

SUMMARY
The first cultivated representative of the enigmatic phylum Saccharibacteria (formerly TM7) was isolated from humans and revealed an ultra-small cell size (200–300 nm), a reduced genome with limited biosynthetic capabilities, and a unique parasitic lifestyle. TM7x was the only cultivated member of the candidate phyla radiation (CPR), estimated to encompass 26% of the domain Bacteria. Here we report on divergent genomes from major lineages across the Saccharibacteria phylum in humans and mammals, as well as from ancient dental calculus. These lineages are present at high prevalence within hosts. Direct imaging reveals that all groups are ultra-small in size, likely feeding off commensal bacteria. Analyses suggest that multiple acquisition events in the past led to the current wide diversity, with convergent evolution of key functions allowing Saccharibacteria from the environment to adapt to mammals. Ultra-small, parasitic CPR bacteria represent a relatively unexplored paradigm of prokaryotic interactions within mammalian microbiomes.
Graphical Abstract
In Brief
McLean et al. show that humans are inhabited by a broad diversity of nanosized bacteria with highly reduced genomes within the Saccharibacteria phylum. They are related to the candidate phyla radiation predominately found in the environment. Saccharibacteria show adaptations and diversification in mammals during their transition from the environment.
INTRODUCTION
The “Candidatus Saccharibacteria” phylum, designated initially as candidate division TM7, has long been an enigma, having remained uncultivated for nearly two decades since the first SSU ribosomal sequence was initially recovered (Rheims et al., 1996). We recently discovered (He et al., 2015), through directed cultivation from human oral samples, that a member of the phylum has an extremely reduced genome (705 CDS), has an ultra-small cell size (200–300 nm), and replicates as an obligate epibiont on the surface of another commonly found oral commensal species, a subspecies of Actinomyces odontolyticus (McLean et al., 2016). Strain TM7x (proposed designation Ca. Nanosynbacter lyticus TM7x) also exhibits what is now defined as a parasitic phase during which it disrupts the bacterial host cell (basibiont), resulting in cell lysis (He et al., 2015). However, unattached TM7x cells remain viable and have been shown to re-infect new bacterial host cells when available (Bor et al., 2018). This discovery and co-culture marked the first concrete evidence of how these ultra-small organisms survive and persist despite missing capacities for de novo biosynthesis of many essential compounds, including all amino acids and vitamins. This model dual-species system has now allowed a deeper understanding of host-bacterium dependence and dynamics in this unique bacteria-bacteria relationship (Bedree et al., 2018; Bor et al., 2016, 2018).
After the first 16S rRNA gene sequence was published, TM7 was designated as a candidate phylum (Hugenholtz et al., 1998), and the first genomic insights were derived from partial genomes (Marcy et al., 2007; Podar et al., 2007), with the history having been recently reviewed (Bor et al., 2019). Candidatus Saccharibacteria is the recently proposed (Albertsen et al., 2013), unofficial, but now generally accepted name for this lineage. For brevity, we will use the term “Saccharibacteria.” In addition to being the only cultivated member of the Saccharibacteria phylum at that time, strain TM7x was reported as the only cultivated member of a recently discovered group of related bacteria known as the candidate phyla radiation (CPR) (Figures 1A and 1B; Brown et al., 2015; Hug et al., 2016). The CPR is a description of a large monophyletic radiation of phyla and superphyla that includes the group currently referred to as Patescibacteria (Castelle and Banfield, 2018), although a consensus on taxonomic classifications of this lineage as a phylum or class within the domain Bacteria is still controversial (Parks et al., 2018). Currently, only three members of the CPR are routinely detected in the human body, “Candidatus Gracilibacteria” (GN02), “Ca. Absconditabacteria” (SR-1), and Saccharibacteria.
Figure 1. Expanded Coverage of the Saccharibacteria Phylum.

(A) Current view of the tree of life highlighting the Saccharibacteria and candidate phyla radiation (tree inferred from concatenated ribosomal gene dataset provided by Hug et al., 2016).
(B) Phylum-level maximum likelihood concatenated ribosomal gene tree of the CPR indicating that Saccharibacteria share a common ancestor with “Ca. Berkelbacteria” and “Ca .Kazan” groups.
(C) Fast tree 16S rRNA gene phylogeny of the major Saccharibacteria groups derived from public ribosomal databases and available genomes. The complete tree is available in a circular format with full bootstrap values in Figure S1 and in Newick format in Data S1.
(D and E) Phylogenetic relationships within the Saccharibacteria phylum using maximum likelihood 16S rRNA and concatenated ribosomal marker gene inference. (D) Maximum likelihood 16S rRNA gene phylogeny expanded tree, sequences from the Human Oral Microbiome Database, and those extracted from genomic assemblies are shown. (E) Concatenated ribosomal RAxML gene tree for new and previously published complete and draft genomes including first representative sequences from the G5 (Ca. Nanoperiomorbus periodonticus), G6 (Ca. Nanogingivalis gingivalicus), and G3 (Ca. Nanosyncoccus nanoralicus, Ca. Nanosyncoccus alces) lineages. Purple filled star and purple filled circle are assemblies from this study. Green filled hexagon and empty circle are assemblies from previous studies. Genomic data source material are indicated with icons. Genome properties and names for the provisionally proposed novel classes and orders are explained in Tables S1 and S2.
Although TM7x was isolated from the human oral cavity, Saccharibacteria/TM7 are widespread in their distribution. The human oral Saccharibacteria have been reported to be associated with inflammatory mucosal diseases (Brinig et al., 2003; Fredricks et al., 2005; Kuehbacher et al., 2008). With few sequenced genomes and the very recent confirmation that most members of the CPR contain only a single copy of the 16S rRNA gene, they may be more difficult to detect than species with multiple 16S rRNA genes. Furthermore, without 16S copy number correction used in these earlier studies, their relative abundance will appear lower than that of other species that contain multiple copies. Their potential role in disease pathogenesis could, therefore, be more important than previously recognized.
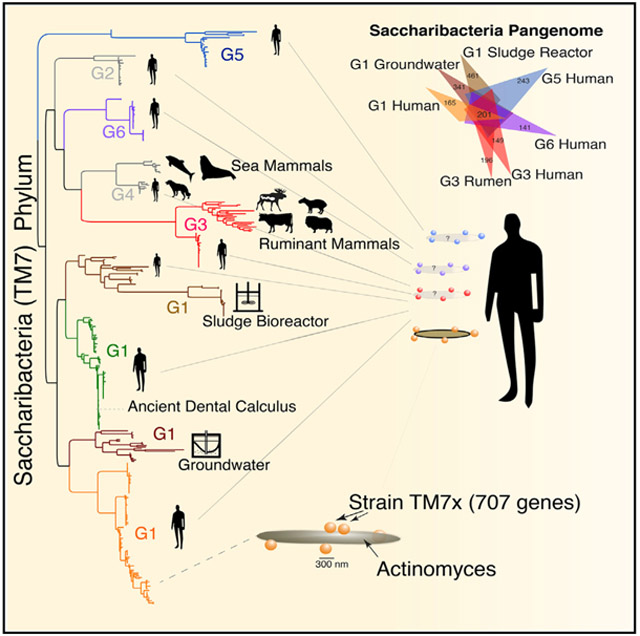
Despite being found in many environments and part of the human microbiome, recognition that Saccharibacteria are ultra-small, parasitic bacteria with reduced genomes and that they are phylogenetically related to the CPR is a very recent discovery. We currently lack information on their genomic diversity, ecology, and evolution as well as their role in human health and disease. Diverse, full-length 16S rRNA sequences have only recently become available to various databases. Cumulative curation and phylogenetic analysis of the available 16S rRNA gene sequences indicated at least six distinct lineages of Saccharibacteria sequences, currently designated groups G1 through G6. Importantly, at the time of this study, the only genomes available belong to group G1. Metagenomic studies, therefore, have been limited by a complete lack of genomes outside of the highly related G1 group (He et al., 2015), currently composed of both mammalian host-associated (MHA) and environmental representatives recovered from activated sludge bioreactors and groundwater. To date, 16S rRNA gene surveys and metagenomics studies therefore have not yet been able to resolve the members in these samples to the taxonomic level of the individual groups. Recognizing that there is an immediate need to describe and determine the encoded metabolic and virulence potential of the Saccharibacteria groups, we specifically sought to capture reference genomes from all of the MHA groups in order to cover the spectrum of known diversity within the phylum. Here we report that the G3, G5, and G6 groups are highly prevalent in humans and, like G1 group strain TM7x, are also ultra-small bacteria with reduced genomes. Despite having streamlined genomes with low intergenic spacing, there is remarkable genomic variation across the phylum. Our results suggest that key adaptations within different Saccharibacteria groups may have occurred during the transition from the environment to the eventual survival and persistence within human hosts. Collectively, our analyses indicate that mammals likely acquired many of these groups through independent acquisition events throughout history, which has resulted in high diversity in humans with several adaptations that are likely to have occurred in a convergent manner.
RESULTS
Phylum Saccharibacteria Genomes Recovered beyond the G1 Group
Current knowledge of the diversity in the Saccharibacteria/TM7 phylum has been limited to 16S rRNA gene SSU sequences and genomes from only the G1 group. However, using available SSU sequences (Figure 1C), the inferred tree reveals additional diverse members designated G1–G6; these lineages are monophyletic with branching sub-lineages. Major sources of the sequences are highlighted in a condensed SSU tree (Figure 1C), showing the mixed environmental and host-associated sources previously observed in the G1 group. The G1 group, to which the cultivated strain TM7x belongs, has closely related members from both environmental sources and the human microbiome. Intriguingly, mammalian rumen and human oral sources formed distinct related lineages within the larger G3 group. To expand genomic knowledge across the entire phylum, we focused on finding and assembling sequences for the missing uncultivated, typically low-abundance groups outside of G1, as well as additional G1 genomes to expand on this group. Assembled scaffolds derived from single samples across various sources were binned into genomes. This work resulted in new genomic data for MHA groups G3, G5, and G6 (Table S1; includes a detailed summary of genome bin sources, curation, and properties). An inferred maximum likelihood 16S rRNA gene tree (Figure S1; Data S1 as a Newick tree file) incorporating sequences in existing databases, as well as extracted 16SrRNA gene sequences from new and previously available assemblies, highlights the expanded coverage and sources of these different groups within this phylum.
Two genomes for the G5 group, the most distantly related by SSU sequencing (“Ca. Nanoperiomorbus periodonticus”; TM7_EAM_G51 and G52) (Table S2), as well as an additional G1 bin (“Ca. Nanosynsacchari sp.”; TM7_G1_3_12lb), were derived from patients with severe periodontitis (McLean et al., 2015). Human Microbiome Project (HMP) datasets (1,149 assemblies, 15 body sites, ~3.5 Tb) were examined for 16S rRNA gene hits to the CPR. Those samples with relatively long contigs were then re-assembled and binned, which yielded a representative G6 genome (“Ca. Nanogingivalis gingivalicus”; TM7_CMJM_G61) from a keratinized gingiva metagenomic sample. A G3 oral genome bin (“Ca. Nanosyncoccus nanoralicus”; TM7_KMM_G31) was recovered from oral-derived lab enrichment cultures by sequencing and genome binning, similar to the approach used for isolating TM7x. A second G3 genome (“Ca. Nanosyncoccus alces”; TM7_G3_Rum) was recovered by mining a metagenomic sample derived from moose rumen. Finally, an ancient G1 group genome (“Ca. Nanosynsacchari sp.”; ANC 38.39) was derived from whole-DNA shotgun sequencing performed on dental calculus from adult human skeletons with evidence of mild to severe periodontal disease (B61 skeleton; ca. 950–1200 CE) (Warinner et al., 2014). CPR members all share reduced genomes and are also missing many single-copy marker genes used to estimate genome completeness within such tools as CheckM (Parks et al., 2015; Table S1 and S3).
Unexplored Diversity of the Phylum Saccharibacteria
We further investigated the phylogenetic and phylogenomic relatedness across the phylum and placed all new genomes in relation to existing sequences. Thus, we compared the SSU gene tree, collapsed into major groups (Figure 1D), with a single-copy marker gene tree generated from a concatenation of 42 genes from the individual Saccharibacteria genomes (Figure 1E). For the majority of the groups, the 16S rRNA gene tree was highly congruent with the marker gene tree. In addition, genome-wide pairwise average amino acid identity (AAI) analysis between orthologous genes was performed across all genomes along with the outgroups (Figure 2A; Table S4). These results were consistent with the phylogenetic relationships observed in the SSU and concatenated ribosomal gene trees (Figures 1D and 1E). Sets of gene orthologs among the Saccharibacteria (15 genomes, 11,615 total genes) were then determined. Consistent with a wide genetic variation at the amino acid level across the Saccharibacteria phylum, AAI within and between groups was generally low, the highest being within the G1. A more detailed look comparing each AAI pair between human oral strain TM7x and the other genomes highlighted the range of gene identity found across the groups as well as the similarities between the oral and environmental genomes in the G1 group (Figure 2B). In agreement with the pairwise AAI values, the pangenome analysis highlighted the surprisingly large genetic variation across this phylum, with a range of 149, 141, and 243 new genes added from the G3, G5, and G6 assemblies, respectively (Figure 2C). Comparing representative genomes of each group with the environmental G1 genomes yielded a set of 201 shared core genes (Figure 2C; Table S6) between human oral and environmental sources. Comparing only MHA oral groups produced a core of 208 genes. A total of 777 genes were found to be unique within the oral genomes. The diversity across the phylum, and steady discovery of new genes with each additional genome (Figure 2D), indicates that the Saccharibacteria pangenome remains open at this stage of genomic discovery.
Figure 2. Unexplored Diversity of the Phylum Saccharibacteria.

(A) Relationships between phylogeny with average amino acid identities and number of shared orthologous genes between Saccharibacteria and other divergent phyla highlight the small number of orthologous genes and percentage homology between groups in these reduced genomes (percentage identity cutoff ≥ 20%). Each row is a genome, and each circle is the AAI value between a pair of genomes colored by the genome in comparison. Size of the circle indicates the number of orthologous genes for the pair. (Full table available in Table S4.)
(B) Comparing the percentage identities of the different genomes with the genes found in the cultivated G1 oral strain TM7x, highlighting the overall distribution of amino acid identities across the genome, with higher average percentage identities with the environmental G1 genomes than other oral-derived groups outside of G1. Each dot is an amino acid identity value for the best hit protein in the corresponding genome.
(C) Pangenome analysis of environmental and mammalian host-associated (MHA) groups (excluding the G3 rumen genome). Oral genome comparisons only using the available genomes from each group (G1, G3, G5, and G6) share 208 core genes, with unique genes ranging from 159 to 267 (Tables S5 and S6).
(D) Number of new and unique genes as genomes are added to the Saccharibacteria pangenome.
In order to begin to bring taxonomic structure to this currently uncharacterized phylum, we used pairwise average values of the amino acid identities and phylogenetic relatedness of these genomesto propose provisional taxonomic rankings and indication of potential type material for the Saccharibacteria phylum members among the recognized lineages at this time (Table S2; Data S2). Previous studies have examined and proposed the use of AAI to determine taxonomic rank, with <45% AAI the cutoff for a prokaryotic phylum (Luo et al., 2014; Rodriguez-R et al., 2018), which is consistent with the AAI found between the Saccharibacteria and the CPR phyla outgroups in the present study. AAI between G5 and the other groups was less than 55%, with the fraction of shared genes reaching only 45% of the genome, and therefore was consistent with a class-level representative designated “Candidatus Nanoperiomorbia class nov.” (Figure 2A; Table S4). Intriguingly, mammalian rumen and human oral sources formed distinct lineages within the monophyletic G3 group, even though closely related (56% of genes shared at 63% AAI). The oral and rumen G3 could represent a new class (“Ca. Nanosyncoccalia class nov.”) because of the low AAI and low fraction of shared genes compared with other groups in the phylum. Similarly, G6, with a slightly higher AAI but similar fraction of shared genes with other groups, was conservatively placed within the “Ca. Nanosyncoccalia” class and was considered a representative member of a new order (“Ca. Nanogingivales ord. nov.”).
We propose that the Saccharimona, a class-level designation based on the previously proposed phylum name (Table S2), include the published G1 genomes from environmental sources, including deep subsurface groundwater (RAAC3, GWC2) and sludge bioreactor (“Ca. Saccharimonas aalborgensis”), as well as oral genomes from single-cell approaches and the cultivated strain TM7x. “Ca. Saccharimona” forms two possible orders, “Ca. Saccharimonales,” containing both oral and environmental representatives, and “Ca. Nanosynbacterales ord. nov.,” which is monophyletic and populated with oral genomes. However, it remains unclear if these environmental genomes are a coherent group. Some variation was observed when comparing the SSU and concatenated marker genes trees. Although all three environmental genomes grouped together in the SSU tree, within the concatenated gene tree, the two groundwater derived members, GWC2 and RAAC3, were together, but “Ca. S. aalborgensis” appeared in a separate lineage. All proposed provisional operational taxonomic classifications are depicted in Figure 1E and presented in Table S2 and Data S2, which describe the provisional etymology and proposals for designation of sequenced representatives as type material at this time.
New Groups from the Human Oral Cavity Are Ultra-small Epibionts
Strain TM7x was discovered to be an ultra-small cell, 200–300 nm in diameter, with an extremely reduced genome, that lives as an obligate epibiont on the surface of an Actinomyces odontolyticus basibiont, providing an explanation for its long-term recalcitrance to cultivation (He et al., 2015). Reduced genomes and limited biosynthetic capacity are common features found across the CPR (Brown et al., 2015). Thus, it has been assumed that many CPR organisms share similar epibiont lifestyles. Given that all of the genomes from G3, G5, and G6 found in the present study, as well as a previous oral-derived genome for CPR phylum SR1 (Campbell et al., 2013), have a reduced genome size, the question arises as to whether having reduced genomes is correlated to being ultra-small in size. Fresh human saliva and tongue surface samples were collected from 12 individuals. Fluorescence in situ hybridization (FISH) results from these confirmed both a small size and propensity to decorate larger host bacteria by the Saccharibacteria (G1, G3, G5, and G6) and SR1 (Figure 3A). As previously observed with the G1 member TM7x, G3 and G6 were bound on long rod-shaped cells, whereas G5 and SR-1 were attached to small cocci, possibly indicating their preferred hosts in vivo. Samples were then filtered through a 0.45 mm filter and again subjected to group-specific PCR probes. Filtered samples were positive for the presence of G3, G5, G6, and SR1 groups. This supported the imaging data and further confirmed ultra-small cell sizes, consistent with the reduced genomes of these bacterial parasites (Figure 3B).
Figure 3. New Groups from the Human Oral Cavity Are Ultra-small Epibionts.

(A) Human saliva and tongue samples were stained using fluorescence in situ hybridization (FISH) probes that specifically targeted the G1, G3, G5, and G6 lineages of TM7 phylum, as well as the SR1 phylum. Column 1 is TM7 staining with lineage-specific probes (red), column 2 is total DNA stain with Syto9 dye (green), and column 3 is merged images (magnified images indicate ultra-small bacteria). All scale bars are 5 μm.
(B) PCR amplification of G3, G5, G6, and SR1 16S rRNA with lineage-specific primers, using template DNA isolated from original saliva samples, saliva supernatants, and filtrate passing through 0.45 μm filters (see STAR Methods) confirming ultra-small cell sizes.
Human Biogeography and Ecology from Neanderthal to Modern Humans
Saccharibacteria (TM7), “Ca. Absconditabacteria” (SR1), and “Ca. Gracilibacteria” (GN02) are unique among the lineages of the CPR in that they have been detected in mammalian body sites. The overall prevalence and distribution of these parasitic bacteria with reduced genomes within body sites from human subjects is an intriguing question. Identifying the presence of Saccharibacteria but not resolving which class, order, or species levels were present has limited our understanding. With the availability of new sequences, the oligotyping results from HMP datasets (Eren et al., 2014) were re-analyzed using the best hit to some of the known Saccharibacteria groups across nine oral cavity sampling sites and stool (Figure S2A). The distribution varied significantly among phylum members, though none showed appreciable levels in stool. Although sequencing depth is generally higher with amplicon sequencing, short SSU rRNA gene sequences have lower phylogenetic resolution. Therefore, we examined each Saccharibacteria group as well as GN02 and SR-1 CPR members’ distribution and prevalence across body sites using the HMP metagenome assemblies (Figure 4A; Figure S2B). Saccharibacteria and CPR lineages were first identified as present within the assembled contigs using hits to the SSU rRNA gene (>300 bp cutoff). We found that the GN02 and SR-1 distributions were predominant in the oral cavity, with few hits in other body sites (Figure S2B). Collectively, using the HMP assemblies, the Saccharibacteria phylum showed presence in 20%–89% of samples across the oral cavity sites, including 84% of the supragingival plaque metagenomes of healthy humans and 89% of the tongue dorsum metagenomes. In particular, sequences related to strain TM7x were found at a prevalence of 59% in supragingival plaque and 61% in tongue dorsum samples from subjects. Outside of the human digestive tract, there is evidence for the presence of the Saccharibacteria phylum on skin (retroauricular crease), mid-vagina, and in stool, although with much lower prevalence. Notably, Saccharibacteria G5 was highly represented in the posterior fornix of the vagina, while GN02 was detected in the mid-vagina body site (Figure S2B).
Figure 4. Human Biogeography and Ecology from Neanderthal to Modern Humans.

(A) Prevalence and distribution of Saccharibacteria groups from 16S rRNA gene hits within body site-specific assembled contigs from the Human Microbiome Project (16S rRNA gene > 300 bp hit cutoff, n = 1,100 assemblies).
(B) Total hit distributions across body sites (Figure S2 includes supporting oligotyping data for these groups and additional SR-1 and GN02 group distributions).
(C) Percentage of genome coverage from mapped metagenomic read sets to genome assemblies from this study and previously available G1 assemblies for Saccharibacteria groups from ancient Neanderthal dental calculus (48,000 years) through to modern body sites in health and disease. Neanderthal calculus from the best preserved Neanderthal, El Sidrón 1, which suffered from a dental abscess, is included, as well as SPYNEWWL8 9 (SPYI), which had extensive DNA damage (Table S7).
In the present study, we provide increased phylogenomic resolution within the Saccharibacteria phylum by identifying new genomic references within the diverging Saccharibacteria groups G1, G3, G5, and G6. Thus, we sought to re-investigate the human biogeography and ecology of these CPR lineages. Samples from Neanderthal and modern humans were analyzed by mapping shotgun reads from available datasets to the available reference genomes in order to determine the percentage of each genome recovered, an indication of presence and a proxy for prevalence among different body sites (Figure 4C). Analysis showed that multiple Saccharibacteria groups were present even in ancient humans. Neanderthal dental calculus, estimated at 48,000 years, allowed the detection of G5 and many G1, though with little to no coverage of G3, G6, or SR-1 (Figure 4C; Table S7). Coverage statistics on the reference genome of N. lyticus strain TM7x alone were first reported in the original study (Weyrich et al., 2017). Our additional analyses have now indicated high genome coverage of G3 and SR1 in medieval dental calculus (~1200 CE).
The Neanderthal and medieval samples were mineralized calculus formed from supragingival plaque. The high coverage of G1 groups in these samples was consistent with the high G1 presence in modern supragingival plaque. The presence of the G5 group, with the genome recovered from severe periodontitis metagenomic samples (McLean et al., 2015), also confirmed that it has long been part of the human oral microbiome. Multiple lines of evidence now strongly confirm that all major groups of the Saccharibacteria are highly prevalent in humans and should be considered part of the core oral microbiome.
Saccharibacteria Epibionts Are Unique Compared with Other Host-Dependent Bacteria
Current knowledge of the physiology and functions of these ultra-small parasites of bacteria from the CPR is extremely limited given the fact that at the time of this study, N. lyticus strain TM7x, from the oral G1 group, is the only cultivated and physiologically characterized member from the CPR. In order to explore the diversity and functional capacity within the Saccharibacteria, and to compare these groups with other known bacteria, we examined the gene functions using Clusters of Orthologous Groups (COGs). Merhej et al. (2009) previously reported on the functional differences in COGs among free-living bacteria, facultative host-associated (FHA) bacteria, obligate intracellular (OI) parasites, and OI mutualists. We performed a similar analysis, including the Saccharibacteria and select CPR genomes (Figures 5A and 5B; Table S8). These CPR epibionts turned out to be quite distinct from traditional parasites and mutualists. There was a clear separation of functional capacity between the epibiont lifestyle and previously investigated host-associated organisms, both FHA and free-living (Figure 5A). For the most part, members of the CPR were tightly clustered with the exception of the G5s, consistent with their deeper branching phylogeny (Figure 1C). Overall, the select CPR members and Saccharibacteria clustered most closely to the OI bacteria, which include both OI parasites and OI mutualists. Saccharibacteria contain a higher proportion of genes that encode defense, replication, recombination, and repair mechanisms than free-living or any of the other host-associated groups (Figure 5B). Interestingly, there were distinct differences between environmental G1 genomes and those from oral and rumen samples. Environmental G1s showed a higher proportion of genes for cell wall, membrane, and envelope biogenesis compared with MHA Saccharibacteria. In contrast, oral and rumen Saccharibacteria had less capacity for nucleotide transport and metabolism than observed within the environmental G1s or the intracellular mutualists or parasites. These results are consistent with the genome reduction process expected when moving from the environment to mammalian host association, indicating that there may be distinct biological changes between the two groups.
Figure 5. Comparative Analyses of Predicted Gene Functions and Conservation of T4SS with Novel Secreted Protein Array.

(A) Principal-component analysis (PCA) clustering of COGs distribution percentages averaged across oral and environmental Saccharibacteria groups for functionally annotated genes within free-living, facultative host-associated, and host-dependent as well as obligate intracellular (mutualistic and parasitic) bacteria.
(B) COGs distribution percentage across genomes for functionally annotated genes within these different lifestyle groups compared with environmental and oral Saccharibacteria groups (additional analyses in Table S8 and Figure S5). Rows are centered and unit variance scaled.
(C) Both environmental and mammalian-associated Saccharibacteria genomes maintained a region containing homologs of the type IV secretion system (VirB4, B6, and B2) and a novel array of small hypothetical proteins (4–6) containing N-terminal signal peptides targeting them for secretion through the sec pathway. This unique array of proteins was found across other reduced bacterial genomes and CPR (Figure S3).
(D) The arrangement of the six small secreted proteins in Nanosynbacter lyticus strain TM7x and T4SS homologs compared with known T4SS systems revealed similarities to the ptx/ptl locus of Bordetella pertussis. The effector proteins secreted by the Ptl machinery in B. pertussis form a large complex called the pertussis toxin (Ptx).
(E) Gapped alignment of pertussis toxin S1 protein with four of the six proteins in the TM7x array. The Ptx subunit, similar to the four TM7x proteins, has an N-terminal signal sequence and is translocated across the inner membrane by the general SecYEG secretory pathway.
Maintenance of Machinery for Parasitism
Despite lacking functional capacity for de novo biosynthesis of all essential amino acids and most vitamins, these diverse groups of bacteria have managed to persist in many environments. We therefore closely examined common features for transport machinery that should be maintained within Saccharibacteria in order to sustain their parasitic lifestyle and persist in the oral and groundwater environments. The COGs analysis revealed a large proportion of genes involved with defense mechanisms, including membrane-associated ABC type transporters (transporter complex LolCDE), involved in the translocation of mature outer membrane-directed lipoproteins, and MacAB, involved in macrolide export. TM7x contains a large percentage of CDS with transmembrane domains (~30%), but with a relatively small proportion of coding regions (<3%, 30 CDS) predicted to have signal peptides targeting them for secretory machinery (He et al., 2015). Through this analysis, a putative type IV secretion system (T4SS) region in the TM7x genome was identified. Further homology searches of this region revealed hits to genes encoding VirB4 and VirB6, which span the cell membrane in Gram-positive bacterial envelopes, as well as the type IV related functional uncharacterized protein Pgrl. This region also contains the competence-related genes encoding the protein Gntx, which is involved with the use of DNA as a substrate in other bacteria (Palchevskiy and Finkel, 2006). This region was also confirmed to be present across a majority of the Saccharibacteria (Figure 5C), other CPR lineages, and lineages with reduced genomes outside the CPR, such as TM6 (Candidatus Dependentiae), an obligate amoebae symbiont (McLean et al., 2013; Yeoh et al., 2016); Figure S3). Bacteria use T4SSs for various purposes: to aid in survival, proliferation in eukaryotic hosts (Gonzalez-Rivera et al., 2016), killing other bacteria such as in Xanthomonas (Souza et al., 2015), and in conjugal transfer of plasmid DNA, transporting a wide range of components, as well as directly injecting effectors into host cells (Bartonella) (Padmalayam et al., 2000).
Of the described T4SS loci, the region within the Saccharibacteria bears the highest resemblance to the characterized T4SS in Bordetella pertussis, which is inherently involved with pertussis toxin secretion. Both Saccharibacteria and B. pertussis have a small gene array with four to six genes that contain coding genes with signal peptidase II (Sec/SPII) signal peptides flanked by T4SS apparatus genes. In B. pertussis, it is believed that these individual pertussis toxin subunits (S1–S5) initially cross the inner membrane through a Sec-like pathway and enter the periplasm, where the leader peptides are removed. In the second step, the assembled toxin traverses the outer membrane with the assistance of a set of nine accessory transport proteins, also known as the Ptl proteins (Farizo et al., 2002). The Ptl proteins belong to a family of T4SSs that are involved in the transport of proteins and/or DNA from bacterial cells (Ptl homologs are shown in Figure 5D). The S1 protein of B. pertussis is a NAD-dependent ADP-ribosyltransferase, which plays a crucial role in the pathogenesis of B. pertussis, disrupting normal host cellular regulation. It has been shown to catalyze the ADP-ribosylation of a cysteine in the alpha subunit of host heterotrimeric G proteins (Fields and Casey, 1997). Alignment of this B. pertussis S1 with four of the six proteins in TM7x highlights the similarity in the arrangement of these signal peptides and domains (Figure 5E). The TM7x_00021 protein (Figure S4A), which bears the highest similarity to B. pertussis S1 (11% AAI), is not likely to have the same exact function in TM7x. At this time, we can only speculate that the TM7x proteins are targeted for export through a T4SS apparatus and on their overall role. This uncharacterized small protein TM7x_00021 seems to be a conserved gene and was identified across the majority of bacterial candidate phyla, mainly the CPR (28%–84% AAI) (Figure S4B). Interestingly, the top hits (8%–21% AAI) to this small protein outside of the CPR were all found in the Actinomycetales order, to which the known TM7x basibiont belongs (Figures S4C and S4D).
In addition to the T4SS, groups G3, G5, and G6 contain a region of homologous genes for type II secretion biosynthesis machinery, whereas TM7x and other G1s include two non-redundant regions. Even though Saccharibacteria are biosynthetically limited because of their reduced genomes, they all have maintained T4SSs with an array of potential secreted proteins. Taking all this evidence into account, we hypothesize these secretion modalities are likely key in the translocation of essential nutrients and/or effectors that may mediate their production and release between Saccharibacteria and their basibiont host.
Lack of Major Genomic Changes between Mammalian Host-Associated G1 and Environmental G1
Uniquely, the Saccharibacteria phylum possesses both MHA and environmental counterparts. This represents an opportunity to investigate the transition of the Saccharibacteria from the environment to human hosts. The phylogenetic relationships from the 16S rRNA and concatenated ribosomal genes strongly support the close relationship of the G1 group members apart from the G2-G6 members, with the G1 group branching earlier (Figure 1E), with further agreement in a tree of 211 concatenated orthologous genes (Figure S5A). Furthermore, there is low shared gene content and variable GC content among the G2–G6 groups in contrast to the range within the G1s. The G1 genomes have a narrow GC content (47% ± 3%), whereas these additional groups showed distinct and highly varied GC, with G5 at the highest (51%), G3 at a slightly lower range (40% for the oral, 43% for the rumen), and G6 at the lowest GC content (32%) in the phylum. When comparing the functional profiles using SEED families, the environmental G1 and oral G1 also clustered together and are most similar to the CPR outgroup Kazan bacteria (Figure S5C). There are logical arguments that the environmental GWC2 and RAAC3 are likely the most related to the ancestral state. The environmental-derived groups possess a larger genome sizes than the oral genomes, which fits the trend of gene reduction seen in bacteria that have become eukaryotic host dependent, such as seen in endosymbionts of insects. Most relevant to the discussion of the closest genomes to the ancestral state, however, is that the GWC2 and RAAC3 G1 group genomes were recovered in deep groundwater within the same samples as the large number of newly discovered reduced genome CPR phyla were also recovered (Brown et al., 2015; Kantor et al., 2013).
One of the most unforeseen aspects of the comparative genomic analysis was the very high genomic synteny maintained across the entire G1 group genomes (Figure 6A). From our comparisons, it is evident that genome reduction is occurring during the transition from the environment to the human oral cavity, but synteny is unexpectedly maintained. This maintenance of synteny is demonstrated by aligning each genomic assembly against the complete reference environmental genome GWC2 (Figure 6A). In contrast, it is evident for the G3, G5, and G6 groups; there is no significant synteny maintained with the G1 genomes (Figure 6A). To our knowledge, there are no other examples of a bacterium that is highly prevalent in human hosts that has an environmental counterpart with so few genetic changes occurring between them.
Figure 6. Mammalian Host and Environmental G1 Are Highly Conserved, and Shared Functions Are Independently Acquired in Mammalian Host-Associated Groups.

(A) All assembled contigs aligned against reference groundwater Saccharibacteria genome GWC2 reveals the synteny maintained across the environmental and human oral G1 group members. Large syntenic blocks are present and maintained in order when comparing full genomes of G1. In contrast, the G3 group containing human oral and rumen members has maintained smaller syntenic blocks with multiple re-arrangements.
(B) A total of 21 unique genes are shared with four or more of the oral genomes and are not found in environmental genomes, which indicates they may have been acquired during mammalian host adaptation. Colored squares indicate presence. Color indicates the phylum with the closest homolog. Several of the unique genes that were potentially acquired show mixed taxonomy across groups, indicating convergent evolution by horizontal gene transfer.
(C) Maximum likelihood phylogenetic protein tree of anaerobic ribonucleoside-triphosphate reductase (NrdD) displaying divergent phylogenetic relatedness among Saccharibacteria groups (full trees in Figure S6).
(D) The L-lactate dehydrogenase gene was identified as being a unique gene among the mammalian associated groups that was not present in the environmental genomes. The taxonomic best hits in (B) and the maximum likelihood protein tree shown here indicate divergent phylogenetic relatedness between the acquired genes, supporting independent acquisition from different bacteria.
In order to further explore the genomic differences between the Saccharibacteria groups and the observed lack of genetic changes within the G1s, we investigated the unique MHA genes shared among the groups but absent in the environmental genomes. A shared set of 22 select genes unique to the MHA groups were compared with known sequences in order to establish the closest taxonomic hit for each gene (Figure 6B). Among these genes, we found that enolase-encoding gene (phosphopyruvate hydratase, COG0148), which is thought to be universally present in bacterial genomes, is found only in the oral groups. The best taxonomic hit for this gene across the oral groups did not extend outside the Saccharibacteria phylum and was not considered a strong candidate for horizontal gene transfer. Several of the other unique MHA genes showed high phylogenetic diversity, which may potentially be indicative of independently acquired gene transfers. For example, the MHA groups have an anaerobic version of the ribonucleoside-triphosphate reductase gene (nrdD) that would enable functioning under the low oxygen conditions in the oral cavity and rumen (Figure 6C). Protein trees were constructed to investigate these genes further. In groups G3 and G5, NrdD phylogenetically clustered together with the Firmicutes phyla from the class Clostridia, but apart from G6, which fell within a distant cluster of predominately Firmicutes phyla from the class Bacilli (Figures 6C and S6). In comparison, the G1 TM7x and S2 G1 NrdD proteins fell within a group of Proteobacteria and Elusimicrobia. An adenosylcobalamin-dependent ribonucleoside-triphosphate reductase can be found in RAAC3 environmental genomes and other members of the CPR, but it is very distant from the anaerobic version (15% AAI) found in MA genomes.
Another notable gene unique to the MHA groups (G5, G3 oral and rumen, and G1 TM7x) within the oral cavity and missing completely in environmental G1 genomes was lactate dehydrogenase (ldh). This is a signature gene found in many oral bacteria that enables the production of a common metabolite found in high abundance in dental plaque. Here, a similar mixture of possible horizontal transfer from disparate groups was found for each of the genes in G1, G3, and G5 (Figures 6D and S6), indicating that varied evolutionary events led to the convergent acquisition of this function.
In addition to single genes unique to MHA groups, CRISPR loci were determined to be divergent between G1 and G5. CRISPR systems are depleted in most CPR (Burstein et al., 2016); however, we discovered cas genes in Saccharibacteria (Figure S7A). G1 12alb and G1 S2 had the gene arrangement (csn2-cas2-cas1-cas9), indicative of class 2 subtype II-A (Shmakov et al., 2017). So far, this subtype has been reported only in animal pathogens and commensals, supporting the argument that these Saccharibacteria groups also acquired them within their respective mammalian hosts. The G5 genome contained a class 2 subtype II-C-like gene order similar to that found in Neisseria, but absent the csn2 protein (Shmakov et al., 2017). The G1 S7 genome contained a very different truncated class 1-like arrangement (cas2-cas1-cas6). Despite this different arrangement, the cas2 gene from S7 clustered with the other G1 (Figure S7C). Overall, the varying classes and subtypes found supported the gain of CRISPR loci by horizontal transfer from varied groups of bacteria and again convergence of an important bacterial immunity for these already minimally functional organisms with reduced genomes.
DISCUSSION
The majority of CPR members are found in groundwater; however, several phyla, “Candidatus Gracilibacteria” (GN02), “Candidatus Absconditabacteria” (SR-1), and Saccharibacteria, can also be found in humans and other mammals. With Saccharibacteria N. lyticus strain TM7x being the only cultivated and physiologically described member of the vast CPR at the time of this study, much remains to be learned about this newly discovered branch of the tree of life. At the time of this study, Saccharibacteria genome sequences were limited to a single group, designated G1 (Figure 1). In order to expand our knowledge of this phylum, we have successfully sought out, reconstructed, and curated Saccharibacteria genomes belonging to the highly prevalent (Figure 4), equally enigmatic, and monophyletic groups apparent from 16S rRNA sequences (groups G2–G6). In the present study, we have successfully reconstructed class-order level groups G3, G5, and G6 from humans. We also discovered that human oral G3 share a close relative in the mammalian rumen, an observation which was also supported in the SSU rRNA sequences deposited from studies including many ruminant mammals (Figure S1). Additional group G1 genomes were also obtained from modern oral plaque as well as ancient dental calculus. Overall, the MHA members share genomic properties similar to currently known Saccharibacteria. All groups across the Saccharibacteria phylum share reduced genomes and reduced capacities for many de novo biosynthetic pathways.
Furthermore, the human oral G3, G5, and G6 were confirmed as ultra-small in size and found in association with other oral bacteria (Figure 3), strongly supporting their epibiotic lifestyle, similar to the cultivated TM7x strain. Global comparative phylogenetic and phylogenomic analyses, along with genomic, functional, and taxonomic comparisons at the gene level, highlight the large diversity across this phylum. Within the human oral cavity alone, variation in as many as 70% of the genes from the nearest oral lineage (50% AAI) as well as wide GC content variation (32%–51% GC) is evident in these newly captured divergent MHA members (G3, G5, and G6). Compared with the members derived from the groundwater environment, they are further streamlined, with a reduced number of genes, which is consistent with other MHA genomes that have adapted to mammalian hosts from the environment.
Along with the observed phylogenomic and functional separation between the G1 and G3–G6 groups, there is a remarkably low genetic variation and maintenance of high levels of synteny across the G1 group between MHA and environmental-derived members (Figure 6). This is in stark contrast to the high GC and gene variation across the G3, G5, and G6 groups. It should be noted that the GC content variation is not entirely consistent with the reduction process seen in other small, eroded genomes of obligate endosymbionts within restricted host compartments, which often result in very AT-rich genomes. To our knowledge, there are no known human host-associated bacteria that have such a high degree of genomic similarity with an environmental counterpart. Our evidence suggests that very few changes have occurred in these reduced genomes when transitioning to living in a human host environment. Further investigation of the unique genes and loci shared between these Saccharibacteria MHA lineages, which may have aided in human host adaptation, indicated that many genes appear to have been acquired from divergent independent sources via horizontal gene transfer. This is consistent with convergent gain of functions. Such variation between groups or lack thereof across the G1 group points to a complex evolutionary history within this phylum that has resulted in the wide diversity seen currently in humans alone. The environmental genomes RAAC3 and GWC2 are found in deep groundwater, where the vast majority of other new CPR members occur (Brown et al., 2015). As such, our working assumption of an environmental ancestral state would imply a species most related to the RAAC3 and GWC2 genomes. But if so, why then is the genomic variation between human oral cavity and environmental genomes within the G1 group so low compared with the other groups (G3–G6)?
A hypothetical model of past events that may have led to the current diversity of Saccharibacteria found in mammals, particularly Homo sapiens, is presented in Figure 7. There are several possibilities. Was the common ancestor of the CPR already associated with a eukaryotic host and then transitioned to the environment or an environmental organism that transitioned into hosts, and if so, was there a single transition giving rise to all the MHA lineages or multiple transitions? In one plausible scenario, the last common ancestor (LCA) of the Saccharibacteria phylum, present in the environment and similar to the other CPR present, was acquired by a eukaryotic host at some point in the past. Diversification over time within and across mammalian hosts led to the lineages G3, G5, and G6, as well as G2 and G4, which appear to be found exclusively in larger eukaryotes and mammals to date. The G3 group, in particular, has diverged to an extent in hominids (Neanderthal and Homo sapiens) compared with ruminant mammals, but they clearly share a common ancestor. Therefore, it is likely that this acquisition predates the divergence of hominids and ruminant mammals. A separate and more recent acquisition event (or even multiple recent events) (Figure 7) could potentially explain why humans have many representatives within the G1 group, a group with members that have not changed their genomic content to the same extent as other Saccharibacteria groups and have maintained a high level of synteny with existing environmental members found in groundwater. Because mammals have consumed groundwater continuously throughout history, there is a strong likelihood that the acquisition of these parasitic epibionts onto resident bacteria harbored by various mammalian hosts could have occurred with high frequency. Concerning acquisition, we recognize that Saccharibacteria either presumably attached directly to a basibiont (epibiont bacterium host) that had already become adapted to its eukaryotic host or co-transferred with its basibiont bacterium from groundwater. Current evidence is insufficient to provide a compelling answer. However, recent work by our group has indicated that the G1 Saccharibacteria strain TM7x can be isolated from its basibiont host bacterium, survive in isolation for some time, and eventually re-infect either a naive basibiont or other compatible bacteria (Bor et al., 2018). It was also shown in this work that new symbiotic pair goes through a parasitic phase in which strain TM7x initially inhibits the basibiont; however, there is co-growth and stabilization after six to nine passages, with the basibiont adapting to its epibiont (Bor et al., 2018). It is unknown if this process can occur outside of the laboratory, but it hints at the possibility that binding and stability may happen when these ultra-small parasites encounter bacteria within a mammalian host. In summary, although difficult to prove, if evolutionary time is the governing factor for genome-wide gene divergence and genome re-arrangements in these extremely reduced genomes, the cumulative evidence is consistent with this model of multiple acquisition events, with G1 acquisition being more recent. A secondary and less likely explanation for the observed maintenance of synteny is that all oral G1s have either lost or have a lower capacity to re-arrange or recombine their genes. However, compared with the environmental genomes, they appear to be reducing their genomes by individual gene loss across the genome, as well as experiencing homologous recombination by gaining individual genes as well as entire CRISPR systems. These oral G1 genomes have predominately undergone smaller variations at the nucleotide level across many genes as opposed to large-scale re-arrangements, resulting in AAI variation of ~55%–65% between MHA and environmental G1 while maintaining overall synteny (Figures 2A and 6A). Interestingly, there is a lack of genetic evidence for long-term evolutionary history between G1 member TM7x and its known Actinomyces basibiont. TM7x does not have a predominance of genes closely related to Actinomyces or a strong indication of horizontally transferred genes. Identification of additional Saccharibacteria strains and basibiont hosts across the phylum may ultimately reveal if there is a conserved signature in the basibiont lineages that dictate the interaction. This may also indicate if the prokaryotic basibionts in non-mammalian environments were related to new basibionts they found in mammals.
Figure 7. Hypothetical Schematic Representation of the Temporally Separated Acquisition Events that May Have Led to The Observed Diversity of Saccharibacteria within Mammals.

Shown near the bottom is the Saccharibacteria branch of the CPR tree, and near the top are the branches in the tree of Eukaryota, with the class Mammalia branch expanded for presentation purposes. Time is indicated at the bottom of the figure by an arrow from left to right. Potential acquisition events in which a member of the Saccharibacteria originally present in the environment was transferred to a mammalian host are also indicated on the Saccharibacteria branch. The expansion in diversity and the time from acquisition for major groups of Saccharibacteria found in mammals (both oral and rumen) are shown within the Mammalia branch. The differences in genomic diversity between the G1 and G2–G6 groups across the Saccharibacteria phylum at the present time in Mammalia could potentially be explained with temporally separated acquisition events. The lack of major genomic variation in gene content and gene order (synteny) between the mammalian associated G1 and environmental G1 (found in groundwater with the majority of the CPR) suggests that mammalian acquisition of the Saccharibacteria G1 from an environmental source was likely a more recent event than that of the G2–G6 groups.
Significant questions remain unanswered regarding these highly prevalent, ultra-small parasitic bacteria with unique lifestyles. What mechanisms enable the dynamics of their host association? How have bacteria lacking many core capabilities survived and persisted within mammals throughout their evolutionary history? Given their ability to infect commensal bacteria, what is their impact on human health and disease states? Investigations into these intriguing questions will be greatly facilitated with this body of work that has increased the available genomic information spanning the major groups within the Saccharibacteria phylum.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Jeffrey McLean (jsmclean@uw.edu).
Materials Availability
This study did not generate new unique reagents
Data and Code Availability
The accession numbers for the genomes reported in this paper are Genbank: Genbank: PRLK00000000, Genbank: PRLL00000000, Genbank: PRLM00000000, Genbank: PRLN00000000, Genbank: PRLO00000000, Genbank: PQNZ00000000, Genbank: PQOA00000000. under Bioproject PRJNA384792. Related information is available via https://www.jsmcleanlab.com/deposited-genomes. All the other considered genomes and metagenomes are publicly available in NCBI and their information listed in Table S1.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All FISH experiments were performed using the original low passage Nanosynbacter lyticus strain TM7x from (He et al., 2015). Healthy subjects aged ≥ 18 y were recruited to provide saliva samples for FISH and PCR analysis. The study had Institutional Review Board approval (13-001075, University of California Los Angeles).
METHOD DETAILS
Genome recovery from In vitro growth enrichments and metagenomic datasets
The assembled genomes were derived from existing publicly available assemblies with reference ID’s (some manually curated, see description below), or from assembled and binned metagenomics reads (public or from in-house sequencing) (see Table S1). MegaBLAST against HOMD 16S rRNA database V14.5 (Chen et al., 2010) was used to find assemblies with hits to TM7 sequences within the 1100 HMP assemblies. Raw reads from in-house sequencing of enrichment samples or public SRA files were assembled after quality-trimming and filtering via BBDuk (Bushnell, 2015) and assembly with St. Petersburg genome assembler (SPAdes) (Nurk et al., 2013). We used CONCOCT (Alneberg et al., 2014) for binning scaffolds. Manual curation of binned genomes was performed by screening for contigs with duplicated genes, GC content, and for contigs that had deviating Kmer frequencies and validated visually with VizBin in an iterative manner (Laczny et al., 2015). Manual curation of publicly available draft genomes using Kmer frequency check (IMG tool) was performed before further analyses, and contaminating contigs were manually removed from the assembly (See Table S1). The resulting assemblies were listed in the tree within Figure 1 and Table S1. In addition, CheckM (Parks et al., 2015) was used to assess the quality and estimate the coverage of a set of single marker genes for each genome. A clear distinct set for estimating the CPR group completeness has not yet been validated. To date, four of the previously recovered G1 genomes are complete circular genomes, designated here TM7x (He et al., 2015), GWC2 (Brown et al., 2015), RAAC3 (Kantor et al., 2013), and S. aal (‘Ca.Saccharimonas aalborgensis’) (Albertsen et al., 2013) (Table S1). When comparing these known closed genomes against CheckM, completeness scores ranged from 65 to 67 percent, with only 117 of the single-copy marker genes of the core set of 171 marker genes used by CheckM (Table S1). All but one of the new genomes, TM7_G1_3_12Alb, scored 57 percent or higher. Given the consistent 117 marker set in complete Saccharibacteria genomes, this implied 90% or higher levels of actual completeness for the majority of the new genomes. New G1 group TM7_G1_3_12Alb and the ancient calculus assembly TM7_ANC_38.39_G1_1 had the lowest percentage of single-copy ribosomal genes when compared to the complete Saccharibacteria genomes (Table S3). In general, the recovered genomes were consistent with high levels of completeness, and they indeed share the reduced genome characteristic prevalent throughout the CPR.
In order to understand the relationships between the Saccharibacteria, a phylogenetic analysis was carried out on the 15 assemblies using three publicly available CPR genomes (SR1, Kazan, Berkelbacteria from Brown et al., 2015; Table S1) as outgroups. PhyloSift (Darling et al., 2014) and the associated genome database as well as CheckM were used to extract, align and concatenate single-copy ribosomal marker genes from assemblies representing genomes/bins. Concatenated alignments were masked with 10% of the gaps removed. Concatenated trees were inferred using RAXML (version 8.2.7, taking the best-scoring tree from GTR CAT rapid bootstrap; bootstrap with 1000 replicates and parsimony random seed 1 with 800 columns). The likelihood of the final tree was evaluated and optimized under the GAMMA model (Stamatakis, 2014). For the phylogenomic placement, both PhyloSift, which places the concatenated alignments onto a comprehensive tree, and CheckM, which only generates a concatenated alignment using the genome set as input, were congruent. Individual protein alignments to investigate phylogenic origins of potentially horizontally acquired genes were also aligned, masked, and trees inferred with RAxML with the same settings as the concatenated trees. Phylogenetic trees were visualized and annotated with FigTree v1.2.2 (Rambaut, 2009). A vast number of new genomes derived from metagenomic shotgun sequencing are rapidly populating databases, exponentially adding to the breadth of reference genomes and expanding our knowledge of the microbial universe. Although not always well-curated for contamination, this exponential growth has led to many unnamed and unclassified groups within various phyla, resulting in complex unknown phylogenetic relationships. At this time, efforts are ongoing to propose the use of genome sequences as the type material for naming prokaryotic taxa (Whitman, 2015), as well as proposals for implementation of an independent nomenclatural system for uncultivated taxa (Konstantinidis et al., 2020) (Konstantinidis et al., 2017). All proposed provisional operational taxonomic classifications are placed on Figure 1E and presented in supplemental materials Table S2 and Data S2 which describe the provisional etymology and proposals for designation of sequenced representatives as type material at this time.
SSU analysis
For SSU analysis, full-length 16S rRNA genes were extracted from genomes where available. These sequences, along with the full-length 16S rRNA genes from the TM7 phylum Groups 1-6 available in HOMD, were aligned with reference sequences with the SINA aligner (Pruesse et al., 2012) using the SILVA web interface (Pruesse et al., 2007) with default parameters. Nearest neighbors references (n-20; 89% cutoff) were extracted for each sequence. Unique sequences from this set were extracted in Geneious version 11 (https://www.geneious.com/) (Kearse et al., 2012). The resulting 400 sequences in the SILVA alignment were masked as above with > 10% gaps removed. Trees were inferred by RAxML, as described above (Figure 1; Figure S1). The complete 16S rRNA tree is available in nexus format as Data S1.
Annotation and metabolic reconstruction
Open reading frames (ORFs) were predicted and annotated for all previously published genomes and genomic scaffolds using Prokka to enable genomics comparisons (Seemann, 2014). CompareM was employed to perform comparative genomic analyses, including finding orthologous genes and the calculation of pairwise amino acid identity (AAI) values between genomes (Parks, 2014). The default parameters, e-value 1e-5, percent sequence identity 30%, and percent alignment length 70%, were used. The Saccharibacteria phylum pangenome was determined using BLAST-based all versus all comparisons with Prokka-annotated genomes using Roary (Page et al., 2015) (minimum identity set to 20%), which clusters proteins using MCL-edge (Enright et al., 2002). General agreement was found between CompareM and Roary for the percentage of orthologous genes found. A core gene alignment and inferred tree (RAXML GTR-CAT, 1000 bootstrap) was constructed with 211 concatenated orthologous genes within four complete and five near-complete Saccharibacteria genomes. GhostKOALA webserver (Kanehisa et al., 2016) was used to derive taxonomy for each gene in the genomes. Each query gene was assigned a taxonomic category according to the besthit gene in the Cd-hit cluster supplemented version of their non-redundant dataset.
TM7 lineage-specific detection using FISH staining of oral samples
Human oral saliva and tongue-surface bacterial samples were collected and processed immediately. Samples were centrifuged at 1500 rpm for 3 minutes to remove large food particles and other debris. The supernatant was collected and centrifuged at 4600 rpm for 15minutes, and the pellets were resuspended and fixed in 4% formaldehyde for 3 hours and permeabilized by 2 mg/mL lysozyme in 20 mM Tris pH7.0 for 9 minutes at 37°C. Fixed cells were washed in 50, 80, and 90% of ethanol, resuspended in 100-300 μL of hybridization buffer (20mM Tris·Cl, pH8.0, 0.9M NaCl, 0.01% SDS, 30% deionized formamide) and incubated at 37°C for 30 minutes. TM7 lineage or SR1 specific probes (see Key Resources Table) were used to stain the cells for 3 hours at 42°C. Cells were then washed three times with 0.1x saline-sodium citrate buffer, 15 minutes each, and mounted on the coverslip with SlowFade Gold antifade reagent (Invitrogen). During the second wash step, Syto9 universal DNA stain (Invitrogen) was added to the samples. Cells were visualized with an LSM 880 inverted confocal microscope equipped with a 100x/1.15 oil immersion objective. We repeated each experiment multiple times and acquired multiple FISH images. Only representative images are shown.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Nanosynbacter lyticus strain TM7x | (He et al., 2015) | TM7x |
| Biological Samples | ||
| Human saliva | This study | N/A |
| Human tongue-surface swabs | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SlowFade Gold antifade mountant | Thermo Fisher Scientific | Cat#: S36936 |
| Syto-9 Wheat germ agglutinin, Alexa Fluor 488 conjugate | Thermo Fisher Scientific | Cat#: W11261 |
| Deposited Data | ||
| Ca. Nanosynsacchari sp. TM7_ANC_38.39_G1_1 | This paper | GenBank: PQNZ00000000 |
| Ca. Nanosynsacchari sp. TM7_G1_3_12Alb | This paper | GenBank: PQOA00000000 |
| Ca. Nanosyncoccus alces TM7_G3_2_Rum_HOT_351B | This paper | GenBank: PRLM00000000 |
| Ca. Nanosyncoccus nanoralicus TM7_KMM_G3_1_HOT_351 | This paper | GenBank: PRLL00000000 |
| Ca. Nanoperiomorbus periodonticus TM7_EAM_G5_1_HOT_356 | This paper | GenBank: PRLO00000000 |
| Ca. Nanoperiomorbus periodonticus TM7_EAM_G5_2_HOT_356 | This paper | GenBank: PRLN00000000 |
| Ca. Nanogingivalis gingivitcus TM7_CMJM_G6_1_HOT_870 | This paper | GenBank: PRLK00000000 |
| Nanosynbacter lyticus strain TM7x HOT_952 | (He et al., 2015) | GenBank: CP007496.1 |
| GWC2 | (Brown et al., 2015) | N/A |
| RAAC3 | (Kantor et al., 2013) | GenBank: CP006915.1 |
| S_aal | (Albertsen et al., 2013) | GenBank: CP005957.1 |
| TM7c | (Marcy et al., 2007) | GenBank: ABBX00000000 |
| TM7_S7 | IMG public release | IMG Id: 2236661026 |
| Oligonucleotides | ||
| G1-specific Cy5-labeled probe: CCTACGCAACTCTTTACGCC | (He et al., 2015) | N/A |
| G3-specific Cy3-labeled probe: ACTTGGCTATTATGCGAGT | This paper | N/A |
| G5-specific Cy3-labeled probe: GCTCTTCGGAGTACACGAGA | This paper | N/A |
| G6-specific Cy3-labeled probe: GCATCGAAAGGTGTGC | This paper | N/A |
| SR1-specific Cy5-labeled probe: TTAACYRGACACCTTGCG | This paper | N/A |
| G3-specific forward primer: ACTTGGCTATTATGCGAGT | This paper | G3-F73 |
| G3-specific reverse primer: GGATACCTTGTTACGAC | This paper | G3-R1469 |
| G5-specific forward primer: GCTCTTCGGAGTACACGAGA | This paper | G5-F47 |
| G5-specific reverse primer: AAATAAATCCGGACGTCGGGTGCTCC | This paper | G5-R1336 |
| G6-specific forward primer: GCATCGAAAGGTGTGC | This paper | G6-F180 |
| G6-specific reverse primer: CCTTGTTACGACTTAAC | This paper | G6-R1469 |
| SR1-specific forward primer: GATGAACGCTAGCGRAAYG | (Camanocha and Dewhirst, 2014) | SR1-AF31-X1 |
| SR1-specific forward primer: CTTAACCCCAGTCACTGATT | (Camanocha and Dewhirst, 2014) | SR1-AF32 |
| Software and Algorithms | ||
| BBDuk | (Bushnell, 2015) | https://sourceforge.net/projects/bbmap/ |
| SPAdes Assembler | (Nurk et al., 2013) | RRID:SCR_000131; https://github.com/ablab/spades |
| CONCOCT | (Alneberg et al., 2014) | https://github.com/BinPro/CONCOCT |
| VizBin | (Laczny et al., 2015) | https://github.com/claczny/VizBin |
| CheckM | (Parks et al., 2015) | http://ecogenomics.github.io/CheckM/ |
| PhyloSift | (Darling et al., 2014) | https://github.com/gjospin/PhyloSift |
| RAxML v8.2.7 | (Stamatakis, 2014) | RRID:SCR_006086; https://cme.h-its.org/exelixis/web/software/raxml/ |
| FigTree v1.2.2 | (Rambaut, 2009) | RRID:SCR_008515; http://tree.bio.ed.ac.uk/software/figtree/ |
| Prokka v | (Seemann, 2014) | RRID:SCR_014732; https://github.com/tseemann/prokka |
| CompareM | (Parks, 2014) | https://github.com/dparks1134/CompareM |
| Roary | (Page et al., 2015) | https://github.com/sanger-pathogens/Roary |
| MCL-edge | (Enright et al., 2002) | https://micans.org/mcl/ |
| GhostKOALA | (Kanehisa et al., 2016) | https://www.kegg.jp/ghostkoala/ |
| EggNOG | (Huerta-Cepas et al., 2016) | RRID:SCR_002456; http://eggnog5.embl.de/ http://eggnog5.embl.de/ |
| ClustVis | (Metsalu et al., 2015; Sievers et al., 2011) | https://biit.cs.ut.ee/clustvis/ |
| SINA aligner | (Pruesse et al., 2012) | RRID:SCR_005067; https://www.arb-silva.de/aligner |
| Geneious v11 | RRID:SCR_010519 | |
| Other | ||
| HOMD 16 s rRNA database v14.5 | (Chen et al., 2010) | http://www.homd.org/?name=seqDownload&type=R |
Previous studies have found non-specific detection of TM7 by fluorescence in situ hybridization (FISH) DNA probes (Sizova, 2015 #4). We sought to ensure specific staining in the current study by designing group-specific probes that target the 16S rRNA for each group respectively (Key Resources Table). To ensure the lineage-specificity of the DNA probes, we performed FISH analysis on the TM7x (G1) strain using different TM7 lineage-specific probes. Only the G1 specific probe stained TM7x cells.
We screened for the presence of TM7 lineages in 12 individuals using PCR.Two individuals showing positive identification for TM7-G3, G5, G6, and SR1 were studied further. 10 mL of saliva samples were collected from each of the two subjects in the morning before brushing their teeth. Saliva samples were mixed 1:1 with cold PBS and kept on ice. Samples were vortexed vigorously for 10 minutes and then centrifuged for 15 minutes at 2600 x g. Pellets were discarded, and the supernatant was filtered through a 41 μm filter to further remove large particles. Resulting samples were filtered through 0.45 μm filters and centrifuged at 120, 000 x g for 90 minutes at 4°C to concentrate the ultra-small bacteria. The supernatant was discarded and the pellet was resuspended in 300 μL of PBS. The genomic DNA was isolated from these samples and the presence of specific TM7 and SR1 were detected by PCR using lineage-specific primers (see STAR Methods). The PCR reaction mixture (25 μL) contained 0.5 mM primers, buffer and Taq polymerase. The reactions were incubated at an initial denaturation at 95°C for 5 min, followed by a 30-cycle amplification consisting of denaturation at 95°C for 1 min, annealing at different temperature for varying duration depending on different primer sets (see table below), and extension at 72°C for 2 min. Initial screening for G2 and G4 using group-specific probes revealed no positives in our sample collections, further supporting the idea that these genomes are rare in the human oral cavity and/or are mainly restricted to other mammalian hosts.
Read mapping to new reference genomes
Warinner et al. sequenced dental calculus using whole DNA shotgun methods from adult human skeletons found in a Medieval monastic site (Warinner et al., 2014). Reads from 4 samples (7 to 13M reads) were mapped to all the reference TM7 genomes. Unique reads were allowed to map and reads that mapped to multiple locations were mapped randomly across all hits so as not to inflate the coverage. Ancient, dental calculus was also deeply sequenced (> 147 million reads) from the best-preserved Neanderthal, El Sidrón 1 sample from the study, which suffered from a dental abscess (Weyrich et al., 2017), as well as more degraded samples from SPYII. Percentage of genome coverage across these datasets as well as periodontal disease samples, (McLean et al., 2015), and HMP datasets (Accession numbers indicated in Table S4) were used to assess the presence of near relatives in the samples chosen (Figure 2; Table S4). For the read mapping data using ancient DNA samples we observed no influence of soil contamination, the oral derived sequences recruited the majority of the reads across the groups with the environmental genomes resulting in less than 0.4% reference coverage with reads mainly mapping to 16S rRNA gene regions of the environmental genomes which should not be considered due to the highly conserved sequence similarity between genomes. In contrast, the coverage using ELSIDRON and the Warriner dataset across the oral genomes are substantial for several of the groups (from 15 to 37% of the reference and between 98%–99% read identity).
Functional comparisons
Gene functions within Clusters of Orthologous Groups (COGs) were determined using EggNOG-mapper (Huerta-Cepas et al., 2016) and the results compared between Saccharibacteria and select CPR and known free-living, eukaryotic host-dependent, and obligate intracellular host-dependent bacteria. This comparison is limited to genomes of bacteria that interact with eukaryotic hosts since minimal information is available for genomes of bacteria that interact with bacterial hosts such as TM7x. Merhej and colleagues (Merhej et al., 2009) determined the mean number of genes assigned to each COG for bacteria with different lifestyles. They compared the mean number of genes assigned to each COG function between free-living bacteria (125 organisms) and all eukaryotic host-dependent bacteria (125 organisms), then between free-living and eukaryotic and obligate intracellular bacteria (40 organisms), and between mutualists (13 organisms) and parasites (27 organisms). We performed the same analyses with the new CPR genomes to determine how its functional repertoire at the COG level compares to sequenced bacterial mutualists and other bacteria considered parasites of eukaryotic hosts. SEED family annotations were also compared across genomes to investigate similarities and differences in functional genes. ClustVis (Metsalu et al., 2015; Sievers et al., 2011) was used to generate clustered heatmaps and PCA plots of the COG and SEED data.
QUANTIFICATION AND STATISTICAL ANALYSIS
CheckM (Parks et al., 2015) was used to assess the quality and estimate the coverage of a set of single marker genes for each genome. All other computational analyses were performed with the open source software tools referenced in the STAR Methods along with the described procedures.
Supplementary Material
Highlights.
Saccharibacteria are ultra-small parasitic bacteria recently discovered in humans
Novel lineages have high genomic diversity within mammalian hosts
Novel lineages are ultra-small, with reduced genomes (<1,000 genes) and a single 16S copy
One group displays minimal genomic changes since transition from environment to humans
ACKNOWLEDGMENTS
We would like to thank Phil Hugenholtz for helpful discussions regarding taxonomic classification for the members of the Saccharibacteria phylum. K.C.W. and L.M.S. were supported by an Early Career Award from the National Science Foundation under Award Number 1750189. A portion of this research was performed under the Facilities Integrating Collaborations for User Science (FICUS) exploratory effort and used resources at the US Department of Energy (DOE) Joint Genome Institute (proposal ID 503154) and the Environmental Molecular Sciences Laboratory (proposal ID 49495), which are DOE Office of Science User Facilities. Facilities are operated under contract numbers DE-AC02-05CH11231 (Joint Genome Institute) and DE-AC05-76RL01830 (EMSL). This research was funded in part by NIH National Institute of Dental and Craniofacial Research (NIDCR) awards DE023810 (W.S., X.H., and J.S.M.), DE020102 (W.S., X.H., and J.S.M.), F32DE025548-01 (B.B.), T90DE021984 (T.T.T. and K.A.K.), and DE026186 (W.S., X.H., and J.S.M.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107939.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, and Nielsen PH (2013). Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol 31, 533–538. [DOI] [PubMed] [Google Scholar]
- Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, Lahti L, Loman NJ, Andersson AF, and Quince C (2014). Binning metagenomic contigs by coverage and composition. Nat. Methods 11,1144–1146. [DOI] [PubMed] [Google Scholar]
- Bedree JK, Bor B, Cen L, Edlund A, Lux R, McLean JS, Shi W, and He X (2018). Quorum sensing modulates the epibiotic-parasitic relationship between Actinomyces odontolyticus and its Saccharibacteria epibiont, a Nanosynbacter lyticus strain, TM7x. Front. Microbiol 9, 2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bor B, Poweleit N, Bois JS, Cen L, Bedree JK, Zhou ZH, Gunsalus RP, Lux R, McLean JS, He X, and Shi W (2016). Phenotypic and physiological characterization of the epibiotic interaction between TM7x and its basibiont actinomyces. Microb. Ecol 71, 243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bor B, McLean JS, Foster KR, Cen L, To TT, Serrato-Guillen A, Dewhirst FE, Shi W, and He X (2018). Rapid evolution of decreased host susceptibility drives a stable relationship between ultrasmall parasite TM7x and its bacterial host. Proc. Natl. Acad. Sci. U S A 115, 12277–12282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bor B, Bedree JK, Shi W, McLean JS, and He X (2019). Saccharibacteria (TM7) in the human oral microbiome. J. Dent. Res 98, 500–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinig MM, Lepp PW, Ouverney CC, Armitage GC, and Relman DA (2003). Prevalence of bacteria of division TM7 in human subgingival plaque and their association with disease. Appl. Environ. Microbiol 69, 1687–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CT, Hug LA, Thomas BC, Sharon I, Castelle CJ, Singh A, Wilkins MJ, Wrighton KC, Williams KH, and Banfield JF (2015). Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 523,208–211. [DOI] [PubMed] [Google Scholar]
- Burstein D, Sun CL, Brown CT, Sharon I, Anantharaman K, Probst AJ, Thomas BC, and Banfield JF (2016). Major bacterial lineages are essentially devoid of CRISPR-Cas viral defence systems. Nat. Commun 7, 10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell B (2015). BBMap (version 35.14). https://sourceforge.net/projects/bbmap/. [Google Scholar]
- Camanocha A, and Dewhirst FE (2014). Host-associated bacterial taxa from Chlorobi, Chloroflexi, GN02, Synergistetes, SR1, TM7, and WPS-2 Phyla/candidate divisions. J. Oral Microbiol 6, 25468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JH, O’Donoghue P, Campbell AG, Schwientek P, Sczyrba A, Woyke T, Söll D, and Podar M (2013). UGA is an additional glycine codon in uncultured SR1 bacteria from the human microbiota. Proc. Natl. Acad. Sci. U S A 110, 5540–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelle CJ, and Banfield JF (2018). Major new microbial groups expand diversity and alter our understanding of the tree of life. Cell 172, 1181–1197. [DOI] [PubMed] [Google Scholar]
- Chen T, Yu W-H, Izard J, Baranova OV, Lakshmanan A, and Dewhirst FE (2010). The Human Oral Microbiome Database: a Web accessible resource for investigating oral microbe taxonomic and genomic information. Database 2010, baq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Jospin G, Lowe E, Matsen FA 4th, Bik HM, and Eisen JA (2014). PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ 2, e243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, and Ouzounis CA (2002). An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30,1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren AM, Borisy GG, Huse SM, and Mark Welch JL (2014). Oligotyping analysis of the human oral microbiome. Proc. Natl. Acad Sci. U S A 111, E2875–E2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farizo KM, Fiddner S, Cheung AM, and Burns DL (2002). Membrane localization of the S1 subunit of pertussis toxin in Bordetella pertussis and implications for pertussis toxin secretion. Infect. Immun 70, 1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields TA, and Casey PJ (1997). Signalling functions and biochemical properties of pertussis toxin-resistant G-proteins. Biochem. J 321, 561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredricks DN, Fiedler TL, and Marrazzo JM (2005). Molecular identification of bacteria associated with bacterial vaginosis. N. Engl. J. Med 353, 1899–1911. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Rivera C, Bhatty M, and Christie PJ (2016). Mechanism and function of type IV secretion during infection of the human host. Microbiol. Spectr 4 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, McLean JS, Edlund A, Yooseph S, Hall AP, Liu SY, Dorrestein PC, Esquenazi E, Hunter RC, Cheng G, et al. (2015). Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. Proc. Natl. Acad. Sci. U S A 112, 244–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Forslund K, Szklarczyk D, Jensen LJ, von Mering C, and Bork P (2016). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. bioRxiv. 10.1101/076331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, Butterfield CN, Hernsdorf AW, Amano Y, Ise K, et al. (2016). A new view of the tree of life. Nat. Microbiol 1, 16048. [DOI] [PubMed] [Google Scholar]
- Hugenholtz P, Goebel BM, and Pace NR (1998). Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol 180, 4765–4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Sato Y, and Morishima K (2016). BlastKOALA and Ghost-KOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol 428, 726–731. [DOI] [PubMed] [Google Scholar]
- Kantor RS, Wrighton KC, Handley KM, Sharon I, Hug LA, Castelle CJ, Thomas BC, and Banfield JF (2013). Small genomes and sparse metabolisms of sediment-associated bacteria from four candidate phyla. MBio 4, e00708–e00713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis KT, Rosselló-Móra R, and Amann R (2017). Uncultivated microbes in need of their own taxonomy. ISME J. 11, 2399–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis KT, Rosselló-Móra R, and Amann R (2020). Advantages outweigh concerns about using genome sequence as type material for prokaryotic taxonomy. Environ. Microbiol 22, 819–822. [DOI] [PubMed] [Google Scholar]
- Kuehbacher T, Rehman A, Lepage P, Hellmig S, Fölsch UR, Schreiber S, and Ott SJ (2008). Intestinal TM7 bacterial phylogenies in active inflammatory bowel disease. J. Med. Microbiol 57, 1569–1576. [DOI] [PubMed] [Google Scholar]
- Laczny CC, Sternal T, Plugaru V, Gawron P, Atashpendar A, Margossian HH, Coronado S, der Maaten Lv.., Vlassis N, and Wilmes P (2015). VizBin–an application for reference-independent visualization and human-augmented binning of metagenomic data. Microbiome 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Rodriguez-R LM, and Konstantinidis KT (2014). MyTaxa: an advanced taxonomic classifier for genomic and metagenomic sequences. Nucleic Acids Res. 42, e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcy Y, Ouverney C, Bik EM, Lösekann T, Ivanova N, Martin HG, Szeto E, Platt D, Hugenholtz P, Relman DA, and Quake SR (2007). Dissecting biological “dark matter” with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc. Natl. Acad. Sci. U S A 104, 11889–11894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean JS, Liu Q, Thompson J, Edlund A, and Kelley S (2015). Draft genome sequence of “Candidatus Bacteroides periocalifornicus,” a new member of the Bacteriodetes phylum found within the oral microbiome of periodontitis patients. Genome Announc. 3, e01485–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean JS, Liu Q, Bor B, Bedree JK, Cen L, Watling M, To TT, Bumgarner RE, He X, and Shi W (2016). Draft genome sequence of Actinomyces odontolyticus subsp. actinosynbacter strain XH001, the basibiont of an oral TM7 epibiont. Genome Announc. 4, e01685–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean JS, Lombardo MJ, Badger JH, Edlund A, Novotny M, Yee-Greenbaum J, Vyahhi N, Hall AP, Yang Y, Dupont CL, et al. (2013). Candidate phylum TM6 genome recovered from a hospital sink biofilm provides genomic insights into this uncultivated phylum. Proc. Natl. Acad Sci. U S A 110, E2390–E2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merhej V, Royer-Carenzi M, Pontarotti P, and Raoult D (2009). Massive comparative genomic analysis reveals convergent evolution of specialized bacteria. Biol. Direct 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metsalu T, Vilo J, Prchal-Murphy M, Putz EM, Holcmann M, Schlederer M, Heller G, Bago-Horvath Z, Witalisz-Siepracka A, Cumaraswamy AA, et al. (2015). ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 43 (W1), W566–W570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, et al. (2013). Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol 20, 714–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmalayam I, Karem K, Baumstark B, and Massung R (2000). The gene encoding the 17-kDa antigen of Bartonella henselae is located within a cluster of genes homologous to the virB virulence operon. DNA Cell Biol. 19,377–382. [DOI] [PubMed] [Google Scholar]
- Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, Fookes M, Falush D, Keane JA, and Parkhill J (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palchevskiy V, and Finkel SE (2006). Escherichia coli competence gene homologs are essential for competitive fitness and the use of DNA as a nutrient. J. Bacteriol 188, 3902–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks D (2014). CompareM. https://github.com/dparks1134/CompareM. [Google Scholar]
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, and Tyson GW (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil PA, and Hugenholtz P (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol 36, 996–1004. [DOI] [PubMed] [Google Scholar]
- Podar M, Abulencia CB, Walcher M, Hutchison D, Zengler K, Garcia JA, Holland T, Cotton D, Hauser L, and Keller M (2007). Targeted access to the genomes of low-abundance organisms in complex microbial communities. Appl. Environ. Microbiol 73, 3205–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, and Glöckner FO (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Peplies J, and Glöckner FO (2012). SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A (2009). FigTree v1.3.1: tree figure drawing tool. http://tree.bio.ed.ac.uk/software/figtree. [Google Scholar]
- Rheims H, Rainey FA, and Stackebrandt E (1996). A molecular approach to search for diversity among bacteria in the environment. J. Ind. Microbiol 17, 159–169. [Google Scholar]
- Rodriguez-R LM, Gunturu S, Harvey WT, Rosselló-Mora R, Tiedje JM, Cole JR, and Konstantinidis KT (2018). The Microbial Genomes Atlas (MiGA) webserver: taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 46 (W1), W282–W288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30,2068–2069. [DOI] [PubMed] [Google Scholar]
- Shmakov S, Smargon A, Scott D, Cox D, Pyzocha N, Yan W, Abudayyeh OO, Gootenberg JS, Makarova KS, Wolf YI, et al. (2017). Diversity and evolution of class 2 CRISPR-Cas systems. Nature reviews Microbiology 15, 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza DP, Oka GU, Alvarez-Martinez CE, Bisson-Filho AW, Dunger G, Hobeika L, Cavalcante NS, Alegria MC, Barbosa LR, Salinas RK, et al. (2015). Bacterial killing via a type IV secretion system. Nat. Commun 6, 6453. [DOI] [PubMed] [Google Scholar]
- Stamatakis A (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warinner C, Rodrigues JF, Vyas R, Trachsel C, Shved N, Grossmann J, Radini A, Hancock Y, Tito RY, Fiddyment S, et al. (2014). Pathogens and host immunity in the ancient human oral cavity. Nat. Genet 46, 336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyrich LS, Duchene S, Soubrier J, Arriola L, Llamas B, Breen J, Morris AG, Alt KW, Caramelli D, Dresely V, et al. (2017). Neanderthal behaviour, diet, and disease inferred from ancient DNA in dental calculus. Nature 544,357–361. [DOI] [PubMed] [Google Scholar]
- Whitman WB (2015). Genome sequences as the type material for taxonomic descriptions of prokaryotes. Syst. Appl. Microbiol 38, 217–222. [DOI] [PubMed] [Google Scholar]
- Yeoh YK, Sekiguchi Y, Parks DH, and Hugenholtz P (2016). Comparative Genomics of Candidate Phylum TM6 Suggests That Parasitism Is Widespread and Ancestral in This Lineage. Mol. Biol. Evol 33, 915–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the genomes reported in this paper are Genbank: Genbank: PRLK00000000, Genbank: PRLL00000000, Genbank: PRLM00000000, Genbank: PRLN00000000, Genbank: PRLO00000000, Genbank: PQNZ00000000, Genbank: PQOA00000000. under Bioproject PRJNA384792. Related information is available via https://www.jsmcleanlab.com/deposited-genomes. All the other considered genomes and metagenomes are publicly available in NCBI and their information listed in Table S1.