

Abstract
Calcium and cyclic AMP form the cornerstones of two ancient signaling systems represented in nearly every kingdom of life. Not surprisingly, these old and ubiquitous messenger molecules have co-evolved multiple means to regulate one another. Zhang et al. describe a new twist on this theme related to the intimate union between the calcium-activated adenylyl cyclase, AC8, and the store-operated Ca2+ channel, Orai1.
Keywords: Adenylyl cyclase, Cyclic AMP, Calcium channels, Electrophysiology, Patch clamp
The idea that store-operated Ca2+ entry pathways provide a privileged conduit for calcium ions to enter the cell and locally regulate the enzymes that generate cAMP, the adenylyl cyclases (ACs), has its foundations in decades of careful experimentation [1]. Mammalian cells express nine transmembrane isoforms of adenylyl cyclase (AC1-AC9) and one “soluble” isoform (AC10 or “sAC). Among these, AC1, AC8, and sAC are activated by Ca2+ (via calmodulin in the case of AC1 and AC8), while AC5 and AC6 are directly inhibited by Ca2+. A given cell type will generally express a particular complement of several different ACs. ACs may also have specific tissue distributions, e.g. AC1 and AC8 are typically regarded as “neuronal ACs”.
The special ability of AC8 to sense and respond to Ca2+ entering specifically via store-operated “Orai” channels (a name coined by Feske and colleagues meaning “keeper of the gate” [2]) is particularly well established [3]. In a convincing series of studies from the lab of Dermot Cooper, AC8 expressed in HEK cells was shown to physically interact with Orai1 via a region in its terminal 49 amino acid tail [1,3]. Furthermore, genetically encoded Ca2+ reporters conjugated to AC8 revealed that the cyclase sees markedly elevated local Ca2+ concentrations during Ca2+ entry through Orai1. Additionally, AC8 tagged with FRET-based cAMP biosensors reported high levels of cAMP in the vicinity of activated Orai channels compared to the bulk cytosol. So clearly Ca2+ signals, particularly those derived from Ca2+ entering the cell via store-operated channels such as Orai1, are capable of potently stimulating cAMP production through AC8 in a fashion independent of the conventional G-protein coupled receptor pathway.
New observations published this year in Nature Communications by Zhang et al. [4] throw an intriguing curveball at this paradigm. These investigators took note of a naturally occurring truncated variant of Orai1 known as Orai1β that lacks 63 amino acids at its N-terminus [5]. This channel has normal currents but differs from the full-length channel in its ability to be inhibited by Ca2+. Specifically, Orai1β exhibits attenuation of the channel property known as “fast Ca2+-dependent inactivation” (“CDI”), a rapid form of negative feedback that serves to put the brakes on the entry of potentially toxic calcium ions through fully activated Orai complexes. The group reasoned that the absence of locally anchored AC8 at the Orai1β N-terminal could be connected to the reduction in fast CDI of the truncated channel.
Using an impressive array of electrophysiological, biophysical and biochemical approaches in the HEK 293 cell model, Zhang and colleagues provided evidence consistent with an effect of local cAMP near the mouth of the Orai channel causing activation of nearby PKA holoenzyme. This allowed for phosphorylation of Orai1 at serine-34 by PKA catalytic subunits and alteration in gating properties of the channel (Fig. 1). Addition of cAMP to the patch pipette or treatment of cells with forskolin, a direct activator of ACs, caused enhanced CDI in HEK cells expressing the full-length Orai1, but not the short variant. Additional experiments using pharmacological and genetic manipulation of PKA activity and PKA holoenzyme localization showed a clear role in this process for PKA tethered to AKAP79.
Fig. 1.
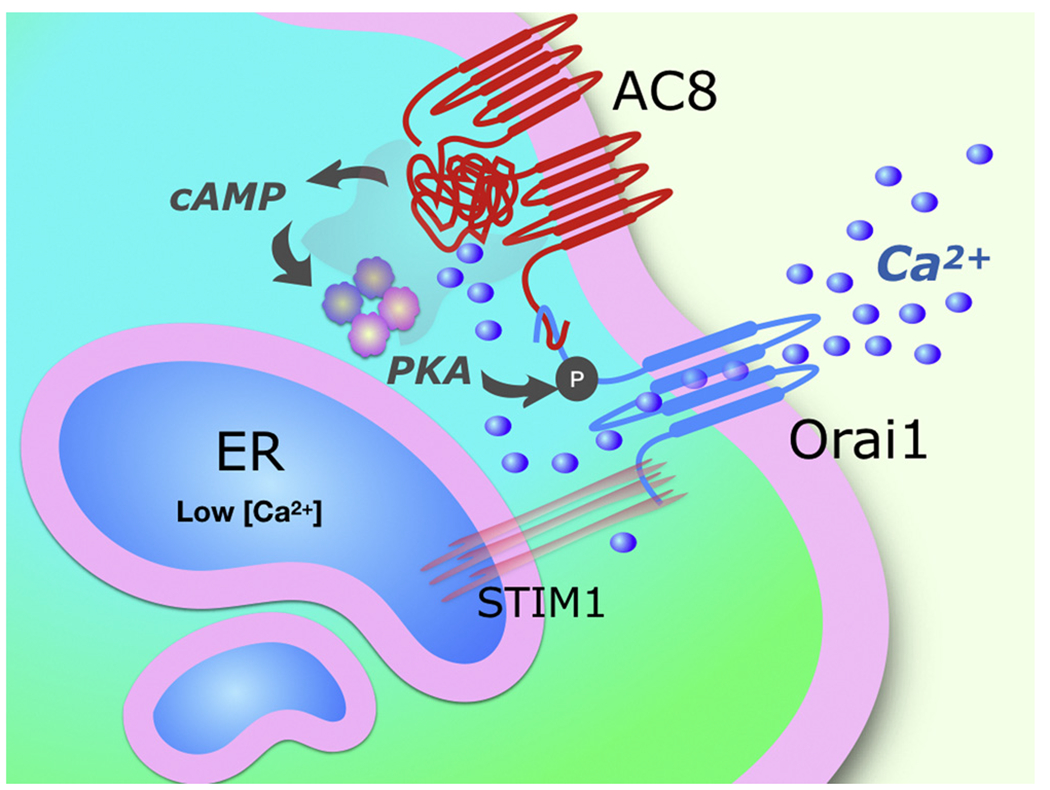
Scheme proposed by Trebak and colleagues for regulation of fast Ca2+-dependent inactivation (CDI) of store-operated Ca2+ entry channels (Orai1). Lowering of free [Ca2+] in the endoplasmic reticulum (ER) causes clustering of STIM1 in the ER membrane and recruitment of Orai1 in the plasma membrane to form membrane contact sites. Ca2+ entering through stimulated Orai1 channels from the extracellular space increases the turnover of adenylyl cyclase 8 (AC8), a Ca2+-activated AC. Orai1 and AC8 are linked via a direct interaction in their N-terminal domains. Zhang et al. add to this model by showing that the cyclic AMP (cAMP) produced by AC8 binds to locally tethered Protein Kinase A (PKA), leading to phosphorylation of Orai1 on serine 34. This phosphorylation in turn provides an important form of negative feedback to acutely limit Ca2+ entry through Orai1 (fast CDI).
The evidence for the involvement of AC8 in regulating fast CDI includes the fact that treatment with siRNA directed at AC8 caused a reduction in CDI in the HEK cell model, whereas overexpression of AC8 produced a small but significant augmentation of CDI. Pull-down experiments indicated that an endogenous species recognized by their AC8 antibody could co-IP with Orai1 but not Orai1β.
As the name would suggest, “fast CDI” is, in fact, fast; the negative feedback regulation on the Ca2+ conductance is initiated after only 5 msec and develops fully over the time scale of ~100 msec. It is distinguished from a slower type of CDI that takes place over tens of seconds and is related to gross elevations of [Ca2+] in the bulk cytosol. Meanwhile, cAMP signaling via PRA is generally considered to be much slower than fast CDI. The whole sequence starting from the production of cAMP by ACs, binding of cAMP to the PRA holoenzyme, dissociation of PRA catalytic subunits, and phosphorylation of soluble PRA target proteins, can take several tens of seconds as measured in the bulk cytosol. The regulation of CDI by AC8, cAMP and PRA described in this manuscript would therefore be particularly notable for its extreme rapidity. The explanation for this apparent discrepancy might lie in the close physical interactions amongst all the players, which Zhang et al. show to be coordinated by ARAP79 and direct binding interactions. Such higher order structures may not necessarily obey the classical rules of enzymes kinetics, conceivably permitting rapid and targeted regulation of Orai1 conductance.
The authors acknowledge that the mechanism they describe probably doesn’t fully account for how fast CDI is initiated or maintained since there was significant residual inactivation of CRAC channels in the absence of cAMP-dependent regulation. However, a key aspect of this article is that subtle tuning of CRAC (Ca2+ release-activated Ca2+; e.g. Orai) channels can nevertheless have significant impact on downstream signaling. Specifically, the authors show that modulation of CDI produces alterations in patterns of Ca2+ oscillations that were translated into significant effects on the efficiency of NFAT4 translocation into the nucleus.
During their current measurements, CRAC channels were pre-activated by maneuvers that deplete the ER of Ca2+. It’s noteworthy that lowering of free [Ca2+] in internal ER Ca2+ stores per se has also been reported to produce a relatively slow (minutes) and modest elevation of cellular cAMP in certain cell types (including HER 293). This phenomenon, called “store-operated cAMP signaling”, works through an incompletely understood STIMl-dependent (but Orai-independent) mechanism [6–9]. AC3 and AC6 have been implicated in this pathway, which is not activated by changes in cytosolic [Ca2+] and is in fact inhibited by Ca2+ entry through Orai1. Because fast CDI takes place so quickly, it is highly unlikely that store-operated cAMP production could participate in CDI, but could however, potentially factor into the regulation of Orai1 on a slower time scale.
The generalizability of this form of AC8-dependent regulation of CDI described in this article also remains to be seen since AC8 has a rather limited tissue distribution. In fact, a quibble with the study of Zhang and colleagues regards whether HER cells actually express AC8 endogenously. Reports from several independent labs would indicate that these cells do not [10,3,6]. The tissue origin of HER cells is not entirely clear, but it’s likely that they are not of epithelial derivation. The explanations for this discrepancy include non-specificity of the antibody used for AC8, changes in the properties of the CRISPR Orai1 knockout cells over many passages, or mistaken identity with respect to cell line. Clarification of this point would be helpful. Future studies might also benefit from a direct assessment of local cAMP production or PRA activity using biosensors targeted near the mouth of the Orai channel. Isoform-specific drugs that target the cyclases would also be of great value for this study and others, although still under development. In any case, the clear actions of cAMP/PKA in regulating Orai1 unveiled by Zhang and colleagues are notable and worthy of follow-up. The potential downstream effects on NFAT4 transcriptional activity and other physiological functions driven by this subtle regulation of CRAC channels also deserve further consideration in the future.
Acknowledgments
Work in the author’s lab was generously supported by grants from NIH/NIDCR (R21 DE025921), Harvard Catalyst (NIH 1Ul1 TR00102-01), and the Department of Veteran’s Affairs (I21 BX004093 and 1IS1BX00478601).
References
- [1].Cooper DM, Store-operated Ca(2)(+)-entry and adenylyl cyclase, Cell Calcium 58 (4) (2015) 368–375. [DOI] [PubMed] [Google Scholar]
- [2].Feske S, et al. , A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function, Nature 441 (7090) (2006) 179–185. [DOI] [PubMed] [Google Scholar]
- [3].Willoughby D, et al. , Direct binding between Orai1 and AC8 mediates dynamic interplay between Ca2+ and cAMP signaling, Sci. Signal 5 (219) (2012) ra29. [DOI] [PubMed] [Google Scholar]
- [4].Zhang X, et al. , A calcium/cAMP signaling loop at the ORAI1 mouth drives channel inactivation to shape NFAT induction, Nat. Commun 10 (1) (2019) 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Fukushima M, Tomita T, Janoshazi A, Putney JW, Alternative translation initiation gives rise to two isoforms of Orai1 with distinct plasma membrane mobilities, J. Cell. Sci 125 (Pt 18) (2012) 4354–4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lefkimmiatis K, et al. , Store-operated cyclic AMP signalling mediated by STIM1, Nat. Cell Biol 11 (4) (2009) 433–442. [DOI] [PubMed] [Google Scholar]
- [7].Maiellaro I, Lefkimmiatis K, Moyer MP, Curci S, Hofer AM, Termination and activation of store-operated cyclic AMP production, J. Cell. Mol. Med 16 (11) (2012) 2715–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Motiani RK, et al. , STIM1 activation of adenylyl cyclase 6 connects Ca(2+) and cAMP signaling during melanogenesis, EMBO J. 37 (5) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Spirli C, et al. , Altered store operated calcium entry increases cyclic 3′,5′-adenosine monophosphate production and extracellular signal-regulated kinases 1 and 2 phosphorylation in polycystin-2-defective cholangiocytes, Hepatology 55 (3) (2012) 856–868. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- [10].Soto-Velasquez M, Hayes MP, Alpsoy A, Dykhuizen EC, Watts VJ, A novel CRISPR/Cas9-based cellular model to explore adenylyl cyclase and cAMP signaling, Mol. Pharmacol 94 (3) (2018) 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]