

Abstract
Background
Vascular smooth muscle cell phenotypic change and consequential intimal hyperplasia (IH) cause arterial stenosis and posttreatment restenosis. Smad3 is a master transcription factor, yet its underlying functional mechanisms in this disease context are not well defined.
Methods and Results
In cultured smooth muscle cells, Smad3 silencing and overexpression respectively reduced and increased the mRNA and protein of NRP2 (neuropilin 2), a recently reported pro‐IH signaling factor. Smad3 silencing attenuated pro‐IH smooth muscle cell phenotypes including proliferation, migration, and dedifferentiation (reduced smooth muscle α‐actin). While increased Smad3 enhanced these phenotypes, NRP2 silencing abolished this enhancement. Interestingly, the 5′ untranslated region but not the promoter of NRP2 was indispensable for Smad3‐enhanced transcriptional activity (luciferase assay); both chromatin immunoprecipitation and electrophoretic mobility shift assay showed predominant Smad3 binding in the +51 to +78 bp region of NRP2′s 5′ untranslated region. In vivo, Smad3 haploinsufficiency reduced NRP2 (immunostaining) and IH (by 47%) in wire‐injured mouse femoral arteries.
Conclusions
Smad3 controls NRP2 expression by occupying its 5′ untranslated region in promoting smooth muscle cell phenotypic change in vitro. This and in vivo results shed new light on the long‐debated role of Smad3 in IH.
Keywords: 5′ untranslated region, intimal hyperplasia, NRP2, Smad3, Smad3‐haploinsufficient mice
Subject Categories: Pathophysiology, Vascular Biology, Basic Science Research
Clinical Perspective
What Is New?
We identified a previously unrecognized mechanism underlying vascular smooth muscle cell proliferative phenotypic changes, ie, Smad3 controls neuropilin 2′s transcription by binding to its 5′ untranslated region.
Whereas the role of Smad3 in neointima has been debated, our in vivo results indicate that Smad3 haploinsufficiency reduces neointimal hyperplasia by ≈50% in wire‐injured mouse femoral arteries.
What Are the Clinical Implications?
Given the contextual complexity of Smad3 functions, the Smad3/neuropilin 2 axis confers a potential target for precision intervention of neointimal pathologies.
Nonstandard Abbreviations and Acronyms.
5′UTR 5′ untranslated region
AoSMC
aortic smooth muscle cell
ChIP chromatin immunoprecipitation
EEL external elastic lamina
EV
empty vector
IH
intimal hyperplasia
NRP neuropilin
qRT‐PCR quantitative real‐time polymerase chain reaction
SBE Smad‐binding element
SMC smooth muscle cell
TGF β transforming growth factor β
TSS transcriptional start site
WT wild‐type
Introduction
Intimal hyperplasia (IH) causes vascular lumen narrowing and hence flow obstruction. It is a critical component of atherosclerosis, the most common cause of cardiovascular disease. It also occurs following vascular reconstructions leading to failure of these procedures, including angioplasty/stenting, bypass vein grafting, arteriovenous fistula, and allograft transplantation. IH is primarily formed by activated vascular smooth muscle cells (SMCs), which lose bona fide SMC identity (contractile protein) while transitioning to various cell states.1 Consequentially, SMCs acquire pro‐IH new phenotypes, such as proliferation, migration, and dedifferentiation.
Elucidation of IH‐associated molecular mechanisms is key to precise and effective targeting for IH mitigation. In spite of years of effort, these mechanisms, particularly Smad3‐directed regulations, remain obscure. The complication is at least 2‐fold. First, while Smad3 is well established as a canonical signaling protein of transforming growth factor β (TGFβ), the functional consequence of Smad3 activation by TGFβ is highly conceptual. For example, TGFβ1 is commonly used to enhance progenitor cell differentiation into SMCs.2 However, elevated TGFβ1/Smad3 dedifferentiate mature SMCs.3, 4 Second, reports on the role of Smad3 in IH have been contradictory. Our group showed that adenoviral expression of Smad7, an antagonizing factor for Smad3, attenuated IH in rats.4 However, another group reported that IH increased rather than decreased in Smad3‐deficient (versus wild‐type [WT]) mice.5 These paradoxical results necessitate further investigation into the Smad3 regulations of pro‐IH SMC phenotypes in vitro and its role in IH.
NRPs (neuropilins, including isoforms 1 and 2) are primarily semaphorin 3 and vascular endothelial growth factor receptors6 originally discovered as regulators of neural patterning. Recently, they were found to increase in balloon‐injured rat carotid arteries and promote IH.7 Furthermore, in cultured SMCs, NRP2 (over NRP1) showed a more prominent function of enhancing pro‐IH phenotypes of activated SMCs. Interestingly, our exploratory microarray experiment suggested that the transcription of NRP2 (not NRP1) was potently activated by TGFβ1 treatment of SMCs. Among a myriad of TGFβ1‐activated factors, Smad3 as a master transcription factor represents the canonical signaling pathway.8 Upon TGFβ1 stimulation, Smad3 is phosphorylated and translocated to the nucleus to regulate transcription. As such, whether and how Smad3 governs NRP2′s transcription became intriguing questions. Moreover, given the reported IH‐promoting role of NRP2,7 addressing these questions would help define the long‐debated role of Smad3 in IH formation.
We found that Smad3 positively regulated NRP2 expression and the pro‐IH SMC activation, which was reverted by NRP2 silencing. Somewhat surprising, this Smad3 transcriptional activity was primarily accounted for by its occupancy at the 5′ untranslated region (5′UTR) rather than promoter of the NRP2 gene. Consistently, in wire‐injured femoral arteries, NRP2 was reduced and IH was mitigated in Smad3‐haploinsufficient mice versus WT control. Thus, this study revealed a previously unidentified molecular mechanism underlying Smad3 transcriptional control of NRP2, providing new evidence for a positive role of Smad3 in the pro‐IH SMC activation and IH formation.
Methods
The data, methods used in the analysis, and materials used to conduct the research will be made available to any researcher upon request to the corresponding author for purposes of reproducing the results or replicating the procedure. All supporting data are available within the article (and its online supplementary files).
Materials
Human aortic SMCs (AoSMCs, CC‐2571), SMC basal medium (CC‐3181), and SMC basal medium plus SingleQuots of supplements (CC‐3182) were purchased from Lonza. Recombinant Human TGFβ1 240‐B was purchased from R&D Systems. A Cell Titer‐Glo 2.0 Assay Kit was purchased from Promega (G9242). Scrambled control and Smad3‐ or NrP2‐specific small interfering RNA were from Thermo Fisher Scientific (Scrambled: AM4635; SMAD3: 4392420, ID‐s8402; NrP2: 4390771, ID‐n319555). Opti‐MEM I Reduced Serum Medium, Lipofectamine3000, and Lipofectamine RNAiMAX Transfection Reagent were from Thermo Fisher Scientific (31985062, L3000008 and 13778150). Pierce Fast Western Blot Kit was from Thermo Fisher Scientific (35050). A 12 mm Transwell with 3.0 μm Pore Polycarbonate Membrane Insert was from Corning (3402). ImmPRESS HRP Anti‐Rabbit IgG (Peroxidase) Polymer Detection Kit (MP‐7451‐15) and ImmPACT DAB Peroxidase (HRP) Substrate (SK‐4105) were purchased from Vector Laboratories.
Animals
All animal studies conformed to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health [NIH]) and protocols were approved by the Institutional Animal Care and Use Committee at The Ohio State University (Columbus, OH). Smad3 +/− breeder mice9 were kindly provided by Dr Sushil Rane at NIH (National Institute of Diabetes and Digestive and Kidney Diseases). Individually genotyped and age‐matched littermate male mice (Smad3 +/+ and Smad3 +/−) were used for experiments. Animals were euthanized in a chamber slowly filled with CO2.
Vascular SMC Culture and Transfection
Human AoSMCs were cultured in SMC basal medium with supplements (full medium) in a humidified incubator with 5% CO2 at 37°C and used at passage 5 to 7. For Smad3 plasmid (Addgene, 11742) transfection, AoSMCs were cultured in full medium until 80% to 90% confluence and changed to basal medium (0% fetal bovine serum) 2 hours before transfection. The cells were transfected with the Smad3 plasmid using Lipofectamine 3000 (following the manufacturer's instructions) for 12 hours and cultured with fresh basal medium (no Lipofectamine) for another 12 hours. TGFβ1 (10 ng/mL) was then added and cells were harvested for assays 20 hours after treatment. Transfection with small interfering RNA and treatment with TGFβ1 followed the same procedures and conditions except that the RNAi Max transfection reagent was used (following manufacturer's protocol) at 70% to 80% AoSMC confluency.
Quantitative Real‐Time Polymerase Chain Reaction
Total RNA was extracted from cell lysates using the TRIzol reagent following the manufacturer's instructions (Thermo Fisher Scientific, 15596026) and used for cDNA synthesis with the High‐Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific, 4368814). In each 20 μL reaction, 10 ng of cDNA was amplified through quantitative real‐time polymerase chain reaction (qRT‐PCR) using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, A25778), and mRNA expression was determined using a 7500 Fast Real‐Time PCR System (Applied Biosystems). mRNA levels were normalized to glyceraldehyde 3‐phosphate dehydrogenase using the ∆∆Ct method. qRT‐PCR was performed in triplicate reactions. The primers used are listed in Table S1.
Western Blotting
Cells were lysed in radioimmunoprecipitation assay buffer (50 mmol/L Tris, 150 mmol/L NaCl, 1% Nonidet P‐40, and 0.1% sodium dodecyl sulfate) containing a Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, 78440). Protein concentration was determined using a Pierce BCA Protein Assay kit (Thermo Fisher Scientific, 23227). Whole‐cell lysates were mixed with Laemmli loading buffer and boiled at 95°C for 5 minutes. Proteins were separated by 12% SDS‐PAGE and transferred to a polyvinylidene fluoride membrane. Immunoblotting was performed using specific primary antibodies and a horseradish peroxidase–conjugated secondary antibody. Specific protein bands on blots were illuminated using enhanced chemiluminescent reagents and recorded with an Azure C600 imager (Azure Biosystems). Band intensity was quantified using ImageJ 64 software (https://imagej.nih.gov/ij/). Densitometry data were normalized to GAPDH (loading control). The information for the antibodies used is presented in Table S2.
Proliferation and Migration Assays
Following transfection and TGFβ1 (or solvent control) treatment, AoSMCs were washed once with PBS and then 50 μL of PBS plus 50 μL of CellTiter‐Glo reagent (per well) were added. Cell proliferation (viability) was analyzed using a FlexStation 3 Multi‐Mode Microplate Reader (Molecular Devices, LLC) to read 96‐well plates.
For migration assay, transfected cells were seeded in the Transwell insert with 3 μm pore membrane (Corning Incorporated) containing serum‐free medium (basal medium), and the lower chamber was filled with full medium (with or without TGFβ1). After 24 hours of incubation, the cells inside the insert were removed, and the cells that migrated to the lower surface of the insert were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet.
Luciferase Assay for the Constructs Containing the NRP2 Promoter and/or 5′UTR
The 2.3 kb NRP2 gene promoter (with 5′UTR) (−1500 to +791) or 1.5 kb promoter (without 5′UTR) (−1500 to transcriptional start site) were amplified from human AoSMC genomic DNA and then subcloned into an EMPTY_PROM (switchgear genomics, S790005) promoter reporter vector. The 5′UTR (transcriptional start site to +791) of NRP2 was amplified and subcloned into an EMPTY_5′UTR (switchgear genomics, S690005) 5′UTR Reporter Vector. The primer sequences used for cloning insertion are listed in Table S3. All clones were verified by DNA sequencing. Luciferase assay was performed using the Lightswitch luciferase assay reagent (Switchgear Genomics, LS010) following the manufacturer's instructions. In brief, 24 hours after transfection with luciferase assay plasmid, 5000 AoSMC cells per well were seeded in white 96‐well plates and cultured for 12 hours (no Lipofectamine). Cells were then transfected with an empty vector or Smad3‐overexpressing plasmid (Smad3‐OE) for 24 hours. The cell culture was changed to fresh basal medium and incubated for 12 hours, treated with TGFβ1 (10 ng/mL) for 2 hours, and then used for luciferase assay by adding 100 μL Assay Solution Promega. The plate was incubated at room temperature for 30 minutes before reading in Luminometer System (Applied Biosystems). Luciferase activity was normalized to cell number from duplicate plates.
ChIP Assay
Chromatin immunoprecipitation (ChIP) was performed using the Pierce Magnetic ChIP Kit (Thermo Fisher Scientific, 26157). Briefly, AoSMCs transfected with empty vector or Smad3‐OE (and treated with 10 ng/mL TGFβ1 for 2 hours) were cross‐linked with 1% formaldehyde for 10 minutes at room temperature. Cross‐linking reactions were stopped by the addition of a 1/10 volume of 10× glycine and incubated at room temperature for 5 minutes. The cells were washed with ice‐cold PBS, collected, and then nuclei were extracted after cell lysis. Micrococcal nuclease was added to the nuclei suspension to digest the DNA for 15 minutes at 37°C, and MNase stop solution was added to stop the reaction. The nuclei were recovered and resuspended in IP Dilution Buffer (Thermo Fisher Scientific), and then sonicated (four 5‐second pulses at 20 W for 1×106 cells) to break the nuclear membrane. Chromatin extracts containing DNA fragments with an average size of 500 base pairs were immunoprecipitated overnight at 4°C using an Anti‐Phospho‐Smad3 antibody (Thermo Fisher Scientific, MA5‐14936) or IgG control (included in kit). ChIP‐grade Protein A/G Magnetic Beads were added and incubated for ≈2 to 4 hours at 4°C. RNAse A and Proteinase K were used to digest RNA and protein. The purified DNA was used for qRT‐PCR using Applied Biosystems 7500 Fast Real‐Time PCR System (Applied Biosystems) and PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, A25778). The primers designed to amplify selected regions of the NRP2 promoter or 5′UTR are listed in Table S1.
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assay was performed using 5′‐biotinylated double‐stranded DNA probes (Thermo Fisher Scientific). NRP2 DNA oligos with or without biotin label were used. The sequences (including one with a 5′UTR mutation) are listed in Table S4. Equimolar complementary strands were mixed and heated to 95°C followed by gradual cooling to ambient temperature for at least 5 hours to anneal the probes. Double‐stranded DNA probes (40 fmol) were mixed with 2 μg of nuclear extract from TGFβ1‐treated AoSMCs in 20 μL reaction buffer containing 10 mmol/L Hepes, pH 7.9, 50 mmol/L KCl, 10 mmol/L NaCl, 5 mmol/L MgCl2, 2.5% glycerol, 1 mmol/L DTT, and 0.4 mmol/L ethylenediaminetetraacetic acid. After 20‐minute reaction at room temperature, the protein:DNA complexes were separated at 10°C on a prerun 6% polyacrylamide gel in 0.5X TBE, transferred to a positively charged nylon membrane, and illustrated using a LightShift Chemiluminescent EMSA Kit (Thermo Fisher Scientific, 20148). For competition experiments, unlabeled oligonucleotides in 1‐, 2‐, 5‐, or 10‐fold molar excess were added before incubation with the labeled oligonucleotides. For the super‐shift assay, IgG or Anti‐Phospho‐Smad3 antibody (Thermo Fisher Scientific, MA5‐14936) was added to the reaction mixture and incubated at 4°C for 30 minutes before the addition of the labeled oligonucleotides.
Mouse Femoral Artery Wire Injury Model
Wire injury was performed as previously described.10 Briefly, the mouse was initially anesthetized with 5% of isoflurane and then maintained with 1% to 1.5% at 1 L/min of oxygen flow throughout the surgery. Buprenorphine was subcutaneously injected (0.08 mg/kg; ≈2.4 μg/mouse) before creating a groin skin incision. A 0.015‐inch diameter guide wire (Cook Medical) was inserted through the left deep femoral artery into the common femoral artery, kept still for 1 minute, and then retrieved. The left deep femoral artery was permanently ligated, the skin incision was closed, and the mouse was placed on a warming pad until recovery from anesthesia.
Morphometric Analysis of IH
Mice were euthanized 28 days after wire injury, and the left common femoral arteries were collected after perfusion fixation with 4% paraformaldehyde. The collected arteries were further fixed overnight in 4% paraformaldehyde and then paraffin‐embedded for morphometric analysis or immunohistochemistry. Cross‐sections of 5 μm each were prepared and Verhoeff–van Gieson‐stained. Planimetric measurements were performed using ImageJ by a student blinded to the experimental conditions. Intima area was calculated as internal elastic lamina area minus lumen area. Media area was calculated as external elastic lamina area minus internal elastic lamina area. IH was quantified as a ratio of intima area versus media area. Lumen perimeter rather than area was measured because lumen area quantification succumbs to high variability as a result of deformed (eg, squashed) artery morphology. The mean for each animal was calculated by averaging data from 3 sections. The means from all of the animals in each treatment group were then averaged to produce mean±SEM.
Immunohistochemistry
Cross‐sections were deparaffinized and rehydrated through xylenes and graded alcohol series (Figure S1). Antigen retrieval was performed using citrate buffer for 2 hours at 80°C in a high‐pressure cooker. Endogenous peroxidase was blocked by incubation with 0.3% H2O2 for 10 minutes. ImmPRESS HRP Anti‐Rabbit IgG (Peroxidase) Polymer Detection Kit (Vector Laboratories, MP‐7451‐15) was used to perform Smad3 (Abcam, ab40845) or NRP2 (Thermo Fisher Scientific, PA5‐47274) immunostaining. The staining protein was visualized using ImmPACT DAB Peroxidase (HRP) Substrate. Six different fields on each section were imaged at ×400.
Statistical Analysis
Data are generally presented as mean±SEM derived from 3 independent repeat experiments. Differences between 2 groups were analyzed by 2‐tailed Student t test. For comparison between >2 sets of experimental conditions, we applied 1‐way ANOVA followed by Bonferroni post hoc test. P<0.05 was considered statistically significant. Significance in all figures is indicated as follows: *P<0.05, **P<0.01, ***P<0.001; NS.
Results
Smad3 Loss‐ and Gain‐of‐Function Reduces and Increases NRP2 Expression, Respectively
In pursuit of novel mediators of Smad3 signaling, our pilot microarray data (Figure S2) lent interesting evidence that NRP2 (but not NRP1) was prominently upregulated in TGFβ1–activated SMCs in vitro. We then investigated whether Smad3 regulates NRP2 expression. As shown in Figure 1, our data validated the microarray result by showing that both NRP2 mRNA and protein levels significantly increased in response to TGFβ1 activation. More importantly, Smad3 silencing repressed NRP2 expression, in the absence or presence of TGFβ1. Further confirming Smad3 regulation of NRP2, Smad3 overexpression, as indicated by total and phosphorylated (active) forms, potently upregulated NRP2 mRNA and protein.
Figure 1.
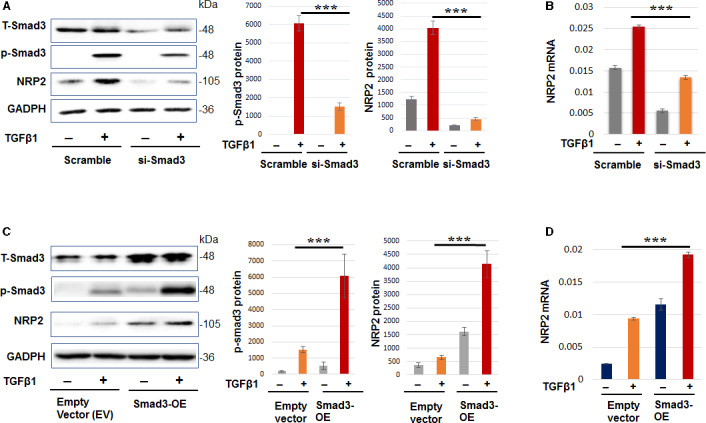
Smad3 regulates neuropilin 2 (NRP2) expression in cultured smooth muscle cells (SMCs).
A–B, Smad3 loss‐of‐function reduces NRP2 expression levels. C–D, Smad3 gain‐of‐function increases NRP2 expression levels. Human primary aortic SMCs were transfected with scrambled small interfering RNA, Smad3‐specific small interfering RNA, empty vector, or Smad3 overexpression plasmid for 12 hours in basal medium (no fetal bovine serum). The cells were cultured for another 12 hours in fresh basal medium (no Lipofectamine) to recover, and then treated with 10 ng/mL of transforming growth factor β for 20 hours before harvest for Western blot and quantitative real‐time polymerase chain reaction (qRT‐PCR) analyses. Quantification: densitometry of Western blots (similar enhanced chemiluminescent exposure) from independent repeat experiments was normalized (to GAPDH) and then averaged to calculate mean±SEM (n=3). Readings of triplicate qRT‐PCR reactions were normalized (to GAPDH) and averaged to calculate mean±SD (n=3). Statistics: 1‐way ANOVA followed by Bonferroni post hoc test; ***P<0.001. TSS indicates transcriptional start site; and WT wild‐type.
Silencing Smad3 Mitigates Pro‐IH SMC Phenotypes and NRP2 Silencing Abolishes Smad3's Enhancement of These Phenotypes
Since it is well established that IH is primarily formed by activated SMCs,7 we intended to confirm the functional role of Smad3 in SMC activation in vitro. The human primary aortic SMCs are known to best preserve properties of mature/differentiated SMCs before activation by a stimulant. Indeed, Smad3 silencing reverted the proliferative and migratory phenotypes of TGFβ1‐activated SMCs (Figure 2A through 2C). Moreover, Smad3 silencing increased α‐smooth muscle actin (a marker of SMC identity) and reduced CD68 (a macrophage marker) in the context of cholesterol stimulation, which is known to transform SMCs to a dedifferentiated and macrophage‐like state.1
Figure 2.
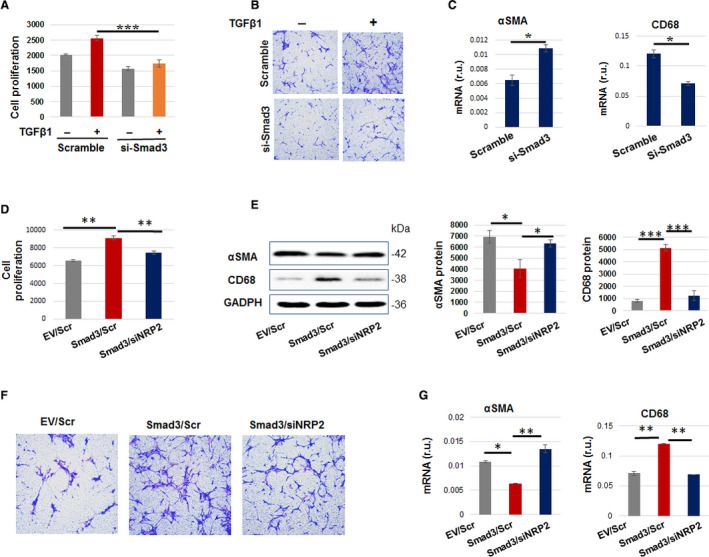
While silencing Smad3 mitigates pro–intimal hyperplasia (IH) smooth muscle cell (SMC) phenotypes, neuropilin 2 (NRP2) silencing abolishes Smad3’s enhancement of these phenotypes.
A–C, Smad3 silencing mitigates pro‐IH SMC phenotypes. D–G, NRP2 silencing abolishes Smad3 overexpression (Smad3‐OE)–enhanced pro‐IH SMC phenotypes. For loss‐ or gain‐of‐function, transfection of human aortic SMCs was performed as described for Figure 2. In proliferation experiments, transfected cells were treated with transforming growth factor β (TGFβ1) (10 ng/mL) for 48 hours before CellTiter‐Glo viability assay. We started with more cells in (D) (vs A), considering 2‐step manipulations (transfection for Smad3 expression followed by that for NRP2 silencing). To monitor migration, transfected cells were seeded in the Transwell insert, the lower chamber filled with full medium (and 10 ng/mL TGFβ1). Cells that migrated to the lower surface of the insert were imaged after 24 hours of incubation. To determine SMC dedifferentiation, transfected cells were induced with cholesterol (10 ng/mL) for 72 hours in basal medium and then harvested for Western blot determination of α‐smooth muscle actin and CD68 protein levels. Quantification (mean±SEM, n=3) was performed as described for Figure 2. Statistics: 1‐way ANOVA followed by Bonferroni post hoc test; *P<0.05, **P<0.01, ***P<0.001. Scramble indicates scrambled small interfering RNA.
We then determined whether NRP2 mediates Smad3's function in SMC activation. We found that whereas Smad3 overexpression enhanced the above‐tested pro‐IH SMC phenotypes, ie, proliferation, migration, and dedifferentiation, silencing NRP2 abolished the enhancement of these phenotypes caused by Smad3 overexpression (Figure 2D through 2G). Thus, NRP2 mediates the promotive role of Smad3 in the pro‐IH SMC activation, a finding not previously reported.
5′UTR of NRP2 is Indispensable for Smad3‐Enhanced Transcriptional Activity
Motivated by the new finding that NRP2 is an effector of Smad3 mediating its function in SMC activation, we next sought to delineate how the transcription factor Smad3 regulates NRP2 expression. Interestingly, while it is most commonly known that transcription factor regulates target gene expression by binding to the gene promoter region, we identified (using software) Smad‐binding elements (SBEs) not only in the NRP2 gene promoter but also in its 5′UTR. Using several luciferase reporter constructs (Figure 3A), we distinguished the relative importance of the NRP2 promoter and 5′UTR for Smad3‐activated luciferase signal. Surprisingly, whereas the NRP2 promoter alone did not significantly enhance transcriptional activation following Smad3 overexpression, the NRP2 5′UTR alone (driven by the generic β‐actin promoter, which Smad3 does not bind) was equally effective in facilitating Smad3‐enhanced transcriptional activation compared with the construct containing both the NRP2 promoter and 5′UTR (Figure 3B). These results indicate that the 5′UTR is necessary and sufficient for Smad3‐regulated NRP2 transcription.
Figure 3.
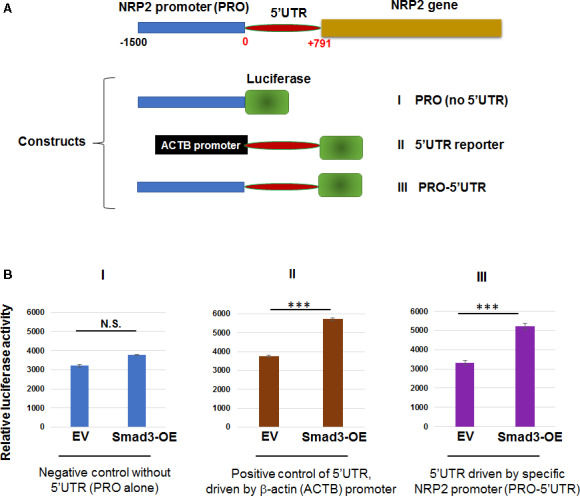
The 5′ untranslated region (5′UTR) but not promoter of the neuropilin 2 (NRP2) gene is responsible for Smad3‐enhanced luciferase reporter activity.
A, Schematic of constructs containing the NRP2 gene promoter and/or 5′UTR. The β‐actin (ACTB) promoter is contained in the original Addgene luciferase reporter vector where no Smad‐binding elements are found, thus serving as a negative promoter control. B, Luciferase assay using aortic smooth muscle cells transfected with constructs listed in (A). Cells transfected with empty vector (EV) or Smad3‐overexpression (Smad3‐OE) were seeded in 96‐well plates and cultured for 12 hours. The cells were then transfected with the indicated luciferase constructs for 36 hours in basal medium and treated with transforming growth factor β (TGFβ1) (10 ng/mL) for 2 hours before adding the luciferase assay solution. Luciferase‐catalyzed bioluminescence reading was normalized to cell numbers in duplicate plates. Data are presented as mean±SD. Statistics: 2‐tailed Student t test, n=8; ***P<0.001; NS. The construct of NRP2 promoter alone without 5′UTR (PRO) served as a negative control for the 5′UTR activity. The construct of 5′UTR driven by the generic ACTB promoter where Smad3 does not bind served as a positive control. The similar results from this positive control and the construct of 5′UTR driven by the specific NRP2 promoter indicate a major contribution of the 5′UTR (vs the NRP2 promoter) to the Smad3‐mediated transcriptional activity, as measured through the luciferase reporter.
5′UTR Region of +51 to +78 bp After the Transcription Start Site Accounts for a Strong Association With Smad3
We then determined the Smad3 transcriptional mechanism in greater depth. Through ChIP followed by qRT‐PCR, we found that among the 4 SBE‐containing sites (chosen based on software LASAGNA‐Search 2.0 and literature11) spread in the NPR2 promoter and 5′UTR (Figure 4A), only the 5′UTR site at +51 to +78 after the transcriptional start site exhibited a strong association with p‐Smad3 (Figure 4B). Electrophoretic mobility shift assay confirmed the p‐Smad3 binding with the biotin‐tagged oligo containing this 5′UTR DNA sequence (denoted as S3; Figure 4C and 4D). Furthermore, this p‐Smad3/S3 binding was abolished by a mutation in the S3 sequence, indicating the specificity of this interaction (Figure 4E). In addition, including a p‐Smad3 antibody in the binding reaction further hindered the motility of the p‐Smad3/S3‐oligo complex (Figure 4F). These and the ChIP–qPCR results demonstrate that Smad3 regulates NRP2 transcription primarily via an occupancy at its 5′UTR rather than the promoter. Taken together, these in vitro results indicate that the expression of NRP2, a recently known pro‐IH signaling factor,7 is regulated by the master transcription factor Smad3.
Figure 4.
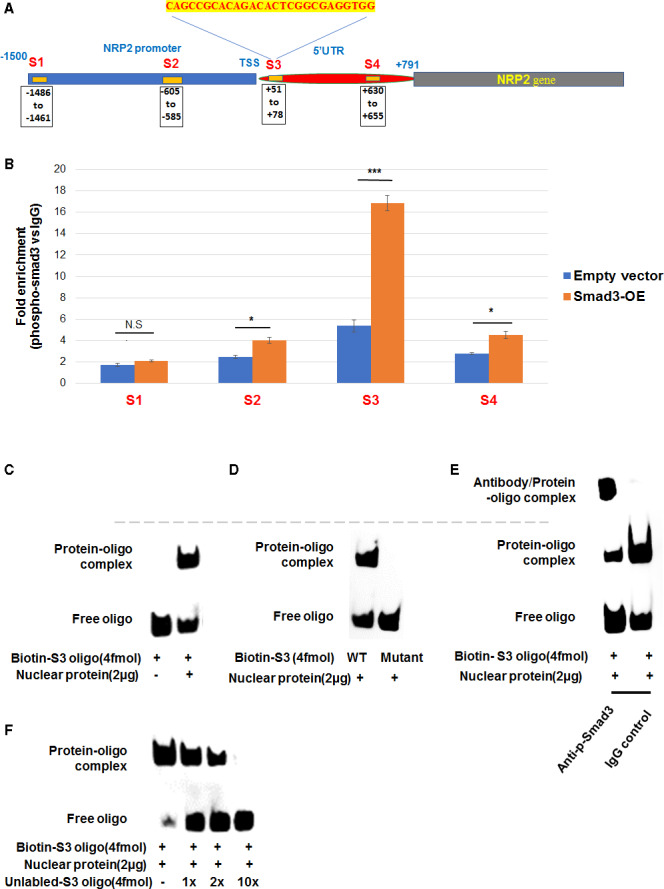
The (+51 to +78) site of 5′ untranslated region (5′UTR) of the neuropilin 2 (NRP2) gene shows the strongest association with Smad3.
A and B, Chromatin immunoprecipitation (ChIP)–quantitative real‐time polymerase chain reaction (qRT‐PCR) assay. Aortic smooth muscle cells transfected with empty vector or Smad3‐overexpression (Smad3‐OE) plasmid were treated with solvent or transforming growth factor β (TGFβ1) (10 ng/mL) for 2 hours and then fixed for ChIP. Precipitated and purified DNA samples were used for quantitative real‐time polymerase chain reaction to detect the Smad‐binding element–containing sites specified in (A) as S1–S4. Data are presented as mean±SD. Statistics: 2‐tailed Student t test, n=8; *P<0.05, ***P<0.001; NS. C through F, Electrophoretic mobility shift assay. Cells were treated with TGFβ1 (10 ng/mL) for 2 hours before nuclear extraction and biotin‐labeled oligo detection (C), as detailed in the Methods section. For the competition experiments (D), unlabeled oligonucleotides in 1×, 2×, 5×, or 10× molar excess were added before incubation with the labeled oligonucleotides. To indicate the protein/oligo binding specificity, a mutant version of the S3 oligo was used (E). For super‐shift assay (F), IgG or anti‐phospho‐Smad3 antibody was incubated with the reaction mixture before the addition of labeled oligonucleotides. Shown each is 1 of 3 similar independent experiments. Dashed line marks the same mobility shift position. TSS indicates transcriptional start site; and WT, wild‐type.
NRP2 Protein Levels are Lower in Arteries of Smad3‐Haploinsufficient Mice Than That in WT Mice
Given the above results, we looked for in vivo evidence for the Smad3 regulation of NRP2 expression. Indeed, based on immunostaining on cross‐sections of wire‐injured arteries, we observed reduced NRP2 protein levels (≈60%) in Smad3‐haploinsufficient mice compared with that of WT control (Figure 5). The specificity of the assay was verified by nearly no NRP2 staining in the negative control (no primary antibody). Based on this result and the recently recognized role of NRP2 as a pro‐IH factor in vivo,7 we anticipated that reduced Smad3 should lead to less IH.
Figure 5.
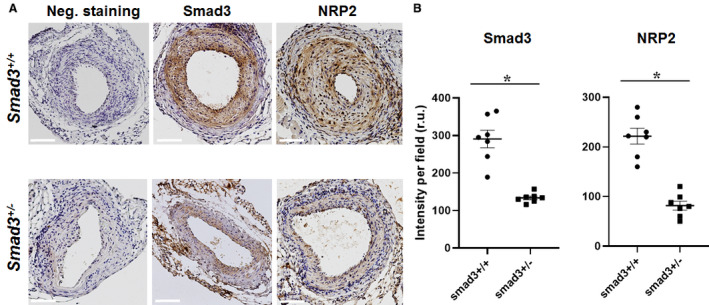
Neuropilin 2 (NRP2) is reduced in arteries of Smad3‐haploinsufficient (vs wild‐type) mice.
Wire injury experiments were performed with Smad3 +/+ and Smad3 +/− mice as described in the Methods section. Immunohistochemistry was performed on cross‐sections of injured arteries collected 28 days after wire injury. A, Representative immunostained cross‐sections. Negative staining: no primary antibody. Scale bar: 50 μm. B, Quantification of immunohistochemistry analysis. Colorimetric intensity (per image field) on a total of 7 cross‐sections from 4 animals was averaged to calculate mean±SEM (n=4 mice). Statistics: unpaired Student t test; *P<0.05. 5′UTR indicates 5′ untranslated region; ACTB, β‐actin; and Smad3‐OE, Smad3 overexpression.
IH in Wire‐Injured Femoral Arteries is Mitigated in Smad3‐Haploinsufficient Versus WT Mice
As introduced earlier, conflicting reports showed that IH increased either after Smad3 ablation5 or overexpression.4 We used Smad3‐haploinsufficient mice to avoid: (1) potential artificial effects accompanying virus‐mediated transgene expression in our previous studies,4, 12 (2) variability (heterogeneous baseline) resulting from abnormal development of homozygous Smad3 KO mice,9 and (3) possible confounding secondary (eg, compensatory) effects due to the extreme condition of complete systemic Smad3 depletion. Individually genotyped littermate Smad3 +/+ and Smad3 +/− mice were used. To induce IH, we performed an authentic, widely accepted model of wired‐injured femoral artery.10 Morphometric quantification indicated 47±17% less IH (measured as intima/media area ratio) in femoral arteries collected at postinjury 28 days from Smad3 +/− mice in comparison to that in Smad3 +/+ mice (Figure 6). Accordingly, lumen size was enlarged (Smad3 +/− versus Smad3 +/+) by ≈40% albeit without reaching statistical significance. This result provided strong evidence that Smad3 was pro‐IH in the experimental setting used herein, consistent with our previous reports using a rat model of carotid artery angioplasty–induced IH.4, 12
Figure 6.
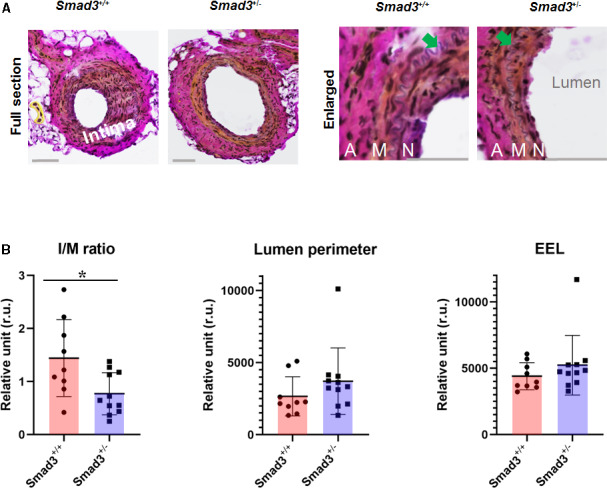
Intimal hiperplasia (IH) is reduced in wire‐injured arteries of Smad3‐haploinsufficient (vs wild‐type [WT]) mice.
A, Representative Verhoeff–van Gieson‐stained artery cross‐sections showing (neo)intima. Wire injury of mouse femoral artery was performed as described in the Methods section. Injured arteries were collected from Smad3 +/+ (WT) and Smad3 +/− (haploinsufficient) mice at 28 days postinjury. Scale bar: 50 μm; arrow points to the internal elastic lamina. A indicates adventitia; M, media; N, neointima. B, Quantitative morphometric analysis. Planimetric measurements are described in the Methods section. IH is evaluated by the intima/media area ratio (I/M). Data from 3 sections were pooled to generate the mean for each animal. The means from all animals in each group were then averaged to calculate a final mean±SEM. Statistics: unpaired Student t test; *P<0.05, n=11 WT or 9 haploinsufficient mice. EEL indicates external elastic lamina.
Discussion
In view of the critical importance of IH in cardiovascular diseases, we have made 2 significant findings: (1) Smad3 controls NRP2 transcription and binds to its 5′UTR––a molecular regulation involved in pro‐IH SMC phenotypes in vitro; and (2) injury‐induced IH and NRP2, a known pro‐IH factor in vivo,7 are both substantially reduced as a result of Smad3 haploinsufficiency in mice. Thus, this study provides unique mechanistic insight into Smad3‐enhanced pro‐IH SMC phenotypes, and also genetic evidence for the role of Smad3 in IH–a long‐held puzzle.
Inasmuch as IH is a key pathological process in major (re)stenotic vascular diseases,1, 7 it is critically important to understand the underlying molecular mechanisms. We identified NRP2 as a novel mediator of Smad3 signaling in effectuating the pro‐IH phenotypes of activated SMCs. During the experiments leading to this finding, we first observed that NRP2 (but not NRP1) mRNA increased in response to TGFβ1 activation of SMCs. We then distinguished that it was Smad3 not Smad2 that was responsible for TGFβ1‐induced NRP2 upregulation (Figure S3). Moreover, pro‐IH SMC phenotypes were enhanced by increasing Smad3 expression, and this enhancement was abrogated by NRP2 silencing. Importantly, we further uncovered that Smad3 as a transcription factor governed NRP2 transcription nearly entirely through its occupancy at the NRP2 gene's 5′UTR. Specifically, although software‐predicted SBEs exist in the NRP2 gene promoter, our data indicated that it was the 5′UTR rather than the promoter that was indispensable for Smad3‐enhanced NRP2 transcription. Moreover, among the tested SBEs, only the one at the beginning (+51 to +78 bp after transcriptional start site) of the 5′UTR exhibited marked specific binding with the Smad3 protein. Although not rare,13 that Smad3 assumes its transcription factor function by binding with the 5′UTR of its target gene is somewhat unexpected given numerous SBE sites identified in promoters of known Smad3 target genes.11
Consistent with the in vitro results, NRP2 protein levels were indeed reduced in Smad3‐haploinsufficient mouse arteries compared with WT control. NRP2 was recently recognized as an IH‐promoting factor, as supported by the result that loss and gain of NRP2 function significantly reduced and enhanced IH, respectively, in the IH model of balloon angioplasty of rat carotid artery.7 Therefore, our finding of Smad3 controlling NRP2 expression lends important evidence to the interpretation of the in vivo role of Smad3 in IH.
Smad3 is a master transcription factor involved in many cellular functions and disease conditions.8 However, whether Smad3 is a positive or negative regulator in IH has been debatable, imposing a dilemma to therapeutic targeting. Early studies suggested that tuning down TGFβ1 or its receptor function mitigates IH,14, 15 although the role of Smad3 was not specifically addressed. Via adenoviral transgenic studies in the model of balloon‐injured rat carotid arteries, we observed that while increasing Smad3 exacerbated IH,12 nullifying its function by expressing its antagonizer Smad7 mitigated IH.4 However, opposing this result, a study by Kobayashi et al5 using knockout mice showed exacerbated IH caused by Smad3 ablation.
To revisit this conundrum, herein we used Smad3‐haploinsufficient mice (instead of homozygotes) subjected to femoral wire injury, a widely accepted IH model,10, 14 so as to minimize compensatory or other secondary effects (eg, abnormal development) possibly caused by the extreme condition of complete Smad3 depletion. The results support a positive role of Smad3 in IH, consistent with our previous reports using the rat angioplasty model.4, 12 Although seemingly contradictory, the conclusions from 2 research groups both using Smad3 knockout mice may bear rationales as follows: (1) IH is complex and context‐dependent, thus its attenuation or exacerbation can be strongly influenced by different experimental models. In the model used by Kobayashi et al,5 laser‐induced photochemical reactions damage the endothelial layer; hence, endothelial cell apoptosis, inflammation, and immune responses may play a major pathogenic role in IH. By contrast, in the rat and mouse artery injury models used by many others,10 it is the physical insult that denudes the endothelium and overstretches the artery wall where SMCs respond by changing to migro‐proliferative and dedifferentiated phenotypes. (2) The consequences of activated TGFβ1/Smad3 signaling are also context‐dependent.8 For example, TGFβ1/Smad3 signaling is deemed immune‐repressive in some inflammatory conditions. However, in cultured SMCs, elevated TGFβ1/Smad3 signaling stimulates the expression of proinflammatory cytokines.3, 12 (3) Even under the same stimulation, SMCs may undergo changes toward opposite directions depending on its differentiated (or dedifferentiated) state to start with. For example, TGFβ1/Smad3 activation is commonly applied to promote progenitor cell differentiation into SMCs.2 However, in the case of highly differentiated SMCs, such as primary human aortic SMCs used in this study, elevated TGFβ1/Smad3 signaling reprograms SMCs toward dedifferentiation exhibiting proliferative/migratory behaviors.3, 4 Whether the above suggested possible reasons underlie the disparate outcomes from different studies remains unclear at present. Moreover, our data presented here cannot distinguish whether Smad3 was solely activated by TGFβ in vivo, given that angiotensin II could also activate the Smad pathway independently of TGFβ in rat aortic arteries.16 In addition, conditional Smad3 knockout specifically in SMCs will benefit further delineation of Smad3‐directed regulations in vivo. Nevertheless, our new study using Smad3‐haploinsufficient mice together with the evidence from previous rat models4, 12 support that Smad3 promotes mechanical injury–induced IH. This conclusion is further enhanced by a recent report where in vitro and in vivo data indicated NRP2 as a pro‐IH factor,7 and also by our data consistently identifying NRP2 as a mediator of Smad3 signaling.
Conclusions
Our results together with the literature sketch out a coherent picture. That is, while the Smad3 transcriptional control of NRP2 represents a novel signaling axis mediating pro‐IH SMC activation in vitro, the in vivo results provide clear genetic evidence for a positive role of Smad3 in injury‐induced IH. Considering the multifaceted, contextual nature of Smad3 functions, identification of the specific role of Smad3/NRP2 in the pro‐IH SMC activation is expected to benefit precise and effective anti‐IH therapeutic targeting.
Sources of Funding
This work was supported by NIH grants R01HL‐068673 (to Kent) and R01HL129785 (to Kent and Guo), and R01HL133665 (to Guo).
Disclosures
None.
Supporting information
Tables S1–S4
Figures S1–S3
Acknowledgments
We thank Dr Sushil Rane for providing Smad3 +/− breeder mice and Dr Paul Tang for assistance in obtaining this mouse strain. We also thank Dr Matt Stratton for discussions.
(J Am Heart Assoc. 2020;9:e015487 DOI: 10.1161/JAHA.119.015487.)
Dr Xie and Dr Urabe are co‐first authors.
For Sources of Funding and Disclosures, see page 12.
Contributor Information
Lian‐Wang Guo, Email: lianwang.guo@osumc.edu.
K. Craig Kent, Email: kc.kent@osumc.edu.
References
- 1. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, et al. KLF4‐dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dong K, Guo X, Chen W, Hsu AC, Shao Q, Chen JF, Chen SY. Mesenchyme homeobox 1 mediates transforming growth factor‐beta (TGF‐beta)‐induced smooth muscle cell differentiation from mouse mesenchymal progenitors. J Biol Chem. 2018;293:8712–8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shi X, DiRenzo D, Guo LW, Franco SR, Wang B, Seedial S, Kent KC. TGF‐beta/Smad3 stimulates stem cell/developmental gene expression and vascular smooth muscle cell de‐differentiation. PLoS One. 2014;9:e93995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsai S, Hollenbeck ST, Ryer EJ, Edlin R, Yamanouchi D, Kundi R, Wang C, Liu B, Kent KC. TGF‐beta through Smad3 signaling stimulates vascular smooth muscle cell proliferation and neointimal formation. Am J Physiol Heart Circ Physiol. 2009;297:H540–H549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kobayashi K, Yokote K, Fujimoto M, Yamashita K, Sakamoto A, Kitahara M, Kawamura H, Maezawa Y, Asaumi S, Tokuhisa T, et al. Targeted disruption of TGF‐beta‐Smad3 signaling leads to enhanced neointimal hyperplasia with diminished matrix deposition in response to vascular injury. Circ Res. 2005;96:904–912. [DOI] [PubMed] [Google Scholar]
- 6. Cao S, Yaqoob U, Das A, Shergill U, Jagavelu K, Huebert RC, Routray C, Abdelmoneim S, Vasdev M, Leof E, et al. Neuropilin‐1 promotes cirrhosis of the rodent and human liver by enhancing PDGF/TGF‐beta signaling in hepatic stellate cells. J Clin Invest. 2010;120:2379–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pellet‐Many C, Mehta V, Fields L, Mahmoud M, Lowe V, Evans I, Ruivo J, Zachary I. Neuropilins 1 and 2 mediate neointimal hyperplasia and re‐endothelialization following arterial injury. Cardiovasc Res. 2015;108:288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. David CJ, Massague J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat Rev Mol Cell Biol. 2018;19:419–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yadav H, Quijano C, Kamaraju AK, Gavrilova O, Malek R, Chen W, Zerfas P, Zhigang D, Wright EC, Stuelten C, et al. Protection from obesity and diabetes by blockade of TGF‐beta/Smad3 signaling. Cell Metab. 2011;14:67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takayama T, Shi X, Wang B, Franco S, Zhou Y, DiRenzo D, Kent A, Hartig P, Zent J, Guo LW. A murine model of arterial restenosis: technical aspects of femoral wire injury. J Vis Exp. 2015;(97). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Martin‐Malpartida P, Batet M, Kaczmarska Z, Freier R, Gomes T, Aragon E, Zou Y, Wang Q, Xi Q, Ruiz L, et al. Structural basis for genome wide recognition of 5‐bp GC motifs by SMAD transcription factors. Nat Commun. 2017;8:2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shi X, Guo LW, Seedial S, Takayama T, Wang B, Zhang M, Franco SR, Si Y, Chaudhary MA, Liu B, et al. Local CXCR4 upregulation in the injured arterial wall contributes to intimal hyperplasia. Stem Cells. 2016;34:2744–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cordova G, Rochard A, Riquelme‐Guzman C, Cofre C, Scherman D, Bigey P, Brandan E. SMAD3 and SP1/SP3 transcription factors collaborate to regulate connective tissue growth factor gene expression in myoblasts in response to transforming growth factor beta. J Cell Biochem. 2015;116:1880–1887. [DOI] [PubMed] [Google Scholar]
- 14. Liao M, Yang P, Wang F, Berceli SA, Ali YH, Chan KL, Jiang Z. Smooth muscle cell‐specific Tgfbr1 deficiency attenuates neointimal hyperplasia but promotes an undesired vascular phenotype for injured arteries. Physiol Rep. 2016;4:e13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Khan R, Agrotis A, Bobik A. Understanding the role of transforming growth factor‐beta1 in intimal thickening after vascular injury. Cardiovasc Res. 2007;74:223–234. [DOI] [PubMed] [Google Scholar]
- 16. Rodriguez‐Vita J, Sanchez‐Lopez E, Esteban V, Ruperez M, Egido J, Ruiz‐Ortega M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor‐beta‐independent mechanism. Circulation. 2005;111:2509–2517. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S4
Figures S1–S3