

Lung epithelial cell–specific loss of function of Miz1 causes spontaneous COPD-like phenotype and up-regulates Ace2 in mice.
Abstract
Cigarette smoking, the leading cause of chronic obstructive pulmonary disease (COPD), has been implicated as a risk factor for severe disease in patients infected with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Here we show that mice with lung epithelial cell-specific loss of function of Miz1, which we identified as a negative regulator of nuclear factor κB (NF-κB) signaling, spontaneously develop progressive age-related changes resembling COPD. Furthermore, loss of Miz1 up-regulates the expression of Ace2, the receptor for SARS-CoV-2. Concomitant partial loss of NF-κB/RelA prevented the development of COPD-like phenotype in Miz1-deficient mice. Miz1 protein levels are reduced in the lungs from patients with COPD, and in the lungs of mice exposed to chronic cigarette smoke. Our data suggest that Miz1 down-regulation–induced sustained activation of NF-κB–dependent inflammation in the lung epithelium is sufficient to induce progressive lung and airway destruction that recapitulates features of COPD, with implications for COVID-19.
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is a growing public health concern. Mortality rates attributable to COPD have steadily risen over the past two decades, and COPD has now surpassed stroke as the third leading cause of death in the United States (1–3). Furthermore, a history of cigarette smoke exposure, the major risk factor for the development of COPD, has been associated with more severe disease in patients infected with a novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent in the ongoing coronavirus disease 2019 (COVID-19) pandemic (4). Pathologically, COPD is characterized by a combination of chronic airway inflammation (chronic bronchitis) and a progressive loss of alveolar structures. The resulting airway thickening and mucus secretion combined with the loss of lung elastic recoil causes excessive airway narrowing during exhalation, limiting ventilation, and increasing the risk of infection (1–3, 5). Observational studies suggest that exposure of susceptible individuals to cigarette smoke or other toxins leads to inflammation in multiple lung cell populations including alveolar macrophages (AMs) and epithelial cells, but several questions remain. First, while cigarette smoke is the most environmental risk factor for COPD, most people who smoke (~75%) never develop COPD, and conversely, a substantial population of patients with COPD never smoke. Second, smokers usually begin to smoke in the second decade of life and continue for decades, but clinical manifestations of COPD are unusual before the seventh decade of life. Third, while smoking cessation slows the progression of COPD, the risk of disease is substantially increased even in individuals who stop smoking relatively early in life (1–3). These observations suggest that in some individuals, inflammation induced by cigarette smoke or other exposures early in life becomes independent of ongoing exposure, leading to progressive age-related lung dysfunction and clinical manifestations of COPD.
Clinical observations suggest a link between emphysema and normal aging (6, 7). Emphysema is observed in the lungs of healthy elderly individuals, the risk of COPD increases exponentially with age, and some of the increased mortality from COPD can be attributed to aging in the population (6, 7). One of the most consistently observed changes in aging humans and rodents is an increase in expression of genes reflecting activation of the innate immune system (8, 9). Specifically, there is an age-related increase in the expression of proinflammatory cytokines including tumor necrosis factor (TNF) and interleukin-6 (IL-6), combined with an attenuated immune response to infectious challenge (9–11). In a population-based cohort, we found that increased circulating levels of C-reactive protein, a transcriptional target of IL-6, in younger individuals were associated with an increased risk of developing airflow obstruction characteristic of COPD later in life (12).
Persistent inflammation in the lung epithelium has been linked to the development of COPD, but factors that regulate this inflammation remain unknown (1–3). We previously reported that the transcription factor Miz1 (also known as c-Myc–interacting zinc finger protein-1 and Zbtb17) is critical for the resolution of pulmonary inflammation through transcriptional repression of nuclear factor κB (NF-κB)–dependent proinflammatory gene Cebpd (13). Here, we report that Miz1 protein levels are reduced in the lungs, predominantly in lung epithelial cells, from patients with COPD requiring lung transplantation compared with the donor lungs and are also reduced in the lungs of mice exposed chronically to cigarette smoke. We show that mice with lung epithelial cell–specific, but not myeloid-specific, deletion of the Miz1 poxvirus and zinc-finger (POZ) domain, which is required for Miz1-mediated transcriptional activation or repression, spontaneously develop age-related progressive changes resembling emphysema and bronchitis, cardinal pathologic features of COPD. Mice with heterozygous epithelial cell–specific loss of function of Miz1 have increased expression of proinflammatory genes in the lung epithelium throughout the life span. While these mice show normal lung morphology in young adulthood, they develop emphysema and chronic bronchitis by 1 year of age. Mice with homozygous loss of Miz1 showed up-regulation in the expression of angiotensin-converting enzyme 2 (Ace2), the receptor for SARS-CoV-2, which is the causative organism in the current COVID-19 pandemic (14). Inhibition of inflammation by NF-κB/RelA haploinsufficiency largely rescues the COPD-like phenotype in these knockout mice, suggesting that proinflammatory signaling induced by the loss of Miz1 is responsible for the COPD-like phenotype in these mice.
RESULTS
MIZ1 protein levels are reduced in human COPD lungs and cigarette smoke–exposed mouse lungs or cigarette smoke extract–treated lung epithelial cells
Examination of the lungs of naïve mice and normal human lungs revealed a similar pattern of MIZ1 expression. Miz1 mRNA was highly expressed in flow-sorted alveolar epithelial type 2 (AT2) cells, AMs, neutrophils, and monocytes, with lower expression in endothelial cells and dendritic cells in the murine lungs (fig. S1, A to C). Immunoblotting confirmed the expression of MIZ1 proteins in flow-sorted AT2 cells and AMs from mouse lungs (fig. S1D). Immunofluorescence (IF) staining of murine lung sections showed signals of MIZ1 proteins and surfactant protein C (SPC), a marker for AT2 cells, in the same cells (fig. S1E). MIZ1 was also expressed in the murine airway epithelium (fig. S1F). A similar pattern of MIZ1 expression was observed in human lungs (fig. S1G).
Inflammation in the lungs of patients with COPD persists long after the inciting stimulus, most commonly cigarette smoke, is removed, suggesting failure to normally terminate lung inflammation (1–3). We previously found that the transcription factor MIZ1 is critical for the resolution of lipopolysaccharide (LPS)– or TNF-induced pulmonary inflammation through epigenetic repression of NF-κB–dependent proinflammatory gene expression including C/EBPδ (CCAAT/enhancer binding protein δ) (13). We therefore sought to determine whether Miz1 might be involved in regulation of the pathogenesis of COPD. We measured MIZ1 expression in the lungs from patients with COPD requiring lung transplantation and in biopsy specimens from donor lungs both obtained at the time of transplant. We found reduced MIZ1 protein expression in the lungs of individuals with COPD compared with the normal lungs (Fig. 1A). MIZ1 protein expression was reduced predominantly in alveolar epithelial cells and the airway epithelium from patients with COPD but was readily detectable in AMs (Fig. 1, A and B). Miz1 may be posttranslationally down-regulated in human COPD lungs as Miz1 mRNA levels were similar between the lungs from patients with COPD and donor lungs (Fig. 1C, left). We have previously reported that the E3 ubiquitin ligase MULE (HUWE1) targets Miz1 for ubiquitination and proteasome-dependent degradation (15). We found that the mRNA and protein levels of MULE were increased in the lungs from patients with COPD compared with those from normal lungs [Fig. 1, C (right) and D]. All of these patients with COPD had stopped smoking for more than 6 months before transplantation, suggesting that the changes we observed in MIZ1 expression do not require ongoing direct exposure to cigarette smoke (CS). To determine whether CS exposure is sufficient to alter MIZ1 levels in the lung, we used a mouse model of emphysema that develops following intermittent exposure to CS for 6 months (16, 17). Consistent with our observations in the lungs from patients with COPD, we observed a progressive decrease in MIZ1 protein levels in the lungs of wild-type mice exposed to CS for 7 days, 3 months, or 6 months compared to room air (RA)–exposed controls as analyzed by immunoblotting (Fig. 1E). IF staining also showed reduced MIZ1 protein expression in the alveolar and airway epithelium from CS-exposed lungs as compared to RA-exposed lungs (Fig. 1, F and G). On the other hand, MULE protein levels were increased in both alveoli and airways of the lungs from wild-type mice exposed to CS for 6 months (Fig. 1H). In vitro, exposure of murine lung epithelial MLE-12 cells to CS extracts also increased Mule expression while decreased Miz1 protein expression at days 1, 2, and 3 (Fig. 1I). Silencing of Mule by tetracycline-inducible short hairpin RNA (shRNA), as we previously reported (15), prevented CS-induced Miz1 down-regulation (Fig. 1J). Together, these data suggest that CS induces up-regulation of Mule, which results in Miz1 down-regulation.
Fig. 1. MIZ1 protein levels are reduced in human COPD lungs and cigarette smoke–exposed mouse lungs or cigarette smoke extract–treated lung epithelial cells.
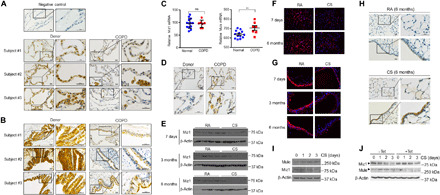
(A and B) Miz1 immunohistochemistry (IHC) staining of normal human lungs or lungs from patients with COPD at the time of lung transplantation (three subjects from each group are shown, which were representative of seven subjects from each group). Scale bars, 20 μm. (C) mRNA levels of Miz1 and Mule from normal human lungs or lungs from patients with COPD analyzed by RNA sequencing (RNA-seq) (normal, n = 13; COPD, n = 7). **P < 0.01; ns, not significant. (D) Mule IHC from human lungs mentioned in (A). (E to H) Western blot (E) or IF staining (F and G) of Miz1 or Mule IHC (H) in mouse lungs exposed to RA or CS for 7 days, 3 months, and 6 months. Representatives of n ≥ 4. (I and J) Western blot of Mule and Miz1 in CS extract–treated MLE-12 cells (I) or cells expressing tetracycline-inducible shRNA for Mule in the absence or presence of tetracycline (Tet) (J). Data are representative of at least three independent experiments.
Lifelong heterozygous lung epithelial deletion of the Miz1 POZ domain results in progressive emphysema that is only evident in middle to late life
To determine whether there is a causal link between the reduced MIZ1 protein expression we observed in lung sections from patients with COPD and CS-exposed mice and the development of COPD phenotypes, we crossed transgenic mice expressing Cre driven by the human SPC promoter (SPC-Cre mice) (18) with Miz1(POZ)fl/fl mice, in which loxP sites flank the N-terminal POZ domain of Miz1 [Miz1(POZ)] that is essential for Miz1-mediated transcriptional activation or repression (13). As SPC is expressed early in lung epithelial progenitors, developmental expression of SPC-Cre results in Cre-mediated recombination in the lung epithelium that persists over the life span of the animal (18). Quantitative polymerase chain reaction (qPCR) using genomic DNA revealed almost complete deletion of the Miz1(POZ) in primary AT2 cells flow-sorted from homozygous SPC-Cre+/Miz1(POZ)fl/fl mice and partial deletion (~ 50%) from heterozygous SPC-Cre+/Miz1(POZ)wt/fl littermates (fig. S2A). Note that there was ~25% reduction in the abundance of Miz1(POZ) in flow-sorted AMs from the SPC-Cre+/Miz1(POZ)fl/fl homozygotes, but not the SPC-Cre+/Miz1(POZ)wt/fl heterozygotes (fig. S2A), consistent with the observations by our group and others that SPC is expressed in AMs, although at relatively low levels (19, 20). There was no deletion of Miz1(POZ) in the other myeloid populations from the homozygous or heterozygous mutant mice (fig. S2A), indicating the specificity of the Miz1(POZ) deletion in lung epithelial cells. Deletion of the Miz1(POZ) did not grossly affect epithelial cell numbers as the percentage of epithelial cells in the CD45− cells from the lungs of the knockout mice was similar to that in the control Miz1(POZ)fl/fl mice (fig. S2B).
Compared with control mice of 4 months old [SPC-Cre/Miz1(POZ)wt/wt or Miz1(POZ)fl/fl)], mice with homozygous epithelial cell–specific deletion of Miz1(POZ) [SPC-Cre+/Miz1(POZ)fl/fl] showed diffuse bullous emphysema-like changes as evident from gross photograph of the lung (fig. S2C). Histology revealed a severe emphysema-like phenotype, characterized by heterogeneous alveolar enlargement and severe lung inflammation (Fig. 2, A and B). We examined alveolar size by measuring the mean linear intercept (MLI) of alveolar septa with computer-assisted stereology (16, 21). Young homozygous SPC-Cre+/Miz1(POZ)fl/fl mice had increased MLI and decreased alveolar surface-to-volume ratio compared to the control Miz1(POZ)fl/fl mice of similar age (Fig. 2C). Consistent with a gene-dose effect, we could not detect any histologic abnormalities in mice with heterozygous epithelial cell–specific loss of Miz1(POZ) [SPC-Cre+/Miz1(POZ)wt/fl] at 4 months of age (Fig. 2D). By 1 year of age, however, we observed alveolar airspace enlargement consistent with emphysema in the heterozygous SPC-Cre+/Miz1(POZ)wt/fl mice compared with the control Miz1(POZ)fl/fl or SPC-Cre+/Miz1(POZ)wt/wt mice of similar age (Fig. 2, D to G, and fig. S2D). At 1 year of age, emphysema severity in the homozygous SPC-Cre+/Miz1(POZ)fl/fl mice was worse when compared with age-matched heterozygous SPC-Cre+/Miz1(POZ)wt/fl mice and was progressive when compared with homozygous SPC-Cre+/Miz1(POZ)fl/fl mice harvested at 4 months of age as determined by histology and stereologic assessments of MLI (Fig. 2, A and E to G, and fig. S2D).
Fig. 2. Lung epithelial cell–specific deletion of the Miz1 POZ domain causes spontaneous emphysema in mice.
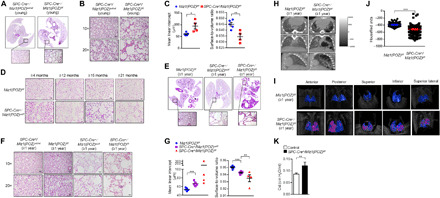
(A and E) Representative whole lung histology from ≤4-month-old control SPC-Cre+/Miz1(POZ)wt/wt or homozygous SPC-Cre+/Miz1(POZ)fl/fl mice (A) or ≥1-year-old control Miz1(POZ)fl/fl, heterozygous SPC-Cre+/Miz1(POZ)wt/fl, or homozygous SPC-Cre+/Miz1(POZ)fl/fl mice (E). (B, D, and F) Representative histological sections (10× or 20× objective) of lungs from ≤4-month-old control SPC-Cre+/Miz1(POZ)wt/wt or homozygous SPC-Cre+/Miz1(POZ)fl/fl mice (B), control Miz1(POZ)fl/fl or heterozygous SPC-Cre+/Miz1(POZ)wt/fl mice of different ages (D), or ≥1-year-old control SPC-Cre+/Miz1(POZ)wt/wt, Miz1(POZ)fl/fl, heterozygous SPC-Cre+/Miz1(POZ)wt/fl, or homozygous SPC-Cre+/Miz1(POZ)fl/fl mice (F). Scale bars, 20 μm. n = 4 to 8 for (A, B, and D to F). (C and G) MLI (micrometers ± SEM) or the ratio of surface area to alveolar air space volume (surface-to-volume ratio; means ± SEM) of alveolar septae measured in the lungs of ≤4-month-old control Miz1(POZ)fl/fl (n = 5) or homozygous SPC-Cre+/Miz1(POZ)fl/fl mice (n = 4) (C) or ≥ 1-year-old control Miz1(POZ)fl/fl (n = 6), heterozygous SPC-Cre+/Miz1(POZ)wt/fl (n = 5), or homozygous SPC-Cre+/Miz1(POZ)fl/fl mice (n = 4) (G). (H and I) Representative micro–computed x-ray tomography (micro-CT) images of the lung cross sections (H) or the three-dimensional lungs (I) of ≥1-year-old control Miz1(POZ)fl/fl or homozygous SPC-Cre+/Miz1(POZ)fl/fl mouse. In (I), the low attenuation area (below −700 HU) is colored in pink, and the whole lung field is colored in blue. (J) Representative HU of the lung parenchyma from ≥1-year-old control Miz1(POZ)fl/fl or homozygous SPC-Cre+/Miz1(POZ)fl/fl mouse. (K) Static lung compliance (Cst) of ≥1-year-old control Miz1(POZ)fl/fl (n = 8) or homozygous SPC-Cre+/Miz1(POZ)fl/fl mice (n = 3). *P < 0.05, **P < 0.01, and ***P < 0.001. Data are representative of at least three independent experiments.
Mice with lung epithelial deletion of the Miz1 POZ domain have lower radiographic pulmonary density and increased lung compliance
Emphysema is characterized in computed tomography (CT) scans by the presence of areas of abnormally low attenuation, which can be easily contrasted with surrounding normal lung parenchyma (22). In vivo imaging of the lungs in living animals with micro–computed x-ray tomography (micro-CT) revealed lower radiographic density (Fig. 2H) and lesions (Fig. 2I, indicated by pink areas) in the lungs of 1-year-old homozygous SPC-Cre+/Miz1(POZ)fl/fl littermate compared to the control Miz1(POZ)fl/fl mouse of similar age. We quantified lung density by measuring Hounsfield units (HU) on three-dimensional micro-CT images. The density of lung parenchyma was substantially reduced in 1-year-old homozygous SPC-Cre+/Miz1(POZ)fl/fl littermate than in the control Miz1(POZ)fl/fl mouse of similar age (Fig. 2J). Physiologically, emphysema results in a loss of lung elastic recoil that manifests as increased static lung compliance. Static lung compliance was increased in the homozygous SPC-Cre+/Miz1(POZ)fl/fl mice (≥1 year old) compared to the control Miz1(POZ)fl/fl mice of similar age (Fig. 2K).
Mice with lung epithelial cell–specific loss of function of Miz1 have inflammatory cell infiltrates and increased apoptosis in the lung
Our genetic strategy specifically deleted the Miz1 POZ domain from the lung epithelium; nevertheless, we consistently observed progressive inflammatory cell infiltration in the lungs of young homozygous SPC-Cre+/Miz1(POZ)fl/fl mice and aged (12 months) homozygous SPC-Cre+/Miz1(POZ)fl/fl and heterozygous SPC-Cre+/Miz1(POZ)wt/fl mice. We sought to better characterize the nature of these infiltrates. Immunohistochemistry (IHC) staining demonstrated the recruitment of large vacuolated AMs, neutrophils, and eosinophils within the airspaces of the ≤4-month-old and 12-month-old homozygous SPC-Cre+/Miz1(POZ)fl/fl and ≥1-year-old heterozygous SPC-Cre+/Miz1(POZ)wt/fl but not in the control SPC-Cre+/Miz1(POZ)wt/wt mice of similar age (fig. S3A). Many of these cells were apoptotic as assessed by terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) staining, suggesting a continuous process of inflammatory cell recruitment and death (fig. S3B). Increased TUNEL+ cells were also observed in the alveolus and airway wall (epithelium) of young and aged homozygous SPC-Cre+/Miz1(POZ)fl/fl and aged heterozygous SPC-Cre+/Miz1(POZ)wt/fl mice compared with controls (fig. S3B). Bronchoalveolar lavage fluid (BALF) from homozygous SPC-Cre+/Miz1(POZ)fl/fl and/or heterozygous SPC-Cre+/Miz1(POZ)wt/fl mice had greater leukocyte counts, including giant, multinucleated, and vacuolated macrophages, as well as smaller numbers of eosinophils and neutrophils (fig. S3C). Similar changes in AM numbers and morphology have been reported in patients with COPD and mice with emphysema (23, 24). Charcot-Leyden crystals, which are formed by fusion of eosinophil granules that contain large amounts of Charcot-Leyden protein or lysophospholipase, have been reported to be present within or associated with giant AMs of patients and mice with chronic inflammation (25–27). We observed similar structures in severely affected lung regions from the Miz1 knockout mice (fig. S3D). These data are reminiscent of histologic findings in the lungs of patient with COPD in which macrophage and neutrophil numbers are consistently increased and eosinophilic granules are associated with severe disease, and increased apoptosis of epithelial and inflammatory cells were also observed (28, 29).
Mice with lung epithelial cell–specific loss of function of Miz1 also exhibit features of chronic bronchitis
Hyperplasia of mucin-secreting goblet cells and associated mucus hypersecretion are common features of chronic bronchitis in COPD. We observed greater thickness of the airways, increased numbers of goblet cells, and increased expression of the mucin gene MUC5AC in the airways of young (≤4-month-old) homozygous SPC-Cre+/Miz1(POZ)fl/fl, ≥1-year-old heterozygous SPC-Cre+/Miz1(POZ)wt/fl, and homozygous SPC-Cre+/Miz1(POZ)fl/fl mice, as compared to the control Miz1(POZ)fl/fl or SPC-Cre+/Miz1(POZ)wt/wt mice of similar age (Fig. 3, A to D). In mice with complete epithelial deletion of the Miz1(POZ) [SPC-Cre+/Miz1(POZ)fl/fl], we observed increased numbers of eosinophils, macrophages, and/or neutrophils within the airways and TUNEL+ cells in the airways (Fig. 3, E to G).
Fig. 3. Mice with lung epithelial cell–specific loss of function of Miz1 also exhibit features of chronic bronchitis.
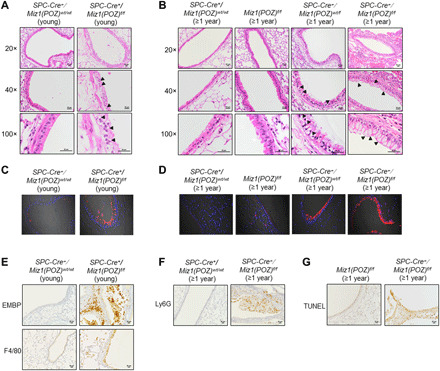
(A and B) Representative histological sections (20×, 40×, or 100× objective) of pulmonary airways from ≤4-month-old control SPC-Cre+/Miz1(POZ)wt/wt or homozygous SPC-Cre+/Miz1(POZ)fl/fl mice (A) or ≥ 1-year-old control SPC-Cre+/Miz1(POZ)wt/wt, Miz1(POZ)fl/fl, heterozygous SPC-Cre+/Miz1(POZ)wt/fl, or homozygous SPC-Cre+/Miz1(POZ)fl/fl mice (B). Arrowheads indicate goblet cells. (C and D) MUC5AC IF staining from mice in (A) and (B). (E to G) Representative IHC staining of EMBP (eosinophils) (20× objective), F4/80 (macrophages) (20× objective) of lungs from young control SPC-Cre+/Miz1(POZ)wt/wt or homozygous SPC-Cre+/Miz1(POZ)fl/fl mice (E), Ly6G (PMNs) (20× objective) (F), or TUNEL (20× objective) (G) staining of ≥1-year-old control Miz1(POZ)fl/fl, SPC-Cre+/Miz1(POZ)wt/wt, or homozygous SPC-Cre+/Miz1(POZ)fl/fl mice. n ≥ 5. Data are representative of at least three independent experiments.
AM- and dendritic cell–specific loss of function of Miz1 is not sufficient to cause a COPD-like phenotype in mice
Collectively, our data suggest that even partial loss of function of Miz1 in lung epithelial cells is sufficient to cause a spontaneous age-related COPD-like phenotype in mice. In these mice, we also observed increased numbers of AMs and other inflammatory cells. To determine whether loss of function of Miz1 in these cells also leads to COPD-like phenotypes we observed with epithelial cell–specific loss of Miz1, we crossed Miz1(POZ)fl/fl mice with transgenic CD11c-Cre mice. CD11c-Cre induces efficient Cre-mediated recombination in AMs and dendritic cells (20). Using qPCR of genomic DNA, we found deletion of the Miz1(POZ) in flow-sorted AMs and dendritic cells and partial deletion of the Miz1(POZ) in lung monocytes and neutrophils from the lungs of CD11c-Cre+/Miz1(POZ)fl/fl mice (fig. S4A). The Miz1(POZ) domain was intact in AT2 cells from CD11c-Cre+/Miz1(POZ)fl/fl mice (fig. S4A). While young (≤4-month-old) CD11c-Cre+/Miz1(POZ)fl/fl mice showed normal lung structure (Fig. 4A), they had increased numbers of macrophages and neutrophils in the BALF compared to the control mice (Fig. 4B). When they were over 1 year old, CD11c-Cre+/Miz1(POZ)fl/fl mice had marked spontaneous regional inflammation in the alveoli and airways (Fig. 4, C to E) and showed a progressive increase in the number of macrophages and neutrophils in the BALF (Fig. 4B). These data are consistent with our previous report that Miz1 in both lung epithelial cells and inflammatory cells is important to restrain pulmonary inflammation (13). However, we were unable to identify COPD-like changes in CD11c-Cre+/Miz1(POZ)fl/fl mice compared with controls, including alveolar enlargement (Fig. 4D) or goblet cell hyperplasia (Fig. 4E) or lower radiographic density (fig. S4B). These data indicate that loss of function of Miz1 in inflammatory cells, including AMs and dendritic cells, is not sufficient to cause spontaneous COPD-like phenotype in mice. Combined with our findings in SPC-Cre+/Miz1(POZ)fl/fl mice, these data strongly suggest that loss of function of Miz1 in epithelial cells but not inflammatory cells drives the age-related progressive emphysema-like phenotype in mice. These data coincide with the observations in human COPD lungs where Miz1 is predominantly down-regulated in lung epithelial but not inflammatory cells (Fig. 1A), implicating the central role of epithelial cell–specific Miz1 in regulation of the pathogenesis of COPD.
Fig. 4. AM- and dendritic cell–specific loss of function of Miz1 is not sufficient to cause a COPD-like phenotype in mice.
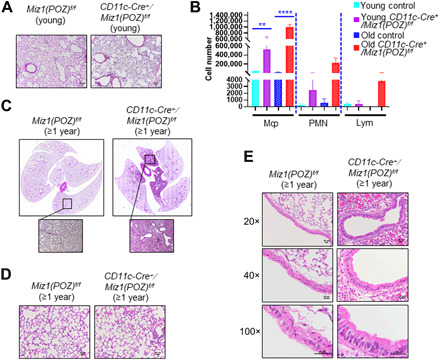
(A, C, and D) Representative lung histological sections (A and D) or whole lung histology (C) of ≤4-month-old (A) or ≥ 1-year-old control Miz1(POZ)fl/fl or CD11c-Cre+/Miz1(POZ)fl/fl mice (C and D). (B) BALF cell differentials from young or ≥1-year-old control Miz1(POZ)fl/fl or CD11c-Cre+/Miz1(POZ)fl/fl mice (n = 4 to 11). Mȹ, macrophages; PMN, neutrophils; Lym, lymphocytes. **P < 0.01, ***P < 0.0001. (E) Representative histological sections (20×, 40×, or 100× objective) of pulmonary airways from ≥1-year-old control Miz1(POZ)fl/fl or CD11c-Cre+/Miz1(POZ)fl/fl mice. Data are representative of at least three independent experiments.
Mice with heterozygous epithelial cell–specific loss of function of Miz1 have increased expression of proinflammatory genes in the lung epithelium throughout the life span
To explore the mechanisms by which lung epithelial cell–specific deletion of Miz1(POZ) drives the development of COPD-associated changes in the aging lung, we flow-sorted AT2 cells from homozygous SPC-Cre+/Miz1(POZ)fl/fl or heterozygous SPC-Cre+/Miz1(POZ)wt/fl mice at different ages (2, 6, or 12 months) and performed RNA sequencing (RNA-seq) and/or quantitative shotgun proteomics. RNA-seq identified 1721 genes up-regulated and 1646 genes down-regulated (P < 0.05) in AT2 cells isolated from 6-month-old homozygous SPC-Cre+/Miz1(POZ)fl/fl mice as compared to the control Miz1(POZ)fl/fl mice of similar age (Fig. 5A). These genes included several known Miz1 targets (30), including Cebpd (encoding C/EBPδ) (fig. S5A), which we previously identified as a Miz1 direct target gene through repressing which Miz1 suppresses NF-κB–dependent inflammation (13). The expression of Ace2, the receptor for SARS-CoV-2, was induced in AT2 cells from SPC-Cre+/Miz1(POZ)fl/fl mice as compared to the control Miz1(POZ)fl/fl mice of similar age (Fig. 5B). In addition, there was a trend of increased expression of Tmprss2, the serine protease for S protein priming essential for SARS-CoV-2 entry (31), in AT2 cells from SPC-Cre+/Miz1(POZ)fl/fl mice as compared to the control Miz1(POZ)fl/fl mice of similar age, although it did not reach statistical significance (P = 0.0769) probably due to limited sample size (Fig. 5B). Gene Ontology (GO) analysis of up-regulated genes identified “innate immune system” as the top significantly enriched function for Miz1 (Fig. 5A), and Kyoto Encyclopedia of Genes and Genomes (KEGG) PATHWAY mapping revealed up-regulated genes involved in NF-κB or TNF or Toll-like receptor (TLR) signaling, including NF-κB/RelA, Jun, IL-6, and IL-1β (Fig. 5C). These data are consistent with our previous report that Miz1 suppresses TNF- or TLR4-induced NF-κB–dependent inflammatory response (13). Similarly, the top three GO processes from up-regulated genes in AT2 cells isolated from 6-month-old heterozygous SPC-Cre+/Miz1(POZ)wt/fl mice included “innate immune system,” “innate immune response,” and “inflammatory response” (Fig. 5D). Similar pathways and genes were identified when we compared 12-month-old heterozygous SPC-Cre+/Miz1(POZ)wt/fl or homozygous SPC-Cre+/Miz1(POZ)fl/fl mice to Miz1(POZ)fl/fl controls (fig. S5, B and C). We confirmed the RNA-seq results by measuring the levels of mRNA for certain NF-κB–dependent inflammatory genes using quantitative reverse transcription PCR (qRT-PCR) (fig. S5D). Note that heterozygous SPC-Cre+/Miz1(POZ)wt/fl mice had increased expression of proinflammatory genes starting at younger age (2 to 6 months), which preceded the COPD-like pathological changes in middle to late life (≥1 year old) and remained throughout the life span (≥1 year old). To determine whether the changes we observed in gene expression resulted in changes in inflammatory proteins, we performed shotgun proteomics. Using a 1.5-fold change (P < 0.05) as a cutoff for significance, we identified 374 Miz1-regulated proteins that were differentially expressed between AT2 cells from homozygous SPC-Cre+/Miz1(POZ)fl/fl mice and Miz1(POZ)fl/fl controls. These included known Miz1 targets including integrin β1 (ITGB1), ankyrin 3 (ANK3), programmed cell death 5 (PDCD5), small nuclear ribonucleoprotein polypeptide A (SNRPA1), and TAO kinase 3 (TAOK3) (fig. S5E) (32), thus validating the system. GO analysis of these differentially expressed proteins revealed “regulation of mRNA stability” and “innate immunity” as the two most significant GO terms (fig. S5F). Collectively, these data suggest that Miz1 functions primarily to suppress innate immunity in lung epithelial cells.
Fig. 5. Loss of the Miz1 POZ domain results in deregulated expression profiles enriched in innate immunity/inflammation.
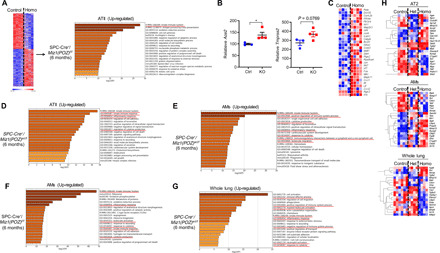
(A and D to G) GO analysis of differentially expressed genes (analyzed by RNA-seq) in AT2 (A and D), AMs (E and F), or whole lungs (G) from 6-month-old control Miz1(POZ)fl/fl, heterozygous SPC-Cre+/Miz1(POZ)wt/fl, or homozygous SPC-Cre+/Miz1(POZ)fl/fl mice. (B) Ace2 and Tmprss2 mRNA levels analyzed by RNA-seq from control Miz1(POZ)fl/fl or homozygous SPC-Cre+/Miz1(POZ)fl/fl mice of similar age [control, n = 3; knockout (KO), n = 4]. (C and H) Heatmaps of differentially expressed genes involved in NF-κB, TNF, or TLR signaling by KEGG PATHWAY mapping from (A) (C) or differentially expressed genes of those identified as COPD susceptibility genes by GWAS from (A) to (G) (H), respectively. n ≥ 4 for (A) to (H). Red underlines indicate pathways involved in innate immunity. Control, Miz1(POZ)fl/fl mice; Het, heterozygous SPC-Cre+/Miz1(POZ)wt/fl mice; Homo, homozygous SPC-Cre+/Miz1(POZ)fl/fl mice.
Loss of lung epithelial function of Miz1 results in augmented inflammatory response in AMs
Increased numbers and abnormal morphology of AMs were observed in homozygous SPC-Cre+/Miz1(POZ)fl/fl mice at 4 and 12 months of age and heterozygous SPC-Cre+/Miz1(POZ)wt/fl mice after 12 months of age (fig. S3, A and C). As AMs have been reported to be important in the pathogenesis of COPD (33), we sought to determine whether there is a possible link between epithelial inflammation induced by the loss of Miz1 and activation of AMs within the lung. We compared gene expression using RNA-seq in flow-sorted AMs and whole lung tissue from mice with homozygous or heterozygous epithelial cell–specific deletion of Miz1(POZ) and control animals. GO analysis of up-regulated genes in AMs or whole lung tissue from 6-month-old heterozygous SPC-Cre+/Miz1(POZ)wt/fl or homozygous SPC-Cre+/Miz1(POZ)fl/fl mice implicated processes involved in inflammatory response and innate immunity (Fig. 5, E to G). In AMs, up-regulated genes include various matrix metalloproteinases (MMPs) (fig. S5, G and H), which have been reported to be central to COPD pathogenesis (34, 35).
Ninety-seven genetic loci that have met stringent standards for statistical significance in large-scale genome-wide association study (GWAS) have been associated with COPD susceptibility, some of which have been replicated in independent cohorts (36–41). Among these genes, 42 were differentially expressed in AT2, AMs, or whole lung tissue from heterozygous SPC-Cre+/Miz1(POZ)wt/fl or homozygous SPC-Cre+/Miz1(POZ)fl/fl mice compared with control Miz1(POZ)fl/fl mice, including Tns1, Ager, Thsd4, Gstcd, Hhip, Mmp15, Tgfb2, and Itga1 (Fig. 5H).
Activation of NF-κB is necessary for the COPD-like phenotypes induced by lung epithelial cell–specific loss of function of Miz1
As augmented NF-κB–dependent inflammation proceeded the COPD-like pathological changes in heterozygous SPC-Cre+/Miz1(POZ)wt/fl mice, we sought to determine whether the enhanced NF-κB–dependent inflammatory response is causally linked to the COPD-like phenotype observed in the Miz1 mutant mice. We crossed SPC-Cre+/Miz1(POZ)fl/fl mice with RelAfl/fl mice, in which the exon 1 of the gene encoding the major NF-κB transactivation subunit, RelA, also named p65, is flanked by loxP sites. Analysis of genomic DNA using qPCR revealed partial deletion of Miz1(POZ) in flow-sorted ATII cells of SPC-Cre+/Miz1(POZ)wt/fl and SPC-Cre+/Miz1(POZ)wt/fl/RelAwt/fl (fig. S6A, left). qRT-PCR showed partial deletion of RelA in ATII cells from SPC-Cre+/Miz1(POZ)wt/fl/RelAwt/fl mice (fig. S6, right). Consistent with our phenotypic, transcriptomic, and proteomic data, concomitant RelA haploinsufficiency almost completely prevented the development of emphysema and bronchitis in ~12-month-old mice with heterozygous epithelial deletion of Miz1(POZ) (comparison of SPC-Cre+/Miz1(POZ)wt/fl and SPC-Cre+/Miz1(POZ)wt/fl/RelAwt/fl) (Fig. 6, A to C).
Fig. 6. RelA haploinsufficiency rescues the phenotype of Miz1-deficient mice, and lung epithelial cell–specific loss of function of Miz1 in the adulthood is also sufficient to induce emphysema.
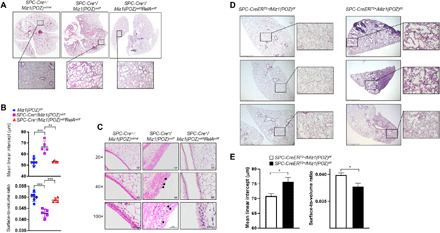
(A and C) Representative whole lung histology (A) or histological sections of pulmonary airways (C) in the lungs of ~1-year-old control SPC-Cre+/Miz1(POZ)wt/wt, heterozygous SPC-Cre+/Miz1(POZ)wt/fl, or SPC-Cre+/Miz1(POZ)wt/flRelAwt/fl mice. (B) MLI (micrometers ± SEM) or surface-to-volume ratio (means ± SEM) of alveolar septae measured in the lungs of ~1-year-old control Miz1(POZ)fl/fl (n = 6), heterozygous SPC-Cre+/Miz1(POZ)wt/fl (n = 5), or SPC-Cre+/Miz1(POZ)wt/flRelAwt/fl mice (n = 4). **P < 0.01 and ***P < 0.001. (D) Representative lung histological sections of control SPC-CreERT2−/Miz1(POZ)f/f or SPC-CreERT2+/Miz1(POZ)f/f mice after treatment with tamoxifen for 14 months. Three subjects from each group were shown, which were the representative of five to eight subjects from each group. Scale bars, 500 μm. (E) MLI (micrometers ± SEM) or the ratio of surface area to alveolar air space volume (surface-to-volume ratio; means ± SEM) of alveolar septae measured in the lungs of control SPC-CreERT2−/Miz1(POZ)f/f or SPC-CreERT2+/Miz1(POZ)f/f mice after treatment with tamoxifen for 14 months. n = 5 to 8. Data are representative of at least three independent experiments.
Lung epithelial cell–specific loss of function of Miz1 in the adulthood is also sufficient to induce emphysema
Our observation that heterozygous SPC-Cre+/Miz1(POZ)wt/fl mice have completely normal lung histology at a young age but spontaneously develop a COPD-like phenotype by 1 year of age argues strongly against a developmental contribution. To confirm this, we crossed Miz1(POZ)fl/fl mice with SPC-CreERT2 mice [available at JAX (Jackson Laboratory)—stock #028054 through B. Hogan, Duke University] (42), in which a tamoxifen-inducible Cre recombinase (CreERT2) is under the control of the human SPC promoter. Adult SPC-CreERT2+/Miz1(POZ)f/f and the control SPC-CreERT2−/Miz1(POZ)f/f mice were administered with tamoxifen in chow (Envigo) for 14 months. qPCR using genomic DNA revealed deletion of Miz1(POZ) in ATII cells but not endothelial cells or AMs of tamoxifen-treated SPC-CreERT2+/Miz1(POZ)f/f mice (fig. S6B). Enlarged alveolar airspace and inflammatory infiltrates were observed in tamoxifen-treated SPC-CreERT2+/Miz1(POZ)f/f but not the control SPC-CreERT2−/Miz1(POZ)f/f mice (Fig. 6, D and E).
DISCUSSION
Our findings suggest that lung epithelial cell–specific loss function of Miz1, a transcriptional repressor of NF-κB signaling, contributes to the pathogenesis of COPD. We show that Miz1 is down-regulated in the alveolar and airway epithelium of some patients with COPD and also in the lungs of mice chronically exposed to CS. Lung epithelial cell–specific loss of function of Miz1 in mice results in age-related, progressive alveolar destruction and airway remodeling accompanied by an increased number of inflammatory cells in the airway and alveolar airspace reminiscent of the pathologic changes in patients with COPD. Using unbiased transcriptomic analysis of Miz1-deficient mice compared with wild-type mice, we found the loss of Miz1 up-regulated expression of Ace2, the receptor for the novel SARS-CoV-2 coronavirus that is the cause of the ongoing COVID-19 pandemic. Loss of Miz1 also altered the expression of 42 of 97 validated genes that have been genetically associated with COPD in GWAS. The COPD-like phenotype in the Miz1 mutant mice is rescued by NF-κB/RelA haploinsufficiency, genetically confirming loss of Miz1-induced NF-κB–dependent inflammation as the central driver of the COPD phenotypes we observed in Miz1 mutant mice. Thus, our data support the notion that reduced Miz1 expression contributes to chronic NF-κB–dependent inflammation and disease progression in COPD. Our findings further suggest that chronic lung epithelial inflammation induced by the loss of Miz1 might contribute to the observed clinical association between cigarette smoke exposure and disease severity after SARS-CoV-2 infection.
Previous work has demonstrated that inducible hyperactivation of NF-κB by overexpression of the constitutively active inhibitor of nuclear factor κB kinase β in Scgb1-expressing airway epithelial cells induces acute lung injury with neutrophil infiltration, exudation of protein-rich fluid into the airspaces, and hypoxemia (43). Chronic activation of NF-κB in this model results in the development of severe airway remodeling and airspace enlargement similar to emphysema and the spontaneous development of bronchial adenomas (44). The overexpression of TNF driven by the SPC promoter in the lung epithelium resulted in the development of large lymphocytic aggregates with associated evidence of emphysema (45). In contrast, Miz1 down-regulation does not directly activate NF-κB but instead results in prolonged signaling in response to physiologically endogenous activation. This more physiologic enhancement of NF-κB signaling was sufficient to induce typical airway epithelial changes of chronic bronchitis (mucin hypersecretion and goblet cell hyperplasia) and emphysema (airspace enlargement) in the absence of an inciting stimulus to activate NF-κB and without evidence of the acute lung injury induced by NF-κB hyperactivation. Furthermore, there appeared to be a strong gene dose effect associated with the loss of Miz1—complete epithelial cell–specific loss of Miz1 resulted in severe premature emphysema, whereas heterozygous loss was associated with no detectable phenotype in early life, instead developing progressive age-related changes of emphysema and chronic bronchitis. Even when the loss of Miz1 was limited to alveolar type II cells in adult mice, we observed changes consistent with emphysema.
The loss of Miz1 resulted in altered abundance of 42 of 97 genes rigorously associated with COPD risk in GWASs. In addition, several groups reported that a single nucleotide polymorphism in MIZ1 is associated with idiopathic dilated cardiomyopathy (46–49) and osteoporosis (50–52), both of which are often related to cigarette smoke and associated with the development of COPD. Furthermore, we found that Miz1 is reduced in the airway and alveolar epithelium in lung sections from some patients with COPD, in the lungs of mice chronically exposed to cigarette smoke and in lung epithelial cells exposed to cigarette smoke extracts, directly implicating a loss of MIZ1 in the lung epithelium in the pathogenesis of COPD. We have previously shown that MIZ1 suppresses inflammation through repressing the NF-κB–C/EBPδ signaling axis and is necessary to terminate TNF- and LPS-induced inflammation (13). Unbiased transcriptomic and proteomic analyses of epithelial cell–specific Miz1-deficient mice in the current study were entirely consistent with this mechanism. We observed increased apoptosis in the lung epithelial and inflammatory cells from the Miz1 knockout mice, which is likely due to the inflammatory damage induced by Miz1 loss, instead of a direct effect from Miz1 loss. The reason is that we did not observe obvious evidence of apoptosis in heterozygous SPC-Cre+/Miz1(POZ)wt/fl mice in early life when inflammation is already evident. That is to say, inflammation precedes and most likely contributes to apoptosis similar to the other manifestations of COPD in our model. More studies are needed, however, to determine how environmental exposures, particularly cigarette smoke, modulate Miz1 levels, how frequently Miz1 is down-regulated in patients with COPD, and whether other components of the transcriptional complex involved in Miz1-mediated repression of NF-κB signaling (e.g., C/EPBδ) are implicated in COPD pathogenesis. A loss of MIZ1-mediated repression of endogenous NF-κB signaling might explain why inflammation persists after the cessation of cigarette smoking in patients susceptible to COPD.
Airway and, to a lesser extent, alveolar inflammation are characteristic features of COPD and have been suggested to play a role in disease pathogenesis (53). We found that the loss of Miz1 in the lung epithelium was sufficient to increase the number of inflammatory cells, particularly AMs in the airspaces. In contrast, deletion of Miz1 from AMs and dendritic cells markedly increased the number and size of AMs but did not induce changes of airspace enlargement, goblet cell hyperplasia, or mucin hypersecretion characteristic of COPD. These results seem to localize signals initiating emphysema to the lung epithelium but do not exclude a role for macrophages, neutrophils, or other inflammatory cells recruited in response to epithelial activation of NF-κB in the development of COPD phenotypes. For example, our RNA-seq analysis of flow-sorted AMs from epithelial Miz1-deficient mice showed up-regulation of inflammatory and matrix remodeling genes including MMPs implicated in emphysema pathogenesis. In a murine model of immunoglobulin A (IgA) deficiency, neutrophils recruited in response to bacterial commensals are necessary for the development of emphysema (54, 55).
In summary, our studies demonstrate that chronic inflammation in the lung epithelium induced by down-regulation of Miz1 is sufficient to drive age-related progressive changes that recapitulate many features of human COPD. Our studies and others in human lungs and GWASs in patients with COPD-associated comorbidities directly implicate MIZ1 in COPD pathogenesis. We show a causal link between chronic subacute activation of NF-κB in the lung epithelium and the tissue destruction and physiologic dysfunction characteristic of COPD via a mechanism that explains the persistence of inflammation in patients with COPD even after smoking cessation.
METHODS
Mice
Miz1(POZ)fl/fl mice on the C57BL/6 background have been described previously (13). SPC-Cre and SPC-CreERT2 mice were provided by B. Hogan (SPC-Cre+ERT2 mice are available at JAX—stock #028054 through B. Hogan, Duke University) (42). CD11c-Cre and RelAfl/fl mice were from the Jackson laboratories. The animal care and experiments were performed in compliance with the institutional and U.S. National Institutes of Health (NIH) guidelines and were approved by the Northwestern University Animal Care and Use Committee.
Antibodies for Western blot
Miz-1 antibody (H-190; 1:500): sc-22837 was used until discontinued by the vendor, and Miz-1 antibody (B-10; 1:250): sc-136985 was then used in replacement of Miz-1 antibody (H-190): sc-22837. β-Actin antibody (A5441, Sigma-Aldrich; 1:20,000) and Mule antibody (ab70161, Abcam) were also used.
IHC and IF staining
Human normal and COPD lung tissues, which were obtained from lung transplant or lung resection donors (who gave informed consent), were fixed in 4% paraformaldehyde (24 hours), processed, and embedded in paraffin. The experimental protocol was approved by the Institutional Review Board at Northwestern University (IRB no. STU00056197-MOD0003 General Biorepository Development for Solid Organ and Stem Cell Transplant Specimens). Miz1 and Mule IHC of human lungs was performed by the Northwestern University Mouse Histology and Phenotyping Laboratory. Briefly, deparaffinized slides were incubated with Miz1 antibody (ab121232, Abcam; 1:500) and Mule antibody (ab70161, Abcam) overnight at 4°. Detection was performed with the biotin-streptavidin peroxidase system (Vector Labs). Nuclear profiles were outlined concomitantly with 4′,6-diamidine-2-phenylindole (DAPI) using Aqua Mount. IHC staining of macrophages, eosinophils, and neutrophils from mouse lungs was performed in deparaffinized slides with F4/80 antibody (14-4801, eBioscience; 1:500), eosinophil major basic protein (EMBP) antibody (sc-33938, Santa Cruz Biotechnology; 1:1500), or Ly6G antibody (#551459, BD Biosciences; 1:500), respectively. TUNEL staining was performed in deparaffinized slides using the ApopTag Peroxidase In Situ Apoptosis Detection Kit (S71000, EMD Millipore). For IF staining, human and mouse lung sections were deparaffinized with xylene, rehydrated with graded ethanol, and subjected to heat-induced antigen retrieval in sodium citrate buffer (10 mM) (pH 6.0). After blocking in 1% bovine serum albumin (w/v) in phosphate-buffered saline (PBS) for 1 hour, lung sections were incubated overnight with rabbit polyclonal Miz1 antibodies [sc-22837, Santa Cruz Biotechnology for mouse lungs (1:200) and ab121232, Abcam for human lungs (1:200)] or MUC5AC antibody (sc-20118, Santa Cruz Biotechnology). Colabeling with SPC (AT2 cell marker) in mouse lungs was achieved by the addition of goat polyclonal SPC antibody (sc-7706, Santa Cruz Biotechnology). Lung sections were then washed with PBS and incubated with Alexa Fluor 555–labeled anti-rabbit IgG and Alexa Fluor 488–labeled anti-goat IgG (1 μg/ml; Thermo Fisher Scientific). Nonimmune rabbit or goat IgGs (Santa Cruz Biotechnology) were used as negative controls. Nuclei were visualized with DAPI. Slides were mounted with Gel Mount (Biomeda, Foster City, CA). Fluorescent images were obtained using a fluorescence microscope Axioplan 2 (Carl Zeiss Meditec Inc.).
RNA sequencing
RNA-seq of primary AT2, AMs, and whole lung tissues was performed by the next-generation sequencing (48) core in the Division of Pulmonary at Northwestern University or the Laboratory of Xiangdong Fu at University of California, San Diego, as previously described (56, 57). Briefly, total RNA was extracted and purified using NucleoSpin RNA kit. mRNA was then enriched from total RNA (50 ng) using NEBNext Poly(A) mRNA Magnetic Isolation Module, and complementary DNA (cDNA) libraries were generated using NEBNext Ultra RNA Library Prep Kit for Illumina. The quantity and quality of the cDNA libraries were assessed using an Agilent 4200 Tapestation (Agilent Technologies). cDNA (∼1 ng) was fragmented using high temperature (94° for 15 min) and amplified with a dual-index (i7 and i5; Illumina; 10 cycles), and individual libraries were purified with magnetic beads. Indexed sequence libraries were pooled for multiplexing (∼40 samples per lane), and paired-end sequencing (75 base pairs) were performed on NextSeq 500 using dual-index sequencing primers (Illumina). Reads were aligned using TopHat2 to the mm10 reference genome, differential expression was assessed using edgeR, and enrichment analysis was performed using curated databases including GO or KEGG PATHWAY mapping.
Quantitative polymerase chain reaction
qPCR was performed using iQ SYBR Green Supermix (Bio-Rad) on a CFX Connect Real-Time PCR Detection System (Bio-Rad). mRNA expression of a particular gene was normalized to hypoxanthine-guanine phosphoribosyltransferse. Primer sequences were listed in table S1. For qRT-PCR, total RNA was extracted from lung tissues or isolated cells by NucleoSpin RNA kit (MACHEREY-NAGEL), followed by cDNA synthesis using M-MuLV Reverse Transcriptase (New England Biolabs) according to the manufacturer’s instructions.
Histological analysis
Tissue samples were fixed in 4% paraformaldehyde (24 hours), processed, and embedded in paraffin. Sections (5 μm) were prepared and mounted on coverslips for staining with hematoxylin and eosin, and tissue images were captured using Nikon Eclipse E800 or using the TissueGnostics Tissue/Cell High Throughput Imaging Analysis System (Vienna, Austria) and TissueFAXS software (TissueGnostics, Los Angeles, CA) at the Northwestern University Cell Imaging Facility.
Micro-CT scan
In vivo mouse lung micro-CT scans were conducted with the individuals anesthetized with 1.5 to 2% isoflurane in 100% oxygen. Micro-CT datasets were acquired on a dedicated small animal micro-positron emission tomography (PET)/CT scanner (NanoScan8, Mediso, Budapest Hungary). Scan parameters were as follows: 50–kilovolt peak x-ray source voltage, 320-ms exposure time/projection, 720 projections with a field of view of ~1 to 2 cm covering the lung. A total acquisition time of ~4 min, yielding three-dimensional datasets with isotropic voxel size of 250 μm, corresponding to four different phases of the breathing cycle.
Static lung compliance analysis
Mice were anesthetized and tracheotomized, followed by mechanically ventilated on a computer-controlled piston ventilator (flexiVent, SCIREQ, Montre’al, Que’bec, Canada) with a tidal volume of 10 ml/kg at a frequency of 150 breaths/min against 2- to 3-cm H2O-positive end-expiratory pressure. A standard volume history was obtained with two total lung capacity maneuvers. A perturbation maneuver was imposed to collect pressure and flow data (reflective of airway and tissue dynamics). Static compliance was calculated as the change in volume divided by the change in pressure.
Alveolar size measurement
The lungs were inflated with 0.5% agarose under 25-cm water pressure, fixed in buffered 10% formalin for 24 hours and embedded in paraffin. Sections (5 μm) were stained with hematoxylin and eosin. Images (35 per lung section) from whole lungs were acquired with a ×20 lens. MLI and alveolar surface-to-volume ratio were determined by computer-assisted morphometry with the MetaMorph Microscopy Automation & Image Analysis software (Molecular Devices).
Proteomics
Protein lysates were precipitated by trichloroacetic acid and then tryptically digested following the procedure described previously (58). Briefly, protein precipitates were resolved by 8 M urea, followed by treatment with 5 mM tris(2-carboxyethyl)phosphine (TCEP) and 10 mM indole-3-acetic acid. The mixture was digested for 16 hours at 37°C by trypsin at an enzyme-to-substrate ratio of 1:50 (w/w). The tryptic digested peptides were desalted and loaded on an in-house packed capillary reverse-phase C18 column (15-cm length, 100-μm inside diameter × 360-μm outer diameter, 3-μm particle size, and 100-Å pore diameter) connected to an Easy LC 1000 system. The samples were analyzed with a 180-min high-performance liquid chromatography gradient from 0 to 100% of buffer B (buffer A, 0.1% formic acid in water; buffer B, 0.1% formic acid in acetonitrile) at 300 nl/min. The eluted peptides were ionized and directly introduced into a Q Exactive or Fusion mass spectrometer using a nanospray source. Survey full-scan mass spectrometry spectra [from a mass/charge ratio (m/z) of 300 to 1800] were acquired in the Orbitrap analyzer with a resolution r = 70,000 at m/z 400. Protein identification was performed with MaxQuant (59). The tandem mass spectra were searched against the UniProt mouse protein database, and sequences of known contaminants such as keratin and porcine trypsin concatenated to a decoy database in which the sequence for each entry in the original database was reversed. Search space included all fully and half-tryptic peptide candidates with missed cleavage restrictions. Under the filtering conditions, the estimated false discovery rate was below ~1% at the protein level in all analysis.
Fluorescence-activated cell sorting
The lungs were digested with dispase (Corning) and deoxyribonuclease I (0.1 mg/ml). Red blood cells were removed using 1× BD Pharm Lyse solution (BD Biosciences). Cells were washed in MACS buffer (Miltenyi Biotech) and counted using a Countess automated cell counter (Invitrogen). For isolation of AT2 cells and AMs, single-cell suspension was incubated in 0.5 μg of Fc block (BD Biosciences) for 10 min at 4°C, followed by staining with fluorescein isothiocyanate (FITC)–conjugated anti-mouse CD45 (eBioscience), allophycocyanin (APC)–conjugated anti-mouse epithelial cell adhesion molecule (EpCAM) (eBioscience), phycoerythrin (PE)–conjugated anti-mouse CD31 (eBioscience), PE-CF594–conjugated anti-mouse SiglecF (BD Biosciences), and eFluor450-conjugated anti-mouse CD11b (eBioscience). The fluorescence-activated cell sorting (FACS) experiments were performed using a BD FACSAria SORP 5-laser instrument (BD Immunocytometry Systems) equipped with 355-, 405-, 488-, 561-, and 640-nm excitation lasers located at the Northwestern University Flow Cytometry Core Facility. All data collection and sorting were performed using BD FACSDiva software (BD Biosciences), and data analyses were performed using FlowJo software (Tree Star, Ashland, OR). Fluorescence minus one controls were used for gating analyses to distinguish positively from negatively staining cell populations. Compensation was performed using single-color controls prepared from BD CompBeads (BD Biosciences). Compensation matrices were calculated and applied using FlowJo software (Tree Star). Biexponential transformation was adjusted manually when necessary. For simultaneous isolation of AT2 and other myeloid cells, after blocking, cells were incubated with biotin-conjugated anti-mouse CD45 antibody for 10 min at room temperature. Cells then were separated using MagniSort Streptavidin Positive selection beads (eBioscience) according to the manufacturer’s instructions. AT2 cells will be isolated from the CD45− fraction by flow sorting using FITC-conjugated anti-mouse CD45 (eBioscience), APC-conjugated anti-mouse EpCAM (eBioscience), and PE-conjugated anti-mouse CD31 (eBioscience). The myeloid cell populations will be isolated from the CD45+ fraction by flow sorting using FITC-conjugated anti-mouse CD45 (eBioscience), PerCP-Cy5.5–conjugated anti-mouse major histocompatibility complex II (BioLegend), eFluor450-conjugated anti-mouse Ly6C (eBioscience), APC-conjugated anti-mouse CD24 (eBioscience), Alexa Fluor 700–conjugated anti-mouse Ly6G (BD Biosciences), APC-Cy7–conjugated anti-mouse CD11b (BioLegend), PE-conjugated anti-mouse CD64 (BioLegend), PE-CF594–conjugated anti-mouse SiglecF (BD Biosciences), and PE-Cy7–conjugated anti-mouse CD11c (eBioscience) (60).
Tamoxifen treatment of mice
Mice were fed with tamoxifen chow (Envigo; ~40 to 80 mg/kg of body weight per day).
Statistical analysis
Data were analyzed by an unpaired Student’s t test, with the assumption of normal distribution of data and equal sample variance. Sample sizes were selected on the basis of preliminary results to ensure an adequate power.
Supplementary Material
Acknowledgments
We thank L, Li and the Northwestern University Mouse Histology and Phenotyping Laboratory for the assistance with IHC and histopathology. Funding: Imaging was performed at the Northwestern University Cell Imaging Facility, supported by NCI CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center. J.L. is supported by the U.S. NIH (HL114763 and HL141459 to J.L.). G.R.S.B. is supported by NIH grants ES013995, HL071643, and AG049665, Veterans Administration grant BX000201, Department of Defense grant PR141319, and NIH/NIAID grant AI135964. P.H.S.S. is supported by NIH grant R01HL131745. K.R.A. is supported by David W. Cugell and Christina Enroth-Cugell Fellowship. A.B. is supported by NIH grants HL145478, HL147290, and HL147575. P.A.R. is supported by NIH K08HL146943, Parker B. Francis Fellowship, and ATS Foundation/Boehringer Ingelheim Pharmaceuticals Inc. Research Fellowship in IPF. Author contributions: H.C.D.-U., C.C., Q.Z., M.A.S., and S.M.C.-M. performed the experiments. A.V.M. helped with flow cytometry and cell sorting. H.A.-V., P.A.R., K.R.A., and X.F. helped with RNA-seq. J.Y. and X.F. helped with RNA-seq analysis. F.J.G.-G. assessed the lung compliance. C.P., P.W., and C.C.L.W. performed the proteomics. A.B. provided the reagents. R.K., H.P., P.H.S.S., K.M.R., N.S.C., I.P., R.T., and J.I.S. provided reagents and suggestions. G.R.S.B. and J.L. contributed to manuscript preparation, hypothesis generation, and experimental design. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/33/eabb7238/DC1
REFERENCES AND NOTES
- 1.Tuder R. M., Petrache I., Pathogenesis of chronic obstructive pulmonary disease. J. Clin. Invest. 122, 2749–2755 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnes P. J., New anti-inflammatory targets for chronic obstructive pulmonary disease. Nat. Rev. Drug Discov. 12, 543–559 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Brusselle G. G., Joos G. F., Bracke K. R., New insights into the immunology of chronic obstructive pulmonary disease. Lancet 378, 1015–1026 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Zhao Q., Meng M., Kumar R., Wu Y., Huang J., Lian N., Deng Y., Lin S., The impact of COPD and smoking history on the severity of Covid-19: A systemic review and meta-analysis. J. Med. Virol. 10.1002/jmv.25889 , (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Decramer M., Janssens W., Miravitlles M., Chronic obstructive pulmonary disease. Lancet 379, 1341–1351 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnes P. J., Senescence in COPD and its comorbidities. Annu. Rev. Physiol. 79, 517–539 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Mercado N., Ito K., Barnes P. J., Accelerated ageing of the lung in COPD: New concepts. Thorax 70, 482–489 (2015). [DOI] [PubMed] [Google Scholar]
- 8.López-Otín C., Blasco M. A., Partridge L., Serrano M., Kroemer G., The hallmarks of aging. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaw A. C., Goldstein D. R., Montgomery R. R., Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 13, 875–887 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morrisette-Thomas V., Cohen A. A., Fülöp T., Riesco É., Legault V., Li Q., Milot E., Dusseault-Bélanger F., Ferrucci L., Inflamm-aging does not simply reflect increases in pro-inflammatory markers. Mech. Ageing Dev. 139, 49–57 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ershler W. B., Interleukin-6: A cytokine for gerontologists. J. Am. Geriatr. Soc. 41, 176–181 (1993). [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg S. R., Kalhan R., Biomarkers in chronic obstructive pulmonary disease. Transl. Res. 159, 228–237 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Do-Umehara H. C., Chen C., Urich D., Zhou L., Qiu J., Jang S., Zander A., Baker M. A., Eilers M., Sporn P. H. S., Ridge K. M., Sznajder J. I., Budinger G. R. S., Mutlu G. M., Lin A., Liu J., Suppression of inflammation and acute lung injury by Miz1 via repression of C/EBP-δ. Nat. Immunol. 14, 461–469 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Wan Y., Shang J., Graham R., Baric R. S., Li F., Receptor recognition by the novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 94, e00127–e00120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y., Do H. C., Tian X., Zhang C., Liu X., Dada L. A., Sznajder J. I., Liu J., E3 ubiquitin ligase Mule ubiquitinates Miz1 and is required for TNFα-induced JNK activation. Proc. Natl. Acad. Sci. U.S.A. 107, 13444–13449 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshida T., Mett I., Bhunia A. K., Bowman J., Perez M., Zhang L., Gandjeva A., Zhen L., Chukwueke U., Mao T., Richter A., Brown E., Ashush H., Notkin N., Gelfand A., Thimmulappa R. K., Rangasamy T., Sussan T., Cosgrove G., Mouded M., Shapiro S. D., Petrache I., Biswal S., Feinstein E., Tuder R. M., Rtp801, a suppressor of mTOR signaling, is an essential mediator of cigarette smoke-induced pulmonary injury and emphysema. Nat. Med. 16, 767–773 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clauss M., Voswinckel R., Rajashekhar G., Sigua N. L., Fehrenbach H., Rush N. I., Schweitzer K. S., Yildirim A. Ö., Kamocki K., Fisher A. J., Gu Y., Safadi B., Nikam S., Hubbard W. C., Tuder R. M., Twigg H. L. III, Presson R. G., Sethi S., Petrache I., Lung endothelial monocyte-activating protein 2 is a mediator of cigarette smoke-induced emphysema in mice. J. Clin. Invest. 121, 2470–2479 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okubo T., Knoepfler P. S., Eisenman R. N., Hogan B. L. M., Nmyc plays an essential role during lung development as a dosage-sensitive regulator of progenitor cell proliferation and differentiation. Development 132, 1363–1374 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Heng T. S., Painter M. W.; Immunological Genome Project Consortium , The Immunological Genome Project: Networks of gene expression in immune cells. Nat. Immunol. 9, 1091–1094 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Misharin A. V., Morales-Nebreda L., Reyfman P. A., Cuda C. M., Walter J. M., McQuattie-Pimentel A. C., Chen C.-I., Anekalla K. R., Joshi N., Williams K. J. N., Abdala-Valencia H., Yacoub T. J., Chi M., Chiu S., Gonzalez-Gonzalez F. J., Gates K., Lam A. P., Nicholson T. T., Homan P. J., Soberanes S., Dominguez S., Morgan V. K., Saber R., Shaffer A., Hinchcliff M., Marshall S. A., Bharat A., Berdnikovs S., Bhorade S. M., Bartom E. T., Morimoto R. I., Balch W. E., Sznajder J. I., Chandel N. S., Mutlu G. M., Jain M., Gottardi C. J., Singer B. D., Ridge K. M., Bagheri N., Shilatifard A., Budinger G. R. S., Perlman H., Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 214, 2387–2404 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rangasamy T., Cho C. Y., Thimmulappa R. K., Zhen L., Srisuma S. S., Kensler T. W., Yamamoto M., Petrache I., Tuder R. M., Biswal S., Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Invest. 114, 1248–1259 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hruban R. H., Meziane M. A., Zerhouni E. A., Khouri N. F., Fishman E. K., Wheeler P. S., Dumler J. S., Hutchins G. M., High resolution computed tomography of inflation-fixed lungs. Pathologic-radiologic correlation of centrilobular emphysema. Am. Rev. Respir. Dis. 136, 935–940 (1987). [DOI] [PubMed] [Google Scholar]
- 23.Yoshida M., Korfhagen T. R., Whitsett J. A., Surfactant protein D regulates NF-κB and matrix metalloproteinase production in alveolar macrophages via oxidant-sensitive pathways. J. Immunol. 166, 7514–7519 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Hirama N., Shibata Y., Otake K., Machiya J.-i., Wada T., Inoue S., Abe S., Takabatake N., Sata M., Kubota I., Increased surfactant protein-D and foamy macrophages in smoking-induced mouse emphysema. Respirology 12, 191–201 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Guo L., Johnson R. S., Schuh J. C., Biochemical characterization of endogenously formed eosinophilic crystals in the lungs of mice. J. Biol. Chem. 275, 8032–8037 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Baldán A., Gomes A. V., Ping P., Edwards P. A., Loss of ABCG1 results in chronic pulmonary inflammation. J. Immunol. 180, 3560–3568 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Ackerman S. J., Corrette S. E., Rosenberg H. F., Bennett J. C., Mastrianni D. M., Nicholson-Weller A., Weller P. F., Chin D. T., Tenen D. G., Molecular cloning and characterization of human eosinophil Charcot-Leyden crystal protein (lysophospholipase). Similarities to IgE binding proteins and the S-type animal lectin superfamily. J. Immunol. 150, 456–468 (1993). [PubMed] [Google Scholar]
- 28.Di Stefano A., Capelli A., Lusuardi M., Balbo P., Vecchio C., Maestrelli P., Mapp C. E., Fabbri L. M., Donner C. F., Saetta M., Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am. J. Respir. Crit. Care Med. 158, 1277–1285 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Postma D. S., Rabe K. F., The asthma-COPD overlap syndrome. N. Engl. J. Med. 373, 1241–1249 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Wolf E., Gebhardt A., Kawauchi D., Walz S., von Eyss B., Wagner N., Renninger C., Krohne G., Asan E., Roussel M. F., Eilers M., Miz1 is required to maintain autophagic flux. Nat. Commun. 4, 2535 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N.-H., Nitsche A., Müller M. A., Drosten C., Pöhlmann S., SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gebhardt A., Frye M., Herold S., Benitah S. A., Braun K., Samans B., Watt F. M., Elsässer H.-P., Eilers M., Myc regulates keratinocyte adhesion and differentiation via complex formation with Miz1. J. Cell Biol. 172, 139–149 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barnes P. J., Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 138, 16–27 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Hu J., Van den Steen P. E., Sang Q.-X. A., Opdenakker G., Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 6, 480–498 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Tuder R. M., Yoshida T., Arap W., Pasqualini R., Petrache I., State of the art. Cellular and molecular mechanisms of alveolar destruction in emphysema: An evolutionary perspective. Proc. Am. Thorac. Soc. 3, 503–510 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilk J. B., Chen T.-H., Gottlieb D. J., Walter R. E., Nagle M. W., Brandler B. J., Myers R. H., Borecki I. B., Silverman E. K., Weiss S. T., O’Connor G. T., A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLOS Genet. 5, e1000429 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho M. H., Boutaoui N., Klanderman B. J., Sylvia J. S., Ziniti J. P., Hersh C. P., De Meo D. L., Hunninghake G. M., Litonjua A. A., Sparrow D., Lange C., Won S., Murphy J. R., Beaty T. H., Regan E. A., Make B. J., Hokanson J. E., Crapo J. D., Kong X., Anderson W. H., Tal-Singer R., Lomas D. A., Bakke P., Gulsvik A., Pillai S. G., Silverman E. K., Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat. Genet. 42, 200–202 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cho M. H., Castaldi P. J., Wan E. S., Siedlinski M., Hersh C. P., Demeo D. L., Himes B. E., Sylvia J. S., Klanderman B. J., Ziniti J. P., Lange C., Litonjua A. A., Sparrow D., Regan E. A., Make B. J., Hokanson J. E., Murray T., Hetmanski J. B., Pillai S. G., Kong X., Anderson W. H., Tal-Singer R., Lomas D. A., Coxson H. O., Edwards L. D., Nee W. M., Vestbo J., Yates J. C., Agusti A., Calverley P. M. A., Celli B., Crim C., Rennard S., Wouters E., Bakke P., Gulsvik A., Crapo J. D., Beaty T. H., Silverman E. K.; ICGN Investigators; ECLIPSE Investigators; COPDGene Investigators , A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Hum. Mol. Genet. 21, 947–957 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soler Artigas M., Wain L. V., Repapi E., Obeidat M., Sayers I., Burton P. R., Johnson T., Zhao J. H., Albrecht E., Dominiczak A. F., Kerr S. M., Smith B. H., Cadby G., Hui J., Palmer L. J., Hingorani A. D., Wannamethee S. G., Whincup P. H., Ebrahim S., Smith G. D., Barroso I., Loos R. J. F., Wareham N. J., Cooper C., Dennison E., Shaheen S. O., Liu J. Z., Marchini J.; Medical Research Council National Survey of Health, Development (NSHD) Respiratory Study Team, Dahgam S., Naluai A. T., Olin A.-C., Karrasch S., Heinrich J., Schulz H., McKeever T. M., Pavord I. D., Heliövaara M., Ripatti S., Surakka I., Blakey J. D., Kähönen M., Britton J. R., Nyberg F., Holloway J. W., Lawlor D. A., Morris R. W., James A. L., Jackson C. M., Hall I. P., Tobin M. D.; SpiroMeta Consortium , Effect of five genetic variants associated with lung function on the risk of chronic obstructive lung disease, and their joint effects on lung function. Am. J. Respir. Crit. Care Med. 184, 786–795 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hobbs B. D., de Jong K., Lamontagne M., Bossé Y., Shrine N., Artigas M. S., Wain L. V., Hall I. P., Jackson V. E., Wyss A. B., London S. J., North K. E., Franceschini N., Strachan D. P., Beaty T. H., Hokanson J. E., Crapo J. D., Castaldi P. J., Chase R. P., Bartz T. M., Heckbert S. R., Psaty B. M., Gharib S. A., Zanen P., Lammers J. W., Oudkerk M., Groen H. J., Locantore N., Tal-Singer R., Rennard S. I., Vestbo J., Timens W., Paré P. D., Latourelle J. C., Dupuis J.; George T O’Connor, Wilk J. B., Kim W. J., Lee M. K., Oh Y.-M., Vonk J. M., de Koning H. J., Leng S., Belinsky S. A., Tesfaigzi Y., Manichaikul A., Wang X.-Q., Rich S. S., Barr R. G., Sparrow D., Litonjua A. A., Bakke P., Gulsvik A., Lahousse L., Brusselle G. G., Stricker B. H., Uitterlinden A. G., Ampleford E. J., Bleecker E. R., Woodruff P. G., Meyers D. A., Qiao D., Lomas D. A., Yim J.-J., Kim D. K., Hawrylkiewicz I., Sliwinski P., Hardin M., Fingerlin T. E., Schwartz D. A., Postma D. S., Nee W. M., Tobin M. D., Silverman E. K., Boezen H. M., Cho M. H.; COPDGene Investigators; ECLIPSE Investigators; LifeLines Investigators; SPIROMICS Research Group; International COPD Genetics Network Investigators, UK BiLEVE Investigators; International COPD Genetics Consortium , Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat. Genet. 49, 426–432 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wain L. V., Shrine N., Artigas M. S., Erzurumluoglu A. M., Noyvert B., Bossini-Castillo L., Obeidat M., Henry A. P., Portelli M. A., Hall R. J., Billington C. K., Rimington T. L., Fenech A. G., John C., Blake T., Jackson V. E., Allen R. J., Prins B. P.; Understanding Society Scientific Group, Campbell A., Porteous D. J., Jarvelin M.-R., Wielscher M., James A. L., Hui J., Wareham N. J., Zhao J. H., Wilson J. F., Joshi P. K., Stubbe B., Rawal R., Schulz H., Imboden M., Probst-Hensch N. M., Karrasch S., Gieger C., Deary I. J., Harris S. E., Marten J., Rudan I., Enroth S., Gyllensten U., Kerr S. M., Polasek O., Kähönen M., Surakka I., Vitart V., Hayward C., Lehtimäki T., Raitakari O. T., Evans D. M., Henderson A. J., Pennell C. E., Wang C. A., Sly P. D., Wan E. S., Busch R., Hobbs B. D., Litonjua A. A., Sparrow D. W., Gulsvik A., Bakke P. S., Crapo J. D., Beaty T. H., Hansel N. N., Mathias R. A., Ruczinski I., Barnes K. C., Bossé Y., Joubert P., van den Berge M., Brandsma C.-A., Paré P. D., Sin D. D., Nickle D. C., Hao K., Gottesman O., Dewey F. E., Bruse S. E., Carey D. J., Kirchner H. L.; Geisinger-Regeneron DiscovEHR Collaboration, Jonsson S., Thorleifsson G., Jonsdottir I., Gislason T., Stefansson K., Schurmann C., Nadkarni G., Bottinger E. P., Loos R. J. F., Walters R. G., Chen Z., Millwood I. Y., Vaucher J., Kurmi O. P., Li L., Hansell A. L., Brightling C., Zeggini E., Cho M. H., Silverman E. K., Sayers I., Trynka G., Morris A. P., Strachan D. P., Hall I. P., Tobin M. D., Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat. Genet. 49, 416–425 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rock J. R., Barkauskas C. E., Cronce M. J., Xue Y., Harris J. R., Liang J., Noble P. W., Hogan B. L. M., Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. U.S.A. 108, E1475–E1483 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng D. S., Han W., Chen S. M., Sherrill T. P., Chont M., Park G. Y., Sheller J. R., Polosukhin V. V., Christman J. W., Yull F. E., Blackwell T. S., Airway epithelium controls lung inflammation and injury through the NF-κB pathway. J. Immunol. 178, 6504–6513 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Zaynagetdinov R., Sherrill T. P., Gleaves L. A., Hunt P., Han W., McLoed A. G., Saxon J. A., Tanjore H., Gulleman P. M., Young L. R., Blackwell T. S., Chronic NF-κB activation links COPD and lung cancer through generation of an immunosuppressive microenvironment in the lungs. Oncotarget 7, 5470–5482 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vuillemenot B. R., Rodriguez J. F., Hoyle G. W., Lymphoid tissue and emphysema in the lungs of transgenic mice inducibly expressing tumor necrosis factor-α. Am. J. Respir. Cell Mol. Biol. 30, 438–448 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Buyandelger B., Mansfield C., Kostin S., Choi O., Roberts A. M., Ware J. S., Mazzarotto F., Pesce F., Buchan R., Isaacson R. L., Vouffo J., Gunkel S., Knöll G., McSweeney S. J., Wei H., Perrot A., Pfeiffer C., Toliat M. R., Ilieva K., Krysztofinska E., López-Olañeta M. M., Gómez-Salinero J. M., Schmidt A., Ng K. E., Teucher N., Chen J., Teichmann M., Eilers M., Haverkamp W., Regitz-Zagrosek V., Hasenfuss G., Braun T., Pennell D. J., Gould I., Barton P. J. R., Lara-Pezzi E., Schäfer S., Hübner N., Felkin L. E., O’Regan D. P., Brand T., Milting H., Nürnberg P., Schneider M. D., Prasad S., Petretto E., Knöll R., ZBTB17 (MIZ1) is important for the cardiac stress response and a novel candidate gene for cardiomyopathy and heart failure. Circ. Cardiovasc. Genet. 8, 643–652 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Li X., Luo R., Mo X., Jiang R., Kong H., Hua W., Wu X., Polymorphism of ZBTB17 gene is associated with idiopathic dilated cardiomyopathy: A case control study in a Han Chinese population. Eur. J. Med. Res. 18, 10 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Villard E., Perret C., Gary F., Proust C., Dilanian G., Hengstenberg C., Ruppert V., Arbustini E., Wichter T., Germain M., Dubourg O., Tavazzi L., Aumont M. C., DeGroote P., Fauchier L., Trochu J. N., Gibelin P., Aupetit J. F., Stark K., Erdmann J., Hetzer R., Roberts A. M., Barton P. J., Regitz-Zagrosek V.; Cardiogenics Consortium, Aslam U., Duboscq-Bidot L., Meyborg M., Maisch B., Madeira H., Waldenström A., Galve E., Cleland J. G., Dorent R., Roizes G., Zeller T., Blankenberg S., Goodall A. H., Cook S., Tregouet D. A., Tiret L., Isnard R., Komajda M., Charron P., Cambien F., A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur. Heart J. 32, 1065–1076 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bernasovská J., Boroňová I., Petrejčiková E., Kmec J., ZBTB17 gene rs10927875 polymorphism in Slovak patients with dilated cardiomyopathy. Int. Scholar. Sci. Res. Innov. 8, 562–565 (2014). [Google Scholar]
- 50.Morris J. A., Kemp J. P., Youlten S. E., Laurent L., Logan J. G., Chai R. C., Vulpescu N. A., Forgetta V., Kleinman A., Mohanty S. T., Sergio C. M., Quinn J., Nguyen-Yamamoto L., Luco A.-L., Vijay J., Simon M.-M., Pramatarova A., Medina-Gomez C., Trajanoska K., Ghirardello E. J., Butterfield N. C., Curry K. F., Leitch V. D., Sparkes P. C., Adoum A.-T., Mannan N. S., Komla-Ebri D. S. K., Pollard A. S., Dewhurst H. F., Hassall T. A. D., Beltejar M.-J. G.; Me Research Team, Adams D. J., Vaillancourt S. M., Kaptoge S., Baldock P., Cooper C., Reeve J., Ntzani E. E., Evangelou E., Ohlsson C., Karasik D., Rivadeneira F., Kiel D. P., Tobias J. H., Gregson C. L., Harvey N. C., Grundberg E., Goltzman D., Adams D. J., Lelliott C. J., Hinds D. A., Ackert-Bicknell C. L., Hsu Y.-H., Maurano M. T., Croucher P. I., Williams G. R., Duncan Bassett J. H., Evans D. M., Richards J. B., An atlas of genetic influences on osteoporosis in humans and mice. Nat. Genet. 51, 258–266 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim S. K., Identification of 613 new loci associated with heel bone mineral density and a polygenic risk score for bone mineral density, osteoporosis and fracture. PLOS ONE 13, e0200785 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kichaev G., Bhatia G., Loh P. R., Gazal S., Burch K., Freund M. K., Schoech A., Pasaniuc B., Price A. L., Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 104, 65–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Isajevs S., Taivans I., Svirina D., Strazda G., Kopeika U., Patterns of inflammatory responses in large and small airways in smokers with and without chronic obstructive pulmonary disease. Respiration 81, 362–371 (2011). [DOI] [PubMed] [Google Scholar]
- 54.Richmond B. W., Brucker R. M., Han W., Du R.-H., Zhang Y., Cheng D.-S., Gleaves L., Abdolrasulnia R., Polosukhina D., Clark P. E., Bordenstein S. R., Blackwell T. S., Polosukhin V. V., Airway bacteria drive a progressive COPD-like phenotype in mice with polymeric immunoglobulin receptor deficiency. Nat. Commun. 7, 11240 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Richmond B. W., Du R.-H., Han W., Benjamin J. T., van der Meer R., Gleaves L., Guo M., McKissack A., Zhang Y., Cheng D. S., Polosukhin V. V., Blackwell T. S., Bacterial-derived neutrophilic inflammation drives lung remodeling in a mouse model of chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 58, 736–744 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fox-Walsh K., Davis-Turak J., Zhou Y., Li H., Fu X.-D., A multiplex RNA-seq strategy to profile poly(A+) RNA: Application to analysis of transcription response and 3′ end formation. Genomics 98, 266–271 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou Y., Li H.-R., Huang J., Jin G., Fu X.-D., Multiplex analysis of polyA-linked sequences (MAPS): An RNA-seq strategy to profile poly(A+) RNA. Methods Mol. Biol. 1125, 169–178 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peng C., Lu Z., Xie Z., Cheng Z., Chen Y., Tan M., Luo H., Zhang Y., He W., Yang K., Zwaans B. M. M., Tishkoff D., Ho L., Lombard D., He T. C., Dai J., Verdin E., Ye Y., Zhao Y., The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteomics 10, M111.012658 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cox J., Mann M., MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 60.Misharin A. V., Morales-Nebreda L., Mutlu G. M., Budinger G. R. S., Perlman H., Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol. 49, 503–510 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/33/eabb7238/DC1