal (P0-P22) male mice
1TG females homozygous for the TRE-Lep transgene but non-carriers for the R26-rtTA allele were crossed to 1TG males heterozygous for R26-rtTA but non-carriers for the TRE-Lep insert. This strategy resulted in all offspring carrying the TRE-Lep/+ gene and about half segregating with rtTA/+. Live offspring were born with the expected 1:1 ratio of 1TG and 2TG. At parturition, mothers were exposed to 200 μg/ml of dox in drinking water for 3 weeks with ad libitum access to chow. During nursing period (P0-P22), pups primarily drink mother’s milk and not the dox-supplemented water; hence the effective dox dose in postnatal pups is diluted in mother’s breast milk. Plasma was collected on P15 and P22. At weaning, mice were separated by genotype (1TG, n=15; 2TG, n=14) into home cages (group-housed, 2–3 per cage) with ad libitum access to chow and dox-free water. At 10 weeks, mice were given ad libitum access to 60% HFD and maintained on this diet until the end of the study. Body weight and food intake per cage were collected twice per week. Body composition was measured biweekly throughout the experiment.
Two mice (genotypes = 2TG) in the postnatal hyperleptinemia experiment were sacrificed prior to termination of the planned study period due to skin lesions (on weeks 10 and 24, respectively, of HFD feeding the two mice showed early signs of illness). These mice were excluded from analyses. The first mouse was sacrificed relatively early in the study and its exclusion decreased the difference in body weight and fat mass between genotypes by 1% and 2%, respectively, in the first 10 weeks of HFD feeding. Exclusion of the second mouse reduced the difference in body weight and fat mass by 1% at the end of the study.
Statistical analysis
Data are expressed as means ± SEM. GraphPad PRISM 8.0 software was used for statistical analyses. 2-tailed Student’s t-tests were used to compare 1TG and 2TG groups on the final datapoints for each phenotype. P < 0.05 was taken as significant. The data were also analyzed using a two-way repeated measure ANOVA (RM ANOVA) or mixed effect model (for datasets with any missing values) followed by a post hoc comparisons using a Fisher’s least significant difference (LSD). GraphPad Prism 8.0 (https://www.graphpad.com/) offers a mixed effect model which is specifically designed to analyze the data sets instead of RM ANOVA when values are missing. This mixed model uses a compound symmetry covariance matrix and is fit using restricted maximum likelihood (REML). When the mixed effect model method is used on data sets without missing values, the p-values and post-hoc test results are the same as when analyzed with RM ANOVA. Red asterisks in figures indicate significance detected by Student’s t-test, whereas black asterisks denote the significance detected by the Fisher’s LSD. Data were tested for normality prior to use of RM ANOVA, t-tests and mixed effect model.
Supplementary Material
Fig. S1. Validation of leptin-overexpressing ES cells and leptin-overexpressing mice.
Fig. S2. Bioactivity of leptin.
Fig. S3. Circulating leptin concentrations during postnatal dox exposure.
Fig. S4. Dox-induced chronic hyperleptinemia (P63-P203) in adult mice with concurrent HFD feeding.
Fig. S5. Release of adult mice from dox-induced chronic (P63-P203) hyperleptinemia and HFD feeding.
Fig. S6. Energy expenditure assessment (indirect calorimetry) at 26 weeks of age in postnatally (P0-P22) hyperleptinemic female mice after 16 weeks of HFD feeding.
Supplementary Excel Data file S1 [Primary data file used to generate main figures].
Supplementary Excel Data file S2 [Primary data file used to generate supplementary figures].
Acknowledgments
Funding: This work was supported by research grants from National Institutes of Health: RO1 DK52431 to RLL, T32 HL007343 to AAS and F30DK108564 to YG. NY Obesity Research Center: P30 DK026687 to RLL. The Russell Berrie Foundation Program in the Neurobiology of Body Weight Regulation to CAL;
Footnotes
Competing Interests: The authors declare that they have no competing interests;
Data and materials availability: All data associated with this study can be found in the paper or supplementary materials. The raw data that support the findings of this study are available in a supplemental data file. The TRE-Lep/Rosa26-rtTA double transgenic mouse is available from the corresponding author upon request.
References and Notes
- 1.Freedman DS, Khan LK, Serdula MK, Dietz WH, Srinivasan SR, Berenson GS, The relation of childhood BMI to adult adiposity: the Bogalusa Heart Study. Pediatrics 115, 22–27 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Geserick M, Vogel M, Gausche R, Lipek T, Spielau U, Keller E, Pfaffle R, Kiess W, Korner A, Acceleration of BMI in Early Childhood and Risk of Sustained Obesity. N Engl J Med 379, 1303–1312 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Silventoinen K, Rokholm B, Kaprio J, Sorensen TI, The genetic and environmental influences on childhood obesity: a systematic review of twin and adoption studies. Int J Obes (Lond) 34, 29–40 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JR, Elliott KS, Lango H, Rayner NW, Shields B, Harries LW, Barrett JC, Ellard S, Groves CJ, Knight B, Patch AM, Ness AR, Ebrahim S, Lawlor DA, Ring SM, Ben-Shlomo Y, Jarvelin MR, Sovio U, Bennett AJ, Melzer D, Ferrucci L, Loos RJ, Barroso I, Wareham NJ, Karpe F, Owen KR, Cardon LR, Walker M, Hitman GA, Palmer CN, Doney AS, Morris AD, Smith GD, Hattersley AT, McCarthy MI, A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316, 889–894 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armitage JA, Poston L, Taylor PD, Developmental origins of obesity and the metabolic syndrome: the role of maternal obesity. Front Horm Res 36, 73–84 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Cohen-Cole E, Fletcher JM, Is obesity contagious? Social networks vs. environmental factors in the obesity epidemic. J Health Econ 27, 1382–1387 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Ogden CL, Carroll MD, Kit BK, Flegal KM, Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 311, 806–814 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenbaum M, Leibel RL, The physiology of body weight regulation: relevance to the etiology of obesity in children. Pediatrics 101, 525–539 (1998). [PubMed] [Google Scholar]
- 9.Reynolds RM, Osmond C, Phillips DI, Godfrey KM, Maternal BMI, parity, and pregnancy weight gain: influences on offspring adiposity in young adulthood. J Clin EndocrinolMetab 95, 5365–5369 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Owen CG, Martin RM, Whincup PH, Smith GD, Cook DG, Effect of infant feeding on the risk of obesity across the life course: a quantitative review of published evidence. Pediatrics 115, 1367–1377 (2005). [DOI] [PubMed] [Google Scholar]
- 11.von Kries R, Koletzko B, Sauerwald T, von Mutius E, Barnert D, Grunert V, von Voss H, Breast feeding and obesity: cross sectional study. BMJ 319, 147–150 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levin BE, Metabolic imprinting: critical impact of the perinatal environment on the regulation of energy homeostasis. Philos TR Soc B 361, 1107–1121 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mann T, Tomiyama AJ, Westling E, Lew AM, Samuels B, Chatman J, Medicare’s search for effective obesity treatments: diets are not the answer. Am Psychol 62, 220–233 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Caprio S, Daniels SR, Drewnowski A, Kaufman FR, Palinkas LA, Rosenbloom AL, Schwimmer JB, Influence of race, ethnicity, and culture on childhood obesity: implications for prevention and treatment. Obesity (Silver Spring) 16, 2566–2577 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Amstalden M, Garcia MR, Williams SW, Stanko RL, Nizielski SE, Morrison CD, Keisler DH, Williams GL, Leptin gene expression, circulating leptin, and luteinizing hormone pulsatility are acutely responsive to short-term fasting in prepubertal heifers: relationships to circulating insulin and insulin-like growth factor I(1). Biol Reprod 63, 127–133 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Jequier E, Leptin signaling, adiposity, and energy balance. Ann N YAcadSci 967, 379–388 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Bouret SG, Draper SJ, Simerly RB, Trophic action of leptin on hypothalamic neurons that regulate feeding. Science 304, 108–110 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Bouret SG, Draper SJ, Simerly RB, Formation of projection pathways from the arcuate nucleus of the hypothalamus to hypothalamic regions implicated in the neural control of feeding behavior in mice. JNeurosci 24, 2797–2805 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morabito MV, Ravussin Y, Mueller BR, Skowronski AA, Watanabe K, Foo KS, Lee SX, Lehmann A, Hjorth S, Zeltser LM, LeDuc CA, Leibel RL, Weight Perturbation Alters Leptin Signal Transduction in a Region-Specific Manner throughout the Brain. PLoS One 12, e0168226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vogt MC, Paeger L, Hess S, Steculorum SM, Awazawa M, Hampel B, Neupert S, Nicholls HT, Mauer J, Hausen AC, Predel R, Kloppenburg P, Horvath TL, Bruning JC, Neonatal insulin action impairs hypothalamic neurocircuit formation in response to maternal high-fat feeding. Cell 156, 495–509 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ravussin Y, LeDuc CA, Watanabe K, Mueller BR, Skowronski A, Rosenbaum M, Leibel RL, Effects of chronic leptin infusion on subsequent body weight and composition in mice: Can body weight set point be reset? MolMetab 3, 432–440 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barker DJ, The fetal and infant origins of adult disease. BMJ 301, 1111 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kral JG, Biron S, Simard S, Hould FS, Lebel S, Marceau S, Marceau P, Large maternal weight loss from obesity surgery prevents transmission of obesity to children who were followed for 2 to 18 years. Pediatrics 118, e1644–1649 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Bayol SA, Farrington SJ, Stickland NC, A maternal ‘junk food’ diet in pregnancy and lactation promotes an exacerbated taste for ‘junk food’ and a greater propensity for obesity in rat offspring. Br JNutr 98, 843–851 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Samuelsson AM, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EH, Piersma AH, Ozanne SE, Twinn DF, Remacle C, Rowlerson A, Poston L, Taylor PD, Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension 51, 383–392 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Nivoit P, Morens C, Van Assche FA, Jansen E, Poston L, Remacle C, Reusens B, Established diet-induced obesity in female rats leads to offspring hyperphagia, adiposity and insulin resistance. Diabetologia 52, 1133–1142 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Tamashiro KL, Terrillion CE, Hyun J, Koenig JI, Moran TH, Prenatal stress or high-fat diet increases susceptibility to diet-induced obesity in rat offspring. Diabetes 58, 1116–1125 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carmody JS, Wan P, Accili D, Zeltser LM, Leibel RL, Respective contributions of maternal insulin resistance and diet to metabolic and hypothalamic phenotypes of progeny. Obesity (Silver Spring) 19, 492–499 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirk SL, Samuelsson AM, Argenton M, Dhonye H, Kalamatianos T, Poston L, Taylor PD, Coen CW, Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PLoS One 4, e5870 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beard C, Hochedlinger K, Plath K, Wutz A, Jaenisch R, Efficient method to generate single-copy transgenic mice by site-specific integration in embryonic stem cells. Genesis 44, 23–28 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Eggan K, Akutsu H, Loring J, Jackson-Grusby L, Klemm M, Rideout WM 3rd, Yanagimachi R, Jaenisch R, Hybrid vigor, fetal overgrowth, and viability of mice derived by nuclear cloning and tetraploid embryo complementation. Proc Natl Acad Sci U S A 98, 6209–6214 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahren B, Diurnal variation in circulating leptin is dependent on gender, food intake and circulating insulin in mice. Acta Physiol Scand 169, 325–331 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Burnett LC, Skowronski AA, Rausch R, LeDuc CA, Leibel RL, Determination of the half-life of circulating leptin in the mouse. Int JObes (Lond) 41, 355–359 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ravussin Y, Gutman R, Diano S, Shanabrough M, Borok E, Sarman B, Lehmann A, LeDuc CA, Rosenbaum M, Horvath TL, Leibel RL, Effects of chronic weight perturbation on energy homeostasis and brain structure in mice. Am J Physiol Regul Integr Comp Physiol 300, R1352–1362 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, Eckel RH, Takahashi JS, Bass J, Obesity and metabolic syndrome in circadian Clock mutant mice. Science 308, 1043–1045 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morabito MV, Ravussin Y, Mueller BR, Skowronski AA, Watanabe K, Foo KS,Lee SX, Lehmann A, Hjorth S, Zeltser LM, LeDuc CA, Leibel RL, Weight Perturbation Alters Leptin Signal Transduction in a Region-Specific Manner throughout the Brain. Plos One 12, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skowronski AA, Morabito MV, Mueller BR, Lee S, Hjorth S, Lehmann A, Watanabe K, Zeltser LM, Ravussin Y, Rosenbaum M, LeDuc CA, Leibel RL, Effects of a Novel MC4R Agonist on Maintenance of Reduced Body Weight in Diet-Induced Obese Mice. Obesity 22, 1287–1295 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ravussin Y, Edwin E, Gallop M, Xu LM, Bartolome A, Kraakman MJ, Leduc CA, Ferrante AW, Evidence for a Non-leptin System that Defends against Weight Gain in Overfeeding. CellMetab 28, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahima RS, Prabakaran D, Flier JS, Postnatal leptin surge and regulation of circadian rhythm of leptin by feeding. Implications for energy homeostasis and neuroendocrine function. J Clin Invest 101, 1020–1027 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Casabiell X, Pineiro V, Tome MA, Peino R, Dieguez C, Casanueva FF, Presence of leptin in colostrum and/or breast milk from lactating mothers: a potential role in the regulation of neonatal food intake. J Clin Endocrinol Metab 82, 4270–4273 (1997). [DOI] [PubMed] [Google Scholar]
- 41.Cottrell EC, Mercer JG, Ozanne SE, Postnatal development of hypothalamic leptin receptors. Vitam Horm 82, 201–217 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Savino F, Liguori SA, Petrucci E, Lupica MM, Fissore MF, Oggero R, Silvestro L, Evaluation of leptin in breast milk, lactating mothers and their infants. Eur J Clin Nutr 64, 972–977 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Proulx K, Richard D, Walker CD, Leptin regulates appetite-related neuropeptides in the hypothalamus of developing rats without affecting food intake. Endocrinology 143, 4683–4692 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Mistry AM, Swick A, Romsos DR, Leptin alters metabolic rates before acquisition of its anorectic effect in developing neonatal mice. Am J Physiol 277, R742–747 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Patterson CM, Bouret SG, Park S, Irani BG, Dunn-Meynell AA, Levin BE, Large litter rearing enhances leptin sensitivity and protects selectively bred diet-induced obese rats from becoming obese. Endocrinology 151, 4270–4279 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Juan De Solis A, Baquero AF, Bennett CM, Grove KL, Zeltser LM, Postnatal undernutrition delays a key step in the maturation of hypothalamic feeding circuits. Mol Metab 5, 198–209 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ravussin Y, Gutman R, LeDuc CA, Leibel RL, Estimating energy expenditure in mice using an energy balance technique. Int JObes (Lond) 37, 399–403 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qiu J, Ogus S, Lu R, Chehab FF, Transgenic mice overexpressing leptin accumulate adipose mass at an older, but not younger, age. Endocrinology 142, 348–358 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Ogawa Y, Masuzaki H, Hosoda K, Aizawa-Abe M, Suga J, Suda M, Ebihara K, Iwai H, Matsuoka N, Satoh N, Odaka H, Kasuga H, Fujisawa Y, Inoue G, Nishimura H, Yoshimasa Y, Nakao K, Increased glucose metabolism and insulin sensitivity in transgenic skinny mice overexpressing leptin. Diabetes 48, 1822–1829 (1999). [DOI] [PubMed] [Google Scholar]
- 50.Ogus S, Ke Y, Qiu J, Wang B, Chehab FF, Hyperleptinemia precipitates diet-induced obesity in transgenic mice overexpressing leptin. Endocrinology 144, 2865–2869 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Pico C, Oliver P, Sanchez J, Miralles O, Caimari A, Priego T, Palou A, The intake of physiological doses of leptin during lactation in rats prevents obesity in later life. Int J Obes (Lond) 31, 1199–1209 (2007). [DOI] [PubMed] [Google Scholar]
- 52.de Oliveira Cravo C, Teixeira CV, Passos MC, Dutra SC, de Moura EG, Ramos C, Leptin treatment during the neonatal period is associated with higher food intake and adult body weight in rats. Horm Metab Res 34, 400–405 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Vickers MH, Gluckman PD, Coveny AH, Hofman PL, Cutfield WS, Gertler A, Breier BH, Harris M, The effect of neonatal leptin treatment on postnatal weight gain in male rats is dependent on maternal nutritional status during pregnancy. Endocrinology 149, 1906–1913 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Plagemann A, Harder T, Brunn M, Harder A, Roepke K, Wittrock-Staar M, Ziska T, Schellong K, Rodekamp E, Melchior K, Dudenhausen JW, Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: an epigenetic model of obesity and the metabolic syndrome. J Physiol 587, 4963–4976 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Licholai JA, Nguyen KP, Fobbs WC, Schuster CJ, Ali MA, Kravitz AV, Why Do Mice Overeat High-Fat Diets? How High-Fat Diet Alters the Regulation of Daily Caloric Intake in Mice. Obesity (Silver Spring) 26, 1026–1033 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fulton S, Appetite and reward. Front Neuroendocrinol 31, 85–103 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Berthoud HR, Metabolic and hedonic drives in the neural control of appetite: who is the boss? Curr Opin Neurobiol 21, 888–896 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, Zhao X, Sarruf DA, Izgur V, Maravilla KR, Nguyen HT, Fischer JD, Matsen ME, Wisse BE, Morton GJ, Horvath TL, Baskin DG, Tschop MH, Schwartz MW, Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest 122, 153–162 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nikodemova M, Kimyon RS, De I, Small AL, Collier LS, Watters JJ, Microglial numbers attain adult levels after undergoing a rapid decrease in cell number in the third postnatal week. JNeuroimmunol 278, 280–288 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao Y, Ottaway N, Schriever SC, Legutko B, Garcia-Caceres C, de la Fuente E, Mergen C, Bour S, Thaler JP, Seeley RJ, Filosa J, Stern JE, Perez-Tilve D, Schwartz MW, Tschop MH, Yi CX, Hormones and diet, but not body weight, control hypothalamic microglial activity. Glia 62, 17–25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rottkamp DM, Rudenko IA, Maier MT, Roshanbin S, Yulyaningsih E, Perez L, Valdearcos M, Chua S, Koliwad SK, Xu AW, Leptin potentiates astrogenesis in the developing hypothalamus. MolMetab 4, 881–889 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim JG, Suyama S, Koch M, Jin S, Argente-Arizon P, Argente J, Liu ZW, Zimmer MR, Jeong JK, Szigeti-Buck K, Gao Y, Garcia-Caceres C, Yi CX, Salmaso N, Vaccarino FM, Chowen J, Diano S, Dietrich MO, Tschop MH, Horvath TL, Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat Neurosci 17, 908–910 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hardie L, Trayhurn P, Abramovich D, Fowler P, Circulating leptin in women: a longitudinal study in the menstrual cycle and during pregnancy. Clin Endocrinol (Oxf) 47, 101–106 (1997). [DOI] [PubMed] [Google Scholar]
- 64.Sattar N, Greer IA, Pirwani I, Gibson J, Wallace AM, Leptin levels in pregnancy: marker for fat accumulation and mobilization? Acta Obstet Gynecol Scand 77, 278–283 (1998). [PubMed] [Google Scholar]
- 65.Highman TJ, Friedman JE, Huston LP, Wong WW, Catalano PM, Longitudinal changes in maternal serum leptin concentrations, body composition, and resting metabolic rate in pregnancy. Am J Obstet Gynecol 178, 1010–1015 (1998). [DOI] [PubMed] [Google Scholar]
- 66.Mukherjea R, Castonguay TW, Douglass LW, Moser-Veillon P, Elevated leptin concentrations in pregnancy and lactation: possible role as a modulator of substrate utilization. Life Sci 65, 1183–1193 (1999). [DOI] [PubMed] [Google Scholar]
- 67.Lage M, Garcia-Mayor RV, Tome MA, Cordido F, Valle-Inclan F, Considine RV, Caro JF, Dieguez C, Casanueva FF, Serum leptin levels in women throughout pregnancy and the postpartum period and in women suffering spontaneous abortion. Clin Endocrinol (Oxf) 50, 211–216 (1999). [DOI] [PubMed] [Google Scholar]
- 68.Sivan E, Whittaker PG, Sinha D, Homko CJ, Lin M, Reece EA, Boden G, Leptin in human pregnancy: the relationship with gestational hormones. Am J Obstet Gynecol 179, 1128–1132 (1998). [DOI] [PubMed] [Google Scholar]
- 69.Clapp JF, Kiess W, Cord blood leptin reflects fetal fat mass. JSoc Gynecol Invest 5, 300–303 (1998). [PubMed] [Google Scholar]
- 70.Lepercq J, Challier JC, Guerre-Millo M, Cauzac M, Vidal H, Hauguel-de Mouzon S, Prenatal leptin production: evidence that fetal adipose tissue produces leptin. J Clin Endocrinol Metab 86, 2409–2413 (2001). [DOI] [PubMed] [Google Scholar]
- 71.Cardoso-Moreira M, Halbert J, Valloton D, Velten B, Chen C, Shao Y, Liechti A, Ascencao K, Rummel C, Ovchinnikova S, Mazin PV, Xenarios I, Harshman K, Mort M, Cooper DN, Sandi C, Soares MJ, Ferreira PG, Afonso S, Carneiro M, Turner JMA, VandeBerg JL, Fallahshahroudi A, Jensen P, Behr R, Lisgo S, Lindsay S, Khaitovich P, Huber W, Baker J, Anders S, Zhang YE, Kaessmann H, Gene expression across mammalian organ development. Nature 571, 505–509 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grayson BE, Allen SE, Billes SK, Williams SM, Smith MS, Grove KL, Prenatal development of hypothalamic neuropeptide systems in the nonhuman primate. Neuroscience 143, 975–986 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Tasic B, Hippenmeyer S, Wang C, Gamboa M, Zong H, Chen-Tsai Y, Luo L, Site-specific integrase-mediated transgenesis in mice via pronuclear injection. Proc Natl Acad Sci U S A 108, 7902–7907 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harris RB, Zhou J, Redmann SM Jr., Smagin GN, Smith SR, Rodgers E, Zachwieja JJ, A leptin dose-response study in obese (ob/ob) and lean (+/?) mice. Endocrinology 139, 8–19 (1998). [DOI] [PubMed] [Google Scholar]
- 75.Hochedlinger K, Yamada Y, Beard C, Jaenisch R, Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell 121, 465–477 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Validation of leptin-overexpressing ES cells and leptin-overexpressing mice.
Fig. S2. Bioactivity of leptin.
Fig. S3. Circulating leptin concentrations during postnatal dox exposure.
Fig. S4. Dox-induced chronic hyperleptinemia (P63-P203) in adult mice with concurrent HFD feeding.
Fig. S5. Release of adult mice from dox-induced chronic (P63-P203) hyperleptinemia and HFD feeding.
Fig. S6. Energy expenditure assessment (indirect calorimetry) at 26 weeks of age in postnatally (P0-P22) hyperleptinemic female mice after 16 weeks of HFD feeding.
Supplementary Excel Data file S1 [Primary data file used to generate main figures].
Supplementary Excel Data file S2 [Primary data file used to generate supplementary figures].