

Abstract
Genetic testing for hypertrophic cardiomyopathy (HCM) is an established clinical technique, supported by 30 years of research into its genetic etiology. Although pathogenic variants are often detected in patients and used to identify at‐risk relatives, the effectiveness of genetic testing has been hampered by ambiguous genetic associations (yielding uncertain and potentially false‐positive results), difficulties in classifying variants, and uncertainty about genotype‐negative patients. Recent case‐control studies on rare variation, improved data sharing, and meta‐analysis of case cohorts contributed to new insights into the genetic basis of HCM. In particular, although research into new genes and mechanisms remains essential, reassessment of Mendelian genetic associations in HCM argues that current clinical genetic testing should be limited to a small number of validated disease genes that yield informative and interpretable results. Accurate and consistent variant interpretation has benefited from new standardized variant interpretation guidelines and innovative approaches to improve classification. Most cases lacking a pathogenic variant are now believed to indicate non‐Mendelian HCM, with more benign prognosis and minimal risk to relatives. Here, we discuss recent advances in the genetics of HCM and their application to clinical genetic testing together with practical issues regarding implementation. Although this review focuses on HCM, many of the issues discussed are also relevant to other inherited cardiac diseases.
Keywords: genetic association, genetic testing, hypertrophic cardiomyopathy
Subject Categories: Genetic, Association Studies; Cardiomyopathy; Hypertrophy; Diagnostic Testing
Nonstandard Abbreviations and Acronyms
- ACMG
American College of Medical Genetics
- AMP
Association of Molecular Pathologists
- CNV
copy number variant
- DCM
dilated cardiomyopathy
- DEff
diagnostic effectiveness
- ESP
National Heart, Lung and Blood Institute Exome Sequencing Project
- ExAC
Exome Aggregation Consortium
- GWAS
genome‐wide association study
- HCM
hypertrophic cardiomyopathy
- LMM
Partners Healthcare Laboratory for Molecular Medicine
- LV
left ventricular
- LVOTO
left‐ventricular outflow tract obstruction
- LVWT
left‐ventricular wall thickness
- NCBI
National Center for Biotechnology Information
- NGS
next‐generation sequencing
- SCD
sudden cardiac death
- VUS
variant of uncertain significance
- WES
whole‐exome sequencing
- WGS
whole‐genome sequencing
Hypertrophic cardiomyopathy (HCM) is an inherited disease of cardiac muscle characterized by substantial heterogeneity in morphology, clinical manifestation, genetic etiology, and outcome.1 HCM is diagnosed in the presence of a maximum left‐ventricular wall thickness of at least 15 mm when this is not solely explained by abnormal loading conditions (such as hypertension and aortic stenosis), with other typical features including myocyte disarray and interstitial fibrosis.2 Although HCM is considered a Mendelian disease with largely autosomal dominant inheritance, incomplete penetrance and variable expressivity are commonly observed in affected families.
As well as being a relatively common (estimated to affect 1 in 500 people)2, 3, 4 and medically important condition, HCM has been an important model disease for studying issues that are shared among many inherited cardiac diseases. Research into the genetic basis of HCM has been ongoing for almost 30 years and early implementation of clinical genetic testing has yielded extensive data sets for use in research studies. However, several issues have limited the impact of HCM genetic testing in clinical practice:
Over half of patients tested have a negative genetic test result (ie, no definitive or putative pathogenic variant is detected).
The significance of many detected variants has been difficult to ascertain.
The limited correlation between genotype and clinical phenotype has constrained the use of genetic knowledge in directing clinical management.
The ever‐increasing number of genes apparently associated with disease has clouded our understanding of the genetic architecture of HCM.
Despite these open challenges, recent findings have transformed our understanding of the genetic basis of HCM and offered the potential for more accurate, sensitive, and impactful application of genetic testing. These findings have been driven by the development of population genetic databases that have revolutionized our understanding of rare genetic variation in health and disease and by the accumulation of large patient cohorts and disease registries. Collaborative models of data sharing, together with the development of consensus guidelines for variant interpretation and validation of disease‐gene associations, have proven invaluable for improving our knowledge of rare genetic diseases.
Here, we discuss the achievements earned in understanding the genetics of HCM in the past 30 years, the clinical implications of the current reassessment of genetic associations, the need to manage the genotype‐negative HCM patient in a coherent and effective manner, and the challenges that need to be addressed before we can adopt a patient‐specific, individualized approach in HCM. While focusing on HCM, the issues discussed are also broadly relevant to our understanding of other cardiomyopathies and inherited cardiac diseases.
Genetic Associations
HCM was first recognized over 50 years ago as a familial myocardial disease with increased risk for sudden death, variable disease expressivity, and natural history.5, 6 After more than 2 decades during which the cause of HCM remained unknown, the dawn of a molecular era arrived in 1989, when the first genetic locus (14q1) associated with the condition was detected by means of a linkage analysis on a large kindred group comprising 96 individuals and spanning 4 generations.7 Shortly after, the associated genomic region was narrowed further by additional linkage studies on the same family, restricting the region containing the genetic defect to locus 14q11‐12,8 and consequently defining a potential causal role for one of the two highly homologous genes encoding myosin heavy chains: MYH6 and MYH7. The responsible gene was identified the same year as MYH7, with the identification of the causative missense substitution p.Arg403Glu.9 The same month, another study from the same research group was published to demonstrate the genetic heterogeneity of HCM, as co‐segregation of variants at the MYH7 locus with HCM was observed in only 2 of 4 affected pedigrees, highlighting the existence of alternative genetic causes at other loci.10
After these pivotal discoveries, the genetic repertoire of HCM was gradually expanded throughout the decade to include seven additional sarcomeric genes (MYBPC3, TNNT2, TPM1, MYL2, MYL3, TNNI3, ACTC1)7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 (Table 1), incontrovertibly associated with HCM by studies based on linkage analyses in large and/or multiple affected families and functional experiments. We refer to these eight “core” disease genes with the term “validated genes” in this review. Simultaneously, definitive genetic associations with genocopies of HCM (ie, metabolic diseases characterized by a phenotype mimicking HCM but caused by genetic variants in different genes) were demonstrated, starting with Pompe disease20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33 (Table 2). Besides Pompe disease, Fabry disease, PRKAG2‐cardiomyopathy, Danon disease, and TTR‐amyloidosis, other conditions (due to mitochondrial dysfunctions) can seldom present resembling isolated HCM, such as Friedreich's ataxia and Leigh syndrome, caused by variants in the FXN and COX15 genes, respectively.
Table 1.
Breakdown of the Original Linkage Studies Demonstrating the Co‐Segregation of Genetic Variants and Hypertrophic Cardiomyopathy in Large Pedigrees and Incontrovertibly Associating Sarcomeric Genes With HCM
| Gene | Protein | Demonstrated Associations | Year | Reference No. | Inheritance | N Pedigrees—(Total Size) | Max LOD | Notes |
|---|---|---|---|---|---|---|---|---|
| MYH7 | Beta‐myosin heavy chain | Locus 14q1 | 1989 | 7 | AD | 1 (96) | 9.37 | ··· |
| Locus 14q11‐12 | 1990 | 8 | 1 (96) | 4.62 | ··· | |||
| Gene | 1990 | 9 | 1 (96) | 15.9 | ··· | |||
| Genetic heterogeneity of HCM | 1990 | 10 | 4 (173) | 10.85 | ··· | |||
| TNNT2 | Cardiac troponin T | Locus 1q3 | 1993 | 11 | AD | 3 (97) | 8.47 | ··· |
| Gene | 1994 | 12 | 1 (70) | 6.3 | ··· | |||
| MYBPC3 | Myosin‐binding protein C | Locus 11p13‐q13 | 1993 | 13 | AD | 1 (54) | 4.98 | ··· |
| Gene | 1995 | 14 | 2 (46) | 3.74 | ··· | |||
| TPM1 | Alpha tropomyosin | Locus 15q2 | 1993 | 15 | AD | 2 (87) | 6.02 | ··· |
| Gene | 1994 | 12 | 2 (87) | 6.94 | ··· | |||
| MYL3 | Essential myosin light chain 3 | Gene | 1996 | 16 | AD | 1 (53) | 6.2 | ··· |
| TNNI3 | Cardiac troponin I | Gene | 1997 | 17 | AD | 1 (18) | 3.1 | ··· |
| MYL2 | Regulatory myosin light chain 2 | Gene | 1998 | 18 | AD | 3 (47) | 2.41 (estimated) | a |
| ACTC1 | Alpha actin (cardiac muscle) 1 | Gene | 1999 | 19 | AD | 1 (22) | 3.6 | ··· |
AD indicates autosomal dominant; and HCM, hypertrophic cardiomyopathy.
Although an LOD score of 2.41 is below the universally accepted threshold of LOD=3 for co‐segregation to be considered unequivocal, in the years following this original association with HCM, further evidence about the gene's disease‐causing role in HCM gradually accumulated20, 21, 22, and collectively made the association incontrovertible. A larger family with HCM due to a pathogenic variant in MYL2 was reported recently, with a LOD score for co‐segregation of 4.51.23
Table 2.
List of the Genetic Associations With Metabolic/Infiltrative Genocopies of HCM, Originally Demonstrated Through the Collation of Different Types of Evidence (See References and Notes), Alongside the Main Currently Available Treatment Options
| Gene | Protein | Disease | Year(s) | Reference No. | Inheritance | Main Treatment Options | Notes |
|---|---|---|---|---|---|---|---|
| GAA | Glucosidase alpha | Pompe disease | 1986–1990 | 24, 25, 26 | AR | Enzyme‐replacement therapy, noninvasive ventilation | a |
| GLA | Galactosidase alpha | Anderson‐Fabry disease | 1989–1994 | 27, 28, 29 | X | Antiplatelet/anticoagulant agents, enzyme‐replacement therapy, analgesic drugs to relieve neuropathic pain | b |
| LAMP2 | Lysosome‐associated membrane protein 2 | Danon disease | 2004–2007 | 30, 31, 32 | X | ICD implantation | c |
| PRKAG2 |
Protein kinase AMP‐activated Non‐catalytic subunit gamma 2 |
Wolff‐Parkinson‐White syndrome | 2001 | 33 | AD | Antiarrhythmic drugs, ablation | d |
| TTR | Transthyretin | Transthyrethin amyloidosis | 1991–2002 | 34, 35, 36, 37 | AD | Liver/kidney/heart transplantation | e |
Of note, 15 other genes (including RASopathy genes such as PTPN11 and RAF1) have been classified as with ≥ moderate evidence by ClinGen for syndromic conditions where HCM can be seen. RASopathy genes are not included in the “genocopies of HCM” category as diagnostic discrimination between RASopathies and HCM is usually easier, due to the systemic features of the former (although in rare cases they may still resemble isolated HCM). AD indicates autosomal dominant; AR, autosomal recessive; HCM, hypertrophic cardiomyopathy; ICD, implantable cardioverter‐defibrillator; and X, X‐linked.
Other reports38, 39, 40 determined the gene sequence and contributed to show that the disease was due to a lack of alpha‐glucosidase.
Other reports41, 42 determined the gene sequence and contributed to show that the disease was due to a lack of alpha‐galactosidase.
Reports by Danon et al43 and Nishino et al44 described Danon disease as a distinct lysosomal glycogen storage disease and showed that the cause was a deficiency of lysosome‐associated membrane protein 2.
The reported study (Gollob et al33) consisted of a linkage analysis on two pedigrees including 70 individuals, with a LOD score for co‐segregation of variants in PRKAG2 and disease of 9.82.
Improvements in sequencing technologies and their throughput, particularly with the advent of next‐generation sequencing (NGS), revolutionized the ability to investigate the genetic architecture of Mendelian diseases such as HCM. Case‐control rare variant comparisons became the design of choice for most studies, where usually relatively small groups of patients and controls were sequenced on one or a few genes of interest, encoding proteins within or outside the sarcomere, implicated in HCM with varying levels of supportive evidence48, 49 (Figure 1).
Figure 1. Examples of molecular complexes and proteins of the cardiomyocyte encoded by genes associated with sarcomeric HCM, its mimics and syndromic conditions featuring HCM with different levels of supporting evidence.
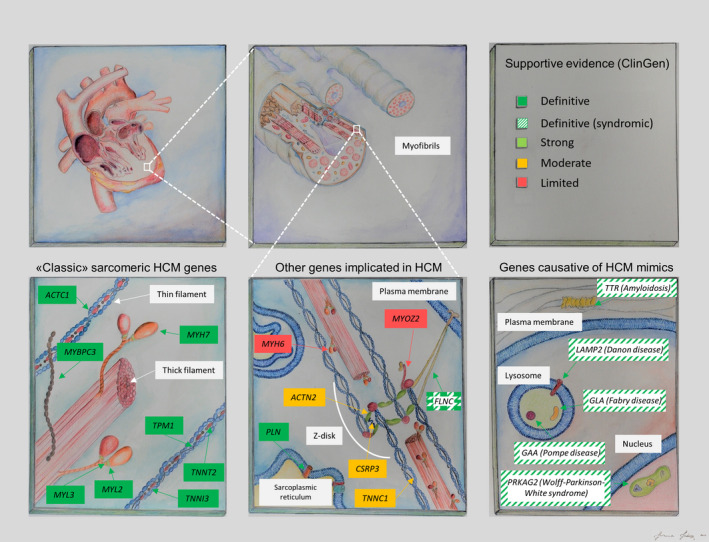
Strength of evidence in favor of pathogenicity (top right panel) is coded as in the curation effort by the ClinGen consortium.48 Details about genetic associations for definitive genes displayed in the bottom left and bottom right panel, and genes with ≥ moderate supportive evidence are provided in Tables 1, 2 through 3. HCM, hypertrophic cardiomyopathy.
Whenever a protein‐altering variant was observed uniquely in patients, the absence of such variants in controls was often used as stand‐alone evidence in support of the claim for pathogenicity, with little convincing segregation or functional evidence in support. Although at the time these approaches were generally considered sufficient to implicate genes in Mendelian disease, it is now recognized that more substantial evidence is necessary to demonstrate association between variants in a given gene and disease.
The consequence of these efforts was an exponential increase in the number of proposed genetic associations in the years between 2000 and 2015 (Figure 2). By 2016, 64 genes had been claimed to be causative in HCM (Human Gene Mutation Database v.2016.3, Table S1) with few of the additional genes being supported by sufficiently comprehensive and robust evidence50, 51, 52, 53, 54, 55, 56, 57 (Table 3).
Figure 2. Number of genes implicated in HCM in the literature with different levels of supporting evidence (black line, left y axis) ranging from fully validated genes (darker green line) to those without substantial evidence supporting pathogenicity.
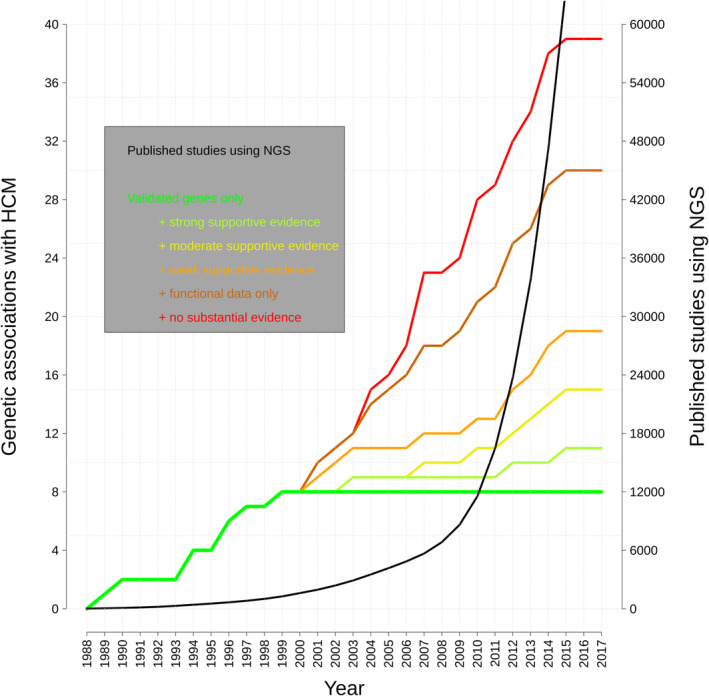
The black line indicates the number of scientific publications in PubMed featuring next‐generation sequencing (colored lines, right y axis). The classification criteria for evidence in favor of pathogenicity (as detailed in the legend) and the genes included (n=39) reflect the analysis and curation effort by Walsh et al.49 HCM, hypertrophic cardiomyopathy; and NGS, next‐generation sequencing.
Table 3.
Other Genes Classified as With Moderate/Strong/Definitive Evidence for Isolated HCM (or Multiple Conditions Including Isolated HCM) by ClinGen,48 or With Convincing Evidence for HCM Causation Published After the ClinGen Curation Effort
| Gene | Protein | Disease | Year | Reference No. | Inheritance | ClinGen Classification | Notes |
|---|---|---|---|---|---|---|---|
| TNNC1 | Troponin C type 1 (slow) | Isolated HCM | 2001 | 36 | AD | Moderate | ··· |
| PLN | Phospholamban | HCM, DCM, and ARVC | 2003 | 37 | AD | Definitive | ··· |
| CSRP3 | Cysteine and glycine‐rich protein 3 (cardiac LIM protein) | Isolated HCM | 2003 | 38 | AD | Moderate | ··· |
| JPH2 | Junctophilin 2 | Isolated HCM | 2007 | 39 | AD | Moderate | ··· |
| ACTN2 | Actinin, alpha 2 | HCM, LVH, LVNC, DCM, idiopathic VF | 2010 | 40 | AD | Moderate | ··· |
| FLNC | Filamin C, gamma | HCM, myofibrillar myopathy | 2014 | 41 | AD | Definitive | ··· |
| ALPK3 | Alpha‐kinase 3 | HCM, DCM (infant‐onset) | 2016 | 42 | AR | Strong | ··· |
| FHOD3 | Formin homology 2 domain containing 3 | HCM | 2018 | 43 | AD | ··· | a |
The year and reference reported refer to the main publication in which a disease‐causing role in HCM was proposed. AD indicates autosomal dominant; ARVC, arrhythmogenic right‐ventricular cardiomyopathy; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; LVH, left‐ventricular hypertrophy; LVNC, left‐ventricular non compaction; and VF, ventricular fibrillation.
FHOD3 was not curated by ClinGen due to the later publication of the reported study (Ochoa et al57). Authors demonstrate a significant excess of rare variants in FHOD3 compared with controls and provide evidence for co‐segregation of FHOD3 variants with HCM using 17 pedigrees (combined LOD score=7.92).
Population databases of whole‐exome sequencing (WES)—and in part, whole‐genome sequencing (WGS)—such as the 1000 Genomes Project58 (≈2500 individuals) and National Heart, Lung, and Blood Institute Exome Sequencing Project59 (ESP, ≈6500 individuals) enabled early observations that many variants previously associated with cardiomyopathies were instead likely benign, as their population frequencies were incompatible with the prevalence of disease.60, 61 The definitive proof of this arrived in October 2014 with the release of the Exome Aggregation Consortium (ExAC) data set, a large collation of WES data from over 60 000 individuals,62 in which the average individual was shown to carry ≈54 variants previously reported as pathogenic in widely used databases of disease‐causing variants.
In the case of HCM and dilated cardiomyopathy (DCM), large‐scale analysis based on ExAC demonstrated that the frequency of rare variants found in many genes implicated in the condition was comparable to background population variation,49, 63, 64 casting doubts on the veracity of many purported associations (Figure 2). In addition, a number of independent disease‐ and gene‐specific approaches have recently been developed to aid decisions on which genes to include in clinical diagnostic panels. One recent example is the diagnostic effectiveness (DEff),65 a gene‐ and disease‐specific metric combining measures of actionability for cascade screening and prior likelihood of pathogenicity of variation detected in patients. DEff scores of validated and putative disease genes can be compared, in order to assist the decision on which of the latter should be routinely assessed in clinical practice and which would yield, at most, inconclusive results (variants of uncertain significance).
Rigorous, disease‐specific gene curation efforts that assess all available lines of evidence for disease association are now essential for genetically heterogeneous Mendelian diseases like HCM. In spite of the lack of a standardized approach to weight different types of evidence for gene‐disease associations, results obtained by curation/classification efforts are characterized by general concordance, with the 8 validated genes unanimously considered as incontrovertibly associated with HCM, and genes such as PLN and CSRP3 supported by strong evidence in favor of HCM pathogenicity.48, 49, 65 The ClinGen initiative66 (https://clinicalgenome.org/)—founded to define the clinical relevance of genes and genetic variants—has recently established a framework to systematically assess all gene‐disease claimed associations’ veracity and their clinical validity based on published evidence. The curation effort for HCM, assessing >50 genes, has recently classified evidence in support of their role in HCM as “definitive” for the 8 validated sarcomeric genes, PLN and FLNC (also associated with myofibrillar myopathy), “strong” for ALPK3 and “moderate” for ACTN2, CSRP3, TNNC1 and JPH2 (Tables 1 and 3).48 Other 20 genes were assigned a ≥ moderate evidence classification for their role in genocopies of HCM (Table 2) or syndromes involving HCM.
Genetic Testing Strategies
The primary purpose of genetic testing in HCM is to facilitate cascade screening in relatives of the index patient in order to identify those at risk and those not at risk of developing disease. As guidelines currently recommend regular clinical evaluation for first‐degree relatives of all HCM patients,2 discharging those relatives lacking identified pathogenic variants offers obvious psychological benefits for these individuals and significant cost savings for healthcare providers. Genetic testing for HCM has been shown to be more cost effective than clinical screening alone in UK67 and Australian68 studies, and the Partners Laboratory for Molecular Medicine (LMM) estimated savings of USD 700 000 (based on Medicare rates) from discharging genotype‐negative relatives of 2912 HCM probands.69 Ongoing reductions in the costs of DNA sequencing, and improvements in variant analysis as described subsequently, will further support the economic case for HCM genetic testing. It has also been recognized that genetic testing in probands can increase the often low take‐up in clinical screening from at‐risk relatives.70
In contrast, knowledge of the causative genetic variant does not currently affect the clinical management for most HCM patients and is not included in sudden cardiac death (SCD) risk assessment models.71, 72 The exception to this is identifying patients with pathogenic variants in a small group of genes—such as GAA, GLA, LAMP2, PRKAG2 and TTR—that are incontrovertibly associated with metabolic diseases that mimic HCM (Figure 1 and Table 2) but have distinctive clinical profiles, inheritance patterns, and treatment options.73 The inclusion of these genes in diagnostic panels for HCM is advantageous given their pronounced phenotypic similarity with the classic sarcomeric form74 and the importance of a prompt differential diagnosis, enabling correct treatment decisions and optimal patient management and de facto representing a significant step toward more personalized approaches.
Despite limited evidence for a causal role in HCM, many of the non‐sarcomeric genes implicated in the disease have been progressively included in panels developed by clinical laboratories and commercial providers75 (listed in the National Center for Biotechnology Information Genetic Testing Registry: https://www.ncbi.nlm.nih.gov/gtr/). The uncertainty around the pathogenicity of these “genes of uncertain significance” in HCM has led to difficulty for clinical diagnostic laboratories in choosing which of the ambiguous genes to include in routinely used diagnostic panels. The choice ultimately lies on the spectrum between the extremes of including only the validated genes at one end (bottom left and bottom right panels in Figure 1) and systematically screening all genes implicated in HCM at the other, irrespective of the strength of evidence in support of their causal role for HCM. The former, conservative choice plausibly leads to the possibility of false negative genetic tests but the latter, inclusive option, bears greater potential for false positive results, which can be detrimental for the patients and their family members, and increases the uncertainty associated with testing through an epidemic of variants of unknown significance (VUS). Unsurprisingly, expanding HCM panels to include genes of questionable pathogenicity has had little effect on the sensitivity of genetic testing. For example, LMM reported identifying only one pathogenic variant in 632 cases (in the PLN gene) outside of the 8 core sarcomeric genes and metabolic cardiomyopathy genes.69
Ongoing reductions in the cost of sequencing have meant that WGS (complete sequencing of the entire genome in an individual) and WES (sequencing of the 1% of the genome that encodes proteins) may become feasible options as first‐line sequencing assays in the near future. However, there is limited evidence that WGS and WES are currently cost effective in clinical practice.76 For example, a recent study that was part of the Medseq initiative to evaluate the clinical potential of WGS77 compared the performance of a targeted gene panel and WGS in 41 HCM patients. WGS identified 19 of the 20 variants of interest detected by panel sequencing, but missed a 18 bp duplication in MYBPC3 due to low coverage and only additionally identified a causative variant in a RASopathy gene (not in the panel) as well as several VUS and secondary findings. The limited value of WGS, WES, or super‐sized gene panels for standard HCM genetic testing, that is, adult‐onset and/or autosomal dominant disease in the absence of large family pedigrees, is clear when considering current variant interpretation guidelines for clinical testing and the requirements for defining a variant as actionable (see next section). It will be impossible to classify a variant in a novel gene as clinically actionable in the vast majority of cases, regardless of how plausible that candidate gene may be, as the guidelines require as a minimum that the gene or variant class has a proven role in the disease.
In contrast, WGS/WES can offer some advantages as diagnostic sequencing assays if testing is restricted to genes and regions that have been validated for the disease in question. In this “virtual panel testing” approach, analysis is restricted to only those genes that can yield interpretable variants, but the extensive sequencing enables reanalysis of archived data as new and validated gene‐disease associations are published, as well as potentially generating comprehensive and useful data sets for research purposes. One advantage of WGS compared with approaches targeting only protein‐coding regions of genes (eg, targeted gene panel and WES) is its enhanced ability to identify variants classes such as copy number variants (CNVs)—deletions or duplications of whole exons, genes, or chromosomal regions—and variants in noncoding regions of the genome such as introns. Studies exploring the role of CNVs in HCM have found such variation in ≈1% of cases,78, 79, 80 although detecting, validating, and interpreting CNVs remains a challenging and nonstandardized task. A recent study used WGS to identify deep intronic variants in MYBPC3 in 4 HCM patients who were negative for standard genetic testing, and these variants were shown to affect splicing and produce truncated transcripts.81 The contribution of these noncoding variants that affect gene dosage is likely to be particularly relevant to genes where the underlying disease mechanism is haploinsufficiency (ie, where the loss of half of the physiological amount of functional protein causes disease). This is often the case for genes in which protein‐truncating variants are the most prevalent pathogenic variant class although, in the case of MYBPC3, haploinsufficiency has also been proposed as the molecular mechanism for several missense variants.82 For most of the other major sarcomeric HCM genes where variants act in a dominant negative manner, such noncoding variants are expected to be less relevant.
In addition to these scientific pros and cons, other factors such as affordability and the quality and technical features of generated data are also important factors in choosing a sequencing platform for genetic testing.83, 84 As highlighted previously, depth of coverage for affordable WGS/WES in the diagnostic setting may currently be inferior to panel sequencing and lead to false negative findings—this is particularly relevant for WES assays as some exons, especially in GC‐rich regions, are not adequately captured.85 It is also important to recognize the extra burden of data processing, analysis, and storage associated with WGS/WES, as well as ethical issues relating to secondary and incidental findings if analysis is not restricted to valid disease genes for the primary condition.
At present, the primary role for unbiased WGS/WES lies in the research setting in order to identify putative causative variants outside of known HCM genes in cases with an inheritance pattern that enables triage and prioritization of rare variants and where the genetic cause of disease remains elusive after initial genetic testing. These cases include autosomal dominant HCM in large and clinically defined family pedigrees, although such cases are rarely encountered during routine clinical genetic testing. More commonly, novel genes underlying recessive or de novo inheritance in pediatric cases can be identified through sequencing of the patient and their unaffected parents (trio analysis) as well as other family members if available. If a definitive genetic cause is identified, such a research finding is of direct diagnostic utility. These approaches have recently identified recessive truncating variants in the ALPK3 gene as pathogenic in several pediatric cardiomyopathy cases.56 Taking all of these issues into consideration, an effective tiered approach to genetic testing in HCM is shown in Figure 3.
Figure 3. Proposal for a tiered approach to genetic testing in HCM. Initial testing is restricted to validated HCM genes using either a targeted gene panel or a virtual panel by focused analysis of WGS or WES data.
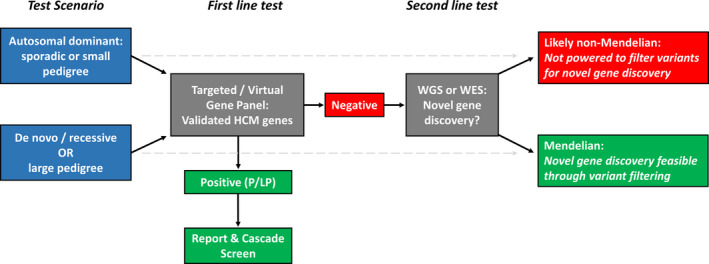
The latter allows for reanalysis of genes subsequently linked to HCM, and WGS can detect additional variant classes including CNVs and deep intronic splice variants, though sequencing coverage is likely to be inferior to targeted panel sequencing. In the case of negative results, variant prioritization in the research setting can aid identification of the causative variant, directly translating into a diagnostic finding. The likelihood of detecting variation in novel genes with broader WGS/WES analysis depends on the profile of the patient and family being tested. In general, large pedigrees informative for segregation and trios (affected child with unaffected parents, indicating likely de novo variant occurrence or recessive inheritance) can enable filtering to a manageably small number of potentially causative variants for detailed evaluation. CNV, copy number variant; HCM, hypertrophic cardiomyopathy; WES, whole‐exome sequencing; and WGS, whole‐genome sequencing.
Analysis should be initially focused on validated HCM genes, regardless of the sequencing assay applied (gene panel or focused analysis of WGS/WES). If negative, the decision to expand analysis to novel genes (with WGS or WES) would depend on the profile of the patient being tested and the likelihood of isolating the causative variant from genome‐wide sequencing. For most adult probands, it is unlikely that pathogenic variants will be identified in genes not previously associated with HCM. In any case, as described subsequently, it is increasingly likely that most such patients represent non‐Mendelian and nonfamilial HCM.
Interpretation of Genetic Findings
Determination of the clinical significance of genetic variants is a complex process involving the evaluation and weighting of several lines of evidence. Accurate interpretation is key, given the potentially detrimental consequences that an erroneous interpretation can have on the proband and their relatives.86, 87 The American College of Medical Genetics and the Association of Molecular Pathologists (ACMG/AMP) first issued standardized recommendations on the interpretation of sequence variation almost 2 decades ago,88 and a series of updated and increasingly articulated guidelines have been issued in the following years, in parallel with the establishment and the exponential growth of clinical and population sequence variation databases (Figure 4).
Figure 4. Variant interpretation guidelines issued by the ACMG/AMP and independent laboratories/societies/associations (below the timeline) and some of the main resources contributing to a finer interpretation of sequence variants (above the timeline) as they became available.
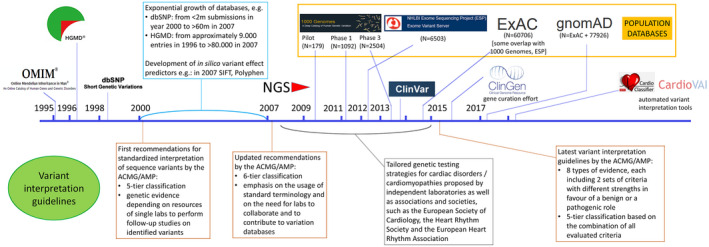
Several sequence variation databases with a focus on variants’ role in disease were released in time, particularly in the late 1990s (eg, the Human Gene Mutation Database in 1996) but also in more recent years (eg, ClinVar in 2013). The advent of cost‐effective, high‐throughput NGS in the years between 2005 and 2010 enabled the establishment of progressively large genome‐wide population variation databases, starting with the Pilot data set of the 1000 Genomes Project in 2009. All these increasingly complex and large‐scale resources on one hand allow a much more detailed characterization of genetic variants, but on the other contribute to the constant growth in the variability and quantity of data types and information on single variants, variant classes, and genes and require increasingly articulated guidelines for variant interpretation. Several efforts are in place to finely tune variant interpretation in a gene‐ and disease‐specific manner (eg, gene curation efforts such as ClinGen,80 established in 2015) and to render interpretation easier and quicker for the geneticist with automated variant interpretation tools (eg, CardioClassifier92 and CardioVAI,93 that interpret variants in the context of cardiomyopathies). ACMG, American College of Medical Genetics; AMP, Association for Molecular Pathology; and NGS, next‐generation sequencing.
The increasing complexity of variant interpretation and the extent of misclassification in previous years underscored the need for more stringent and comprehensive approaches to variant classification, prompting the release of the current ACMG/AMP guidelines in 201589 (Figure 4). These guidelines, considerably more complex than previous recommendations, are designed to be applicable to variants in all Mendelian genes, and to serve as a basis for more detailed and focused implementation for specific genes and diseases. As a result, several rules embedded in the current interpretation framework avoid providing specific instructions on how to weight certain classes of evidence, inevitably giving rise to differences in approaches adopted by different laboratories and discordant variant classification.90, 91 For this reason, besides the development of automated, guideline‐based variant interpretation tools,92, 93 the implementation of consensus guideline adaptations is now an active area of research, both for different evidence categories and for specific disorders or gene‐disease pairs. Examples include proposals for weighting the evidence of co‐segregation of variants with disease in pedigrees94 and for a more finely tuned interpretation of loss‐of‐function variants,95 quantitative scores estimating the likelihood of pathogenicity based on the variant location within the protein sequence,96 an algorithm to define a disease‐specific population frequency cut‐off for potentially pathogenic variants.97 More complex approaches taking into account the variant position within the three‐dimensional structure of the protein (as opposed to its one‐dimensional sequence) have also been used to identify protein regions enriched in HCM‐causing variants,98 and could augment variant interpretation accuracy if embedded in the variant interpretation framework. For specific genes/diseases, tailored recommendations to interpret genetic variation have been developed for RASopathies99 and for MYH7 variants in HCM,100 with ongoing efforts for other genes and diseases by the ClinGen initiative and other groups. A description of ACMG/AMP guidelines’ rule‐specific application to variants detected in HCM patients, current issues, and ongoing/future developments toward an improved application are provided in Table 4.
Table 4.
Summary of the Main Lines of Evidence Used to Assess the Pathogenicity of Genetic Variants as Described by the ACMG/AMP Variant Interpretation Guidelines, Including Their Specific Application to Variants Detected in HCM Patients, Alongside Current Criteria‐Specific Issues and Ongoing/Future Developments for a More Refined Application
| ACMG/AMP Rule/Evidence Class | Application to HCM | Issues and Future Developments |
|---|---|---|
| PVS1—null/truncating variant in gene with loss‐of‐function mechanism for disease. | Applied to truncating variants in MYBPC3, detected in ≈10% of HCM patients. |
|
| PS1/PM5—same amino acid change/change at same residue as an established pathogenic variant | Numerous established pathogenic HCM variants and many examples of different variants affecting same residues. |
|
| PS3—proven deleterious effect with functional studies. | Animal or cell‐based studies can be used to assess the phenotypic effect of a variant detected in a patient. |
|
| PS4—variant is significantly enriched in cases compared with controls. | There are numerous founder and recurrent pathogenic HCM variants that are observed in multiple HCM probands/families. Comparison with control or population data sets can identify significantly enriched variants. |
|
| PM1/PP2—relative frequency of variants in cases and controls for genes or gene regions. | All sarcomeric genes enriched for rare variants in HCM, with several mutation hotspots, eg, MYH7 head domain. |
|
| PM2—variant is rare enough in the population to be plausibly pathogenic (also BA1, BS1, BS2). | Population frequency data from gnomAD and disease‐specific threshold based on HCM characteristics provide stringent variant rarity threshold.97 |
|
| PP1—segregation of variant with disease in family pedigrees. | Segregation evidence is available for many HCM‐causing variants (in literature and ClinVar). Strength applied to evidence depends on the number of informative meioses.94, 100 |
|
| PP3—computational evidence to support a deleterious effect. | As with most other Mendelian diseases, predictions from several different algorithms are used to provide supportive evidence for pathogenicity. |
|
ACMG indicates American College of Medical Genetics; AMP, Association of Molecular Pathologists; and HCM, indicates hypertrophic cardiomyopathy.
The current guidelines are designed to minimize false‐positive results (ie, over‐calling of variant pathogenicity) as it is usually less detrimental for patients and their relatives to receive an inconclusive result in presence of a pathogenic variant than it is to receive a positive result in absence of a real genetic cause. While assigning variants a categorical classification, guidelines essentially follow an underlying Bayesian framework, with a variant considered “likely pathogenic” if its probability of pathogenicity is ≥90%, and “pathogenic” if ≥99%.101 However, the lack of variant‐specific information available to diagnostic laboratories for many rare novel variants often translates into the inability to correctly identify those with a probability of pathogenicity ≥90%, de facto rendering the application of the guidelines very stringent and causing a general under‐calling of pathogenicity.
Although this conservative approach offers the advantage of minimizing false‐positive variant classifications, this inevitably comes at the cost of an increased rate of false negative results (ie, where pathogenic variants are classified as VUS). This drawback is particularly relevant in HCM because of its marked genetic heterogeneity with many variants private to affected families, the fact that most causative variants are missense substitutions which are inherently more difficult to interpret than protein‐truncating variants and occur with higher background rates in controls, and the limitations of functional assays to effectively evaluate their molecular effects unlike, for example, those in genes encoding ion channels. Recent evidence has quantified this false negative rate for HCM, suggesting that ≈8% of HCM patients who undergo clinical genetic testing will receive a reported VUS that is likely to be a pathogenic variant63 (ie, a VUS in one of the 8 sarcomeric genes was detected in 12.4% of HCM patients compared with a background rate of rare variation of 4.3% in the ExAC population data set). In support of this, analysis of the SHaRe registry (https://theshareregistry.org/), comprising genetic data and cardiac morphofunctional parameters for >9000 HCM patients), showed that variants classified as VUS have a measurable clinical impact on outcome,102 highlighting how these include misclassified pathogenic variants.
In order to overcome these obstacles to variant interpretation, knowledge sharing between different centers through databases such as ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) is critical. As an example, the MYH7 p.Arg869His variant had conflicting interpretations in ClinVar (VUS or likely pathogenic) based on the variant characteristics and some published reports. However, this variant is observed at locally high frequencies in northern Tuscany (detected in 30 of 1198 unrelated HCM probands tested at Careggi University Hospital compared with 0 of 356 non‐HCM patients affected with other cardiovascular conditions, P=6.22×10−4), including co‐segregation with disease in 7 HCM patients in a three‐generation family. Based on this evidence (detailed in ClinVar accession SCV001147002.1), the MYH7 p.Arg869His variant can now be classified as definitively pathogenic for HCM and highlights the benefits of sharing findings from different populations and geographic regions.
The role of both public and private diagnostic laboratories is key, as they represent the “hubs” where many novel variants are observed and where patients and their families are referred when a genetic cause of overt or suspected disease is investigated. Many diagnostic laboratories (such as Invitae and GeneDX) comprehensively publish variant interpretation summaries in ClinVar, based on published data and in‐house findings, which helped ClinVar recently reach the milestone of 1 million submissions. Nevertheless, some diagnostic companies still refuse to share data in this manner and instead produce proprietary variant interpretation that takes advantage of public resources such as ClinVar and gnomAD in addition to their own databases. Given the arduous challenge of variant interpretation for genetically heterogeneous diseases like HCM, and the importance of accurate classification in clinical practice, the ethics of such practices are dubious. One possible solution would be for healthcare providers and cardiologists/clinical geneticists to insist on submission to ClinVar when engaging the services of diagnostic laboratories or only employ companies committed to knowledge sharing.
Data sharing initiatives are at the basis of several approaches that aim to tackle the high rate of inconclusive findings in clinical genetic testing. The availability of increasingly large patient and control cohorts may enable the development of quantitative statistical approaches to identify variant classes with high likelihood of pathogenicity. For example, case‐control analyses can estimate these prior probabilities of pathogenicity through metrics such as the odds ratio and etiological fraction.63 A recent study using this approach, which defined regions of genes where case variants are significantly enriched and incorporated this evidence to the ACMG/AMP framework, increased the relative yield of clinically actionable variants in sarcomeric genes by up to 20% in HCM.96 More comprehensive classification algorithms based on ensemble‐ or machine‐learning techniques that integrate different classes of evidence may also plausibly improve the accuracy of variant classification and contribute to lowering the proportion of inconclusive findings in HCM and other diseases.103, 104 Advanced computational techniques like these may face considerable hurdles though with integration into current guidelines as well as the workflows of clinical genetics laboratories.
Assays that accurately assess the functional effects of specific genetic variants, through cell‐based or in vivo models, also offer the possibility of informing the pathogenicity of variants without the need for segregation evidence from large families. However, the lack of effective and standardized translational assays has limited the role for this class of evidence for cardiomyopathies, with the cost and time to implement such assays also rendering them impractical in the context of clinical genetic screening. Novel techniques, in particular those using the CRISPR/Cas9 genome editing technology, are now being applied for distinct alleles in HCM105, 106, 107, 108 and DCM.109 The further development of such approaches into more comprehensive and high‐throughput assays, such as a recent study that functionally evaluated thousands of alleles in BRCA1 110 simultaneously, may be required for such evidence to be routinely incorporated into clinical genetic testing.
Improvements in variant classification are especially relevant to non‐white populations, which are characterized by significantly lower detection rates of (likely) pathogenic variants in cardiomyopathy patients compared with white patients.111 This is likely to result from the fact that the vast majority of research efforts and genetic tests for cardiomyopathies have been carried out on patients and controls of European descent (and therefore recurrent and founder variants occurring in whites are more likely to have been previously observed and characterized). This is emblematic of a wider problem in genetics research, exemplified by the proportion of genome‐wide association studies (GWAS) performed on individuals of European descent (≈85% of the total) being vastly disproportionate to their proportion of the world population (≈15%).112 These observations underscore the urgent need for more genetic research to be undertaken in non‐European populations as well as the further development of functional and quantitative approaches that reduce reliance on the prior observation and characterization of rare pathogenic variants.
The accurate interpretation of variants in HCM genes is equally critical when they have been detected as secondary or incidental findings by WGS or WES in individuals undergoing genetic testing for noncardiac conditions. Each of the 8 core sarcomeric HCM genes are on the list of genes recommended by the ACMG to be analyzed as secondary findings.113 Given the relatively high rate of rare variation in these genes in the population, only those variants with strong evidence of pathogenicity should be considered actionable. However, as many HCM variants display reduced penetrance, clinical evaluation to detect the cardiomyopathy phenotype is essential, including ongoing surveillance screening where appropriate.114
Interpretation of Genotype‐Negative HCM
When variants in sarcomeric genes were first implicated in HCM, there were indications that knowledge of the causative variant might be prognostic for patients and potentially useful for clinical decision‐making. For example, TNNT2 variants were initially shown to be associated with poor prognosis and high incidence of sudden cardiac death (SCD),115 whereas variants in MYBPC3 were related with delayed onset of disease and more favorable outcomes.116 Subsequently, genotype‐phenotype analyses on ever‐larger cohorts of HCM patients have been undertaken to clarify these initial findings and attempt to identify clear and consistent correlations between the pathogenic variant class and the clinical profile and outcomes in patients.
Recent cohort studies have identified some statistically significant differences in phenotype between patients carrying different variant classes. In particular, HCM patients with variants in thin filament genes (TNNT2, TNNI3, TPM1, ACTC1) were shown to have distinctive patterns of left‐ventricular hypertrophy and diastolic profile, as well as lower prevalence of left‐ventricular outflow tract obstruction (LVOTO) and late gadolinium enhancement (a marker of myocardial fibrosis) compared with those with variants in thick filament genes (MYH7, MYBPC3).117 Although patients with variants in thin filament genes were more likely to develop advanced LV dysfunction, no differences were observed in mortality or SCD between thin and thick filament variant‐positive patient groups in this study.117
Studies comparing outcomes between patients with MYH7 and MYBPC3 pathogenic variants (the predominant genes associated with HCM) have yielded conflicting results—no significant differences were observed for 2 moderately sized cohorts118, 119 but in the larger SHaRe registry those with MYH7 variants had a higher risk of advanced heart failure and overall composite outcome.102 Furthermore, patients with complex genotypes carrying 2 pathogenic mutations were at greater risk of adverse outcome and sudden cardiac death, irrespective of the genes involved. Despite these broad correlations, all of these studies have revealed extensive heterogeneity in symptoms, disease severity, and outcome, even among patients with the same or similar pathogenic variants. As a result, the individual patient's genetic status has limited impact on clinical management and is largely limited to cascade screening and to the small subsets of patients with rare storage or metabolic diseases mimicking HCM.
In contrast, clear and consistent differences in demographics, phenotype, and outcomes have been observed when comparing genotype‐positive and genotype‐negative patients, that is, those with and without causative variants in sarcomeric genes102, 118, 120, 121, 122, 123, 124, 125, 126 (Table 5).
Table 5.
Summary of Demographic, Baseline Clinical and Outcome Data for Major Published HCM Studies Comparing Genotype‐Positive and Genotype‐Negative Patients
| Center | SHaRe Registry | Mayo Clinic, USA | UCL, UK | Toronto General Hospital, Canada | Erasmus Medical Centre, Netherlands | Centenary Institute, Australia | Meta‐Analysis (13 Cohorts) |
|---|---|---|---|---|---|---|---|
| Study | Ho et al102 | Bos et al120 | Lopes et al121 | Li et al118 | van Velzen et al125 | Ingles et al126 | Lopes et al124 |
| Cohort size | 4591 | 1053 | 874 | 558 | 512 | 265 | 2459 |
| Genes tested | 8 | 9 | 8 | 8 | Large panel | 10 | ··· |
| Genotype positive, % | 46.3% | 34.1% | 43.8% | 35.5% | 45.7% | 52.1% | ··· |
| Demographics | |||||||
| Age at inclusion | 45.8±14.7 vs 53.1±14.9 | 46±15 vs 55±15 | 47±16 vs 56±17 | ||||
| P<0.001 | P<0.001 | P<0.001 | |||||
| Age at diagnosis | 37.3±17.1 vs 49.0±17.4 | 36.4±17 vs 48.5±18 | 39.5±15.2 vs 48.5±14.8 | 34±17 vs 44±18 | 38.4±10.3 vs 46.0±10.4 | ||
| P<0.001 | P<0.001 | P<0.001 | P<0.001 | P<0.001 | |||
| Sex (% male) | 60.6% vs 66.0% | 58.5% vs 60.4% | 56.1% vs 70.3% | 67.9% vs 61.5% | 53.6% vs 70.9% | 57.5% vs 61.5% | |
| P<0.01 | P=0.6 | P=0.001 | P=0.13 | P=0.005 | P=0.422 | ||
| FH HCM (%) | 57.9% vs 24.5% | 50.4% vs 22.9% | 39.8% vs 15.6% | 52.5% vs 20.0% | 73.9% vs 30.7% | 50.6% vs 23.1% | |
| P<0.001 | P<0.001 | P<0.001 | P<0.001 | P<0.001 | P<0.001 | ||
| FH SCD (%) | 27.0% vs 15.0% | 28.5% vs 15.2% | 16.7% vs 7.2% | 19.7% vs 5.4% | 41.3% vs 6.3% | 27.0% vs 14.9% | |
| P<0.001 | P<0.001 | P=0.002 | P<0.001 | P<0.001 | P<0.001 | ||
| Baseline characteristics | |||||||
| Max LVWT, mm | 19.7±6.2 vs 18.1±5.2 | 22.6±6 vs 20.1±5 | 18.8±4.4 vs 18.1±4.1 | 20.8±4.8 vs 19.6±4.9 | 20±5 vs 18±4 | 22±6 vs 21±5 | 21.0±4.1 vs 19.3±3.5 |
| P<0.001 | P<0.001 | P=0.004 | P=0.16 (adjusted) | P<0.001 | P=0.03 | P=0.03 | |
| Hypertension (%) | 19.2% vs 43.2% | 25.3% vs 46.9% | 22.8% vs 41.7% | ||||
| P<0.001 | P<0.001 | P=0.004 | |||||
| Clinical outcomes—hazard ratio (CI/log rank P value) | |||||||
| Mean follow‐up years | 5.4±6.9 | 6.6±6.3/6.2±5.6 | 12±9 | ||||
| CV mortality | 2.41 (1.73–3.35) (all death) | 3.99 (P=0.001) | 2.82 (P=0.002) | ||||
| HF‐related mortality | 6.33 (P=0.004) | ||||||
| SCD/aborted SCD | 3.44 (P=0.028) | 2.88 (P=0.015) | |||||
| Combined HF events | 1.87 (1.55–2.25) | 4.51 (P<0.001) | |||||
| Overall composite | 1.98 (1.72–2.28) | ||||||
Genotype‐positive status is defined by the presence of a putatively pathogenic variant—the vast majority of these occur in one of the 8 core sarcomeric genes (MYH7, MYBPC3, TNNT2, TNNI3, TPM1, MYL2, MYL3, ACTC1). The meta‐analysis by Lopes et al124 involved 13 previously published cohorts—this included subsets of the cohorts from the Mayo Clinic and Toronto General Hospital that had been published prior to the larger versions of those cohorts described here. The SHaRe registry may include cases from the Erasmus Medical Centre study and the meta‐analysis by Lopes et al124. CI indicates confidence interval; FH, family history; HCM, hypertrophic cardiomyopathy; HF, heart failure; LVWT, left‐ventricular wall thickness; SCD, sudden cardiac death; and UCL, University College London.
Genotype‐positive patients present approximately a decade earlier and unsurprisingly have a greater proportion of family history of both HCM and SCD. Maximum left‐ventricular wall thickness (LVWT) is significantly greater in genotype‐positive patients, who are characterized by more asymmetrical hypertrophy than genotype‐negative cases. Most important, these differences translate into poorer outcomes for genotype‐positive patients across a range of measures and composites in a number of studies, including the 4591 cases of the SHaRe registry.102
In these studies, genotype‐negative cases were defined as lacking pathogenic (or potentially pathogenic, ie, rare and protein‐altering) variants in the tested HCM genes (for the most part the 8 validated sarcomeric genes). The expectation that at least some of these cases may have novel genetic causes drove the many attempts to discover novel HCM genes, as described previously. However, the non‐sarcomeric genes whose role in HCM has been validated thus far account for only a small proportion of HCM patients, with cumulative weight of evidence now suggesting that genotype‐negative HCM is often nonfamilial and most likely to represent non‐Mendelian disease.
Ingles et al123 recently defined “non‐familial HCM” as the condition that lacks both a potentially pathogenic sarcomeric variant (whether classified as pathogenic or VUS) and any prior family history for HCM. Nonfamilial cases (which accounted for 40% of the cohort in the study) were phenotypically distinct from genotype‐positive cases (though in a similar manner to overall genotype‐negative patients) and with very low yield from cardiac clinical screening on first‐degree relatives. Consistently, Ko et al127 found that only 3% of first‐degree relatives of “nonfamilial” probands were diagnosed with HCM by clinical screening, compared with 17% of relatives of probands with a positive genetic test and/or prior family history of HCM. As familial occurrence of HCM is still nonnegligible in pedigrees without an identified causative variant, current clinical recommendations state that regular clinical screening of family members should be considered for relatives in such families. However, the actual risk of disease in families without prior family history of HCM, and the consequent necessity for ongoing clinical screening in phenotype‐negative relatives, remains uncertain. In this respect, it is worth emphasizing that history taking in HCM is a complex task due to the high variability in terms of age of onset and phenotype severity characterizing this condition. In contrast to other conditions in which the phenotype usually manifests clearly and penetrance is not age related, in the case of HCM the adoption of specific strategies to maximize precision and completeness of the information gathered from the proband and his/her relatives may help to reconstruct a reliable clinical history of the family. As an example, in addition to applying the principles detailed in internationally accepted guidelines,114 at first clinical appointment the cardiologist could also ask for information regarding family history, so to complement and confirm those gathered by the genetic counsellor. Furthermore, it may be good practice to ask patients to share a copy of all clinical documents regarding important clinical events such as hospitalizations or cardiovascular events, due to the fact that the knowledge of family members on the cause underlying such events is generally limited.
Further large‐scale studies are required to definitively assess the risk in relatives of index cases with apparently nonfamilial HCM and to accurately define the criteria to establish family history of the disease. Such studies may lead to modification of current guidelines on the extent and frequency of clinical screening recommended for relatives of HCM patients, based on more accurate assessments of their risk of developing disease depending on genetic status and family history. This may offer the opportunity for additional significant savings in healthcare provision and will further underscore the utility and cost‐effectiveness of genetic testing for HCM.
Future Directions
Almost 30 years after the first definitive evidence of the genetic etiology of HCM,7 great progress has been achieved and the genes underlying the majority of Mendelian monogenic cases of HCM have now been identified. It is clear that HCM remains essentially a disease of the sarcomere, characterized by reduced penetrance and highly variable expressivity. Although additional causative genes may be found, these will most likely explain only a minor proportion of cases, as demonstrated by the few non‐sarcomeric genes now deemed validated for HCM.
Recent findings and recommendations now point to a more focused application of genetic testing in HCM. Genes of unknown significance, whose association with the disease remains unproven, should be excluded from diagnostic testing as they offer a negligible increase in diagnostic yield but considerably increase the background noise of VUS.25, 29, 33, 103 Conversely, more comprehensive and focused analysis of known disease genes is likely to yield greater results than attempts to discover novel disease genes. This will include improved classification techniques for coding variants and detection of variant classes historically considered to be of low pathogenic potential, including intronic variants affect splicing, which recent research has highlighted are sometimes irrefutably pathogenic and could therefore contribute to resolving an additional proportion of unexplained disease cases.81, 128
A large proportion of HCM cases still remain without an identified causative variant, with 40% to 60% of screened individuals receiving an inconclusive or a negative test result.49, 65, 102, 129, 130 More complex genetic models for HCM are increasingly believed to hold the answer to the majority of these genetically elusive cases. These are still largely unexplored and could include a polygenic model, where multiple rare variants cause disease when carried by the same individual but are benign in isolation, and complex disease model combining the effects of common variants of low effect size with nongenetic factors such as hypertension and obesity. However, investigations into such models have so far been limited by the lack of availability of case and control cohorts large enough to reach satisfactory statistical power. To date, the only GWAS reported for HCM comprised 174 patients and 823 controls, and detected a significant association between a common intronic variant in the FHOD3 gene on chromosome 18 and the presence of disease.131 Such efforts will be increasingly possible as single centers are more easily able to sequence large numbers of patients, and through publicly accessible (though with variable data access policies) phenotyped and sequenced population cohorts such as those by the National Institutes of Health (the ongoing “All of us” research program that aims at 1 million genomes, and the dbGap database132), the UK Biobank,133 Genomics England134 and the Cooperative Health Research in South Tyrol.135 Such data sets, coupled with data sharing efforts such as the SHaRe Registry, will plausibly enable investigations on multi‐genic and other complex disease models.
The lack of clear correlation between the genetic and clinical status of patients has until now dented hopes for the development of personalized medicine approaches, with genetic testing results still being used in a strictly diagnostic (rather than predictive) fashion. The incomplete penetrance of pathogenic sarcomeric variants, and the variable phenotypic expression in variant carriers, indicates that other genetic and non‐genetics factors are likely to influence the development of disease. It has been observed that the presence of multiple rare sarcomeric variants has a cumulative effect on disease onset and incidence of events, regardless of the variants’ diagnostic classification (ie, even when the variants could not be classified as disease‐causing),136 even though previous studies on multiple co‐occurring sarcomeric mutations in HCM likely overestimated the pathogenicity of the individual variants.137, 138 This suggests that variants that are not pathogenic in isolation may act to modify and exacerbate the disease phenotype in presence of a disease‐causing allele. Research into the full range of genetic factors that may interact with primary sarcomeric mutations to influence phenotype is at an early stage but is promising—such factors could include common, low frequency, and rare variants that add to the mutational burden, act as protective variants, or regulate the ratio of expression of mutated and wild‐type alleles.139 Identifying these factors through large‐scale genome‐wide association studies and WGS, and integrating such a genetic profile of common and rare variants with known nongenetic modifying factors such as hypertension and obesity, offers the promise in the near future of improved risk prediction for HCM patients and their relatives and a more personalized approach to clinical care.
Conclusions
The path to a complete and detailed understanding of how genetic variants cause HCM and determine its severity will be long and challenging. Three major challenges remain unresolved and need to be addressed:
The still suboptimal accuracy of variant interpretation strategies,
The scarcity of clear genotype‐phenotype correlations, and
The necessity to expand research on genetic causes of HCM beyond the monogenic Mendelian inheritance model.
Advances in our understanding of population genetics, the development of new functional genomics techniques and quantitative approaches to variant interpretation, data sharing of cohort data and evidence from clinical genetics laboratories, and the introduction of new standards for assessing gene and variant pathogenicity have all greatly improved our understanding of the Mendelian genetics of HCM.
Genetic testing can now be more rigorously and consistently applied for HCM patients, enabling the accurate identification of primary causative variants and at‐risk individuals.
Our understanding of the more complex genetics that may underlie both the highly variable phenotypes observed in sarcomeric variant carriers and the genetic basis of currently genotype negative and likely non‐Mendelian HCM is still at an early stage. It is likely that significant investment in sequencing and phenotyping, and further initiatives in developing international registries and consortia, will be essential for allowing us to make similar progress in understanding the complex genetics underlying HCM.
Sources of Funding
This work was supported by the Horizon 2020 Framework Programme (GA777204 – SILICOFCM); the Italian Ministry of Health (RF‐2013‐02356787); the Italian Ministry of Education, University and Research; the Cardiovascular Research Centre – Royal Brompton & Harefield NHS Trust and the NIHR Imperial College Biomedical Research Centre; and from Amsterdam Cardiovascular Sciences.
Disclosures
None.
Supporting information
Table S1
Acknowledgments
We thank Giovanna Gadenz (F.M.'s mother) for drawing the watercolor painting displayed in Figure 1, and Dr Franco Cecchi for providing clinical history information for individuals of the family carrying the MYH7:p.Arg869His variant (ClinVar accession SCV001147002.1).
(J Am Heart Assoc. 2020;9:e015473 DOI: 10.1161/JAHA.119.015473.)
For Sources of Funding and Disclosures, see page 15.
References
- 1. Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381:242–255. [DOI] [PubMed] [Google Scholar]
- 2. Authors/Task Force Members , Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35:2733–2779. [DOI] [PubMed] [Google Scholar]
- 3. Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785–789. [DOI] [PubMed] [Google Scholar]
- 4. Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65:1249–1254. [DOI] [PubMed] [Google Scholar]
- 5. Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;20:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Braunwald E, Lambrew CT, Rockoff SD, Ross J, Morrow AG. Idiopathic hypertrophic subaortic stenosis. I. A description of the disease based upon an analysis of 64 patients. Circulation. 1964;30(suppl 4):3–119. [DOI] [PubMed] [Google Scholar]
- 7. Jarcho JA, McKenna W, Pare JA, Solomon SD, Holcombe RF, Dickie S, Levi T, Donis‐Keller H, Seidman JG, Seidman CE. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med. 1989;321:1372–1378. [DOI] [PubMed] [Google Scholar]
- 8. Solomon SD, Geisterfer‐Lowrance AA, Vosberg HP, Hiller G, Jarcho JA, Morton CC, McBride WO, Mitchell AL, Bale AE, McKenna WJ. A locus for familial hypertrophic cardiomyopathy is closely linked to the cardiac myosin heavy chain genes, CRI‐L436, and CRI‐L329 on chromosome 14 at q11‐q12. Am J Hum Genet. 1990;47:389–394. [PMC free article] [PubMed] [Google Scholar]
- 9. Geisterfer‐Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006. [DOI] [PubMed] [Google Scholar]
- 10. Solomon SD, Jarcho JA, McKenna W, Geisterfer‐Lowrance A, Germain R, Salerni R, Seidman JG, Seidman CE. Familial hypertrophic cardiomyopathy is a genetically heterogeneous disease. J Clin Invest. 1990;86:993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Watkins H, MacRae C, Thierfelder L, Chou YH, Frenneaux M, McKenna W, Seidman JG, Seidman CE. A disease locus for familial hypertrophic cardiomyopathy maps to chromosome 1q3. Nat Genet. 1993;3:333–337. [DOI] [PubMed] [Google Scholar]
- 12. Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, Seidman JG, Seidman CE. Alpha‐tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–712. [DOI] [PubMed] [Google Scholar]
- 13. Carrier L, Hengstenberg C, Beckmann JS, Guicheney P, Dufour C, Bercovici J, Dausse E, Berebbi‐Bertrand I, Wisnewsky C, Pulvenis D. Mapping of a novel gene for familial hypertrophic cardiomyopathy to chromosome 11. Nat Genet. 1993;4:311–313. [DOI] [PubMed] [Google Scholar]
- 14. Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ, Maron BJ, Seidman JG, Seidman CE. Mutations in the cardiac myosin binding protein‐C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet. 1995;11:434–437. [DOI] [PubMed] [Google Scholar]
- 15. Thierfelder L, MacRae C, Watkins H, Tomfohrde J, Williams M, McKenna W, Bohm K, Noeske G, Schlepper M, Bowcock A. A familial hypertrophic cardiomyopathy locus maps to chromosome 15q2. Proc Natl Acad Sci USA. 1993;90:6270–6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, Rayment I, Sellers JR, Fananapazir L, Epstein ND. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996;13:63–69. [DOI] [PubMed] [Google Scholar]
- 17. Kimura A, Harada H, Park JE, Nishi H, Satoh M, Takahashi M, Hiroi S, Sasaoka T, Ohbuchi N, Nakamura T, et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet. 1997;16:379–382. [DOI] [PubMed] [Google Scholar]
- 18. Flavigny J, Richard P, Isnard R, Carrier L, Charron P, Bonne G, Forissier JF, Desnos M, Dubourg O, Komajda M, et al. Identification of two novel mutations in the ventricular regulatory myosin light chain gene (MYL2) associated with familial and classical forms of hypertrophic cardiomyopathy. J Mol Med (Berl). 1998;76:208–214. [DOI] [PubMed] [Google Scholar]
- 19. Mogensen J, Klausen IC, Pedersen AK, Egeblad H, Bross P, Kruse TA, Gregersen N, Hansen PS, Baandrup U, Borglum AD. Alpha‐cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J Clin Invest. 1999;103:R39–R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Szczesna D, Ghosh D, Li Q, Gomes AV, Guzman G, Arana C, Zhi G, Stull JT, Potter JD. Familial hypertrophic cardiomyopathy mutations in the regulatory light chains of myosin affect their structure, Ca2+ binding, and phosphorylation. J Biol Chem. 2001;276:7086–7092. [DOI] [PubMed] [Google Scholar]
- 21. Kabaeva Z, Perrot A, Wolter B, Dietz R, Cardim N, Correia JM, Schulte HD, Aldashev AA, Mirrakhimov MM, Osterziel KJ. Systematic analysis of the regulatory and essential myosin light chain genes: genetic variants and mutations in hypertrophic cardiomyopathy. Eur J Hum Genet. 2002;10:741–748. [DOI] [PubMed] [Google Scholar]
- 22. Grey C, Méry A, Pucéat M. Fine‐tuning in Ca2+ homeostasis underlies progression of cardiomyopathy in myocytes derived from genetically modified embryonic stem cells. Hum Mol Genet. 2005;14:1367–1377. [DOI] [PubMed] [Google Scholar]
- 23. Renaudin P, Janin A, Millat G, Chevalier P. A novel missense mutation p.Gly162Glu of the gene MYL2 involved in hypertrophic cardiomyopathy: a pedigree analysis of a proband. Mol Diagnosis Ther. 2018;22:219–223. [DOI] [PubMed] [Google Scholar]
- 24. Martiniuk F, Mehler M, Pellicer A, Tzall S, La Badie G, Hobart C, Ellenbogen A, Hirschhorn R. Isolation of a cDNA for human acid alpha‐glucosidase and detection of genetic heterogeneity for mRNA in three alpha‐glucosidase‐deficient patients. Proc Natl Acad Sci USA. 1986;83:9641–9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Van der Ploeg AT, Hoefsloot LH, Hoogeveen‐Westerveld M, Petersen EM, Reuser AJ. Glycogenosis type II: protein and DNA analysis in five South African families from various ethnic origins. Am J Hum Genet. 1989;44:787–793. [PMC free article] [PubMed] [Google Scholar]
- 26. Martiniuk F, Bodkin M, Tzall S, Hirschhorn R. Identification of the base‐pair substitution responsible for a human acid alpha glucosidase allele with lower “affinity” for glycogen (GAA 2) and transient gene expression in deficient cells. Am J Hum Genet. 1990;47:440–445. [PMC free article] [PubMed] [Google Scholar]
- 27. Bernstein HS, Bishop DF, Astrin KH, Kornreich R, Eng CM, Sakuraba H, Desnick RJ. Fabry disease: six gene rearrangements and an exonic point mutation in the alpha‐galactosidase gene. J Clin Invest. 1989;83:1390–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Davies JP, Winchester BG, Malcolm S. Mutation analysis in patients with the typical form of Anderson‐Fabry disease. Hum Mol Genet. 1993;2:1051–1053. [DOI] [PubMed] [Google Scholar]
- 29. Eng CM, Desnick RJ. Molecular basis of Fabry disease: mutations and polymorphisms in the human alpha‐galactosidase A gene. Hum Mutat. 1994;3:103–111. [DOI] [PubMed] [Google Scholar]
- 30. Charron P, Villard E, Sébillon P, Laforêt P, Maisonobe T, Duboscq‐Bidot L, Romero N, Drouin‐Garraud V, Frébourg T, Richard P, et al. Danon's disease as a cause of hypertrophic cardiomyopathy: a systematic survey. Heart. 2004;90:842–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arad M, Maron BJ, Gorham JM, Johnson WH, Saul JP, Perez‐Atayde AR, Spirito P, Wright GB, Kanter RJ, Seidman CE, et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med. 2005;352:362–372. [DOI] [PubMed] [Google Scholar]
- 32. Taylor MRG, Ku L, Slavov D, Cavanaugh J, Boucek M, Zhu X, Graw S, Carniel E, Barnes C, Quan D, et al. Danon disease presenting with dilated cardiomyopathy and a complex phenotype. J Hum Genet. 2007;52:830–835. [DOI] [PubMed] [Google Scholar]
- 33. Gollob MH, Green MS, Tang AS, Gollob T, Karibe A, Ali Hassan AS, Ahmad F, Lozado R, Shah G, Fananapazir L, et al. Identification of a gene responsible for familial Wolff‐Parkinson‐White syndrome. N Engl J Med. 2001;344:1823–1831. [DOI] [PubMed] [Google Scholar]
- 34. Benson MD. Inherited amyloidosis. J Med Genet. 1991;28:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saraiva MJ. Transthyretin mutations in health and disease. Hum Mutat. 1995;5:191–196. [DOI] [PubMed] [Google Scholar]
- 36. Saraiva MJ. Transthyretin mutations in hyperthyroxinemia and amyloid diseases. Hum Mutat. 2001;17:493–503. [DOI] [PubMed] [Google Scholar]
- 37. Lachmann HJ, Booth DR, Booth SE, Bybee A, Gilbertson JA, Gillmore JD, Pepys MB, Hawkins PN. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786–1791. [DOI] [PubMed] [Google Scholar]
- 38. Zellweger H, Brown BI, McCormick WF, Tu JB. A mild form of muscular glycogenosis in two brothers with alpha‐1, 4‐glucosidase deficiency. Ann Paediatr. 1965;205:413–437. [PubMed] [Google Scholar]
- 39. Hoefsloot LH, Hoogeveen‐Westerveld M, Kroos MA, van Beeumen J, Reuser AJ, Oostra BA. Primary structure and processing of lysosomal alpha‐glucosidase; homology with the intestinal sucrase‐isomaltase complex. EMBO J. 1988;7:1697–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuo WL, Hirschhorn R, Huie ML, Hirschhorn K. Localization and ordering of acid alpha‐glucosidase (GAA) and thymidine kinase (TK1) by fluorescence in situ hybridization. Hum Genet. 1996;97:404–406. [DOI] [PubMed] [Google Scholar]
- 41. Romeo G, Migeon BR. Genetic inactivation of the α‐galactosidase locus in carriers of fabry's disease. Science. 1970;170:180–181. [DOI] [PubMed] [Google Scholar]
- 42. Kornreich R, Desnick RJ, Bishop DF. Nucleotide sequence of the human α‐galactosidase a gene. Nucleic Acids Res. 1989;17:3301–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Danon MJ, Oh SJ, DiMauro S, Manaligod JR, Eastwood A, Naidu S, Schliselfeld LH. Lysosomal glycogen storage disease with normal acid maltase. Neurology. 1981;31:51–57. [DOI] [PubMed] [Google Scholar]
- 44. Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, Sue CM, Yamamoto A, Murakami N, Shanske S, Byrne E, Bonilla E, Honaka I, DiMauro S, Hirano M. Primary LAMP‐2 deficiency causes X‐linked vacoular cardiomyopathy and myopathy (Danon disease). Nature. 2000;406:906–910. [DOI] [PubMed] [Google Scholar]
- 45. Kanda Y, Goodman DS, Canfield RE, Morgan FJ. The amino acid sequence of human plasma prealbumin. J Biol Chem. 1974;249:6796–6805. [PubMed] [Google Scholar]
- 46. Episkopou V, Maeda S, Nishiguchi S, Shimada K, Gaitanaris GA, Gottesman ME, Robertson EJ. Disruption of the transthyretin gene results in mice with depressed levels of plasma retinol and thyroid hormone. Proc Natl Acad Sci USA. 1993;90:2375–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jinno Y, Matsumoto T, Kamei T, Kondoh T, Maeda S, Araki S, Shimada K. Niikawa N. Localization of the human prealbumin gene to 18p11.1‐q12.3 by gene dose effect study of Southern blot hybridization. Jpn J Hum Genet. 1986;31:243–248. [DOI] [PubMed] [Google Scholar]
- 48. Ingles J, Goldstein J, Thaxton C, Caleshu C, Corty EW, Crowley SB, Dougherty K, Harrison SM, McGlaughon J, Milko LV, et al. Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ Genom Precis Med. 2019;12:e002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Walsh R, Buchan R, Wilk A, John S, Felkin LE, Thomson KL, Chiaw TH, Loong CCW, Pua CJ, Raphael C, et al. Defining the genetic architecture of hypertrophic cardiomyopathy: re‐evaluating the role of non‐sarcomeric genes. Eur Heart J. 2017;38:3461–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hoffmann B, Schmidt‐Traub H, Perrot A, Osterziel KJ, Gessner R. First mutation in cardiac troponin C, L29Q, in a patient with hypertrophic cardiomyopathy. Hum Mutat. 2001;17:524. [DOI] [PubMed] [Google Scholar]
- 51. Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan G‐C, Tsiapras D, Hahn HS, Adamopoulos S, et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Geier C, Perrot A, Ozcelik C, Binner P, Counsell D, Hoffmann K, Pilz B, Martiniak Y, Gehmlich K, van der Ven PFM, et al. Mutations in the human muscle LIM protein gene in families with hypertrophic cardiomyopathy. Circulation. 2003;107:1390–1395. [DOI] [PubMed] [Google Scholar]
- 53. Landstrom AP, Weisleder N, Batalden KB, Bos JM, Tester DJ, Ommen SR, Wehrens XHT, Claycomb WC, Ko J‐K, Hwang M, et al. Mutations in JPH2‐encoded junctophilin‐2 associated with hypertrophic cardiomyopathy in humans. J Mol Cell Cardiol. 2007;42:1026–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chiu C, Bagnall RD, Ingles J, Yeates L, Kennerson M, Donald JA, Jormakka M, Lind JM, Semsarian C. Mutations in alpha‐actinin‐2 cause hypertrophic cardiomyopathy: a genome‐wide analysis. J Am Coll Cardiol. 2010;55:1127–1135. [DOI] [PubMed] [Google Scholar]
- 55. Valdés‐Mas R, Gutiérrez‐Fernández A, Gómez J, Coto E, Astudillo A, Puente DA, Reguero JR, Álvarez V, Morís C, León D, et al. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat Commun. 2014;5:5326. [DOI] [PubMed] [Google Scholar]
- 56. Almomani R, Verhagen JMA, Herkert JC, Brosens E, van Spaendonck‐Zwarts KY, Asimaki A, van der Zwaag PA, Frohn‐Mulder IME, Bertoli‐Avella AM, Boven LG, et al. Biallelic truncating mutations in ALPK3 cause severe pediatric cardiomyopathy. J Am Coll Cardiol. 2016;67:515–525. [DOI] [PubMed] [Google Scholar]
- 57. Ochoa JP, Sabater‐Molina M, García‐Pinilla JM, Mogensen J, Restrepo‐Córdoba A, Palomino‐Doza J, Villacorta E, Martinez‐Moreno M, Ramos‐Maqueda J, Zorio E, et al. Formin homology 2 domain containing 3 (FHOD3) is a genetic basis for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2018;72:2457–2467. [DOI] [PubMed] [Google Scholar]
- 58. Consortium T 1000 GP . An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Exome Variant Server . Available at: http://evs.gs.washington.edu/EVS/. Accessed December 3, 2018.
- 60. Golbus JR, Puckelwartz MJ, Fahrenbach JP, Dellefave‐Castillo LM, Wolfgeher D, McNally EM. Population‐based variation in cardiomyopathy genes. Circ Cardiovasc Genet. 2012;5:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Andreasen C, Nielsen JB, Refsgaard L, Holst AG, Christensen AH, Andreasen L, Sajadieh A, Haunsø S, Svendsen JH, Olesen MS. New population‐based exome data are questioning the pathogenicity of previously cardiomyopathy‐associated genetic variants. Eur J Hum Genet. 2013;21:918–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell‐Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mazzarotto F, Tayal U, Buchan RJ, Midwinter W, Wilk A, Whiffin N, Govind R, Mazaika E, de Marvao A, Dawes TJW, et al. Re‐evaluating the genetic contribution of monogenic dilated cardiomyopathy. Circulation. 2020;141:387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mazzarotto F, Girolami F, Boschi B, Barlocco F, Tomberli A, Baldini K, Coppini R, Tanini I, Bardi S, Contini E, et al. Defining the diagnostic effectiveness of genes for inclusion in panels: the experience of two decades of genetic testing for hypertrophic cardiomyopathy at a single center. Genet Med. 2019;21:284–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, Ledbetter DH, Maglott DR, Martin CL, Nussbaum RL, et al. ClinGen–the clinical genome resource. N Engl J Med. 2015;372:2235–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wordsworth S, Leal J, Blair E, Legood R, Thomson K, Seller A, Taylor J, Watkins H. DNA testing for hypertrophic cardiomyopathy: a cost‐effectiveness model. Eur Heart J. 2010;31:926–935. [DOI] [PubMed] [Google Scholar]
- 68. Ingles J, McGaughran J, Scuffham PA, Atherton J, Semsarian C. A cost‐effectiveness model of genetic testing for the evaluation of families with hypertrophic cardiomyopathy. Heart. 2012;98:625–630. [DOI] [PubMed] [Google Scholar]
- 69. Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB, Shen J, McLaughlin HM, Clark EH, Babb LJ, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. 2015;17:880–888. [DOI] [PubMed] [Google Scholar]
- 70. Batte B, Sheldon JP, Arscott P, Huismann DJ, Salberg L, Day SM, Yashar BM. Family communication in a population at risk for hypertrophic cardiomyopathy. J Genet Couns. 2015;24:336–348. [DOI] [PubMed] [Google Scholar]
- 71. O'Mahony C, Jichi F, Pavlou M, Monserrat L, Anastasakis A, Rapezzi C, Biagini E, Gimeno JR, Limongelli G, McKenna WJ, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk‐SCD). Eur Heart J. 2014;35:2010–2020. [DOI] [PubMed] [Google Scholar]
- 72. Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:e783–e831. [DOI] [PubMed] [Google Scholar]
- 73. Towbin JA, Jefferies JL. Cardiomyopathies due to left ventricular noncompaction, mitochondrial and storage diseases, and inborn errors of metabolism. Circ Res. 2017;121:838–854. [DOI] [PubMed] [Google Scholar]
- 74. Sankaranarayanan R, J Fleming E, J Garratt C. Mimics of hypertrophic cardiomyopathy—diagnostic clues to aid early identification of phenocopies. Arrhythm Electrophysiol Rev. 2013;2:36–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jamuar SS, Tan E‐C. Clinical application of next‐generation sequencing for Mendelian diseases. Hum Genomics. 2015;9:10 DOI: 10.1186/s40246-015-0031-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schwarze K, Buchanan J, Taylor JC, Wordsworth S. Are whole‐exome and whole‐genome sequencing approaches cost‐effective? A systematic review of the literature. Genet Med. 2018;20:1122–1130. [DOI] [PubMed] [Google Scholar]
- 77. Cirino AL, Lakdawala NK, McDonough B, Conner L, Adler D, Weinfeld M, O'Gara P, Rehm HL, Machini K, Lebo M, et al. A comparison of whole genome sequencing to multigene panel testing in hypertrophic cardiomyopathy patients. Circ Cardiovasc Genet. 2017;10:e001768 DOI: 10.1161/CIRCGENETICS.117.001768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lopes LR, Murphy C, Syrris P, Dalageorgou C, McKenna WJ, Elliott PM, Plagnol V. Use of high‐throughput targeted exome‐sequencing to screen for copy number variation in hypertrophic cardiomyopathy. Eur J Med Genet. 2015;58:611–616. [DOI] [PubMed] [Google Scholar]
- 79. Mademont‐Soler I, Mates J, Yotti R, Espinosa MA, Pérez‐Serra A, Fernandez‐Avila AI, Coll M, Méndez I, Iglesias A, del Olmo B, et al. Additional value of screening for minor genes and copy number variants in hypertrophic cardiomyopathy. PLoS One. 2017;12:e0181465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ceyhan‐Birsoy O, Pugh TJ, Bowser MJ, Hynes E, Frisella AL, Mahanta LM, Lebo MS, Amr SS, Funke BH. Next generation sequencing‐based copy number analysis reveals low prevalence of deletions and duplications in 46 genes associated with genetic cardiomyopathies. Mol Genet Genomic Med. 2016;4:143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bagnall RD, Ingles J, Dinger ME, Cowley MJ, Ross SB, Minoche AE, Lal S, Turner C, Colley A, Rajagopalan S, et al. Whole genome sequencing improves outcomes of genetic testing in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2018;72:419–429. [DOI] [PubMed] [Google Scholar]
- 82. Marston S, Copeland O, Gehmlich K, Schlossarek S, Carrier L, Carrrier L. How do MYBPC3 mutations cause hypertrophic cardiomyopathy? J Muscle Res Cell Motil. 2012;33:75–80. [DOI] [PubMed] [Google Scholar]
- 83. Pua CJ, Bhalshankar J, Miao K, Walsh R, John S, Lim SQ, Chow K, Buchan R, Soh BY, Lio PM, et al. Development of a comprehensive sequencing assay for inherited cardiac condition genes. J Cardiovasc Transl Res. 2016;9:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Walsh R, Cook SA. Issues and challenges in diagnostic sequencing for inherited cardiac conditions. Clin Chem. 2017;63:116–128. [DOI] [PubMed] [Google Scholar]
- 85. Manase D, D'Alessandro LC, Manickaraj AK, Al Turki S, Hurles ME, Mital S. High throughput exome coverage of clinically relevant cardiac genes. BMC Med Genomics. 2014;7:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Manrai AK, Funke BH, Rehm HL, Olesen MS, Maron BA, Szolovits P, Margulies DM, Loscalzo J, Kohane IS. Genetic misdiagnoses and the potential for health disparities. N Engl J Med. 2016;375:655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ackerman JP, Bartos DC, Kapplinger JD, Tester DJ, Delisle BP, Ackerman MJ. The promise and peril of precision medicine: phenotyping still matters most. Mayo Clin Proc. 2016;91:1606–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. ACMG recommendations for standards for interpretation of sequence variations. Genet Med. 2000;2:302–303. [DOI] [PubMed] [Google Scholar]
- 89. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Harrison SM, Dolinsky JS, Knight Johnson AE, Pesaran T, Azzariti DR, Bale S, Chao EC, Das S, Vincent L, Rehm HL. Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genet Med. 2017;19:1096–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Harrison SM, Dolinksy JS, Chen W, Collins CD, Das S, Deignan JL, Garber KB, Garcia J, Jarinova O, Knight Johnson AE, et al. Scaling resolution of variant classification differences in ClinVar between 41 clinical laboratories through an outlier approach. Hum Mutat. 2018;39:1641–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Whiffin N, Walsh R, Govind R, Edwards M, Ahmad M, Zhang X, Tayal U, Buchan R, Midwinter W, Wilk AE, et al. CardioClassifier: disease‐ and gene‐specific computational decision support for clinical genome interpretation. Genet Med. 2018;20:1246–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Nicora G, Limongelli I, Gambelli P, Memmi M, Malovini A, Mazzanti A, Napolitano C, Priori S, Bellazzi R. CardioVAI: an automatic implementation of ACMG‐AMP variant interpretation guidelines in the diagnosis of cardiovascular diseases. Hum Mutat. 2018;39:1835–1846. [DOI] [PubMed] [Google Scholar]
- 94. Jarvik GP, Browning BL. Consideration of cosegregation in the pathogenicity classification of genomic variants. Am J Hum Genet. 2016;98:1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, Harrison SM; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI) . Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018;39:1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Walsh R, Mazzarotto F, Whiffin N, Buchan R, Midwinter W, Wilk A, Li N, Felkin L, Ingold N, Govind R, et al. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: the case of hypertrophic cardiomyopathy. Genome Med. 2019;11:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Whiffin N, Minikel E, Walsh R, O'Donnell‐Luria AH, Karczewski K, Ing AY, Barton PJR, Funke B, Cook SA, MacArthur D, et al. Using high‐resolution variant frequencies to empower clinical genome interpretation. Genet Med. 2017;19:1151–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Homburger JR, Green EM, Caleshu C, Sunitha MS, Taylor RE, Ruppel KM, Metpally RPR, Colan SD, Michels M, Day SM, et al. Multidimensional structure‐function relationships in human β‐cardiac myosin from population‐scale genetic variation. Proc Natl Acad Sci USA. 2016;113:6701–6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Gelb BD, Cavé H, Dillon MW, Gripp KW, Lee JA, Mason‐Suares H, Rauen KA, Williams B, Zenker M, Vincent LM. ClinGen's RASopathy Expert Panel consensus methods for variant interpretation. Genet Med. 2018;20:1334–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kelly MA, Caleshu C, Morales A, Buchan J, Wolf Z, Harrison SM, Cook S, Dillon MW, Garcia J, Haverfield E, et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7‐associated inherited cardiomyopathies: recommendations by ClinGen's Inherited Cardiomyopathy Expert Panel. Genet Med. 2018;20:351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Tavtigian SV, Greenblatt MS, Harrison SM, Nussbaum RL, Prabhu SA, Boucher KM, Biesecker LG. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. 2018;20:1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, Cirino AL, Fox JC, Lakdawala NK, Ware JS, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy. Circulation. 2018;138:1387–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, Bernstein JA, Bejerano G. M‐CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48:1581–1586. [DOI] [PubMed] [Google Scholar]
- 104. Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Da'as SI, Fakhro K, Thanassoulas A, Krishnamoorthy N, Saleh A, Calver BL, Safieh‐Garabedian B, Toft E, Nounesis G, Lai FA, et al. Hypertrophic cardiomyopathy‐linked variants of cardiac myosin‐binding protein C3 display altered molecular properties and actin interaction. Biochem J. 2018;475:3933–3948. [DOI] [PubMed] [Google Scholar]
- 106. Mosqueira D, Mannhardt I, Bhagwan JR, Lis‐Slimak K, Katili P, Scott E, Hassan M, Prondzynski M, Harmer SC, Tinker A, et al. CRISPR/Cas9 editing in human pluripotent stem cell‐cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur Heart J. 2018;39:3879–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yang K‐C, Breitbart A, De Lange WJ, Hofsteen P, Futakuchi‐Tsuchida A, Xu J, Schopf C, Razumova MV, Jiao A, Boucek R, et al. Novel adult‐onset systolic cardiomyopathy due to MYH7 E848G mutation in patient‐derived induced pluripotent stem cells. JACC Basic Transl Sci. 2018;3:728–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ma N, Zhang JZ, Itzhaki I, Zhang SL, Chen H, Haddad F, Kitani T, Wilson KD, Tian L, Shrestha R, et al. Determining the pathogenicity of a genomic variant of uncertain significance using CRISPR/Cas9 and human‐induced pluripotent stem cells. Circulation. 2018;138:2666–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Lv W, Qiao L, Petrenko N, Li W, Owens AT, McDermott‐Roe C, Musunuru K. Functional annotation of TNNT2 variants of uncertain significance with genome‐edited cardiomyocytes. Circulation. 2018;138:2852–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, Janizek JD, Huang X, Starita LM, Shendure J. Accurate classification of BRCA1 variants with saturation genome editing. Nature. 2018;562:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Landry LG, Rehm HL. Association of racial/ethnic categories with the ability of genetic tests to detect a cause of cardiomyopathy. JAMA Cardiol. 2018;3:341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 2019;51:584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2. 0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–255. [DOI] [PubMed] [Google Scholar]
- 114. Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, Morales A, Taylor MRG, Vatta M, Ware SM, et al. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2018;20:899–909. [DOI] [PubMed] [Google Scholar]
- 115. Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O'Donoghue A, Spirito P, Matsumori A, Moravec CS, Seidman JG. Mutations in the genes for cardiac troponin T and alpha‐tropomyosin in hypertrophic cardiomyopathy. N Engl J Med. 1995;332:1058–1064. [DOI] [PubMed] [Google Scholar]
- 116. Niimura H, Bachinski LL, Sangwatanaroj S, Watkins H, Chudley AE, McKenna W, Kristinsson A, Roberts R, Sole M, Maron BJ, et al. Mutations in the gene for cardiac myosin‐binding protein C and late‐onset familial hypertrophic cardiomyopathy. N Engl J Med. 1998;338:1248–1257. [DOI] [PubMed] [Google Scholar]
- 117. Coppini R, Ho CY, Ashley E, Day S, Ferrantini C, Girolami F, Tomberli B, Bardi S, Torricelli F, Cecchi F, et al. Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin‐filament gene mutations. J Am Coll Cardiol. 2014;64:2589–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Li Q, Gruner C, Chan RH, Care M, Siminovitch K, Williams L, Woo A, Rakowski H. Genotype‐positive status in patients with hypertrophic cardiomyopathy is associated with higher rates of heart failure events. Circ Cardiovasc Genet. 2014;7:416–422. [DOI] [PubMed] [Google Scholar]
- 119. Weissler‐Snir A, Hindieh W, Gruner C, Fourey D, Appelbaum E, Rowin E, Care M, Lesser JR, Haas TS, Udelson JE, et al. Lack of phenotypic differences by cardiovascular magnetic resonance imaging in MYH7 (β‐myosin heavy chain)‐ versus MYBPC3 (myosin‐binding protein C)‐related hypertrophic cardiomyopathy. Circ Cardiovasc Imaging. 2017;10:e005311 DOI: 10.1161/CIRCIMAGING.116.005311. [DOI] [PubMed] [Google Scholar]
- 120. Bos JM, Will ML, Gersh BJ, Kruisselbrink TM, Ommen SR, Ackerman MJ. Characterization of a phenotype‐based genetic test prediction score for unrelated patients with hypertrophic cardiomyopathy. Mayo Clin Proc. 2014;89:727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Lopes LR, Syrris P, Guttmann OP, O'Mahony C, Tang HC, Dalageorgou C, Jenkins S, Hubank M, Monserrat L, McKenna WJ, et al. Novel genotype‐phenotype associations demonstrated by high‐throughput sequencing in patients with hypertrophic cardiomyopathy. Heart. 2014;101:294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. van Velzen HG, Schinkel AFL, Oldenburg RA, van Slegtenhorst MA, Frohn‐Mulder IME, van der Velden J; Michels M. Clinical characteristics and long‐term outcome of hypertrophic cardiomyopathy in individuals with a MYBPC3 (myosin‐binding protein C) founder mutation. Circ Cardiovasc Genet. 2017;10:e001660 DOI: 10.1161/CIRCGENETICS.116.001660. [DOI] [PubMed] [Google Scholar]
- 123. Ingles J, Burns C, Bagnall RD, Lam L, Yeates L, Sarina T, Puranik R, Briffa T, Atherton JJ, Driscoll T, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet. 2017;10:e001620 DOI: 10.1161/CIRCGENETICS.116.001620. [DOI] [PubMed] [Google Scholar]
- 124. Lopes LR, Rahman MS, Elliott PM. A systematic review and meta‐analysis of genotype‐phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart. 2013;99:1800–1811. [DOI] [PubMed] [Google Scholar]
- 125. van Velzen HG, Vriesendorp PA, Oldenburg RA, van Slegtenhorst MA, van der Velden J, Schinkel AFL, Michels M. Value of genetic testing for the prediction of long‐term outcome in patients with hypertrophic cardiomyopathy. Am J Cardiol. 2016;118:881–887. [DOI] [PubMed] [Google Scholar]
- 126. Ingles J, Sarina T, Yeates L, Hunt L, Macciocca I, McCormack L, Winship I, McGaughran J, Atherton J, Semsarian C. Clinical predictors of genetic testing outcomes in hypertrophic cardiomyopathy. Genet Med. 2013;15:972–977. [DOI] [PubMed] [Google Scholar]
- 127. Ko C, Arscott P, Concannon M, Saberi S, Day SM, Yashar BM, Helms AS. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med. 2018;20:69–75. [DOI] [PubMed] [Google Scholar]
- 128. Singer ES, Ingles J, Semsarian C, Bagnall RD. Key value of RNA analysis of MYBPC3 splice‐site variants in hypertrophic cardiomyopathy. Circ Genom Precis Med. 2019;12:e002368. [DOI] [PubMed] [Google Scholar]
- 129. Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol. 2012;60:705–715. [DOI] [PubMed] [Google Scholar]
- 130. Seidman CE, Seidman JG. Identifying sarcomere gene mutations in HCM: a personal history. Circ Res. 2011;108:743–750. DOI: 10.1161/CIRCRESAHA.110.223834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Wooten EC, Hebl VB, Wolf MJ, Greytak SR, Orr NM, Draper I, Calvino JE, Kapur NK, Maron MS, Kullo IJ, et al. Formin homology 2 domain containing 3 variants associated with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2013;6:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Tryka KA, Hao L, Sturcke A, Jin Y, Wang ZY, Ziyabari L, Lee M, Popova N, Sharopova N, Kimura M, et al. NCBI's database of genotypes and phenotypes: dbGaP. Nucleic Acids Res. 2014;42:D975–D979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Turnbull C, Scott RH, Thomas E, Jones L, Murugaesu N, Pretty FB, Halai D, Baple E, Craig C, Hamblin A, et al. The 100 000 Genomes Project: bringing whole genome sequencing to the NHS. BMJ. 2018;361:k1687. [DOI] [PubMed] [Google Scholar]
- 135. Pattaro C, Gögele M, Mascalzoni D, Melotti R, Schwienbacher C, De Grandi A, Foco L, D'Elia Y, Linder B, Fuchsberger C, et al. The Cooperative Health Research in South Tyrol (CHRIS) study: rationale, objectives, and preliminary results. J Transl Med. 2015;13:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Burns C, Bagnall RD, Lam L, Semsarian C, Ingles J. Multiple gene variants in hypertrophic cardiomyopathy in the era of next‐generation sequencing. Circ Cardiovasc Genet. 2017;10:e001666 DOI: 10.1161/CIRCGENETICS.116.001666. [DOI] [PubMed] [Google Scholar]
- 137. Fourey D, Care M, Siminovitch KA, Weissler‐Snir A, Hindieh W, Chan RH, Gollob MH, Rakowski H, Adler A. Prevalence and clinical implication of double mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2017;10:e001685. [DOI] [PubMed] [Google Scholar]
- 138. Fatkin D, Johnson R. Are double mutations double trouble? Circ Cardiovasc Genet. 2017;10:e001749. [DOI] [PubMed] [Google Scholar]
- 139. Castel SE, Cervera A, Mohammadi P, Aguet F, Reverter F, Wolman A, Guigo R, Iossifov I, Vasileva A, Lappalainen T. Modified penetrance of coding variants by cis‐regulatory variation contributes to disease risk. Nat Genet. 2018;50:1327–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1