

Abstract
Sphingosine-1-phosphate (S1P) plays important roles as a signaling lipid in a variety of physiological and pathophysiological processes. S1P signals via a family of G protein-coupled receptors (S1P1–5) and intracellular targets. Here, we report on photoswitchable analogs of S1P and its precursor sphingosine, respectively termed PhotoS1P and PhotoSph. PhotoS1P enables optical control of S1P1–3, shown through electrophysiology and Ca2+ mobilization assays. We evaluated PhotoS1P in vivo, where it reversibly controlled S1P3-dependent pain hypersensitivity in mice. The hypersensitivity induced by PhotoS1P is comparable to that induced by S1P. PhotoS1P is uniquely suited for the study of S1P biology in cultured cells and in vivo because it exhibits prolonged metabolic stability compared to the rapidly metabolized S1P. Using lipid mass spectrometry analysis, we constructed a metabolic map of PhotoS1P and PhotoSph. The formation of these photoswitchable lipids was found to be light-dependent, providing a novel approach to optically probe sphingolipid biology.
Graphcial Abstract
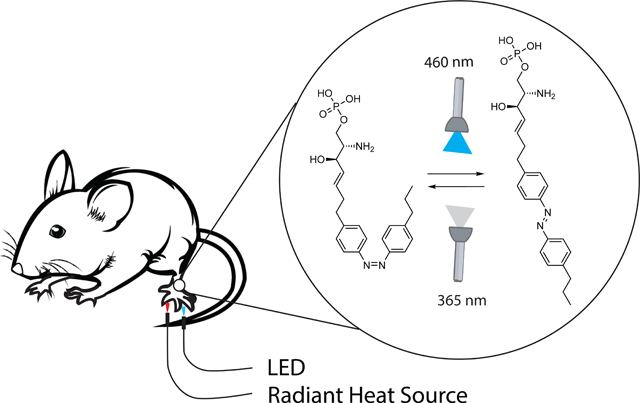
Introduction
Sphingosine and sphingosine-1-phosphate (S1P) are bioactive lipids that play key physiological roles in health and disease.1,2 S1P targets the lysophospholipid receptors S1P1–5, a class of GPCRs that is expressed across most tissues and has important roles in the immune, vascular, and nervous systems.3,4 S1P has emerged as an important modulator of intracellular targets, including histone deacetylases (HDACs), TNF receptor-associated factor 2 (TRAF2), and protein kinase C δ (PKCδ). In addition, S1P receptor signaling has recently been shown to mediate both acute and inflammatory pain.5–8 Studying the physiological function and therapeutic targets of S1P is challenging due to its low metabolic stability (t1/2 < 5 min) and complex systemic mechanisms governing its synthesis and transport.9,10 And as such, metabolically stable synthetic analogs of S1P (e.g. Fingolimod-phosphate11,12) have shown more promise in the clinic. However, these analogs do not activate all S1P receptor subtypes, and are metabolized differently than native S1P.
In general, the study of lipid biology relies largely on functionalized synthetic lipid analogs such as isotope labeled lipids,13,14 fluorescent lipids,15 and photocrosslinking lipids.16 Photocages can precisely control lipid function in space and time, as release (‘uncaging’) of endogenous lipids can be induced by light.17,18 More recently, photoswitchable lipids have emerged as useful tools to control cell signaling in a reversible manner. They rely on the incorporation of an azobenzene photoswitch in the hydrophobic tail and mediate optical control of lipid function by reversible, light-induced isomerization between a cis-isomer (bent) and trans-isomer (straight).19 In comparison to caged lipids, photoswitchable lipids require less intense irradiation and do not lead to the formation of side-products. To date, applications of photoswitchable lipids include the modulation of ion channels,20–23 the fatty acid receptor GPR40,22 lipid rafts,24 lipid vesicle budding and fission,25 and lipid-protein interactions in canonical lipid signaling.26
Motivated by the importance of sphingosine and S1P to basic physiological processes, and the success of recently published photoswitchable lipids27, here we developed synthetic derivatives of S1P that allow for precise spatiotemporal control of S1P signaling and metabolism. Using these photoswitchable lipids, termed PhotoSph (1) and PhotoS1P (2), we demonstrated their ability to rapidly and reversibly control sphingolipid biology in vitro and in vivo.
Results
Design and synthesis of photoswitchable sphingolipids
The molecular design was guided by our recent studies on photoswitchable fatty acids and diacylglycerols.20,26 While the logic behind these photoswitchable lipids largely depended on the modulation of biophysical properties mimicking a change in lipid saturation, highly unsaturated forms of sphingoid bases are not known, and therefore have not been explored systematically. Nevertheless, we thought that photoswitchable analogs of sphingosine and S1P could facilitate optical control over these lipids, because both metabolic enzymes (e.g. sphingosine kinases) and S1P receptors exhibit marked differences in affinity towards substrates or ligands with chemical modifications.28 For example, S1P receptor activation is strongly dependent on sphingoid base chain length.29 In our design of PhotoSph and PhotoS1P, we planned to incorporate the azobenzene moiety near the middle of the lipid tail, and mimic the predominant form of sphingosine, d-erythro-sphingosine (18:1), and the phosphorylated version thereof (Fig. 1A).
Figure 1 |. Design, synthesis, and photophysical properties of PhotoSph and PhotoS1P.

(A) Design of PhotoSph and PhotoS1P through incorporation of an azobenzene photoswitch into the lipid tail of sphingosine and S1P. (B) Chemical synthesis of PhotoSph and PhotoS1P. (C,D) The UV-Vis spectra of PhotoSph (C) and PhotoS1P (D) in the dark-adapted (black, trans), 365 nm adapted (grey, cis) and 460 nm adapted (blue, trans) photostationary states. Reversible cycling between photoisomers with alternating illumination at 365/460 nm. The experiment was repeated two times with similar results.
The synthesis of PhotoSph and PhotoS1P (Fig. 1B) commenced by conversion of 4-propylaniline (3) to the corresponding nitrosocompound followed by condensation with 4-iodoaniline under Baeyer–Mills conditions to yield iodoazobenzene 4.20 A Heck reaction with allyl alcohol and subsequent Wittig reaction yielded olefin 5.30 Cross metathesis with a Garner’s aldehyde-derived allylic alcohol 6 gave protected PhotoSph, which was subjected to global deprotection under acidic conditions.31 PhotoSph was then converted to PhotoS1P by Boc-protection, followed by formation of the protected phosphate ester and global deprotection.32 Both photoswitchable lipids exhibited photophysical properties similar to classical azobenzenes, including effective switching at 365/460 nm and bistability (Fig. 1C–D). Additional NMR experiments for the photophysical characterization are included in Supplementary Fig. 6 and 7. The photophysical properties of PhotoSph and PhotoS1P are very similar to other photoswitchable lipids.20,24,26
PhotoS1P enables fast reversible control of the S1P1
We evaluated the ability of PhotoS1P to optically control S1P1 receptors using whole-cell electrophysiology in human embryonic kidney (HEK 293T) cells transiently expressing the S1P1 receptor and a mutant version of the G protein-coupled inwardly rectifying GIRK1 potassium channel (GIRK1-F137S,33 Fig. 2 and Supplementary Fig. 1). The GIRK1-F137S mutant was examined due to its enhanced cell surface expression that markedly increases GIRK currents.33 GIRK channels are effectors of Gi/o signaling, and their activation provided a time-resolved report of S1P1 receptor activation. While HEK293T cells exhibit some endogenous S1P1 receptor expression34, we observed negligible GIRK current in HEK293T cells exclusively transfected with GIRK1-F137S upon addition of PhotoS1P (100 nM), which had been maintained for a long period in the dark and so allowed to relax to the trans state. Cells transfected with both S1P1 receptor and GIRK1-F137S exhibited a large inward current upon addition of trans-PhotoS1P (7) (100 nM), and the current was reversed by illumination with 365 nm light (10.8 ± 3.7% of activity at 450 nm; n = 7 cells). The current could almost be completely switched off under 365 nm illumination, suggesting a large advantage to activation by the trans isomer of PhotoS1P and a photostationary state >9:1 at 365 nm (Supplementary Fig. 7). The S1P1/3 receptor antagonist VPC23019 completely inhibited the trans-PhotoS1P induced current. Finally, switching back and forth between illumination at 450 nm and 365 nm repeatedly cycled the current on and off, indicating robust and reversible photo-activation of the S1P1 receptor.
Figure 2 |. Optical control of S1P1-GIRK coupling.

(A) Schematic of selective binding to and activation of S1P1 receptor by PhotoS1P in trans state, resulting in activation and opening of GIRK channel. (B) A representative single current trace of PhotoS1P evaluated using whole-cell patch clamp recording in HEK293T cells transiently expressing S1P1 receptors and GIRK channels. Switching illumination between 365 and 450 nm does not induce current in the absence of PhotoS1P. Application of PhotoS1P (100 nM) in trans state induces an inward current, which is reversed by illumination at 365 nm and re-activated by illumination at 450 nm. The current induced by PhotoS1P under 450 nm light is inhibited by the S1P1 receptor antagonist VPC23019. Currents measured at constant membrane potential of −80 mV. This experiment was repeated 4 times with similar results.
Optical control of S1P1–5 receptors in HTC4 cells
Next, we systematically evaluated the potential of PhotoS1P to modulate each of the five S1P receptor subtypes using a Ca2+-mobilization assay. HTC4 cells, which were found to be non-responsive to S1P, were used. Cells were stably expressing one of the five S1P receptors (S1P1–5) and Gαq or chimeric Gαq proteins to elicit Ca2+ release from ER stores (Fig. 3A).35 In agreement with the above electrophysiological experiments in HEK 293T cells, trans-PhotoS1P activated the S1P1 receptor, with similar potency to S1P, while cis-PhotoS1P was considerably less potent. The trans-PhotoS1P was also the more potent photoisomer for activation of the S1P3, receptor and with the S1P5 receptor we observed the same photoisomer preference at low concentrations with no clear trend observed for this receptor at concentrations >EC50. Interestingly, we observed the opposite trend with the S1P2 receptor where cis-PhotoS1P (8) was the more potent photoisomer. The S1P4 receptor responded almost equally to cis- and trans-PhotoS1P. Thus, with the exception of the S1P4 receptor, whose natural ligand is phytosphingosine-1-phosphate36, S1P receptors showed differential responses to cis and trans photoisomers of PhotoS1P. We also found that pre-treatment of S1P1-receptor expressing HTC4 cells with PhotoS1P (300 nM) completely desensitized the cells and inhibited a Ca2+ response elicited by S1P (Fig. 3B–C). This result suggests that PhotoS1P triggers receptor internalization similar to S1P. In an effort to understand the molecular basis of the preferred trans-PhotoS1P activation of the S1P1 receptor and cis-PhotoS1P activation of the S1P2 receptor, we constructed homology models of the S1P1–5 receptors and docked S1P, trans-PhotoS1P, and cis-PhotoS1P into each. Fig. 3 panels D and E, respectively, show results of docking into the S1P1 and S1P2 receptors. These models suggest that the polar headgroups of both S1P and trans-PhotoS1P interact with a common set of amino acids in TM segments 3 and 7 in the S1P1 receptor model, but that cis-PhotoS1P shows fewer interactions between the polar headgroup and the same residues, consistent with the lower potency observed for cis-PhotoS1P in S1P1-expressing HTC4 cells. In contrast, only S1P shows a critical set of complementary interactions between the polar headgroup of a low-energy ligand conformation and the S1P2 receptor, consistent with its substantially better potency than either trans-PhotoS1P (only one hydrogen bond observed) or cis-PhotoS1P (high energy conformation with poor electron delocalization required to occupy S1P2 receptor binding pocket). More results for docking into S1P1–5 are shown in the Supplementary Fig. 8–12.
Figure 3 |. Optical control of S1P1–5 receptor-mediated calcium release and homology modelling of S1P1–2 receptors.

(A) Fura2-AM calcium imaging in HTC4 cells stably transfected with S1P1–5 receptors. Dose response of S1P, trans-PhotoS1P and cis-PhotoS1P in HTC4, HTC4 S1P1, HTC4 S1P2, HTC4 S1P3, HTC4 S1P4, and HTC4 S1P5 cells. Samples (N = 8) were run in quadruplicates and at two independent experiments. Data points were normalized to maximal S1P response for each receptor. Error bars represent mean ± S.D. (B,C) Calcium mobilization in HTC4 cells stably transfected with S1P1 receptor after prior treatment with S1P (300 nM), cis-PhotoS1P (300 nM), trans-PhotoS1P (300 nM), or vehicle (CS-BSA, 300 nM). Data points were normalized to maximum S1P response. Error bars represent mean ± S.D. Representative traces after application of 300 nM S1P are shown in (C). Samples (N = 3) were run in triplicates. (D,E) Computationally-predicted binding poses for S1P (blue), trans-PhotoS1P (cyan) and PhotoS1P (orange) docked into S1P1 (D) and S1P2 (E). Receptor pocket surface is highlighted in green (hydrophobic)/ violet(hydrophilic).
Optical control of S1P3 dependent nociception in vivo
S1P induces acute pain behaviors and heat hypersensitivity in mice via S1P3 receptor signaling in dorsal root ganglia (DRG) neurons. DRG neurons are a heterogeneous population of primary sensory neurons that respond differentially to thermal, chemical, and/or mechanical stimuli.37 For example, heat nociceptors can be identified by their response to capsaicin, which targets the heat-activated ion channel TRPV1. Cold nociceptors are selectively activated by menthol which targets the cold-activated ion channel TRPM8. We previously showed that S1P activates a subset of capsaicin-responsive heat nociceptors, comprising approximately 30% of all dorsal root ganglia (DRG) neurons.6,8,38 Hence, we investigated the potential of PhotoS1P to optically control S1P3-mediated Ca2+ influx in capsaicin-responsive thermal nociceptors in cultured DRG neurons isolated from wild-type (WT) or S1P3 knockout (KO) mice loaded with the ratiometric Ca2+ indicator Fura2-AM. Photoisomerization of cis- to trans-PhotoS1P triggered Ca2+influx in WT DRG neurons (Fig. 4A). The neuronal responses to S1P (representative traces in Fig. 4B) and trans-PhotoS1P (representative traces in Fig. 4C) were similar in both the amplitude of the Ca2+responses (Fig. 4B–C) and the percentage of responsive neurons (Fig. 4E). In contrast, treatment with the less potent photoisomer cis-PhotoS1P showed significantly fewer responsive cells (Fig. 4D–E), and the cells that did respond showed decreased response amplitude (Fig. 4F). We observed no significant responses to PhotoS1P in neurons from S1P3 KO mice (Fig. 4G), indicating selectivity of this new tool.
Figure 4 |. Optical control of nociception in DRG neurons and mice.

(A) Representative images depicting Fura2-AM Ca2+ imaging of WT DRG neurons after addition of cis-PhotoS1P (500 nM, upper and lower left) and after illumination with 460 nm light to trigger isomerization to trans-PhotoS1P (upper right) or illumination with 365 nm light (lower right). Rainbow scale indicates F340/F380 ratio; scale = 50 μm. (B,C,D) Representative traces (light grey, dark grey, back) of three DRG neurons each displaying the F340/F380 signal from Fura2-AM Ca2+ imaging with addition of S1P (500 nM; B) or cis-PhotoS1P (500 nM; C-D), with illumination at 460 nm to trigger isomerization to trans-PhotoS1P (C), or illumination at 365 nm (D). At the end of all experiments, capsaicin (1 μM) was added to trigger activation of TRPV1+ nociceptors and high K+ Ringer’s solution was added to activate excitable neurons. (E) Quantification of % of total wild-type (WT) mouse DRG somatosensory neurons responding to S1P (500 nM), trans-PhotoS1P (500 nM), trans-PhotoS1P (250 nM), trans-PhotoS1P (100 nM), cis-PhotoS1P (500 nM), vehicle control (VH), capsaicin (1 μM) and high K+ Ringer’s solution (***p < 0.0001; F(7,100) = 165.5; one-way ANOVA). Tukey’s multiple comparisons p-values (left to right): n.s. > 0.999; *** = 0.0002; n.s. = 0.8659; n.s. = 0.9715. Experiments were performed on a total of 2,322 neurons from N = 3 animals, with n = number 50 of imaging experiments of 20–100 DRG neurons each. Experiment was repeated a total of three times (once per animal used), yielding similar results. (F) Max change in F340/F380 ratio after addition of S1P (500 nM), trans-PhotoS1P (500 nM), cis-PhotoS1P (500 nM), or vehicle control (VH) (***p < 0.0001; F(3,1848) = 4.41; one-way ANOVA). Tukey’s multiple comparisons p-values (left to right): n.s. = 0.9841; *** < 0.0001; *** < 0.0001; n.s. = 0.8213. n = number of DRG cells from N = 3 animals. (G) Quantification of total S1P3 receptor KO DRG neurons responding to trans-PhotoS1P (500 nM), vehicle control (VH), capsaicin (1 μM), and high K+ Ringer’s solution (p = 0.081; t = 1.84; d.f. = 20; two-tailed unpaired t-test). Experiments were performed on a total of 1,114 neurons from N = 3 animals, with n = number of imaging experiments of 20–100 DRG neurons each. Experiment was repeated a total of three times (once per animal used), yielding similar results. (H) 5 mM cis-PhotoS1P was injected into one hind paw of WT mice. Paw withdrawal latency was recorded 20 min before and 10 min after injection. The paw was irradiated at 460 nm (***p < 0.0001; F (2, 12) = 31.16; one-way ANOVA, N = 5 mice); ***p < 0.0001 (Tukey’s multiple comparisons). (I) Paw withdrawal latency was recorded 10 min after injection of S1P (10 mM), after irradiation at 460 nm for 3 min, and after irradiation at 365 nm for 3 min (**p = 0.0028; F (2, 9) = 12.09; one-way ANOVA, N = 4 mice); n.s. = 0.92 (Tukey’s multiple comparisons). (J) The contralateral paws of mice in (H) were injected with cis-PhotoS1P as before, and irradiated at 365 nm for 3 min each (p = 0.5108; F(2,12) = 0.7107; one-way ANOVA, N = 5 mice). Paw withdrawal latency was recorded directly after irradiation. (K) Paw withdrawal latency was recorded in a non-injected control group before irradiation, after irradiation at 460 nm for 3 min, and after irradiation at 365 nm for 3 min (p = 0.7553; F (2, 9) = 0.2896; one-way ANOVA, N = 4 mice). (L) Experiment was conducted as in (H, J), but withdrawal latency was recorded after a subsequent cycle of irradiation at 365 nm for 3 min, and after a cycle of irradiation at 460 nm for 3 min (**p = 0.0013; F (1.478, 4.435) = 45.21; one-way ANOVA, N = 4 mice). Tukey’s multiple comparisons p-values (left to right): ** = 0.0047; * = 0.0208; * = 0.0220. Unless otherwise indicated, error bars represent mean ± S.D. and statistics were performed using one-way ANOVA followed by Tukey-Kramer post hoc test or two-tailed t-test, where appropriate.
We subsequently investigated the potential of PhotoS1P for the optical control of heat hypersensitivity in vivo. Light was delivered to the hindpaw of mice by transdermal illumination using a LED flashlight (21×5 mm LED flashlight array with λmax = 460 ± 10 nm). PhotoS1P was injected as inactive cis-PhotoS1P into each hindpaw. One paw was irradiated with 460 nm light to drive photoswitching to trans-PhotoS1P, while the other paw was irradiated with 365 nm light to prevent photoisomerization. We measured the latency to paw withdrawal from a noxious heat stimulus immediately before and after illumination with 460 nm or 365 nm light using the Hargreaves assay (Fig. 4H). Illumination with 460 nm light to trigger photoisomerization to trans-PhotoS1P (Fig. 4I) evoked hypersensitivity similar to injection of S1P (Fig. 4J). In contrast, heat sensitivity was unchanged by illumination with 365 nm light (Fig. 4K), or by illumination with either 460 nm or 365 nm light in un-injected paws (Fig. 4L).
Notably, the degree of heat hypersensitivity could be modulated in the same animal by alternating illumination with 460 and 365 nm light (Fig. 4M). Following induction of heat hypersensitivity with 460 nm light, illumination with 365 nm partially, but significantly, increased the latency to respond to noxious heat stimuli (indicative of reduced hypersensitivity). Following the 365 nm illumination, illumination with 460 nm could again decrease response latencies, indicative of increased hypersensitivity. Taken together, our results show that PhotoS1P can reversibly and robustly manipulate S1P signaling in vitro and in vivo.
Light-dependent metabolism of PhotoSph and PhotoS1P
The long duration of the responses we observed in our in vivo experiments suggested that PhotoS1P could exhibit markedly improved metabolic stability compared to S1P. This motivated us to explore the metabolism of these synthetic lipids systematically. Additionally, PhotoSph and PhotoS1P could be valuable tools for the control of sphingolipid metabolism. We focused this study on metabolic processes catalyzed by S1P-lyase (Sgpl1), sphingosine kinase 1/2 (SPHK1/2), and S1P-phosphatase 1/2 (Sgpp1/2). The capacity of SPHK1/2 to phosphorylate PhotoSph and a potential photoisomer-dependence of this catalytic process was first evaluated in vitro (Fig. 5). Interestingly we observed opposite selectivities: SPHK1 phosphorylated cis-PhotoSph (9) more readily than trans-PhotoSph (10), while SPHK2 phosphorylated trans-PhotoSph more readily than cis-PhotoSph. In each case the more readily phosphorylated photoisomer was transformed with similar catalytic efficiency compared to d-erythro-sphingosine (18:1). The opposing selectivities observed could potentially make PhotoSph a useful tool for the determination of relative activity between the SPHK isoforms in biological samples.
Figure 5 |. In vitro SPHK Assay.

100 μM Sphingosine, trans-PhotoSph and cis-PhotoSph were incubated with SPHK1 (A) or SPHK2 (B) with ATP as the limiting reagent for 2 h. ATP was converted into a luminescence signal proportional to the amount of remaining ATP. Samples were run in triplicates. Samples (N = 3) were run in triplicates. Error bars represent SEM. **p = 0.0031; ***p = 0.0002; ****p < 0.0001; n.s. = 0.8662; two tailed student’s t-test.
We subsequently investigated how PhotoSph and PhotoS1P are taken-up and metabolized in living cells (Fig. 6). WT HeLa MZ cells treated with trans-PhotoSph exhibited higher levels of PhotoSph and lower levels of PhotoS1P after 15 min when compared to treatment with cis-PhotoSph (Fig. 6B). WT HeLa MZ cells treated with trans-PhotoS1P exhibited higher levels of PhotoS1P and PhotoSph compared to treatment with cis-PhotoSph (Fig. 6C). These results could be influenced by a variety of factors, including isomer-dependent cell uptake, bioavailability (especially for partially cytosolic PhotoS1P), and competing metabolic pathways (Fig. 6A). We took advantage of various KO cell lines generated in parallel to previous studies18,39 to investigate the light-dependence of individual metabolic processes and cell uptake. We first used HeLa MZ SPHK1/2 double KO cells together with fumonisin B1, a potent inhibitor of ceramide synthases, to inhibit the metabolism of PhotoSph and study the direct cellular uptake of PhotoSph (Fig. 6D). PhotoSph was rapidly taken up by cells with no significant isomer-dependence. Additionally, the level of PhotoS1P was markedly reduced to the detection limit, indicating that PhotoSph is a selective substrate for SPHK1/2 and was not phosphorylated by other kinases present in the same cell. We next employed McA-RH7777 Sgpl1/Sgpp1/Sgpp2 triple KO cells to limit PhotoS1P metabolism and study its direct uptake in cells (Fig. 6E). This experiment showed that the trans isomer of PhotoS1P was taken up more by cells more readily than the cis isomer. This experiment further showed a marked reduction in PhotoSph levels compared to wild type cells suggesting at least partial substrate selectivity of PhotoS1P for the depleted phosphatases in this cell type. Finally, we evaluated the cellular metabolic equilibrium between PhotoSph and PhotoS1P in McA-RH7777 Sgpl1 KO cells in the presence of fumonisin B1 (Fig. 6F), and found the equilibrium was not isomer-dependent. It is most likely that the expression level and activity of SphK isoforms are cell-type dependent, which in turn affect the rates of PhotoSph isomer phosphorylation. This experiment also revealed a shift of the PhotoSph/PhotoS1P equilibrium to the phosphorylated form, suggesting that phosphorylation of PhotoSph is more efficient than the dephosphorylation of PhotoS1P. As our previous in vivo experiments suggested that PhotoS1P is a metabolically stable analog of S1P, we decided to assess PhotoS1P levels over an extended time course in WT HeLa MZ cells (Fig. 6G). After an initial metabolic equilibration, the levels of trans-PhotoS1P remained nearly constant from 15–60 minutes, demonstrating markedly improved metabolic stability compared to S1P. From the whole dataset we could conclude that PhotoSph and PhotoS1P are taken up by cells and interconverted by sphingosine kinases and S1P phosphatases with a bias towards the phosphorylated form, which might also contribute to the metabolic stability of PhotoS1P. The markedly increased metabolic stability observed suggests that a pool of PhotoS1P is not quickly accessible to degradation by S1P lyase. The observation that PhotoSph and PhotoS1P are metabolized by the same set of enzymes as endogenous sphingoid bases shows that these lipid analogs are useful modulators of sphingolipid metabolism. These processes are at least in part isomer-dependent, providing opportunities for the optical control of this metabolic network.
Figure 6 |. Lipid mass spectrometry analysis of PhotoSph/PhotoS1P metabolism.

(A) Metabolic network of photoswitchable sphingolipids. (B) WT HeLa MZ cells were treated with photoisomers of PhotoSph for 15 min. PhotoSph (left) and PhotoS1P (right) were extracted and quantified (**p < 0.0055 and **p = 0.0050 (left to right); two-tailed student’s t-test, N = 3 biologically independent experiments). (C) WT HeLa MZ cells were treated with photoisomers of PhotoS1P for 15 min. PhotoSph (left) and PhotoS1P (right) were extracted and quantified (****p < 0.0001; ***p = 0.0002; two-tailed student’s t-test, N = 3 biologically independent experiments). (D) HeLa MZ SPHK1/2 dKO cells were treated with photoisomers of PhotoSph for 15 min in the presence of fumonisin B1 (10 μM) to block ceramide synthases. PhotoSph (left) and PhotoS1P (right) were extracted and quantified (n.s. = 0.0716 and n.s. = 0.1752 (left to right); two-tailed student’s t-test, N = 3 biologically independent experiments). (E) McA-RH7777 Sgpl1/Sgpp1/Sgpp2 triple KO cells were treated with photoisomers of PhotoS1P for 15 min. PhotoSph (left) and PhotoS1P (right) were extracted and quantified (**p = 0.0048; *p = 0.0211; two-tailed student’s t-test, N = 3 biologically independent experiments). (F) McA-RH7777 Sgpl1 KO cells were treated with photoisomers of PhotoSph for 15 min in the presence of fumonisin B1 (10 μM) to block ceramide synthases. PhotoSph (left) and PhotoS1P (right) were extracted and quantified (n.s. = 0.4989 and n.s. = 0.7910 (left to right); two-tailed student’s t-test, N = 3 biologically independent experiments). (G) Metabolic stability of trans-PhotoS1P. WT HeLa cells were treated with trans-PhotoS1P and PhotoS1P was extracted and quantified after 0, 15, 30, and 60 min, respectively. Error bars represent SEM.
The degradation of sphingolipids generates fatty aldehydes that are further metabolized into fatty acids, which can be incorporated into various lipid species.40 If this happens too extensively with PhotoSph and PhotoS1P, many undesirable photoswitchable lipids might be produced in the cells. To investigate this possibility, we used various scan modes of mass spectrometry to detect different classes of sphingolipids and glycerophospholipids selectively.41 By performing a scan specific for ceramides and hexosylceramides bearing the azobenzene photoswitch (Supplementary Fig. 4A), we found that PhotoSph is converted into various ceramide species, the signals of which reached comparable levels to endogenous ones (Supplementary Fig. 4B). On the other hand, conversion into hexosylceramides was undetectable under these conditions (Supplementary Fig. 4C). Importantly, on incubation with cells PhotoS1P was neither converted into ceramides nor into hexosylceramides (Supplementary Fig. 4A–C), as could be imagined from the very low levels of PhotoSph produced under these conditions (Fig. 6C). We then performed a scan specific for lipids bearing a phosphocholine head group, namely phosphatidylcholine (PC) and sphingomyelin (SM) (Supplementary Fig. 5). While endogenous PC and SM species were robustly detected, signals that would correspond to PC or SM bearing the azobenzene photoswitch were very weak and unchanged between samples. Thus, these signals were interpreted as mixtures of noise and isotopic peaks of endogenous PC and/or SM, and not coming from metabolites of PhotoSph. Similar experiments were performed to detect phosphatidylethanolamine (PE), phosphatidylserine, and phosphatidylinositol. Similar to PC, no specific increase in lipids bearing the azobenzene photoswitch was detected (data not shown, except for PE, which quantification results are shown in Supplementary Fig. 5C, as an example). Thus, our results showed that at least upon short incubation, the metabolism of PhotoSph is limited to ceramides, while PhotoS1P is not converted into other species in significant quantities. Thus, the effects of PhotoS1P on cellular phenotypes are likely to be caused by PhotoS1P itself, and not its metabolites.
Discussion
Here we show that photoswitchable analogs of sphingosine and S1P, PhotoSph and PhotoS1P, are versatile tools for the optical control of sphingolipid metabolism and signaling in vitro and in vivo. These molecules were synthesized by incorporation of an azobenzene photoswitch into the lipid tail. This approach preserves the integrity of the lipid headgroup, allowing these molecules to function in cells similarly to the endogenous lipids, while enabling light-dependent modulation of lipid function. We show that PhotoS1P provides precise optical control of S1P receptor 1, 2 & 3 function. Moreover, we used PhotoS1P to reversibly induce S1P3 receptor-mediated nociceptor activation in cultured DRG neurons and to modulate S1P-evoked pain hypersensitivity in mice.
S1P signaling in DRG neurons is particularly well-suited to our photopharmacological approach for several reasons. First, PhotoS1P elicits a comparable nociceptive response in behavioral studies by activating the same signaling pathways as native S1P, which cannot be achieved using chemogenetic or optogenetic approaches. This is particularly important for the study of pain because nociceptors are polymodal and utilize distinct receptor-linked transduction pathways in response to distinct inflammatory ligands.37 All modes of depolarization are not equal. Second, unlike PhotoS1P, application of S1P receptor agonists and antagonists to cells or injection in animals does not allow for temporal control over S1P signaling. We have shown that such agonists/antagonists trigger long-lasting effects on neuronal function and animal behavior.6 In contrast, here we show we can turn the S1P pathway on and off within seconds (in neurons) and minutes (in vivo behavior). This level of control is key to unraveling both the acute effects of S1P signaling (e.g. depolarization and action potential firing) and the more long-term effects on Ca2+-dependent release of inflammatory mediators from nociceptors.37 Third, photoswitching can be used to examine ON/OFF dynamics of the downstream signal transduction pathways (e.g. coupling of S1P receptors to ion channels, G protein pathways, etc.) by regulating the activity of native S1P receptors. This is especially useful given that it was recently discovered that GPCRs display sequence selectivity for distinct G protein coupled pathways.42 Their analysis revealed at least one S1P receptor, S1P3 (the primary S1P receptor in the nervous system6,43), was shown to display a uniquely promiscuous pattern of G protein coupling, which is supported by recent studies of S1P3 signaling mechanisms in neurons38. Unlike our experiments using photoswitchable S1P, substitution of native receptors with a DREADD or a light-activated GPCR would not allow for mechanistic study of the endogenous S1P signaling pathways. The ability of tools such as ours to rapidly modulate endogenous receptor activity may provide a unique advantage in biomedical studies and translational studies over transgenesis, viral induction, and other approaches that may take days to months to take effect.
PhotoS1P may prove useful in combination with in vivo recording of neuronal activity of peripheral and central neurons in awake mice to better unravel the cells and circuitry of pain sensation. The transdermal illumination employed here is minimally invasive and convenient but potentially limits reversibility because of limited tissue penetration of the UV/A light that photoisomerizes PhotoS1P to the less potent cis form for the S1P3 receptor. Implanted light delivery devices could overcome this limitation.
Our study indicates that PhotoS1P is not quickly degraded by S1P lyase and is therefore significantly more stable in cells and in vivo than S1P. As the headgroup of S1P is retained in PhotoS1P, it closely mimics the native ligand. As a metabolically stable and photoswitchable derivative, PhotoS1P is uniquely and ideally suited to study S1P biology with opportunities for unprecedented spatial and temporal control without the rapid degradation that has limited the direct study of S1P action in vivo. It would be interesting to explore whether incorporation of conformationally restricted aromatic rings in lipid tails translates to increased metabolic stability in other lipids. Such metabolically stable analogs could be useful for applications in research and medicine.
Furthermore, mass spectrometry analysis revealed that phosphorylation and dephosphorylation of PhotoS1P are exclusively catalyzed by sphingolipid-metabolizing enzymes, providing further evidence for how closely our photoswitchable lipids mimic endogenous sphingolipids and opening up new opportunities for these lipids in the modulation of sphingolipid metabolism.
In addition to the applications reported herein, which rely on the S1P receptor family and its metabolism, PhotoS1P could be useful for the modulation of intracellular S1P targets. These include HDACs for the optical control of epigenetic processes5, TRAF2 for the optical control of immunity44, prohibitin 2 for the optical control of mitochondrial biology, and BACE145. While we have only explored PhotoSph in its metabolic context, this tool opens up a wide range of opportunities in the optical control of sphingosine biology.
Online Methods
Electrophysiology
HEK293T cells were maintained in DMEM (Invitrogen) with 10% fetal bovine serum on poly-L-lysine-coated coverslips. Cells were seeded onto 18 mm coverslips, transiently transfected overnight with 0.5 μg/well S1P1 receptor and GIRK1-F137S, along with 0.1 μg/well tdTomato as a transfection marker using Lipofectamine 2000 (Invitrogen). Whole cell patch clamp recordings were performed 16–24 h after transfection in high potassium solution containing 120 mM KCl, 25 mM NaCl, 10 mM HEPES, 2 mM CaCl2, and 1 mM MgCl2, pH 7.4. Cells were voltage clamped to −80 mV using an Axopatch 200A (Molecular Devices) amplifier. All compounds were applied using a gravity-driven perfusion system. Illumination was applied to the entire field of view using a Polychorme V monochromator (TILL Photonics) through a 20x objective (4 mW/mm2 at 460 nm or 0.5 mW/mm2 at 360 nm). pClamp software was used for both data acquisition and control of illumination.
Calcium Mobilization
Ca2+ mobilization assays were carried out as previously described.46 Briefly, HTC4 cells stably expressing S1P1–5 receptors were plated in poly-L-lysine coated 96 well microplates (25,000 cells/well) and cultured overnight. The culture medium was replaced with Krebs buffer for 2 – 3 h before assays. The transfected cells were loaded with Fura-2/AM in Krebs buffer containing 0.01% pluronic acid for 30 min, and rinsed with Krebs buffer before measuring Ca2+ mobilization. The Ca2+ responses were measured using a Flex Station III fluorescent plate reader (Molecular Devices, Sunnyvale, CA). The ratio of peak emissions at 510 nm after 2 min of ligand addition was determined for excitation wavelengths of 340 nm/380 nm. All samples were run in at least in triplicate, and assays were performed at least two times for each receptor. The calcium response of trans-PhotoS1P was recorded in the dark-adapted state without prior illumination. The calcium response of cis-PhotoS1P was recorded in the same plate from the same stock solution. Between measurements for trans-PhotoS1P and cis-PhotoS1P, the compound addition well plate was removed from the plate reader shortly and illuminated for 90 s at 365 nm light. The responses were measured and reported in terms of maximal activation (Emax) and potency (EC50) in the Supporting Information. For receptor desensitization, HTC4 cells stably expressing S1P1 receptor were prepared analogously and treated with S1P (300 nM) or charcoal-stripped BSA (300 nM) for 2 h. Cells were loaded with Fura-2/AM in Krebs buffer containing 0.01% pluronic acid for 30 min, and rinsed with Krebs buffer before measuring Ca2+ mobilization.
Modeling
Models of S1P1-S1P5 receptors were constructed by homology modeling using the MOE version 2018.01 software47 using the crystal structure of S1P1 receptor (PDB48 entry 3V2Y49) as a template. Structural models of ligands were constructed with phosphate groups in both −1 and −2 ionization states as both are expected to be present at the pH values used in functional assays. Other functional groups were modeled in the ionization state expected at pH 7.4. Receptors and ligands were geometry optimized using the MMFF94x forcefield before docking. Docking was performed using default settings except that 400 ligand poses from the placement phase were subjected to refinement during which the ligand and surrounding receptor sidechains were freely mobile. The five top poses were retained and reviewed after the refinement phase. The top pose from both ionization states of each ligand in each receptor was selected based on best complementation of ligand/receptor polar functional groups and good surface contact between nonpolar ligand segments and the receptor. These procedures provided S1P complexes with S1P1,2,4,5 receptors in which the phosphate group of S1P interacts with the conserved R3.28 residue as previous mutagenesis studies indicate is essential for S1P receptor activation.50,51 S1P3 receptor docking results were inconsistent with observed experimental results due to poor complementation of S1P polar functional groups by S1P3 receptor in the original model (results not shown). A second S1P3 receptor model was constructed using the S1P1 receptor model complex with S1P in which the S1P position was used as an environment for induced fit during S1P3 receptor model construction. The ligands were docked into the second S1P3 receptor model (results shown in the supplementary data).
Calcium Imaging
Neuron dissection and Ca2+ imaging experiments were carried out as previously described.52 Neurons from dorsal root ganglia (2–8 week old male adult mice, either C56bl/6 (WT, Jackson Laboratory) or S1pr3tm1Rlp (S1PR3 KO, MGI)) were dissected and incubated for 10 min in 2 mg/ml Collagenase P (Roche) in Hanks’ calcium-free balanced salt solution (Gibco), followed by incubation in 0.25% trypsin (vol/vol) STV versene-EDTA solution (Gibco) for 2 min with gentle agitation. Cells were then triturated, plated onto poly D-lysine (Sigma) and Laminin (Corning) coated glass coverslips and used within 24 h. Media: MEM Eagle’s with Earle’s BSS medium, supplemented with 10% horse serum (vol/vol), MEM vitamins, penicillin/streptomycin and L-glutamine (Gibco). Cells were loaded for 60 min at room temperature with 10 mM Fura-2AM supplemented with 0.01% Pluronic F-127 (wt/vol, Life Technologies) in a physiological Ringer’s solution containing (in mM) 140 NaCl, 5 KCl, 10 HEPES, 2 CaCl2, 2 MgCl2 and 10 D-(+)-glucose, pH 7.4. All chemicals were purchased from Sigma. Acquired images were displayed as the ratio of 340 nm/380 nm. Cis-PhotoS1P was added to cells. Images were acquired once every three seconds with exposure times ranging between 10–100 ms. Light stimuli were manually applied to cells using LED light source in between image acquisition. Cells were identified as heat nociceptors via capsaicin addition (1 μM) and as neurons by eliciting depolarization with high potassium Ringer’s solution (75 mM) at the end of each experiment. Responding neurons were defined as those having a > 20% increase from mean ratio prior to compound addition. Fura-2 ratios were normalized to the baseline ratio, defined as F340/F380 = (Ratio)/(Ratio t = 0). Images were acquired using an Olympus IX71 microscope with a Lambda LS-xl light source (Sutter Instruments), and data were analyzed using IgorPro 6 (Wavemetrics).
Behavioral Studies and Mice
Age-matched male C57bl/6 mice (The Jackson Laboratory) were used for all behavioral experiments. Mice (20–25 g) were housed in a 12 h light-dark cycle at 21 °C in a specific pathogen-free facility with unrestricted access to food and water. Mice were singly housed one week prior to all behavioral experiments and were between 8–10 weeks of age at the time of the experiment. Behavioral experiments were performed starting in the late morning through early afternoon. All mice were acclimated in Plexiglass behavioral chambers (IITC Life Sciences) on two subsequent days for 1 h prior to all behavioral experiments. Pain behavioral measurements were performed as previously described, using the Hargreaves radiant heat assay (IITC).53 Three trials were recorded for each mouse and time point, and the average of the three trials was used. For behavioral experiments, “N” was defined as an individual mouse. Compounds injected: 10 μM S1P (Tocris, Avanti Polar Lipids) in 1% methanol-PBS vehicle, and 5 μM PhotoS1P with 0.5% ethanol-PBS vehicle. cis-PhotoS1P was injected intradermally into the hind paws of mice using a 31 g insulin syringe. LED light source was manually applied to hindpaw for 3 minutes to activate or inactivate Photo-S1P. All injections were 20 μL delivered intradermally. No randomisation was employed, since mice received control treatment in one paw and the experimental treatment in the other paw. Experimenter was blinded to compounds injected and mice were acclimated, injected, and tested in the dark with a red light source. The radiant heat source raised the platform temperature to 39.8 °C within 5 seconds, and to 60 °C within 10 seconds, as measured by a fast temperature probe (Physitemp). All experiments were performed under the policies and recommendations of the International Association for the Study of Pain and approved by the University of California, Berkeley Animal Care and Use Committee.
In Vitro SphK Assays
The activity of sphingosine kinase was quantified by using a commercial sphingosine kinase activity assay kit (Echelon Biosciences, Salt Lake City, UT) as the manufacturer instructed. Trans-PhotoS1P was tested in the dark-adapted state without prior illumination and cis-PhotoS1P was obtained through pre-illumination by 365 nm light for 90 s and tested in the same experiment. All compounds were tested at least in triplicates and incubated with the respective isoform of sphingosine kinase for 2 h.
Lipid Mass Spectrometry
In each experiment, the lipids in DMSO stock were illuminated with 365 nm or 460 nm light for 3 min at room temperature to obtain trans-PhotoS1P and cis-PhotoS1P. The lipids were then dissolved in HBSS solution (physiological buffer) with a final concentration of 2 μM during all experiments. Fully confluent cells in 60 mm dishes were treated with photoswitchable lipids for 15 min at 37 °C. Lipids were extracted and measured by LC-MS using protocols published previously.54–56 Briefly, cells were washed with cold PBS and scraped off in 500 μl cold PBS on ice. The suspension was transferred to a 1.5 ml Eppendorf tube in which it was spin down at 2500 rpm for 5 min at 4 °C. After taking off the PBS, samples were stored at −20 °C or directly used for further extraction. Samples were resuspended in 150 μL extraction buffer (ethanol, water, diethyl ether, pyridine, and 4.2 N ammonium hydroxide (15:15:5:1:0.018, v/v)). A mixture of the internal standards (0.04 nmol of C17 sphingosine, 0.4 nmol of C17 sphingosine-1-phosphate) was added. The samples were vigorously vortexed using a Cell Disruptor Homogenizer (Disruptor Genie, Scientific Industries) for 10 min at 4 °C and incubated on ice for 20 min. Cell debris were pelleted by centrifugation at 14,000 rpm for 2 min at 4 °C, and the supernatant was collected. The extraction was repeated once more without ice incubation. The supernatants were combined and dried under vacuum in a CentriVap (Labconco, Kansas City, MO). The samples were re-suspended in a mixture of solvents composed of 70 μl of borate buffer (200 mM boric acid pH 8.8, 10 mM tris (2-carboxyethyl)-phosphine, 10 mM ascorbic acid and 33.7 mM 15N13C-valine), and 10 μl of formic acid solution (0.1% aqueous solution), derivatized by reacting with 20 μl 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC) solution (2.85 mg/ml in acetonitrile) for 15 min at 55 °C. After overnight incubation at 24 °C, samples were analyzed by LC- MS in an Accela HPLC system (ThermoFisher Scientific, Waltham, USA) coupled to a TSQ Vantage (ThermoFisher Scientific, Waltham, USA). MRM-MS was used to identify and quantify sphingoid bases. The raw data were normalized by C17 sphingolipid standards and cell population, as “pmol / 10^6 cells”. For the extended time-course of PhotoS1P degradation, cells were washed with HBSS after 15 minutes of PhotoS1P incubation and were scraped off for centrifugation and extraction at the indicated time points. Data represent the average of three independent experiments. Error bars represent standard error of the mean (SEM) as indicated. Statistical significance was calculated based on two-tailed unpaired student’s t-test. To detect the incorporation of the azobenzene photoswitch into various lipid classes, cells were treated with trans-PhotoSph or trans-PhotoS1P, and pelleted as described above. Total lipids were extracted using the methyl-tert-butyl ether method57, after addition of internal standards (0.4 nmol 24:0 phosphatidylcholine, 1 nmol 31:1 phosphatidylethanolamine, 1 nmol 31:1 phosphatidylinositol, 3.3 nmol 31:1 phosphatidylserine, 0.7 nmol 56:0 cardiolipin, 2.5 nmol C12 sphingomyelin, 0.5 nmol C17 ceramide, 0.1 nmol glucosylceramide, and 8 nmol ergosterol). Lipids from selected classes were detected by specific scans using a triple-stage quadrupole TSQ Vantage mass spectrometer (Thermo Scientific) receiving nanoflow infusion from a robotic ion source, Nanomate HD (Advion Biosciences, Ithaca, NY). Ceramides + hexosylceramides, Photoceramides + photohexosylceramides, phosphatidylcholine + sphingomyelin, phosphatidylethanolamine, phosphatidylserine, and phosphatidylinositol were detected by performing precursor ion scans for m/z 264.3 (positive mode, collision energy 30 eV), precursor ion scans for m/z 332.3 (positive mode, collision energy 30 eV), precursor ion scans for m/z 184.1 (positive mode, collision energy 37 eV), neutral loss scans for m/z 141.0 (positive mode, collision energy 20 eV), neutral loss scans for m/z 87.0 (negative mode, collision energy 23 eV), and precursor ion scans for m/z 241 (negative mode, collision energy 44 eV), respectively. Peak intensities were analyzed using the XCalibur software (Thermo Scientific), and normalized with those of appropriate internal standards.
Generation of knockout cells using CRISPR/Cas9
SPHK1/2 double knockout HeLa MZ cells (a HeLa cell subclone gifted by Marino Zerial, Max Planck Institute) were generated previously56. Sgpl1 knockout and Sgpl1/Sgpp1/Sgpp2 triple knockout McA-RH7777 cells were generated by Hprt co-targeting58, using guide RNAs that were previously designed59. Sequences of the guide RNAs and the plasmid backbones used for their expression (pX330, addgene plasmid #42230 deposited by Feng Zhang, Broad Institute; pUC-U6-sg59) are listed in supplementary table. Plasmids encoding both Cas9 and the target guide RNA(s) were co-transfected with plasmids encoding the guide RNA for the Hprt gene using Lipofectamine 3000 (ThermoFisher Scientific, Waltham, USA). Seven days later, cells were selected for 1 week with 4 μg/mL 6-thioguanine, which enabled a strong enrichment of target-mutated cells, due to the resistance conferred by loss-of-function in the co-targeted Hprt gene. The resulting mutation rate was based on the loss of WT signal calculated by TIDE analysis60, which was 100% for Sgpl1 in Sgpl1 knockout cells, and 96.1%, 100%, and 83.8% for Sgpl1, Sgpp1, and Sgpp2, respectively in Sgpl1/Sgpp1/Sgpp2 triple knockout cells. Details about optimization of the methodology will be published elsewhere.
Synthetic protocols
Unless otherwise stated, all reactions were performed with magnetic stirring under a positive pressure of nitrogen or argon gas. Dry tetrahydrofuran (THF), diethyl ether (Et2O), dichloromethane (CH2Cl2), triethylamine (Et3N), N,N-dimethylformamide (DMF), toluene (PhMe), dioxane and methanol (MeOH) were obtained by passing the previously degassed solvents through activated alumina columns. Solvents and reagents were used as received from commercial sources (Sigma-Aldrich, Tokyo Chemical Industry Co., Alfa Aesar, Acros Organics, Strem Chemicals). Reactions were monitored by thin-layer chromatography (TLC) using silica gel F254 pre-coated glass plates (Merck) and visualized by exposure to ultraviolet light (λ = 254 nm) or by staining with aqueous potassium permanganate (KMnO4) solution (7.5 g KMnO4, 50 g K2CO3, 6.25 mL aqueous 10% NaOH, 1000 mL distilled H2O), aqueous acidic ceric ammonium molybdate (IV) (CAM) solution (2.0 g Ce(NH4)4(SO4)4·2H2O, 48 g (NH4)6Mo7O24·4H2O, 60 mL concentrated sulfuric acid, 940 mL distilled H2O) or a butanolic ninhydrin solution (13.5 g ninhydrin, 900 mL n-BuOH, 27 mL acetic acid) followed by heating with a heat gun (150–600 °C). Flash column chromatography was performed using silica gel (60 Å, 40–63 μm, Merck). Proton (1H) and carbon (13C) nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance III HD 400 MHz spectrometer equipped with a CryoProbe™ or a Bruker Avance III HD 600 MHz spectrometer equipped with a CryoProbe™. Proton chemical shifts are expressed in parts per million (ppm, δ scale) and referenced to residual undeuterated solvent signals. Carbon chemical shifts are expressed in parts per million (ppm, δ scale) and referenced to the central carbon resonance of the solvent. The reported data is represented as follows: chemical shift in parts per million (ppm, δ scale) (multiplicity, coupling constants J in Hz, integration intensity). Abbreviations used for analysis of multiplets are as follows: s (singlet), br s (broad singlet), d (doublet), t (triplet), q (quartet), p (pentet), h (hextet), and m (multiplet) or combinations thereof. Variable temperature NMR spectroscopy was performed on a Bruker AV-400 High Performance Digital NMR Spectrometer (400 MHz). NMR spectra were acquired at 25 °C unless stated otherwise. NOTE: Due to the trans/cis equilibrium of some compounds containing an azobenzene functionality, more signals were observed in the 1H and 13C spectra than would be expected for the pure trans-isomer. Only signals for the major trans-isomer are reported, however the identities of the remaining peaks were verified by 2D COSY, HSQC and HMBC experiments. UV-Vis spectra were recorded using a Varian Cary 50 Bio UV-Visible Spectrophotometer with Helma SUPRASIL precision cuvettes (10 mm light path). All compounds were dissolved at a concentration of 20 μM in DMSO. An initial spectrum was recorded (dark-adapted state, black) and then again following illumination at λ = 365 nm (cis-adapted state, gray). A third spectrum was recorded after irradiation at λ = 460 nm for 30 s (trans-adapted state, blue). For illumination, a 12 × 5 mm 365 nm LED flashlight array (λmax = 373 ± 17 nm) and a 21 × 5 mm 460 nm LED flashlight array (λmax = 460 ± 10 nm) were used. High-resolution mass spectrometry (HRMS) experiments were performed on an Agilent 6224 Accurate-Mass TOF/LC/MS spectrometer.
Statistical Analysis
Unless stated otherwise, data in the present publication are expressed as mean ± standard error of the mean (s.e.m.) and data were analyzed using ANOVA or paired Student’s t-test (see section).
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data Availability
The authors declare that all relevant data supporting the findings in this study are available within this paper and the supplementary information files.
Supplementary Material
ACKNOWLEDGMENTS
J.M. thanks German Academic Scholarship Foundation (Studienstiftung) for a PhD Fellowship. J.M. and A.J.E.N. thank New York University for MacCracken PhD fellowships. T.H. was supported by the Japan Society for the Promotion of Science (JSPS) Postdoctoral Fellowships for Research Abroad. D. D. N. and G. Y. T. were supported by the NCI grant CA092160. H.R. was supported by Sinergia, the Swiss National Science Foundation (CRSII3-154405) and the NCCR Chemical Biology funded by the Swiss National Science Foundation (51NF40-160589). D.M.B was supported by NIH grants AR059385 and NS077224, and an HHMI Faculty Scholar Award. We thank Belinda Hetzler for critical discussion of the photophysical characterization and Dr. Chin Lin for assistance with NMR experiments. Dr. Sue Chin Lee is acknowledged for performing TNAα assays with PhotoS1P on S1P receptor subtypes (data not included).
Footnotes
COMPETING INTERESTS
The authors declare not competing interests.
References
- (1).Fyrst H; Saba JD An Update on Sphingosine-1-Phosphate and Other Sphingolipid Mediators. Nat. Chem. Biol. 2010, 6 (7), 489–497. 10.1038/nchembio.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hannun YA; Obeid LM Principles of Bioactive Lipid Signalling: Lessons from Sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9 (2), 139–150. 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- (3).Maceyka M; Harikumar KB; Milstien S; Spiegel S Sphingosine-1-Phosphate Signaling and Its Role in Disease. Trends Cell Biol. 2012, 22 (1), 50–60. 10.1016/j.tcb.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Proia RL; Hla T Emerging Biology of Sphingosine-1-Phosphate: Its Role in Pathogenesis and Therapy. J. Clin. Invest. 2015, 125 (4), 1379–1387. 10.1172/JCI76369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hait NC; Allegood J; Maceyka M; Strub GM; Harikumar KB; Singh SK; Luo C; Marmorstein R; Kordula T; Milstien S; et al. Regulation of Histone Acetylation in the Nucleus by Sphingosine-1-Phosphate. Science 2009, 325 (5945), 1254–1257. 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hill RZ; Hoffman BU; Morita T; Campos SM; Lumpkin EA; Brem RB; Bautista DM The Signaling Lipid Sphingosine 1-Phosphate Regulates Mechanical Pain. eLife 2018, 7, e33285 10.7554/eLife.33285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Mair N; Benetti C; Andratsch M; Leitner MG; Constantin CE; Camprubí-Robles M; Quarta S; Biasio W; Kuner R; Gibbins IL; et al. Genetic Evidence for Involvement of Neuronally Expressed S1P1 Receptor in Nociceptor Sensitization and Inflammatory Pain. PLOS ONE 2011, 6 (2), e17268 10.1371/journal.pone.0017268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Camprubí-Robles M; Mair N; Andratsch M; Benetti C; Beroukas D; Rukwied R; Langeslag M; Proia RL; Schmelz M; Montiel AVF; et al. Sphingosine-1-Phosphate-Induced Nociceptor Excitation and Ongoing Pain Behavior in Mice and Humans Is Largely Mediated by S1P3 Receptor. J. Neurosci. 2013, 33 (6), 2582–2592. 10.1523/JNEUROSCI.4479-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kim RH; Takabe K; Milstien S; Spiegel S Export and Functions of Sphingosine-1-Phosphate. Biochim. Biophys. Acta BBA - Mol. Cell Biol. Lipids 2009, 1791 (7), 692–696. 10.1016/j.bbalip.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Książek M; Chacińska M; Chabowski A; Baranowski M Sources, Metabolism, and Regulation of Circulating Sphingosine-1-Phosphate. J. Lipid Res. 2015, 56 (7), 1271–1281. 10.1194/jlr.R059543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Brinkmann V; Davis MD; Heise CE; Albert R; Cottens S; Hof R; Bruns C; Prieschl E; Baumruker T; Hiestand P; et al. The Immune Modulator FTY720 Targets Sphingosine 1-Phosphate Receptors. J. Biol. Chem. 2002, 277 (24), 21453–21457. 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- (12).Chun J; Hartung H-P Mechanism of Action of Oral Fingolimod (FTY720) in Multiple Sclerosis. Clin. Neuropharmacol. 2010, 33 (2), 91–101. 10.1097/WNF.0b013e3181cbf825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wenk MR Lipidomics: New Tools and Applications. Cell 2010, 143 (6), 888–895. 10.1016/j.cell.2010.11.033. [DOI] [PubMed] [Google Scholar]
- (14).Wenk MR The Emerging Field of Lipidomics. Nat. Rev. Drug Discov. 2005, 4 (7), 594–610. 10.1038/nrd1776. [DOI] [PubMed] [Google Scholar]
- (15).Schwarzmann G; Arenz C; Sandhoff K Labeled Chemical Biology Tools for Investigating Sphingolipid Metabolism, Trafficking and Interaction with Lipids and Proteins. Biochim. Biophys. Acta BBA - Mol. Cell Biol. Lipids 2014, 1841 (8), 1161–1173. 10.1016/j.bbalip.2013.12.011. [DOI] [PubMed] [Google Scholar]
- (16).Haberkant P; Holthuis JCM Fat & Fabulous: Bifunctional Lipids in the Spotlight. Biochim. Biophys. Acta BBA - Mol. Cell Biol. Lipids 2014, 1841 (8), 1022–1030. 10.1016/j.bbalip.2014.01.003. [DOI] [PubMed] [Google Scholar]
- (17).Höglinger D; Nadler A; Schultz C Caged Lipids as Tools for Investigating Cellular Signaling. Biochim. Biophys. Acta BBA - Mol. Cell Biol. Lipids 2014, 1841 (8), 1085–1096. 10.1016/j.bbalip.2014.03.012. [DOI] [PubMed] [Google Scholar]
- (18).Feng S; Harayama T; Montessuit S; David FP; Winssinger N; Martinou J-C; Riezman H Mitochondria-Specific Photoactivation to Monitor Local Sphingosine Metabolism and Function. eLife 2018, 7, e34555 10.7554/eLife.34555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Broichhagen J; Frank JA; Trauner D A Roadmap to Success in Photopharmacology. Acc. Chem. Res. 2015, 48 (7), 1947–1960. 10.1021/acs.accounts.5b00129. [DOI] [PubMed] [Google Scholar]
- (20).Frank JA; Moroni M; Moshourab R; Sumser M; Lewin GR Photoswitchable Fatty Acids Enable Optical Control of TRPV1. 2015, 6, 7118 10.1038/ncomms8118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Leinders-Zufall T; Storch U; Bleymehl K; Schnitzler M. M. y; Frank JA; Konrad DB; Trauner D; Gudermann T; Zufall F PhoDAGs Enable Optical Control of Diacylglycerol-Sensitive Transient Receptor Potential Channels. Cell Chem. Biol. 2017, 0 (0). 10.1016/j.chembiol.2017.11.008. [DOI] [PubMed] [Google Scholar]
- (22).Frank JA; Yushchenko D; Fine NHF; Duca M; Citir M; Broichhagen J; Hodson DJ; Schultz C; Trauner D Optical Control of GPR40 Signalling in Pancreatic β-Cells. Chem. Sci. 2017. 10.1039/C7SC01475A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lichtenegger M; Tiapko O; Svobodova B; Stockner T; Glasnov TN; Schreibmayer W; Platzer D; de la Cruz GG; Krenn S; Schober R; et al. An Optically Controlled Probe Identifies Lipid-Gating Fenestrations within the TRPC3 Channel. Nat. Chem. Biol. 2018, 14 (4), 396–404. 10.1038/s41589-018-0015-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Frank JA; Franquelim HG; Schwille P; Trauner D Optical Control of Lipid Rafts with Photoswitchable Ceramides. J. Am. Chem. Soc. 2016, 138 (39), 12981–12986. 10.1021/jacs.6b07278. [DOI] [PubMed] [Google Scholar]
- (25).Pernpeintner C; Frank JA; Urban P; Roeske CR; Pritzl SD; Trauner D; Lohmüller T Light-Controlled Membrane Mechanics and Shape Transitions of Photoswitchable Lipid Vesicles. Langmuir 2017, 33 (16), 4083–4089. 10.1021/acs.langmuir.7b01020. [DOI] [PubMed] [Google Scholar]
- (26).Frank JA; Yushchenko DA; Hodson DJ; Lipstein N; Nagpal J; Rutter GA; Rhee J-S; Gottschalk A; Brose N; Schultz C; et al. Photoswitchable Diacylglycerols Enable Optical Control of Protein Kinase C. Nat Chem Biol 2016, 12 (9), 755–762. 10.1038/nchembio.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hüll K; Morstein J; Trauner D In Vivo Photopharmacology. Chem. Rev. 2018, 118 (21), 10710–10747. 10.1021/acs.chemrev.8b00037. [DOI] [PubMed] [Google Scholar]
- (28).Bünemann M; Liliom K; Brandts BK; Pott L; Tseng JL; Desiderio DM; Sun G; Miller D; Tigyi G A Novel Membrane Receptor with High Affinity for Lysosphingomyelin and Sphingosine 1-Phosphate in Atrial Myocytes. EMBO J. 1996, 15 (20), 5527–5534. [PMC free article] [PubMed] [Google Scholar]
- (29).Troupiotis-Tsaïlaki A; Zachmann J; González-Gil I; Gonzalez A; Ortega-Gutiérrez S; López-Rodríguez ML; Pardo L; Govaerts C Ligand Chain Length Drives Activation of Lipid G Protein-Coupled Receptors. Sci. Rep. 2017, 7 (1), 2020 10.1038/s41598-017-02104-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Jeffery T Palladium-Catalysed Arylation of Allylic Alcohols: Highly Selective Synthesis of β-Aromatic Carbonyl Compounds or β-Aromatic α,β-Unsaturated Alcohols. Tetrahedron Lett. 1991, 32 (19), 2121–2124. 10.1016/S0040-4039(00)71252–4. [DOI] [Google Scholar]
- (31).Yamamoto T; Hasegawa H; Hakogi T; Katsumura S Versatile Synthetic Method for Sphingolipids and Functionalized Sphingosine Derivatives via Olefin Cross Metathesis. Org. Lett. 2006, 8 (24), 5569–5572. 10.1021/ol062258l. [DOI] [PubMed] [Google Scholar]
- (32).Lim H-S; Oh Y-S; Suh P-G; Chung S-K Syntheses of Sphingosine-1-Phosphate Stereoisomers and Analogues and Their Interaction with EDG Receptors. Bioorg. Med. Chem. Lett. 2003, 13 (2), 237–240. 10.1016/S0960-894X(02)00893-4. [DOI] [PubMed] [Google Scholar]
- (33).Chan KW; Sui J-L; Vivaudou M; Logothetis DE Control of Channel Activity through a Unique Amino Acid Residue of a G Protein-Gated Inwardly Rectifying K+ Channel Subunit. Proc. Natl. Acad. Sci. U. S. A. 1996, 93 (24), 14193–14198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Atwood BK; Lopez J; Wager-Miller J; Mackie K; Straiker A Expression of G Protein-Coupled Receptors and Related Proteins in HEK293, AtT20, BV2, and N18 Cell Lines as Revealed by Microarray Analysis. BMC Genomics 2011, 12, 14 10.1186/1471-2164-12-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Valentine WJ; Tigyi G High-Throughput Assays to Measure Intracellular Ca2+ Mobilization in Cells That Express Recombinant S1P Receptor Subtypes In Sphingosine-1-Phosphate; Methods in Molecular Biology; Humana Press, 2012; pp 77–87. 10.1007/978-1-61779-800-9_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Rios Candelore M; Wright MJ; Tota LM; Milligan J; Shei G; Bergstrom JD; Mandala SM Phytosphingosine 1-Phosphate: A High Affinity Ligand for the S1P4/Edg-6 Receptor. Biochem. Biophys. Res. Commun. 2002, 297 (3), 600–606. 10.1016/S0006-291X(02)02237-4. [DOI] [PubMed] [Google Scholar]
- (37).Basbaum AI; Bautista DM; Scherrer G; Julius D Cellular and Molecular Mechanisms of Pain. Cell 2009, 139 (2), 267–284. 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hill RZ; Morita T; Brem RB; Bautista DM S1PR3 Mediates Itch and Pain via Distinct TRP Channel-Dependent Pathways. J. Neurosci. 2018, 38 (36), 7833–7843. 10.1523/JNEUROSCI.1266-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Harayama T; Riezman H Detection of Genome-Edited Mutant Clones by a Simple Competition-Based PCR Method. PLOS ONE 2017, 12 (6), e0179165. 10.1371/journal.pone.0179165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Harayama T; Riezman H Understanding the Diversity of Membrane Lipid Composition. Nat. Rev. Mol. Cell Biol. 2018, 19 (5), 281–296. 10.1038/nrm.2017.138. [DOI] [PubMed] [Google Scholar]
- (41).Han X; Yang K; Gross RW Multi-Dimensional Mass Spectrometry-Based Shotgun Lipidomics and Novel Strategies for Lipidomic Analyses. Mass Spectrom. Rev. 2012, 31 (1), 134–178. 10.1002/mas.20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Flock T; Hauser AS; Lund N; Gloriam DE; Balaji S; Babu MM Selectivity Determinants of GPCR–G-Protein Binding. Nature 2017, 545 (7654), 317–322. 10.1038/nature22070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Weth-Malsch D; Langeslag M; Beroukas D; Zangrandi L; Kastenberger I; Quarta S; Malsch P; Kalpachidou T; Schwarzer C; Proia RL; et al. Ablation of Sphingosine 1-Phosphate Receptor Subtype 3 Impairs Hippocampal Neuron Excitability In Vitro and Spatial Working Memory In Vivo. Front. Cell. Neurosci. 2016, 10 10.3389/fncel.2016.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Alvarez SE; Harikumar KB; Hait NC; Allegood J; Strub GM; Kim EY; Maceyka M; Jiang H; Luo C; Kordula T; et al. Sphingosine-1-Phosphate Is a Missing Cofactor for the E3 Ubiquitin Ligase TRAF2. Nature 2010, 465 (7301), 1084–1088. 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Takasugi N; Sasaki T; Suzuki K; Osawa S; Isshiki H; Hori Y; Shimada N; Higo T; Yokoshima S; Fukuyama T; et al. BACE1 Activity Is Modulated by Cell-Associated Sphingosine-1-Phosphate. J. Neurosci. 2011, 31 (18), 6850–6857. 10.1523/JNEUROSCI.6467-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Method References
- (46).Valentine WJ; Tigyi G High-Throughput Assays to Measure Intracellular Ca2+ Mobilization in Cells That Express Recombinant S1P Receptor Subtypes In Sphingosine-1-Phosphate; Methods in Molecular Biology; Humana Press, 2012; pp 77–87. 10.1007/978-1-61779-800-9_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group ULC, 1010 Sherbooke St West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2018. [Google Scholar]
- (48).Berman HM; Battistuz T; Bhat TN; Bluhm WF; Bourne PE; Burkhardt K; Feng Z; Gilliland GL; Iype L; Jain S; et al. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 2002, 58 (Pt 6 No 1), 899–907. [DOI] [PubMed] [Google Scholar]
- (49).Hanson MA; Roth CB; Jo E; Griffith MT; Scott FL; Reinhart G; Desale H; Clemons B; Cahalan SM; Schuerer SC; et al. Crystal Structure of a Lipid G Protein-Coupled Receptor. Science 2012, 335 (6070), 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Inagaki Y; Pham TT; Fujiwara Y; Kohno T; Osborne DA; Igarashi Y; Tigyi G; Parrill AL Sphingosine 1-Phosphate Analogue Recognition and Selectivity at S1P4 within the Endothelial Differentiation Gene Family of Receptors. Biochem. J. 2005, 389, 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Wang DA; Lorincz Z; Bautista DL; Liliom K; Tigyi G; Parrill AL A Single Amino Acid Determines Lysophospholipid Specificity of the S1P1 (EDG1) and LPA1 (EDG2) Phospholipid Growth Factor Receptors. J. Biol. Chem. 2001, 276, 49213–49220. [DOI] [PubMed] [Google Scholar]
- (52).Wilson SR; Gerhold KA; Bifolck-Fisher A; Liu Q; Patel KN; Dong X; Bautista DM TRPA1 Is Required for Histamine-Independent, Mas-Related G Protein–Coupled Receptor–Mediated Itch. Nat. Neurosci. 2011, 14, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Hill RZ; Hoffman BU; Morita T; Campos SM; Lumpkin EA; Brem RB; Bautista DM The Signaling Lipid Sphingosine 1-Phosphate Regulates Mechanical Pain. eLife 2018, 7, e33285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Guan XL; Souza CM; Pichler H; Dewhurst G; Schaad O; Kajiwara K; Wakabayashi H; Ivanova T; Castillon GA; Piccolis M; et al. Functional Interactions between Sphingolipids and Sterols in Biological Membranes Regulating Cell Physiology. Mol. Biol. Cell 2009, 20, 2083–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).da Silveira dos Santos AX; Riezman I; Aguilera-Romero M-A; David F; Piccolis M; Loewith R; Schaad O; Riezman H; Lemmon S Systematic Lipidomic Analysis of Yeast Protein Kinase and Phosphatase Mutants Reveals Novel Insights into Regulation of Lipid Homeostasis. Mol. Biol. Cell 2014, 25, 3234–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Feng S; Harayama T; Montessuit S; David FP; Winssinger N; Martinou J-C; Riezman H Mitochondria-Specific Photoactivation to Monitor Local Sphingosine Metabolism and Function. eLife 2018, 7, e34555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Matyash V; Liebisch G; Kurzchalia TV; Shevchenko A; Schwudke D Lipid Extraction by Methyl-Tert-Butyl Ether for High-Throughput Lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Liao S; Tammaro M; Yan H Enriching CRISPR-Cas9 Targeted Cells by Co-Targeting the HPRT Gene. Nucleic Acids Res. 2015, 43, e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Harayama T; Riezman H Detection of Genome-Edited Mutant Clones by a Simple Competition-Based PCR Method. PLOS ONE 2017, 12, e0179165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Brinkman EK; Chen T; Amendola M; van Steensel B Easy Quantitative Assessment of Genome Editing by Sequence Trace Decomposition. Nucleic Acids Res. 2014, 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all relevant data supporting the findings in this study are available within this paper and the supplementary information files.