

Abstract
Objective
We tested the hypothesis that variant repeat interruptions (RIs) within the DMPK CTG repeat tract lead to milder symptoms compared with pure repeats (PRs) in myotonic dystrophy type 1 (DM1).
Methods
We evaluated motor, neurocognitive, and behavioral outcomes in a group of 6 participants with DM1 with RI compared with a case-matched sample of 12 participants with DM1 with PR and a case-matched sample of 12 unaffected healthy comparison participants (UA).
Results
In every measure, the RI participants were intermediate between UA and PR participants. For muscle strength, the RI group was significantly less impaired than the PR group. For measures of Full Scale IQ, depression, and sleepiness, all 3 groups were significantly different from each other with UA > RI > PR in order of impairment. The RI group was different from unaffected, but not significantly different from PR (UA > RI = PR) in apathy and working memory. Finally, in finger tapping and processing speed, RI did not differ from UA comparisons, but PR had significantly lower scores than the UA comparisons (UA = RI > PR).
Conclusions
Our results support the notion that patients affected by DM1 with RI demonstrate a milder phenotype with the same pattern of deficits as those with PR indicating a similar disease process.
Myotonic dystrophy type 1 (DM1; OMIM 160900) is an autosomal dominant, progressive, multisystem disorder caused by expansion of a CTG repeat in the 3′-untranslated region of DMPK.1–4 DM1 affects many organ systems, including skeletal muscle, heart, gastrointestinal, integumentary, endocrine, and CNS.3,5,6 Although the primary symptoms of DM1 are myotonia and muscle weakness, some of the most disabling symptoms of the disease are those arising from CNS involvement.7,8 These include progressive cognitive and behavioral changes, as well as fatigue and excessive daytime sleepiness, which greatly affect overall quality of life.9–11
In 3%–5% of patients with DM1, the CTG repeat tract is interrupted by naturally occurring variant sequences, such as CCG, CTC, or GGC motifs.12,13 Variant repeats most commonly occur at the 3′-end of the DMPK CTG repeat tract.14,15 These are referred to as variant repeat interruptions (RIs). Increasing evidence from case reports suggests that patients with DM1 who carry RI alleles exhibit a later age at symptom onset, milder muscle symptoms, and atypical patterns of symptoms (smaller proportion of cataracts, cardiac problems, and muscle weakness) compared with those with pure repeats (PRs).12–14,16,17 These effects have now been confirmed in 2 large independent DM1 cohorts.18,19 The attenuation of symptoms is hypothesized to result, at least in part, from a stabilizing effect of RI that reduces expansion-biased instability in somatic cells.13,20,21 In this context, we set out to compare motor, neurocognitive, and behavioral outcome measures of participants with adult-onset DM1 with RIs matched to participants with DM1 with PRs, as well as comparison of both groups to participants unaffected by DM1.
Methods
Recruitment of participants
Participants with adult-onset DM1 were recruited to the University of Iowa “DM1 Brain Study” from across the United States by advertisements through the Myotonic Dystrophy Foundation or word of mouth, as described previously.22 Recruitment was targeted to adult-onset DM1 only, with symptom onset at age 18 years or older. Unaffected participants were primarily recruited from the local community through advertisements. Recruitment for baseline assessments took place between September 2014 and July 2017. Inclusion criteria were as follows: (1) between ages 21 and 65 years; (2) clinical diagnosis of DM1 after age 21 years; (3) committed to completing annual evaluations for 2 years following intake; and (4) commitment of an informant to accompany the participant to study visits. Exclusion criteria included (1) unstable psychiatric illness (including current substance abuse) and (2) history of major head trauma with loss of consciousness for longer than a few minutes and including clinically significant sequelae.
Standard protocol approvals, registrations, and patient consents
All participants gave written informed consent before enrolling in the protocol in accordance with the Declaration of Helsinki. The study was approved by the University of Iowa's Institutional Review Board.
Data availability
Anonymized data will be shared by request from any qualified investigator.
Measurement of CTG repeat length and variant repeat identification
For genotyping of CTG repeats in participants with DM1, we used the same methodology as the one used in previous studies.22,23 For variant repeat identification, small-pool PCR products underwent AciI enzyme digestion (New England Biolabs UK Ltd.; restriction site 5′CCGC-3′) and Southern blotting to indicate the presence of CCG interruptions within the CTG repeat array in the expanded allele as previously described.13
Motor testing
Motor, neurocognitive, and behavioral outcome measures of interest were selected a priori to reduce the number of comparisons. Severity of muscle weakness was measured using the Muscle Impairment Rating Scale (MIRS) during examination by a neuromuscular specialist experienced in DM1, blinded to the participants' genetic status.24 This scale evaluates muscular impairment severity according to an ordinal 5-point scale as follows: (1) no muscular impairment, (2) minimal signs, (3) distal weakness, (4) mild to moderate proximal weakness, and (5) severe proximal weakness.
Grip strength was measured using a Lafayette Instruments dynamometer. The stirrup of the dynamometer was adjusted to comfortably fit the participant's hand size, after which they were instructed to squeeze as hard as they possibly could. Strong motivational encouragement was provided by the examiner during each of the 6 trials (3 for the dominant hand and 3 for the nondominant hand) to elicit the participant's maximal effort. Ultimate scores were the means of 3 trials for each hand.
Neurocognitive and behavioral testing
Neurocognitive and behavioral assessments included the Wechsler Adult Intelligence Scale–Fourth Edition (WAIS-IV), Beck Depression Inventory-II (BDI-II), Apathy Evaluation Scale (AES) self-assessment, and Scales for Outcomes in Parkinson's Disease (SCOPA). These measures were administered by a trained examiner experienced in DM1 who was blinded to the patient's clinical condition (CTG expansion length and muscular impairment).
Statistical analysis
All statistical analyses were performed using R (version 3.6.2). Each participant who was determined to have variant repeats was matched by age, sex, and CTG repeat length to 2 participants with pure CTG repeats and 2 unaffected participants. Mixed-effects multivariable linear regression models were used to examine the impact of group, age, sex, and a subject-matching variable on the dependent variables (motor, neurocognitive, and behavioral outcome measures). The coefficient of determination (R2) for the model, semi-partial R2 for each fixed effect (group, age, and sex), and 95% confidence intervals (CIs) were calculated using the Nakagawa & Schielzeth approach.25 Effect sizes were considered very weak (R2 < 0.1), weak (0.1 < R2 < 0.3), moderate (0.3 < R2 < 0.5), and strong (0.5 < R2). Post hoc least-squares means tests were used for pairwise comparisons between UA, RI, and PR groups. All outcome measures of interest were selected a priori to minimize multiple testing considerations.
Results
Sample
From a cohort of 57 adults affected by DM1, 6 participants (11%) were identified as positive for variant RIs by AciI enzyme digest. Each RI participant was equivalently matched to 2 PR participants and 2 unaffected healthy comparison participants (UA) by age, sex, and CTG repeat length for a total sample of 30 participants (6 RI, 12 PR, and 12 UA) (table 1). There was an equal proportion of men (66%) and women (33%) within each group (χ2 = 0.0, df = 2, p = 1.0). Mean age at evaluation was 42.27 years (SD = 11.89) for the UA group, 42.25 years (SD = 12.52) for the RI group, and 40.42 years (SD = 11.03) for the PR group, with no significant group effect (χ2 = 4.32, df = 2, p = 0.115). There were no significant differences in age between the UA and RI groups (t(22) = 0.017, 95% CI [−2.44, 2.48]), the UA and PR groups (t(22) = 1.90, 95% CI [−0.16, 3.85]), and the RI and PR groups (t(22) = 1.53, 95% CI [−0.63, 4.28]) (table 1). Mean age at disease onset was 31.75 years (SD = 5.76) for the RI group and 24.14 years (SD = 9.48) for the PR group, with no significant difference between groups (t(13) = 1.93, 95% CI [−0.86, 15.89]) (table 1). The length of the estimated progenitor CTG allele (ePAL)26 ranged from 12 to 22 in the UA group (mean = 14.43, SD = 3.41), from 157 to 625 in the RI group (mean = 386.17, SD = 149.66), and from 125 to 750 in the PR group (mean = 327.92, SD = 190.13), with a significant group effect in the model (χ2 = 32.91, df = 2, p < 0.001). As expected for CTG length, there were significant differences between the UA group and the RI (t(17.9) = −5.10, 95% CI [−545.39, −227.07]) and PR (t(18.3) = −5.01, 95% CI [−468.87, −192.23]) groups and no significant difference between the RI and PR groups (t(17.5) = 0.82, 95% CI [−86.25, 197.62]) (table 1).
Table 1.
Demographics of the study sample
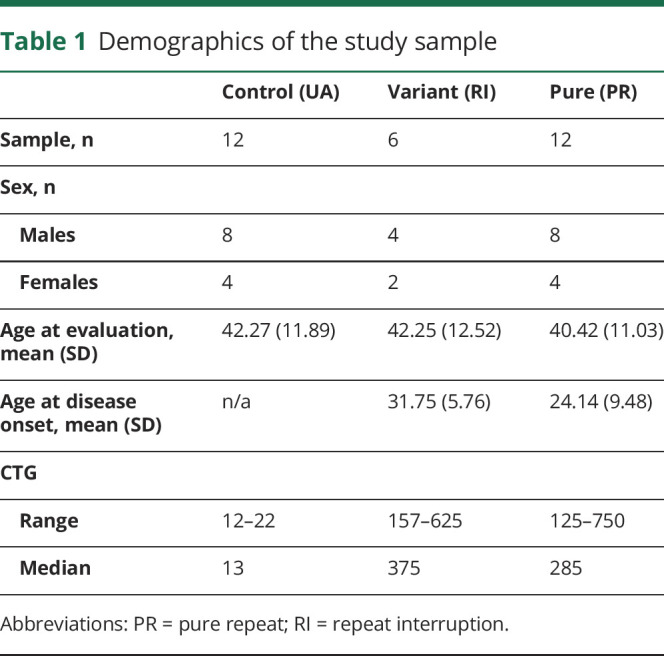
Motor performance
Detailed statistics for each outcome measure from the mixed-effects multivariable linear regression model with post hoc least-squares means tests are shown in table 2 and table 3. The figure summarizes group differences for all outcome measures, which show that the RI group is always intermediate between the UA and PR groups.
Table 2.
Outcome comparisons between groupsa

Table 3.
Mixed-effects multivariate model resultsa

Figure. Patients with DM1 with variant repeats have milder symptoms in motor, neurocognitive, and behavioral domains.

Motor (A–C), cognitive (D–F), and behavioral (G–I) scores (y-axes) are shown across groups (x-axes), including controls, patients with DM1 with variant repeats, and patients with DM1 with pure repeats. There were significant differences in MIRS, FSIQ, processing speed, Beck Depression Inventory, and SCOPA-Sleep between the variant and pure repeat groups. Circles represent the mean value of each group (red = control group, green = variant repeat group, and blue = pure repeat group). The vertical, solid lines represent 95% confidence interval. The horizontal bars with asterisks (*) represent significant differences between groups. DM1 = myotonic dystrophy type 1; FSIQ = Full Scale IQ; MIRS = Muscle Impairment Rating Scale; SCOPA = Scales for Outcomes in Parkinson's Disease.
As shown in figure, A, there was a significant difference between the RI and PR groups in MIRS scores (t(11) = −2.2, 95% CI [−1.95, −0.003]) with an overall moderate effect size of the model (R2 = 0.308, 95% CI [0.090, 0.668]) and a significant group effect (χ2 = 4.87, df = 1, p = 0.027) with a higher proportion of PR participants scoring 3 and 4 (mild to moderate proximal weakness) (mean = 2.92; SD = 1.08) than the RI group (mean 2.00; SD = 0.63). The PR group had 5 participants with a score of 4 (moderate proximal weakness) and no participants with a score of 5 (severe proximal weakness), whereas the RI group had no participants with a score of 4 or 5.
Finger tapping test results for the dominant hand are shown in figure, B. The mean scores were 43.98 (SD = 8.98) for the UA group; 36.83 (SD = 5.54) for the RI group; and 29.16 (SD = 11.78) for the PR group. There was an overall moderate effect size (R2 = 0.459, 95% CI [0.264, 0.694]) and significant group effect in the model (χ2 = 16.84, df = 2, p < 0.001), with no significant difference between the UA and RI groups (t(25) = 1.61, 95% CI [−1.96, 16.25]), a significant difference between the UA and PR groups (t(25) = 4.10, 95% CI [7.41, 22.34]), and no significant difference between the RI and PR groups (t(25) = 1.74, 95% CI [−1.40, 16.86]).
As shown in figure, C, for grip strength (dominant hand), the mean was 41.08 (SD = 11.12) for the UA group, 29.19 (SD = 15.7) for the RI group, and 19.25 (SD = 14.42) for the PR group. The group effect was significant in the model (χ2 = 22.01, df = 2, p < 0.001) with a strong overall effect size (R2 = 0.505, 95% CI [0.312, 0.722]), no significant difference between the UA and RI groups (t(22) = 2.03, 95% CI [−0.25, 24.02]), a significant difference between the UA and PR groups (t(22) = 4.69, 95% CI [12.56, 32.45]), and no significant difference between the RI and PR groups (t(22) = 1.81, 95% CI [−1.54, 22.79]).
Neurocognitive functioning
Figure, D through F shows group differences on the WAIS-IV Full Scale IQ, Working Memory Index (WMI), and Processing Speed Index (PSI), respectively. The mean Full Scale IQ was 120.08 (SD = 13.52) for the UA group; 107.17 (SD = 8.77) for the RI group; and 94.67 (SD = 7.67) for the PR group. There was a significant group effect in the model for Full Scale IQ (χ2 = 33.39, df = 2, p < 0.001), with a strong overall effect size (R2 = 0.537, 95% CI [0.346, 0.741]), a significant difference between the UA and RI groups (t(25) = 2.37, 95% CI [1.72, 24.11]), a significant difference between the UA and PR groups (t(25) = 5.78, 95% CI [16.55, 34.90]), and a significant difference between the RI and PR groups (t(25) = 2.35, 95% CI [1.59, 24.02]).
For WMI, the mean score was 120.17 (SD = 11.57) for the control group, 101.83 (SD = 11.55) for the RI group, and 94.75 (SD = 14.59) for the PR group, with a significant group effect in the model (χ2 = 24.06, df = 2, p < 0.001) and moderate overall effect size (R2 = 0.458, 95% CI [0.262, 0.694]). There was a significant difference between the UA and RI groups (t(25) = 2.79, 95% CI [4.84, 31.84]), a significant difference between the UA and PR groups (t(25) = 4.82, 95% CI [14.82, 36.95]), and no significant difference between the RI and PR groups (t(25) = 1.14, 95% CI [−5.99, 21.08]).
PSI exhibited a similar pattern as WMI, with the UA group having a mean score of 114.5 (SD = 16.71), the RI group having a mean score of 106.00 (SD = 12.88), and the PR group having a mean score of 88.08 (SD = 8.15). There was a significant group effect in the model (χ2 = 24.90, df = 2, p < 0.001) and moderate overall effect size (R2 = 0.468, 95% CI [0.272, 0.700]), with no significant difference between the UA and RI groups (t(25) = 1.27, 95% CI [−5.19, 22.20]), a significant difference between the UA and PR groups (t(25) = 4.93, 95% CI [15.62, 38.06]), and a significant difference between the RI and PR groups (t(25) = 2.75, 95% CI [4.61, 32.06]).
Behavioral outcomes
Figure, G shows scores for the BDI across groups. The mean score was 1.92 (SD = 1.83) for the UA group, 7.17 (SD = 7.05) for the RI group, and 12.00 (SD = 6.63) for the PR group, with a significant group effect (χ2 = 27.04, df = 2, p < 0.001) and strong overall effect size (R2 = 0.523, 95% CI [0.331, 0.733]). Age had a significant effect on the model (χ2 = 5.93, df = 1, p = 0.014) with weak effect size (R2 = 0.170, 95% CI [0.009, 0.440]), and sex had a significant effect on the model (χ2 = 3.83, df = 1, p = 0.051) with weak effect size (R2 = 0.117, 95% CI [0.002, 0.381]). There were significant differences between the UA and RI groups (t(25) = −2.13, 95% CI [−10.33, −0.18]), the UA and PR groups (t(25) = −5.20, 95% CI [−14.66, −6.34]), and the RI and PR groups (t(25) = −2.12, 95% CI [−10.34, −0.16]).
Results for the AES are shown in figure, H. The mean score for the UA group was 22.33 (SD = 3.73), 31.83 (SD = 6.31) for the RI group, and 34.33 (SD = 11.37) for the PR group, with a significant group effect (χ2 = 14.71, df = 2, p < 0.001) and moderate overall effect size (R2 = 0.400, 95% CI [0.208, 0.658]). Age also had a significant effect (χ2 = 5.93, df = 1, p = 0.014) with weak effect size (R2 = 0.170, 95% CI [0.009, 0.440]). There was a significant difference between the UA and RI groups (t(25) = −2.24, 95% CI [−16.30, −0.71]), a significant difference between the UA and PR groups (t(25) = −3.75, 95% CI [−18.04, −5.26]), and no significant difference between the RI and PR groups (t(25) = −0.83, 95% CI [−10.96, 4.68]).
The group scores for the SCOPA-Sleep scale for daytime sleepiness are shown in figure, I. The mean score was 1.08 (SD = 1.38) for the UA group, 3.83 (SD = 2.48) for the RI group, and 6.00 (SD = 2.7) for the PR group, with a significant group effect (χ2 = 41.13, df = 2, p < 0.001) and strong overall effect size (R2 = 0.598, 95% CI [0.419, 0.777]). There were significant differences between the UA and RI groups (t(25) = −2.60, 95% CI [−4.93, −0.57]), UA and PR groups (t(25) = −6.41, 95% CI [−7.35, −3.77]), and RI and PR groups (t(25) = −2.64, 95% CI [−4.99, −0.62]).
Discussion
We investigated the differences in motor, neurocognitive, and behavioral outcomes of patients with PR DM1 compared with patients with DM1 with variant RIs in the DMPK CTG repeat tract. We demonstrated that there are significant differences in all domains for patients with RI, as shown by significantly better scores on the MIRS, Full Scale IQ, processing speed, BDI-II, and SCOPA-Sleep. This is the largest cross-sectional, case-matched control study to examine patients with RIs in DM1.
It should be noted that although the RI group performed significantly better than the PR group on multiple different outcome measures, they still had deficits compared with the unaffected comparison group, consistent with the typical pattern of DM1 pathology.
The putative cellular process by which CTG expansion leads to DM1 pathology is incompletely understood. Furthermore, the true mechanism yielding a milder phenotype in patients with RI is not yet known. It has been hypothesized that the presence of variant repeats within the CTG repeat tract may disrupt the secondary structures formed by mutant DMPK alleles, thus decreasing their affinity for splicing regulators and interfering with the proposed pathologic mechanism. In addition, expanded PRs are characterized by high levels of somatic and germline instability, mediated by a cell division–independent, DNA mismatch repair protein-dependent process.27–29 It has been previously observed in other trinucleotide repeat disorders that the presence of variant repeats results in relative stabilization of simple repeats in both germline and somatic cells.30–33 Variant repeat–mediated suppression of somatic instability in DM1 has also been observed and strongly associated with milder symptoms and later age at onset.13,18–21,34
Patients with DM1 experience a slow progression of muscle weakness and atrophy, initially involving the distal muscles of the extremities and later affecting the proximal musculature. Patients can develop dysphagia and respiratory muscle weakness, with an increased risk for weight loss and aspiration. Consistent with recent findings from the OPTIMISTIC and Saguenay cohorts,18,19 our data confirm a significant difference in muscle power between PR and RI participants, detectable clinically by MIRS assessment,24 under study conditions in which the evaluating clinician is blinded to the participants' genetic status.
Deficits in cognitive functioning are a well-recognized and defined feature of DM1 that contribute significantly to decreased quality of life.9 The mechanisms underlying CNS pathology in DM1 are poorly understood, although evidence favors a significant role of dysregulation of alternative splicing involving key CNS genes including Tau.35 Furthermore, somatic instability is seen to be particularly marked in the cerebral cortex.36 Our findings of a significantly reduced impairment of Full Scale IQ and processing speed in RI vs PR participants is therefore consistent with a protective effect of RI by limiting somatic instability and so the abundance of expanded CUGn in the brain. This observation highlights somatic instability as a potential therapeutic target for CNS involvement in DM1 as well as for peripheral muscle symptoms. Furthermore, with the advent of large clinical trials in DM1,37 this finding also further emphasizes the need to screen and control for the presence of RI in DM1 clinical study cohorts.
Excessive daytime sleepiness (EDS) is common in DM1 and significantly affects quality of life. The etiology of EDS in DM1 is complex. Evidence broadly favors a central cause of somnolence symptoms, although poor sleep hygiene and medication side effects may also be relevant.3 Sleep-disordered breathing (SDB) is a frequent finding in DM1,38 contributing to sleep fragmentation and hence somnolence symptoms, although symptomatic response to nocturnal ventilation is frequently disappointing.39 Evaluation of EDS may be further complicated by impaired symptom awareness, as part of the CNS phenotype.40 We found that participants with RI reported less EDS than those with PR. Although this could represent further evidence of a protective effect in the CNS, further exploration of this observation would benefit from polysomnography, to rule out an effect of SDB associated with peripheral muscle weakness, and objective measures of somnolence such as the multiple sleep latency test.
A limitation of this present study is small sample size, with only 6 participants with variant repeats, 12 participants with PRs, and 12 unaffected participants. This decreased the statistical power with which we could possibly detect significant differences in additional motor, neurocognitive, and behavioral domains. Another limitation is the overall mild nature of symptoms of the study cohort. A lower age at disease onset could have possibly revealed more evident changes between groups. In addition, although the mean age at disease onset for the variant repeat group was greater than 7 years than the PR group (31.75 years vs 24.1 years), it was not statistically significant. However, in larger cohorts, AciI sites are statistically significantly associated with later onset.18,19 A follow-up neuroimaging study in a larger sample could possibly help elucidate the specific brain changes behind this pattern of deficits. Nonetheless, motor, cognitive, and behavioral measures were significantly better in the presence of variant repeats. Our group has demonstrated that cognitive deficits in DM1 are associated with altered brain structure.41 We would expect that the RI group will exhibit a milder neuroanatomic phenotype than their PR counterparts.
Our study supports the hypothesis that variant RIs within the CTG repeat tract of the DMPK gene have a protective effect in multiple systems in DM1, including the CNS. Further exploration of the mechanisms underlying this effect is required to improve prognostic information available to affected patients and may reveal potential targets for novel therapy.
Acknowledgment
The authors are grateful to the participants and their families for their cooperation with the study and also to the team of research assistants who collected the data.
Glossary
- AES
Apathy Evaluation Scale
- BDI-II
Beck Depression Inventory-II
- CI
confidence interval
- DM1
myotonic dystrophy type 1
- EDS
excessive daytime sleepiness
- MIRS
Muscle Impairment Rating Scale
- PR
pure repeat
- PSI
Processing Speed Index
- RI
repeat interruption
- SCOPA
Scales for Outcomes in Parkinson's Disease
- SDB
sleep-disordered breathing
- WAIS-IV
Wechsler Adult Intelligence Scale–Fourth Edition
- WMI
Working Memory Index
Appendix. Authors
Study funding
This work was supported by a grant from the NINDS (Ref: 5R01NS094387-02) and the Wyck Foundation. The funders did not have any role in the study design, data collection, analysis, or in preparing this manuscript.
Disclosure
J.N. Miller, M.J. Hamilton, E. van der Plas, T.R. Koscik, L. Gutmann, and S.J. Cumming report no disclosures. D.G. Monckton in the last 3 years has been a scientific consultant and/or received an honoraria or stock options from AMO Pharma, Charles River, Vertex Pharmaceuticals, Triplet Therapeutics, LoQus23, and Small Molecule RNA; is on the Scientific Advisory Board of the Myotonic Dystrophy Foundation; is a scientific advisor to the Myotonic Dystrophy Support Group; and is a vice president of Muscular Dystrophy UK. P.C. Nopoulos reports no disclosures. Go to Neurology.org/NG for full disclosures.
References
- 1.Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992;69:385. [DOI] [PubMed] [Google Scholar]
- 2.Harper PS. Myotonic Dystrophy. Kent, UK: Bailliere Tindall; 2001. [Google Scholar]
- 3.Smith CA, Gutmann L. Myotonic dystrophy type 1 management and therapeutics. Curr Treat Options Neurol 2016;18:52. [DOI] [PubMed] [Google Scholar]
- 4.Mahadevan MS, Amemiya C, Jansen G. Structure and genomic sequence of the myotonic dystrophy (DM kinase) gene. Hum Mol Genet 1993;2:299–304. [DOI] [PubMed] [Google Scholar]
- 5.Thornton CA. Myotonic dystrophy. Neurol Clin 2014;32:705–719, viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner C, Hilton-Jones D. Myotonic dystrophy: diagnosis, management and new therapies. Curr Opin Neurol 2014;27:599–606. [DOI] [PubMed] [Google Scholar]
- 7.Bosco G, Diamanti S, Meola G; DM-CNS Group. Workshop Report: consensus on biomarkers of cerebral involvement in myotonic dystrophy, 2-3 December 2014, Milan, Italy. Neuromuscul Disord 2015;25:813–823. [DOI] [PubMed] [Google Scholar]
- 8.Bugiardini E, Meola G; DM-CNS Group. Consensus on cerebral involvement in myotonic dystrophy: workshop report: May 24–27, 2013, Ferrere (AT), Italy. Neuromuscul Disord 2014;24:445–452. [DOI] [PubMed] [Google Scholar]
- 9.Antonini G, Soscia F, Giubilei F, et al. Health-related quality of life in myotonic dystrophy type 1 and its relationship with cognitive and emotional functioning [online]. J Rehabil Med 2006;38:181–185. 10.1080/16501970500477967. [DOI] [PubMed] [Google Scholar]
- 10.Axford MM, Pearson CE. Illuminating CNS and cognitive issues in myotonic dystrophy: workshop report. Neuromuscul Disord 2013;23:370–374. [DOI] [PubMed] [Google Scholar]
- 11.Rakocevic-Stojanovic V, Peric S, Madzarevic R, et al. Significant impact of behavioral and cognitive impairment on quality of life in patients with myotonic dystrophy type 1. Clin Neurol Neurosurg 2014;126:76–81. [DOI] [PubMed] [Google Scholar]
- 12.Musova Z, Mazanec R, Krepelova A, et al. Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am J Med Genet A 2009;149A:1365–1374. [DOI] [PubMed] [Google Scholar]
- 13.Braida C, Stefanatos RKA, Adam B, et al. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum Mol Genet 2010;19:1399–1412. [DOI] [PubMed] [Google Scholar]
- 14.Botta A, Rossi G, Marcaurelio M, et al. Identification and characterization of 5’ CCG interruptions in complex DMPK expanded alleles. Eur J Hum Genet 2017;25:257–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dryland PA, Doherty E, Love JM, Love DR. Simple repeat-primed PCR analysis of the myotonic dystrophy type 1 gene in a clinical diagnostics environment [online]. J Neurodegener Dis 2013;2013:857564 10.1155/2013/857564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santoro M, Masciullo M, Silvestri G, Novelli G, Botta A. Myotonic dystrophy type 1: role of CCG, CTC and CGG interruptions within DMPK alleles in the pathogenesis and molecular diagnosis. Clin Genet 2017;92:355–364. [DOI] [PubMed] [Google Scholar]
- 17.Santoro M, Masciullo M, Pietrobono R, et al. Molecular, clinical, and muscle studies in myotonic dystrophy type 1 (DM1) associated with novel variant CCG expansions. J Neurol 2013;260:1245–1257. [DOI] [PubMed] [Google Scholar]
- 18.Cumming SA, Jimenez-Moreno C, Okkersen K, et al. Genetic determinants of disease severity in the myotonic dystrophy type 1 OPTIMISTIC cohort. Neurology 2019;93:e995–e1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Overend G, Légaré C, Mathieu J, Bouchard L, Gagnon C, Monckton DG. Allele length of the DMPK CTG repeat is a predictor of progressive myotonic dystrophy type 1 phenotypes. Hum Mol Genet 2019;28:2245–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cumming SA, Hamilton MJ, Robb Y, et al. De novo repeat interruptions are associated with reduced somatic instability and mild or absent clinical features in myotonic dystrophy type 1. Eur J Hum Genet 2018;26:1635–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pešović J, Perić S, Brkušanin M, Brajušković G, Rakočević-Stojanović V, Savić-Pavićević D. Repeat interruptions modify age at onset in myotonic dystrophy type 1 by stabilizing DMPK expansions in somatic cells. Front Genet 2018;9:601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Plas E, Hamilton MJ, Miller JN, et al. Brain structural features of myotonic dystrophy type 1 and their relationship with CTG repeats. J Neuromuscul Dis 2019;6:321–332. 10.3233/JND-190397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gomes-Pereira M, Bidichandani SI, Monckton DG. Analysis of unstable triplet repeats using small-pool polymerase chain reaction. Methods Mol Biol 2004;277:61–76. [DOI] [PubMed] [Google Scholar]
- 24.Mathieu J, Boivin H, Meunier D, Gaudreault M, Bégin P. Assessment of a disease-specific muscular impairment rating scale in myotonic dystrophy. Neurology 2001;56:336–340. [DOI] [PubMed] [Google Scholar]
- 25.Nakagawa S, Schielzeth H. A general and simple method for obtaining R2 from generalized linear mixed-effects models. Methods Ecol Evol 2013;4:133–142. [Google Scholar]
- 26.Morales F, Couto JM, Higham CF, et al. Somatic instability of the expanded CTG triplet repeat in myotonic dystrophy type 1 is a heritable quantitative trait and modifier of disease severity [online]. Hum Mol Genet 2012;21:3558–3567. 10.1093/hmg/dds185. [DOI] [PubMed] [Google Scholar]
- 27.Gomes-Pereira M, Monckton DG. Chemical modifiers of unstable expanded simple sequence repeats: what goes up, could come down. Mutat Res 2006;598:15–34. [DOI] [PubMed] [Google Scholar]
- 28.Gomes-Pereira M, Hilley JD, Morales F, Adam B, James HE, Monckton DG. Disease-associated CAG·CTG triplet repeats expand rapidly in non-dividing mouse cells, but cell cycle arrest is insufficient to drive expansion. Nucleic Acids Res 2014;42:7047–7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmidt MHM, Pearson CE. Disease-associated repeat instability and mismatch repair. DNA Repair 2016;38:117–126. [DOI] [PubMed] [Google Scholar]
- 30.Eichler EE, Holden JJ, Popovich BW, et al. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat Genet 1994;8:88–94. [DOI] [PubMed] [Google Scholar]
- 31.Chong SS, McCall AE, Cota J, et al. Gametic and somatic tissue–specific heterogeneity of the expanded SCA1 CAG repeat in spinocerebellar ataxia type 1. Nat Genet 1995;10:344–350. [DOI] [PubMed] [Google Scholar]
- 32.Choudhry S, Mukerji M, Srivastava AK, Jain S, Brahmachari SK. CAG repeat instability at SCA2 locus: anchoring CAA interruptions and linked single nucleotide polymorphisms. Hum Mol Genet 2001;10:2437–2446. [DOI] [PubMed] [Google Scholar]
- 33.Gao R, Matsuura T, Coolbaugh M, et al. Instability of expanded CAG/CAA repeats in spinocerebellar ataxia type 17. Eur J Hum Genet 2008;16:215–222. [DOI] [PubMed] [Google Scholar]
- 34.Pešović J, Perić S, Brkušanin M, Brajušković G, Rakočević-Stojanović V, Savić-Pavićević D. Molecular genetic and clinical characterization of myotonic dystrophy type 1 patients carrying variant repeats within DMPK expansions. Neurogenetics 2017;18:207–218. [DOI] [PubMed] [Google Scholar]
- 35.Caillet-Boudin ML, Fernandez-Gomez FJ, Tran H, Dhaenens CM, Buee L, Sergeant N. Brain pathology in myotonic dystrophy: when tauopathy meets spliceopathy and RNAopathy. Front Mol Neurosci 2014;6:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jinnai K, Mitani M, Futamura N, Kawamoto K, Funakawa I, Itoh K. Somatic instability of CTG repeats in the cerebellum of myotonic dystrophy type 1. Muscle Nerve 2013;48:105–108. [DOI] [PubMed] [Google Scholar]
- 37.Okkersen K, Jimenez-Moreno C, Wenninger S, et al. OPTIMISTIC consortium. Cognitive behavioural therapy with optional graded exercise therapy in patients with severe fatigue with myotonic dystrophy type 1: a multicentre, single-blind, randomised trial. Lancet Neurol 2018;17:671–680. [DOI] [PubMed] [Google Scholar]
- 38.Laberge L, Bégin P, Dauvilliers Y, et al. A polysomnographic study of daytime sleepiness in myotonic dystrophy type 1. J Neurol Neurosurg Psychiatry 2009;80:642–646. [DOI] [PubMed] [Google Scholar]
- 39.O'Donoghue FJ, Borel JC, Dauvilliers Y, Levy P, Tamisier R, Pépin JL. Effects of 1-month withdrawal of ventilatory support in hypercapnic myotonic dystrophy type 1: NIV in myotonic dystrophy type 1. Respirology 2017;22:1416–1422. [DOI] [PubMed] [Google Scholar]
- 40.Baldanzi S, Bevilacqua F, Lorio R, et al. Disease awareness in myotonic dystrophy type 1: an observational cross-sectional study. Orphanet J Rare Dis 2016;11:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langbehn KE, van der Plas E, Moser DJ, Long JD, Gutmann L, Nopoulos PC. Cognitive function and its relationship with brain structure in myotonic dystrophy type 1. J Neurosci Res Epub 2020 Feb 13. 10.1002/jnr.24595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data will be shared by request from any qualified investigator.