

Abstract
Triple-negative breast cancer (TNBC) is an aggressive form of breast cancer that does not respond to endocrine therapy or human epidermal growth factor receptor 2 (HER2)-targeted therapies. Individuals with TNBC experience higher rates of relapse and shorter overall survival compared to patients with receptor-positive breast cancer subtypes. Preclinical discoveries are needed to identify, develop, and advance new drug targets to improve outcomes for patients with TNBC. Herein, we report that MYCN, an oncogene typically overexpressed in tumors of the nervous system or with neuroendocrine features, is heterogeneously expressed within a substantial fraction of primary and recurrent TNBC and is expressed in an even higher fraction of TNBCs that do not display a pathological complete response after neoadjuvant chemotherapy. We performed high-throughput chemical screens on TNBC cell lines with varying amounts of MYCN expression and determined that cells with higher expression of MYCN were more sensitive to bromodomain and extra-terminal motif (BET) inhibitors. Combined BET and MEK inhibition resulted in a synergistic decrease in viability, both in vitro and in vivo, using cell lines and patient-derived xenograft (PDX) models. Our preclinical data provide a rationale to advance a combination of BET and MEK inhibitors to clinical investigation for patients with advanced MYCN-expressing TNBC.
One Sentence Summary
This study demonstrates the potential utility of BET and MEK inhibitors for advanced MYCN-expressing triple-negative breast cancer.
INTRODUCTION
Triple-negative breast cancer (TNBC) affects younger women and is characterized by increased rates of relapse, more frequent metastasis, and shorter survival compared to the other breast cancer subtypes (1). Although TNBC only represents ~15% of all breast cancer cases, it accounts for ~25% of all breast cancer-related deaths (2), with treatment options for most patients limited to cytotoxic chemotherapy. Prognosis is unfavorable for patients with metastatic TNBC as >50% of patients with metastatic disease die within one year of diagnosis (2). Development of targeted therapies for TNBC is challenging due to its molecular heterogeneity and lack of therapeutically targetable, high-frequency driver alterations (3). Understanding the heterogeneity within TNBC and molecular mechanisms that contribute to the emergence of treatment-resistant, metastatic disease may inform the development of more effective therapeutics and address an unmet medical need in breast cancer.
Aside from TP53, the majority of mutations found in TNBC are within the phosphoinositide 3-kinase (PI3K) or mitogen-activated protein kinase kinase ½ (MEK) signaling pathways. The most frequent oncogenic mutations in TNBC occur in ‘hotspot’ regions of the PIK3CA gene (E545 helical domain and H1047 kinase domain) (4), and the most frequently amplified oncogene is MYC (5, 6). MYC family members, MYC, MYCN, and MYCL, are transcription factors that regulate the expression of genes involved in normal development, cell growth, proliferation, metabolism, and survival (7). Aberrant expression of MYC family members has been considered tumorigenic in a tissue-specific manner [MYCN in neuronal (8, 9) or neuroendocrine tumors (10, 11) and MYCL in lung (7)]. However, recent reports have shown elevated MYCN expression in non-neuronal tissues, such as ovarian (12) and prostate cancer (13), as well as hematopoietic cells that give rise to acute lymphoblastic (14) and myeloid (15) leukemias. Further, there is increasing evidence that MYCN expression is deregulated in a subset of breast cancers with unfavorable prognostic features and clinical outcomes (16–18). MYCN transcript has been found in circulating breast tumor cell clusters within the blood stream of breast cancer patients (19) and is associated with a stem-cell program found in tumor-initiating metastatic cells (18), implicating a role for MYCN in the recurrence and metastatic spread of breast cancer.
To determine the overall frequency of MYCN-expressing tumors in primary TNBC and whether MYCN expression changes in response to neoadjuvant chemotherapy (NAC), we evaluated TNBC patient cohorts comprised of primary, treatment-naïve tumors or primary, NAC-treated tumors. We also evaluated the quantity of MYCN RNA and protein in the metastatic setting. In parallel, we investigated the biological relevance of MYCN versus MYC expression in TNBC cells and whether MYCN expression was associated with response to compounds currently or previously under clinical development [including the NCI FDA-Approved Oncology Drug (AOD) library]. Top “hits” from the drug screen were examined as single agents and in combination, in vitro and in mice harboring TNBC patient-derived xenografts (PDXs) with differing amounts of MYCN. We discovered that combined bromodomain and extra-terminal motif (BET) and MEK inhibition synergistically inhibited growth of MYCN-expressing PDX TNBC tumors.
RESULTS
A substantial fraction of primary TNBCs express MYCN
To evaluate MYCN expression in TNBC, we first identified TNBC tumors from primary, treatment-naïve cases in The Cancer Genome Atlas (TCGA) Breast Invasive Carcinoma (BRCA) dataset (fig. S1A) (4). MYCN transcript was expressed in all tumors [transcript per million (TPM) >0] and elevated [>12 TPM, >1 standard deviation (SD) above the mean] in 10.2% (20/197) of cases (Fig. 1A). Likewise, we detected elevated MYCN expression in a similar proportion of primary TNBC cases (fig. S1B) in two other datasets, TNBC587 (>0.65 median-centered log2 normalized, n=65/587) (20) and Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) (>7 log2 normalized, n=48/323) (21) (fig. S2, A and B). To gain insight into the biological relevance of MYCN expression in TNBC, we compared the amount of MYCN transcript in primary, treatment-naïve TNBC (source: TCGA, BRCA) to transcript expressed in known MYCN-driven cancers (22) (Fig. 1B). Cancers with MYCN gene amplifications such as neuroblastoma (NB), glioblastoma multiforme (GBM), and acute myeloid leukemia (AML) originate from migrating neural crest cells, neural stem cells, or hematopoietic stem cells, respectively (22). MYCN is also amplified or overexpressed in at least 20% and 60% of adenocarcinoma (Adeno) and neuroendocrine (NE) castration-resistant prostate cancer (CRPC) cases, respectively (10, 13). Although the amount of transcript in TNBC was not as high as in NB (23), AML (source: TCGA, LAML), or GBM (source: TCGA, GBM), MYCN expression was similar to NE-CRPC and signifcantly higher (p<0.0001) than Adeno-CRPC (24, 25) (Fig. 1B and data file S1). Further, elevated MYCN-expressing TNBC cases identified in TCGA (Fig. 1A) had higher MYCN expression than the top MYCN-expressing NE-CRPC tumors (Fig. 1B and data file S1).
Fig. 1. MYCN RNA and MYCN protein expression in primary, treatment-naïve TNBC.

(A) MYCN transcript (TPM) from 197 primary, treatment-naïve TNBCs [source: TCGA (BRCA)]. SD, standard deviation. μ, mean. (B) Violin plot showing MYCN expression in TNBC [source: TCGA (BRCA), n=197] compared to neuroblastoma (NB, n=161) (23), acute myeloid leukemia (AML) [source: TCGA (LAML), n=173], glioblastoma multiforme (GBM) [source: TCGA (GBM), n=156], and castration-resistant prostate cancer (CRPC), including neuroendocrine (NE, n=15) and adenocarcinoma (Adeno, n=123) (24, 25). Wilcoxon rank sum test comparing TNBC to the other cancer types. P-values were adjusted by false discovery rate, ****p<0.0001. ns, not significant. (C) MYCN protein quantities (H-scores) from 191 primary, treatment-naïve TNBCs [source: Vanderbilt University Medical Center (VUMC) and US Biomax]. Int., intermediate. (D) Representative MYCN IHC images in TNBC specimens devoid of MYCN protein expression (H-score=0), that contain intermediate amounts of MYCN (H-score between >0 and ≤30), or that have high MYCN (H-score >30). Scale bar, 20 μm.
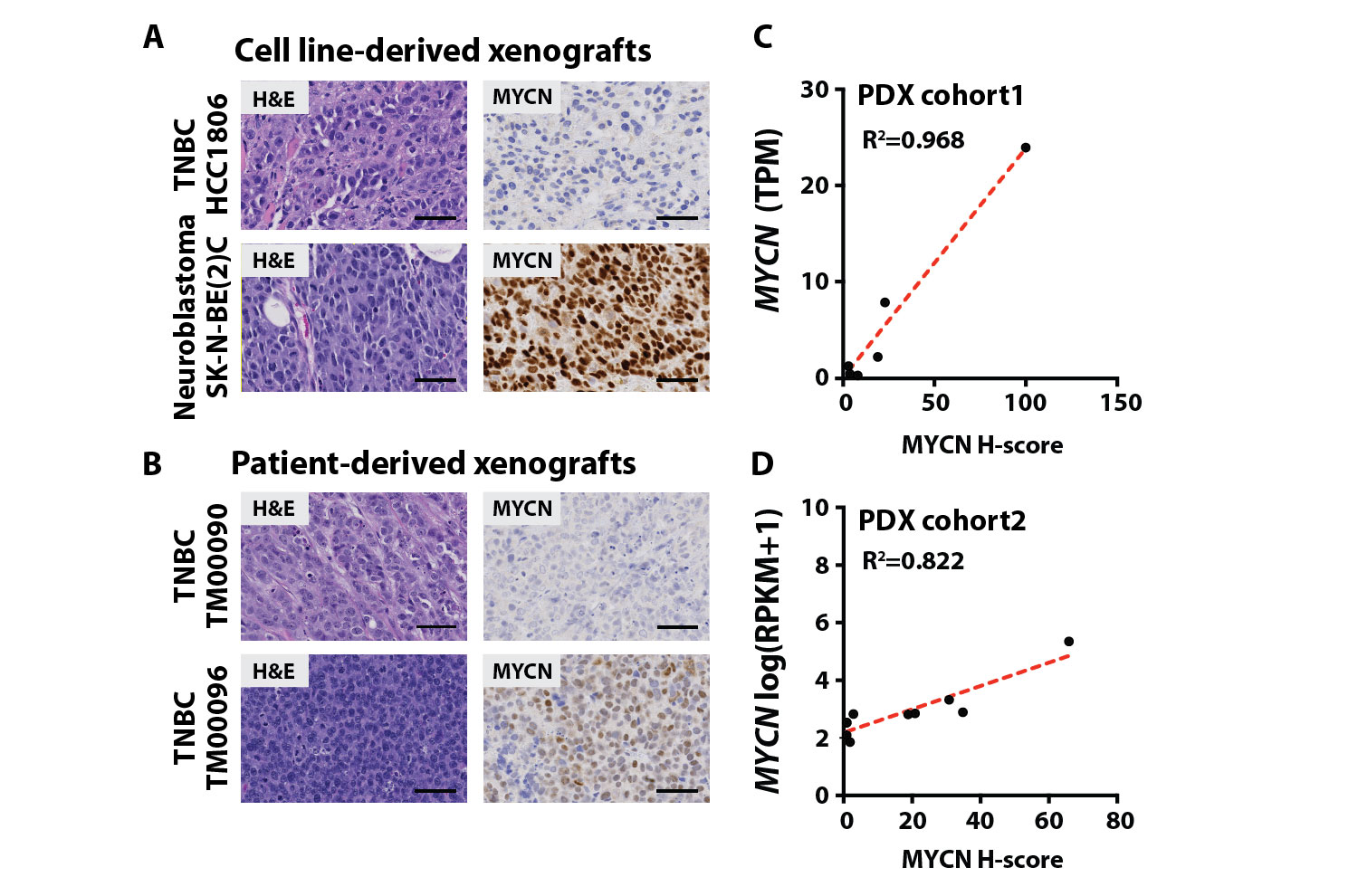
Because the MYCN transcript in clinical specimens could have originated from tumor or tumor-infiltrating immune or stromal cells, we performed MYCN immunohistochemistry (IHC) to identify the cellular distribution of MYCN protein in an independent cohort of 191 primary, treatment-naïve TNBC tumors, curated at Vanderbilt University Medical Center (VUMC) and US Biomax. IHC demonstrated that 45% of specimens contained nuclear MYCN within tumor cells, and similar to our RNA analyses, 11.5% of cases had high expression (H-score >30, >1 SD above the mean) (Fig. 1, C and D, and data file S2). Of note, IHC specificity was confirmed with positive and negative controls from patient-derived xenografts (PDXs) and cell line-derived xenografts (CDXs), including SK-N-BE(2)C, a validated MYCN-amplified neuroblastoma CDX (fig. S3, A and B, table S1A, and data file S1) (26). The relative amounts of MYCN transcript highly correlated with IHC protein quantities (H-score) across two PDX cohorts (cohort1: R2=0.968, cohort2: R2=0.822), further validating antibody specificity (fig. S3, C and D, table S1, A and B, and data file S1). Collectively, these data demonstrated the prevalence of MYCN protein in TNBC tumor cell nuclei and provided rationale to further characterize MYCN-expressing cells in the context of disease etiology.
Increased fraction of MYCN-expressing cells in residual TNBC after neoadjuvant chemotherapy
Due to the lack of therapeutic targets in TNBC, patients are primarily treated with combination chemotherapy, and less than 30% of patients achieve a pathological complete response (pCR) after neoadjuvant chemotherapy (NAC) (27, 28). Patients with residual disease after NAC exhibit poor overall survival due to an enrichment of chemotherapy-resistant tumor cells and a lack of subsequent therapeutic options (29, 30). To evaluate MYCN expression in residual tumor cells after NAC, we performed IHC for MYCN on a primary TNBC cohort (n=115) with residual disease surgically resected after NAC (6) (Fig. 2A, table S2A, and data file S2). MYCN expression was significantly (p=0.001) higher in the post-NAC-treated TNBC cohort (Fig. 2A and data file S2) compared to cases in the treatment-naïve TNBC cohort (Fig. 1C), with 65% vs. 45% of cases having an H-score greater than zero (Fig. 2, A and B, and table S2B). The majority (90%) of patients in the NAC-treated TNBC cohort had stage III disease at the time of diagnosis, while the treatment-naïve cohort consisted primarily of patients with stage I (11%) and stage II (70%) disease (table S2A). To remove a potential bias due to differences in clinical stage between cohorts, we restricted the comparison of MYCN expression to tumors from patients with stage III disease from each cohort; MYCN expression (H-score >0) remained significantly (p=0.014) higher in the residual disease of patients after NAC treatment (65%, 54/83) compared to treatment-naïve patients (40%, 10/25) (table S2B). Since the primary treatment-naïve and NAC-treated TNBC cohorts were independently assembled, we examined MYCN expression in patient-matched TNBC before and after NAC treatment (n=6) (table S2C). Compared to the quantity of MYCN protein before treatment, MYCN protein expression was similar or increased after NAC, demonstrating that MYCN-expressing cells remained after treatment (Fig. 2C and data file S1). These data suggest that either MYCN expression was induced or pre-existing MYCN-expressing tumor cells persisted in the TNBC cell populations after chemotherapy.
Fig. 2. Increased percentage of MYCN-expressing cells in residual disease after neoadjuvant chemotherapy.

(A) MYCN H-scores in residual disease from 115 primary, NAC-treated TNBCs (source: VUMC). Null, H-score=0. Int., intermediate, H-score >0 to ≤30. High, H-score >30. (B) Box plot showing MYCN H-scores in primary, treatment-naïve TNBC cases (n=191, see Fig. 1C) compared to residual disease from primary, NAC-treated TNBC cases (n=115, see Fig. 2A). Wilcoxon rank sum test, ***p=0.0001. (C) MYCN H-scores in patient-matched TNBC cases pre- and post-NAC (n=6, see table S2C for treatments and patient characteristics).
Primary and metastatic TNBC display heterogeneous MYCN and MYC protein expression
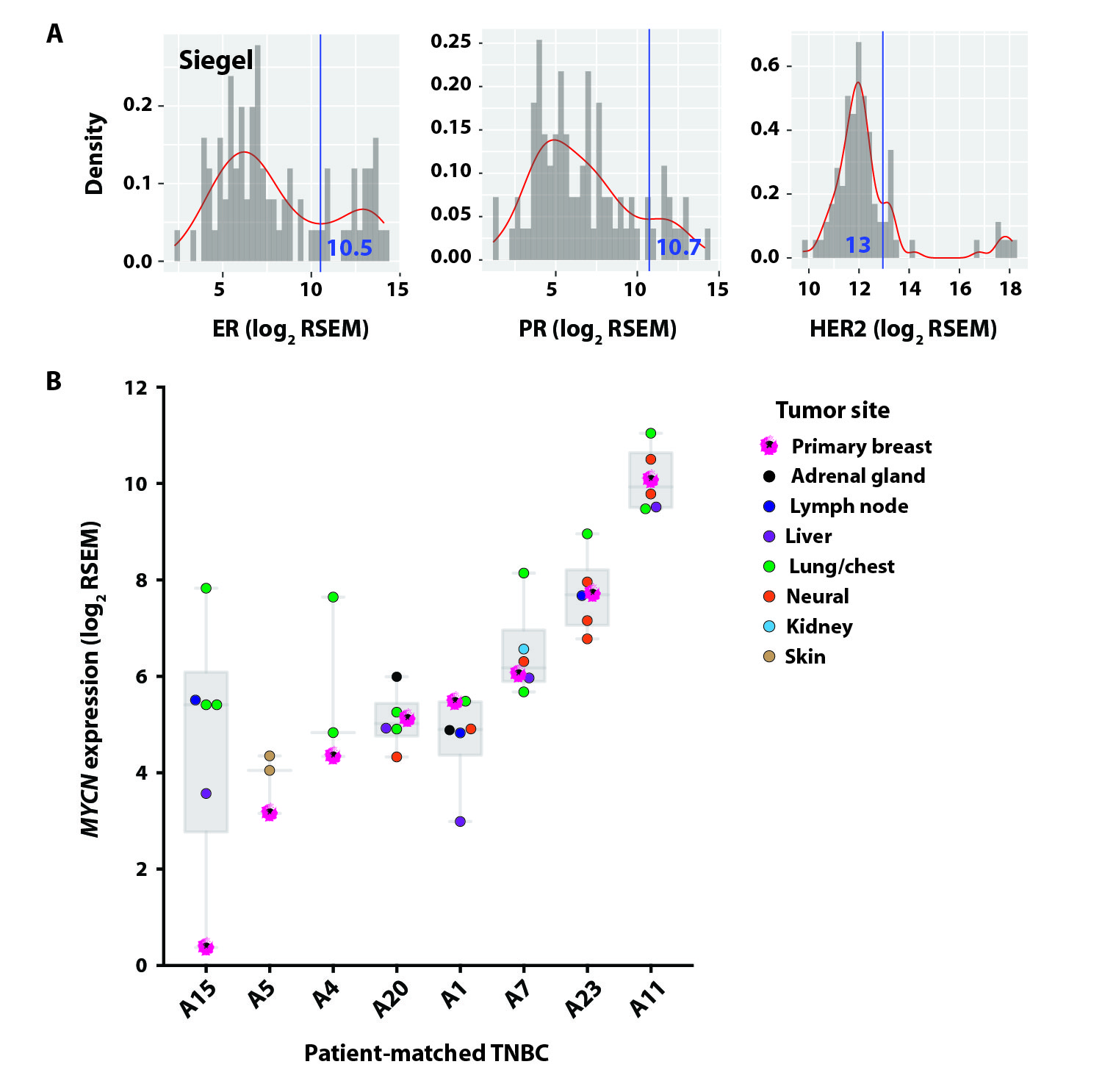
Despite better initial responses to NAC in TNBC compared to the other breast cancer subtypes, patients with TNBC experience higher rates of relapse and a worse overall survival in the metastatic setting (27). Given that nearly all women with metastatic TNBC ultimately die of their disease (31), we evaluated MYCN expression in the context of disease recurrence. We analyzed the TNBC cases from a recent study evaluating transcriptional changes between primary and metastatic breast cancer (32) (fig. S4A). MYCN transcript was increased or similarly expressed in nearly all metastatic specimens compared to matched primary TNBC, and MYCN was expressed at all metastatic sites evaluated [adrenal gland, lymph node, liver, lung, chest (chest wall, rib, pleura, mediastinum), neural tissue (brain, spine), kidney, skin] (fig. S4B and data file S1). Similarly, we performed MYCN IHC on 10 locally recurrent (five chest wall and five skin) and 28 metastatic (five lung and 23 brain) surgically resected TNBC tumors and detected MYCN protein expression (H-score >0) in 55% (21/38) of the recurrent TNBC tumors analyzed [lung: 80% (4/5); skin: 80% (4/5); chest wall: 60% (3/5); brain: 43% (10/23)] (Fig. 3A and data file S2).
Fig. 3. Intratumoral heterogeneity of MYCN and MYC expression in TNBC.

(A) MYCN H-scores from 38 recurrent TNBC cases with quantification of percent positive cases (H-score >0) for each site of recurrence, labeled by color [lung (magenta), skin (blue), chest wall (orange), brain (black)]. (B) MYCN and MYC H-scores for each of the 88 primary, treatment-naïve; 114 primary, NAC-treated; and 38 recurrent TNBC cases. Stacked bar graphs represent the percentages of total cases expressing (H-score) each MYC-family isoform [alone (MYCN Only, MYC Only); both isoforms (MYCN and MYC); or neither isoform (None)]. (C) Representative hematoxylin and eosin (H&E), IHC, and TSA-IF stains of MYCN and MYC in primary and recurrent TNBC. Individual fluorescence images for cell nuclei (blue), MYCN (magenta), and MYC (green) can be found in fig. S5. The dotted line separating a MYCN-amplified neuroblastoma positive control from TNBC cases represents the same exposure times for all samples but a decreased brightness adjustment for MYCN in the neuroblastoma control due to over-expression of MYCN. Tumor images do not represent serial sections. Scale bar, 50 μm (top four rows), 20 μm (bottom row).
Because MYCN expression has been shown to be elevated in newly seeded metastatic TNBC lesions that differentiate into high MYC-expressing proliferative tumors (18), we investigated the relationship between MYC-family isoforms (MYCN and MYC) in both primary and recurrent TNBC. We performed MYC IHC on tissue representing each of our TNBC patient cohorts [primary, treatment-naïve TNBC (Fig. 1C); primary, NAC-treated TNBC (Fig. 2A); and recurrent TNBC (Fig. 3A)] previously analyzed for MYCN. Thirty four percent (30/88) of primary, treatment-naïve TNBC; 49% (56/114) of primary, NAC-treated TNBC; and 50% (19/38) of recurrent TNBC expressed both MYC-family isoforms (Fig. 3B). MYCN and MYC can be expressed both spatially and temporally in a mutually exclusive manner during normal tissue development (33); thus, we assessed the distribution of these proteins in individual cells within a given tumor section using dual MYC-family isoform tyramide signal-amplified immunofluorescence (TSA-IF). We found that both MYCN and MYC were heterogeneously expressed in tumor cells throughout the sections, and the majority of cell nuclei robustly expressed only one MYC family member (Fig. 3C and fig. S5). These data demonstrate the cell-to-cell heterogeneity of MYC-family isoform expression in TNBC and the dynamic distribution of expression of these oncogenes at both primary and metastatic sites.
Preclinical models of MYCN-expressing TNBC
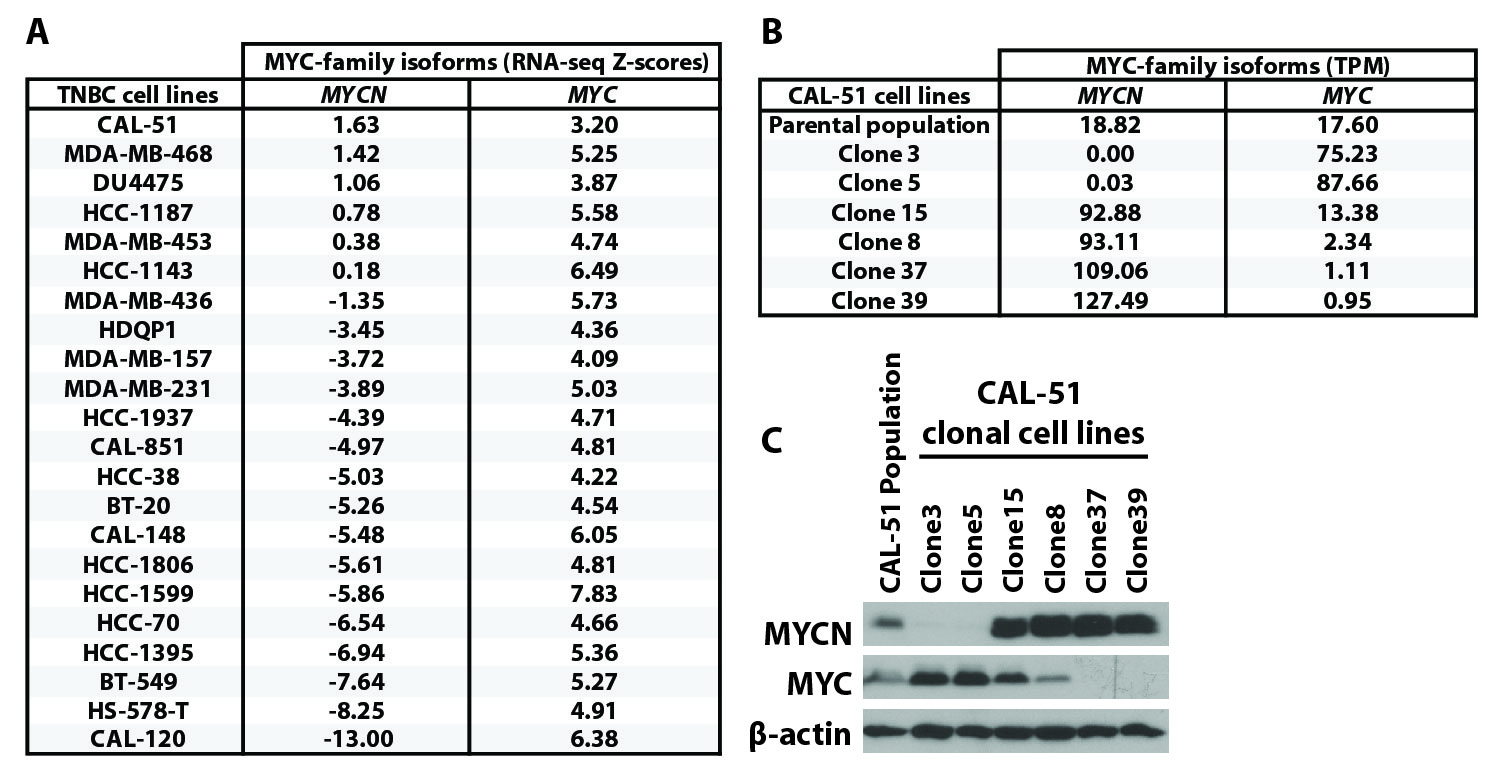
To identify MYCN-expressing TNBC cell line models for preclinical evaluation, we assessed MYCN expression across TNBC cell lines in the Cancer Cell Line Encyclopedia (CCLE) (34). CAL-51 and MDA-MB-468 displayed the highest amounts of MYCN transcript (fig. S6A). Given that TNBC clinical specimens displayed heterogenous MYCN and MYC expression (Fig. 3C), we evaluated whether this heterogeneity existed within TNBC cell line models. We adapted our TSA-IF staining procedure used on FFPE tumor sections to cells fixed in situ after growth as adherent cultures and analyzed cellular MYCN and MYC expression within the CAL-51 and MDA-MB-468 cell populations. Individual cells in either cell line culture robustly expressed either nuclear MYCN or MYC (Fig. 4A), consistent with observed MYC-family isoform heterogeneity in clinical specimens (Fig. 3C). To further evaluate the biological characteristics of MYCN-expressing tumor-derived cells, we isolated single cells from the CAL-51 parental cell line and generated clonally-derived cell lines. Individual clones displayed varying MYCN and MYC protein expression, with 6% (2/33) of cells exhibiting elevated MYCN (Fig. 4B). MYCN and MYC protein quantities were consistent with the relative MYCN and MYC transcript in six of the clonal cell lines evaluated (Cln3, Cln5, Cln8, Cln15, Cln37, Cln39; fig. S6, B and C), and individual MYC-family isoform RNA and protein were expressed at higher quantities in the clonal lines as compared to the CAL-51 cell population (fig. S6, B and C). Thus, the CAL-51 cell line is composed of a heterogeneous population of cells with varying MYC-family isoform expression.
Fig. 4. Evaluation of MYCN-expressing TNBC clonal cell line drug-sensitivity.

(A) Representative TSA-IF stains of MYCN and MYC in the CAL-51 and MDA-MB-468 TNBC cell lines. Colors represent cell nuclei (blue), MYCN (magenta), and MYC (green). Scale bar, 50 μm for overlay fluorescence images at 20X magnification (left panel for each cell line), 20 μm for individual fluorescence images at 40X magnification (right panels for each cell line). (B) Immunoblot analysis of MYCN, MYC, and β-actin in the indicated 33 clonally-derived cell lines established from CAL-51. NB control, MYCN-amplified SK-N-BE(2)C cell lysate. (C) Viability of PI3Ki-resistant (PI3KiR) CAL-51 clonally-derived cell lines after treatment with escalating doses of GDC-0032 or GDC-0941 for 72 hours. Black and red dose-response curves represent the indicated MYCNLow and MYCNHigh clonally-derived cell lines, respectively. Data shown represent the means ± standard error mean (SEM) of three biological replicates. (D) Immunoblot analysis of MYCN, MYC, and β-actin in the 14 indicated CAL-51PI3KiR clonally-derived cell lines. (E) IC50 of 40 compounds used in a secondary screen to treat five MYCNLow and MYCNHigh cell lines for 72 hours. Colors associate with drug class [PI3K (purple), ATR (orange), BRD family (blue), Aurora kinase A (brown), MAPK pathway (green)]. Horizonal red dashed lines represent a separation of compounds that had a greater or less than two-fold difference in IC50 between MYCNLow and MYCNHigh cell lines. (F) IC50 of 31 CAL-51 clonally-derived cell lines after treatment with escalating doses of INCB054329 for 72 hours. Red lines represent means. Student’s t-test, ****p<0.0001. (G) Quantification of crystal violet stained colonies compared to control for ten MYCNLow and four MYCNHigh cell lines treated with 0.5 μM INCB054329 for six days. Red lines represent means. Student’s t-test, ***p<0.001.
CAL-51 cells harbor an activating PIK3CA mutation (E542K), and their growth is dependent on PI3K pathway signaling (35). Given the frequent evolution of tumor cell drug-resistance in response to PI3K-targeted cancer therapies (36), we hypothesized that MYCN-expressing cells in the CAL-51 population (Fig. 4, A and B) would have a growth advantage under selective pressure with PI3K inhibitor (PI3Ki) treatment. To test this hypothesis, we treated CAL-51 with increasing concentrations of the PI3Ki, taselisib (GDC-0032), over time to generate PI3Ki-resistant cells (CAL-51PI3KiR). After six months, single cells from CAL-51PI3KiR were isolated to generate clonally-derived PI3Ki-resistant cell lines. To determine if the individual CAL-51PI3KiR clonal cell lines displayed durable resistance to PI3Ki, we treated CAL-51PI3KiR cells with taselisib or another PI3Ki, pictilisib (GDC-0941), after the lines were cultured for two weeks in the absence of drug (a “drug holiday”). Five of the seven CAL-51PI3KiR clonal cell lines maintained resistance to PI3K inhibition, whereas two of the lines reverted back to a PI3Ki-sensitive state (Fig. 4C and data file S1). CAL-51PI3KiR clonal cell lines were evaluated for MYC-family isoform expression, and those lines that had acquired durable resistance to PI3Ki also displayed higher MYCN protein expression (compare Fig. 4C to 4D). In contrast to 6% (2/33) of the parental clonal cell lines, the majority (86%, 12/14) of CAL-51PI3KiR clonal cell lines expressed MYCN (Fig. 4D), suggesting that MYCN expression conferred a growth advantage to CAL-51 cells under the continuous selective pressure of PI3Ki treatment. For all subsequent description of results presented herein, we refer to the clonally-derived CAL-51 low and high MYCN-expressing cell lines as MYCNLow and MYCNHigh, respectively.
MYCN-expressing TNBC cells have increased sensitivity to bromodomain and extra-terminal motif inhibitors
The heterogeneity of MYC-family isoform expression in the CAL-51 and MDA-MB-468 cell lines, which is consistent with the heterogeneity observed in TNBC clinical specimens (Fig. 3C), supports the use of these two cell lines as preclinical tools to investigate differential drug sensitivity of MYCN-expressing cells. Since the MYC-family members are basic helix-loop-helix (bHLH) transcription factors lacking catalytic domains, strategies to inhibit their activity have been limited to indirect targeting of proteins that regulate MYC-family isoform stability or expression; these include the bromodomain (BRD)-containing family of transcriptional regulators, PIM1, MEK½, and Aurora kinase A (9, 37–40). To gain insight into potential strategies for targeting MYCN-expressing TNBC, we performed a high-throughput drug sensitivity screen on two MYCNLow and two MYCNHigh clonally-derived cell lines (described in the previous section) for sensitivity to a library of 158 compounds, containing the 114 compounds in the NCI FDA-Approved Oncology Drug (AOD) library and 44 additional compounds of interest. Analysis of half-maximal inhibitory concentrations (IC50) demonstrated similar drug sensitivities between each clonal cell line set [MYCNLow (R2=0.9476) and MYCNHigh (R2=0.9439)], with MYCNHigh cell lines having greater sensitivity to compounds that target the BRD family, Aurora kinase A, and MAPK pathway proteins (fig. S7 and data file S3). We performed a secondary screen on MYCNLow (n=5) and MYCNHigh (n=5) cell lines with inhibitors that demonstrated a >2-fold increase or decrease in IC50, plus additional related compounds of interest. Again, MYCNHigh cell lines displayed greater sensitivity to compounds previously shown to regulate MYC-isoform expression or activity (Fig. 4E and data file S4), including compounds targeting the BRD family of transcriptional regulators (JQ1, INCB054329, and OTX-015) (41–43).
Bromodomain and extra-terminal motif inhibitors (BETis) are a class of compounds currently under clinical development that broadly target the BRD family (predominantly BRD2, BRD3, and BRD4) (44). Preclinical studies have demonstrated that BETis are a promising strategy to target MYCN-amplified neuroblastoma because BRD4 regulates the transcription of MYCN and occupies MYCN target-gene enhancers and super-enhancers (8, 9). Since BETi sensitivity has been reported to have a stronger positive correlation with MYCN expression than with MYC expression in both hormonally (12) and non-hormonally regulated malignancies (8, 9), we investigated BETis further using our MYCN-expressing TNBC preclinical models. By treating additional CAL-51 clonally-derived cell lines that had differing expression of MYCN (n=26) with BETi, we validated results from our earlier drug screens showing that high MYCN-expressing cells were more sensitive (p<0.0001) to BETi (Fig. 4F and data file S1). Further, we performed longer-term drug treatments and evaluated the colony-forming ability of a subset of clonal cell lines (n=14) differing in MYCN and MYC expression. Again, high MYCN-expressing cells were more sensitive to BETi, and longer-term treatments resulted in more profound differential sensitivity (p<0.001) (Fig. 4G and data file S1). MYCNHigh cell lines had a ≥5-fold decrease in cell growth compared to MYCNLow cell lines in both short-term metabolic and long-term colony formation assays, demonstrating an association between MYCN expression and BETi sensitivity in TNBC.
Changes in MYC-family isoform expression in response to BETi treatment
To determine if the increased sensitivity of MYCN-expressing cells to BETi was MYCN-dependent, MYCNLow and MYCNHigh lines were subjected to MYCN siRNA-mediated knockdown. siRNAs targeting MYCN RNA decreased MYCN protein and decreased viability in a dose-dependent manner only in the MYCNHigh cell lines, without altering the amount of MYC in MYCNLow cells (Fig. 5A). Of note, MYC expression increased with MYCN knockdown in MYCNHigh cells (Fig. 5A), suggesting a feedback signaling mechanism between the MYC-family members to ensure cell survival under normal growth conditions. To determine if MYCN is a downstream target of BRD-mediated transcriptional regulation, we performed precision nuclear run-on sequencing (PRO-seq) on two MYCNHigh and two MYCNLow cell lines treated with BETi (0.5 μM INCB054329) for 15 minutes. Nascent RNA at the MYCN locus was observed only in MYCNHigh cells, and MYCN transcripts were reduced after BETi treatment (Fig. 5B). Nascent RNA at the MYC locus decreased in the MYCNLow cell lines after BETi treatment, consistent with reported responses to BETi in previous studies (37, 45) (Fig. 5B). However, the MYC RNA increased to basal quantities by four hours (RNA-Seq; Fig. 5C) in the MYCNLow cells, and protein amounts were increased at 24 hours (immunoblot; Fig. 5D) in the MYCNHigh cells, in parallel experiments. Gene set enrichment analyses (GSEA) performed on RNA samples harvested after four hours of BETi treatment demonstrated that MYC target genes were significantly downregulated in response to BETi treatment in the MYCNHigh cells (Hallmark MYC targets V1, p<0.0001, FDR q<0.0001; Hallmark MYC targets V2, p<0.0001, FDR q<0.0001); fig. S8), consistent with BETi-mediated downregulation of MYCN-mediated transcriptional activity.
Fig. 5. Evaluation of MYC-family isoform expression after BETi treatment.

(A) Top: Viability of MYCNLow and MYCNHigh cell lines after siRNA-mediated knockdown using non-targeting (siNT) or anti-MYCN (siMYCN) siRNAs for 96 hours. Data shown represent the mean of three technical replicates. Bottom: Immunoblot analysis of MYCN, MYC, and β-actin in MYCNLow and MYCNHigh cell lines after the described knockdown with 25 nM siRNAs. (B) Genome viewer showing sequencing alignment tracks of nascent transcript PRO-seq mapping at the MYCN and MYC gene loci for the two indicated MYCNLow and MYCNHigh cell lines after treatment with DMSO control (Unt, blue) or 0.5 μM INCB054329 (BETi, red) for 15 minutes. (C) MYCN and MYC expression (TPM) in the two indicated MYCNLow (Cln3 and Cln5) and four MYCNHigh (Cln8, Cln15, Cln37, and Cln39) cell lines after treatment with 0.5 μM INCB054329 for four hours. (D) Immunoblot analysis of MYCN, MYC, and β-actin in three of the cell lines described in C after treatment with 0.5 and 1.0 μM INCB054329 or JQ1 for 24 hours. (E) MYC-family isoform TSA-IF on two MYCN-expressing TNBC cell lines (MDA-MB-468 and CAL-51) after 0, 0.25, 0.5, or 1.0 μM INCB054329 or JQ1 for 24 hours. Colors represent cell nuclei (blue), MYCN (magenta), and MYC (green). Scale bar, 50 μm. (F) Quantification of fluorescence intensity per nucleus for MYCN and MYC after BETi treatments described in E. Data shown represent the median ± SEM of three biological replicates. Student’s t-test between untreated and BETi-treated cells, *p<0.05, **p<0.01.
To evaluate MYC-family isoform dynamics in individual cells after BETi exposure, CAL-51 and MDA-MB-468 were treated with increasing doses of BETi (INCB054329 or JQ1) for 24 hours and TSA-IF performed for MYCN and MYC detection. Similar to MYC-family isoform expression changes observed in the CAL-51 clonal cell lines (Fig. 5, C and D), BETi treatment decreased MYCN expression in a dose-dependent manner in the heterogeneous CAL-51 and MDA-MB-468 parental populations (Fig. 5, E and F, and data files S5 and S6). In addition to the activating PIK3CA mutation in CAL-51, both CAL-51 and MDA-MB-468 lack PTEN protein, a negative regulator of PI3K pathway signaling (35). BETi treatment resulted in little to no change in MYC expression in both CAL-51 and MDA-MB-468 (Fig. 5, E and F, and data files S5 and S6), which is consistent with a previous study demonstrating that BETi treatment had little effect on MYC expression in PI3K pathway-mutant breast cancer (46).
Combination BETi and MEKi treatment in MYCN-expressing TNBC cell lines
Given that the majority of MYCN-expressing TNBCs also contain MYC-expressing tumor cells (Fig. 3B), we identified drug combinations that would decrease expression of both isoforms and thereby inhibit cell proliferation and tumor development. We performed differential gene expression analyses using TNBC tumors from the TNBC587 dataset (20) with high MYCN expression and low MYC (MYCNRatioHigh) compared to tumors with high MYC expression and low MYCN (MYCRatioHigh). Selecting tumors on the basis of expression ratios allowed us to minimize the inclusion of heterogeneous tumors co-expressing both isoforms that would confound the results. The optimal number of tumors used for comparative analyses was determined by comparing the number of differentially expressed genes for different percentages of MYCNRatioHigh and MYCRatioHigh tumors compared to random samplings (fig. S9, A and B). To determine the degree of variance among all MYCNHighRatio and MYCHighRatio tumors selected for analysis, we performed a principal component analysis (PCA). MYCNHighRatio and MYCHighRatio tumors clustered apart from each other, indicating that tumors within each respective group have a greater similarity (fig. S9C). GSEA comparing MYCNHighRatio and MYCHighRatio tumors demonstrated a positive association between MYCN expression and MEK signaling (El-Ashry MEK Up V1 Up, p<0.001, FDR q=<0.001; fig. S9D), providing rationale to explore the regulation of MYCN and MYC expression by MAPK pathway signaling.
MYC protein stability can be regulated by the MAPK pathway (47), and inhibition of MAPK pathway signaling can cause MYC instability and proteasomal degradation (48). Given that MAPK pathway inhibitors are also under preclinical investigation to treat aggressive relapsed MYCN-driven neuroblastoma (49, 50) and were among the top “hits” in our previously described drug screens (fig. S7 and Fig. 4E), we evaluated whether MAPK pathway inhibition would alter MYCN protein quantities and/or be effective at decreasing MYC expression when combined with BRD inhibition. MYCNHigh and MYC-expressing MYCNLow CAL-51 clonal cell lines were treated with inhibitors targeting proteins in the MAPK pathway, including EGFR (erlotinib), RAF (TAK-632), MEK½ (trametinib and GDC-0973), and ERK½ (SCH772984). MEK inhibitors (MEKis) were most effective at inhibiting MAPK pathway signaling, as evidenced by decreased ERK½ phosphorylation, and decreased MYC and MYCN in each line that expressed a given isoform (Fig. 6A). Since the FDA-approved MEKi, trametinib, demonstrated the greatest decrease in MYC and MYCN, we evaluated the effects of trametinib treatment alone or in combination with BETi. MYCN decreased while MYC increased in CAL-51 MYCNHigh clonal cell lines treated with either BETi agent alone (INCB054329 or JQ1, Fig. 6B). However, trametinib in combination with either BETi attenuated MYC upregulation, thereby decreasing the amount of both MYC-family isoforms (Fig. 6B).
Fig. 6. Effect of BETi and MEKi combination treatment on MYC-family isoform expression and cell viability of MYCN-expressing TNBC cell lines.

(A) Immunoblot analysis for pERK½, total ERK½, MYCN, MYC, and β-actin in MYCNLow and MYCNHigh cell lines after treatment with MAPK pathway inhibitors at 0.25 μM for 24 hours. (B) Immunoblot analysis of pERK½, total ERK½, MYCN, MYC, and β-actin in four MYCNHigh CAL-51 cell lines after treatment with 0.25 μM trametinib, 0.5 μM INCB054329, 0.5 μM JQ1, or the combination of trametinib with either BETi for 48 hours. All immunoblot experiments shown are representative of at least two biological replicates. (C) MYC-family isoform TSA-IF on two MYCN-expressing TNBC cell lines (MDA-MB-468 and CAL-51) after 0.25 μM trametinib, 0.5 μM INCB054329, 0.5 μM JQ1, or the combination of trametinib with either BETi for 48 hours. Colors represent cell nuclei (blue), MYCN (magenta), and MYC (green). Scale bar, 50 μm. (D) Violin plots showing quanfication of fluorescence intensity for MYCN and MYC per nucleus after BETi treatments described in C. TSA-IF images and quantification are representative of three biological replicates. (E) Crystal violet colony formation assays for MDA-MB-468 and CAL-51 after treatment with the indicated concentrations of trametinib, INCB054329, or JQ1 alone, or either BETi in combination with trametinib, for six days. Values in red represent mean delta Bliss synergy for combination treatments of three biological replicates.
To expand our analysis of effects of BETi and MEKi combination treatment on heterogeneous populations of MYCN-expressing TNBC, we treated CAL-51 and MDA-MB-468 cells with trametinib, INCB054329, or JQ1 as single agents, or with either BETi in combination with trametinib, for 48 hours and examined MYC and MYCN expression. Treatment with either BETi alone decreased MYCN expression in both TNBC cell lines (Fig. 6, C and D, and data file S7), consistent with previous single agent results (Fig. 5, E and F). Whereas BETi treatment resulted in little to no change in MYC, single agent trametinib decreased MYC expression to a greater extent than MYCN in both cell lines; and, when trametinib was combined with either BETi, MYC and MYCN decreased to a larger extent than with either agent alone (Fig. 6, C and D, and data file S7). MDA-MB-468 and CAL-51 cell populations were treated with a range of low-dose BETi and MEKi concentrations to evaluate growth and viability in response to BETi and MEKi treatment. Both TNBC cell lines were treated with escalating doses of INCB054329 or JQ1, as single agents, or in combination with increasing doses of trametinib, and colony-forming ability was assessed after six days (Fig. 6E and data file S1). MDA-MB-468, the higher MYCN-expressing cell line (Fig. 5F and Fig. 6D), displayed greater sensitivity to single agent BETi treatments compared to CAL-51, and the combination of BETi and MEKi resulted in a synergistic decrease in cell growth, as determined by Bliss independence analyses (51, 52), in both MYCN-expressing lines (Fig. 6E and data file S1). These data demonstrate that low-dose BETi and MEKi combinations are effective in MYCN-expressing TNBC cell populations and provide rationale to further evaluate the combination using in vivo model systems of MYCN-expressing TNBC.
Combination BETi and MEKi treatment is effective at inhibiting in vivo growth of MYCN-expressing TNBC PDXs
To evaluate the preclinical efficacy of BET and MEK inhibition in vivo, we first confirmed MYCN and MYC protein expression in three TNBC PDX models with differing MYCN and MYC RNA expression (Fig. 7A and table S1A). The TM00096 PDX model was derived from a TNBC metastatic lung lesion (table S1A) (53) and expresses MYCN and MYC in ~37% and ~51% of the tumor cells, respectively (Fig. 7B). PDX models TM01273 and TM00090 both have a low percentage of MYCN-expressing cells (~2% and <1%, respectively) relative to MYC-expressing cells (~63% and ~32%, respectively) (Fig. 7B). For all three models, a 2 mm3 tumor was subcutaneously implanted into NOD scid gamma (NSG) mice, and when xenograft tumor volumes reached ~150 mm3, mice were treated with vehicle control, trametinib (0.1 mg/kg, QD), INCB054329 (50 mg/kg, BID), or the combination of the two agents at the indicated doses for 14 days. Compared to vehicle-treated controls, combined BET and MEK inhibitor treatment resulted in a synergistic and significant (p<0.01) reduction in tumor growth only in the high MYCN-expressing PDX model [delta Bliss synergy (Syn) and tumor growth inhibition (TGI): TM00096, Syn=38, TGI=97%; TM01273, Syn=−4, TGI=58%; TM00090, Syn=−8, TGI=35%] (Fig. 7C and data file S1). These in vivo results were consistent with our in vitro observations and further confirmed an association between MYCN expression and efficacy of BETi and MEKi combination treatment.
Fig. 7. Evaluation of tumor growth after BETi and MEKi combination treatment in vivo.

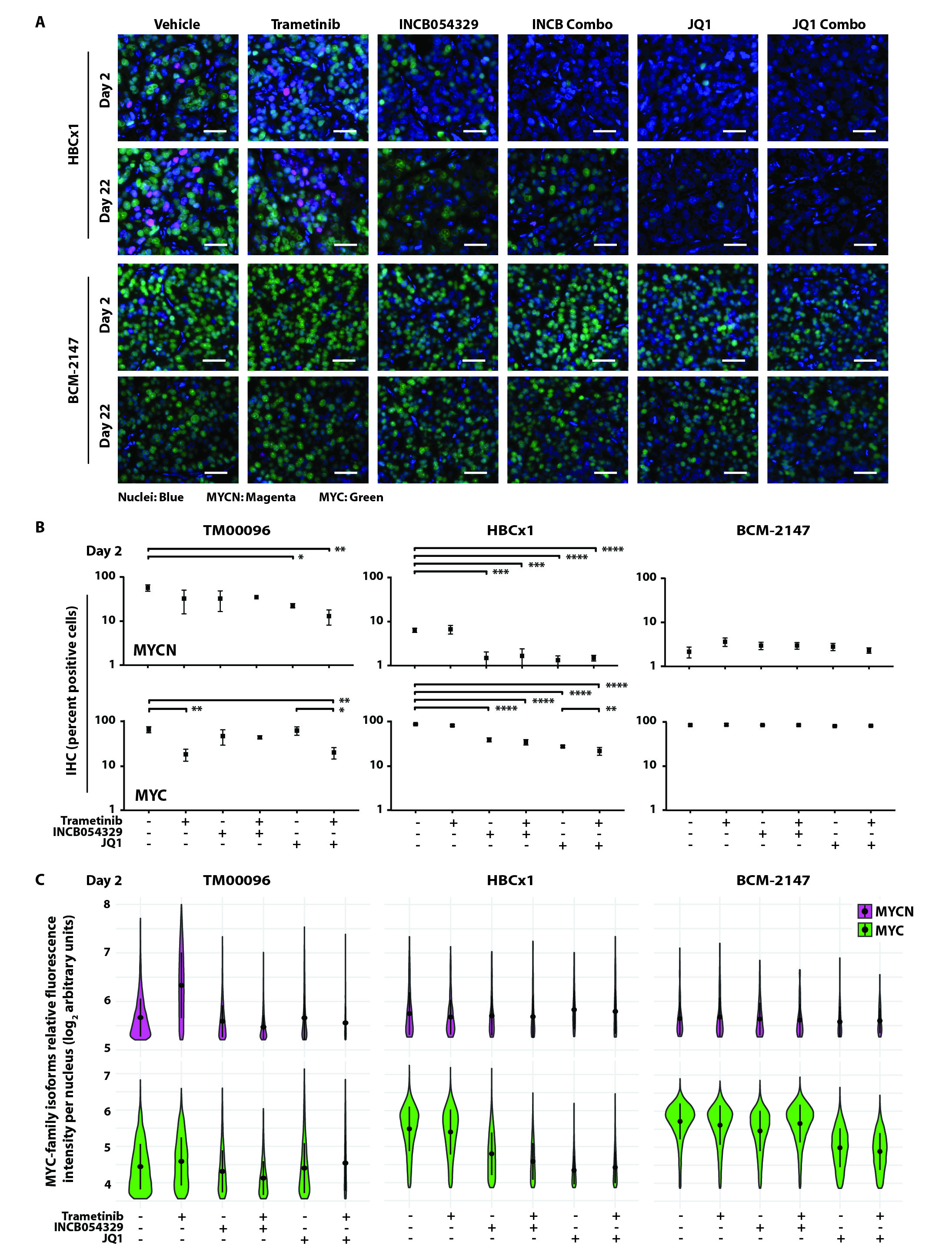
(A) MYCN and MYC expression (TPM) in three TNBC PDX models (TM00096, TM01273, and TM00090). (B) Representative IHC and quantification of percent positive cells for MYCN and MYC in TM00096, TM01273, and TM00090 sections. Scale bar, 20 μm. (C) Tumor volume (mm3) of TM00096, TM01273, and TM00090 treated with trametinib (0.1 mg/kg, QD) or INCB054329 (50 mg/kg, BID) alone, or in combination for 14 days. Red bars represent means. (D) Representative IHC and quantification of percent positive cells for MYCN and MYC in HBCx1 and BCM-2147 sections. Scale bar, 20 μm. (E) Tumor volume (mm3) of TM00096, HBCx1, and BCM-2147 treated with trametinib, INCB054329, or JQ1 (50 mg/kg, BID) alone, or either BETi in combination with trametinib for 22 days. The dashed line represents the mean initial tumor volume at time of first treatment. Avg, average. TGI, tumor growth inhibition. QD, once daily. BID, twice daily. Data shown represent the means ± SEM. Student’s t-test between vehicle, BETi, and corresponding combination treated tissue, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
To expand and reproduce our in vivo findings, we performed another PDX “trial” with TM00096 (MYCNHigh) alongside two additional TNBC PDX models, HBCx1 and BCM-2147, that have an intermediate (MYCNIntermediate) or low (MYCNLow) percentage of MYCN-expressing cells (~20% and ~2%, respectively) relative to MYC-expressing cells (~80% and ~95%, respectively) (Fig. 7D). All three models were treated for 22 days with trametinib, INCB054329, or JQ1 (50 mg/kg, BID) as single agents, or with the indicated BETi combined with trametinib. All compounds administered were well tolerated, and all animals completed the study without excess weight loss (fig. S10 and data file S1) or limiting morbidities. In response to either single agent BETi treatment, we observed the greatest statistical difference from vehicle in the MYCNHigh model (TM00096), with a 63% TGI in response to INCB054329 treatment (compared to 40% and 38% in the MYCNIntermediate and MYCNlow models, respectively) and an 83% TGI in response to JQ1 (compared to 75% and 57% in the MYCNIntermediate and MYCNlow models, respectively; Fig. 7E and data file S1). Combined MEKi and BETi resulted in a synergistic TGI in mice harboring either MYCNHigh or MYCNIntermediate tumors (INCB054329 and trametinib: Syn=21 and 15, respectively; JQ1 and trametinib: Syn=18 and 16, respectively; Fig. 7E and data file S1) and an 11% and 85% reduction in tumor volume, compared to the starting treatment-naïve tumor volume, in the MYCNHigh PDX model when trametinib was combined with either INCB054329 or JQ1, respectively (left panel, below the gray dashed line; Fig. 7E and data file S1).
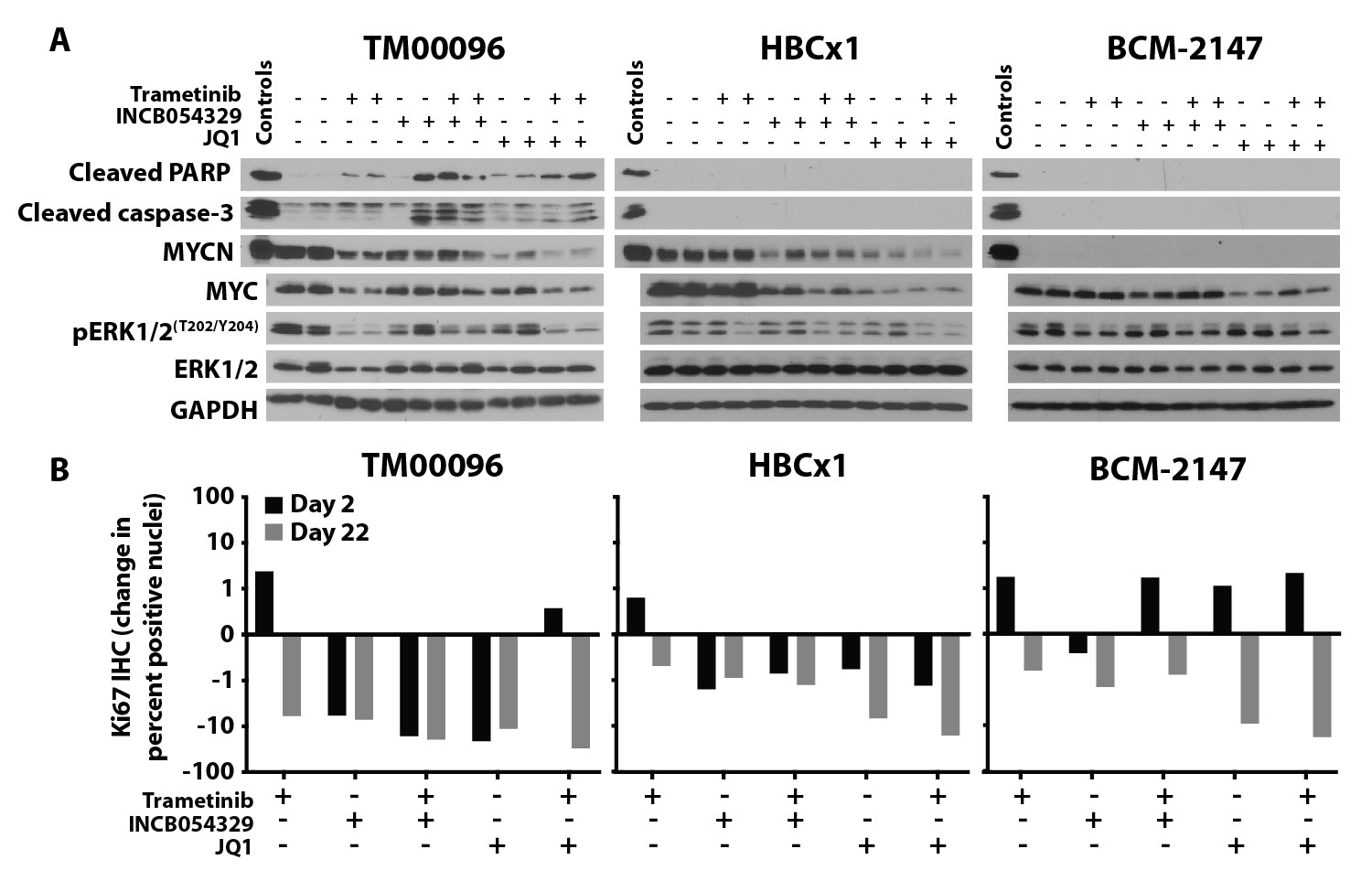
To determine the effects of the agents on pharmacodynamic markers in vivo, tumors were resected and protein extracted after the initial (two days) and final (22 days) treatments during the PDX study. Through immunoblot analyses, we observed that trametinib decreased pERK½ and both BETis decreased MYC and MYCN in all three PDX models, consistent with the agents’ predicted biochemical activities (fig. S11A). To determine whether decreased cell proliferation or increased apoptosis contributed to the observed decrease in tumor growth in the MYCNHigh and MYCNIntermediate models treated with the combination, we evaluated markers of proliferation (Ki67) and apoptosis (cleaved PARP and cleaved caspase-3) by IHC and immunoblot, respectively. Unlike the MYCNLow PDX model, Ki67 decreased in tissue from the MYCNHigh and MYCNIntermediate models treated with BETi, as a single agent or in combination with MEKi, after two days of treatment and to a greater extent at the end of treatment (fig. S11B and data file S1). Only the MYCNHigh model displayed markers of apoptosis after two days of treatment with each single agent alone or in combination (fig. S11A). These data suggest that BETis decreased the quantity of both MYCN and MYC in tumor cells grown in vivo and the combination treatments that resulted in a decrease in tumor volume in both MYCN-expressing TNBC models (Fig. 7E) was due to pro-apoptotic mechanisms in the MYCNHigh model and anti-proliferative effects in the MYCNHigh and MYCNIntermediate models.
Changes in MYC-family isoform expression in vivo after BETi and MEKi combination treatment
To evaluate changes in cellular expression of MYCN and MYC during treatment, we performed IHC and dual MYC-family isoform TSA-IF on PDX tissue collected after initial and final doses. Similar to immunoblot results at the early treatment timepoint (fig. S11A), single agent BETis decreased MYC in the MYCNLow PDX model and both MYC and MYCN in the MYCNHigh and MYCNIntermediate models compared to vehicle-treated MYC-family isoform expression (Fig. 8A, fig. S12, and data files S1 and S7). At the late treatment timepoint, MYCN expression was inhibited to a greater extent than MYC after treatment with either single agent BETi in both MYCN-expressing PDX models compared to vehicle-treated tissue (Fig. 8, B and C, and data files S1 and S7). However, trametinib combined with either BETi decreased MYCN and MYC to a greater extent than with either BETi alone throughout the time course of treatment in both the MYCNHigh and MYCNIntermediate models (Fig. 8, B and C, and data files S1 and S7). Taken together, treatment with either structurally distinct BETi, INCB054329 or JQ1, when combined with MEKi, continuously inhibited MYC-family isoform expression and resulted in synergistic TGI in the MYCNHigh and MYCNIntermediate TNBC PDX models and tumor regression in the MYCNHigh model.
Fig. 8. Evaluation of MYC-family isoform expression after BETi and MEKi combination treatment in vivo.

(A) Representative TSA-IF of MYCN and MYC in TM00096 after two or 22 days of treatment with trametinib (0.1 mg/kg, QD), INCB054329 (50 mg/kg, BID), or JQ1 (50 mg/kg, BID) alone, or either BETi in combination with trametinib. Colors represent cell nuclei (blue), MYCN (magenta), and MYC (green). Scale bar, 20 μm. (B) Quantification of IHC (percent positive cells) and (C) violin plots showing the distribution of TSA-IF intensity per nucleus for MYCN and MYC in TM00096, HBCx1, and BCM-2147 sections after treatments described in A for 22 days. Student’s t-test between vehicle and treatment arms and between single agent BETi and corresponding combination treated tissue, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
DISCUSSION
The lack of therapeutically targetable, high-frequency driver alterations across TNBC creates a challenge for developing strategies to treat patients with this cancer. Herein, we evaluate the expression of MYCN, a transcription factor associated with increased stemness, EMT, survival, and dormancy phenotypes in TNBC cells (18). Through the use of IHC, we assessed MYCN protein expression in several TNBC patient cohorts, including both primary tumors and metastatic disease, and report that a substantial fraction (45–64%) of tumors heterogeneously express MYCN. Further, MYCN-expressing cells are present in residual disease after NAC treatment, as well as in TNBC cell line cultures that acquired resistance to PI3Ki, suggesting that induction or maintenance of MYCN expression confers a survival advantage for cells treated with compounds that target microtubule structure (taxanes), induce DNA damage (anthracyclines), or cause metabolic stress (PI3Ki). NE prostate cancer, a tumor type considered to be driven by MYCN expression (54), is associated with castration- and androgen inhibitor-resistance and a poor prognosis (54, 55). Unlike MYCN-amplified NB, AML, and GBM, which are tumors that have retained a same-cell lineage, NE prostate cancers are thought to have differentiated from castration-resistant prostate adenocarcinoma through MYCN-mediated mechanisms and lineage switching (13, 54). Herein, we found MYCN transcript in primary, treatment-naïve TNBC to be comparable to MYCN expression in NE-CRPC, suggesting that MYCN-expressing TNBC could represent a similar altered differentiation state.
In addition to TNBC tumors lacking therapeutic targets, the development of effective drug-treatment strategies for TNBC patients has also been hindered by the presence of highly heterogeneous intratumoral cell populations with differing biological properties within an individual patient’s tumor. Through the use of dual MYC-isoform TSA-IF, we report that MYCN and its family member MYC are heterogeneously expressed in separate cell nuclei within a given tumor in at least 40% of TNBC tumors. Previous studies have demonstrated MYCN and MYC preferentially regulate the same set of core genes involved in metabolism and cell growth, and while the MYCN allele can functionally replace MYC in murine development (56), MYCN and MYC have separate temporal regulation over organogenesis in early vertebrate development (33). MYCN expression is essential for initial establishment of stem and progenitor populations; over the course of organ system development, MYCN expression switches to low MYC expression to support stem and progenitor cell maintenance, and during cell lineage commitment and expansion, increased MYC drives highly proliferative cells until they reach terminal differentiation (33). We observed similar MYC-family isoform switching in our clonally-derived TNBC cell line models, indicating that tumor cells have retained the ability to transition between MYCN and MYC, which may account for the large range (2–100%) of MYCN expression within heterogenous TNBC cell populations.
By isolating and expanding single cells from heterogeneous TNBC tumor-derived cell line populations, we generated distinct MYCN- and MYC-expressing cell cultures with a similar genetic background, thus allowing us to assign the biological relevance of MYCN versus MYC expression to sensitivity of compounds under preclinical or clinical investigation. We conducted a high-throughput 158-drug screen that included compounds from the NCI FDA-AOD library and identified inhibitors of the BRD-family of transcriptional regulators (BETi) that were preferentially effective in inhibiting MYCN-expressing tumor cell growth. BETis are a class of compounds currently under early stage clinical development that broadly target the BRD family (predominantly BRD2, BRD3, and BRD4) of transcriptional regulators (44). These compounds were of particular interest given previous reports that MYC-family isoform signaling, including contributions from MYCN, is enriched in TNBC (57) and that TNBC has preferential sensitivity to BETis compared to other breast cancer subtypes (45). Further, efficacy of BETis has been predominantly attributed to selective disruption of super-enhancer-associated genes that deregulates transcription factor activity (45, 58, 59). BRD4 regulates transcription of MYCN as well as occupies MYCN-associated target genes, enhancers, and super-enhancers (22), and preclinical studies have suggested BETis as a promising strategy to target MYCN-driven neuronal [neuroblastoma (8, 9), medulloblastoma (60), embryonal tumors with multilayered rosettes (61)] and non-neuronal [ovarian cancer (12), alveolar rhabdomyosarcomas (62)] tumor cell growth. Whereas prior studies have focused on BRD-mediated targeting of MYC, we show that TNBC tumors are heterogeneously composed of MYC- and MYCN-expressing cells and MYCN-expressing cells have differential sensitivity to BETis in select tumor cells and model systems.
We acknowledge that limitations exist in regard to this study. As previously mentioned, MYCN-expressing cells exist within highly heterogeneous intratumoral cell populations. Our assessment of MYCN expression in TNBC tumors is limited to the tissue sections under investigation and may not be representative of the entire tumor. Thus, the number of MYCN-expressing TNBC tumors may be higher than reported herein. We also demonstrate the presence of MYCN-expressing cells in residual disease after NAC and PI3Ki treatment. Whether MYCN-expressing cells were pre-existing and selected for with treatment or whether epigenetic events upregulated MYCN expression in cells initially devoid of MYCN remains unclear. Lastly, the restricted availability of MYCN-expressing TNBC models for in vitro and in vivo preclinical evaluation limits analyses of the effects of combined MEKi and BETi treatment across a larger cohort of MYCN-expressing TNBC.
Currently, BETis are in the initial stages of clinical assessment and have had their greatest single agent clinical efficacy in hematopoietic and nuclear protein in testis (NUT) midline malignancies (44); however, favorable preclinical investigations with BETi combination treatments have catalyzed interventional trials to improve hematopoietic malignancy and solid tumor patient responses (44, 63). In our study, we discovered that single agent BETi and MEKi treatments decreased both MYCN and MYC expression and had a greater effect when used in combination. Importantly, combined low-dose BETi and MEKi displayed a synergistic decrease in tumor cell viability, both in the setting of in vitro cell cultures and in mice harboring TNBC PDXs with heterogeneous expression of both MYCN and MYC. Synergies between BETi and MEKi have been attributed to an upregulation of MAPK pathway signaling in response to BETi treatment (64) and the ability of BETis to disrupt adaptive bypass mechanisms induced by MEKi treatment (65). Although we did not observe an upregulation of MAPK pathway signaling after BETi treatment in either our TNBC cell lines or PDX tissue, we cannot rule out chromatin modulation or enhancer remodeling in response to treatment with either single agent given the rebound/upregulation of MYC expression in response to BETi treatment in the CAL-51 clonal cell lines. Aurora kinase inhibitors, which are also used to target MYCN-driven tumors (10, 24), were a top “hit” in our screens against MYCN-expressing TNBC. Given preferential effects of cyclin-dependent kinase (CDK) inhibitors on MYC-family isoform pathway signaling (57), CDK and aurora kinase inhibitors could also be evaluated as a means for targeting MYCN-expressing TNBC.
In summary, we have identified MYCN-expressing TNBC cell populations within a substantial fraction of evaluated tumors that have the ability to survive various forms of drug-induced cellular stress, have survival advantages in vitro under selective anti-proliferative treatments, and transition between differentiation states (as defined by MYC-family expression status). Based on our preclinical results using in vitro and in vivo TNBC models, we posit that BETi and MEKi combination treatment will induce regression of MYCN-expressing TNBC tumors. Given that patients with TNBC primarily receive systemic cytotoxic chemotherapies that frequently result in unfavorable outcomes, we propose the clinical development of combination BET and MEK inhibitors for patients with advanced TNBC, with parallel evaluation of MYCN as a potential marker for patient selection.
MATERIALS AND METHODS
Study design
The study was designed to identify the proportion of treatment-naïve (n=191), NAC-treated (n=115), and recurrent TNBC tumors (n=38) that express MYCN. Clinical specimens for IHC analyses were collected at VUMC in Nashville, TN; Instituto Nacional de Enfermedades Neoplásicas in Lima, Peru; or in conjunction with a commercial source, US Biomax. All clinical and pathologic data were retrieved under institutionally approved protocols. Protein expression (H-scores) resulting from IHC for MYCN and MYC was determined by a pathologist (P.I.G-E.), and analyses were performed by researchers blinded to the patients’ medical background and treatments received.
We also designed the study to investigate whether compounds identified through in vitro assays would induce TNBC cell growth inhibition and/or apoptosis in vivo. Mice were housed and treated in accordance with protocols approved by the Institutional Care and Use Committee for animal research at Vanderbilt University. To sufficiently power the studies at 90% (β=0.2) and a significance level of α=0.05, assuming normal distributions, equal SD, and an expected effect size of 50%, five to nine mice were used for tumor measurements per arm, depending on the growth kinetics of each PDX model. Once the PDX tumors reached approximately 150–250 mm3, mice were randomized into single agent or combination treatment groups that consisted of a MEKi (trametinib) and/or BETi (INCB054329 or JQ1). Two additional mice per arm were included in the study for early PDX molecular analyses and were removed after two days of treatment. The MYCNHigh PDX model (TM00096) was evaluated twice; first, in a four-arm study with trametinib and INCB054329 treatments for 14 days and again, in a six-arm study with all described compounds for 22 days. No data exclusion criteria were applied or outliers excluded. Early and late molecular analyses (after two and 22 days of treatment, respectively) on PDX tumors were performed. MYCN and MYC expression (H-scores) were quantified by a pathologist (P.I.G-E.) and analyses performed blinded to treatments received.
In vivo patient-derived xenograft (PDX) experiments
Mice were housed and treated in accordance with protocols approved by the Institutional Care and Use Committee for animal research at Vanderbilt University. Female 6- to 8-week-old NOD scid gamma (NSG) or athymic nude mice (Jackson Laboratory) were anesthetized with isoflurane and subjected to subcutaneous engraftment of a 2 mm3 TNBC PDX [Jackson Laboratory (TM00096, TM00090, TM01273), Baylor University (BCM-2147), Xentech (HBCx1)] fragment into the lateral dorsal side of each mouse. After surgical implantation, the mice were monitored daily for 10–14 days. Once wound clips were removed and tumors reached approximately 150–250 mm3, mice were randomized into single agent and combination treatment groups. Mice were treated with the MEKi, trametinib (0.1 mg/kg, once daily), in 0.5% methylcellulose with 0.2% Tween-80, and/or a BETi, INCB054329 or JQ1 (50 mg/kg, twice daily), in 0.5% methylcellulose with 5% N,N-dimethylacetamide, through orogastric gavage for either 14 or 22 days. Tumor volumes were calculated twice a week by caliper measurements (width2 X length/2) and body weight measured once a week. Tumors used for subsequent molecular analyses were snap-frozen and deposited in a liquid nitrogen storage tank.
Statistical methods
Statistical analyses were performed using GraphPad Prism software (GraphPad) and R (Version 3.6, https://www.R-project.org/). As indicated in the figure legends, the SD, SEM, or boxplot is shown. Wilcoxon rank sum test was used to compare the amount of MYCN transcript between TNBC and the other MYCN-expressing cancer types. P-values were adjusted by false discovery rate (Fig. 1B). Wilcoxon rank sum test was also used to determine the difference in MYCN expression between treatment-naïve and NAC-treated MYCN-expressing tumors (Fig. 2B). Student’s t-tests were used to determine differential BETi sensitivity between MYCNLow and MYCNHigh CAL-51 clonal cell lines (Fig. 4, F and G) and changes in MYC-family isoform relative fluorescence intensity per nucleus before and after BETi treatments (Fig. 5F). Student’s t-tests were also used to determine significance of differences in tumor volumes and MYC-family isoform expression between MEK and/or BETi treatments in the PDX experiments (Fig. 7, C and E, and Fig. 8B). P <0.05 was considered statistically significant.
Supplementary Material
Fig. S1. Identification of TNBC breast cancers in TCGA and METABRIC datasets.
{kind=link}
Fig. S2. Distribution of MYCN expression across TNBC.
{kind=link}
Fig. S3. IHC detection of MYCN in cell line- and patient-derived xenograft tissue.
{kind=link}
Fig. S4. MYCN expression in primary TNBC and patient-matched metastases.
{kind=link}
Fig. S5. MYCN and MYC intratumoral heterogeneity in primary and recurrent TNBC.
{kind=link}
Fig. S6. MYCN and MYC expression in TNBC cell populations and CAL-51 clonally-derived cell lines.
{kind=link}
Fig. S7. BETi sensitivity of CAL-51 MYCNHigh cell lines.
Fig. S8. Changes in MYC target gene expression in CAL-51 MYCNHigh cell lines after BETi treatment.
{kind=link}
Fig. S9. Differential gene expression analyses between MYCNRatioHigh and MYCRatioHigh TNBC.
{kind=link}
Fig. S10. Effect of BETi and MEKi combination treatments on weight of treated mice.
{kind=link}
Fig. S11. Evaluation of apoptosis and proliferation after BETi and MEKi treatment in TNBC PDX models.
{kind=link}
Fig. S12. Evaluation of MYC-family isoform expression after BETi and MEKi combination treatment in vivo.
{kind=link}
Table S1. MYCN and MYC expression in breast cancer PDX models.
Table S2. Characteristics of patients with treatment-naïve and NAC-treated primary TNBC.
Data file S1. Tabular data points for experiments with a sample size of n < 20.
Data file S2. IHC results for MYCN and MYC in primary, treatment-naïve; primary, NAC-treated; and recurrent TNBC cases.
Data file S3. Primary drug screen results using CAL-51 MYCNLow and MYCNHigh cell lines.
Data file S4. Secondary drug screen results using CAL-51 MYCNLow and MYCNHigh cell lines.
Data file S5. Tabular data points for MYC-family isoform TSA-IF in CAL-51 after single agent BETi treatment.
Data file S6. Tabular data points for MYC-family isoform TSA-IF in MDA-MB-468 after single agent BETi treatment.
Data file S7. Tabular data points for MYC-family isoform TSA-IF in TNBC cell lines and PDX tissue after BETi and MEKi single agent and combination treatment.
Acknowledgments
We thank the patients whom contributed tissue used in this study, the clinical providers at Vanderbilt University Medical Center (Nashville, TN) and the Instituto Nacional de Enfermedades Neoplásicas (Lima, Perú) for processing of tumor samples, and B.C., B.C.M., and J.M.B. for construction of TMA11-4-09, TMA111, and TMAP1,P2,P3, respectively. We thank Michael T. Lewis for generating and supplying the BCM-2147 TNBC PDX model and S.W.H for his expertise and resources to conduct PRO-Seq. SK-N-BE(2)C and VU661013 were kindly provided by Dr. Dai H. Chung and Dr. Stephen W. Fesik, respectively.
Funding: This research was supported by: a grant from Incyte Corporation (J.A.P., S.W.H) as part of the Incyte-Vanderbilt Alliance; NCI grants CA068485 (J.A.P.), CA098131 (J.A.P.), and CA211206 (J.A.B.); and Susan G. Komen grants SAC110030 (J.A.P.) and CCR13262005 (B.D.L). We thank Vanderbilt Technologies for Advanced Genomics (VANTAGE) and the Translational Pathology Shared Resources (TPSR), supported by the Vanderbilt-Ingram Cancer Center (P30 CA068485); and the Pathology and Tissue Informatics Core of the Specialized Program of Research Excellence (SPORE) in Breast Cancer (P50 CA098131) for providing the histopathological analyses.
Footnotes
Competing Interests: M.C.S. is a current employee of Incyte Corporation. P.C.C.L. and P.S. are former employees of Incyte Corporation; current affiliations are Kymera Therapeutics and Prelude Therapeutics, respectively.
Data and materials availability: All data associated with this study are present in the main text or Supplementary Materials. The CAL-51 clonal cell lines are available from the corresponding author’s laboratory and require a Material Transfer Agreement.
SUPPLEMENTARY MATERIALS
Materials and methods
REFERENCES AND NOTES
- 1.Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S, Deming SL, Geradts J, Cheang MCU, Nielsen TO, Moorman PG, Earp HS, Millikan RC, Race, Breast Cancer Subtypes, and Survival in the Carolina Breast Cancer Study, JAMA 295, 2492 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA, Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence, Clin. Cancer Res 13, 4429–4434 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, Burleigh A, Yap D, Bernard V, McPherson A, Shumansky K, Crisan A, Giuliany R, Heravi-Moussavi A, Rosner J, Lai D, Birol I, Varhol R, Tam A, Dhalla N, Zeng T, Ma K, Chan SK, Griffith M, Moradian A, Cheng SWG, Morin GB, Watson P, Gelmon K, Chia S, Chin S-F, Curtis C, Rueda OM, Pharoah PD, Damaraju S, Mackey J, Hoon K, Harkins T, Tadigotla V, Sigaroudinia M, Gascard P, Tlsty T, Costello JF, Meyer IM, Eaves CJ, Wasserman WW, Jones S, Huntsman D, Hirst M, Caldas C, Marra MA, Aparicio S, The clonal and mutational evolution spectrum of primary triple-negative breast cancers, Nature 486, 1–5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, Fulton LL, Dooling DJ, Ding L, Mardis ER, Wilson RK, Ally A, Balasundaram M, Butterfield YSN, Carlsen R, Carter C, Chu A, Chuah E, Chun HJE, Coope RJN, Dhalla N, Guin R, Hirst C, Hirst M, Holt RA, Lee D, Li HI, Mayo M, Moore RA, Mungall AJ, Pleasance E, Robertson AG, Schein JE, Shafiei A, Sipahimalani P, Slobodan JR, Stoll D, Tam A, Thiessen N, Varhol RJ, Wye N, Zeng T, Zhao Y, Birol I, Jones SJM, Marra MA, Cherniack AD, Saksena G, Onofrio RC, Pho NH, Carter SL, Schumacher SE, Tabak B, Hernandez B, Gentry J, Nguyen H, Crenshaw A, Ardlie K, Beroukhim R, Winckler W, Getz G, Gabriel SB, Meyerson M, Chin L, Kucherlapati R, Hoadley KA, Auman JT, Fan C, Turman YJ, Shi Y, Li L, Topal MD, He X, Chao HH, Prat A, Silva GO, Iglesia MD, Zhao W, Usary J, Berg JS, Adams M, Booker J, Wu J, Gulabani A, Bodenheimer T, Hoyle AP, Simons JV, Soloway MG, Mose LE, Jefferys SR, Balu S, Parker JS, Hayes DN, Perou CM, Malik S, Mahurkar S, Shen H, Weisenberger DJ, Triche T, Lai PH, Bootwalla MS, Maglinte DT, Berman BP, Van Den Berg DJ, Baylin SB, Laird PW, Creighton CJ, Donehower LA, Noble M, Voet D, Gehlenborg N, Di Cara D, Zhang J, Zhang H, Wu CJ, Yingchun Liu S, Lawrence MS, Zou L, Sivachenko A, Lin P, Stojanov P, Jing R, Cho J, Sinha R, Park RW, Nazaire MD, Robinson J, Thorvaldsdottir H, Mesirov J, Park PJ, Reynolds S, Kreisberg RB, Bernard B, Bressler R, Erkkila T, Lin J, Thorsson V, Zhang W, Shmulevich I, Ciriello G, Weinhold N, Schultz N, Gao J, Cerami E, Gross B, Jacobsen A, Sinha R, Aksoy BA, Antipin Y, Reva B, Shen R, Taylor BS, Ladanyi M, Sander C, Anur P, Spellman PT, Lu Y, Liu W, Verhaak RRG, Mills GB, Akbani R, Zhang N, Broom BM, Casasent TD, Wakefield C, Unruh AK, Baggerly K, Coombes K, Weinstein JN, Haussler D, Benz CC, Stuart JM, Benz SC, Zhu J, Szeto CC, Scott GK, Yau C, Paull EO, Carlin D, Wong C, Sokolov A, Thusberg J, Mooney S, Ng S, Goldstein TC, Ellrott K, Grifford M, Wilks C, Ma S, Craft B, Yan C, Hu Y, Meerzaman D, Gastier-Foster JM, Bowen J, Ramirez NC, Black AD, White P, Zmuda EJ, Frick J, Lichtenberg TM, Brookens R, George MM, Gerken MA, Harper HA, Leraas KM, Wise LJ, Tabler TR, McAllister C, Barr T, Hart-Kothari M, Tarvin K, Saller C, Sandusky G, Mitchell C, Iacocca MV, Brown J, Rabeno B, Czerwinski C, Petrelli N, Dolzhansky O, Abramov M, Voronina O, Potapova O, Marks JR, Suchorska WM, Murawa D, Kycler W, Ibbs M, Korski K, Spychała A, Murawa P, Brzeziński JJ, Perz H, Łaźniak R, Teresiak M, Tatka H, Leporowska E, Bogusz-Czerniewicz M, Malicki J, Mackiewicz A, Wiznerowicz M, Van Le X, Kohl B, Viet Tien N, Thorp R, Van Bang N, Sussman H, Phu BD, Hajek R, Hung NP, Phuong TVT, Thang HQ, Khan KZ, Penny R, Mallery D, Curley E, Shelton C, Yena P, Ingle JN, Couch FJ, Lingle WL, King TA, Gonzalez-Angulo AM, Dyer MD, Liu S, Meng X, Patangan M, Waldman F, Stöppler H, Rathmell WK, Thorne L, Huang M, Boice L, Hill A, Morrison C, Gaudioso C, Bshara W, Daily K, Egea SC, Pegram MD, Gomez-Fernandez C, Dhir R, Bhargava R, Brufsky A, Shriver CD, Hooke JA, Campbell JL, Mural RJ, Hu H, Somiari S, Larson C, Deyarmin B, Kvecher L, Kovatich AJ, Ellis MJ, Stricker T, White K, Olopade O, Luo C, Chen Y, Bose R, Chang LW, Beck AH, Pihl T, Jensen M, Sfeir R, Kahn A, Chu A, Kothiyal P, Wang Z, Snyder E, Pontius J, Ayala B, Backus M, Walton J, Baboud J, Berton D, Nicholls M, Srinivasan D, Raman R, Girshik S, Kigonya P, Alonso S, Sanbhadti R, Barletta S, Pot D, Sheth M, Demchok JA, Shaw KRM, Yang L, Eley G, Ferguson ML, Tarnuzzer RW, Zhang J, Dillon LAL, Buetow K, Fielding P, Ozenberger BA, Guyer MS, Sofia HJ, Palchik JD, Comprehensive molecular portraits of human breast tumours, Nature 490, 61–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N, The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1., Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, Owens P, Sanders ME, Kuba MG, Sánchez V, Kurupi R, Moore PD, Pinto JA, Doimi FD, Gómez H, Horiuchi D, Goga A, Lehmann BD, Bauer JA, Pietenpol JA, Ross JS, Palmer GA, Yelensky R, Cronin M, Miller VA, Stephens PJ, Arteaga CL, Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets., Cancer Discov. 4, 232–45 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tansey WP, Mammalian MYC Proteins and Cancer, New J. Sci 2014, 1–27 (2014). [Google Scholar]
- 8.Henssen A, Althoff K, Odersky A, Beckers A, Koche R, Speleman F, Scha fers S, Bell E, Nortmeyer M, Westermann F, De Preter K, Florin A, Heukamp L, Spruessel A, Astrahanseff K, Lindner S, Sadowski N, Schramm A, Astorgues-Xerri L, Riveiro ME, Eggert A, Cvitkovic E, Schulte JH, Targeting MYCN-Driven Transcription By BET-Bromodomain Inhibition, Clin. Cancer Res 22, 2470–2481 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, Garnett MJ, McDermott U, Benes CH, Kung AL, Weiss WA, Bradner JE, Stegmaier K, Targeting MYCN in neuroblastoma by BET bromodomain inhibition., Cancer Discov. 3, 308–23 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, Cyrta J, Sboner A, Noorzad Z, MacDonald T, Cheung C, Yuen KS, Gao D, Chen Y, Eilers M, Mosquera J-M, Robinson BD, Elemento O, Rubin MA, Demichelis F, Rickman DS, N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer., Cancer Cell 30, 563–577 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, Baertsch R, Sokolov A, Meyerowitz JG, Mathis C, Cheng D, Stuart JM, Shokat KM, Gustafson WC, Huang J, Witte ON, N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells, Cancer Cell 29, 536–547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baratta MG, Schinzel AC, Zwang Y, Bandopadhayay P, Bowman-Colin C, Kutt J, Curtis J, Piao H, Wong LC, Kung AL, Beroukhim R, Bradner JE, Drapkin R, Hahn WC, Liu JF, Livingston DM, An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma, Proc. Natl. Acad. Sci 112, 232–237 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berger A, Brady NJ, Bareja R, Robinson B, Conteduca V, Augello MA, Puca L, Ahmed A, Dardenne E, Lu X, Hwang I, Bagadion AM, Sboner A, Elemento O, Paik J, Yu J, Barbieri CE, Dephoure N, Beltran H, Rickman DS, N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer., J. Clin. Invest 130, 3924–3940 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, McCastlain K, Edmonson M, Pounds SB, Shi L, Zhou X, Ma X, Sioson E, Li Y, Rusch M, Gupta P, Pei D, Cheng C, Smith MA, Auvil JG, Gerhard DS, V Relling M, Winick NJ, Carroll AJ, Heerema NA, Raetz E, Devidas M, Willman CL, Harvey RC, Carroll WL, Dunsmore KP, Winter SS, Wood BL, Sorrentino BP, Downing JR, Loh ML, Hunger SP, Zhang J, Mullighan CG, The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia, Nat. Genet 49, 1211–1218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawagoe H, Kandilci A, Kranenburg TA, Grosveld GC, Overexpression of N-Myc rapidly causes acute myeloid leukemia in mice., Cancer Res. 67, 10677–85 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Beltran H, The N-myc Oncogene: Maximizing its Targets, Regulation, and Therapeutic Potential, Mol. Cancer Res 12, 815–822 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Mizukami Y, Nonomura A, Takizawa T, Noguchi M, Michigishi T, Nakamura S, Ishizaki T, N-myc protein expression in human breast carcinoma: prognostic implications., Anticancer Res. 15, 2899–905. [PubMed] [Google Scholar]
- 18.Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang C-Y, Yaswen P, Goga A, Werb Z, Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells., Nature 526, 131–5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, Brannigan BW, Kapur R, Stott SL, Shioda T, Ramaswamy S, Ting DT, Lin CP, Toner M, Haber DA, Maheswaran S, Circulating Tumor Cell Clusters Are Oligoclonal Precursors of Breast Cancer Metastasis, Cell 158, 1110–1122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA, Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies., J. Clin. Invest 121, 2750–2767 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Gräf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, Aparicio S, Brenton JD, Ellis I, Huntsman D, Pinder S, Murphy L, Bardwell H, Ding Z, Jones L, Liu B, Papatheodorou I, Sammut SJ, Wishart G, Chia S, Gelmon K, Speers C, Watson P, Blamey R, Green A, MacMillan D, Rakha E, Gillett C, Grigoriadis A, De Rinaldis E, Tutt A, Parisien M, Troup S, Chan D, Fielding C, Maia AT, McGuire S, Osborne M, Sayalero SM, Spiteri I, Hadfield J, Bell L, Chow K, Gale N, Kovalik M, Ng Y, Prentice L, Tavaré S, Markowetz F, Langerød A, Provenzano E, Purushotham A, Børresen-Dale AL, Caldas C, The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups, Nature 486, 346–352 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rickman DS, Schulte JH, Eilers M, The Expanding World of N-MYC-Driven Tumors., Cancer Discov. 8, 150–163 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, Kim J, Lawrence MS, Lichenstein L, McKenna A, Pedamallu CS, Ramos AH, Shefler E, Sivachenko A, Sougnez C, Stewart C, Ally A, Birol I, Chiu R, Corbett RD, Hirst M, Jackman SD, Kamoh B, Khodabakshi AH, Krzywinski M, Lo A, Moore RA, Mungall KL, Qian J, Tam A, Thiessen N, Zhao Y, Cole KA, Diamond M, Diskin SJ, Mosse YP, Wood AC, Ji L, Sposto R, Badgett T, London WB, Moyer Y, Gastier-Foster JM, Smith MA, Auvil JMG, Gerhard DS, Hogarty MD, Jones SJM, Lander ES, Gabriel SB, Getz G, Seeger RC, Khan J, Marra MA, Meyerson M, Maris JM, The genetic landscape of high-risk neuroblastoma, Nat. Genet 45, 279–284 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BVSK, Varambally S, Tomlins SA, Nanus DM, Tagawa ST, Van Allen EM, Elemento O, Sboner A, Garraway LA, Rubin MA, Demichelis F, Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer, Nat. Med 22, 298–305 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, Cieslik M, Benelli M, Robinson D, Van Allen EM, Sboner A, Fedrizzi T, Mosquera JM, Robinson BD, De Sarkar N, Kunju LP, Tomlins S, Wu YM, Nava Rodrigues D, Loda M, Gopalan A, Reuter VE, Pritchard CC, Mateo J, Bianchini D, Miranda S, Carreira S, Rescigno P, Filipenko J, Vinson J, Montgomery RB, Beltran H, Heath EI, Scher HI, Kantoff PW, Taplin M-E, Schultz N, deBono JS, Demichelis F, Nelson PS, Rubin MA, Chinnaiyan AM, Sawyers CL, Genomic correlates of clinical outcome in advanced prostate cancer, Proc. Natl. Acad. Sci 116, 11428–11436 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harenza JL, Diamond MA, Adams RN, Song MM, Davidson HL, Hart LS, Dent MH, Fortina P, Reynolds CP, Maris JM, Transcriptomic profiling of 39 commonly-used neuroblastoma cell lines, Sci. Data 4, 170033 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guarneri V, Broglio K, Kau S-W, Cristofanilli M, Buzdar AU, Valero V, Buchholz T, Meric F, Middleton L, Hortobagyi GN, Gonzalez-Angulo AM, Prognostic value of pathologic complete response after primary chemotherapy in relation to hormone receptor status and other factors., J. Clin. Oncol 24, 1037–44 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Liedtke C, Mazouni C, Hess KR, André F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M, Cristofanilli M, Hortobagyi GN, Pusztai L, Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer., J. Clin. Oncol 26, 1275–81 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, Crosetto N, Foukakis T, Navin NE, Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing., Cell 173, 879–-893.e13. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balko JM, Cook RS, Vaught DB, Kuba MG, Miller TW, Bhola NE, Sanders ME, Granja-Ingram NM, Smith JJ, Meszoely IM, Salter J, Dowsett M, Stemke-Hale K, González-Angulo AM, Mills GB, Pinto JA, Gómez HL, Arteaga CL, Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance., Nat. Med 18, 1052–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonotto M, Gerratana L, Poletto E, Driol P, Giangreco M, Russo S, Minisini AM, Andreetta C, Mansutti M, Pisa FE, Fasola G, Puglisi F, Measures of Outcome in Metastatic Breast Cancer: Insights From a Real-World Scenario, Oncologist 19, 608–615 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siegel MB, He X, Hoadley KA, Hoyle A, Pearce JB, Garrett AL, Kumar S, Moylan VJ, Brady CM, Van Swearingen AED, Marron D, Gupta GP, Thorne LB, Kieran N, Livasy C, Mardis ER, Parker JS, Chen M, Anders CK, Carey LA, Perou CM, Integrated RNA and DNA sequencing reveals early drivers of metastatic breast cancer, J. Clin. Invest 128, 1371–1383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hurlin PJ, Control of vertebrate development by MYC., Cold Spring Harb. Perspect. Med 3, a014332 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jané-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P, de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA, The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity, Nature 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lehmann BD, Bauer JA, Schafer JM, Pendleton CS, Tang L, Johnson KC, Chen X, Balko JM, Gómez H, Arteaga CL, Mills GB, Sanders ME, Pietenpol JA, PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors., Breast Cancer Res. 16, 406 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klempner SJ, Myers AP, Cantley LC, What a tangled web we weave: emerging resistance mechanisms to inhibition of the phosphoinositide 3-kinase pathway., Cancer Discov. 3, 1345–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, Mitsiades CS, BET bromodomain inhibition as a therapeutic strategy to target c-Myc., Cell 146, 904–17 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horiuchi D, Camarda R, Zhou AY, Yau C, Momcilovic O, Balakrishnan S, Corella AN, Eyob H, Kessenbrock K, Lawson DA, Marsh LA, Anderton BN, Rohrberg J, Kunder R, V Bazarov A, Yaswen P, McManus MT, Rugo HS, Werb Z, Goga A, PIM1 kinase inhibition as a targeted therapy against triple-negative breast tumors with elevated MYC expression, Nat. Med 22, 1321–1329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sears R, Leone G, DeGregori J, Nevins JR, Ras Enhances Myc Protein Stability, Mol. Cell 3, 169–179 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Dauch D, Rudalska R, Cossa G, Nault J-C, Kang T-W, Wuestefeld T, Hohmeyer A, Imbeaud S, Yevsa T, Hoenicke L, Pantsar T, Bozko P, Malek NP, Longerich T, Laufer S, Poso A, Zucman-Rossi J, Eilers M, Zender L, A MYC–aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer, Nat. Med 22, 744–753 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE, Selective inhibition of BET bromodomains, Nature 468, 1067–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stubbs MC, Burn TC, Sparks R, Maduskuie T, Diamond S, Rupar M, Wen X, Volgina A, Zolotarjova N, Waeltz P, Favata M, Jalluri R, Liu H, Liu XM, Li J, Collins R, Falahatpisheh N, Polam P, DiMatteo D, Feldman P, Dostalik V, Thekkat P, Gardiner C, He X, Li Y, Covington M, Wynn R, Ruggeri B, Yeleswaram S, Xue C-B, Yao W, Combs AP, Huber R, Hollis G, Scherle P, Liu PCC, The Novel Bromodomain and Extraterminal Domain Inhibitor INCB054329 Induces Vulnerabilities in Myeloma Cells That Inform Rational Combination Strategies, Clin. Cancer Res., 1–13 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Boi M, Gaudio E, Bonetti P, Kwee I, Bernasconi E, Tarantelli C, Rinaldi A, Testoni M, Cascione L, Ponzoni M, Mensah AA, Stathis A, Stussi G, Riveiro ME, Herait P, Inghirami G, Cvitkovic E, Zucca E, Bertoni F, The BET bromodomain inhibitor OTX015 affects pathogenetic pathways in preclinical B-cell tumor models and synergizes with targeted drugs, Clin. Cancer Res 21, 1628–1638 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Stathis A, Bertoni F, BET Proteins as Targets for Anticancer Treatment., Cancer Discov. 8, 24–36 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, Doherty E, Mohammed H, Guo H, Stover DG, Ekram MB, Brown J, D’Santos C, Krop IE, Dillon D, McKeown M, Ott C, Qi J, Ni M, Rao PK, Duarte M, Wu S-Y, Chiang C-M, Anders L, Young RA, Winer E, Letai A, Barry WT, Carroll JS, Long H, Brown M, Liu XS, Meyer CA, Bradner JE, Polyak K, Polyak K, Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer., Nature 529, 413–417 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stratikopoulos EE, Dendy M, Szabolcs M, Khaykin AJ, Lefebvre C, Zhou M-M, Parsons R, Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy., Cancer Cell 27, 837–51 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sears RC, The life cycle of C-myc: from synthesis to degradation., Cell Cycle 3, 1133–7 (2004). [PubMed] [Google Scholar]
- 48.Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR, Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability., Genes Dev. 14, 2501–14 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eleveld TF, Oldridge DA, Bernard V, Koster J, Daage LC, Diskin SJ, Schild L, Bentahar NB, Bellini A, Chicard M, Lapouble E, Combaret V, Legoix-Né P, Michon J, Pugh TJ, Hart LS, Rader J, Attiyeh EF, Wei JS, Zhang S, Naranjo A, Gastier-Foster JM, Hogarty MD, Asgharzadeh S, Smith MA, Guidry Auvil JM, Watkins TBK, Zwijnenburg DA, Ebus ME, van Sluis P, Hakkert A, van Wezel E, van der Schoot CE, Westerhout EM, Schulte JH, Tytgat GA, Dolman MEM, Janoueix-Lerosey I, Gerhard DS, Caron HN, Delattre O, Khan J, Versteeg R, Schleiermacher G, Molenaar JJ, Maris JM, Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations, Nat. Genet 47, 864–871 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hart LS, Rader J, Raman P, Batra V, Russell MR, Tsang M, Gagliardi M, Chen L, Martinez D, Li Y, Wood A, Kim S, Parasuraman S, Delach S, Cole KA, Krupa S, Boehm M, Peters M, Caponigro G, Maris JM, Preclinical Therapeutic Synergy of MEK½ and CDK4/6 Inhibition in Neuroblastoma., Clin. Cancer Res 23, 1785–1796 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Borisy AA, Elliott PJ, Hurst NW, Lee MS, Lehar J, Price ER, Serbedzija G, Zimmermann GR, Foley MA, Stockwell BR, Keith CT, Systematic discovery of multicomponent therapeutics., Proc. Natl. Acad. Sci. U. S. A 100, 7977–82 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wallin JJ, Guan J, Prior WW, Lee LB, Berry L, Belmont LD, Koeppen H, Belvin M, Friedman LS, Sampath D, GDC-0941, a Novel Class I Selective PI3K Inhibitor, Enhances the Efficacy of Docetaxel in Human Breast Cancer Models by Increasing Cell Death In Vitro and In Vivo, Clin. Cancer Res 18, 3901–3911 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Kim H, Kumar P, Menghi F, Noorbakhsh J, Cerveira E, Ryan M, Zhu Q, Ananda G, George J, Chen HC, Mockus S, Zhang C, Yang Y, Keck J, Karuturi RKM, Bult CJ, Lee C, Liu ET, Chuang JH, High-resolution deconstruction of evolution induced by chemotherapy treatments in breast cancer xenografts., Sci. Rep 8, 17937 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davies AH, Beltran H, Zoubeidi A, Cellular plasticity and the neuroendocrine phenotype in prostate cancer, Nat. Rev. Urol 15, 271–286 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Kirby M, Hirst C, Crawford ED, Characterising the castration-resistant prostate cancer population: a systematic review, Int. J. Clin. Pract 65, 1180–1192 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Malynn BA, De Alboran IM, O’Hagan RC, Bronson R, Davidson L, DePinho RA, Alt FW, N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation, Genes Dev. (2000). [PMC free article] [PubMed] [Google Scholar]
- 57.Horiuchi D, Kusdra L, Huskey NE, Chandriani S, Lenburg ME, Gonzalez-Angulo AM, Creasman KJ, V Bazarov A, Smyth JW, Davis SE, Yaswen P, Mills GB, Esserman LJ, Goga A, MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition., J. Exp. Med 209, 679–96 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MGM, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, Reichert E, Kung AL, Rodig SJ, Young RA, Shipp MA, Bradner JE, Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma., Cancer Cell 24, 777–90 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA, Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers, Cell 153, 320–334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher SE, Zeid R, Masoud S, Yu F, Vue N, Gibson WJ, Paolella BR, Mitra SS, Cheshier SH, Qi J, Liu K-W, Wechsler-Reya R, Weiss WA, Swartling FJ, Kieran MW, Bradner JE, Beroukhim R, Cho Y-J, BET Bromodomain Inhibition of MYC-Amplified Medulloblastoma, Clin. Cancer Res 20, 912–925 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sin-Chan P, Mumal I, Suwal T, Ho B, Fan X, Singh I, Du Y, Lu M, Patel N, Torchia J, Popovski D, Fouladi M, Guilhamon P, Hansford JR, Leary S, Hoffman LM, Mulcahy Levy JM, Lassaletta A, Solano-Paez P, Rivas E, Reddy A, Gillespie GY, Gupta N, Van Meter TE, Nakamura H, Wong T-T, Ra Y-S, Kim S-K, Massimi L, Grundy RG, Fangusaro J, Johnston D, Chan J, Lafay-Cousin L, Hwang EI, Wang Y, Catchpoole D, Michaud J, Ellezam B, Ramanujachar R, Lindsay H, Taylor MD, Hawkins CE, Bouffet E, Jabado N, Singh SK, Kleinman CL, Barsyte-Lovejoy D, Li X-N, Dirks PB, Lin CY, Mack SC, Rich JN, Huang A, A C19MC-LIN28A-MYCN Oncogenic Circuit Driven by Hijacked Super-enhancers Is a Distinct Therapeutic Vulnerability in ETMRs: A Lethal Brain Tumor., Cancer Cell 36, 51–67.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Gryder BE, Yohe ME, Chou H-C, Zhang X, Marques J, Wachtel M, Schaefer B, Sen N, Song Y, Gualtieri A, Pomella S, Rota R, Cleveland A, Wen X, Sindiri S, Wei JS, Barr FG, Das S, Andresson T, Guha R, Lal-Nag M, Ferrer M, Shern JF, Zhao K, Thomas CJ, Khan J, PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability., Cancer Discov. 7, 884–899 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cochran AG, Conery AR, Sims RJ, Bromodomains: a new target class for drug development, Nat. Rev. Drug Discov 18, 609–628 (2019). [DOI] [PubMed] [Google Scholar]
- 64.Wyce A, Matteo JJ, Foley SW, Felitsky DJ, Rajapurkar SR, Zhang X-P, Musso MC, Korenchuk S, Karpinich NO, Keenan KM, Stern M, Mathew LK, McHugh CF, McCabe MT, Tummino PJ, Kruger RG, Carpenter C, Barbash O, MEK inhibitors overcome resistance to BET inhibition across a number of solid and hematologic cancers., Oncogenesis 7, 35 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zawistowski JS, Bevill SM, Goulet DR, Stuhlmiller TJ, Beltran AS, Olivares-Quintero JF, Singh D, Sciaky N, Parker JS, Rashid NU, Chen X, Duncan JS, Whittle MC, Angus SP, Velarde SH, Golitz BT, He X, Santos C, Darr DB, Gallagher K, Graves LM, Perou CM, Carey LA, Earp HS, Johnson GL, Enhancer Remodeling during Adaptive Bypass to MEK Inhibition Is Attenuated by Pharmacologic Targeting of the P-TEFb Complex., Cancer Discov. 7, 302–321 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dodt M, Roehr J, Ahmed R, Dieterich C, FLEXBAR—Flexible Barcode and Adapter Processing for Next-Generation Sequencing Platforms, Biology (Basel). 1, 895–905 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner., Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, Barnes I, Bignell A, Boychenko V, Hunt T, Kay M, Mukherjee G, Rajan J, Despacio-Reyes G, Saunders G, Steward C, Harte R, Lin M, Howald C, Tanzer A, Derrien T, Chrast J, Walters N, Balasubramanian S, Pei B, Tress M, Rodriguez JM, Ezkurdia I, van Baren J, Brent M, Haussler D, Kellis M, Valencia A, Reymond A, Gerstein M, Guigo R, Hubbard TJ, GENCODE: The reference human genome annotation for The ENCODE Project, Genome Res. 22, 1760–1774 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L, Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation., Nat. Biotechnol. 28, 511–5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2., Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kolde R, pheatmap: Pretty Heatmaps. R package version 1.0.1 (2019) (available at https://cran.r-project.org/package=pheatmap).
- 72.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles., Proc. Natl. Acad. Sci. U. S. A 102, 15545–50 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP, Molecular signatures database (MSigDB) 3.0., Bioinformatics 27, 1739–40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, Lis JT, Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq)., Nat. Protoc 11, 1455–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schmieder R, Edwards R, Quality control and preprocessing of metagenomic datasets., Bioinformatics 27, 863–4 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li H, Toward better understanding of artifacts in variant calling from high-coverage samples., Bioinformatics 30, 2843–51 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup, The Sequence Alignment/Map format and SAMtools, Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang J, Zhao Y, Zhou X, Hiebert SW, Liu Q, Shyr Y, Nascent RNA sequencing analysis provides insights into enhancer-mediated gene regulation., BMC Genomics 19, 633 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM, CellProfiler: image analysis software for identifying and quantifying cell phenotypes., Genome Biol. 7, R100 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Identification of TNBC breast cancers in TCGA and METABRIC datasets.
Fig. S2. Distribution of MYCN expression across TNBC.
Fig. S3. IHC detection of MYCN in cell line- and patient-derived xenograft tissue.
Fig. S4. MYCN expression in primary TNBC and patient-matched metastases.
Fig. S5. MYCN and MYC intratumoral heterogeneity in primary and recurrent TNBC.
Fig. S6. MYCN and MYC expression in TNBC cell populations and CAL-51 clonally-derived cell lines.
Fig. S7. BETi sensitivity of CAL-51 MYCNHigh cell lines.
Fig. S8. Changes in MYC target gene expression in CAL-51 MYCNHigh cell lines after BETi treatment.
Fig. S9. Differential gene expression analyses between MYCNRatioHigh and MYCRatioHigh TNBC.
Fig. S10. Effect of BETi and MEKi combination treatments on weight of treated mice.
Fig. S11. Evaluation of apoptosis and proliferation after BETi and MEKi treatment in TNBC PDX models.
Fig. S12. Evaluation of MYC-family isoform expression after BETi and MEKi combination treatment in vivo.
Table S1. MYCN and MYC expression in breast cancer PDX models.
Table S2. Characteristics of patients with treatment-naïve and NAC-treated primary TNBC.
Data file S1. Tabular data points for experiments with a sample size of n < 20.
Data file S2. IHC results for MYCN and MYC in primary, treatment-naïve; primary, NAC-treated; and recurrent TNBC cases.
Data file S3. Primary drug screen results using CAL-51 MYCNLow and MYCNHigh cell lines.
Data file S4. Secondary drug screen results using CAL-51 MYCNLow and MYCNHigh cell lines.
Data file S5. Tabular data points for MYC-family isoform TSA-IF in CAL-51 after single agent BETi treatment.
Data file S6. Tabular data points for MYC-family isoform TSA-IF in MDA-MB-468 after single agent BETi treatment.
Data file S7. Tabular data points for MYC-family isoform TSA-IF in TNBC cell lines and PDX tissue after BETi and MEKi single agent and combination treatment.