

Abstract
C–H activation reactions enable chemists to unveil new retrosynthetic disconnections and streamline conventional synthetic approaches. A longstanding challenge in C–H activation is the inability to distinguish electronically and sterically similar C–H bonds. Although numerous synergistic combinations of transition-metal complexes and chelating directing groups have been utilized to distinguish C–H bonds, undirected regioselective C–H functionalization strategies remain elusive. Herein, we report a regioselective C–H activation/amination reaction of various unsymmetrical dialkyl-substituted alkenes. The regioselectivity of C–H activation is correlated to the electronic properties of allylic C–H bonds indicated by the corresponding 1JCH coupling constants. A linear relationship between the difference of 1JCH coupling constants of the two competing allylic C–H bonds (Δ1JCH) and the C–H activation barriers (ΔΔG‡) has also been determined.
The development of synthetic strategies to diversify molecular frameworks through site-selective functionalization of ubiquitous C–H bonds has been an overarching goal in synthetic chemistry1,2,3,4 However, C–H bonds with nearly identical chemical environments give rise to an enormous challenge for achieving site-selectivity (Fig. 1a). The inherently difficult discrimination of these C–H bonds has been achieved by the synergistic combination of transition-metal complexes and chelating directing groups5,6,7,8, which exploits the differences in conformational energies of the in-situ generated metallacycles in the transition state, so that a particular C–H bond is favored. However, its synthetic applications are limited to substrates with preinstalled directing groups, which mandate additional synthetic steps for removal or further manipulation. A more direct and versatile approach may involve a catalyst system that recognizes the subtle differences in C–H bond strengths, ultimately enabling a site-selective C–H bond activation9,10,11. Electronic effects have been utilized as a powerful tool for the site-selective functionalization of arenes12,13,14. Nevertheless, the application of electronic factors in a broader context of C(sp3)–H functionalizations is extremely challenging as the inductive effect gets weakened significantly through saturated bonds15,16,17.
Figure 1. Site-selective allylic C–H amination.

a, Distinguishing electronically and sterically similar C–H bonds has been a longstanding challenge. b, Previous examples of intermolecular regioselective allylic C–H amination of alkenes possessing two sets of allylic protons. c, A summary of this work, illustrating successful regioselective C–H activation/amination for 1,1- and trans-1,2-disubstituted alkenes as well as a linear correlation between Δ1JCH coupling constants of the two competing C–H bonds and the difference of C–H activation barriers (ΔΔG‡) [Ir], iridium complex; [N], nitrogen source.
Selective allylic C–H functionalizations provide a platform for the construction of valuable building blocks from chemical feedstocks18,19,20,21,22. Currently, intermolecular allylic C–H amination reactions are mostly limited to alkenes with only one distinct set of allylic protons23,24,25,26,27,28,29, due to the lack of methods to distinguish similar allylic positions. Two important exceptions are Dauban’s work30,31 of Rh(II)-catalyzed outer-sphere nitrene insertion preferring methylene over methyl C–H bonds and Tambar’s two-step protocol32 where an asymmetric ene-type transformation of cis-olefins was demonstrated to distinguish between two allylic positions (Fig. 1b). In complementary work, Katsuki has described a Ru-salen catalyst capable of discriminating benzylic and allylic positions on the basis of size, with the consequence that it aminates only methyl and ethyl groups33. However, selective C–H activation/amination of alkenes possessing two similar sets of allylic protons has yet to be disclosed, presumably due to the following two issues: 1) two similar allylic C–H bonds are competing for C–H activation; and 2) two marginally distinguishable reactive sites of the resulting metal-allyl species could potentially lead to a mixture of four regioisomers, especially for substrates containing trans-1,2-disubstituted alkenes (Fig. 1c). Herein, we report a regioselective C–H activation/amination reaction of unsymmetrical 1,1- and trans-1,2-disubstituted alkenes. We further demonstrate that the exquisite selectivity is electronically controlled through the inherent inductive effect of a remote electron-withdrawing group. We propose that C–H activation selectivity can be predicted using 1JCH coupling constants at the allylic positions, based on a linear relationship between the difference of 1JCH coupling constants (Δ1JCH) and C–H activation barriers (ΔΔG‡).
Results and discussion
1,1-disubstituted alkene 1a was selected as the model substrate to initiate our study. We postulated that the homoallylic trifluoromethyl group could electronically differentiate between potentially reactive β and δ allylic C–H bonds through the inductive effect. Following the selective C–H activation, the resultant π-allyl-metal species may undergo selective C–N bond formation at the internal position. At the outset, we first tested the reaction with [Cp*MCl2]2 (M = Co, Rh, Ir; Cp* = pentamethylcyclopentadienyl) as the precatalyst, silver tetrafluoroborate (AgBF4) as the additive, lithium acetate (LiOAc) as the base, and p-toluenesulfonyl azide (TsN3) as the nitrene precursor. Although no amination products were detected with either [Cp*CoCl2]2 or [Cp*RhCl2]2, we realized the formation of δ amination 2a as a major product with [Cp*IrCl2]2 in moderate yield. In line with our proposal, the reaction proceeds through the selective C–H activation of the distal δ C–H bond, followed by C–N bond formation at the internal position of the corresponding π-allyl-Ir species. After an extensive evaluation of the reaction conditions, the optimal conditions were achieved by making three crucial changes: 1) switching the ligand from Cp* to Cp™ (Cp™ = tetramethylcyclopentadienyl), which significantly improves the yield; 2) replacing lithium acetate with silver trifluoroacetate (AgTFA); 3) adding cesium carbonate (Cs2CO3) as a co-base (see Supplementary Section 4 for details). Control experiments further revealed that both [Cp™IrCl2]2 and AgBF4 are necessary components for the reaction. Moreover, [Cp™Ir(TFA)2] was also tested as a replacement for [Cp*IrCl2]2 and AgTFA, which leads to a comparable yield and regioselectivity, but no reactivity is observed in the absence of AgBF4.
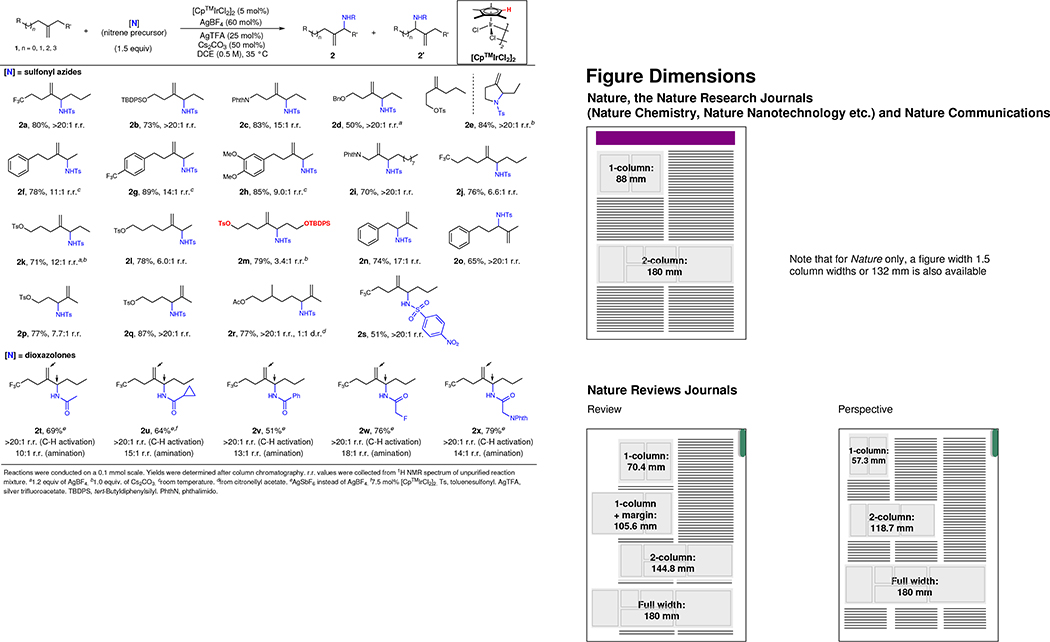
Having established the optimal conditions, we investigated the scope of the reaction by examining a diverse array of unsymmetrical 1,1-dialkylsubstituted alkenes (Table 1). Various electron-withdrawing groups (EWG) are tolerated providing excellent yields and regioselectivities (2a–2e). Notably, when a suitably placed toluenesulfonate group (OTs) is present, a pyrrolidine ring is formed in situ from the corresponding amination product (2e). High selectivity is also observed with a weakly electron-withdrawing phenyl group, and the electron density of the arene affects the regioselectivity (2f–2h). With respect to the distance between the EWG and olefin, this method tolerates tethers ranging from one to four methylene units while maintaining good reactivity and regioselectivity (2i–2l). Surprisingly, a substrate bearing marginally different allylic C–H bonds, which are remotely influenced by OTs vs OTBDPS, reacts with measurable selectivity (2m). Moreover, the reaction is exquisitely selective for secondary C–H bonds over methyl groups under the standard conditions, and the regioselectivity increases as the EWG moves away from the reactive center (2n–2q). In terms of trisubstituted olefins, citronellyl acetate provides the allylic amine with migration of the double bond to the terminal position (2r). p-Nitrobenzene-sulfonyl azide can also be employed, which allows facile deprotection (2s). Besides sulfonyl azides, a variety of dioxazolones were utilized under slightly modified conditions. Various amides, possessing substitutions that include phenyl, cyclopropyl, α-fluoro, or α-amino groups, are effectively incorporated in the distal allylic position (2t–2x).
Table 1.
Scope of unsymmetrical 1,1-disubstituted alkenes and nitrene precursors
 |
Based on the success of selective C–H activation of 1,1-disubstituted alkenes, we envisioned that similar selectivity could also be achieved with unsymmetrical trans-1,2-disubstituted alkenes containing a remote electron-withdrawing group (EWG). However, the π-allyl-metal intermediates derived from C–H activation of 1,2-dialkylsubstituted alkenes bearing two similar internal positions have rarely been differentiated34. We speculated that the electronic difference between the two reactive sites, produced by the inductive effect, could also lead to regioselective nitrene insertion. To set the stage, we examined the Ir-catalyzed C–H activation/amination of trans-3-hexene, a symmetrical substrate that would lead to an unsymmetrical π-allyl-Ir intermediate. In the event, under standard reaction conditions, amination occurs unselectively to give two products (Fig. 2a). In contrast, reaction of substrate 3a bearing p-toluenesulfonate (OTs) as the EWG leads to a moderate selectivity favoring the more electron-rich side of the π-allyl-Ir intermediate, which results from C–H activation of the distal allylic C–H bond (Fig. 2b). Regioisomers derived from C–H activation of the proximal C–H bonds were not detected.
Figure 2. Study of regioselectivities in C–H amination of unsymmetrical trans-1,2-disubstituted alkenes.

a, Non-selective C–H amination of trans-3-hexene. b, Remote electronically controlled C–H activation and amination. c, Electronic and steric effects of Cp ligands with substrate 3a. d, Scope of unsymmetrical trans-1,2-disubstituted alkenes catalyzed by Ir-9. Unless otherwise noted, all reactions were conducted on a 0.1 mmol scale using alkene (1.0 equiv.), TsN3 (1.5 equiv.), [Ir-n] (15 mol% of monomer), AgBF4 (60 mol%), LiOAc (1.0 equiv.). Yields were determined after column chromatography. Yields* and r.r. values (β/α) were collected from 1H NMR spectrum of unpurified reaction mixutre with mesitylene as the internal standard. a5 mol% [Cp™IrCl2]2 (Ir-6) was used. bReaction was conducted at 50 °C.
The electronic effect of the Cp ligand35,36 was investigated by comparing catalysts Ir-2, Ir-3, and Ir-4, bearing electronically variant aryl rings on the Cp core. These catalysts all lead to inferior regioselectivities, suggesting that the electron density of the Cp ligand has little effect on the regioselectivity while the steric properties seem to have a profound influence (Fig. 2c). Indeed, when the Cp ligand contains fewer methyl groups, the regioselectivity is dramatically improved (Ir-6, Ir-7, and Ir-8). Additionally, by comparing the outcomes of Ir-5 and Ir-8, both mono-alkyl Cp derivatives, the same steric effect on the regioselectivity is observed. Eventually, an electron-donating hyperconjugative interaction was found to improve the productive reactivity, which delivers Ir-9 as the optimal catalyst.
The scope of trans-1,2-disubstituted alkenes was then explored (Fig. 2d). Substrate 3b bearing an n-butyl group is tolerated, indicating that the regioselectivity is not related to the steric bias between two sides of the π-allyl-Ir intermediate. Other electron-withdrawing groups may be used, including one with chloride substitution (4c–4e). Allylic substitutions that are known to deactivate the alkene also perform well (4f, 4g). Even those substrates with conjugated carbonyl groups are reactive at elevated temperature, leading to the desired amination products (4h, 4i). Additionally, β-alkyl styrene 3j was found to give the conjugated amination product 4j selectively, which is complementary to Blakey’s report using Cp*Rh-catalyst and tert-butyldioxazolone29.
To gain further insight into the origin of the observed regioselectivities, we conducted several studies probing the mechanism and the impact of substrate electronics. Several experiments proved particularly enlightening. On the basis of a deuterium labeling experiments, we conclude that the allylic C–H activation step is irreversible for both alkene classes (see Supplementary Section 7 for details). 1JCH coupling constants37, which are closely correlated with the s character of the C–H bonding orbital and also influenced by the nature of substituents, were utilized to understand the electronic properties of allylic C–H bonds. Inspired by a previous study where a positive linear correlation between 1JCH and Hammett σ constants was unveiled38, we propose that the electronic difference between two allylic C–H bonds could be indicated by the corresponding difference of 1JCH (Δ1JCH). It’s worth noting that most allylic C–H bonds of the substrates investigated in Figure 2 have very similar steric environment and are electronically influenced by a remote (non-α-substitution) EWG, which excludes the influence of angular distortion effects and α-substitution effects. Therefore, the differences of 1JCH between two allylic positions are mainly induced by the substituent inductive effect. Comparing the data of substrates 1a, 1j, and 1y, we noticed that the Δ1JCH coupling constant between two sides of the olefin decreases as the CF3 group becomes more distal to the double bond, in line with our hypothesis (Fig. 3a). Additionally, as the electron density of the aromatic ring two methylenes away from the alkene is varied, the Δ1JCH between two sides of the olefin also varies accordingly. Moreover, we found that the electronic differences indicated by Δ1JCH likely correlates to the regioselectivities. Indeed, we graphed the results for twelve substrates involving a methylene vs methylene competition chosen from Table 1, and found a linear relationship between Δ1JCH coupling constant and the difference of C–H activation barriers (ΔΔG‡) (Fig. 3b). Namely, the regioselectivity of C–H activation could be quantitatively predicted by calculating the difference of 1JCH coupling constants of two allylic C–H bonds (Δ1JCH).
Figure 3. Study of the origin of regioselectivities.

a, Inductive effect induced changes in 1JCH coupling constants and regioselectivities. b, The analysis of twelve substrates involving a methylene vs methylene competition and identification of a linear relationship between Δ1JCH coupling constants of the two competing C–H bonds and the difference of C–H activation barriers (ΔΔG‡). Error bars indicate a ±0.1 Hz error in the Δ1JCH and ±0.058 kcal/mol in the ΔΔG‡.
In all these cases, C–H abstraction occurs selectively at the carbon bearing the lower 1JCH coupling which corresponds to greater p-character at that position, and a weaker C–H bond. Given that C–H activation is irreversible as evidence by the isotope labelling experiment, this effect is almost certainly kinetic in nature. It is also worth noting that an outlier 1c was found probably due to the interfering chelation to the Ir by Lewis basic carbonyl, which suggests potential limitation of the correlation in some special cases. In general, we believe that the electronic difference between allylic C–H bonds, which is introduced by the inductive effect, is the primary factor contributing to regioselectivity.
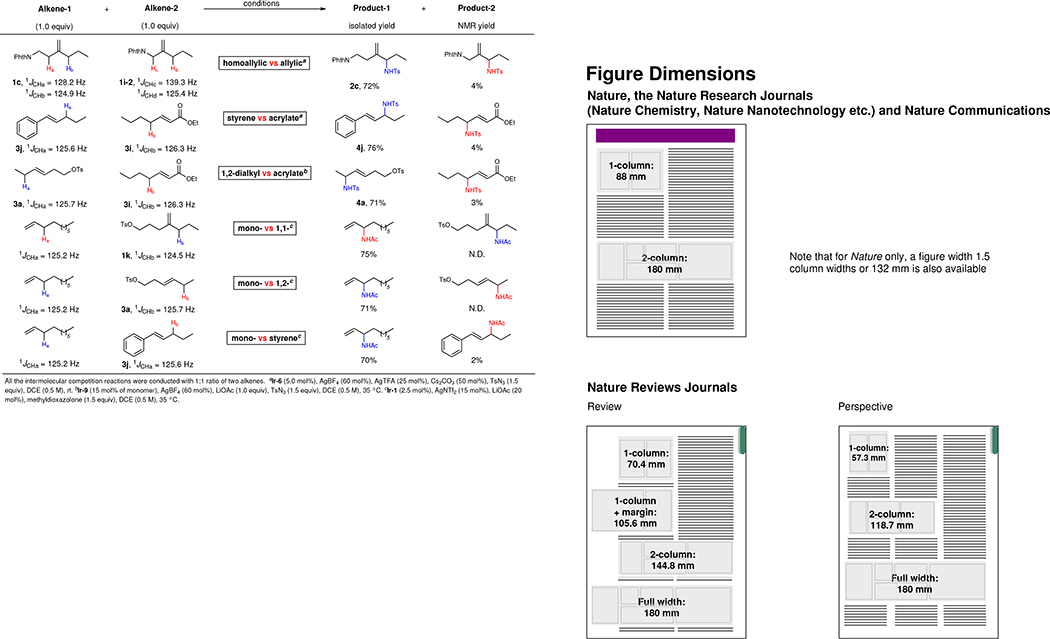
Besides the selectivities between two sets of allylic C–H bonds or two positions of the π-allyl-Ir intermediates, intermolecular competition reactions were studied (Table 2). For example, a competition reaction between 1:1 ratio of substrates 1c and 1i-2 produces 2c as the major product, which suggests the preference of the allylic C–H bonds with the smallest 1JCH coupling constant among total four sets of allylic protons. Moreover, β-alkyl styrene 3j and 1,2-trans-alkene 3a are more reactive than α,β-unsaturated ester 3i because of the electronic deactivation by the conjugated carbonyl group (1JCHa < 1JCHb). Additionally, accessibility of the alkene plays an important role in the intermolecular competition reactions. For instance, 1-decene undergoes allylic amination completely selectively over either 1,1- or 1,2-disubstituted alkenes, regardless of the electronic properties of the allylic C–H bonds.
Table 2.
Intermolecular competition reactions
 |
Conclusion
In summary, we have developed an intermolecular regioselective allylic C–H amination of unsymmetrical disubstituted alkenes. This method exploits subtle electronic differences induced by remote electron withdrawing groups to effect a selective C–H activation of allylic C–H bonds. The selectivity can be predicted based on the linear relationship between the Δ1JCH coupling constants of two competing C–H bonds and the difference of C–H activation barriers (ΔΔG‡). The key findings also include the successful differentiation of two internal positions of the π-allyl-Ir intermediates with the assistance of the novel monosubstituted Cp ligand. We further provide a rubric by which to understand C–H activation in more complex systems resulting from competition experiments. More broadly, we envision that this protocol could also benefit other allylic C–H functionalizations of unsymmetrical internal olefins.
Methods
General procedure for 1,1-disubstituted alkenes with TsN3
To an oven-dried screw-capped vial with a magnetic stir bar was sequentially added alkene (0.1 mmol, 1.0 equiv.), tosyl azide (23 μl, 1.5 equiv.), [Cp™IrCl2]2 (Ir-6) (3.9 mg, 5.0 mol%), cesium carbonate (16.3 mg, 50 mol%), silver trifluoroacetate (5.5 mg, 25 mol%), silver tetrafluoroborate (11.7 mg, 60 mol%), and 1,2-dichloroethane (200 μl, 0.5 M). The cap was screwed on, and the reaction was stirred at 35 °C for 20 hours. The reaction mixture was filtered through a plug of Celite, and the filtrate was concentrated under vacuum. The crude mixture was analyzed via 1H NMR spectroscopy with mesitylene (12.0 mg, 0.1 mmol) as an internal standard. The sample was further purified by column chromatography to give the desired amination product.
General procedure for 1,1-disubstituted alkenes with dioxazolones
To an oven-dried screw-capped vial with a magnetic stir bar was sequentially added alkene (0.1 mmol, 1.0 equiv.), [Cp™IrCl2]2 (Ir-6) (3.9 mg, 5.0 mol%), cesium carbonate (16.3 mg, 50 mol%), silver trifluoroacetate (5.5 mg, 25 mol%), and silver hexafluoroantimonate (20.6 mg, 60 mol%). In a separated vial, dioxazolone (1.5 equiv.) was dissolved in 1,2-dichloroethane (200 μl), which was then transferred to the first vail. The cap was screwed on, and the reaction was stirred at 35 °C for 20 hours. The reaction mixture was filtered through a plug of Celite, and the filtrate was concentrated under vacuum. The crude mixture was analyzed via 1H NMR spectroscopy with mesitylene (12.0 mg, 0.1 mmol) as an internal standard. The sample was further purified by column chromatography to give the desired amination product.
General procedure for 1,2-disubstituted alkenes
To an oven-dried screw-capped vial with a magnetic stir bar was sequentially added Ir-9 (9.0 mg, 15 mol% of monomer), lithium acetate (6.6 mg, 1.0 equiv.), and silver tetrafluoroborate (11.7 mg, 60 mol%). In a separated vial alkene (0.1 mmol, 1.0 equiv.) and tosyl azide (23 μl, 1.5 equiv.) were dissolved in 1,2-dichloroethane (200 μl, 0.5 M), and the resultant solution was transferred to the first vial. The cap was screwed on, and the reaction was stirred at 35 °C for 40 hours. The reaction mixture was filtered through a plug of Celite, and the filtrate was concentrated under vacuum. The crude mixture was analyzed via 1H NMR spectroscopy with mesitylene (12.0 mg, 0.1 mmol) as an internal standard. The sample was further purified by column chromatography to give the desired amination product.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information file.
Supplementary Material
Acknowledgements
We thank NIGMS (GM80442) for support. We thank John Decatur for assistance with determining 1JCH coupling constants. Correspondence and requests for materials should be directed to TR.
Footnotes
Competing interests
The authors declare no competing interests.
Refernces
- 1.McMurray L, O’Hara F & Gaunt MJ Recent developments in natural product synthesis using metal-catalysed C–H bond functionalisation. Chem. Soc. Rev 40, 1885–1898 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Gutekunst WR & Baran PS C–H functionalization logic in total synthesis. Chem. Soc. Rev 40, 1976–1991 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Yamaguchi J, Yamaguchi AD & Itami K C–H bond functionalization: emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem. Int. Ed 51, 8960–9009 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Cernak T, Dykstra KD, Tyagarajan S, Vachal P & Krska SW The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev 45, 546–576 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Colby DA, Bergman RG & Ellman JA Rhodium-catalyzed C–C bond formation via heteroatom-directed C–H bond activation. Chem. Rev 110, 624–655 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lyons TW & Sanford MS Palladium-catalyzed ligand-directed C–H functionalization reactions. Chem. Rev 110, 1147–1169 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He J, Wasa M, Chan KSL, Shao Q & Yu JQ Palladium-catalyzed transformations of alkyl C–H bonds. Chem. Rev 117, 8754–8786 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sambiagio C et al. A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry. Chem. Soc. Rev 47, 6603–6743 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newhouse T & Baran PS If C–H bonds could talk: selective C–H bond oxidation. Angew. Chem. Int. Ed 50, 3362–3374 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartwig JF & Larsen MA Undirected, homogeneous C–H bond functionalization: challenges and opportunities. ACS Cent. Sci 2, 281–292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xue XS, Ji P, Zhou B & Cheng JP The essential role of bond energetics in C–H activation/functionalization. Chem. Rev 117, 8622–8648 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Romero NA, Margrey KA, Tay NE & Nicewicz DA Site-selective arene C–H amination via photoredox catalysis. Science 349, 1326–1330 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Paudyal MP et al. Dirhodium-catalyzed C–H arene amination using hydroxylamines. Science 353, 1144–1147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berger F et al. Site-selective and versatile aromatic C–H functionalization by thianthrenation. Nature 567, 223–228 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Chen MS & White MC Combined effects on selectivity in Fe-catalyzed methylene oxidation. Science 327, 566–571 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Schmidt VA, Quinn RK, Bruscoe AT & Alexanian EJ Site-selective aliphatic C–H bromination using N-bromoamides and visible light. J. Am. Chem. Soc 136, 14389–14392 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Sharma A & Hartwig JF Metal-catalysed azidation of tertiary C–H bonds suitable for late-stage functionalization. Nature 517, 600–604 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eames J & Watkinson M Catalytic allylic oxidation of alkenes using an asymmetric Kharasch–Sosnovsky reaction. Angew. Chem. Int. Ed 40, 3567–3571 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Sharma A & Hartwig JF Enantioselective functionalization of allylic C–H bonds following a strategy of functionalization and diversification. J. Am. Chem. Soc 135, 17983–17989 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cuthbertson JD & MacMillan DWC The direct arylation of allylic sp3 C–H bonds via organic and photoredox catalysis. Nature 519, 74–77 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu W, Ali SZ, Ammann SE & White MC Asymmetric allylic C–H alkylation via palladium(II)/cis-ArSOX catalysis. J. Am. Chem. Soc 140, 10658–10662 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J et al. Site-specific allylic C–H bond functionalization with a copper-bound N-centred radical. Nature 574, 516–521 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Reed SA & White MC Catalytic intermolecular linear allylic C–H amination via heterobimetallic catalysis. J. Am. Chem. Soc 130, 3316–3318 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu G, Yin G & Wu L Palladium-catalyzed intermolecular aerobic oxidative amination of terminal alkenes: efficient synthesis of linear allylamine derivatives. Angew. Chem., Int. Ed 47, 4733–4736 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Bao H & Tambar UK Catalytic enantioselective allylic amination of unactivated terminal olefins via an ene reaction/[2,3]-rearrangement. J. Am. Chem. Soc 134, 18495–18498 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buman JS & Blakey SB Regioselective intermolecular allylic C–H amination of disubstituted olefins via rhodium/π-allyl intermediates. Angew. Chem., Int. Ed 56, 13666–13669 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Lei H & Rovis T Ir-catalyzed intermolecular branch-selective allylic C-H amidation of unactivated terminal olefins. J. Am. Chem. Soc 141, 2268–2273 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knecht T, Mondal S, Ye JH, Das M & Glorius F Intermolecular, branch-selective, and redox-neutral Cp*IrIII-catalyzed allylic C–H amidation. Angew. Chem., Int. Ed 58, 7117–7121 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Burman JS, Harris RJ, Farr CMB, Bacsa J & Blakey SB Rh(III) and Ir(III)Cp* complexes provide complementary regioselectivity profiles in intermolecular allylic C–H amidation reactions. ACS Catal. 9, 5474–5479 (2019). [Google Scholar]
- 30.Liang C et al. Toward a synthetically useful stereoselective C–H amination of hydrocarbons. J. Am. Chem. Soc 130, 343–350 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Lescot C, Darses B, Collet F, Retailleau P & Dauban P Intermolecular C–H amination of complex molecules: insights into the factors governing the selectivity. J. Org. Chem 77, 7232–7240 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Bayeh L, Le PQ & Tambar UK Catalytic allylic oxidation of internal alkenes to a multifunctional chiral building block. Nature 547, 196–200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishioka Y, Uchida T & Katsuki T Enantio- and regioselective intermolecular benzylic and allylic C–H bond amination. Angew. Chem., Int. Ed 52, 1739–1742 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Szabó KJ Nature of the interaction between β-substituents and the allyl moiety in (η3-allyl)palladium complexes. Chem. Soc. Rev 30, 136–143 (2001). [Google Scholar]
- 35.Piou T et al. Correlating reactivity and selectivity to cyclopentadienyl ligand properties in Rh(III)-catalyzed C–H activation reactions: an experimental and computational study. J. Am. Chem. Soc 139, 1296–1310 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piou T & Rovis T Electronic and steric tuning of a prototypical piano stool complex: Rh(III) catalysis for C–H functionalization. Acc. Chem. Res 51, 170–180 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansen PE Carbon–hydrogen spin–spin coupling constants. Prog. Nucl. Magn. Reson. Spectrosc 14, 175–295 (1981). [DOI] [PubMed] [Google Scholar]
- 38.Yoder CH, Tuck RH & Hess RG Nuclear magnetic resonance studies of the bonding in aromatic systems. Correlation of Hammett sigma constants with methyl13C–H coupling constants and chemical shifts. J. Am. Chem. Soc 91, 539–543 (1969) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article and its supplementary information file.