

SUMMARY
Transcription by RNA polymerase II (RNAPII) is pervasive in the human genome. However, the mechanisms controlling transcription at promoters and enhancers remain enigmatic. Here, we demonstrate that Integrator subunit 11 (INTS11), the catalytic subunit of the Integrator complex, regulates transcription at these loci through its endonuclease activity. Promoters of genes require INTS11 to cleave nascent transcripts associated with paused RNAPII and induce their premature termination in the proximity of the +1 nucleosome. The turnover of RNAPII permits the subsequent recruitment of an elongation-competent RNAPII complex, leading to productive elongation. In contrast, enhancers require INTS11 catalysis not to evict paused RNAPII but rather to terminate enhancer RNA transcription beyond the +1 nucleosome. These findings are supported by the differential occupancy of negative elongation factor (NELF), SPT5, and tyrosine-1-phosphorylated RNAPII. This study elucidates the role of Integrator in mediating transcriptional elongation at human promoters through the endonucleolytic cleavage of nascent transcripts and the dynamic turnover of RNAPII.
Graphical Abstract
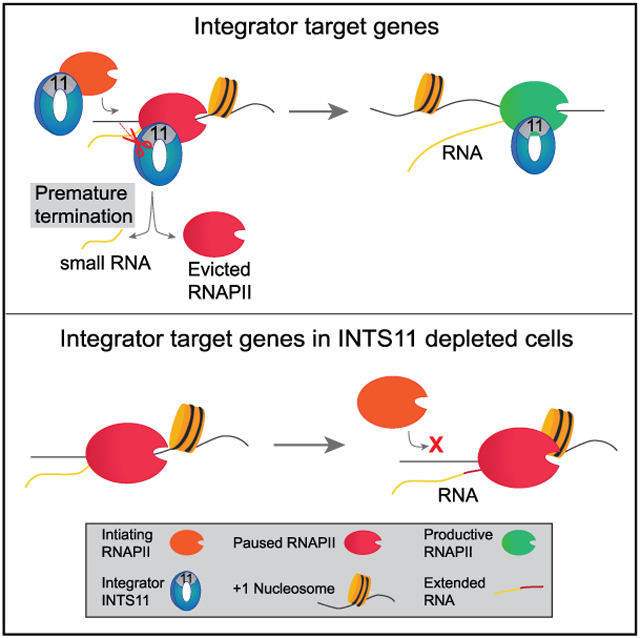
In Brief
In this study, Beckedorff et al. demonstrate that the human Integrator complex associates with paused RNA polymerase II and mediates productive transcriptional elongation through its RNA endonuclease activity. This work supports the dynamic turnover model of paused RNA polymerase II complexes and is contrary to observations described in Drosophila.
INTRODUCTION
Transcription by RNA polymerase II (RNAPII) in metazoans is governed at multiple stages, allowing for the precise execution of gene expression programs critical to cellular development and homeostasis. During formation of the preinitiation complex, general transcription factors coalesce around the promoter and orient RNAPII at transcription start sites (TSSs). The XPB helicase, a component of TFIIH, unwinds the local chromatin and enables RNAPII to initiate transcription from single-stranded template DNA (Saunders et al., 2006). RNAPII undergoes promoter escape, where it breaks contacts with promoter elements and preinitiation machinery to begin elongation. A critical regulatory step occurs after RNAPII transcribes 20–120 nt from the TSS, where it transiently stalls in the vicinity of the +1 nucleosome (Chen et al., 2018). This phenomenon, known as RNAPII promoter-proximal pausing, was initially described at heat-shock-inducible genes in Drosophila and has since been the subject of intense investigation in the genome-wide era (Rougvie and Lis, 1988). Surprisingly, these studies revealed that most RNAPII-transcribed genes display greatest RNAPII occupancy in promoter regions, establishing that RNAPII proximal pausing is ubiquitous in the genome (Muse et al., 2007; Rahl et al., 2010; Zeitlinger et al., 2007). The involvement of these genes in development, stimulus responsiveness, and cellular reproduction suggests that multicellular organisms evolved RNAPII proximal pausing as an additional regulatory layer in the expression of critical genes (Zeitlinger et al., 2007).
Numerous factors contribute to the establishment and maintenance of paused RNAPII, including sequence-specific and general transcription factors, negative elongation factor (NELF), DRB-sensitivity inducible factor (DSIF), polymerase-associated factor 1 (PAF1), and Gdown1 (Chen et al., 2018). In addition, DNA/RNA hybrids and the +1 nucleosome have also been shown to promote RNAPII pausing by imposing an energetic barrier that obstructs RNAPII entry into gene bodies (Bintu et al., 2012; Eddy et al., 2011; Teves et al., 2014; Voong et al., 2016; Weber et al., 2014). Release of RNAPII and its nascent RNA into productive elongation relies on the kinase activity of P-TEFb, which phosphorylates NELF, SPT5 (a subunit of DSIF), and serine-2 on the RNAPII C-terminal domain (Peterlin and Price, 2006).
Our previous studies demonstrate that the Integrator complex mediates RNAPII pause-release through its recruitment of P-TEFb and AFF4, two components of the super elongation complex (SEC) (Gardini et al., 2014). Other groups have shown that Integrator also associates with NELF and DSIF, suggesting that Integrator may also functionally coordinate with factors that promote pausing (Stadelmayer et al., 2014; Yamamoto et al., 2014). The engagement of Integrator and the SEC is critical for activating the mitogen-activated protein kinase (MAPK) gene expression program, though less is known about the mechanisms by which Integrator regulates transcription and RNAPII pause-release genome-wide (Gardini et al., 2014; Yue et al., 2017).
Integrator is a metazoan-restricted multisubunit protein complex that is intimately associated with the phosphorylated form of RNAPII (Baillat et al., 2005; Ebmeier et al., 2017; Egloff et al., 2007; Shah et al., 2018). Integrator harbors a RNA endonuclease subunit, Integrator subunit 11 (INTS11), and our initial characterization of the complex revealed its role in the 3′ end processing of small nuclear RNAs involved in spliceosome formation (Baillat et al., 2005). It is now understood that the enzymatic activity of Integrator requires functional association of the INTS11/INTS9 heterodimer with INTS4 to collectively form the catalytically active cleavage module (Albrecht et al., 2018; Wu et al., 2017). In an effort to identify novel RNA substrates of Integrator, we recently found that Integrator is critical for the biogenesis and 3′ end processing of enhancer RNAs (eRNAs), thus expanding the scope of Integrator function and enhancer regulation (Gardini et al., 2014; Lai et al., 2015).
Promoters and enhancers share conserved structural and functional features including DNase I hypersensitivity and nucleosome positioning, transcription factor occupancy, histone acetylation, and bidirectional transcription (Kim and Shiekhattar, 2015). We sought to understand how Integrator regulates transcription at these loci, with a specific interest in the role of its catalytic activity. We find that Integrator regulates RNAPII recruitment genome-wide, independent of its enzymatic activity. At numerous human genes, INTS11 mediates RNAPII pause-release through endonucleolytic cleavage of nascent transcripts, allowing for the reinitiation of an elongation-competent RNAPII complex. At enhancers, INTS11 catalysis is needed strictly for the termination of eRNA transcription. Such mechanistic distinctions are reflected in the increased occupancy of pause factors (NELF and SPT5) following Integrator depletion at promoters. Our results highlight fundamental differences in the regulation of transcription at human promoters and enhancers. This work places Integrator as a critical determinant in the recruitment and dynamic turnover of RNAPII, where it initiates and terminates numerous rounds of transcription to promote productive elongation through the +1 nucleosome.
RESULTS
Transcriptional Circuitry of the Human Integrator Core Catalytic Module
To gain insight into the transcriptional targets regulated by INTS11, INTS9, and INTS4, the core subunits of the Integrator catalytic module, we generated HeLa cells with stable doxycycline-inducible (Dox) short hairpin RNA against each subunit in addition to a short hairpin RNA (shRNA) against green fluorescent protein (control shRNA) (Figure S1A). Interestingly, while depletion of INTS4 did not affect the stability of INTS11 or INTS9, reduction of either INTS11 or INTS9 leads to the loss of both proteins, reflecting their association as obligate partners (Figure S1A). We performed total RNA sequencing (RNA-seq) to assess their genome-wide effect on 24,832 expressed genes in HeLa cells (Table S1). All genome-wide results presented are a merge of at least two independent biological replicates. Knockdown of INTS11, INTS9, or INTS4 resulted in the differential expression of 5,347, 7,423, and 5,692 genes respectively, and nearly half in each condition were down- or upregulated (Figures 1A-1C; Table S2). Moreover, we found 1,898 core genes whose expression was collectively regulated by the three subunits where ~950 genes were commonly down- or upregulated (Figures 1D and 1E; Table S2). Transcriptional repressors and heterochromatin regulators were the predominant Gene Ontology categories of downregulated genes, pointing to a role of the catalytic module in regulating genes involved in transcriptional repression. Upregulated pathways included those related to epidermal growth factor signaling, consistent with reports in Drosophila (Table S3) (Elrod et al., 2019).
Figure 1. Integrator Regulates Steady-State Gene Expression.

(A–C) Volcano plot depicting differentially expressed genes after (A) INTS11 shRNA, (B) INTS9 shRNA, and (C) INTS4 shRNA determined by RNA-seq (n = 24,832 genes, fold change = 1.5, q-value > 0.05).
(D and E) Venn diagram of common (D) downregulated genes and (E) upregulated genes among cells depleted of INTS11, INTS9, or INTS4.
Nascent Transcript Sequencing Reveals a Predominant Role for Integrator in Mediating the Transition to Productive Elongation
To mechanistically dissect the direct functions of Integrator in transcription, we first performed chromatin immunoprecipitation sequencing (ChIP-seq) of INTS11 and RNAPII and found strong correlation in their genome-wide occupancy, in agreement with biochemical evidence (Figure 2A) (Baillat et al., 2005). Next, we depleted INTS11 and performed precision run-on and sequencing (PRO-seq), which provides single-nucleotide resolution of the leading edge of RNAPII engaged in transcription (Figure S2A) (Kwak et al., 2013). We also performed cap analysis of gene expression (CAGE) and assay for transposase-accessible chromatin using sequencing (ATAC-seq) to determine the positions of transcriptional initiation and the +1 nucleosome, respectively. Multiple bioinformatic filters were used to assess transcriptionally engaged RNAPII, including detectable signal from RNA-seq, PRO-seq, and CAGE, and a positioned +1 nucleosome within 500 nt from the CAGE signal (Figure S2B). These cutoffs yielded 8,000 genes (Figure S2B). We used PRO-seq to calculate the RNAPII traveling ratio (TR) as the density of RNAPII in the promoter region (defined here as −50 nt from CAGE to +300 nt) over the region corresponding to the gene body (+301 nt from CAGE to TES), which allows for genome-wide assessment of RNAPII behavior (Rahl et al., 2010). We detected an increase in the TR after depletion of INTS11, suggesting that Integrator is required for RNAPII pause-release (Figures S2C and S2D). However, an increased TR does not provide sufficient mechanistic evidence that Integrator is required to license transcriptional elongation. To more thoroughly examine transcriptional dynamics, we separated the PRO-seq signal into promoter and gene body regions (defined above), as these loci correspond to transcriptional initiation and elongation, respectively. We also centered the PRO-seq promoter profiles on the +1 nucleosome dyad to assess the response of RNAPII as it encounters the +1 nucleosome barrier (Figures S2E and S2F). Depletion of INTS11 led to an increase and shift of RNAPII toward 3′ end of the promoter region (Figure S2F). Moreover, we observed reduced read density through the gene body, reflecting impaired elongation (Figure S2F).
Figure 2. Integrator Mediates Transcriptional Elongation.

(A) Spearman correlation of RNAPII and INTS11 ChIP-seq occupancy centered on the TSS (CAGE peak ±200 bp) of 8,000 genes.
(B) Hypothetical schematic of control condition “A” whereby treatments producing conditions “B” and “C” generate identical traveling ratio (TR) values by distinct transcriptional mechanisms.
(C and D) TM of control shRNA cells (C) and INTS11 shRNA cells (D).
(E) Differential TM of INTS11 shRNA cells.
(F and G) ChIP-seq promoter profiles of INTS11 (F) and RNAPII (G) centered on the +1 nucleosome dyad at gene classes defined by the differential TM. Kolmogorov-Smirnov (KS) test p value < 0.001 for classes paired against class IV.
(H and I) PRO-seq examples of class IV genes SEL1L (H) and MED4 (I) in control shRNA cells and INTS11 shRNA cells without and with shRNA induction.
The Traveling Matrix Defines a Transcriptional Initiation to Elongation Continuum
Measurements of the TR pose a major limitation in that the same value can result from reduced signal in the gene body or increased reads in the promoter, obscuring transcriptional dynamics (Figure 2B, compare conditions B and C). Assessment of the TR and RNAPII average profiles provide a representative insight into global gene expression changes, though we strived to understand the response of individual genes to INTS11 depletion. Therefore, we devised a two-dimensional Gaussian distribution of RNAPII, which we termed the traveling matrix (TM), to simultaneously dissect the alterations at promoters and gene bodies of INTS11-responsive genes (Figure 2B). The TM resolves changes in the promoter and gene body regions of individual genes, which are then represented as a two-dimensional topographic map. The color intensity in the TM displays the concentration of genes at a particular position. Induction of control shRNA minimally changed its TM, as most genes were centered close to the origin of the two-dimensional graph (Figures 2C and S2G, which depict individual genes). In contrast, depletion of INTS11 induced an extensive transcriptional effect, as the TM coordinates of numerous genes were altered (Figures 2D and S2H). Consistent with the average profiles, the genes in the INTS11 TM showed reduced gene body read density (Figure 2D, note the downward shift of the matrix).
We set the control TM as the baseline expected null dispersion and excluded these genes from the INTS11 TM to identify genes that significantly (false discovery rate [FDR] < 0.05) differ from the expected control dispersion. We named this matrix the differential TM (Figures 2E and S2I). We then divided the differential TM into four gene class quadrants (class I–IV), representing differentially expressed genes with coincident changes in promoter and gene body signal (Table S4). While INTS11 depletion altered many gene coordinates in the differential TM (compare Figures 2C and 2D; also see Figures S2G and S2H), the majority of INTS11-responsive genes accumulated in class III and IV quadrants, signifying a loss of transcriptional elongation (Figures 2E and S2I). Moreover, the pronounced enrichment of genes in class IV, where augmented promoter signal is coupled with reduced gene body reads, implicates a requirement for INTS11 in mediating RNAPII pause-release and productive elongation at these loci. Consistent with their responsiveness to INTS11 depletion, class IV genes show the highest levels of INTS11 and RNAPII occupancy at their promoters (Figures 2F and 2G). Figures 2H and 2I depict single-nucleotide PRO-seq tracks for two representative class IV genes following control or INTS11 shRNA induction.
The Requirement of Integrator at Human Genes with a Highly Positioned +1 Nucleosome
To understand how INTS11 regulates transcription of the four gene classes determined by the differential TM, we displayed the promoter profile of engaged RNAPII centered on the +1 nucleosome and depicted the gene body profile from its first 25% to the TES (Figures 3A-3D). Class III and IV genes represent the majority of INTS11-responsive genes (n = 2,104 or 67%), which showed decreased read density in their gene bodies upon depletion of INTS11 (Figure 3D). As seen in the differential TM, class IV genes exhibited a substantial increase in paused RNAPII in proximity of +1 nucleosome, and the peak of RNAPII was also shifted toward the center of the dyad (Figure 3D). This behavior was also noted in class II genes (Figure 3B). In contrast, class III and I genes showed decreased read density close to +1 nucleosome without a change in the position of RNAPII (Figures 3B and 3D). Analysis of the TR illuminated elongation defects in class II, III, and IV genes, while this impairment was most pronounced at class IV genes (Figures S3A-S3D). Importantly, these alterations in nascent transcription were largely recapitulated in steady-state gene expression (Figure S3E). The PRO-seq results were then validated using ChIP-seq of total RNAPII, which showed comparable profiles of RNAPII at promoters and gene bodies of the four classes of genes (Figures 3E and 3F). Interestingly, while depletion of INTS11 did not impact the position of the +1 nucleosome in the four classes of genes, the two gene classes (II and IV) that featured increased RNAPII pausing showed highest ATAC-seq read density of the +1 nucleosome (Figures 3G-3J). The enhanced positioning of the +1 nucleosome at class IV genes was coupled with high-occupancy histone H3 lysine 4 trimethylation (H3K4me3) and histone H3 lysine 36 trimethylation (H3K36me3) and low enrichment of histone H3 lysine 4 monomethylation (H3K4me1) (Figures S3F-S3H). Notably, class I genes displayed the lowest levels of H3K4me3 and H3K36me3 compared to the other classes, consistent with their low expression (Figures S3F-S3I). Taken together, we find that at genes with a highly positioned +1 nucleosome decorated in H3K4me3, INTS11 depletion elicits a substantial increase in RNAPII pausing in conjunction with reduced elongation, as exemplified by class IV genes in the differential TM.
Figure 3. Integrator Relieves RNAPII Pausing Adjacent to the +1 Nucleosome Dyad.

(A and B) PRO-seq promoter-proximal read density and gene body profile at class I and II genes in control shRNA cells (A) and INTS11 shRNA cells (B) without and with shRNA induction. KS test p value = 0.01 of class I promoter and gene body profiles (−Dox versus +Dox). KS test p value <0.001 of class II promoter and gene body profiles (−Dox versus +Dox).
(C and D) PRO-seq promoter-proximal read density and gene body profile at class III and IV genes in control shRNA cells (C) and INTS11 shRNA cells (D) without and with shRNA induction. KS test p value < 0.001 for class III and IV (−Dox versus +Dox) promoter and gene body profiles.
(E) RNAPII ChIP-seq promoter and gene body profiles at class I and II genes without and with INTS11 shRNA induction. KS test p value < 0.05 and 0.01 for class I and II (−Dox versus +Dox) promoter profiles, respectively, and KS test p value < 0.05 for gene body profiles.
(F) RNAPII ChIP-seq promoter and gene body profiles at class III and IV genes without and with INTS11 shRNA induction. KS test p value < 0.0001 and 0.05 for class III and IV (−Dox versus +Dox) promoter profiles, respectively, and KS test p value < 0.001 for gene body profiles.
(G and H) Mononucleosome profile at class I and II genes in control shRNA cells (G) and INTS11 shRNA cells (H) without and with shRNA induction.
(I and J) Mononucleosome profile at class III and IV genes in control shRNA cells (I) and INTS11 shRNA cells (J) without and with shRNA induction.
Integrator Catalytic Activity Relieves RNAPII Pausing and Contributes to Transcriptional Elongation
Few studies have examined the direct contribution of INTS11 catalytic activity to gene expression and Integrator function. Accordingly, we depleted INTS11 and performed a rescue experiment by ectopically expressing wild-type INTS11 (WT-INTS11) or its catalytic mutant INTS11 (E203Q-INTS11) from a sequence refractory to the shRNA (Figure S4A). Exogenous expression of WT-INTS11 rescued the changes incurred by INTS11 depletion at all genes and the four gene classes, as seen by the average profiles and the TR (Figures 4A-4F and S4B-S4I). In contrast, rescue with INTS11-E203Q produced a similar average profile and TR to that of INTS11 depletion at all 8,000 genes, indicating increased RNAPII pausing and decreased elongation (Figures S2C-S2F and S4B-S4E). Examination of the average profiles at the four gene classes revealed that at class II and IV genes, INTS11 catalysis is required for the release of paused RNAPII into productive elongation (Figures 4B and 4D; see examples in Figures 4E and 4F). Class III and IV genes highlighted the requirement of INTS11 catalysis for transcriptional elongation, as the reduction in gene body read density was not restored by E203Q-INTS11 expression (Figures 4D, S4H, and S4I). Strikingly, the impaired recruitment of RNAPII to class I and III genes was rescued by E203Q-INTS11 expression, thus delineating a catalytic-independent role of Integrator in the recruitment of RNAPII to these genes (Figures 4B and 4D). At class III genes, while E203Q-INTS11 expression corrected RNAPII recruitment defects, the reduction in gene body read density persisted, affirming the essentiality of INTS11 catalysis in the transition of RNAPII to productive elongation (Figure 4D). Notably, the read density in the bodies of the class I and II genes increased to a lesser extent compared to that of INTS11 depletion (Figure 3B versus Figure 4B). In summary, we find that INTS11 catalytic activity is needed to release RNAPII pausing in the proximity of the +1 nucleosome and promote transcriptional elongation at numerous human genes (Figures 4E and 4F).
Figure 4. INTS11 Catalysis Facilitates RNAPII Elongation.

(A and B) PRO-seq promoter-proximal read density and gene body profile at class I and II genes in INTS11 shRNA cells expressing WT-INTS11 (A) and INTS11 shRNA cells expressing E203Q-INTS11 (B) without and with shRNA induction. KS test p value < 0.0001 for class II (−Dox versus +Dox) promoter profile and KS test p value < 0.05 and 0.01 for class I and II (−Dox versus +Dox) gene body profiles, respectively.
(C and D) PRO-seq promoter-proximal read density and gene body profile at class III and IV genes in INTS11 shRNA cells expressing WT-INTS11 (C) and INTS11 shRNA cells expressing E203Q-INTS11 (D) without and with shRNA induction. KS test p value < 0.0001 for class IV (−Dox versus +Dox) promoter profile and KS test p value < 0.001 for class III and IV (−Dox versus +Dox) gene body profiles. PRO-seq promoter profiles are centered on the +1 nucleosome dyad. PRO-seq examples of class IV genes.
(E and F) SEL1L (E) and MED4 (F) in INTS11 shRNA cells expressing WT-INTS11 and INTS11 shRNA cells expressing E203Q-INTS11 without and with shRNA induction.
INTS11 Induces Premature Termination of Stalled RNAPII Transcripts at Human Promoters
Our experiments demonstrate that in the proximity of +1 nucleosome, the catalytic activity of INTS11 is required for the release of paused RNAPII into productive elongation. We surmised that nascent transcripts protruding from the active site of RNAPII undergo endonucleolytic cleavage by INTS11 and prematurely terminate, allowing for new rounds of initiation (Shao and Zeitlinger, 2017). To address this hypothesis, we performed small RNA (smRNA) sequencing. We detected an enrichment of smRNA in the 5′ end of all genes and subsequently analyzed the reads that mapped to the first 150 nt from the TSS (Figure 5A). The presence of smRNA was observed at all classes of genes, indicating that the generation of RNAPII-associated smRNA is pervasive throughout the genome (Figures 5B, 5C, S5A, and S5B). This is in line with previous studies that showed the production of smRNA was not exclusive to genes with paused RNAPII, despite their enrichment at paused sites (Nechaev et al., 2010; Taft et al., 2009). We found that the amount of smRNA at class II and IV genes was unchanged in all conditions, while at class I and III genes, the level of smRNA in INTS11-depleted cells was reduced due to impaired RNAPII recruitment (Figures 5B, 5C, S5A, and S5B; see Figure 3B).
Figure 5. INTS11 Catalyzes the Termination of Small RNA Associated with Paused RNAPII.

(A) smRNA whole gene profile of all expressed genes (n = 8000) in INTS11 shRNA cells.
(B and C) Boxplot of log2(+Dox/−Dox) smRNA counts per million (CPM) at (B) Class IV and (C) Class III genes, t test; ***p value < 0.001.
(D) Boxplot of smRNA read length at all genes in INTS11 shRNA cells expressing WT-INTS11 and INTS11 shRNA cells expressing E203Q-INTS11. t test; ***p value < 0.001.
(E and F) smRNA mean size distribution centered on the TSSs of class IV (E) and class III (F) genes in control shRNA cells and INTS11 shRNA cells without and with shRNA induction.
(G and H) smRNA mean size distribution centered on the TSSs of class IV (G) and class III (H) genes in INTS11 shRNA cells expressing WT-INTS11 and INTS11 shRNA cells expressing E203Q-INTS11 without and with shRNA induction.
(I–L) smRNA read length distribution at all genes in control shRNA cells (I), INTS11 shRNA cells (J), INTS11 shRNA cells expressing WT-INTS11 (K), and INTS11 shRNA cells expressing E203Q-INTS11 (L) without and with shRNA induction.
(M and N) smRNA examples of class IV genes SEL1L (M) and MED4 (N) in control shRNA cells, INTS11 shRNA cells, INTS11 shRNA cells expressing WT-INTS11, and INTS11 shRNA cells expressing E203Q-INTS11 without and with shRNA induction.
We previously established that depletion of INTS11 or overexpression of E203Q-INTS11 compromises UsnRNA (uridylaterich small nuclear RNA) termination, resulting in RNAPII read-through and transcript lengthening (Baillat et al., 2005). Interestingly, analysis of smRNA read length illuminated the extension of transcripts in E203Q-INTS11 cells at baseline, suggesting that E203Q-INTS11 exerted a dominant-negative effect on RNA processing prior to the depletion of endogenous INTS11 (Figure 5D). Evidence from the aforementioned studies implicate that defects in premature termination would be accentuated at class II and IV genes, as they display pronounced stalling of RNAPII in the absence of INTS11 or its catalytic activity (Nechaev et al., 2010; Taft et al., 2009). Indeed, the mean read length of smRNA at these classes increased following depletion of INTS11, consistent with the augmented pausing of RNAPII and its 3′-shift toward the center of the +1 nucleosome dyad (Figures 5E and S5C; see Figures 3D and 3F). At class III genes, knockdown of INTS11 generated shorter smRNA extensions compared to that of class II and IV genes, owing to the reduction in RNAPII recruitment (Figure 5F; see Figures 3D and 3F). Importantly, ectopic expression of WT-INTS11 rescued the lengthening of smRNA transcripts at all classes, while expression of E203Q-INTS11 failed to do so (Figures 5G, 5H, S5E, and S5F). We noted that the magnitude of smRNA lengthening was subdued by the dominant-negative activity of E203Q-INTS11; however, the importance of INTS11 catalytic activity in smRNA processing was underscored at all gene classes (Figures 5E-5H and S5C-S5F).
Further dissection of smRNA by the read length distribution revealed two populations of transcripts that originate from TSSs with average sizes of 21 and 41 nt, respectively (Figures 5I-5L and S5G-S5J). After INTS11 knockdown, we observed an accumulation and extension of the 41-nt smRNA population concomitant with a depletion of the 21-nt smRNA population (Figures 5J and S5H). This processing defect was rescued by WT-INTS11 yet persisted in cells expressing E203Q-INTS11, indicating that the population of 21-nt transcripts correspond to the cleavage product of INTS11 catalysis (Figures 5K, 5L, and S5I-S5J). Taken together, these data demonstrate a role for Integrator and the catalytic activity of INTS11 in the termination of RNAPII-associated nascent transcripts at genes with high levels of paused RNAPII (Figures 5M and 5N).
Integrator Terminates Transcription at Human Enhancers
We asked whether our findings at promoters were generalizable to enhancer regions given the known requirement for Integrator in the biogenesis and 3′ end processing of eRNA. We evaluated RNAPII profiles at 2,293 enhancers by finding common intergenic H3K27ac and H3K4me1 peaks and excluded regions that overlap within 2 kb of an annotated TSS. We then centered the reads of the bidirectional eRNA transcripts on the maximum PRO-seq signal (Figures 6A-6D and S6A). Consistent with previous observations at epidermal-growth-factor-responsive enhancers, depletion of INTS11 produced a global defect in eRNA termination, as demonstrated by the extension of RNAPII reads and accumulation of transcripts beyond their canonical 3′ end (Figures 6A-6D and S6B). This was particularly evident in cells expressing E203Q-INTS11 following the knockdown of INTS11 (compare Figures 6C and 6D). We then centered PRO-seq reads on the +1 nucleosome as promoters and enhancers share remarkably conserved transcriptional architecture, including well-positioned +1 and −1 nucleosomes (Core et al., 2014). Knockdown of INTS11 also decreased recruitment of RNAPII to enhancers, similar to that observed at class I and III promoters (Figures 6E, 6F, and 3A-3F). Strikingly, expression of both WT-INTS11 and E203Q-INTS11 restored defects in RNAPII occupancy incurred by the loss of INTS11, further attesting to the catalytic-independent role of Integrator in the recruitment of RNAPII (Figures 6G and 6H). We next determined the TR and TM at enhancers. In agreement with the PRO-seq average profiles, the TR at enhancers displayed augmented transcriptional elongation resulting from decreased RNAPII density at the TSS and an accumulation of reads within enhancer bodies (Figures S6C-S6F). The TM at enhancers was entirely different from that of promoters (Figures 6I, 6J, S6G, S6H, 2C, 2D, S2G, and S2H). While INTS11 depletion lead to reduced reads in the bodies of most genes as shown in Figure 2D, enhancers featured enhanced reads in their bodies beyond the +1 nucleosome, reflecting impaired termination (Figure 6B and 6J). The differential TM at enhancers also depicted a pattern considerably different from the differential TM at promoters (compare Figures 6K, S6I, 2E, and S2I). Notably, only a few enhancers showed increased RNAPII-stalling after INTS11 knockdown, suggesting that Integrator plays little role in RNAPII pause-release at enhancers. These data indicate that Integrator is required for the global recruitment of RNAPII to enhancers and promoters independent of its catalytic activity. At promoters, INTS11 catalysis facilitates transcriptional elongation through the premature termination of nascent mRNA adjacent to the +1 nucleosome, while at enhancers, the endonuclease activity of INTS11 is needed strictly for the termination of eRNA transcripts beyond the +1 nucleosome.
Figure 6. Enhancer RNA Transcriptional Termination Requires INTS11 Catalysis.

(A–D) PRO-seq enhancer profile (n = 2,293) in control shRNA cells (A), INTS11 shRNA cells (B), INTS11 shRNA cells expressing WT-INTS11 (C), and INTS11 shRNA cells expressing E203Q-INTS11 (D) without and with shRNA induction.
(E–H) PRO-seq enhancer TSS profile in control shRNA cells (E), INTS11 shRNA cells (F), INTS11 shRNA cells expressing WT-INTS11 (G), and INTS11 shRNA cells expressing E203Q-INTS11 (H) without and with shRNA induction. KS test p value < 0.001 for all conditions (−Dox versus +Dox).
(I and J) Enhancer TM of control shRNA cells (I) and INTS11 shRNA cells (J).
(K) Differential enhancer TM of INTS11 shRNA cells.
Factors Associated with RNAPII Pausing Display a Differential Behavior at Promoters and Enhancers
Finally, we analyzed factors associated with RNAPII pause-release at promoters and enhancers following depletion of INTS11. In agreement with the increased RNAPII pausing observed at promoters of class II and IV genes, knockdown of INTS11 increased occupancy of the NELF subunit NELF-E and the DSIF subunit SPT5 (Figures 7A, 7B, S7A, and S7B). At enhancers, NELF-E levels were reduced, in line with decreased RNAPII recruitment (Figure 7C). Moreover, while SPT5 levels were unchanged around eRNA start sites, there was increased occupancy in enhancer bodies (Figure 7D). We evaluated levels of tyrosine-1-phosphorylated RNAPII (RNAPII-Y1), which was shown to be required for transcriptional termination and functionally associated with Integrator by mass spectroscopy (Mayer et al., 2012; Shah et al., 2018). After INTS11 depletion, elevated levels of RNAPII-Y1 were found at enhancers and at promoters of class II and IV genes, consistent with the pronounced defects in RNA processing at these sites (Figures 7E, 7F, S7C, 5A, and S5A). In contrast, promoters of class I and III genes were reduced in RNAPII-Y1 occupancy due to impaired recruitment of RNAPII (Figures 7E and S7C). This was further confirmed through analysis of serine-5-phosphorylated RNAPII (RNAPII-S5), which was similarly diminished at these classes (Figures S7D and S7E). Moreover, occupancy of serine-2-phosphorylated RNAPII (RNAPII-S2) closely mirrored the PRO-seq gene body profiles at the four classes of genes (Figures S7F and S7G). To ascertain whether these results could be visualized in cells, we performed STORM (stochastic optical reconstruction microscopy) imaging of INTS1 and RNAPII-Y1, which revealed highly colocalized signals in large clusters throughout the nucleus (Figures 7G and S7D). Strikingly, depletion of INTS11 significantly increased the density of these clusters (Figures 7H and S7E), in agreement with the elevated ChIP-seq profile of RNAPII-Y1 at paused genes and enhancers. In summary, these findings link the catalytic activity of Integrator to premature termination and RNAPII pause-release at promoters and transcriptional termination at enhancers.
Figure 7. Pausing Factors and Tyrosine-1-Phosphorylated RNAPII Convey Integrator Functions.

(A) NELF-E ChIP-seq promoter profile at class III and -IV genes without and with shRNA induction in INTS11 shRNA cells. KS test p value < 0.0001 for class III and IV (−Dox versus +Dox), respectively.
(B) SPT5 ChIP-seq promoter profile at class III and IV genes without and with shRNA induction in INTS11 shRNA cells. KS test p value < 0.0001 for class III and IV (−Dox versus +Dox), respectively.
(C) NELF-E ChIP-seq enhancer profile without and with shRNA induction in INTS11 shRNA cells.
(D) SPT5 ChIP-seq enhancer profile without and with shRNA induction in INTS11 shRNA cells.
(E) RNAPII-Y1 ChIP-seq promoter profile at class III and IV genes without and with shRNA induction in INTS11 shRNA cells. KS test p value < 0.0001 for class III and IV (−Dox versus +Dox), respectively.
(F) RNAPII-Y1 ChIP-seq enhancer profile without and with shRNA induction in INTS11 shRNA cells.
(G) STORM images of INTS1, RNAPII-Y1, and a merge without and with shRNA induction in INTS11 shRNA cells.
(H) Quantification of RNAPII-Y1 cluster density without and with shRNA induction in INTS11 shRNA cells (n = 8, **p < 0.01).
(I) Model depicting Integrator mediating premature termination of nascent transcripts and subsequent recruitment of elongation-competent RNAPII complex.
DISCUSSION
Here, we show that depletion of Integrator subunits that compose the catalytic module (INTS11, INTS9, and INTS4) leads to alterations in steady-state gene expression, consistent with previous reports (Gardini et al., 2014; Lai et al., 2015; Stadelmayer et al., 2014; Yamamoto et al., 2014; Yue et al., 2017). While recent studies indicate that Drosophila Integrator primarily attenuates gene expression, our data demonstrate that Integrator plays a role in the repression and activation of human genes (Tatomer et al., 2019; Elrod et al., 2019). For example, we found more than half of 7,425 differentially expressed genes were downregulated after knockdown of human INTS9, while depletion of Drosophila INTS9 reduced expression of <18% of genes (Tatomer et al., 2019; Elrod et al., 2019). Interestingly, upregulated Gene Ontology pathways were the most conserved between these species, suggesting that throughout evolution the scope of Integrator expanded from a set of repressed target genes to include those that require its function for activation. We previously showed that Integrator is stably associated with the C-terminal domain of RNAPII, and ChIP-seq analysis of INTS11 and RNAPII showed highly correlated genomic occupancy at 8,000 genes (r = 0.79). In Drosophila, only a 39% correlation was found between INTS1 and PRO-seq signals at 9,499 genes, suggesting differences in Integrator occupancy, RNAPII association, and regulatory function in lower metazoans.
To address the direct contribution of Integrator to transcription at promoters and enhancers in human cells, we depleted INTS11 and performed PRO-seq. We utilized a two-dimensional TM to project the positional changes in RNAPII at promoters and gene bodies incurred by the loss of INTS11. Strikingly, more than two-thirds of differentially regulated genes exhibited a loss of RNAPII signal in gene bodies, indicating that Integrator promotes transcriptional elongation in human cells. Half of these genes displayed deficient RNAPII recruitment, whereas the other half showed increased RNAPII pausing. This reduction in elongating RNAPII was evident in the steady-state levels of RNA, which were largely diminished in these classes. Critically, ectopic expression of E203Q-INTS11 restored recruitment of RNAPII, while defects in pause-release persisted. Thus, utilization of catalytic-dead INTS11 in conjunction with the TM illuminated the mechanistically distinct roles of Integrator in the recruitment of RNAPII and its catalytic activity in the transition of RNAPII to productive elongation.
Class IV genes determined by the TM featured Integrator-dependent RNAPII pausing, a strongly positioned +1 nucleosome decorated in H3K4me3, gene bodies enriched in H3K36me3, and were most occupied by INTS11 and RNAPII compared to the other classes. These results contrast a recent report in Drosophila that shows a repressive function of Integrator at lowly expressed genes and its anticorrelation with H3K4me3 and H3K36me3 (Elrod et al., 2019). Furthermore, we found an inverse correlation of Integrator occupancy and H3K4me1, also in opposition to this work. Interestingly, high levels of H3K4me1 and low levels of H3K4me3 and H3K36me3 were characteristic of class I genes, which featured an epigenetic landscape most closely attributed to enhancer elements. Notably, this class comprised the fewest Integrator-responsive genes in our study (327 of 3,125 genes), which is consistent with a small set of lowly expressed genes in Drosophila that display de-repression upon Integrator depletion (Elrod et al., 2019). These data suggest that throughout evolution, Integrator acquired a predominant function in the transcriptional activation of genes that require INTS11 catalysis for pause-release and elongation.
We found that loss of INTS11 or ectopic expression of E203Q-INTS11 resulted in not only an accumulation of paused RNAPII but also a 3′-shift of RNAPII peak toward the center of the +1 nucleosome dyad. Remarkably, smRNA transcripts concomitantly lengthened over this region, demonstrating the requirement of INTS11 catalysis for their termination. Importantly, the levels of smRNA were unchanged at all gene classes in cells expressing E203Q-INTS11, which argues against a role for Integrator in regulating smRNA stability. Closer examination of smRNA revealed two populations that emanate from TSSs: a 21-nt cleavage product that reduced upon loss of INTS11 or its catalytic activity and another 41-nt population that extended as a consequence of defective processing. While a recent report in Drosophila attributes numerous promoter-associated transcripts >80 nt as Integrator dependent, the smRNA in the present study is consistent with the length reported in previous literature and demonstrated conditional size changes that mechanistically reflect defective endonucleolytic processing (Kapranov et al., 2007; Nechaev et al., 2010; Taft et al., 2009; Tatomer et al., 2019). These results indicate that INTS11 terminates nascent transcripts to rapidly evict paused RNAPII from the chromatin. This allows for the subsequent recruitment of an elongation-competent RNAPII complex to traverse the +1 nucleosome and elongate into the body of genes (Erickson et al., 2018; Krebs et al., 2017; Shao and Zeitlinger, 2017; Steurer et al., 2018).
Genome-wide studies have found numerous commonalities between two classes of transcriptional regulatory elements, promoters, and enhancers (Kim and Shiekhattar, 2015). Indeed, examination of these distinct loci found architectural commonalities, including DNA sequence, transcription factor occupancy, and nucleosome deposition. We wondered whether the functions of Integrator are also conserved among these regions. Consistent with our observations at promoters, enhancers also demonstrate a catalytic-independent requirement of Integrator in RNAPII recruitment. However, it was proposed that RNAPII pausing at enhancers is more transient compared to promoters, as evidenced by a lower RNAPII TR and increased susceptibility to P-TEFb inhibition (Henriques et al., 2018). In support, the enhancer TM revealed that Integrator has a limited contribution to RNAPII pause-release at these regulatory elements. At promoters of human genes, the enzymatic activity of INTS11 is needed to promote elongation through the dynamic turnover of paused RNAPII, while at enhancers, the catalytic activity of INTS11 is strictly utilized for eRNA termination and plays little role in promoting pause-release and elongation. Collectively, these data indicate that Integrator catalyzes the termination of transcripts during distinct stages of transcription at human enhancers and promoters.
ChIP-seq studies examining occupancy of the pause factors NELF-E and SPT5 corroborate the role of Integrator in mediating recruitment of RNAPII and facilitating pause-release at promoters. Moreover, the increased recruitment of pause factors was unique to promoters and was not seen at enhancers. The reduced occupancy of RNAPII-S5 at class III genes substantiates the role of Integrator in RNAPII recruitment, while its unchanged levels at class IV genes signifies that the accumulation of PRO-seq signal at promoters was due to impaired pause-release and not increased initiation. Importantly, the elevation in RNAPII-Y1 occupancy at paused genes and enhancers highlight the requirement for Integrator in the termination of transcripts at these sites. At promoter-proximal sites, Integrator induces the premature termination of nascent transcripts, while at enhancers, Integrator processes the 3′ end of eRNAs.
Recent studies have elegantly shown that RNAPII undergoes dynamic cycles of initiation, pausing, premature termination, and eviction prior to the establishment of a RNAPII complex that transitions to productive elongation (Erickson et al., 2018; Krebs et al., 2017; Steurer et al., 2018). Our data indicate that Integrator is central to this model, as we demonstrate its catalytic-independent recruitment of RNAPII and catalytic-dependent premature termination of nascent transcripts associated with paused RNAPII. We propose that at human genes, Integrator is key for mediating dynamic turnover of RNAPII through these dual functions, thus balancing the cycle of initiation and early termination events to promote productive elongation of RNAPII.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Ramin Shiekhattar: rshiekhattar@med.miami.edu.
Materials Availability
The reagents generated in this study will be readily shared via materials transfer agreement.
Data and Code Availability
All sequencing data generated in this study is made available at the Gene Expression Omnibus (GEO). The accession number for the raw and processed data reported in this paper is GEO: GSE125534, GSE125535
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines were maintained in mycoplasma-free conditions and were routinely tested for infection. HeLa INTS11 and GFP inducible knockdown clones were established as previously described (Lai et al., 2015). HeLa rescue cells were established by cloning the same shINTS11 sequence into Tet-pLKO-neo vector (Addgene) and single clones were selected with G418 (500 μg/ml). shRNA-resistant N-terminal Flag-tagged WT or E203Q mutant INTS11 cDNA (Baillat et al., 2005) were cloned into Cumate-pLenti-Cloning-2A-GFP vector (ABM Inc.), and transfected into a shINTS11-Tet-pLKO-neo single clone. Stable cell lines were maintained in puromycin (2 μg/ml) and G418 (200 μg/ml) containing DMEM medium.
METHOD DETAILS
RNA isolation
Total RNA was extracted using Trizol reagent (Thermo Fisher Scientific, #15596026) according to the manufacturer’s instructions. Genomic DNA was removed by Turbo DNase treatment (Invitrogen, #AM1907).
Library preparation
ChIP-seq libraries were generated using the NEBNext Ultra II DNA library prep kit (New England Biolabs, #E7645S) with at least 10 ng of input DNA. Total RNA-seq libraries were produced using Truseq Stranded Total RNA library prep kit (Illumina, #20020596) with 500 ng of DNase-treated Input RNA. Small RNA libraries were prepared using the SMARTer smRNA-seq kit (Takara, #635030) with 750 ng of Nuclear-enriched total RNA. Genome-wide experiments were performed as two independent biological replicates. To avoid a batch effect in library preparation and sequencing flow cell, these replicates were processed together.
PRO-seq and data analysis
PRO-seq experiments were performed as described previously (Mahat et al., 2016). Nuclei were isolated by Dounce homogenizer with loose pestle. 1×10^7 nuclei were subjected to nuclear run-on (30°C, 3 min) in the presence of 25 μM Biotin-11-ATP/GTP/CTP/UTP (PerkinElmer). Total RNA was extracted and hydrolyzed in 0.2 M NaOH (on ice, 10 min). Biotinylated nascent RNAs were purified by Dynabeads M-280 streptavidin (Invitrogen). Following adaptor ligation, cDNA synthesis, and PCR amplification 140–350 bp libraries were size-selected by Pippin HT with 2% gel cassette 20B (Sage Science) then sequenced by NextSeq 500 system (Illumina) with single-read runs. Raw fastq data were trimmed by Cutadapt 1.14 (Martin, 2011) and Trimmomatic v0.32 (Bolger et al., 2014), and then aligned on hg19 or dm3 genome by bowtie 1.1.2 (Langmead et al., 2009). Strand-specific single nucleotide ends of aligned reads were generated by BEDTools v2.28 with genomecov (Quinlan and Hall, 2010) as bedgraph format. Bedgraph data were normalized by the number of reads mapped to spike-in dm3 genome, and then converted to bigwig data, which were used for downstream analyses.
CAGE library preparation and analysis
CAGE library preparation, sequencing and mapping were performed by DNAFORM (Kanagawa, Japan), the CAGE library prep is based on nAnTiCAGE protocol (Murata et al., 2014). Briefly first strand cDNAs were transcribed to the 5′ end of capped RNAs and were attached to CAGE “bar code„ tags. Multiplex deep sequencing of two cDNA libraries (shGFP without dox) was performed on an Illumina HiSeq2500 sequencer. The sequenced CAGE tags were mapped to the human genome hg19 using BWA (Li and Durbin, 2009) and used the a filter of MAPQ > = 20. Next, we removed the rRNA from the bam files. The reads with MAPQ < 20 in BWA were mapped again by HISAT2 (Kim et al., 2015) we keep only the unique mapped reads (with the tag NH:i:1) and sum with the BWA mapped reads these were utilized in subsequent analyses. For tag clustering, the CAGE-tag were input for RECLU clustering, with a maximum irreproducible discovery rate (IDR) of 0.1 (Ohmiya et al., 2014). We used BedTools command closest to annotated the CAGE peaks against ENSENBL (v87). Peaks with distance > 500nt from an annotated TSS region were discarded to avoid multiple CAGE signals assigned to the same promoter. When multiple peaks were detected in the same promoter, we considered the CAGE peak with lower IDR. The gene isoforms related with the annotated TSS by CAGE was considered for further analysis if we detect a FPKM > 0 in total RNA-seq of INTS11 shRNA with or without dox. We used deepTool2 (Ramírez et al., 2016) to generate normalized bigwig.
ChIP-Seq and data analysis
ChIP-seq was performed as previously described (Gardini et al., 2014; Lai et al., 2015; Yue et al., 2017) with slight modifications. 2 × 107 cells were crossl-inked in 1% formaldehyde for 10 minutes at room temperature and quenched with 125 mM glycine. For INTS11 ChIP, samples were crosslinked with Cross-link Gold (Diagenode C01019027) according to the manufacturer’s instructions prior to formaldehyde fixation. Samples were resuspended in ChIP lysis buffer (20 mM Tris-HCl, 50 mM NaCl, 0.1% SDS, 0.5% Triton X-100, 1 mM EDTA) and sonicated in a Covaris M220 focused sonicator until chromatin fragments of 150-300 bp were produced. Protein A/G magnetic beads were bound with antibody and incubated with 1 mg of sonicated chromatin overnight. The following day, beads were washed twice with Mixed Micelle Buffer (150 mM NaCl, 1% Triton X-100, 0.2% SDS, 20 mM Tris-HCl, 5 mM EDTA, 65% sucrose), Buffer 500 (500 mM NaCl, 1% Triton X-100, 0.1% Na deoxycholate, 25 mM HEPES, 10 mM Tris-HCl, 1 mM EDTA), LiCl/detergent wash (250 mM LiCl, 0.5% Na deoxycholate, 0.5% NP-40, 10 mM Tris-HCl, 1 mM EDTA), and eluted in Decrosslinking Buffer (20 mM Tris-HCl, 300 mM NaCl, 0.5% SDS, 1 mM EDTA) at 65°C. Eluates were treated with RNase A for 1 hour at 37°C and then Proteinase K for 3 hours at 56°C. The DNA was finally purified by phenol/chloroform and eluted in 1x TE. Raw FASTQ data were processed with Trimmomatic v0.32 to remove low-quality reads and then aligned to the human genome hg19 using STAR aligner v2.5.3a (Dobin et al., 2013). We used deepTool2 (Ramírez et al., 2016) to generate normalized bigwig and heatmaps. Peaks were called using MACS2.1.2 (Zhang et al., 2008) with default parameters –shiftsize 75 –nomodel–extsize 200 –p 0.01 for all data. Whole-cell extract input from the corresponding cell lines were used as controls. Peaks with fold change > 3 and a q-value < 0.01 were used for downstream analysis. Homer annotatePeaks v4.9.1-5 (Heinz et al., 2010) was used for peak annotation and intergenic peaks were selected for eRNA identification.
ATAC-seq and data analysis
ATAC-seq experiments were performed as previously described (Corces et al., 2017), with the following modification. DNA libraries were quantified using Qubit dsDNA high sensitivity reagents (Thermo Fisher Scientific) and measured Qubit 2 fluorometer. The samples were pooled and sequenced, paired-end, 75bp on a NextSeq 500. ATAC-seq analysis was performed as described in Chan et al. (2018) for nucleosome position we used the common peaks shINTS11 without dox as input for NucleoATAC (Schep et al., 2015).
RNA-seq data analysis
Raw fastq RNA-seq data were processed with Trimmomatic v0.32 (Bolger et al., 2014) and aligned to human genome (hg19 version) using STAR aligner v2.5.3a (Dobin et al., 2013) with default parameters and RSEM v1.2.31 (Li and Dewey, 2011) to obtain expected gene counts against the human Ensembl (release 87). To select genes for downstream analysis, we extended the TSS positions (defined by the CAGE peaks) by 500 base pair toward to the3′end. We overlapped the nucleosome position defined by NucleoATAC (Schep et al., 2015) and used the closet nucleosome to the CAGE peak to capture the +1 nucleosome dyad center position. The genes with no nucleosome in the 500nt window were excluded from the subsequent analysis. We merged the 2 PRO-Seq biological replicates for each condition, shINTS11 with and without dox, and excluded all TSS region with a CPM < 0.5 in both conditions. The remaining TSS were defined as all genes (n = 8000). Differential expression was determined between INTS11 shRNA +Dox or INTS9 shRNA +Dox or INTS4 shRNA +Dox against GFP shRNA +Dox or INTS11 shRNA (+Dox versus −Dox) and GFP shRNA (+Dox versus −Dox) using DESeq2 (Love et al., 2014) and R v3.2.3 with q-value < 0.05. For visualization on the UCSC Genome Browser, all tracks were CPM (count per million) normalized against the total number of usable reads in that dataset using deep Tools2 (Ramírez et al., 2016). Gene Ontology analysis was performed using DAVID (v6.8) online tool with standard parameters (https://david.ncifcrf.gov/home.jsp).
Small RNA analysis
Reads were then adaptor trimmed (AAAAAAA) as recommend by SMARTer smRNA-seq kit (Takara, #635030) protocol using Cutadapt (v1.14) and reads less than 17 bp were discarded. First, we aligned the reads against human elements in RepBase (v23.08) with STAR (v2.5.3a), repeat-mapping reads were removed all others were then mapped against the full human genome (hg19 version) keeping all uniquely aligned reads. For visualization on the UCSC Genome Browser, all tracks were CPM (count per million) normalized against the total number of usable reads in that dataset using deepTools2 (Ramírez et al., 2016). The average smRNA length profiles were calculated with the first quartile of all gene classes and we used a window from CAGE peak to +150 nt for the calculation.
Differentially paused genes modeling
PRO-seq was used to calculate the traveling ratio (TR) as previously described in Rahl et al. (2010). Briefly, we calculated the ratio between the RPKM values in promoter region (−50 nt from CAGE to + 300 nt) over the region corresponding to the gene body (+301 nt from CAGE to TES) for all transcripts used in the analysis (n = 8000). The traveling matrix (TM) can be visualized as a 2D distribution by using the log2 ratio (RPKM −Dox/ RPKM +Dox) of promoter region (x axis) and gene body (y axis). To access the genes with differential pause after INTS11 depletion, we developed a pause relative metric using a 2D Gaussian distribution model. First, we calculated a null model dispersion based in the Control shRNA (−Dox) and (+Dox) observed changes. The two-dimensional Control shRNA relative distribution was estimated using a 2D Gaussian probabilistic model, with the number of mixture k= 1 using the numerical Expectation Maximization (EM) algorithm with a convergence threshold < 1e-3. To select the genes from INTS11 shRNA condition presenting a significant relative pause value (FDR < 0.01), we exclude those genes that show a high probability to belong to the null Control shRNA. The relative difference generated by this 2D representation can be categorized in four different groups:
Class 1: log2 promoter > 0 and log2 gene body < 0
Class 2: log2 promoter < 0 and log2 gene body < 0
Class 3: log2 promoter > 0 and log2 gene body > 0
Class 4: log2 promoter < 0 and log2 gene body > 0
The model can be summarized as:
p = Log2(−Dox/+Dox) RPKM at the −50 nt CAGE peak up to + 300 nt window region.
c = Log2(−Dox/+Dox) RPKM from the CAGE peak +301 nt up to gene TES.
p = {p1, p2 … pn}, where, P is populated with the p shControl values.
C = {c1, c2 … cn}, where, C is populated with the c shControl values.
μ, Σ = EM (P, C), where the parameters from the multivariate Gaussian distribution μ (mean vector) and Σ (2x2 covariance matrix) are estimated using the Expectation Maximization algorithm.
x = [pshINTs11,cshINTS11], is a vector containing the values p and c for a given gene in the shINTS11 condition
We defined the probability of a gene x be differentially paused by using the following 2D multivariate normal joint probability distribution equation:
After obtaining the differential pause probability for the genes in the INTS11 shRNA condition, we selected as significant those genes that presented an adjusted probability FDR < 0.01. The 2D Gaussian model and prediction was estimated using the python sklearn mixture package version 0.21.1 (Pedregosa et al., 2011).
Genome-wide identification of eRNA loci
For eRNA identification, we first defined a set of intergenic enhancers by using the BEDTools Window (v2.28.0) function to overlap H3K27ac and H3K4me1 intergenic peaks (defined in ChIP-Seq and data analysis section) in a window of ± 200 bp. After identifying intergenic regions containing H3K27ac/H3K4me1 peaks, the BEDtools Intersect function was used with the option –v to discard any peaks that overlap with any gene from the hg19 reference genome (ENSENBL v87), including an additional 2 kb upstream of every TSS. In order to determine which intergenic enhancers presented transcriptional activity, we separated the mapped read from PRO-seq by strand and individually call peaks using MACS2.1.2 (Zhang et al., 2008) with the following parameters –nomodel–extsize 200–broad-cutoff 0.1 and −broad to identify broad peaks. We merge the common peaks from each samples (Control shRNA and INTS11 shRNA) for each stranded peaks and later all the peaks from common plus strand were merge with common minus strand peaks. BEDtools intersect between the intergenic enhancer peaks and the common peaks from PRO-Seq was use to define the enhancer that contain eRNAs. We excluded all transcripts with less than 800nt, in order to minimize any bias in the TR analysis, since these eRNAs are short to precisely separate promoter and gene body regions. Lastly, we define the TSS coordinate for each eRNA based on the position of the highest signal occurrence in the PRO-Seq peak window and discarded those with CPM > 0.5. For eRNA TR calculation we use as promoter region (−100 nt from PRO-seq summit to + 150 nt) and gene body (+151 nt from PRO-seq summit to TES plus 1kb) all transcripts used in the analysis (n = 2293). The output from the TR calculation was used as input for the traveling matrix pipeline described above.
Microscopy STORM imaging
Cells (4x103) were plated onto micro slide 4 well plates (Ibidi), fixed in 4% formaldehyde, and immunostained with appropriate primary and secondary antibodies. Imaging experiments were carried out with a Nikon eclipse Ti2 microscope equipped with Nikon Instruments (NSTORM). For two color imaging dSTORM imaging, Janelia Fluor 646 [Novus Bio NBP1-7-2739JF646 Lot #36481-051615] was used as a secondary staining for INTS1 and Alexa Fluor 568 [Invitrogen A11077 Lot #1917936] was used as a secondary staining for RPB1 Tyr-1. The secondary antibodies were used with MEA STORM imaging buffer and were imaged continuously with 5000 frames collected per filter range at a frequency of 30 ms. The images were acquired using a 100x, 1.49 NA objective, and imaged onto a Hamamatsu C11440 ORCA-flash 4.0. Storm analysis was carried out with Nikon Elements Analysis 5.02.01 for identification of molecules. Molecule list files were then exported from Nikon elements to be further analyzed using software Clus-Doc software in MATLAB R2018b (Pageon et al., 2016). Cluster density analysis, specifically DBSCAN function, was carried out after manually selecting region of interest corresponding to the nucleus. Statistical significance and graphing were performed using Prism software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Significance was determined by either Student’s t test, non-parametric Wilcox test or Kolmogorov–Smirnov test, as indicated. Error bars in figures represent standard deviation (SD) of at least two independent experiments.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal Anti-INTS11 (ChIP and western) |
Atlas Antibodies | Cat# HPA029025; RRID: AB_10600425 |
| Rabbit polyclonal Anti-INTS11 (ChIP) | Abcam | Cat# 75276; RRID: AB_1280958 |
| Rabbit polyclonal Anti-INTS9 | Atlas Antibodies | Cat# HPA051615; RRID: AB_2681552 |
| Rabbit polyclonal Anti-INTS4 | Bethyl Laboratories | Cat# A301-296A; RRID: AB_937909 |
| Mouse monoclonal Anti-INTS1 | Millipore | Cat# MABS1984; RRID: AB_2848204 |
| Mouse monoclonal Anti-Lamin A | Active Motif | Cat# 39962; RRID: AB_2615028 |
| Rabbit monoclonal Anti-GAPDH | Cell Signaling Technology | Cat# 5174; RRID: AB_10622025 |
| Rabbit monoclonal Anti-RPB1 NTD | Cell Signaling Technology | Cat# 14958; RRID: AB_2687876 |
| Rabbit polyclonal Anti-RNA pol II CTD phospho S2 - RPB1 Ser-2 |
Abcam | Cat# ab5095; RRID: AB_304749 |
| Rabbit polyclonal Anti-RNA pol II CTD phospho S5 - RPB1 Ser-5 |
Abcam | Cat# ab5131; RRID: AB_449369 |
| Rat monoclonal Anti-RNA Pol II CTD phospho Tyr1 - RPB1 Tyr-1 |
Active Motif | Cat# 61383; RRID: AB_2793613 |
| Rabbit polyclonal Anti-NELF-E | Santa Cruz Biotechnology | Cat# sc-377052; NA |
| Rat monoclonal Anti-SPT5 | Millipore | Cat# MABE1803; RRID: AB_2848205 |
| Rabbit polyclonal Anti-Histone H3K4me1 | Abcam | Cat# ab8895; RRID: AB_306847 |
| Rabbit polyclonal Anti-Histone H3K4me3 | Diagenode | Cat# pAb-003-050; RRID: AB_2616052 |
| Rabbit polyclonal Anti-Histone H3K36me3 | Abcam | Cat# ab9050; RRID: AB_306966 |
| Rabbit polyclonal Anti-Histone H3K27ac | Diagenode | Cat# C15410196; RRID: AB_2637079 |
| Goat anti-Rat IgG Secondary Antibody, Alexa Fluor 568 |
Thermo Fisher Scientific | Cat# A11077; RRID: AB_2534121 |
| Goat anti-Mouse IgG Secondary Antibody, Janelia Fluor 646 |
Novus Biologicals | Cat# NBP1-72739JF646; RRID: AB_2857337 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Turbo DNase | Invitrogen | cat# AM1907 |
| Biotin-11-NTPs | Perkin Elmer | Cat# NEL54(2/3/4/5)001 |
| Dynabeads M-280 streptavidin | Invitrogen | Cat# 11205 |
| Cross-link Gold | Diagenode | Cat# C01019027 |
| Critical Commercial Assays | ||
| NEBNext Ultra II DNA library prep kit | NEB | Cat# E7645S |
| Truseq Stranded Total RNA library prep kit | Illumina | Cat# 20020596 |
| SMARTer smRNA-seq kit | Takara | Cat# 635030 |
| Nextera DNA Library Prep Kit | Illumina | Cat# FC-121-1030 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE125534 |
| Raw and analyzed data | This paper | GEO: GSE125535 |
| Experimental Models: Cell Lines | ||
| HeLa shGFP | Gardini et al., 2014 | N/A |
| HeLa shINTS11 | Gardini et al., 2014 | N/A |
| HeLa shINTS11 - FLAG - E203Q - INTS11 | This paper | N/A |
| HeLa shINTS11 - FLAG - WT-INTS11 | This paper | N/A |
| Recombinant DNA | ||
| Tet-pLKO-puro vector | addgene | Cat# 21915 |
| Tet-pLKO-neo vector | addgene | Cat# 21916 |
| Cumate-pLenti-Cloning-2A-GFP vector | ABM inc. | Cat# iCu003 |
| Software and Algorithms | ||
| Cutadapt v1.14 | Martin 2011 | N/A |
| Trimmomatic v0.32 | Bolger et al., 2014 | N/A |
| Bowtie v1.1.2 | Langmead et al., 2009 | N/A |
| BEDTools v2.28 | Quinlan and Hall, 2010 | N/A |
| STAR v2.5.3a | Dobin et al., 2013 | N/A |
| deepTools2 | Ramírez et al., 2016 | N/A |
| MACS2 v2.1.2 | Zhang et al., 2008 | N/A |
| Homer annotatePeaks v4.9.1-5 | Heinz et al., 2010 | N/A |
| ATAC-seq pipeline | https://github.com/kundajelab/atac_dnase_pipelines | N/A |
| NucleoATAC | Schep et al., 2015 | N/A |
| RSEM v1.2.31 | Li and Dewey, 2011 | N/A |
| DESeq2 | Love et al., 2014 | N/A |
| RECLU clustering | Ohmiya et al., 2014 | N/A |
| Prism v7.0c | GraphPad | N/A |
| Clus-Doc MATLAB R2018b | Pageon et al., 2016 | N/A |
| MATLAB R2018b | https://www.mathworks.com/products/new_products/release2018b.html | N/A |
| Nikon Elements Analysis 5.02.01 | Nikon | N/A |
Highlights.
Integrator promotes transcription elongation at human genes
The RNA endonuclease of Integrator subunit 11 is critical for gene activation
Integrator functions at human genes decorated with H3K4me3 and H3K36me3
ACKNOWLEDGMENTS
We thank the Shiekhattar lab for constructive discussions and suggestions for experimental design. We thank Lucia Speroni for technical help. We thank the Oncogenomics core facility at Sylvester Comprehensive Cancer Center for performing high-throughput sequencing. This work was supported by funding from the University of Miami Miller School of Medicine, the Sylvester Comprehensive Cancer Center, and the National Institutes of Health (grants R01 GM078455, GM105754, and DP1 CA228041 to R.S.). Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award number P30CA240139. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107917.
REFERENCES
- Albrecht TR, Shevtsov SP, Wu Y, Mascibroda LG, Peart NJ, Huang KL, Sawyer IA, Tong L, Dundr M, and Wagner EJ (2018). Integrator subunit 4 is a ‘Symplekin-like’ scaffold that associates with INTS9/11 to form the Integrator cleavage module. Nucleic Acids Res. 46, 4241–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillat D, Hakimi MA, Näär AM, Shilatifard A, Cooch N, and Shiekhattar R (2005). Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase II. Cell 123, 265–276. [DOI] [PubMed] [Google Scholar]
- Bintu L, Ishibashi T, Dangkulwanich M, Wu YY, Lubkowska L, Kashlev M, and Bustamante C (2012). Nucleosomal elements that control the topography of the barrier to transcription. Cell 151, 738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan HL, Beckedorff F, Zhang Y, Garcia-Huidobro J, Jiang H, Colaprico A, Bilbao D, Figueroa ME, LaCava J, Shiekhattar R, and Morey L (2018). Polycomb complexes associate with enhancers and promote oncogenic transcriptional programs in cancer through multiple mechanisms. Nat. Commun 9, 3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FX, Smith ER, and Shilatifard A (2018). Born to run: control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol 19, 464–478. [DOI] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Martins AL, Danko CG, Waters CT, Siepel A, and Lis JT (2014). Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet 46, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebmeier CC, Erickson B, Allen BL, Allen MA, Kim H, Fong N, Jacobsen JR, Liang K, Shilatifard A, Dowell RD, et al. (2017). Human TFIIH kinase CDK7 regulates transcription-associated chromatin modifications. Cell Rep. 20, 1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy J, Vallur AC, Varma S, Liu H, Reinhold WC, Pommier Y, and Maizels N (2011). G4 motifs correlate with promoter-proximal transcriptional pausing in human genes. Nucleic Acids Res. 39, 4975–4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff S, O’Reilly D, Chapman RD, Taylor A, Tanzhaus K, Pitts L, Eick D, and Murphy S (2007). Serine-7 of the RNA polymerase II CTD is specifically required for snRNA gene expression. Science 318, 1777–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod ND, Henriques T, Huang KL, Tatomer DC, Wilusz JE, Wagner EJ, and Adelman K (2019). The Integrator complex attenuates promoter-proximal transcription at protein-coding genes. Mol. Cell 76, 738–752.e737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson B, Sheridan RM, Cortazar M, and Bentley DL (2018). Dynamic turnover of paused Pol II complexes at human promoters. Genes Dev. 32, 1215–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardini A, Baillat D, Cesaroni M, Hu D, Marinis JM, Wagner EJ, Lazar MA, Shilatifard A, and Shiekhattar R (2014). Integrator regulates transcriptional initiation and pause release following activation. Mol. Cell 56, 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineagedetermining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques T, Scruggs BS, Inouye MO, Muse GW, Williams LH, Burkholder AB, Lavender CA, Fargo DC, and Adelman K (2018). Widespread transcriptional pausing and elongation control at enhancers. Genes Dev. 32, 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermüller J, Hofacker IL, et al. (2007). RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316, 1484–1488. [DOI] [PubMed] [Google Scholar]
- Kim TK, and Shiekhattar R (2015). Architectural and functional commonalities between enhancers and promoters. Cell 162, 948–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B,and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs AR, Imanci D, Hoerner L, Gaidatzis D, Burger L, and Schubeler D (2017). Genome-wide single-molecule footprinting reveals high RNA polymerase II turnover at paused promoters. Mol. Cell 67, 411–422 e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak H, Fuda NJ, Core LJ, and Lis JT (2013). Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339, 950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F, Gardini A, Zhang A, and Shiekhattar R (2015). Integrator mediates the biogenesis of enhancer RNAs. Nature 525, 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, and Lis JT (2016). Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc 11, 1455–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J 17, 10–12. [Google Scholar]
- Mayer A, Heidemann M, Lidschreiber M, Schreieck A, Sun M, Hinter mair C, Kremmer E, Eick D, and Cramer P (2012). CTD tyrosine phosphorylation impairs termination factor recruitment to RNA polymerase II. Science 336, 1723–1725. [DOI] [PubMed] [Google Scholar]
- Murata M, Nishiyori-Sueki H, Kojima-Ishiyama M, Carninci P, Hayashizaki Y, and Itoh M (2014). Detecting expressed genes using CAGE. Methods Mol. Biol 1164, 67–85. [DOI] [PubMed] [Google Scholar]
- Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, Zeitlinger J, and Adelman K (2007). RNA polymerase is poised for activation across the genome. Nat. Genet 39, 1507–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechaev S, Fargo DC, dos Santos G, Liu L, Gao Y, and Adelman K (2010). Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science 327, 335–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmiya H, Vitezic M, Frith MC, Itoh M, Carninci P, Forrest AR, Hayashizaki Y, and Lassmann T; FANTOM Consortium (2014). RECLU: a pipeline to discover reproducible transcriptional start sites and their alternative regulation using capped analysis of gene expression (CAGE). BMC Genomics 15, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pageon SV, Nicovich PR, Mollazade M, Tabarin T, and Gaus K (2016). Clus-DoC: a combined cluster detection and colocalization analysis for single-molecule localization microscopy data. Mol. Biol. Cell 27, 3627–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: machine learning in Python. J. Mach. Learn. Res 12, 2825–2830. [Google Scholar]
- Peterlin BM, and Price DH (2006). Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 23, 297–305. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, and Young RA (2010). c-Myc regulates transcriptional pause release. Cell 141,432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Díndar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44 (W1), W160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougvie AE, and Lis JT (1988). The RNA polymerase II molecule at the 5′ end of the uninduced hsp70 gene of D. melanogaster is transcriptionally engaged. Cell 54, 795–804. [DOI] [PubMed] [Google Scholar]
- Saunders A, Core LJ, and Lis JT (2006). Breaking barriers to transcription elongation. Nat. Rev. Mol. Cell Biol 7, 557–567. [DOI] [PubMed] [Google Scholar]
- Schep AN, Buenrostro JD, Denny SK, Schwartz K, Sherlock G, and Greenleaf WJ (2015). Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res. 25, 1757–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N, Maqbool MA, Yahia Y, El Aabidine AZ, Esnault C, Forne I, Decker TM, Martin D, Schuller R, Krebs S, et al. (2018). Tyrosine-1 of RNA polymerase II CTD controls global termination of gene transcription in mammals. Mol. Cell 69, 48–61.e46. [DOI] [PubMed] [Google Scholar]
- Shao W, and Zeitlinger J (2017). Paused RNA polymerase II inhibits new transcriptional initiation. Nat. Genet 49, 1045–1051. [DOI] [PubMed] [Google Scholar]
- Stadelmayer B, Micas G, Gamot A, Martin P, Malirat N, Koval S, Raffel R, Sobhian B, Severac D, Rialle S, et al. (2014). Integrator complex regulates NELF-mediated RNA polymerase II pause/release and processivity at coding genes. Nat. Commun 5, 5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steurer B, Janssens RC, Geverts B, Geijer ME, Wienholz F, Theil AF, Chang J, Dealy S, Pothof J, van Cappellen WA, et al. (2018). Live-cell analysis of endogenous GFP-RPB1 uncovers rapid turnover of initiating and promoter-paused RNA polymerase II. Proc. Natl. Acad. Sci. USA 115, E4368–E4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taft RJ, Glazov EA, Cloonan N, Simons C, Stephen S, Faulkner GJ, Lassmann T, Forrest AR, Grimmond SM, Schroder K, et al. (2009). Tiny RNAs associated with transcription start sites in animals. Nat. Genet 41, 572–578. [DOI] [PubMed] [Google Scholar]
- Tatomer DC, Elrod ND, Liang D, Xiao MS, Jiang JZ, Jonathan M, Huang KL, Wagner EJ, Cherry S, and Wilusz JE (2019). The Integrator complex cleaves nascent mRNAs to attenuate transcription. Genes Dev. 33, 1525–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves SS, Weber CM, and Henikoff S (2014). Transcribing through the nucleosome. Trends Biochem. Sci 39, 577–586. [DOI] [PubMed] [Google Scholar]
- Voong LN, Xi L, Sebeson AC, Xiong B, Wang JP, and Wang X (2016). Insights into nucleosome organization in mouse embryonic stem cells through chemical mapping. Cell 167, 1555–1570.e1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber CM, Ramachandran S, and Henikoff S (2014). Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol. Cell 53, 819–830. [DOI] [PubMed] [Google Scholar]
- Wu Y, Albrecht TR, Baillat D, Wagner EJ, and Tong L (2017). Molecular basis for the interaction between Integrator subunits IntS9 and IntS11 and its functional importance. Proc. Natl. Acad. Sci. USA 114, 4394–4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto J, Hagiwara Y, Chiba K, Isobe T, Narita T, Handa H, and Yamaguchi Y (2014). DSIF and NELF interact with Integrator to specify the correct post-transcriptional fate of snRNA genes. Nat. Commun 5, 4263. [DOI] [PubMed] [Google Scholar]
- Yue J, Lai F, Beckedorff F, Zhang A, Pastori C, and Shiekhattar R (2017). Integrator orchestrates RAS/ERK1/2 signaling transcriptional programs. Genes Dev. 31, 1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger J, Stark A, Kellis M, Hong JW, Nechaev S, Adelman K, Levine M, and Young RA (2007). RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat. Genet 39, 1512–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data generated in this study is made available at the Gene Expression Omnibus (GEO). The accession number for the raw and processed data reported in this paper is GEO: GSE125534, GSE125535