

Abstract
Background
Hepatocellular carcinoma (HCC) is a major health problem and a primary cause of cancer‐related death worldwide. Although great advances have achieved recently by large‐scale high‐throughput analysis, the precise molecular mechanism underlying HCC progression remains to be clearly elucidated. We investigated the relationship between Tescalcin (TESC), a candidate oncogene, and clinicopathological features of HCC patients and explored the role of TECS in HCC development.
Methods
To identify new genes involved in HCC development, we analyzed The Cancer Genome Atlas liver cancer database, and TESC was selected for further investigation. HCC tissue microarray analysis for TESC and its association with clinicopathological features were performed to investigate its clinical significance. TESC was knocked down by using short‐hairpin RNAs. Cell proliferation was analyzed by WST‐1 assay and cell counting. Cell apoptosis was tested by fluorescence‐activated cell sorting. A subcutaneous xenograft tumor model in nude mice was established to determine the in vivo function of TESC. Affymetrix microarray was used to identify its molecular mechanism.
Results
TESC was significantly increased in HCC tissues compared with the adjacent normal liver tissues. High expression of TESC was detected in 61 of 172 HCC patients by tissue microarray. Large tumor (> 5 cm) and elevated total bilirubin were associated with high TESC expression (both P < 0.050). In multivariate analysis, TESC was identified as an independent prognostic factor for short overall survival of HCC patients. TESC knockdown impaired HCC cell growth in vitro and in vivo. TESC knockdown significantly increased cell apoptosis in HCC cell lines. Furthermore, Affymetrix microarray analysis revealed that TESC knockdown inhibited tumor proliferation‐related pathways while activated cell death‐related pathways.
Conclusion
TESC was identified as an independent prognostic factor for short overall survival of HCC patients, and was critical for HCC cell proliferation and survival.
Keywords: Apoptosis, Cell proliferation, Hepatocellular carcinoma, Oncogene, Tescalcin
Abbreviations
- ALDH1
aldehyde dehydrogenase‐1
- AST
aspartate aminotransferase
- ATCC
American Type Culture Collection
- BCV
biological coefficient of variation
- CI
confidence interval
- FACS
fluorescence‐activated cell sorting
- FDR
false discovery rate
- FLT3‐ITD+ AML
FMS‐like tyrosine kinase 3‐internal tandem duplication positive acute myelocytic leukemia
- GFP
green fluorescent protein
- GLMs
general linear models
- HBV
hepatitis B virus
- HCC
hepatocellular carcinoma
- HR
hazard ratio
- IHC
immunohistochemistry
- NHE1
Na(+)/H(+) exchanger type‐1
- OS
overall survival
- PBS
phosphate buffered saline
- RFS
recurrence‐free survival
- RT‐qPCR
real‐time quantitative polymerase chain reaction
- shRNA
short‐hairpin RNA
- SYSUCC
Sun Yat‐sen University Cancer Center
- TACE
transarterial chemoembolization
- TCGA
The Cancer Genome Atlas
- TESC
Tescalcin
- TMM
trimmed mean of M‐values
1. BACKGROUND
Liver cancer is the 6th most frequent and the 2nd most common death‐causing cancer worldwide, of which hepatocellular carcinoma (HCC) is the predominant type [1, 2, 3]. Liver resection and radiofrequency ablation are currently the curative treatments for early‐stage HCC. For unresectable HCC, transarterial chemoembolization (TACE) [4, 5] and multikinase inhibitors (only sorafenib and lenvatinib at present) have shown clinical benefits as first‐line treatments for advanced HCC [6, 7]. However, even with these two targeted agents, advanced HCC patients only reached a overall survival (OS) of 12.3 months and had a 30% objective response rate [8]. HCC is still a lethal tumor, with limited curative therapeutic methods for advanced cases [9].
The initiation and progression of HCC, a multi‐step process, comprise of liver injury, progressive inflammation, fibrosis, and chromosomal instability [10, 11]. The risk factors of liver injuries, such as chronic hepatitis B virus (HBV) and hepatitis C virus infections, autoimmune hepatitis, alcohol abuse, and some metabolic diseases, have been well‐characterized. However, the cellular and molecular mechanisms that contribute to the progression of liver cancer are still not fully understood. Recently, a large‐scale, multi‐platform analysis had been performed to understand the molecular landscape of HCCs, which generated tons of information requiring further confirmation[12]. Therefore, the mining of new possible targets from these data in an attempt to prevent HCC or its progression is urgent.
Tescalcin (TESC; also known as calcineurin B homologous protein 3, CHP3) belongs to the EF‐hand Ca2+‐binding protein superfamily, acting as a Са2+ sensor with Ca2+‐binding affinity in micromolar range [13]. Since Са2+ is a universal second messenger involved in multiple bio‐processes, TESC is supposed to function in cell growth and differentiation [14]. TESC has also been reported to be highly expressed in the brain and affect neuron development [15, 16, 17, 18]. Recent studies have suggested that TESC can be a regulator of cancer progression. TESC is reported to contribute to the invasive and metastatic activity of colorectal cancer [19] and the survival of gastric cancer cells [20]. TESC may also mediate sorafenib resistance in FMS‐like tyrosine kinase 3‐internal tandem duplication‐positive acute myelocytic leukemia (FLT3‐ITD+ AML) through the Na(+)/H(+) exchanger type‐1(NHE1) [21], as well as enhance cancer stemness and radioresistant properties in non‐small cell lung cancer through aldehyde dehydrogenase‐1 (ALDH1) [22]. In contrast, the involvement of TESC in the regulation of HCC development is yet to be investigated.
In the present study, TESC was screened out using bioinformatic analysis of The Cancer Genome Atlas (TCGA) liver cancer database. We investigated the relationship between TESC, a candidate oncogene, and clinical outcomes of HCC patients. Furthermore, we explored the role of TECS in the development of HCC and the potential molecular mechanism.
2. MATERIALS AND METHODS
2.1. Bioinformatics analysis
The RNA‐sequencing data were obtained from the TCGA liver hepatocellular carcinoma (LIHC) database. Differential expression analysis was conducted using the EdgeR Bioconductor package with R 3.4.3 (https://www.r-project.org/) [23]. Normalization factors were calculated using the trimmed mean of M‐values (TMM) to evaluate the differences in library size resulted from different sequencing depths [24]. To normalize the gene‐level variance, the biological coefficient of variation (BCV) was calculated using the square root of the common dispersion for negative binomial general linear models (GLMs). Samples were fitted to a negative‐binomial log‐linear model and tested for differential expression using a likelihood‐ratio test. The P values of differential expression tests were corrected for multiple‐hypothesis testing using the Benjamini‐Hochberg false discovery rate (FDR) correction. FDR < 0.05 and |log2 (fold change)| > 1 were set as cut‐off values to screen out differentially expressed genes.
2.2. Clinical samples and data
Tumor tissues were obtained from patients who underwent curative liver resection for HCC at Sun Yat‐sen University Cancer Center (SYSUCC) between January 2005 and December 2011.
Inclusion criteria: 1) the patient had pathologically diagnosed HCC; 2) the patient underwent liver resection without any prior treatment, and 3) the absence of distant metastasis was confirmed radiologically. Patients with Child‐Pugh class B or C conditions were excluded.
Tumor grade and stage were classified according to the World Health Organization criteria and the sixth edition of the TNM classification of the Union for International Cancer Control (UICC, 2002). OS was defined as the time interval between the date of resection and the date of death or the last follow‐up. Recurrence‐free survival (RFS) was defined as the time interval between the date of resection and the date of recurrence or the last follow‐up. The patients were followed up 1 month after surgery and every 3 months thereafter. The last follow‐up date was January 10, 2016.
For tissue microarray construction, the tissues were punched and placed into recipient paraffin blocks using a 0.6 mm diameter stylet and cut into 5 mm thick sections. The study protocol was approved by the institutional review board of SYSUCC. Informed consent was signed by the patients or their parents or legal guardians.
2.3. Immunohistochemistry (IHC) and scoring
The HCC tissue microarray slides were kept overnight at 37°C, dewaxed in xylene, rehydrated with graded alcohol, and immersed in 3% hydrogen peroxide for 10 min to block endogenous peroxidase activity. Antigen retrieval was carried out by heating the slides in a pressurized cabin for 15 min in 10 mmol/L sodium citrate, pH 6.0. The slides were then incubated with 10% normal goat serum at room temperature for 30 min to reduce non‐specific reactions. Subsequently, the slides were incubated overnight at 4°C with rabbit polyclonal antibody against TESC (11125‐1‐AP, 1:500, Proteintech, Chicago, MI, USA). Immunohistochemical staining was then carried out by using a two‐step EnVision System (Dako Cytomation, Glostrup, Denmark). Finally, the sections were counterstained with hematoxylin, dehydrated, and mounted. Two independent pathologists blinded to the clinicopathological information performed the IHC analysis for TESC. Immuno‐positive staining was evaluated in five randomly selected areas of the tissue section. The tissue sections were scored based on the staining proportion and intensity. Briefly, a score of 0 was defined as a proportion of 0‐5% of stained cells, 1 as 5‐25%, 2 as 26‐50%, and 3 as more than 50%. For TESC staining intensity, the slides were allocated a score of 0 for negative, 1 for mild, 2 for moderate, and 3 for intense staining, using high‐power (× 400) microscopy. Finally, the expression of TESC was classified based on the product of the intensity score and the proportion score: high expression (intensity multiply proportion scores > 3) or low expression (intensity multiply proportion scores ≤ 3).
2.4. Paired tumor and adjacent tissues
Eight pairs of HCC tissues and matched adjacent non‐tumorous tissues were selected from the Department of Hepatobiliary Oncology, SYSUCC, froze and stored in an ‐80°C refrigerator until use.
2.5. Human cell line cultures
The human cell lines HEK293T, SMMC‐7721, HepG2, BEL‐7402, and BEL‐7404 were obtained from American Type Culture Collection (ATCC). SMMC‐7721 and BEL‐7402 cells were maintained in RPMI‐1640, while BEL‐7404, HepG2 and HEK293T cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 2% penicillin and streptomycin. All cells were grown in a humidified incubator at 37°C with 5% CO2.
2.6. Short‐hairpin RNA (shRNA) knockdown vector construction and transfection
The TESC‐overexpressing plasmid with Flag‐tagged TESC (Flag‐TESC) and three pairs of shRNA against TESC were designed (target sequences in supplementary Tab. S1) and synthesized by Generay Biotech (Shanghai, P. R. China). The oligoes were annealed and cloned into GV115 lentivirus vector (Genechem Biotech, Shanghai, P. R. China) by the AgeI and EcorI (NEB Co., Ipswich, MA, USA) enzyme sites. Selected clones were confirmed by sequencing. Flag‐TESC and shRNAs were co‐transfected into HEK293T cells by lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA). Western blotting analysis of lysates of HEK293T cells at 36 h after antibody transfection to select the superior shRNA. To generate the lentivirus, GV115 vectors containing the shRNA against TESC or the scramble control were transfected into HEK293T by lipofectamine 2000 together with the helper plasmids psPAX2 and pMD2.G (Genechem Biotech). Supernatant containing the lentivirus was collected after 48 hours of transfection. SMMC‐7721 or BEL‐7404 cells were seeded into 6‐well plates at a concentration of 20% overnight and infected with 5 × 105 infectious units of lentiviruses and 8 mg/mL polybrene (Sigma‐Aldrich, St. Louis, MI, USA).
2.7. WST‐1 assay and celigo cell counting
For WST‐1 assay, cells were seeded in 96‐well plates at a concentration of 2000 cells/well. The cells were incubated with 10 μL of 5 mg/mL WST‐1 reagent (Roche, Basel, Switzerland) at 37°C for 1 h, and OD450 was determined by SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA, USA). Celigo cell counting was performed according to the manufacturer's instructions (Nexcelom, Lawrence, MA, USA) to evaluate cell proliferation every 24 h.
2.8. Xenograft model establishment and fluorescence imaging
The 6‐week‐old female nude mice (Guangdong Medical Laboratory Animal Center, Guangzhou, Guangdong, P. R. China) were maintained in the medical experimental animal center of Sun Yat‐sen University. There were 10 mice in each group. All mice were cared according to the guides approved by the China Association of Laboratory Animal Care and the Institutional Animal Care Committee. Cell suspensions in phosphate‐buffered saline (PBS) at a concentration of 4 × 107 cells/mL were injected subcutaneously into a single side of each mouse in a volume of 0.1 mL. Xenografts were observed and measured every three days. The volume of xenografts was calculated as V (mm3) = (length × width2) / 2. Living fluorescence images of the xenografts were taken. The mice were anesthetized with an intraperitoneal injection of 0.7% sodium pentobarbital, and the total radiant efficiency of green fluorescent protein (GFP) was measured with IVIS Lumina LT (Perkin Elmer, Waltham, MA, USA) according to the manufacturer's instructions.
2.9. Microarray and ingenuity pathway analysis
Total RNA was extracted from TESC‐knockdown or control BEL‐7404 cells with Trizol, purified, and amplified with GeneChip 3′IVT Express Kit (Affymetrix‐Thermo Fisher, Waltham, MA, USA). Purified amplified RNAs were analyzed on GeneChip primeview human (Affymetrix‐Thermo Fisher). Differentially expressed genes were identified with EdgeR [23] and subjected to ingenuity pathway analysis (www.qiagenbioinformatics.com) [25, 26]. Ingenuity pathway analysis was performed to identify most significantly suppressed or activated signaling pathways, and to predict potential proteins interacted with TESC through protein interaction network analysis.
2.10. Real‐time quantitative polymerase chain reaction (RT‐qPCR)
Total RNAs were extracted from cultured cells with the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions, and reverse transcription was performed using Superscript III reverse transcriptase (Invitrogen, Waltham, MA, USA). All gene expression levels were quantified by RT‐qPCR with iQ SYBR Green supermix and CFX96 RT‐qPCR system (Bio‐Rad, Hercules, CA, USA). The primers used are listed in supplementary Tab. S2.
2.11. Western blotting
Western blotting was performed as previously described [27]. Briefly, the whole‐cell lysates were separated by sodium dodecyl sulfate‐polyacrylamide gel electrophoresis, transferred onto nitrocellulose membranes (Bio‐Rad), and hybridized to a rabbit polyclonal antibody against TESC (1:2000, Proteintech) or a rabbit monoclonal anti‐GAPDH antibody (1:2000, Bioworld Technology, St Louis Park, MN, USA). After three washes with 1% (vol/vol) Tween‐20 in Tris‐buffered saline, membranes were further incubated for 1 h at room temperature with horseradish peroxidase‐conjugated anti‐rabbit immunoglobulin G. After washing, peroxidase activity was detected by chemiluminescence according to the manufacturer's instructions (Pierce Biotechnology, Waltham, MA, USA).
2.12. Flow cytometry
For cell apoptosis assay, TESC‐knockdown or control cells were collected and washed with pre‐chilled PBS twice. Then, they were stained with 10 μL Annexin V‐APC (eBioscience, San Diego, CA, USA) for 15 min. After washing, cell apoptosis was analyzed by fluorescence‐activated cell sorting (FACS) with Guava easyCyte HT (Millipore Inc., Burlington, MA, USA).
2.13. Caspase 3/7 activity
Caspase 3/7 activities in BEL‐7404 and SMMC‐7721 cells were determined with Caspase‐Glo® 3/7 Assay kit (Promege, Madison, WI, USA) according to the manufacturer's instructions.
2.14. Statistical analysis
Quantitative data are presented as the mean ± standard deviation. Quantitative data with normal distribution were compared between two groups using unpaired Student's t‐tests. Otherwise, rank‐sum test was considered. Analysis of variance with Bonferroni's multiple comparisons was applied for three or more groups of quantitative data. For categorical data, Pearson's chi‐square test was performed. The Kaplan‐Meier method was used to calculate the OS and RFS, and comparisons were performed using the log‐rank test. The prognostic variables used in predicting the OS were assessed by univariate Cox proportional hazards regression analysis. Variables that were significant in the univariate analysis were subsequently tested with the multivariate Cox proportional hazard model. All statistical tests were two‐sided, and a difference with P < 0.050 was considered statistically significant. All analyses were performed using the SPSS software (version 20.0, SPSS Inc., Chicago, IL, USA). In vitro experiments were performed at least in triplicate.
3. RESULTS
3.1. Bioinformatics analysis of TCGA HCC database
To identify new potential genes involved in HCC development, we analyzed TCGA HCC database. Overall, 360 cases with both RNA‐sequencing and pathological data were available. Among them, 50 cases with paired cancer and the adjacent normal tissue data were used for further analysis (supplementary Tab. S3). After standardization by TMM, the cancer tissues could be distinguished from the normal ones as shown by the BCV analysis, suggesting the stability of sequencing data of our selected samples (Figure 1a). Genes expressed with a significant difference between cancer and normal tissues were identified (some are listed in supplementary Tab. S4). Since we were interested in the novel regulators in HCC development, the targets with more than 100 publications were excluded after Pubmed searching. The associations of these candidate genes with clinicopathological data were further analyzed. Among them, TESC was found to be significantly up‐regulated (fold change of cancer/normal > 2.0) in liver cancer tissues compared with adjacent normal tissues in 28 (56.0%) cases (P < 0.001; Figure 1b).
FIGURE 1.
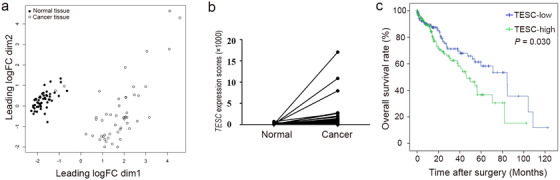
TCGA data mining identified TESC as a potential gene involved in HCC development. a. Biological coefficient of variation analysis showing liver cancer RNA‐sequencing data from TCGA database after standardization by TMM. b. TESC expression scores in cancer and the adjacent normal tissues. c. Kaplan‐Meier analysis shows that high TESC expression was significantly associated with short overall survival of HCC patients (n = 358). TESC: Tescalcin; HCC: hepatocellular carcinoma; TCGA: The Cancer Genome Atlas; TMM: trimmed mean of M‐values; logFC: log (fold change)
Among the 360 cases with both RNA‐sequencing and pathological data, 2 cases with incomplete survival data were excluded from survival analysis. Of the remaining 358 cases, 185 had low TESC expression, and 173 had high TESC expression (supplementary Tab. S5). The median age was 61 years (range: 16‐90 years). OS analysis by Kaplan‐Meier plotter showed that the high expression of TESC was significantly associated with poor outcome in patients with HCC (P = 0.030; Figure 1c).
More information from TCGA database was collected and analyzed to investigate more features of TESC. The methylation level of TESC was significantly lower in tumor tissues than in normal tissues (P = 0.011; supplementary Fig. S1a), and the TESC level was negatively correlated with the methylation level (P < 0.001; supplementary Fig. S1b). In addition, the TESC expression level was positively correlated with infiltration levels of macrophages and B cells (supplementary Fig. S1c).
3.2. The expression and prognostic value of TESC in HCC
To confirm the above mentioned findings, we compared the mRNA expression level of TESC between liver cancer tissues and in paired adjacent normal tissues. TESC mRNA level was shown to be much higher in liver cancer tissues (P < 0.001, Figure 2a). A tissue microarray was performed with tumor samples from 172 HCC patients who were previously treated at SYSUCC. TESC expression was detected by IHC. High expression of TESC was found in 61 (35.5%) samples, while low expression in 111 (64.5%) samples (Figure 2b). Established clinicopathological feature analysis indicated that large tumor (> 5 cm) and elevated total bilirubin level was associated with high TESC expression (both P < 0.050, Table 1). More patients underwent postoperative TACE in the high TESC expression group than in the low TESC expression group (24.6% vs. 11.7%, P = 0.029).
FIGURE 2.
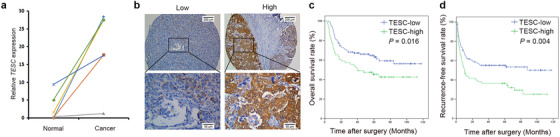
The expression and prognostic value of TESC in HCC. a. RT‐qPCR analysis of the TESC mRNA level in cancer tissues and the paired adjacent normal tissues (n = 8, P < 0.001). b. Representative IHC images (100 × and 400 ×) of low or high expression of TESC in human HCC tissues. c. Kaplan‐Meier analysis shows that TESC expression was significantly associated with overall survival (n = 172, P = 0.016). d. Kaplan‐Meier analysis shows that TESC expression was significantly associated with recurrence‐free survival (n = 172, P = 0.004). TESC: Tescalcin; HCC: hepatocellular carcinoma; RT‐qPCR: real‐time quantitative polymerase chain reaction; IHC: immunohistochemistry
TABLE 1.
Associations of TESC expression and the clinicopathological features of 172 HCC patients
| Feature | TESC expression | P value | |
|---|---|---|---|
| Low | High | ||
| Total (cases) | 111 | 61 | |
| Age [years, median (range)] | 51 (20‐75) | 49 (34‐72) | 0.787 |
| Gender [cases (%)] | 0.583 | ||
| Male | 99 (89.2) | 56 (91.8) | |
| Female | 12 (10.8) | 5 (8.2) | |
| HBV infection [cases (%)] | 0.481 | ||
| Yes | 96 (86.5) | 55 (90.2) | |
| No | 15 (13.5) | 6 (9.8) | |
| Tumor number [cases (%)] | 0.925 | ||
| Single | 79 (71.2) | 43 (70.5) | |
| Multiple | 32 (28.8) | 18 (29.5) | |
| Tumor size [cases (%)] | 0.011 | ||
| ≤5 cm | 51 (45.9) | 16 (26.2) | |
| > 5 cm | 60 (54.1) | 45 (73.8) | |
| Macrovascular invasion [cases (%)] | 0.200 | ||
| Yes | 5 (4.5) | 6 (9.8) | |
| No | 106 (95.5) | 55 (90.2) | |
| Cirrhosis [cases (%)] | 0.801 | ||
| Yes | 55 (49.5) | 29 (47.5) | |
| No | 56 (50.5) | 32 (52.5) | |
| Clinical TNM stage [cases (%)] | 0.322 | ||
| I | 72 (64.9) | 35 (57.4) | |
| II | 13 (11.7) | 8 (13.1) | |
| III | 26 (23.4) | 18 (29.5) | |
| Differentiation grade [cases (%)] | 0.753 | ||
| I+II | 61 (55.0) | 32 (52.5) | |
| III+IV | 50 (45.0) | 29 (47.5) | |
| AFP [cases (%)] | 0.252 | ||
| ≤400 ng/mL | 61 (55.0) | 39 (63.9) | |
| > 400 ng/mL | 50 (45.0) | 22 (36.1) | |
| Postoperative TACE [cases (%)] | 0.029 | ||
| Yes | 13 (11.7) | 15 (24.6) | |
| No | 98 (88.3) | 46 (75.4) | |
| White blood cells (× 109/L, mean ± SD) | 6.53 ± 1.61 | 7.02 ± 2.32 | 0.105 |
| Platelet count (× 109/L, mean ± SD) | 196.86 ± 83.25 | 191.57 ± 82.05 | 0.692 |
| Hemoglobin (g/L, mean ± SD) | 143.39 ± 16.75 | 140.32 ± 15.62 | 0.243 |
| Serum ALT level (U/L, mean ± SD) | 42.08 ± 26.66 | 46.73 ± 32.61 | 0.314 |
| Total bilirubin (μmol/L, mean ± SD) | 41.13 ± 20.17 | 51.63 ± 37.73 | 0.018 |
| Serum AST level (U/L, mean ± SD) | 15.80 ± 5.51 | 14.39 ± 4.95 | 0.098 |
| Albumin (g/L, mean ± SD) | 42.81 ± 3.95 | 41.71 ± 4.12 | 0.086 |
| Prothrombin time (s, mean ± SD) | 12.55 ± 1.38 | 12.43 ± 1.36 | 0.588 |
TESC: Tescalcin; HCC: hepatocellular carcinoma; HBV: hepatitis B virus; AFP: alpha‐fetoprotein; TACE: transarterial chemoembolization; SD: standard deviation; ALT: alanine aminotransferase; AST: aspartate aminotransferase.
Univariate analyses indicated that the tumor number, tumor size, aspartate aminotransferase (AST), albumin, macrovascular invasion, and TESC level were significant impact factors of OS in our cohort (all P < 0.050, Table 2). Multivariate analysis showed that high TESC expression, multiple tumor number, and tumor size > 5 cm were independent prognostic factors for shorter OS, whereas high albumin level was an independent prognostic factor for longer OS (Table 2). OS curves demonstrated that patients with high TESC expression had significantly reduced survival rate (P = 0.016, Figure 2c). The 1‐, 3‐, and 5‐year OS rates were 67.2%, 54.1%, and 45.4% respectively in the high TESC expression group and were 84.7%, 67.2%, and 63.1% respectively in the low TESC expression group. Similarly, high TESC expression was also an independent prognostic factor for poor RFS (Table 3). The recurrence rate of patients with high TESC expression was higher (P = 0.004; Figure 2d). Taken together, TESC was highly expressed in tumor tissues and reversely associated with the overall survival rate of HCC patients.
TABLE 2.
Univariate and multivariate analyses of prognostic factors associated with overall survival of HCC patients
| Univariate analysis | Multivariate analysis | |||
|---|---|---|---|---|
| Variable | P value | HR (95% CI) | P value | HR (95% CI) |
| Gender (female vs. male) | 0.543 | 1.241 (0.619‐2.491) | ||
| Age (> 50 vs. ≤50 years) | 0.303 | 1.270 (0.806‐2.000) | ||
| AFP level (> 400 vs. ≤400 ng/mL) | 0.197 | 1.347 (0.857‐2.119) | ||
| HBV infection (yes vs. no) | 0.826 | 0.928 (0.477‐1.805) | ||
| Tumor number (multiple vs. single) | <0.001 | 4.319 (2.734‐6.822) | <0.001 | 4.174 (2.586‐6.738) |
| Tumor size (> 5 vs. ≤5 cm) | <0.001 | 3.157 (1.817‐5.485) | 0.016 | 2.028 (1.138‐3.614) |
| Serum ALT (> 40 vs. ≤40 U/L) | 0.244 | 1.308 (0.833‐2.054) | ||
| Serum AST (> 40 vs. ≤40 U/L) | 0.049 | 1.575 (1.003‐2.474) | 0.135 | 1.439 (0.893‐2.317) |
| Albumin (> 35 vs. ≤35 g/L) | <0.001 | 0.886 (0.833‐0.943) | 0.001 | 0.897 (0.842‐0.955) |
| Total bilirubin (> 21 vs. ≤21 μmol/L) | 0.092 | 1.673 (0.920‐3.041) | ||
| Prothrombin time (> 13.5 vs. ≤13.5 s) | 0.502 | 1.199 (0.705‐2.039) | ||
| Macrovascular invasion (yes vs. no) | 0.005 | 2.863 (1.368‐5.995) | 0.120 | 1.827 (0.855‐3.902) |
| Differentiation grade (III+IV vs. I+II) | 0.166 | 1.375 (0.876‐2.157) | ||
| Cirrhosis (yes vs. no) | 0.347 | 1.244 (0.790‐1.959) | ||
| TESC level (high vs. low) | 0.018 | 1.729 (1.100‐2.719) | 0.011 | 1.863 (1.155‐3.005) |
HCC: hepatocellular carcinoma; HR: hazard ratio; CI: confidence interval; HBV: hepatitis B virus; AFP: alpha‐fetoprotein; ALT: alanine aminotransferase; AST: aspartate aminotransferase; TESC: Tescalcin.
TABLE 3.
Univariate and multivariate analyses of prognostic factors associated with recurrence‐free survival of HCC patients
| Univariate analysis | Multivariate analysis | |||
|---|---|---|---|---|
| Variable | P value | HR (95% CI) | P value | HR (95% CI) |
| Gender (female vs. male) | 0.506 | 0.782 (0.379‐1.614) | ||
| Age (> 50 vs. ≤50 years) | 0.666 | 1.093 (0.727‐1.644) | ||
| AFP level (> 400 vs. ≤400 ng/mL) | 0.062 | 1.476 (0,981‐2.221) | ||
| HBV infection (yes vs. no) | 0.527 | 1.236 (0.641‐2.384) | ||
| Tumor number (multiple vs. single) | <0.001 | 3.411 (2.253‐5.164) | <0.001 | 3.655 (2.340‐5.707) |
| Tumor size (> 5 vs. ≤5 cm) | <0.001 | 2.734 (1.704‐4.386) | 0.005 | 1.995 (1.227‐3.244) |
| Serum ALT (> 40 vs. ≤40 U/L) | 0.618 | 1.110 (0.736‐1.675) | ||
| Serum AST (> 40 vs. ≤40 U/L) | 0.147 | 1.351 (0.899‐2.029) | ||
| Albumin (> 35 vs. ≤35 g/L) | 0.026 | 2.561 (1.116‐5.878) | 0.029 | 2.606(1.101‐6.170) |
| Total bilirubin (> 21 vs. ≤21 μmol/L) | 0.060 | 1.702 (0.978‐2.963) | ||
| Prothrombin time (> 13.5 vs. ≤13.5 s) | 0.263 | 0.729 (0.418‐1.268) | ||
| Macrovascular invasion (yes vs. no) | 0.102 | 1.903 (0.879‐4.118) | ||
| Differentiation grade (III+IV vs. I+II) | 0.036 | 1.547 (1.029‐2.326) | 0.398 | 1.197 (0.789‐1.816) |
| Cirrhosis (yes vs. no) | 0.878 | 1.032 (0.687‐1.552) | ||
| TESC level (high vs. low) | 0.005 | 1.797 (1.193‐2.706) | 0.002 | 1.948 (1.272‐2.984) |
HCC: hepatocellular carcinoma; HR: hazard ratio; CI: confidence interval; HBV: hepatitis B virus; AFP: alpha‐fetoprotein; ALT: alanine aminotransferase; AST: aspartate aminotransferase; TESC: Tescalcin.
3.3. Association of TESC with HCC cell growth in vitro
We next explored the biological functions of TESC. Its expression levels in several frequently‐used liver cancer cell lines, including HepG2, SMMC‐7721, BEL‐7404, BEL‐7402, were tested by Western blotting. SMMC‐7721 and BEL‐7404 showed relatively high expression of TESC (Figure 3a) and were selected for in vitro and in vivo experiments. We designed three pairs of shRNA against TESC. After screening using HEK293T cells, the #3 shRNA (shTESC‐3) showed the highest efficiency in down‐regulating TESC expression and was used for the following experiments (Figure 3b).
FIGURE 3.
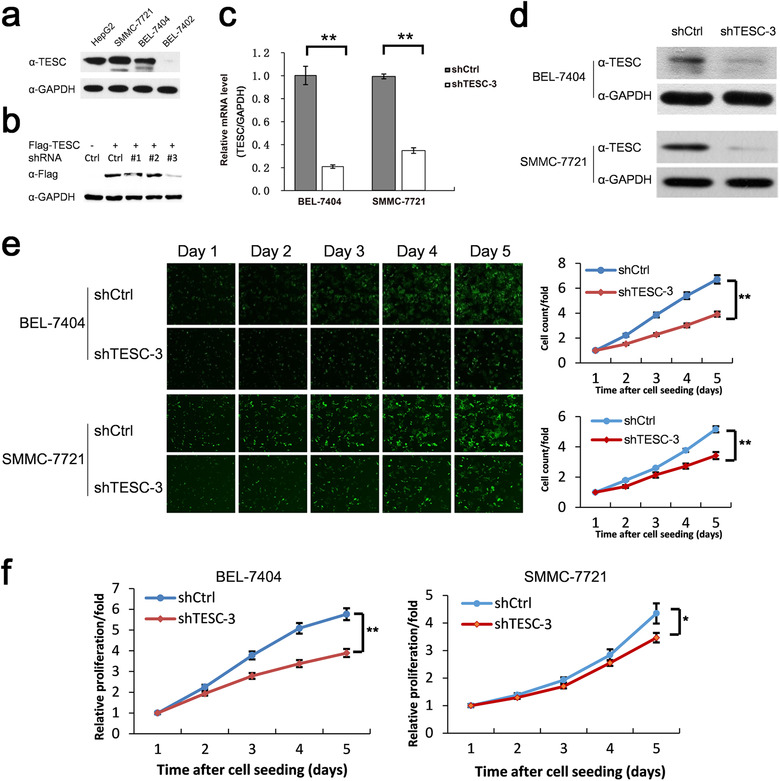
Association of TESC with HCC cell growth. a. Western blotting analysis of TESC expression in different liver cancer cell lines. b. Western blotting analysis of cell lysates of HEK293T transfected with Flag‐tagged TESC and 3 pairs of TESC shRNA (#1‐#3) or the control shRNA (Ctrl) after 36 hours with indicated antibody. c, d. RT‐qPCR (c) and Western blotting (d) analyses of BEL‐7404 and SMMC‐7721 cells infected with lentiviruses containing TESC shRNA (shTESC‐3) or the control shRNA (shCtrl). e. Celigo cell counting of BEL‐7404 and SMMC‐7721 cells infected with lentiviruses containing shTESC‐3 or shCtrl. f. WST‐1 assay of BEL‐7404 and SMMC‐7721 cells infected with lentiviruses containing shTESC‐3 or shCtrl. *: P < 0.050; **: P < 0.010. TESC: Tescalcin; HCC: hepatocellular carcinoma; shRNA: short‐hairpin RNA; RT‐qPCR: real‐time quantitative polymerase chain reaction
In SMMC‐7721 and BEL‐7404 cells, shTESC‐3 knocked down TESC efficiently as shown by using RT‐qPCR (Figure 3c) and Western blotting (Figure 3d). Celigo cell counting showed that TESC knockdown suppressed cell proliferation (Figure 3e). Similar results were obtained by WST‐1 assay (Figure 3f).
Furthermore, cell apoptosis was also tested by FACS, and we found that TESC knockdown significantly increased cell apoptosis in both cell lines (Figure 4a). Consistently, Caspase 3/7 activity was much higher in shTESC‐3‐transfected cells than in shCtrl‐transfected cells (Figure 4b). Collectively, these data indicated that TESC was critical for HCC cell growth by regulating cell proliferation and apoptosis in vitro.
FIGURE 4.
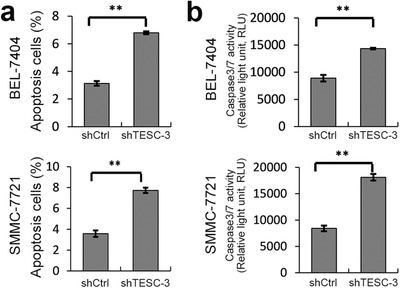
Association of TESC with apoptosis and caspase 3/7 activity in HCC cells. BEL‐7404 and SMMC‐7721 cells were infected with lentiviruses containing TESC shRNA (shTESC‐3) or the control shRNA (shCtrl). a. The apoptosis of shTESC‐3‐transfected BEL‐7404 and SMMC‐7721 cells was increased. b. Caspase 3/7 activity was also increased in shTESC‐3‐transfected BEL‐7404 and SMMC‐7721 cells. **: P < 0.010. TESC: Tescalcin; HCC: hepatocellular carcinoma; shRNA: short‐hairpin RNA
3.4. Association of TESC with HCC growth in vivo
To further confirm the role of TESC in vivo, a subcutaneous xenograft tumor model in nude mice was constructed. The total GFP radiant efficiency was significantly lower in mice inoculated with shTESC‐3‐transfected BEL‐7404 cells than in those with shCtrl‐transfected cells, as detected using living fluorescence imaging (n = 10, P < 0.001, Figure 5a and 5b), indicating that TESC knockdown arrested the tumor growth. The mean tumor volume was also significantly less in the shTESC‐3 group than in the vector control group (P < 0.050, Figure 5c). Collectively, our data demonstrated that TESC had a significant effect on tumor growth in vivo, suggesting that TESC could be a potential oncogene for HCC.
FIGURE 5.
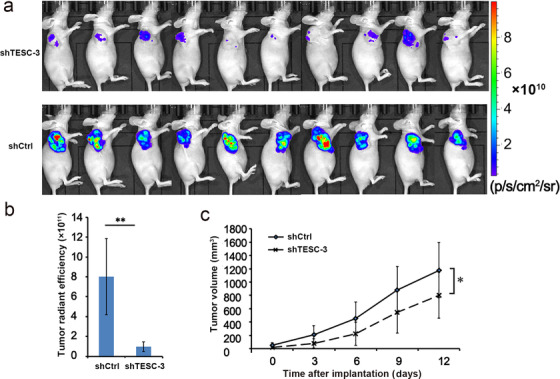
Association of TESC with HCC growth in vivo. a. Living fluorescence imaging monitored xenograft growth in nude mice inoculated with TESC shRNA (shTESC‐3)‐ or the control shRNA (shCtrl)‐transfected BEL‐7404 cells on day 12 (n = 10). b. The statistical diagram based on total radiant efficiency on day 12. c. The tumor volume in mice xenografted with shTESC‐3‐ or shCtrl‐transfected BEL‐7404 cells. *: P < 0.050; **: P < 0.010; TESC: Tescalcin; HCC: hepatocellular carcinoma; shRNA: short‐hairpin RNA
3.5. Molecular mechanism of TESC affecting HCC development
To reveal the molecular mechanism of how TESC affect HCC development, we used Affymetrix microarray to compare the gene expression profile of BEL‐7404 cells with and without TESC knockdown. A total of 2560 differentially expressed genes were identified with 1272 up‐regulated and 1288 down‐regulated in TESC‐knockdown cells comparing with control cells (P < 0.050). After ingenuity pathway analysis, we found that the most suppressed pathways were cell proliferation of tumor cell lines (activation Z score = –2.329, P < 0.001), viral infection (activation Z score = –2.191, P < 0.001), and cell viability of tumor cell lines (activation Z score = –2.107, P < 0.001), while the most activated pathways were neuritogenesis (activation Z score = 3.852, P < 0.001), interphase of connective tissue cells (activation Z score = 2.593, P < 0.001), organization of cytoplasm (activation Z score = 2.294, P < 0.001), and cell death of malignant tumor (activation Z score = 2.013, P < 0.001; Figure 6a). Changes in other signaling pathways, including oxidative stress response, tRNA charging, epithelial adherens junction, protein ubiquitination, and others, were also observed (supplementary Fig. S2).
FIGURE 6.
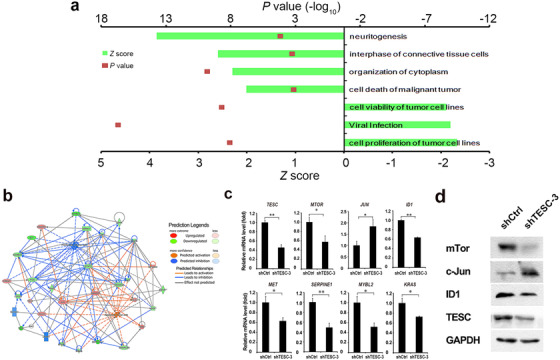
The roles of TESC in tumor cell proliferation and apoptosis pathways. a. Ingenuity pathway analysis shows the pathways suppressed or activated after TESC knockdown. Positive Z scores indicate activation; minus Z scores indicate suppression. b. Protein interaction network analysis of proteins related to TESC in regulating cell proliferation and apoptosis. c. RT‐qPCR detects mRNA levels of TESC and genes that are potentially involved in the TESC‐regulated cell proliferation and apoptosis in TESC shRNA (shTESC‐3)‐ or the control shRNA (shCtrl)‐transfected BEL‐7404 cells. d. Western blotting detects protein levels of TESC, mTor, c‐Jun, and ID1 in TESC shRNA (shTESC‐3)‐ or the control shRNA (shCtrl)‐transfected BEL‐7404 cells. *: P < 0.050; **: P < 0.010; TESC: Tescalcin; shRNA: short‐hairpin RNA; RT‐qPCR: real‐time quantitative polymerase chain reaction
Protein interaction network analysis was applied to visualize the downstream genes. The mammalian target of rapamycin (MTOR), Jun proto‐oncogene (JUN), MET proto‐oncogene (MET), inhibitor of DNA‐binding 1 (ID1), serpin family E member 1 (SERPINE1), MYB proto‐oncogene like 2 (MYBL2), and KRAS proto‐oncogene (KRAS) were demonstrated to be potentially involved in the TESC‐regulated cell proliferation and apoptosis (Figure 6b). The expression of these genes was confirmed by RT‐qPCR (Figure 6c). Knockdown of TESC decreased MTOR, ID1, MET, SERPINE1, MYBL2, and KRAS expression but increased JUN expression. Similar changes in the protein levels of mTor, c‐JUN, and ID1 were also confirmed by Western blotting (Figure 6d). Altogether, these data suggested that TESC was critical for tumor proliferation and apoptosis.
4. DISCUSSION
Here, we identified TESC to be significantly up‐regulated in liver cancer tissues compared with adjacent normal tissues after TCGA data mining and demonstrated that TESC was an independent prognostic factor for HCC and that its expression was associated with tumor size and total bilirubin level.
TESC was first reported to be commonly expressed in mouse testis, heart, and brain [28, 29]. Emerging data suggested that TESC could be a regulator in cell proliferation, methylation, and stemness of some cancers, such as gastric cancer [20], non‐small cell lung cancer [22], and colorectal cancer [30]. TESC expression was reported to be higher in colorectal cancer than in normal mucosa and premalignant dysplastic lesions [30], which was similar to our finding. We found that TESC expression was significantly increased in HCC tissues compared with matched adjacent non‐tumorous tissues. In addition, high expression of TESC was found to be an independent prognostic factor for short OS and RFS. Some previous studies demonstrated that AFP level was one of the prognostic factors of HCC [31, 32]. However, AFP level was not found to be a prognostic factor for HCC in the present study and some other studies [33, 34]. The discrepancy may have been resulted from the relatively small sample size of the present study. In addition, the AFP level may not be powerful enough to predict the prognosis. TESC could be a supplement for prognosis prediction. It is appealing that TESC could mediate sorafenib resistance in FLT3‐ITD+ AML [21]. Since sorafenib is also the first‐line treatment for advanced HCC, whether TESC expression is related to the low effectiveness of sorafenib in HCC needs to be considered and further studied.
TESC was supposed to contribute to tumor cell survival in gastric cancer [20]. This indeed agrees with our results that TESC promoted the in vitro and in vivo cell growth of HCC, which potentially contributed to HCC development. When we analyzed the TESC molecular mechanism by gene microarray, the enriched cluster was related to the suppression of the proliferation and viability of tumor cell lines pathways as well as the activation of the cell death of malignant tumor pathway (Figure 6a). This is consistent with our in vitro and in vivo findings: 1) TESC knockdown impaired HCC cell growth and increased cell apoptosis and Caspase 3/7 activity (Figure 3 and Figure 4); 2) high expression of TESC was associated with tumor size (Table 1); and 3) TESC was critical for xenograft tumor development in nude mice (Figure 5). The pathway related to neuritogenesis was also highlighted in our profile analysis, but may not be associated with tumorigenesis. Actually, the relationship between TESC and the neuritogenesis‐related pathway is quite reasonable, considering that TESC was critical for neuron functions as reported [15, 16, 17, 18].
After confirming the genes related to tumor cell proliferation and apoptosis, such as MTOR, JUN, and ID1, we noted that the mTor protein level exhibited greater change as compared with its mRNA level after TESC knockdown. The mTOR pathway regulated diverse cellular functions, including proliferation, survival, growth, autophagy, metabolism, metastasis, and angiogenesis[35]. Activation of the mTOR pathway is common and pivotal in HCC [36]. Thus, mTor might be a regulatory target of TESC, and the regulation may occur not only in the mRNA level but also in the protein level. It is known that TESC binds to the subunit 4 of the COP9 signalosome and plays a role in negatively regulating COP9 signalosome activity [37]. COP9 signalosome complex is a prominent ubiquitin‐proteasome system, specifically ubiquitinating proteins for proteolytic elimination, and plays a critical role in regulating the mTor pathway [38, 39]. It is possible that TESC regulates the mTor protein stability through the COP9 signalosome complex. However, this hypothesis needs further investigation. Nevertheless, we demonstrated that TESC was involved in HCC development by affecting cell proliferation and apoptosis, which could serve as a promising prognostic predictor and a potential therapeutic target of HCC.
In the present study, a low methylation level of TESC was observed in tumor tissues (supplementary Figure S1). Methylation of TESC may play a role in facilitating tumor development. Furthermore, TESC expression was positively correlated with the infiltration of macrophages and B cells. Generally, tumor‐associated macrophages contribute to HCC progression by promoting angiogenesis, metastasis, and immune suppression through the secretion of cytokines, chemokines, growth factors, and matrix metalloproteases [40, 41]. B cells often constitute of abundant cellular components in human tumors. However, the activation status and functions of these cells in human cancers remain elusive. It was reported that B cells with high expression levels of chemokine (C‐X‐C motif) receptor 3 or programmed cell death‐1 may contribute to tumor progression [42, 43]. Would liver cancer cells with aggressive biological behaviors on the background of high TESC expression recruit more immune cells? To prove the influence of TESC on immune response, more targeted studies are needed.
There were several limitations in the present study. First, we investigated the potential mechanism of how TESC affect tumor cell proliferation and apoptosis but did not investigate how TESC was regulated. Second, we found that TESC was mainly related to the proliferation and apoptosis of tumor cells, but whether it is involved in the regulation of other biological behaviors of malignant tumors, such as tumor invasion and metastasis and tumor stemness, remains to be further studied. Third, initial data indicated that the TESC expression was associated with immune cell infiltration, and its role in immune response needs to be further verified using in vivo and vitro experiments.
In conclusion, TESC was identified as an independent prognostic factor for short survival in HCC patients and was considered to be critical for HCC cell proliferation and survival.
DECLARATIONS
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All human tissues experiments were approved by the institutional review board of Sun Yat‐sen University Cancer Center. Written informed consent form for the experimental studies was obtained from the patients. All animals received care according to the criteria outlined in the guides approved by the China Association of Laboratory Animal Care and the Institutional Animal Care Committee. Written informed consent for publication was obtained from the participants or their guardians at the time of admission, with which the tissue samples might be used for scientific research but did not relate to individual privacy.
CONSENT FOR PUBLICATION
Not applicable
AVAILABILITY OF DATA AND MATERIALS
The key raw data have been deposited into the Research Data Deposit (www.researchdata.org.cn), with the approval number of RDDB2020000821.
COMPETING INTERESTS
The authors made no disclosures.
FUNDING
This study was supported by grants from the National Natural Science Foundation of China (81602143), Sun Yat‐sen University Cancer Center Physician‐scientist Funding (16zxqk04), Guangdong Key Laboratory of Liver Disease Research (GS2017011002), National Science and Technology Major Project of China (2018ZX10302205, 2018ZX10723204), National Natural Science Foundation of Guangdong (2017A030313605) and Guangdong Province Medical Science and Technology Research Foundation (B2019132).
AUTHORS' CONTRIBUTIONS
ZGZ and MSC designed the experiments. ZGZ and RXZ participated in the whole experiment, collected and analyzed the data and wrote the manuscript. JBC, RXZ, JCW, YXP, XHW and WXL participated in the cell culture and animal experiments. JBC performed the IHC staining and clinical data analysis. YJZ and XL contributed to the human specimen data collection and assembly. ZGZ, YL and MSC provided financial support, administrative support, and final approval of the manuscript. JBC, YL and ZGZ revised the manuscript. All authors read and approved the final manuscript for publication.
Supporting information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGEMENTS
We would like to thank Ms. Liyi Zhang for helping to edit our manuscript and critical suggestions.
Zhou Z‐G, Chen J‐B, Zhang R‐X, et al. Tescalcin is an unfavorable prognosis factor that regulats cell proliferation and survival in hepatocellular carcinoma patients. Cancer Commun. 2020;40:355–369. 10.1002/cac2.12069
REFERENCES
- 1. Mortality GBD, Causes of Death C. Global, regional, and national life expectancy, all‐cause mortality, and cause‐specific mortality for 249 causes of death, 1980‐2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1459‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. He W, Zheng Y, Zou R, Shen J, Yang J, Qiu J, et al. Long‐ versus short‐interval follow‐up after resection of hepatocellular carcinoma: a retrospective cohort study. Cancer Commun (Lond). 2018;38(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379(9822):1245‐55. [DOI] [PubMed] [Google Scholar]
- 4. Llovet JM, Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology. 2003;37(2):429‐42. [DOI] [PubMed] [Google Scholar]
- 5. Wei W, Jian PE, Li SH, Guo ZX, Zhang YF, Ling YH, et al. Adjuvant transcatheter arterial chemoembolization after curative resection for hepatocellular carcinoma patients with solitary tumor and microvascular invasion: a randomized clinical trial of efficacy and safety. Cancer Commun (Lond). 2018;38(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378‐90. [DOI] [PubMed] [Google Scholar]
- 7. Ikeda M, Morizane C, Ueno M, Okusaka T, Ishii H, Furuse J. Chemotherapy for hepatocellular carcinoma: current status and future perspectives. Jpn J Clin Oncol. 2018;48(2):103‐14. [DOI] [PubMed] [Google Scholar]
- 8. Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first‐line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non‐inferiority trial. Lancet. 2018;391(10126):1163‐73. [DOI] [PubMed] [Google Scholar]
- 9. He MK, Le Y, Li QJ, Yu ZS, Li SH, Wei W, et al. Hepatic artery infusion chemotherapy using mFOLFOX versus transarterial chemoembolization for massive unresectable hepatocellular carcinoma: a prospective non‐randomized study. Chin J Cancer. 2017;36(1):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zucman‐Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology. 2015;149(5):1226‐39 e4. [DOI] [PubMed] [Google Scholar]
- 11. Dhanasekaran R, Bandoh S, Roberts LR. Molecular pathogenesis of hepatocellular carcinoma and impact of therapeutic advances. F1000Research. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cancer Genome Atlas Research Network . Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017;169(7):1327‐41 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gutierrez‐Ford C, Levay K, Gomes AV, Perera EM, Som T, Kim YM, et al. Characterization of tescalcin, a novel EF‐hand protein with a single Ca2+‐binding site: metal‐binding properties, localization in tissues and cells, and effect on calcineurin. Biochemistry. 2003;42(49):14553‐65. [DOI] [PubMed] [Google Scholar]
- 14. Kolobynina KG, Solovyova VV, Levay K, Rizvanov AA, Slepak VZ. Emerging roles of the single EF‐hand Ca2+ sensor tescalcin in the regulation of gene expression, cell growth and differentiation. J Cell Sci. 2016;129(19):3533‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dannlowski U, Grabe HJ, Wittfeld K, Klaus J, Konrad C, Grotegerd D, et al. Multimodal imaging of a tescalcin (TESC)‐regulating polymorphism (rs7294919)‐specific effects on hippocampal gray matter structure. Mol Psychiatry. 2015;20(3):398‐404. [DOI] [PubMed] [Google Scholar]
- 16. Han KM, Won E, Kang J, Choi S, Kim A, Lee MS, et al. TESC gene‐regulating genetic variant (rs7294919) affects hippocampal subfield volumes and parahippocampal cingulum white matter integrity in major depressive disorder. J Psychiatr Res. 2017;93:20‐9. [DOI] [PubMed] [Google Scholar]
- 17. Stein JL, Medland SE, Vasquez AA, Hibar DP, Senstad RE, Winkler AM, et al. Identification of common variants associated with human hippocampal and intracranial volumes. Nat Genet. 2012;44(5):552‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takamatsu G, Katagiri C, Tomoyuki T, Shimizu‐Okabe C, Nakamura W, Nakamura‐Higa M, et al. Tescalcin is a potential target of class I histone deacetylase inhibitors in neurons. Biochem Biophys Res Commun. 2017;482(4):1327‐33. [DOI] [PubMed] [Google Scholar]
- 19. Kang J, Kang YH, Oh BM, Uhm TG, Park SY, Kim TW, et al. Tescalcin expression contributes to invasive and metastatic activity in colorectal cancer. Tumour Biol. 2016;37(10):13843‐53. [DOI] [PubMed] [Google Scholar]
- 20. Kim TW, Han SR, Kim JT, Yoo SM, Lee MS, Lee SH, et al. Differential expression of tescalcin by modification of promoter methylation controls cell survival in gastric cancer cells. Oncol Rep. 2019;41(6):3464‐74. [DOI] [PubMed] [Google Scholar]
- 21. Man CH, Lam SS, Sun MK, Chow HC, Gill H, Kwong YL, et al. A novel tescalcin‐sodium/hydrogen exchange axis underlying sorafenib resistance in FLT3‐ITD+ AML. Blood. 2014;123(16):2530‐9. [DOI] [PubMed] [Google Scholar]
- 22. Lee JH, Choi SI, Kim RK, Cho EW, Kim IG. Tescalcin/c‐Src/IGF1Rbeta‐mediated STAT3 activation enhances cancer stemness and radioresistant properties through ALDH1. Sci Rep. 2018;8(1):10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA‐seq data. Genome Biol. 2010;11(3):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Felciano RM, Bavari S, Richards DR, Billaud JN, Warren T, Panchal R, et al. Predictive systems biology approach to broad‐spectrum, host‐directed drug target discovery in infectious diseases. Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing. 2013:17‐28. [PubMed] [Google Scholar]
- 26. Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, et al. A network‐based analysis of systemic inflammation in humans. Nature. 2005;437(7061):1032‐7. [DOI] [PubMed] [Google Scholar]
- 27. Shi M, Deng W, Bi E, Mao K, Ji Y, Lin G, et al. TRIM30 alpha negatively regulates TLR‐mediated NF‐kappa B activation by targeting TAB2 and TAB3 for degradation. Nat Immunol. 2008;9(4):369‐77. [DOI] [PubMed] [Google Scholar]
- 28. Bao Y, Hudson QJ, Perera EM, Akan L, Tobet SA, Smith CA, et al. Expression and evolutionary conservation of the tescalcin gene during development. Gene expression Patterns: GEP. 2009;9(5):273‐81. [DOI] [PubMed] [Google Scholar]
- 29. Perera EM, Martin H, Seeherunvong T, Kos L, Hughes IA, Hawkins JR, et al. Tescalcin, a novel gene encoding a putative EF‐hand Ca(2+)‐binding protein, Col9a3, and renin are expressed in the mouse testis during the early stages of gonadal differentiation. Endocrinology. 2001;142(1):455‐63. [DOI] [PubMed] [Google Scholar]
- 30. Kang YH, Han SR, Kim JT, Lee SJ, Yeom YI, Min JK, et al. The EF‐hand calcium‐binding protein tescalcin is a potential oncotarget in colorectal cancer. Oncotarget. 2014;5(8):2149‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xu L, Peng ZW, Chen MS, Shi M, Zhang YJ, Guo RP, et al. Prognostic nomogram for patients with unresectable hepatocellular carcinoma after transcatheter arterial chemoembolization. J Hepatol. 2015;63(1):122‐30. [DOI] [PubMed] [Google Scholar]
- 32. Chen J, Lu S, Zhang Y, Xu L, Chen J, Wang J, et al. Sorafenib Monotherapy Versus Sorafenib Combined with Regional Therapies for Hepatocellular Carcinoma Patients with Pulmonary Oligometastases: A Propensity Score‐matched Analysis. J Cancer. 2018;9(10):1745‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen J, Fang A, Chen M, Tuoheti Y, Zhou Z, Xu L, et al. A novel inflammation‐based nomogram system to predict survival of patients with hepatocellular carcinoma. Cancer Med. 2018;7(10):5027‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen J, Peng K, Hu D, Shen J, Zhou Z, Xu L, et al. Tumor Location Influences Oncologic Outcomes of Hepatocellular Carcinoma Patients Undergoing Radiofrequency Ablation. Cancers. 2018;10(10):378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matter MS, Decaens T, Andersen JB, Thorgeirsson SS. Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J Hepatol. 2014;60(4):855‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Levay K, Slepak VZ. Regulation of Cop9 signalosome activity by the EF‐hand Ca2+‐binding protein tescalcin. J Cell Sci. 2014;127(Pt 11):2448‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gummlich L, Rabien A, Jung K, Dubiel W. Deregulation of the COP9 signalosome‐cullin‐RING ubiquitin‐ligase pathway: mechanisms and roles in urological cancers. Int J Biochem Cell Biol. 2013;45(7):1327‐37. [DOI] [PubMed] [Google Scholar]
- 39. Su H, Li F, Ranek MJ, Wei N, Wang X. COP9 signalosome regulates autophagosome maturation. Circulation. 2011;124(19):2117‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Capece D, Fischietti M, Verzella D, Gaggiano A, Cicciarelli G, Tessitore A, et al. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor‐associated macrophages. Biomed Res Int. 2013;2013:187204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hernandez‐Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology. 2013;144(3):512‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu RX, Wei Y, Zeng QH, Chan KW, Xiao X, Zhao XY, et al. Chemokine (C‐X‐C motif) receptor 3‐positive B cells link interleukin‐17 inflammation to protumorigenic macrophage polarization in human hepatocellular carcinoma. Hepatology. 2015;62(6):1779‐90. [DOI] [PubMed] [Google Scholar]
- 43. Xiao X, Lao XM, Chen MM, Liu RX, Wei Y, Ouyang FZ, et al. PD‐1hi Identifies a Novel Regulatory B‐cell Population in Human Hepatoma That Promotes Disease Progression. Cancer Discov. 2016;6(5):546‐59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The key raw data have been deposited into the Research Data Deposit (www.researchdata.org.cn), with the approval number of RDDB2020000821.