

Abstract
Selecting informative nodes over large-scale networks becomes increasingly important in many research areas. Most existing methods focus on the local network structure and incur heavy computational costs for the large-scale problem. In this work, we propose a novel prior model for Bayesian network marker selection in the generalized linear model (GLM) framework: the Thresholded Graph Laplacian Gaussian (TGLG) prior, which adopts the graph Laplacian matrix to characterize the conditional dependence between neighboring markers accounting for the global network structure. Under mild conditions, we show the proposed model enjoys the posterior consistency with a diverging number of edges and nodes in the network. We also develop a Metropolis-adjusted Langevin algorithm (MALA) for efficient posterior computation, which is scalable to large-scale networks. We illustrate the superiorities of the proposed method compared with existing alternatives via extensive simulation studies and an analysis of the breast cancer gene expression dataset in the Cancer Genome Atlas (TCGA).
Keywords: gene network, generalized linear model, network marker selection, posterior consistency, thresholded graph Laplacian Gaussian prior
1. Introduction
In biomedical research, complex biological systems are often modeled or represented as biological networks (Kitano, 2002). High-throughput technology such as next generation sequencing (Schuster, 2007), mass spectrometry (Aebersold and Mann, 2003) and medical imaging (Doi, 2007) has generated massive datasets related to those biological networks. For example, in omics studies, a biological network may represent the interactions or dependences among a large set of genes/proteins/metabolites; and the expression data are a number of observations at each node of the network (Barabási et al., 2011). In neuroimaging studies, a biological network may refer to the functional connectivity among many brain regions or voxels; and the neural activity can be measured at each node of the network. In many biomedical studies, one important research question is given a known network, to select informative nodes from tens of thousands of candidate nodes that are strongly associated with the disease risk or other clinical outcomes (Greicius et al., 2003). We refer to these informative nodes as network markers (Kim et al., 2012; Peng et al., 2014; Yuan et al., 2017) and the selection procedure as network marker selection. One promising solution is to perform network marker selection under regression framework where the response variable is the clinical outcome and predictors are nodes in the network. The classical variable selection (George and McCulloch, 1993; Fan and Li, 2001) in the regression model can be considered as a special case of the network marker selection, where the variable refers to the nodes in a network without edges.
For variable selection in regression models, many regularization methods have been proposed with various penalty terms, including the least absolute shrinkage and selection operator or the L1 penalty (Tibshirani, 1996; Zou, 2006, LASSO), elastic-net or the L1 plus L2 penalty (Zou and Hastie, 2005), the Smoothly Clipped Absolute Deviation penalty (Fan and Li, 2001, SCAD), the minimax concave penalty (Zhang, 2010, MCP) and so on. Several network constrained regularization regression approaches have been developed to improve selection accuracy and increase prediction power. One pioneering work is the graph-constrained estimation (Li and Li, 2008, Grace), which adopts the normalized graph Laplacian matrix to incorporate the network dependent structure between connected nodes. As an extension of Grace, the adaptive Grace (Li and Li, 2010, aGrace) makes constraints on the absolute values of weighted coefficients between connected nodes. Alternatively, an Lγ norm group penalty (Pan et al., 2010) and a fused LASSO type penalty (Luo et al., 2012) have been proposed to penalize the difference of absolute values of coefficients between neighboring nodes. Instead of imposing constraints on coefficients between neighboring nodes, an L0 loss to penalize their selection indicators (Kim et al., 2013) has been proposed, leading to a non-convex optimization problem for parameter estimation, which can be solved by approximating the non-continuous L0 loss using the truncated lasso penalty (TLP).
In addition to frequentist approaches, Bayesian variable selection methods have received much attention recently with many successful applications. The Bayesian methods are natural to incorporate the prior knowledge and make posterior inference on uncertainty of variable selection. A variety of prior models have been studied, such as the spike and slab prior (George and McCulloch, 1993), the LASSO prior (Park and Casella, 2008), the Horseshoe prior (Polson and Scott, 2012), the non-local prior (Johnson and Rossell, 2012), the Dirichlet Laplace prior (Bhattacharya et al., 2015) and more. To incorporate the known network information, Stingo et al. (2011) employed a Markov random field to capture network dependence and jointly select pathways and genes; and Chekouo et al. (2016) adopted a similar approach for imaging genetics analysis. Zhou and Zheng (2013) proposed rGrace, a Bayesian random graph-constrained model to combine network information with empirical evidence for pathway analysis. A partial least squares (PLS) g-prior was developed in Peng et al. (2013) to incorporate prior knowledge on gene-gene interactions or functional relationship for identifying genes and pathways. Chang et al. (2016) proposed a Bayesian shrinkage prior which smoothed shrinkage parameters of connected nodes to a similar degree for structural variable selection.
The Ising model is another commonly used Bayesian structural variable selection method. It has been used as a prior model for latent selection indicators that lay on an undirected graph characterizing the local network structure. They are especially successful for variable selection over the grid network motivated by some applications, for example, the motif finding problem (Li and Zhang, 2010) and the imaging data analysis (Goldsmith et al., 2014; Li et al., 2015). However, it is very challenging for fully Bayesian inference on the Ising model over the large-scale network due to at least two reasons: 1) The posterior inference can be quite sensitive to the hyperparameter specifications in the Ising priors based on empirical Bayes estimates or subjective prior elicitation in some applications. However, fully Bayesian inference on those parameters is difficult due to the intractable normalizing constant in the model. 2) Most posterior computation algorithms, such as the single-site Gibbs sampler and the Swendsen-Wang algorithm, incur heavy computational costs for updating the massive binary indicators over large-scale networks with complex structures. It worth noting that, different from using the known network structure for variable selection, Dobra (2009); Kundu et al. (2015); Liu et al. (2014) and Peterson et al. (2016) also proposed Bayesian structured variable selection without using a known network structure.
To address limitations of existing methods, we propose a new prior model: the thresholded graph Laplacian Gaussian (TGLG) prior, to perform network marker selection over the large-scale network by thresholding a latent continuous variable attached to each node. To model the selection dependence over the network, all the latent variables are assumed to follow a multivariate Gaussian distribution with mean zero and covariance matrix constructed by a normalized graph Laplacian matrix. The effect size of each node is modeled through an independent Gaussian distribution.
Threshold priors have been proposed for Bayesian modeling of sparsity in various applications. Motivated by the analysis of financial time series data, Nakajima and West (2013a) and Nakajima and West (2013b) proposed a latent threshold approach to imposing dynamic sparsity in the simultaneous autoregressive models (SAR). Nakajima et al. (2017) further extended this type of models for the analysis of Electroencephalography (EEG) data. To analyze neuroimaging data, Shi and Kang (2015) proposed a hard-thresholded Gaussian process prior for image-on-scalar regression; and Kang et al. (2018) introduced a soft-thresholded Gaussian process for scalar-on-image regression. To construct the directed graphs in genomics applications, Ni et al. (2017) adopted a hard threshold Gaussian prior in a structural equation model. However, all the existing threshold prior models do not incorporate the useful network structural information, and thus are not directly applicable to the network marker selection problem of our primary interest.
In this work, we propose to build the threshold priors using the graph Laplacian matrix, which has been used to capture the structure dependence between neighboring nodes (Li and Li, 2008; Zhe et al., 2013; Li and Li, 2010). Most of those frequentist methods directly specify the graph Laplacian matrix from the existing biological network. Liu et al. (2014) has proposed a Bayesian regularization graph Laplacian (BRGL) approach which utilizes the graph Laplacian matrix to specify a priori precision matrix of regression coefficients. However, BRGL is fundamentally different from our method in that it is one type of continuous shrinkage priors for regression coefficients which have quite different prior supports compared with our TGLG priors. BRGL were developed only for linear regression and its computational cost can be extremely heavy for large-scale networks. In addition, there is lack of theoretical justifications for BRGL when the large-scale network has a diverging number of edges and nodes.
Our method is a compelling Bayesian approach to variable selection with a known network structure. The TGLG prior has at least four markable features: 1) Fully Bayesian inference for large-scale networks is feasible in that the TGLG prior does not involve any intractable normalizing constants. 2) Posterior computation can be more efficient, since the TGLG-based inference avoids updating the latent binary selection indicators and instead updates the latent continuous variables, to which many existing approximation techniques can be potentially applied. 3) The graph Laplacian matrix (Chung, 1997; Li and Li, 2008; Zhe et al., 2013) based prior can incorporate the topological structure of the network which has been adopted in genomics. 4) The TGLG prior enjoys the large support for Bayesian network marker selection over large-scale networks, leading to posterior consistency of model inference with a diverging number of nodes and edges under the generalized linear model (GLM) framework.
The remainder of the manuscript is organized as follows. In Section 2, we introduce the TGLG prior and propose our model for network marker selection under the GLM framework. In Section 3, we study the theoretical properties for the TGLG prior and show the posterior consistency of model inference. In Section 4, we discuss the hyper prior specifications and the efficient posterior computation algorithm. We illustrate the performance of our approach via simulation studies and an application on the breast cancer gene expression dataset from The Cancer Genome Atlas (TCGA) in Section 5. We conclude our paper with a brief discussion on the future work in Section 6.
2. The Model
Suppose the observed dataset includes a network with pn nodes, one response variable and q confounding variables. For each node, we have n observations. For observation i, i = 1, … , n, let yi be the response variable, xi = (xi1, ⋯ , xipn)T be the vector of nodes and zi = (zi1, ⋯ , ziq)T be the vector of confounding variables. Denote by the dataset. We write the number of nodes as pn to emphasize on the diverging number of nodes in our asymptotical theory. Drop subscript i to have generic notation for a response variable y, a vector of nodes x and a vector of confounders z. Generalized linear model (GLM) is a flexible regression model to relate a response variable to a vector of nodes and confounding variables. The GLM density function for (y, x, z) with one natural parameter is:
| (1) |
where h* = zT ω* + xT β* is the linear parameter in the model, ω* and β* are true coefficients that generate data, a(h) and b(h) are continuous differentiable functions. The true mean function is
where g−1(·) is an inverse link function, which can be chosen according to the specific type of the response variable. For example, one can choose the identity link for the continuous response and the logit link for the binary response.
In (1), coefficient vector ω is a nuisance parameter to adjust for the confounder effects, for which we assign a Gaussian prior with mean zero and independent covariance, i.e. ω ~ N(0, ) for . Here Id represents an identity matrix of dimension d for any d > 0. Coefficient vector β represents the effects of nodes on the response variable. Here we perform network marker selection by imposing sparsity on β. To achieve this goal, we develop a new prior model for β: the thresholded graph Laplacian Gaussian (TGLG) prior. Suppose the observed network can be represented by a graph G, with each vertex corresponding to one node in the network. Let j ~ k indicate there exists an edge between vertices j and k in G. Let dj represent the degree of vertex j, i.e., the number of nodes that are connected to vertex j in G. Denote by L = (Ljk) a pn × pn normalized graph Laplacian matrix, i.e. Ljk = 1 if j = k and dj ≠ 0, if j ~ k, and Ljk = 0 otherwise. For any d > 0, denote by 0d an all zero vector of dimension d. For any λ, ε, , , we consider an element-wise decomposition of β for the prior specifications:
| (2) |
Here α = (α1, … , αpn)T represents the effect size of nodes. The operator ”ο” is the element-wise product. The vector thresholding function is tλ (γ) = {I(∣γ1∣ > λ), … , I(∣γpn∣ > λ)}T, where is the event indicator with = 1 if occurs and otherwise. The latent continuous vector γ = (γ1, … γpn)T controls the sparsity over graph G. We refer to (2) as the TGLG prior for β, denoted as β ~ TGLG (λ, ε, , ). The TGLG prior implies that for any two nodes j and k, γj and γk are conditionally dependent given others if and only if j ~ k over the graph G. In this case, their absolute values are more likely to be smaller or larger than a threshold value λ together. This further implies that nodes j and k are more likely to be selected as network marker or not selected together if j ~ k. Figure 1 shows an example of a graph and the corresponding correlation matrix of γ for ε = 10−2, where the γ’s of connected vertices are highly correlated.
Figure 1:

An example of the graph and the corresponding correlation matrix of γ that was constructed from the inverse graph Laplacian matrix.
There are four hyperparameters in the TGLG prior model. The threshold λ controls a priori the sparsity. When λ → 0, all the nodes tend to be selected. When λ → ∞, none of them will be selected. The parameter ε determines the impact of the network on the sparsity. When ε → ∞, γ’s of connected vertices tend to be independent while they tend to be perfectly correlated when ε → 0. The two variance parameters and control the prior variability of the latent vectors γ and α respectively. Note that and λ are not completely identifiable, but for some specifications they can affect much the sparsity of βj’s in prior specifications. For example, when 3σγ is much smaller than λ, the prior probability of zero βj can be close to one. On the other hand, for any positive σγ, when λ close to zero, βj is nonzero with a high prior probability.
Now we discuss how to specify the hyperparameters. For variance terms and , we use the conjugate prior model by assigning the Inverse-Gamma distribution IG(aγ, bγ) and IG(aα, bα) respectively. We fix as a large value. We assign the uniform prior to the threshold parameter λ, i.e. λ ~ Unif(0, λu) with upper bound λu > 0. We choose a wide range by set λu = 10 in the rest of manuscript. For parameter ε, we can either assign an log-normal prior (logε ~ N (με, )) or set as a fixed small value.
3. Theoretical Properties
In this section, we examine the theoretical properties of TGLG prior based network marker selection under the GLM framework. In particular, we establish the posterior consistency with a diverging number of nodes in the large-scale networks.
Let ξ ⊂ {1, 2, ⋯ , pn} denote the set of selected node indices, i.e. I(∣γj∣ > λ) = 1, if j ∈ ξ, I(∣γj∣ > λ) = 0, otherwise. The number of nodes in ξ is denoted as ∣ξ∣. For a model ξ = (i1, ⋯ , i∣ξ∣), denote by βξ = (βi1, ⋯ , βi∣ξ∣)T the coefficient of interest, respectively. Let π(ξ, dβξ, dω) represent the joint prior probability measure for model ξ, parameters βξ and confounding coefficients ω. Their joint posterior probability measure conditional on dataset Dn is:
where f(yi, hi) = exp{a(hi)yi + b(hi) + c(yi)} is the density function of yi given xi and zi based on GLM with . We examine asymptotic properties of the posterior distribution of the density function f regarding to the Hellinger distance (Jiang, 2007; Song and Liang, 2015) under some regularity conditions. The Hellinger distance d(f1, f2) between two density functions f1(x, y) and f2(x, y) is defined as
We list all the regularity conditions in the Appendix. We show that the TGLG prior and the proposed model enjoy the following properties:
Theorem 1 (Large Support for Network Marker Selection). Assume a sequence ϵn ∈ (0, 1] with and a sequence of nonempty models ξn. Assume conditions (C1)–(C3) and (C7) hold. Given and , for any sufficiently small η > 0, there exists Nη such that for all n > Nη, we have
| (3) |
| (4) |
There exists Cn > 0, such that for all sufficiently large n and for any j ∈ ξn:
| (5) |
This theorem shows that the TGLG prior has a large support for the network marker selection. Particularly, (3) states that the TGLG prior can select the true network marker with a positive prior probability bounded away from zero, (4) ensures that the prior probability of the coefficients falling within an arbitrarily small neighborhood of the true coefficients with probability bounded away from zero, and (5) indicates a sufficiently small tail probability of the TGLG prior.
Theorem 2 (Posterior Consistency for Network Marker Selection). For the GLM with bounded covariates, i.e. ∣xj∣ ≤ M for all j = 1, ⋯ , pn and ∣zk∣ ≤ M for all k = 1, ⋯ , q, suppose the true node regression coefficients satisfy
Let ϵn ∈ (0, 1] be a sequence such that . Assume conditions (C1)–(C7) hold. Then we have the following results:
- Posterior consistency:
where f is the density function sampled from the posterior distribution and f* is the true density function.(6) For all sufficiently large n:
| (7) |
For all sufficiently large n:
| (8) |
Probability measure P and expectation E are both with respect to data Dn that are generated from the true density f*.
This theorem establishes the posterior consistency of network marker selection. In particular, (6) implies that the posterior distribution of density f concentrates on an arbitrarily small neighborhood of the true density f* under the Hellinger distance with a large probability. This probability converges to one as sample size n → ∞. (7) provides the convergence rate of the posterior distribution indicating how fast the tail probability approaches to zero. (8) indicates the average convergence rate of the posterior distribution of density f concentrating on the arbitrarily small neighborhood of the true density f*.
Please refer to the Supplementary File 1 (Cai et al., 2018a) for proofs of Theorems 1 and 2.
4. Posterior Computation
Our primary goal is to make posterior inference on regression coefficients for network markers, i.e. β. According to the model specification, the sparsity of βj is determined by the sparsity of αj and whether ∣γj∣ is less than λ or not, i.e. I(βj = 0) = I(αj = 0)I(∣γj∣ ≤ λ). Since αj has a non-sparse normal prior, the posterior inclusion probability of node j is just equal to the posterior probability of ∣γj∣ being greater than λ; and given βj ≠ 0, the effect-size can be estimated by E(βj∥γj∣ > λ, Dn). All other parameters in the model can be estimated by its posterior expectations.
To simulate the joint posterior distribution for all parameters, we adopt an efficient Metropolis-adjusted Langevin algorithm (MALA) (Roberts and Rosenthal, 1998) for posterior computation. We introduce a smooth approximation for the thresholding function:
leading to the analytically tractable first derivative:
We choose ε0 = 10−8 in the simulation studies and real data application in this article.
Denote by f(yi ∣ ω, α, γ, λ) the likelihood function for all the parameters of interest for observation i. Let φ(x ∣ μ, Σ) denote the density function of a multivariate normal distribution with mean μ and covariance matrix Σ and φ+(x ∣ μ, μl, μu, τ2) denote the density of a truncated normal distribution N+(μ, μl, μu, τ2) density with mean μ, variance τ2 and interval [μl, μu]. Let be the variance of the prior distribution for ω. Let Λγ = (L + εIpn)−1. To update λ, it is natural to use the random walk with a normal distribution as the proposal distribution. As we have a uniform prior specifications for λ, we use the truncated normal distribution to improve the sample efficiency, since the candidate sample falls in the same range in the prior specifications. Our choices are λl = 0 and λl = 10. The proposal variances , , and are all adaptively chosen by tuning acceptance rates to 30% for random walk and 50% for MALA in simulation studies and 15% for random walk and 30% for MALA in real data analysis. Our choice of the acceptance rate takes into account the general theoretical results on the optimal scaling of random walk (Roberts et al., 1997) and MALA (Roberts and Rosenthal, 1998; Roberts et al., 2001). However, the log-likelihood of our model involves both smooth and discontinuous functions, which do not satisfy the regularity conditions of the general theoretical results. Thus, we have made slight changes in the theoretical optimal acceptance rates according to our numerical experiments. See Algorithm 1 for the details of our posterior updates.
| Algorithm 1 Posterior updates within each iteration. | ||
Denote by the MCMC samples obtained after burn-in. We estimate the posterior inclusion probability for node j(j = 1, ⋯ , pn) by
According to Barbieri et al. (2004), we select the informative nodes with at least 50% inclusion probability, denote by the indices of all the informative nodes. To estimate regression coefficients of informative nodes, we choose the estimated conditional expectation of βj given βj ≠ 0 by
5. Numerical Studies
We conduct simulation studies to evaluate performance of the proposed methods compared with existing methods for different scenarios.
5.1. Small Simple Networks
Following settings in Li and Li (2008), Zhe et al. (2013) and Kim et al. (2013), we simulate small simple gene networks consisting of multiple subnetworks, where each subnetwork contains one transcription factor (TF) gene and 10 target genes that are connected to the TF gene; and two of the subnetworks are set as the true network markers. We consider two types of true network markers. In Type 1 network marker, TF and all 10 target genes are informative nodes; see Figure 2(a). In Type 2 network marker, TF and half of target genes are informative nodes; see Figure 2(b). For each informative node, the magnitude of the effect size, β, is simulated from Unif(1, 3) and its sign is randomly assigned as positive or negative.
Figure 2:
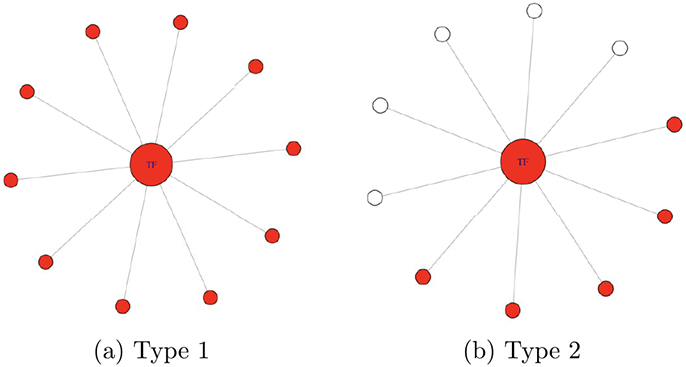
Two types network markers in the simulated small simple networks, where true informative nodes are marked in red. In Type 1 network marker, TF and all target genes are informative nodes. In Type 2 network marker, TF and half of target genes are informative nodes.
In each subnetwork, the covariate variables for 11 nodes, i.e., the expression levels of the TF gene and 10 target gene, are jointly generated from a 11-dimensional multivariate normal distribution with zero mean and unit variance, where the correlation between the TF gene and each target gene is 0.5; and the correlation between any two different target genes is 0.25. We assume the covariate variables are independent across different subnetworks.
To generate the response variable given the true network markers, we consider binary and continuous cases, where the continuous response variable is generated from linear regression, i.e. y ~ N(Xβ, ); and the binary response is generated from logistic regression, i.e. Pr(y = 1) = 1/{1 + exp(−Xβ)}.
We consider two scenarios for the number of subnetworks: 3 and 10; the corresponding numbers of nodes, p = 33 and p = 110 respectively. For the network with Type 1 markers, the number of informative nodes is 22; For the network with Type 2 markers, the number of informative nodes is 12. We generate 50 datasets for each scenario. For linear regression, each dataset contains 100 training samples and 100 test samples; for logistic regression, each dataset contains 200 training samples and 200 test samples.
We compare the proposed TGLG approach with the following existing methods: Lasso (Tibshirani, 1996), Elastic-net (Zou and Hastie, 2005), Grace (Li and Li, 2008), aGrace (Li and Li, 2010), L∞ and aL∞ (Luo et al., 2012), TTLP and LTLP (Kim et al., 2013), BRGL (Liu et al., 2014) and Ising model (Goldsmith et al., 2014; Li et al., 2015). For the hyper priors in the TGLG approach, we assign weakly informative priors: ~ IG(0.01, 0.01), ~ IG(0.01, 0.01). For all the regularized approaches, we adopt three-fold cross validations to choose tuning parameters. For the Ising prior model, we specify the priors as
and βi∣γi = 1 ~ N(0, ), where Ni denotes the neighbor nodes set of node i. For hyper prior specifications in Ising model, we fix a = −2 and choose b from 2, 5, 7 and 10 based on model performance. We implement a single-site Gibbs sampler for Ising model. For BRGL by Liu et al. (2014), the network markers are selected when the posterior probability exceeds 0.5.
To evaluate posterior sensitivity to the prior specification of γ in TGLG, we consider three cases. 1) TGLG-I: assign a network-independent prior for γ, i.e., γ ~ N{0p, ); 2) TGLG-F: fix ε = 10−5 and 3) TGLG-L: assign a log-normal prior to ε, i.e., log ε ~ N(−5, 9).
For all the Bayesian methods, we run 30,000 MCMC iterations with the first 20,000 as burn-in. We also check the MCMC convergences by running five chains and computing the Gelman-Rubin diagnostics. For all the Bayesian methods, the estimated 95% CI of the potential scaled reduction factors for the deviance is [1.0, 1.0], indicating the convergence of the MCMC algorithm. To compare the performance of different methods, we compute true positives, false positives and the area under the curve (AUC) for true network markers recovery, prediction mean squared error (PMSE) for linear regression and classification error (CE) for logistic regression regarding to outcome. We report the mean and standard error over 50 datasets for each metric we choose to compare in the result table.
Table 1 summarizes the results for linear regression under different settings. In most cases, TGLG approaches with incorporating network structure achieve a smaller PMSE, smaller number of false positives with a comparable amount of true positives compared with other methods. For the Ising model, we only report the results in the case of b = 7 since it has an overall best performance among all choices of b values. In fact, the performance of the Ising model varies greatly for different choices of values for b and it may perform very bad with an inappropriate value of b. Table 3 shows the mean computation time over 50 datasets for Ising model and TGLG. It shows that our method is much more computationally efficient than the Ising model, especially for the large-scale networks.
Table 1:
Simulation results for linear regression. PMSE: prediction mean squared error. TP: true positives, FP: false positives; number of informative nodes in Type 1 network is 22; number of informative nodes in Type 2 network is 12.
| Method | PMSE | TP | FP | AUC | PMSE | TP | FP | AUC |
|---|---|---|---|---|---|---|---|---|
| Type 1 p = 33 | Type 1 p = 110 | |||||||
| Lasso | 52.3(1.6) | 20.6(0.2) | 7.3(0.3) | 0.778(0.006) | 71.6(1.9) | 17.2(0.3) | 19.6(1.2) | 0.792(0.005) |
| Elastic-net | 50.9(1.4) | 21.8(0.1) | 10.4(0.2) | 0.788(0.004) | 73.7(1.8) | 19.6(0.3) | 46.6(2.9) | 0.811(0.004) |
| Grace | 56.8(1.5) | 21.6(0.1) | 10.1(0.2) | 0.864(0.007) | 87.5(2.0) | 17.9(0.4) | 37.5(2.5) | 0.897(0.004) |
| aGrace | 53.7(1.5) | 22.0(0.0) | 10.7(0.1) | 0.875(0.007) | 76.4(2.1) | 20.6(0.3) | 65.9(3.6) | 0.899(0.005) |
| L∞ | 51.4(1.5) | 21.8(0.1) | 8.9(0.4) | 0.970(0.006) | 66.5(1.7) | 21.5(0.2) | 22.7(1.5) | 0.973(0.005) |
| aL∞ | 54.2(1.3) | 21.8(0.1) | 8.2(0.6) | 0.669(0.034) | 63.5(1.5) | 21.5(0.2) | 19.6(1.4) | 0.946(0.010) |
| TTLP | 54.3(1.6) | 21.9(0.0) | 10.1(0.4) | 0.834(0.019) | 72.6(2.0) | 20.9(0.4) | 44.2(4.6) | 0.920(0.004) |
| LTLP | 51.3(1.2) | 22.0(0.0) | 8.8(0.6) | 0.933(0.005) | 67.1(1.7) | 21.5(0.2) | 57.6(2.7) | 0.897(0.009) |
| BRGL | 51.0(1.3) | 19.5(0.2) | 4.1(0.3) | 0.883(0.008) | 79.7(1.8) | 17.9(0.2) | 22.1(0.9) | 0.867(0.006) |
| Ising(b=7) | 54.9(3.0) | 19.7(0.7) | 2.9(0.7) | 0.925(0.017) | 94.9(5.9) | 15.1(0.9) | 33.9(2.4) | 0.786(0.023) |
| TGLG-I | 50.1(1.3) | 21.9(0.1) | 10.7(0.2) | 0.863(0.010) | 81.4(2.1) | 14.8(0.5) | 22.6(2.6) | 0.779(0.009) |
| TGLG-F | 45.2(1.2) | 22.0(0.0) | 2.2(0.6) | 0.912(0.032) | 63.9(2.8) | 19.7(0.4) | 17.8(2.9) | 0.899(0.016) |
| TGLG-L | 46.0(1.3) | 21.9(0.1) | 1.7(0.5) | 0.968(0.016) | 74.1(2.4) | 17.1(0.5) | 19.3(2.7) | 0.847(0.013) |
| Type 2 p =33 | Type 2 p = 110 | |||||||
| Lasso | 23.1(0.6) | 11.7(0.1) | 11.8(0.6) | 0.830(0.006) | 30.6(0.8) | 9.5(0.2) | 19.1(1.1) | 0.826(0.007) |
| Elastic-net | 23.4(0.6) | 11.8(0.1) | 15.4(0.6) | 0.802(0.006) | 31.4(0.9) | 10.6(0.2) | 34.0(2.1) | 0.818(0.006) |
| Grace | 25.8(0.6) | 11.4(0.1) | 14.7(0.6) | 0.813(0.005) | 35.2(0.8) | 9.1(0.2) | 25.8(1.9) | 0.855(0.005) |
| aGrace | 25.9(0.7) | 12.0(0.0) | 20.3(0.3) | 0.868(0.006) | 32.8(0.8) | 11.6(0.1) | 73.0(3.5) | 0.895(0.007) |
| L∞ | 23.8(0.6) | 11.9(0.1) | 17.2(0.6) | 0.812(0.005) | 30.3(0.7) | 11.3(0.2) | 28.9(1.9) | 0.928(0.005) |
| aL∞ | 26.1(0.7) | 11.9(0.1) | 16.9(0.6) | 0.643(0.018) | 30.6(0.6) | 11.3(0.2) | 27.1(1.7) | 0.893(0.009) |
| TTLP | 25.9(0.8) | 12.0(0.0) | 20.0(0.5) | 0.801(0.008) | 32.2(0.8) | 11.6(0.2) | 64.3(5.2) | 0.923(0.004) |
| LTLP | 24.7(0.7) | 12.0(0.0) | 20.4(0.4) | 0.825(0.008) | 30.6(0.7) | 11.7(0.2) | 75.1(3.6) | 0.864(0.006) |
| BRGL | 23.7(0.6) | 11.4(0.1) | 7.3(0.4) | 0.938(0.007) | 37.7(0.9) | 9.9(0.1) | 23.8(1.1) | 0.876(0.008) |
| Ising(b=7) | 27.8(1.5) | 9.9(0.5) | 11.6(0.8) | 0.855(0.024) | 45.8(2.6) | 7.6(0.6) | 44.5(2.0) | 0.709(0.032) |
| TGLG-I | 23.7(0.6) | 10.8(0.2) | 8.0(0.9) | 0.918(0.006) | 33.9(0.9) | 7.2(0.3) | 7.6(1.5) | 0.829(0.011) |
| TGLG-F | 22.8(0.6) | 11.4(0.1) | 10.2(0.7) | 0.901(0.015) | 28.7(1.1) | 10.5(0.3) | 14.2(2.1) | 0.922(0.012) |
| TGLG-L | 22.3(0.6) | 11.6(0.1) | 8.9(0.6) | 0.930(0.008) | 28.8(0.9) | 8.8(0.3) | 6.4(1.1) | 0.908(0.011) |
Table 3:
Average computing time with standard error in seconds for Ising model and TGLG based network marker selection. All the computations run on a desktop computer with 3.40 GHz i7 CPU and 16 GB memory.
| Linear regression | Logistic regression | ||||
|---|---|---|---|---|---|
| Ising | TGLG | Ising | TGLG | ||
| p = 33 | Type 1 | 140.1(0.5) | 21.5(0.2) | 230.1(7.6) | 26.7(0.3) |
| Type 2 | 140.1(0.5) | 21.0(0.3) | 229.9(7.6) | 26.4(0.2) | |
| p = 110 | Type 1 | 1191.4(7.1) | 31.7(0.2) | 1210.1(10.1) | 37.7(1.0) |
| Type 2 | 1153.4(8.5) | 30.6(0.1) | 1203.6(8.4) | 36.5(0.9) | |
As for the three cases of adopting TGLG approaches, TGLG-L has the best overall performance regarding to the PMSE and false positives. TGLG-F tends to have a larger false positive than TGLG-L and TGLG-I, since selection variables for connected nodes are highly dependent when fixed ε = 10−5. However, TGLG-F still has a smaller PMSE than TGLG-I. Compared with TGLG-I, TGLG-L has smaller false positives and PMSE in most cases. These facts show that incorporating network structure can improve model prediction performance of TGLG in linear regression.
Table 2 summarizes the results for the logistic regression under different simulation settings. Here the TGLG is only compared with Lasso, Elastic-net and the Ising model. For Type 1 network, the Ising model has a smaller number of false positives than all three TGLG approaches. However, the Ising model has a larger prediction error and a smaller number of true positives. In all other scenarios, TGLG outperforms the Ising model. Table 3 demonstrates the TGLG approach is much more computational efficient than the Ising model in Logistic regression. In addition, TGLG-F and TGLG-L have a smaller number of false positives and classification error than TGLG-I in most cases, which indicates that including network structure could improve model performance in logistic regression.
Table 2:
Simulation results for logistic regression with sample size 200. CE: classification error (number of incorrect classification); TP: true positive; FP: false positive; number of Type 1 true network markers: 22; number of Type 2 true network markers: 12.
| Method | CE | TP | FP | AUC | CE | TP | FP | AUC |
|---|---|---|---|---|---|---|---|---|
| Type 1 p = 33 | Type 1 p = 110 | |||||||
| Lasso | 20.8(0.7) | 21.2(0.1) | 6.9(0.4) | 0.811(0.004) | 30.8(1.1) | 19.1(0.4) | 25.1(1.7) | 0.836(0.004) |
| Elastic-net | 21.0(0.8) | 21.4(0.1) | 8.4(0.4) | 0.818(0.004) | 32.6(0.8) | 19.9(0.2) | 29.4(2.1) | 0.848(0.003) |
| Ising(b=5) | 39.2(3.0) | 15.2(1.2) | 0.0(0.0) | 0.937(0.011) | 47.6(4.1) | 13.5(1.1) | 10.2(2.9) | 0.826(0.031) |
| TGLG-I | 19.2(0.6) | 21.9(0.1) | 10.0(0.2) | 0.877(0.011) | 30.5(0.9) | 17.1(0.3) | 16.0(1.4) | 0.851(0.008) |
| TGLG-F | 19.4(0.7) | 21.8(0.1) | 8.0(0.5) | 0.858(0.021) | 30.8(1.1) | 17.6(0.4) | 13.0(1.1) | 0.870(0.007) |
| TGLG-L | 18.7(0.7) | 21.8(0.1) | 7.5(0.5) | 0.875(0.018) | 30.4(1.0) | 17.3(0.3) | 13.4(1.1) | 0.858(0.008) |
| Type 2 p = 33 | Type 2 p = 110 | |||||||
| Lasso | 25.2(0.9) | 11.7(0.1) | 10.1(0.7) | 0.856(0.004) | 32.7(1.0) | 10.6(0.2) | 22.7(2.2) | 0.872(0.004) |
| Elastic-net | 26.1(0.8) | 11.9(0.0) | 13.2(0.7) | 0.796(0.004) | 36.6(1.2) | 10.5(0.3) | 25.9(2.5) | 0.849(0.004) |
| Ising(b=5) | 27.4(1.4) | 9.5(0.4) | 7.2(0.4) | 0.899(0.016) | 37.7(2.8) | 7.4(0.5) | 9.0(1.7) | 0.820(0.025) |
| TGLG-I | 22.6(0.8) | 11.4(0.1) | 4.8(0.6) | 0.961(0.007) | 29.4(1.2) | 9.7(0.3) | 6.9(0.9) | 0.897(0.0012) |
| TGLG-F | 23.2(0.8) | 11.5(0.1) | 6.3(0.6) | 0.941(0.010) | 29.3(1.0) | 9.9(0.3) | 6.7(0.6) | 0.903(0.010) |
| TGLG-L | 22.1(0.8) | 11.6(0.1) | 5.8(0.7) | 0.959(0.005) | 28.6(1.0) | 10.1(0.2) | 6.2(0.8) | 0.921(0.009) |
5.2. Large Scale-Free Networks
We perform simulation studies on large scale-free networks, which are commonly used network models for gene networks. We simulate scale-free network (Barabási and Albert, 1999) with 1,000 nodes using R function barabasi.game in package igraph. In the simulated scale-free network, we set the true network markers by selecting 10 nodes out of 1,000 as the true informative nodes according to two criteria: 1) all the true informative nodes form a connected component (Hopcroft and Tarjan, 1973) in the network; 2) all the true informative nodes are disconnected, in which case the TGLG model assumption does not hold. For each informative node, the magnitude of the effect size is simulated from Unif(1, 3) and its sign is randomly assigned as positive or negative. Covariates X are generated from a multivariate normal distribution X ~ N(0, 0.3D), where D is the shortest path distance matrix between nodes in the generated scale-free network. Response variable Y is generated using Y ~ N(Xβ, ) for linear regression and Pr(Y = 1) = 1/{1 + exp(−Xβ)} for logistic regression. According to the above procedure, we simulate 50 datasets with sample size 200.
We apply the aforementioned all three TGLG methods (TGLG-I, TGLG-F, TGLG-L) to each dataset compared with Lasso and Elastic-net. In addition, to evaluate the robustness of network structure mis-specifications, for each simulated scale-free network, we randomly select 20% of nodes and permute their labels; and then we apply TGLG-L with this mis-specified network. We refer to this approach as TGLG-M.
Table 4 reports the same performance evaluation metrics as Table 1 and Table 2. When the informative nodes form a connected component in the network, overall TGLG-L achieve the best performance regarding to PMSE or CE, and false positives. When the informative nodes are all disconnected, TGLG-L still has the best performance in linear regression, but is slightly worse than TGLG-I in logistic regression. This fact indicates that TGLG approaches is not sensitive to our model assumption regarding the true network markers. In both cases, TGLG-M performs worse than TGLG-L with correctly specified networks, but still better than Lasso and Elastic-net. This implies that the useful network information can improve the performance of TGLG, while TGLG-L is robust the network misspecification.
Table 4:
Simulation results for scale-free network. The number of true informative nodes is 10. Sample size is 200 and the number of nodes is 1,000.
| Method | PMSE | TP | FP | AUC | CE | TP | FP | AUC |
|---|---|---|---|---|---|---|---|---|
| Linear regression | Logistic regression | |||||||
| True informative nodes form a connected component | ||||||||
| Lasso | 21.7(0.6) | 9.5(0.1) | 54.4(3.8) | 0.936(0.004) | 43.3(1.6) | 8.4(0.2) | 29.6(3.4) | 0.771(0.028) |
| Elastic-net | 23.2(0.7) | 9.6(0.1) | 69.0(3.9) | 0.931(0.004) | 57.9(2.4) | 7.7(0.2) | 22.4(3.2) | 0.928(0.006) |
| TGLG-I | 21.7(0.8) | 9.1(0.1) | 13.5(1.9) | 0.950(0.007) | 37.2(1.3) | 7.7(0.2) | 8.9(0.9) | 0.892(0.011) |
| TGLG-F | 21.8(0.9) | 9.3(0.1) | 14.6(1.5) | 0.968(0.006) | 35.2(1.3) | 8.0(0.2) | 7.8(0.9) | 0.902(0.011) |
| TGLG-L | 20.7(0.7) | 9.1(0.1) | 10.1(1.5) | 0.957(0.006) | 35.4(1.4) | 7.9(0.3) | 8.3(1.0) | 0.893(0.011) |
| TGLG-M | 21.2(0.8) | 9.1(0.1) | 11.3(1.5) | 0.952(0.007) | 37.1(1.3) | 7.8(0.2) | 9.3(1.1) | 0.892(0.012) |
| True informative nodes are all disconnected | ||||||||
| Lasso | 20.8(0.6) | 9.8(0.1) | 55.0(3.7) | 0.940(0.003) | 43.4(1.2) | 8.9(0.2) | 26.8(3.0) | 0.824(0.028) |
| Elastic-net | 22.2(0.7) | 9.8(0.1) | 68.6(3.9) | 0.941(0.003) | 55.7(1.9) | 8.4(0.2) | 27.3(4.0) | 0.939(0.003) |
| TGLG-I | 21.4(0.9) | 9.4(0.1) | 13.4(2.0) | 0.974(0.006) | 35.4(1.3) | 8.6(0.2) | 7.9(0.8) | 0.931(0.009) |
| TGLG-F | 21.7(0.8) | 9.4(0.1) | 16.7(1.9) | 0.971(0.006) | 35.5(1.4) | 8.4(0.2) | 7.8(0.9) | 0.922(0.010) |
| TGLG-L | 20.6(0.8) | 9.6(0.1) | 11.6(2.1) | 0.980(0.004) | 36.9(1.5) | 8.5(0.2) | 9.4(1.1) | 0.925(0.009) |
| TGLG-M | 21.3(0.9) | 9.4(0.1) | 11.4(1.7) | 0.969(0.005) | 35.3(1.2) | 8.5(0.2) | 8.4(0.9) | 0.928(0.008) |
5.3. Application to Breast Cancer Data from the Cancer Genome Atlas
In the real data application, we use the High-quality INTeractomes (HINT) database for the biological network (Das and Yu, 2012). We apply our method to the TCGA breast cancer (BRCA) RNA-seq gene expression dataset with 762 subjects and 10,792 genes in the network. The response variable we consider here is ER status – whether the cancer cells grow in response to the estrogen. The ER status is a molecular characteristic of the cancer which has important implications in prognosis. The purpose here is not focused on prediction. Rather we intend to find genes and functional modules that are associated with ER status, through which biological mechanisms differentiating the two subgroups of cancer can be further elucidated.
We code ER-positive as 1 and ER-negative as 0. We remove subjects with unknown ER status. In total, there are 707 subjects with 544 ER-positive and 163 ER-negative. We remove 348 gene nodes with low count number, which leaves us with 10,444 nodes. To apply our methods, we first standardize the gene nodes and then apply a logistic regression model for network marker selection. For prior settings, we use ~ IG(0.01, 0.01), ~ IG(0.01, 0.01) and = 50. We fix λ at different grid values and choose λ = 0.004 by maximizing the likelihood values. The MCMC algorithms run 100,000 iterations with first 90,000 as burn-in and thin by 10. To perform the Gelman Rubin diagnostics, we run three chains with different initial values randomly drawn from priors and the upper bound of the 95% CI of the potential reduction scale factor for the model deviance is around 1.1, showing an acceptable MCMC convergence in terms of model fitting.
A total of 470 genes are selected as networks marker by our approach. To facilitate data interpretation, we conduct the community detection on the network containing the selected network markers and their one-step neighbors (Clauset et al., 2004). There is a total of eight modules that contain 10 or more selected genes. The plot of the modules, together with their over-represented biological process as identified using the ‘GOstats’ package (Falcon and Gentleman, 2007), are listed in Supplementary File 2 (Cai et al., 2018b).
Figure 3 shows two example network modules. The first example (Figure 3(a)) contains 95 selected gene network markers, including 14 that are connected with other network markers. The top 5 biological processes associated with these 95 genes are listed in Table 5. The most significant biological process that is over-represented by the selected genes in this module is regulation of cellular response to stress (p=0.00016), with 14 of the selected genes involved in this biological process. Besides the general connection between stress response and breast cancer, ER status has some specific interplay with various stress response processes. For example, breast cancer cells up-regulate hypoxia-inducible factors, which cause higher risk of metastasis (Gilkes and Semenza, 2013). Hypoxia inducible factors can influence the expression of estrogen receptor (Wolff et al., 2017). In addition, estrogen changes the DNA damage response by regulating proteins including ATM, ATR, CHK1, BRCA1, and p53 (Caldon, 2014). Thus it is expected that DNA damage response is closely related to ER status.
Figure 3:

Two example modules of selected genes.
Table 5:
Selected Goterm results for the two selected modules shown in Figure 3. The upper part is the Goterm results for Figure 3(a) and the lower part is the Goterm results for Figure 3(b).
| GOBPID | Pvalue | Term |
|---|---|---|
| GO:0080135 | 0.0001618 | regulation of cellular response to stress |
| GO:0044070 | 0.000381 | regulation of anion transport |
| GO:0060969 | 0.0004409 | negative regulation of gene silencing |
| GO:0055013 | 0.000757 | cardiac muscle cell development |
| GO:0030888 | 0.0009629 | regulation of B cell proliferation |
| GOBPID | Pvalue | Term |
| GO:0030097 | 0.00006398 | hemopoiesis |
| GO:1902533 | 0.0003036 | positive regulation of intracellular signal transduction |
| GO:0002250 | 0.0004063 | adaptive immune response |
| GO:0032467 | 0.0004452 | positive regulation of cytokinesis |
| GO:0070229 | 0.0005767 | negative regulation of lymphocyte apoptotic process |
Five other genes in this module are involved in the pathway of regulation of anion transport, which include the famous mTOR gene, which is implicated in multiple cancers (Le Rhun et al., 2017). The PI3K/AKT/mTOR pathway is an anticancer target in ER+ breast cancer (Ciruelos Gil, 2014). The other four genes, ABCB1 (Jin and Song, 2017), SNCA (Li et al., 2018), IRS2 (Yin et al., 2017) and NCOR1 (Lopez et al., 2016) are all involved in some other types of cancer.
In ER- breast cancer cells, the lack of ER signaling triggers the epigenetic silencing of downstream targets (Leu et al., 2004), which explains the significance of the biological process ”negative regulation of gene silencing”. Many genes in the ”cardiac muscle cell development” processes are also part of the growth factor receptor pathway, which has a close interplay with estrogen signaling (Osborne et al., 2005). Four of the genes fall into the process ” regulation of B cell proliferation”. Among them, AHR has been identified as a potential tumor suppressor (Formosa et al., 2017). ERα is recruited in AhR signaling (Matthews and Gustafsson, 2006). IRS2 responds to interleukin 4 treatment, and its polymorphism is associated with colorectal cancer risk (Yin et al., 2017). CLCF1 signal transduction was found to play a critical role in the growth of malignant plasma cells (Burger et al., 2003). It appears that these genes are found due to their functionality in signal transduction, rather than specific functions in B cell proliferation.
The second example is a much smaller module including 14 selected genes. Six of the 14 genes are involved in both hemopoiesis and immune system development (Table 5). They are all signal transducers. Among them, AGER is a member of the immunoglobulin superfamily of cell surface receptors, which also acts as a tumor suppressor (Wu et al., 2018). CD27 is a tumor necrosis factor (TNF) receptor. Treatment with the estrogen E2 modulates the expression of CD27 in the bone marrow and spleen cells (Stubelius et al., 2014). TNFSF18 is a cytokine that belongs to the tumor necrosis factor (TNF) ligand family. Although its relation with estrogen and breast cancer is unclear, its receptor GITR shows increased expression in tumor-positive lymph nodes from advanced breast cancer patients (Krausz et al., 2012), and is targeted by some anti-cancer immunotherapy (Schaer et al., 2012). UBD is a ubiquitin-like protein, which promotes tumor proliferation by stabilizing the translation elongation factor eEF1A1 (Liu et al., 2016).
Interestingly, three of the other top biological processes are also immune processes. In normal immune cells, estrogen receptors regulate innate immune signaling pathways (Kovats, 2015). In addition, some of the selected genes in these pathways have been found to associate with cancer. Examples include AURKB, which belongs to the family of serine/threonine kinases, and contributes to chemo-resistance and poor prognosis in breast cancer (Zhang et al., 2015), and SVIL, which mediates the suppression of p53 protein and enhances cell survival (Fang and Luna, 2013).
Overall, genes selected by TGLG are easy to interpret. Many known links exist between these genes and ER status, or breast cancer in general. Still many of the selected genes are not reported so far to be linked to ER status or breast cancer. Our results indicate they may play important roles.
6. Discussion
In summary, we propose a new prior model: TGLG prior for Bayesian network marker selection over large-scale networks. We show the proposed prior model enjoys large prior support for network marker selection over large-scale networks, leading to the posterior consistency. We also develop an efficient Metropolis-adjusted Langevin algorithm (MALA) for posterior computation. The simulation studies show that our method performs better than existing regularized regression approaches with regard to the selection and prediction accuracy. Also, the analysis of TCGA breast cancer data indicates that our method can provide biologically meaningful results.
This paper leads to some obvious future work. First, we can apply the TGLG prior for network marker selection under other modeling framework such as the survival model and the generalized mixed effects model. Second, the current posterior computation can be further improved by utilizing the parallel computing techniques within each iteration of the MCMC algorithm, for updating the massive latent variables simultaneously. Third, another promising direction is to use the integrated nested laplace approximations (INLA) for Bayesian approximating computation taking advantages of the TGLG prior involving high-dimensional Gaussian latent variables.
Supplementary Material
Acknowledgments
Funding for the project was provided by the NIH grants 1R01MH105561 and 1R01GM124061.
Footnotes
Supplementary Material
Supplementary file 1 for “Bayesian network marker selection via the thresholded graph Laplacian Gaussian prior” (DOI: 10.1214/18-BA1442SUPPA; .pdf). Supplementary materials available at Bayesian Analysis online includes proofs of the theoretical results.
Supplementary file 2 for “Bayesian network marker selection via the thresholded graph Laplacian Gaussian prior” (DOI: 10.1214/18-BA1442SUPPB; .pdf). Supplementary materials available at Bayesian Analysis online includes results for real data analysis.
References
- Aebersold R and Mann M (2003). “Mass spectrometry-based proteomics.” Nature, 422(6928): 198. 79 [DOI] [PubMed] [Google Scholar]
- Barabási A-L and Albert R (1999). “Emergence of scaling in random networks.” Science, 286(5439): 509–512. MR2091634. doi: 10.1126/science.286.5439.509. 92 [DOI] [PubMed] [Google Scholar]
- Barabási A-L, Gulbahce N, and Loscalzo J (2011). “Network medicine: a network-based approach to human disease.” Nature reviews genetics, 12(1): 56. 79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri MM, Berger JO, et al. (2004). “Optimal predictive model selection.” The annals of statistics, 32(3): 870–897. MR2065192. doi: 10.1214/009053604000000238. 88 [DOI] [Google Scholar]
- Bhattacharya A, Pati D, Pillai NS, and Dunson DB (2015). “Dirichlet–Laplace priors for optimal shrinkage.” Journal of the American Statistical Association, 110(512): 1479–1490. MR3449048. doi: 10.1080/01621459.2014.960967. 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger R, Bakker F, Guenther A, Baum W, Schmidt-Arras D, Hideshima T, Tai Y-T, Shringarpure R, Catley L, Senaldi G, Gramatzki M, and Anderson KC (2003). “Functional significance of novel neurotrophin-1/B cell-stimulating factor-3 (cardiotrophin-like cytokine) for human myeloma cell growth and survival.” British Journal of Haematology, 123(5): 869–78. 95 [DOI] [PubMed] [Google Scholar]
- Cai Q, Kang J, and Yu T (2018a). “Supplementary File 1 for “Bayesian Network Marker Selection via the Thresholded Graph Laplacian Gaussian Prior”.” Bayesian Analysis. doi: 10.1214/18-BA1142SUPPA. 86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Kang J, and Yu T (2018b). “Supplementary file 2 for “Bayesian Network Marker Selection via the Thresholded Graph Laplacian Gaussian Prior”.” Bayesian Analysis. doi: 10.1214/18-BA1142SUPPB. 93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldon CE (2014). “Estrogen signaling and the DNA damage response in hormone dependent breast cancers.” Frontiers in Oncology, 4: 106. 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Kundu S, and Long Q (2016). “Scalable Bayesian variable selection for structured high-dimensional data.” arXiv preprint arXiv:1604.07264. MR3130862. 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chekouo T, Stingo FC, Guindani M, Do K-A, et al. (2016). “A Bayesian predictive model for imaging genetics with application to schizophrenia.” The Annals of Applied Statistics, 10(3): 1547–1571. MR3553235. doi: 10.1214/16-AOAS948. 80 [DOI] [Google Scholar]
- Chung FR (1997). Spectral graph theory, volume 92. American Mathematical Society. MR1421568. 82 [Google Scholar]
- Ciruelos Gil EM (2014). “Targeting the PI3K/AKT/mTOR pathway in estrogen receptor-positive breast cancer.” Cancer Treatment Reviews, 40(7): 862–71. 95 [DOI] [PubMed] [Google Scholar]
- Clauset A, Newman ME, and Moore C (2004). “Finding community structure in very large networks.” Physical review E, 70(6): 066111. 93 [DOI] [PubMed] [Google Scholar]
- Das J and Yu H (2012). “HINT: High-quality protein interactomes and their applications in understanding human disease.” BMC Systems Biology, 6: 92. 92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobra A (2009). “Variable selection and dependency networks for genomewide data.” Biostatistics, 10(4): 621–639. 81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi K (2007). “Computer-aided diagnosis in medical imaging: historical review, current status and future potential.” Computerized medical imaging and graphics, 31(4–5): 198–211. 79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon S and Gentleman R (2007). “Using GOstats to test gene lists for GO term association.” Bioinformatics, 23(2): 257–8. MR2138207. doi: 10.2202/1544-6115.1034. 93 [DOI] [PubMed] [Google Scholar]
- Fan J and Li R (2001). “Variable selection via nonconcave penalized likelihood and its oracle properties.” Journal of the American statistical Association, 96(456): 1348–1360. MR1946581. doi: 10.1198/016214501753382273. 80 [DOI] [Google Scholar]
- Fang Z and Luna EJ (2013). “Supervillin-mediated suppression of p53 protein enhances cell survival.” Journal of Biological Chemistry, 288(11): 7918–29. 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formosa R, Borg J, and Vassallo J (2017). “Aryl hydrocarbon receptor (AHR) is a potential tumour suppressor in pituitary adenomas.” Endocrine Related Cancer, 24(8): 445–457. 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George EI and McCulloch RE (1993). “Variable selection via Gibbs sampling.” Journal of the American Statistical Association, 88(423): 881–889. 80 [Google Scholar]
- Gilkes DM and Semenza GL (2013). “Role of hypoxia-inducible factors in breast cancer metastasis.” Future Oncology, 9(11): 1623–36. 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith J, Huang L, and Crainiceanu CM (2014). “Smooth scalar-on-image regression via spatial Bayesian variable selection.” Journal of Computational and Graphical Statistics, 23(1): 46–64. MR3173760. doi: 10.1080/10618600.2012.743437. 81, 89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greicius MD, Krasnow B, Reiss AL, and Menon V (2003). “Functional connectivity in the resting brain: a network analysis of the default mode hypothesis.” Proceedings of the National Academy of Sciences, 100(1): 253–258. 79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopcroft J and Tarjan R (1973). “Algorithm 447: efficient algorithms for graph manipulation.” Communications of the ACM, 16(6): 372–378. 92 [Google Scholar]
- Jiang W (2007). “Bayesian variable selection for high dimensional generalized linear models: convergence rates of the fitted densities.” The Annals of Statistics, 35(4): 1487–1511. MR2351094. doi: 10.1214/009053607000000019 84 [DOI] [Google Scholar]
- Jin S-S and Song W-J (2017). “Association between MDR1 C3435T polymorphism and colorectal cancer risk: A meta-analysis.” Medicine (Baltimore), 96(51): e9428. 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE and Rossell D (2012). “Bayesian model selection in high-dimensional settings.” Journal of the American Statistical Association, 107(498): 649–660. MR2980074. doi: 10.1080/01621459.2012.682536. 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Reich BJ, and Staicu A-M (2018). “Scalar-on-image regression via the soft-thresholded Gaussian process.” Biometrika, 105(1): 165–184. MR3768872. doi: 10.1093/biomet/asx075. 81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Gao L, and Tan K (2012). “Multi-analyte network markers for tumor prognosis.” PLoS One, 7(12): e52973. 79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Pan W, and Shen X (2013). “Network-based penalized regression with application to genomic data.” Biometrics, 69(3): 582–593. MR3106586. doi: 10.1111/biom.12035. 80, 88, 89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano H (2002). “Systems biology: a brief overview.” Science, 295(5560): 1662–1664. 79 [DOI] [PubMed] [Google Scholar]
- Kovats S (2015). “Estrogen receptors regulate innate immune cells and signaling pathways.” Cellular Immunology, 294(2): 63–9. 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krausz LT, Fischer-Fodor E, Major ZZ, and Fetica B (2012). “GITR-expressing regulatory T-cell subsets are increased in tumor-positive lymph nodes from advanced breast cancer patients as compared to tumor-negative lymph nodes.” International Journal of Immunopathology and Pharmacology, 25(1): 59–66. 95 [DOI] [PubMed] [Google Scholar]
- Kundu S, Shin M, Cheng Y, Manyam G, Mallick BK, and Baladandayuthapani V (2015). “Bayesian Variable Selection with Structure Learning: Applications in Integrative Genomics.” arXiv preprint arXiv:1508.02803. 81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Rhun E, Bertrand N, Dumont A, Tresch E, Le Deley M-C, Mailliez A, Preusser M, Weller M, Revillion F, and Bonneterre J (2017). “Identification of single nucleotide polymorphisms of the PI3K-AKT-mTOR pathway as a risk factor of central nervous system metastasis in metastatic breast cancer.” European Journal of Cancer, 87: 189–198. 95 [DOI] [PubMed] [Google Scholar]
- Leu Y-W, Yan PS, Fan M, Jin VX, Liu JC, Curran EM, Welshons WV, Wei SH, Davuluri RV, Plass C, Nephew KP, and Huang TH-M (2004). “Loss of estrogen receptor signaling triggers epigenetic silencing of downstream targets in breast cancer.” Cancer Research, 64(22): 8184–92. 95 [DOI] [PubMed] [Google Scholar]
- Li C and Li H (2008). “Network-constrained regularization and variable selection for analysis of genomic data.” Bioinformatics, 24(9): 1175–1182. 80, 81, 82, 88, 89 [DOI] [PubMed] [Google Scholar]
- Li C and Li H (2010). “Variable selection and regression analysis for graph-structured covariates with an application to genomics.” The annals of applied statistics, 4(3): 1498. MR2758338. doi: 10.1214/10-AOAS332. 80, 81, 89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F and Zhang NR (2010). “Bayesian Variable Selection in Structured High-Dimensional Covariate Spaces With Applications in Genomics.” Journal of the American Statistical Association, 105(491): 1202–1214. MR2752615. doi: 10.1198/jasa.2010.tm08177 81 [DOI] [Google Scholar]
- Li F, Zhang T, Wang Q, Gonzalez MZ, Maresh EL, Coan JA, et al. (2015). “Spatial Bayesian variable selection and grouping for high-dimensional scalar-on-image regression.” The Annals of Applied Statistics, 9(2): 687–713. MR3371331. doi: 10.1214/15-AOAS818. 81, 89 [DOI] [Google Scholar]
- Li Y-X, Yu Z-W, Jiang T, Shao L-W, Liu Y, Li N, Wu Y-F, Zheng C, Wu X-Y, Zhang M, Zheng D-F, Qi X-L, Ding M, Zhang J, and Chang Q (2018). “SNCA, a novel biomarker for Group 4 medulloblastomas, can inhibit tumor invasion and induce apoptosis.” Cancer Science, 109(4): 1263–1275. 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Chakraborty S, Li F, Liu Y, Lozano AC, et al. (2014). “Bayesian regularization via graph Laplacian.” Bayesian Analysis, 9(2): 449–474. MR3217003. doi: 10.1214/14-BA860. 81, 89 [DOI] [Google Scholar]
- Liu X, Chen L, Ge J, Yan C, Huang Z, Hu J, Wen C, Li M, Huang D, Qiu Y, Hao H, Yuan R, Lei J, Yu X, and Shao J (2016). “The Ubiquitin-like Protein FAT10 Stabilizes eEF1A1 Expression to Promote Tumor Proliferation in a Complex Manner.” Cancer Research, 76(16): 4897–907. 95 [DOI] [PubMed] [Google Scholar]
- Lopez SM, Agoulnik AI, Zhang M, Peterson LE, Suarez E, Gandarillas GA, Frolov A, Li R, Rajapakshe K, Coarfa C, Ittmann MM, Weigel NL, and Agoulnik IU (2016). “Nuclear Receptor Corepressor 1 Expression and Output Declines with Prostate Cancer Progression.” Clinical Cancer Research, 22(15): 3937–49. 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Pan W, and Shen X (2012). “A two-step penalized regression method with networked predictors.” Statistics in biosciences, 4(1): 27–46. 80, 89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews J and Gustafsson J-A (2006). “Estrogen receptor and aryl hydrocarbon receptor signaling pathways.” Nuclear Receptor Signaling, 4: e016. 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima J and West M (2013a). “Bayesian analysis of latent threshold dynamic models.” Journal of Business & Economic Statistics, 31(2): 151–164. MR3055329. doi: 10.1080/07350015.2012.747847. 81 [DOI] [Google Scholar]
- Nakajima J and West M (2013b). “Bayesian dynamic factor models: Latent threshold approach.” Journal of Financial Econometrics, 11: 116–153. 81 [Google Scholar]
- Nakajima J, West M, et al. (2017). “Dynamics & sparsity in latent threshold factor models: A study in multivariate EEG signal processing.” Brazilian Journal of Probability and Statistics, 31(4): 701–731. MR3738175. doi: 10.1214/17-BJPS364. 81 [DOI] [Google Scholar]
- Ni Y, Stingo FC, and Baladandayuthapani V (2017). “Bayesian graphical regression.” Journal of the American, Statistical Association, (just-accepted). 81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne CK, Shou J, Massarweh S, and Schiff R (2005). “Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer.” Clinical Cancer Research, 11(2 Pt 2): 865s–70s. 95 [PubMed] [Google Scholar]
- Pan W, Xie B, and Shen X (2010). “Incorporating predictor network in penalized regression with application to microarray data.” Biometrics, 66(2): 474–484. MR2758827. doi: 10.1111/j.1541-0420.2009.01296.x. 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park T and Casella G (2008). “The Bayesian Lasso.” Journal of the American Statistical Association, 103(482): 681–686. MR2524001. doi: 10.1198/016214508000000337. 80 [DOI] [Google Scholar]
- Peng B, Zhu D, Ander BP, Zhang X, Xue F, Sharp FR, and Yang X (2013). “An integrative framework for Bayesian variable selection with informative priors for identifying genes and pathways.” PloS one, 8(7): e67672. 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S, Eidelberg D, and Ma Y (2014). “Brain network markers of abnormal cerebral glucose metabolism and blood flow in Parkinson?s disease.” Neuroscience bulletin, 30(5): 823–837. 79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CB, Stingo FC, and Vannucci M (2016). “Joint Bayesian variable and graph selection for regression models with network-structured predictors.” Statistics in medicine, 35(7): 1017–1031. MR3476525. doi: 10.1002/sim.6792. 81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polson NG and Scott JG (2012). “Local shrinkage rules, Lévy processes and regularized regression.” Journal of the Royal Statistical Society: Series B (Statistical Methodology), 74(2): 287–311. MR2899864. doi: 10.1111/j.1467-9868.2011.01015.x. 80 [DOI] [Google Scholar]
- Roberts GO, Gelman A, Gilks WR, et al. (1997). “Weak convergence and optimal scaling of random walk Metropolis algorithms.” The annals of applied probability, 7(1): 110–120. MR1428751. doi: 10.1214/aoap/1034625254. 87 [DOI] [Google Scholar]
- Roberts GO and Rosenthal JS (1998). “Optimal scaling of discrete approximations to Langevin diffusions.” Journal of the Royal Statistical Society: Series B (Statistical Methodology), 60(1): 255–268. MR1625691. doi: 10.1111/1467-9868.00123. 86, 87 [DOI] [Google Scholar]
- Roberts GO, Rosenthal JS, et al. (2001). “Optimal scaling for various Metropolis-Hastings algorithms.” Statistical science, 16(4): 351–367. MR1888450. doi: 10.1214/ss/1015346320 87 [DOI] [Google Scholar]
- Schaer DA, Murphy JT, and Wolchok JD (2012). “Modulation of GITR for cancer immunotherapy.” Current Opinion in Immunology, 24(2): 217–24. 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster SC (2007). “Next-generation sequencing transforms today’s biology.” Nature methods, 5(1): 16. 79 [DOI] [PubMed] [Google Scholar]
- Shi R and Kang J (2015). “Thresholded multiscale Gaussian processes with application to Bayesian feature selection for massive neuroimaging data.” arXiv preprint arXiv: 1504.06074. 81 [Google Scholar]
- Song Q and Liang F (2015). “A split-and-merge Bayesian variable selection approach for ultrahigh dimensional regression.” Journal of the Royal Statistical Society: Series B (Statistical Methodology), 77(5): 947–972. MR3414135. doi: 10.1111/rssb.12095. 84 [DOI] [Google Scholar]
- Stingo FC, Chen YA, Tadesse MG, and Vannucci M (2011). “Incorporating biological information into linear models: A Bayesian approach to the selection of pathways and genes.” The annals of applied statistics, 5(3). MR2884929. doi: 10.1214/11-AOAS463. 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubelius A, Erlandsson MC, Islander U, and Carlsten H (2014). “Immunomodulation by the estrogen metabolite 2-methoxyestradiol.” Clinical Immunology, 153(1): 40–8. 95 [DOI] [PubMed] [Google Scholar]
- Tibshirani R (1996). “Regression shrinkage and selection via the lasso.” Journal of the Royal Statistical Society. Series B (Methodological), 267–288. MR1379242. 80, 89 [Google Scholar]
- Wolff M, Kosyna FK, Dunst J, Jelkmann W, and Depping R (2017). “Impact of hypoxia inducible factors on estrogen receptor expression in breast cancer cells.” Archives of Biochemistry and Biophysics, 613: 23–30. 94 [DOI] [PubMed] [Google Scholar]
- Wu S, Mao L, Li Y, Yin Y, Yuan W, Chen Y, Ren W, Lu X, Li Y, Chen L, Chen B, Xu W, Tian T, Lu Y, Jiang L, Zhuang X, Chu M, and Wu J (2018). “RAGE may act as a tumour suppressor to regulate lung cancer development.” Gene, 651: 86–93. 95 [DOI] [PubMed] [Google Scholar]
- Yin J, Zhang Z, Zheng H, and Xu L (2017). “IRS-2 rs1805097 polymorphism is associated with the decreased risk of colorectal cancer.” Oncotarget, 8(15): 25107–25114. 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Chen J, Lin Y, Li Y, Xu L, Chen L, Hua H, and Shen B (2017). “Network biomarkers constructed from gene expression and protein-protein interaction data for accurate prediction of Leukemia.” Journal of Cancer, 8(2): 278. 79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C-H (2010). “Nearly unbiased variable selection under minimax concave penalty.” The Annals of statistics, 894–942. MR2604701. doi: 10.1214/09-AOS729. 80 [DOI] [Google Scholar]
- Zhang Y, Jiang C, Li H, Lv F, Li X, Qian X, Fu L, Xu B, and Guo X (2015). “Elevated Aurora B expression contributes to chemoresistance and poor prognosis in breast cancer.” International Journal of Clinical and Experimental Pathology, 8(1): 751–7. 95 [PMC free article] [PubMed] [Google Scholar]
- Zhe S, Naqvi SA, Yang Y, and Qi Y (2013). “Joint network and node selection for pathway-based genomic data analysis.” Bioinformatics, 29(16): 1987–1996. 81, 82, 88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H and Zheng T (2013). “Bayesian hierarchical graph-structured model for pathway analysis using gene expression data.” Statistical applications in genetics and molecular biology, 12(3): 393–412. MR3101037. doi: 10.1515/sagmb-2013-0011. 80 [DOI] [PubMed] [Google Scholar]
- Zou H (2006). “The adaptive lasso and its oracle properties.” Journal of the American statistical association, 101(476): 1418–1429. MR2279469. doi: 10.1198/016214506000000735. 80 [DOI] [Google Scholar]
- Zou H and Hastie T (2005). “Regularization and variable selection via the elastic net.” Journal of the Royal Statistical Society: Series B (Statistical Methodology), 67(2): 301–320. MR2137327. doi: 10.1111/j.1467-9868.2005.00503.x. 80, 89 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.