

Abstract
Background
Although multiple signaling cascades and molecules contributing to the pathophysiological process have been studied, the treatments for stroke against present targets have not acquired significant clinical progress. Although CARD3 (caspase activation and recruitment domain 3) protein is an important factor involved in regulating immunity, inflammation, lipid metabolism, and apoptosis, its role in cerebral stroke is currently unknown.
Methods and Results
Using a mouse model of ischemia‐reperfusion (I‐R) injury based on transient blockage of the middle cerebral artery, we have found that CARD3 expression is upregulated in a time‐dependent manner during I‐R injury. Further animal study revealed that, relative to control mice, CARD3‐knockout mice exhibited decreased inflammatory response and neuronal apoptosis, with reduced infarct volume and lower neuropathological scores. In contrast, neuron‐specific CARD3‐overexpressing transgenic (CARD3‐TG) mice exhibited increased I‐R induced injury compared with controls. Mechanistically, we also found that the activation of TAK1 (transforming growth factor‐β–activated kinase 1) was enhanced in CARD3‐TG mice. Furthermore, the increased inflammation and apoptosis seen in injured CARD3‐TG brains were reversed by intravenous administration of the TAK1 inhibitor 5Z‐7‐oxozeaenol.
Conclusions
These results indicate that CARD3 promotes I‐R injury via activation of TAK1, which not only reveals a novel regulatory axis of I‐R induced brain injury but also provides a new potential therapeutic approach for I‐R injury.
Keywords: apoptosis, caspase activation and recruitment domain 3, inflammation, ischemia reperfusion injury
Subject Categories: Ischemic Stroke, Cerebrovascular Disease/Stroke
Nonstandard Abbreviations and Acronyms
- CARD3
caspase activation and recruitment domain 3
- CBF
cerebral blood flow
- DMSO
dimethyl sulfoxide
- ICA
internal carotid artery
- I‐R
ischemia‐reperfusion
- JNK
c‐Jun N‐terminal kinase
- LPS
lipopolysaccharide
- MAPKs
mitogen‐activated protein kinase
- MCA
middle cerebral artery
- MDP
muramyl dipeptide
- OCT
optimal cutting temperature compound
- OGD
oxygen and glucose deprivation
- PCR
polymerase chain reaction
- PDGF
platelet‐derived growth factor
- qPCR
quantitative PCR
- TAK1
transforming growth factor‐β‐activated kinase 1
- tMCAO
transient middle cerebral artery occlusion model
- TUNEL
terminal dexynucleotidyl transferase(Tdt)‐mediated dutp nick end labeling
Clinical Perspective
What Is New?
CARD3 (caspase activation and recruitment domain 3) promotes ischemia‐reperfusion injury by boosting inflammation and apoptosis.
CARD3 mediates NF‐κB (nuclear factor kappa‐light‐chain‐enhancer of activated B cells) and JNK (C‐Jun N‐terminal kinase)/p38 MAPK (mitogen‐activated protein kinase) signaling to promote neuron death by activating TAK1 (transforming growth factor‐β‐activated kinase 1).
What Are the Clinical Implications?
Targeting CARD3‐TAK1 axis might provide a new approach for exploring drugs and therapeutic strategies against ischemia‐reperfusion injury.
Despite recent advancements in therapeutic approaches, stroke is still one of the top leading causes of death worldwide. According to World Health Organization estimates, about 5.9 million people die from stroke annually, and more than 16.9 million individuals suffer from this malady.1 Approximately 87% of these events represent ischemic stroke.2 Intravascular surgery, including angioplasty/stenting and mechanical thrombectomy, along with pharmacological thrombolysis using t‐PA (tissue plamnipen activator) has been clinically demonstrated as effective interventions for stroke.3, 4, 5 However, ischemia‐reperfusion (I‐R) injury following vascular recanalization is still a critical complication that affects clinical prognosis.4, 6 A plethora of experimental and clinical investigations of I‐R injury have demonstrated that multiple pathophysiological processes may be involved in causing I‐R injury. This list includes excitotoxicity, inflammation, immune response, oxidative and nitrosative stress, endoplasmic reticulum stress, apoptosis, autophagy, and others.5, 7, 8, 9 Nevertheless, treatments based on these mechanisms have yielded less satisfactory clinical outcomes. Thus, additional mechanisms associated with I‐R injury still need to be investigated in the context of treatment for stroke.
CARD3 (caspase activation and recruitment domain 3) protein (also known as Rip2, RIPK, RICK or CARDIAK) is a 62‐kDa adaptor that is extensively expressed in liver, heart, brain, lymph nodes, and lung.10 CARD3 was originally identified as a serine/threonine kinase that provides a vital function in immunity against bacterial infection.11 Upon stimulation by muramyl dipeptide or lipopolysaccharide, CARD3 participates in both “classical” and “alternative” activation of nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB) pathway upon NOD1/2 (an intracellular receptor of bacterial peptidoglycan) stimulation.12 CARD3 can also regulate CD95/Fas receptor signaling through an interaction with CLARP (caspase‐like apoptosis‐regulatory protein)13, 14 that induces epithelial cell death via release of LDH (lactate dehydrogenase). Through activation of VEGFR (vascular endothelial growth factor receptor) and ERK1/2 (extracellular regulated protein kinases), CARD3 is known to enhance myocardial damage.15 Similarly, a previous study from our laboratory demonstrated that CARD3 increases cardiomyocyte apoptosis and ventricular remodeling via activation of NF‐κB and p38 signaling.16 Other recent reports have shown that CARD3 plays critical roles in the central nervous system. For example, mice deficient in CARD3 exhibit markedly reduced dendritic cell recruitment in experimental autoimmune encephalomyelitis.17 Under neurotrophin stimuli, CARD3 also has a prosurvival function in regulating cerebellar granule neuron development via competition with TRAF6 for binding p75 neurotrophin receptor.18, 19 Despite these initial clues, the exact role of CARD3 in cerebral I‐R damage remains unknown.
Our current study delineates the involvement of CARD3 in the process of I‐R injury by regulating inflammation and neuro‐apoptosis. We have observed that the expression of CARD3 is markedly elevated in an I‐R injury model and in neurons subjected to oxygen and glucose deprivation (OGD). Transgenic overexpression of CARD3 increases the extent of infarct areas in the transient middle cerebral artery occlusion (tMCAO) model, and promotes inflammation, and apoptosis following a 6 hours reperfusion. Consistently, CARD3 ablation mitigates the aforementioned processes. We further demonstrate that CARD3 mediates NF‐κB and JNK (C‐Jun N‐terminal kinase)/p38 MAPK signaling by activating the central modulator TAK1 (transforming growth factor‐β–activated kinase 1), leading to neuron death. Therefore, targeting the CARD3/TAK1 axis represents a promising new therapeutic approach to treatment of I‐R damage.
Materials and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Mice
All experimental procedures have been approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University and carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All animal experiments were performed on 10‐ to 12‐week‐old male mice. All mice were on C57BL/6J background. CARD3 KO (knockout) mice were obtained from Jackson Laboratory (IRRID:IMSR_JAX:007017; Bar Harbor, ME), as previously described.16 The CARD3−/− mice were crossed with male C57BL/6 mice to obtain the CARD3 heterozygote mice. Then the CARD3 heterozygote were repeatedly crossed to obtain CARD3−/− and littermate WT (wild type) mice for further study. CARD3 neuron‐specific transgenic mice (TG) were established by inserting cloned full‐length mouse CARD3 cDNA downstream of the neuron‐specific platelet‐derived growth factor B‐chain promoter as previously described.20, 21 This plasmid was microinjected into fertilized embryos (C57BL/6 background). Six transgenic lines were generated, and genomic tail DNA was used for polymerase chain reaction (PCR) analysis. The primers used were forward: 5′‐GCTCTTCGTTGAAGGCACAC‐3′ and reverse: 5′‐TCTGGCTCACAATGGCTTCC‐3′. Based on these analyses, four lines were selected for further study. Throughout the investigation, genotyped mice were identified with earmarks. Investigators were blinded as to mouse genotypes and other cohort details. Totally, 240 mice were involved in this study. Among these mice, two WT mice, one CARD3‐KO mice, one NTG mouse, and three CARD3‐TG mice died after operation and were excluded from this study. Two hundred and thirty‐three mice, including 78 WT mice, 42 CARD3‐KO mice, 56 NTG mice, and 57 CARD3‐TG mice, were used for further experiments.
Transient Middle Cerebral Artery Occlusion Model
We constructed the cerebral I‐R model based on transient blockage of the proximal middle cerebral artery, as previously described.21 Briefly, mice were anesthetized with 3% isoflurane in O2. Following surgical exposure of the left carotid artery, a 6‐0 silicon‐coated monofilament (Doccol, Sharon, MA, 602156PK5) was inserted into the internal carotid artery via the external carotid artery, temporarily blocking blood flow to the middle cerebral artery (MCA). After a 45‐minute blockage, the monofilament was slowly retracted from the internal carotid artery (ICA), and the mouse was kept immobilized for at least 10 minutes. Throughout the procedure, left cerebral blood flow (CBF) was monitored with a Doppler flowmeter (Periflux System 5010, Perimed, Stockholm, Sweden) and the regional CBF change in different stages of the four groups are shown in Figure S1A. We found that a >70% decrease in CBF after occlusion and a >60% restoration after reperfusion showed a consistent I‐R injury phenotype via TTC staining and relatively low mortality. Therefore, a successful tMCAO experiment was defined by a CBF reduction of more than 70% during the ischemic stage and subsequent restoration to at least 60% of the control baseline CBF. Unsuccessful tMCAO models were excluded in this study. For sham controls, the monofilament was withdrawn immediately after the initial onset of reduced CBF. Rectal temperature remained at 37±0.5°C throughout the surgery. Mice were allowed to recover in a 37°C incubator for specified time periods and were allowed unrestricted access to food and water.
Neurological Impairment Scores
After reperfusion for designated periods of time, neurological impairment due to the tMCAO procedure was evaluated according to the following nine‐point scale21, 22 as shown in Table S1. The investigators were blinded to carry out all assessments of neurological deficits.
Measurement of Infarct Size
Following various times of reperfusion, mice were euthanized by an overdose of 3% sodium pentobarbital. Intact brains were carefully collected and then sliced into seven consecutive 1 mm coronal sections from 2 mm posterior to the olfactory bulb. Sections were incubated with 2% TTC (2,3,5‐triphenyltetrazolium chloride, Sigma‐Aldrich, St. Louis, MO, 057K0694) solution for 15 minutes and then fixed with 10% formalin solution for 24 hours. Sections were photographed for analysis with Image‐Pro Plus 6.0 software (Media Cybernetics, Rockville, MD). Normal brain tissue is stained red by this procedure, whereas infarct tissue and penumbra appear pale or gray. Total infarct volumes were computed using a previously described formula23, 24: volume of the contralateral hemisphere−the volume of the nonlesioned ipsilateral hemisphere)/(contralateral volume×2)×100%. All investigators were blinded to the mice genotype.
Immunofluorescence Staining, Fluoro‐Jade B Staining, and TUNEL Assay
For histopathological staining, the mice were euthanized and transcardially perfused with 20 mL 0.1 mol/L PBS pH 7.4, followed by 4% paraformaldehyde solution for 8 minutes. Brains were then carefully collected and immersed in 4% paraformaldehyde solution at room temperature for 2 additional hours. Subsequently, the brains were dehydrated with 20% sucrose solution in PBS, overnight, followed by 30% sucrose for 6 hours. Brains were then embedded in OCT (Optimal Cutting Temperature Compound), and 8‐μm‐thick serial coronal sections were prepared using a cryostat microtome. For antigen retrieval, sections were incubated in pH 9.0 EDTA solution at 86°C for 5 minutes. Prior to labeling procedures, sections were incubated with 10% goat serum at 37°C for 1 hour. For cultured primary neurons, coverslips were fixed in 4% paraformaldehyde for 15 minutes and then incubated in 10% goat serum. For labeling, sections or coverslips were incubated overnight at 4°C with the following primary antibodies: (anti‐CARD3, Cell Signaling Technology Cat# 4142, RRID:AB_2716277; anti‐MAP2, Abcam, Cambridge, UK, Cat# ab5392, RRID:AB_2138153; anti‐NeuN, Millipore, Bedford, MA, Cat# ABN91, RRID:AB_11205760). After washing three times with PBS, sections were incubated with appropriate secondary antibodies for 1 hour at 37°C. Fluorescent‐dye‐labeled second antibodies were as follows: (anti‐rabbit IgG, Thermo Fisher Scientific, Waltham, MA, Cat# A‐21206, RRID:AB_2535792; anti‐mouse IgG, Thermo Fisher Scientific Cat# A10037, RRID:AB_2534013; anti‐chicken, Abcam Cat# ab96947, RRID:AB_10681017). Finally, nuclei were stained with DAPI (DAPI, Invitrogen, Carlsbad, CA, Cat# S36939). Fluoro‐Jade B (AG310, Millipore) according to the manufacturer's instructions. An ApopTag Plus In Situ Apoptosis Fluorescein Detection Kit (S7111; Chemicon) was used to perform TUNEL (Terminal Dexynucleotidyl Transferase(Tdt)‐Mediated Dutp Nick End Labeling) assays according to the manufacturer's protocol. Fluorescence images were acquired using an OLYMPUS DX51 microscope and analyzed with Image‐Pro Plus software.
Sample Preparation for Real‐Time Quantitative PCR and Western Blotting
Mice used for quantitative PCR (qPCR) and Western blotting were anesthetized with sodium pentobarbital and transcardially perfused with 20 mL cold sodium phosphate buffer. Brains were then quickly removed. Both the rostral and caudal 1 mm of the left cerebral hemisphere were excised off. The remaining brain tissue (including the infarct and peri‐infarct areas) was immediately frozen in liquid nitrogen and subsequently stored in −80°C freezer.
Western Blotting Analysis
Proteins extracted from peri‐infarct areas were separated on 8% SDS‐PAGE gels and electrophoretically transferred to polyvinylidene difluoride membranes (Millipore). After immersion in a 5% solution of skim milk at room temperature for 1 hour, membranes were incubated overnight at 4°C with appropriate primary antibodies. After washing to remove primary antibody, membranes were subsequently incubated with corresponding secondary antibodies at room temperature for 1 hour. Finally, the expression of specific proteins was assessed using Bio‐Rad imaging system (ChemiDoc™ XRS+). All antibodies used for Western blotting are listed in Table S2.
Quantitative Real‐Time PCR Analysis
Total RNA was extracted from the snap‐frozen tissue samples using Tripure Isolation Reagent (Roche, Basel, Switzerland, 7950567275) according to the manufacturer's protocol. Reverse transcription of RNA to yield cDNA was performed using a Transcriptor First Strand cDNA Synthesis Kit (Roche, 04896866001). The LightCycler 480 SYBR Green 1 Master Mix (Roche, 04887352001) was used to perform real‐time PCR analysis with the LightCycler 480 qPCR System (Roche). Relative quantities of target mRNAs were normalized to the quantity of control GAPDH mRNA. Primer pairs used for PCR analysis are presented in Table S3.
Culture of Primary Neurons for Mimicking I‐R Injury In Vitro
Primary cortical neurons were prepared from embryos of pregnant Sprague‐Dawley rats (E15‐E18) as previously described.24 In brief, minced cerebral cortex were dissociated with 0.125% trypsin (GIBCO, 25200) at 37°C for 15 minutes, and trypsin was then inactivated by addition of 4 mL of DMEM/F‐12 medium (GIBCO, C11330) containing 20% fetal bovine serum (FBS, GIBCO, C20270). The resulting cell suspension was centrifuged at 150g for 5 minutes and resuspended in DMEM/F‐12 containing 20% FBS. After passage through 100‐mm sterile filters, the cells were seeded on a sterile culture dish coated with poly‐L‐lysine (0.1 mg/mL, Sigma, P7886) and cultured at 37°C in 5% CO2. Three hours later, the medium was replaced by Neurobasal medium (GIBCO, 10888) supplemented with B27 (GIBCO, 17504‐044). AraC (10 mol/L, Sigma, C6645) was added to the medium to inhibit glial cell growth. After 5 days in culture, the cells were subjected to OGD to mimic the I‐R injury. Primary neurons were incubated for 60 minutes in serum‐free, glucose‐free DMEM (GIBCO, 11966025) in an experimental hypoxia chamber containing 95% N2 and 5% CO2. Cells were then returned to normal culture conditions for several specific periods of time. Control neurons were maintained in a humidified atmosphere containing 95% air and 5% CO2.
Administration of 5Z‐7‐Oxozeaenol
The specific TAK1 inhibitor (5Z‐7‐oxozeaenol; Sigma‐Aldrich, O9890) was dissolved in dimethyl sulfoxide (DMSO, Sigma‐Aldrich, D2650) (0.8 μg/μL), as previously described.25 2 μL of 5Z‐7‐oxozeaenol solution was administered into the intracerebroventricular of non‐transgenic and CARD3‐TG mice 30 minutes before tMCAO through stereotaxic apparatus (Stoelting, Wood Dale, IL, 51900). An equal volume of DMSO was administered as control treatment.
Statistical Analysis
Data distributions were analyzed using the Shapiro‐Wilk normality test. Normal distributed data were expressed as mean±SD. Difference between the two groups was compared using the two‐tailed Student t test. One‐way analysis of variance (ANOVA) was used to analyze differences among multiple groups, followed by Bonferroni post hoc analysis or Tamhane'sT2 analysis. Non‐normal distributed data were expressed as median (interquartile range), followed by Mann‐Whitney Tests. P<0.05 was considered to be statistically significant. Sample sizes were predetermined on the basis of preliminary data, using power analysis and sample size (PASS) software. All statistical analysis was performed by IBM SPSS Statistics version 19 software.
Results
CARD3 Expression is Upregulated During Brain I‐R Injury
To begin exploring the potential role of CARD3 in stroke, we used a mouse model of tMCAO as previously described21 to examine CARD3 expression at specific time points during ischemic brain injury. We observed that levels of CARD3 detected by Western blotting were significantly upregulated in a time‐dependent manner following I‐R injury (Figure 1A). In addition, immunofluorescence staining revealed that I‐R increased the number of CARD3‐positive neurons in both the ischemic cortex and striatum compared with the contralateral hemisphere (Figure 1B and 1C). In vitro, primary neurons subjected to OGD and recovery also exhibited upregulation of CARD3 expression (Figure 1D). Once again, immunofluorescence staining of primary cultures indicated an increased number of CARD3‐positive neurons following OGD (Figure 1E). These results support a possible association of CARD3 with I‐R injury both in vivo and in vitro.
Figure 1. Expression of CARD3 (caspase activation and recruitment domain 3) is upregulated in neurons after experiencing ischemia‐reperfusion (I‐R) in vivo and in vitro.
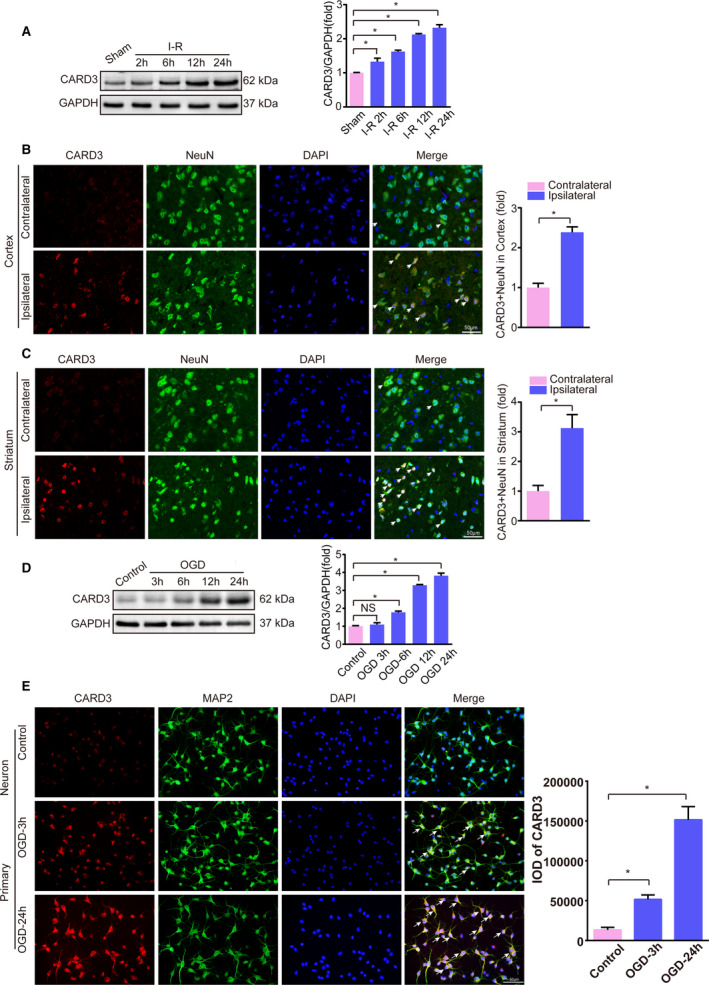
A, Western blotting showed the expression of CARD3 in mice experiencing I‐R injury at indicated time points. Error bar shown as mean±SEM. Statistical analysis was performed by Student t test, *P<0.05 vs sham group. GAPDH served as loading control, n=6 mice per group. B and C, Brain sections were stained for NeuN (neuronal nuclear protein; green), CARD3 (red), and DAPI (blue) in the ischemic cortex (upper) and striatum (lower) after reperfusion for 24 hours, and the white arrows showed CARD3‐positive neurons. Right bar reflected the relative level of CARD3‐positive neurons in the ischemic cortex and striatum. Data were represented as mean±SD. Statistical analysis was performed by Student t test, *P<0.05 vs contralateral, n=5 mice. Scale bars, 50 μm. D, In vitro, the level of CARD3 in primary neurons subjected to OGD (oxygen and glucose deprivation) for 1 hour and reoxygenation for indicated time point was analyzed by Western blotting. Error bar shown as mean±SD. Statistical analysis was performed by Bonferroni post hoc or Tamhane's T2 analysis, *P<0.05 vs the control group., *P<0.05 vs control. GAPDH served as loading control. E, Neurons were also stained with MAP2 (microtubule associated protein 2; green), CARD3 (red) and DAPI (blue) after suffering to OGD reperfusion at indicated time points. All data were presented as mean±SD. Statistical analysis was performed by one‐way analysis of variance (ANOVA), followed by Bonferroni post hoc or Tamhane's T2 analysis, *P<0.05 vs the control group. Three independent experiments were repeated. IOD indicates integrated option density. Scale bars, 50 μm.
CARD3 Promotes Increased Infarct Area and Neurological Deficits Following I‐R Injury
To further determine whether CARD3 plays a specific role in regulating I‐R injury, CARD3‐KO mice were enrolled in tMCAO study and identified by Western blotting (Figure 2A). Following 24‐ and 72‐hour periods of reperfusion after tMCAO, CARD3‐KO mice exhibited marked reductions in both infarct volumes and neurological scores, when compared with littermate controls (Figure 2B). Thus, the CARD3 deficiency appears to protect mice against brain injury during postischemic reperfusion. In order to further validate the apparent effect of CARD3 in aggravating stroke‐related damage, we created four transgenic lines of neuron‐specific CARD3 overexpressing mice (CARD3‐TG) by inserting CARD3 cDNA downstream of the platelet‐derived growth factor B‐chain promoter (Figure 2C). Western blotting revealed greatly increased expression of CARD3 in CARD3‐TG mice, especially in line 3 and line 4. Line 4 mice were chosen for further experiments (hereafter referred to simply as CARD3‐TG) (Figure 2D) and the expression of CARD3 in different tissues from CARD3‐TG mice hardly changed (Figure S1B). As anticipated, the neuron‐specific CARD3‐TG mice exhibited increased infarct volumes and more severe neurological damage than non‐transgenic controls (Figure 2D). Taken together, these findings strongly support a crucial role for CARD3 in exacerbating stroke damage.
Figure 2. CARD3 (caspase activation and recruitment domain 3) promotes I‐R (ischemia‐reperfusion)‐inducing neuron damage.
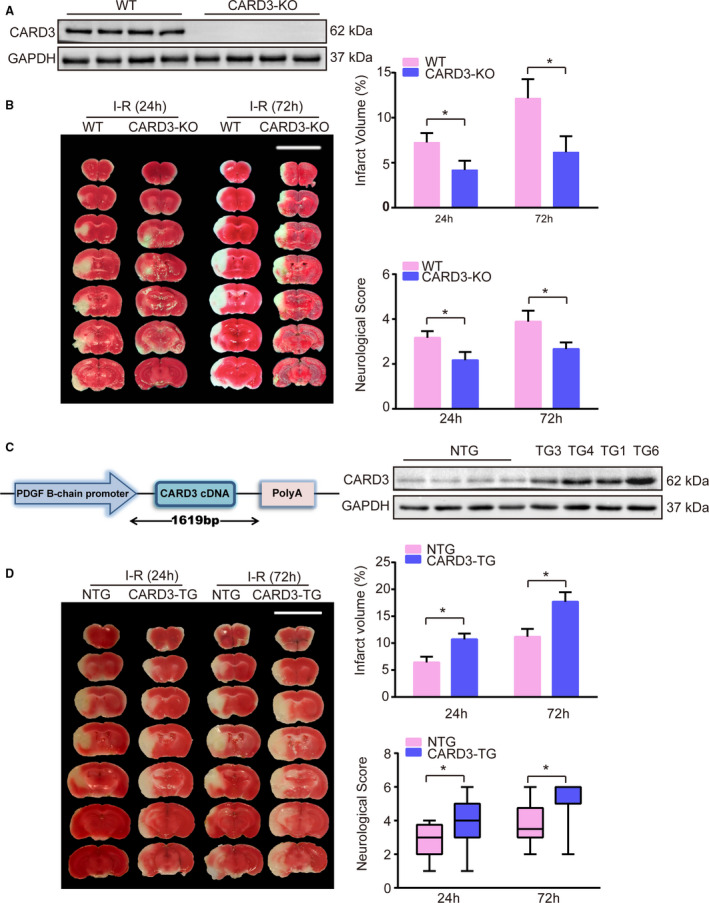
A, CARD3‐KO (knocked‐out) mice were identified by Western blotting. GAPDH served as loading control. B, Representative images of TTC (2,3,5‐triphenyltetrazolium chloride) stained section from both WT (wild type) and CARD3‐KO (CARD3‐knocked‐out) groups after reperfusion for 24 or 72 hours, respectively, and quantitative analysis of infarct volume (upper) and neurological scores (lower) of the two groups were performed at 24 and 72 hours after I‐R injury. Error bar shown as mean±SD. Statistical analysis was compared by Student t test, *P<0.05 vs WT, n=12 mice per group at 24 hours and n=9 mice per group at 72 hours. C, Schematic diagram shows construction of neuron‐specific CARD3‐overexpression mice and Western blotting (right) showed the level of CARD3 from NTG (non‐transgenic) mice and different neuron‐specific CARD3‐TG (CARD3 transgenic) lines. Platelet‐derived growth factor B indicated platelet‐derived growth factor B‐chain. GAPDH served as loading control (right) (D), left panel showed representative images of TTC‐stained section from NTG mice and CARD3‐TG mouse after reperfusion for 24 and 72 hours, respectively, and infarct volume (upper) and neurological scores (lower) of both NTG and neuron‐specific CARD3‐TG groups were performed after reperfusion at indicated time point in the right panel. Error bar shown as mean±SD. Statistical analysis was performed by Student t test or Mann‐Whitney Test, *P<0.05 vs NTG group, n=12 mice per group.
CARD3 Enhances I‐R Induced Neuronal Apoptosis
Due to high metabolic demands and limited capacity for energy storage, neurons are more sensitive to ischemic insult than many other cell types. Previous studies have found neuronal death to be the main pathophysiological cause of brain damage after stroke.21 We therefore investigated the potential role of CARD3 in neuronal death. Brain sections taken from both CARD3‐KO and neuron‐specific CARD3‐TG mice at 24‐hour time points after I‐R injury were evaluated for neuronal death using Fluoro‐Jade B and TUNEL labeling. Compared with the WT control group, CARD3‐KO mice exhibited a sharp reduction in Fluoro‐Jade B‐positive neurons, whreas the number of Fluoro‐Jade B‐positive neurons was increased in neuron‐specific CARD3‐TG mice in comparison with non‐transgenic mice (Figure 3A). The result of TUNEL assays further indicated that neuronal apoptosis was increased in CARD3‐TG mice but reduced in CARD3‐KO mice (Figure 3B). Considering these findings, we further investigated the involvement of proapoptotic and antiapoptotic factors in these phenomena. In CARD3‐KO mice after reperfusion for 6 hours, increases in the levels of the proapoptotic proteins, such as Bax, Bid, and cleaved‐caspase3, were lower than in WT mice. In contrast, levels of the antiapoptotic factors Bcl2 were higher than in the control group (Figure 3C). Conversely, the opposite results were observed in CARD3‐overexpression mice (Figure 3D). Additionally, the transcription level of proapoptotic genes were inhibited in CARD3‐KO mice but enhanced in CARD3‐TG mice after I‐R injury (Figure S1C). Thus, we conclude that CARD3 promotes neuronal death during I‐R injury.
Figure 3. CARD3 (caspase activation and recruitment domain 3) exacerbates neuron apoptosis after I‐R (ischemia‐reperfusion) injury.
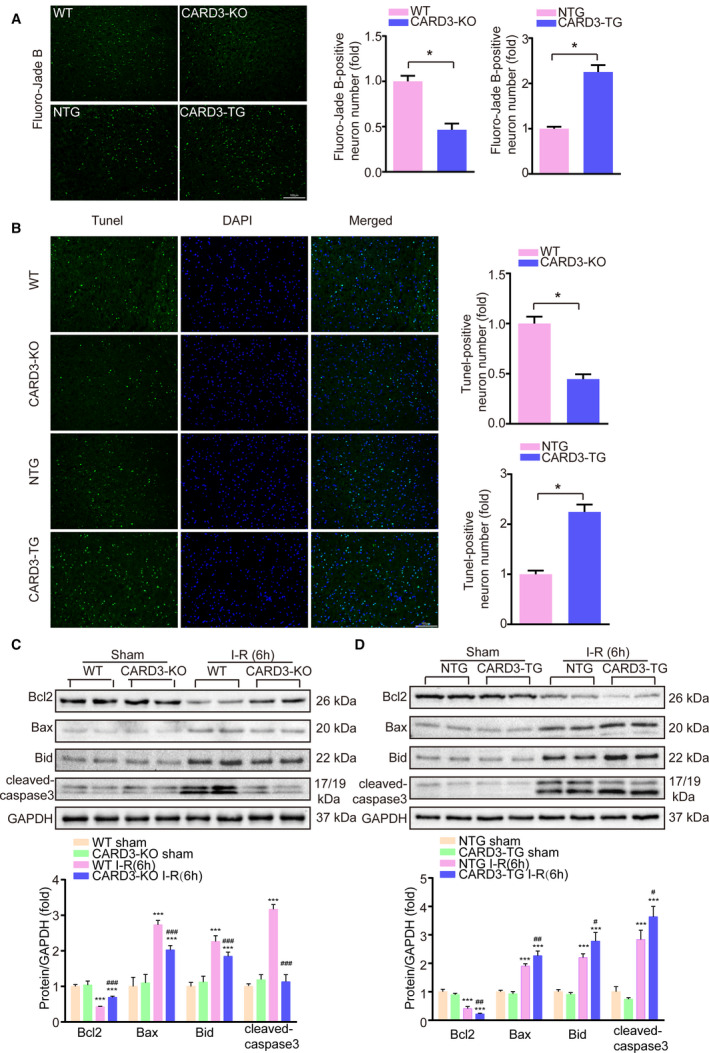
Fluoro‐Jade B staining (A) and TUNEL (terminal dexynucleotidyl transferase (tdt)‐mediated dutp nick end labeling) assays (B) were performed in ischemic brain sections from both CARD3‐KO (CARD3 knocked‐out) and neuron‐specific CARD3‐TG (CARD3‐transgenic) mice at 24 hours after I‐R injury. Nuclei were stained with DAPI. WT, (wild type); NTG, (non‐transgenic). The right panels showed the relative quantification of the positive neurons. All data were shown as mean±SD. Statistical analysis was performed by Student t test, *P<0.05 vs control, n=3 to 5 mice per group. Scale bars, 100 μm. C and D, Brain homogenates from sham‐ or I‐R injury‐ WT, CARD3‐KO, NTG, and neuron‐specific CARD3‐TG mice were collected. Samples of the same group were mixed and then analyzed by Western blotting. The expressions of apoptotic‐related proteins were presented. All data were shown as mean±SD. Statistical analysis was performed by one‐way analysis of variance (ANOVA), followed by Bonferroni post hoc or Tamhane's T2 analysis, ***P<0.001 vs the control group, # P<0.05, ## P<0.01, ### P<0.001 vs the CARD3‐KO or‐TG operated group. GAPDH is served as loading control, n=4 to 5 mice per group. Bcl2 indicates B‐cell lymphoma‐2; and Bax, Bcl2‐Associated X.
CARD3 Exacerbates the Inflammatory Response Induced by I‐R
Inflammation plays a major role in the progression of I‐R injury.26 Therefore, we investigated differences in the inflammatory response between CARD‐KO and WT mice after 6 hours of reperfusion following tMCAO. The mRNA levels for proinflammatory mediators, including interleukin‐(IL)‐1β, IL‐6, tumor necrosis factor ‐ alpha (TNF‐α), and COX2 were reduced in the CARD3‐KO group (Figure 4A). The same results were observed in primary neurons subjected to OGD stimulation (Figure S1D). In addition, Western blotting revealed that the phosphorylation indicates inhibitor of nuclear factor kappa‐B kinase beta (IKKβ) and p65 was significantly decreased in CARD3‐KO mice. CARD3 ablation also suppressed the degradation of inhibitor of nuclear factor kappa‐Bα (IKBα), which regulates translocation of NF‐κB into the nucleus (Figure 4B). Contrasting results of the aforementioned factors were observed in neuron‐specific CARD3‐TG mice, as demonstrated by qPCR and Western blotting (Figure 4C, 4D and Figure S1D). Altogether, these data demonstrate that, following tMCAO, CARD3 contributes to pathological inflammation.
Figure 4. CARD3 (caspase activation and recruitment domain 3) activates inflammation response to mediate I‐R (ischemia‐reperfusion) injury.
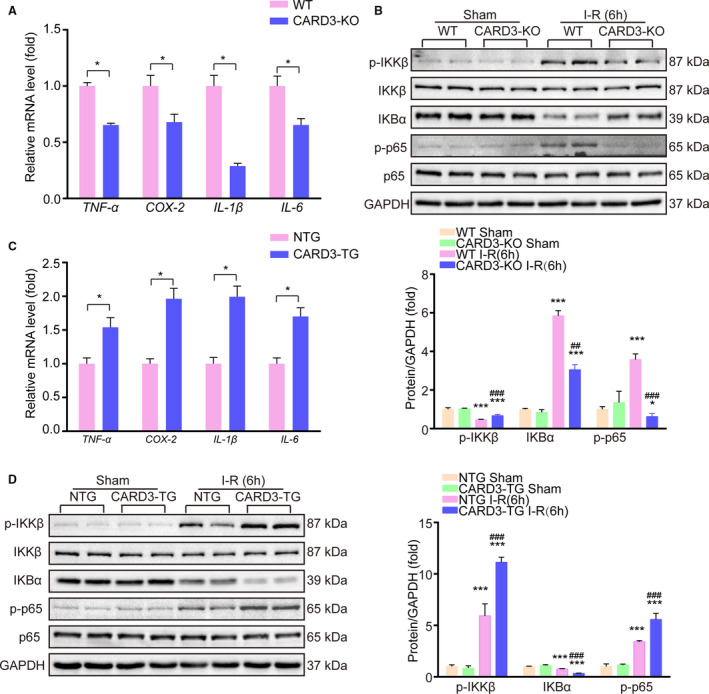
A, The mRNA levels of inflammatory‐related factors in WT (wild type) or CARD3‐KO (CARD3‐knocked‐out) mice after reperfusion for 24 hours were analyzed with quantitative polymerase chain reaction (qPCR). All data were normalized to GAPDH and shown as mean±SD. Comparison between groups were performed with Student t test, *P<0.05 vs the control group, n=4 mice per group. TNF‐α indicates tumor necrosis factor alpha; COX‐2, cyclooxygenase‐2; IL‐1β, interleukin‐1β; IL‐6, interleukin‐6. B, Western blotting showed the expression of indicated proteins from the WT and CARD3‐KO mice after I‐R 6 hours. All data were shown as mean±SD. Statistical analysis was performed by one‐way analysis of variance (ANOVA), followed by Bonferroni post hoc or Tamhane's T2 analysis, *P<0.05 ***P<0.001 vs the control group, ## P<0.01, ### P<0.001 vs the CARD3‐KO operated group. GAPDH served as a loading control, n=4 to 5 mice per group. IKKβ indicates inhibitor of nuclear factor kappa‐B kinase beta; IKBα, inhibitor of nuclear factor kappa‐B α; p65, nuclear factor kappa‐B RelA(p65). C, The mRNA levels of inflammatory‐related factors in NTG (non‐transgenic) or neuron‐specific CARD3‐TG (CARD3‐transgenic) mice after reperfusion for 24 hours were analyzed with qPCR. All data were normalized to GAPDH and shown as mean±SD. Comparison between groups were performed with Student t test, *P<0.05 vs the control group, n=4 mice per group. D, Western blotting reflected the relative level of indicated proteins from NTG mice and neuron‐specific CARD3‐TG mice after I‐R 6 hours. All data were shown as mean±SD. Statistical analysis was performed by one‐way ANOVA, followed by Bonferroni post hoc or Tamhane's T2 analysis, ***P<0.001 vs their control group, ### P<0.001 vs the CARD3‐TG operated group. GAPDH served as a loading control, n=4 to 5 mice per group.
CARD3 Activates the JNK/p38 MAPKs Death Signaling Pathway
Because CARD3 involvement in the pathophysiology of stroke is based on upregulation of neuronal death, we thus hypothesized that CARD3 may influence pro‐death signaling pathways. MAPKs (mitogen‐activated protein kinase) family members, such as JNK, ERK, and p38, are well‐known contributors to increased cell death after stroke. Interestingly, we discovered that CARD3 ablation reduced the phosphorylation of JNK and p38 after 6 hours of reperfusion. In contrast, no change was seen in levels of p‐ERK (Figure 5A), the kinase that is primarily involved in cell proliferation and survival.27, 28 These effects were reversed in CARD3‐TG mice (Figure 5A). Thus, CARD3 may facilitate I‐R damage by increasing the phosphorylation of JNK and p38. Transforming growth factor‐β‐activated kinase 1 (TAK1) is widely accepted as a common regulator for the three conventional MAPKs pathways.29 In response to different stimuli, TAK1 activates different downstream effectors, such as IKKγ, ERK, JNK, and p38.30, 31, 32, 33 Therefore, we reasoned that TAK1 might be involved in the process of CARD3‐mediated cerebral I‐R injury. As expected, activation of TAK1 was significantly enhanced after stroke in neuron‐specific CARD3‐overexpressing mice and reduced in the CARD3‐KO group (Figure 5B).
Figure 5. CARD3 enhances ischemia‐reperfusion (I‐R) injury via activation of MAPKs (mitogen‐activated protein kinase) cascades.
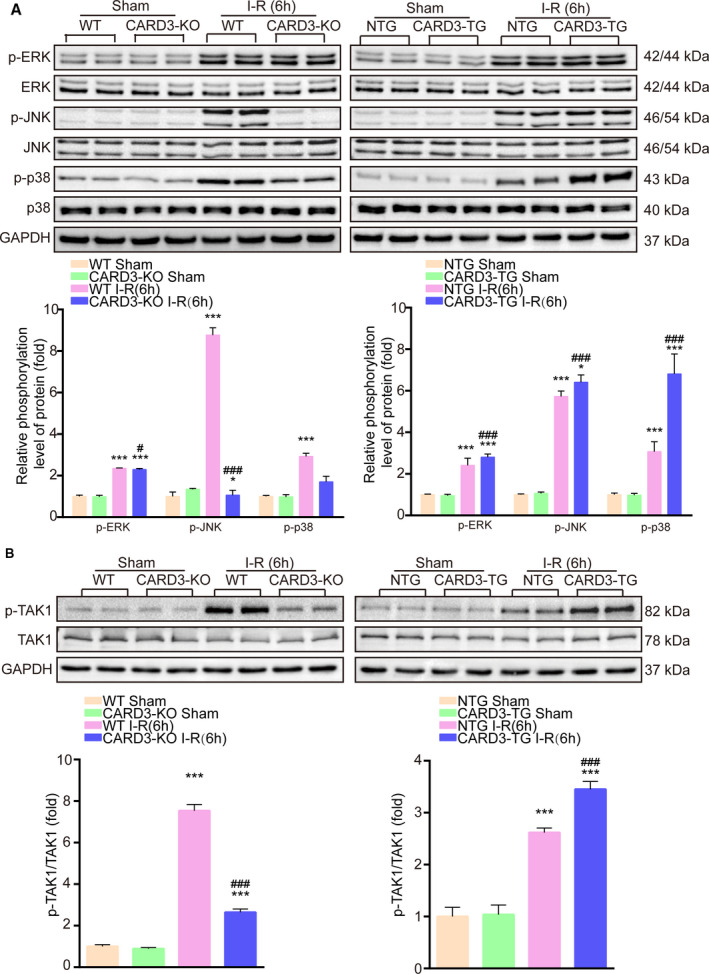
Brain homogenates from sham‐ or I‐R injury‐CARD3‐KO mice (left) and neuron‐specific CARD3‐TG mice (right) were collected, WT mice and NTG mice served as controls, respectively. Western blotting analysis was performed to detect the phosphorylation level of ERK (extracellular regulated protein kinases), JNK (c‐Jun N‐terminal kinase), and p38 (p38 mitogen‐activated protein kinase) (A) and the levels of phosphorylated TAK1 (transforming growth factor‐β‐activated kinase 1) (B). All data were shown as mean±SD. Statistics analysis was performed by one‐way analysis of variance (ANOVA), followed by Bonferroni post hoc or Tamhane's T2 analysis, *P<0.05, ***P<0.001 vs the control group, # P<0.05, ### P<0.001 vs the CARD3‐KO or ‐TG operated group. GAPDH served as a loading control, n=4 to 5 mice per group.
CARD3 Promotes Neuronal Damage Through Phosphorylation of TAK1
To further establish the relationship between CARD3 and TAK1, the specific TAK1 inhibitor, 5Z‐7‐oxozeaenol was administered to CARD3‐TG mice 30 minutes prior to the tMCAO procedure. TAK1 inhibition largely blocked the increase in infarct area size seen in untreated CARD3‐TG mice (Figure 6A). Moreover, phosphorylation levels of p38 and JNK were suppressed by 5Z‐7‐oxozeaenol treatment of neuron‐specific CARD3‐TG mice. Additionally, TAK1 inhibition also prevented the ischemia‐related apoptosis and inflammation, caused by CARD3 overexpression (Figure 6B through 6D). In summary, TAK1 plays a central role in CARD3 mediated neuronal death. And CARD3 might activate NF‐κB signaling and JNK/p38 MAPKs pro‐death signaling pathways via phosphorylation of TAK1 during I‐R injury.
Figure 6. CARD3 induced ischemia‐reperfusion (I‐R) injury via activating TAK1.
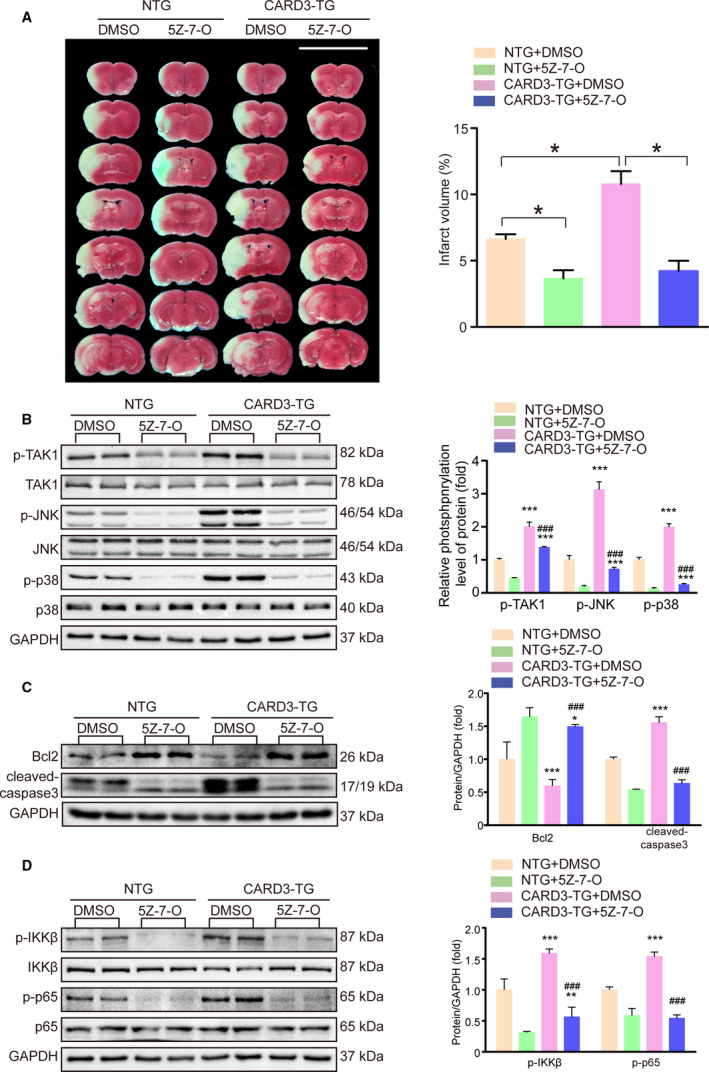
A, TAK1 (transforming growth factor‐β‐activated kinase 1) specific inhibitors, 5Z‐7‐oxozeaenol (5Z‐7‐O) were intravenously administrated into non‐transgenic (NTG) or neuron‐specific CARD3‐TG mice 30 minutes before suffering cerebral ischemia, and dimethyl sulfoxide (DMSO) was injected as control. After reperfusion for 24 hours, brain sections of the indicated group were stained with TTC (2,3,5‐triphenyltetrazolium chloride) (left) and quantitative analysis of infarct volume (right) was performed. Data were exhibited as mean±SD. Statistical analysis was performed by Student t test, *P<0.05 vs their control group, n=6 mice per group. B through D, brain homogenates of the indicated group were obtained after reperfusion for 6 hours. And the level of the indicated proteins were analyzed with Western blotting, n=4 mice per group. Data were exhibited as mean±SD. Statistical analysis was performed by one‐way analysis of variance (ANOVA), followed by Bonferroni post hoc or Tamhane's T2 analysis, *P<0.01, **P<0.01, ***P<0.001 vs the NTG group treated with DMSO or 5Z‐7O, and ##P<0.01, ###P<0.001 vs the CARD3‐TG group treated with DMSO. GAPDH served as a loading control, n=4 mice per group. JNK indicates c‐Jun N‐terminal kinase; p38, p38 mitogen‐activated protein kinase; Bcl2 indicates B‐cell lymphoma‐2; IKKβ, inhibitor of nuclear factor kappaB kinase beta; IKBα, inhibitor of nuclear factor kappa‐B α; and p65, nuclear factor kappa‐B RelA(p65).
Discussion
I‐R injury is considered to be a critical factor in determining the outcome of stroke. Even though targeting a variety of pathological processes can effectively reduce neuronal death in mice, successful translation of these methodologies into clinical practice will require additional insight into the mechanisms underlying I‐R induced damage. In our present study, we have demonstrated that CARD3 serves as an upstream regulator to mediate inflammation, and neuronal cell apoptosis following transient cerebral stroke. Furthermore, we showed the CARD3/TAK1 axis has a potential role in determining cerebral I‐R injury.
The most important finding of our study is that the interaction between CARD3 and TAK1 regulates classical signaling pathways, namely NF‐κB, and JNK/p38, to induce I‐R injury after stroke. TAK1, a member of the MAP3Ks family, has been reported to exert diverse effects on various downstream pathways in different tissues or cells.34 In response to DNA damage, TAK1 was recruited to SUMO‐1 and ubiquitin‐modified RIP1 modified to promote multiple tumor cells survival.34 Inhibiting the kinase activity of TAK1 sensitized cells to TNF‐induced necrosis through enhancing RIP1/RIP3 complex formation.36 Windheim et al37 have also demonstrated that TAK1 is essential for the NOD/CARD3 signaling, exerting a cardioprotective role in myocardial infarction model.30 It has been reported that short‐term inhibition of TAK1 has a protective effect on acute ischemic stroke, mainly via inactivation of classical JNK and p38 signaling,31 whereas prolonged inhibition or deletion of the TAK1 gene lose such protective effect against stroke due to the compensatory activation of ASK1.38 These facts indicate the importance of the precise regulation of MAPK pathways, particularly in stroke. In our current study, we demonstrate that CARD3 deficiency‐medicated TAK1 inhibition protected mouse brain against I‐R injury without alternatively activating ASK1. Furthermore, the success of treatment against I‐R induced brain injury with a specific TAK1 inhibitor demonstrates that it is possible to attenuate the pro‐death function of CARD3 that is seen in CARD3‐TG mice. Therefore, future focus on the interaction of CARD3 with TAK1 will provide a new avenue toward mitigating I‐R injury of brain.
Among various aspects of the postischemic response, the inflammation cascade is initiated within minutes to hours after the occlusion of CBF.39 In this signaling cascade, activated NF‐κB translocates into nuclei to promote transcription of inflammatory genes such as IL‐1β, TNF‐α, ICAM, VCAM, and COX‐2. Although many aspects of inflammation are beneficial in restoring tissue homeostasis, inactivation of NF‐κB by inhibitor or gene loss has the potential to minimize infarct areas in stroke.40, 41 Although several studies have reported that CARD3 promotes NF‐κB activation in the immune system,42, 43 the detail of how CARD3 activates NF‐κB pathway in stroke remains to be investigated. We here not only demonstrated that CARD3 modulates NF‐κB activation and inflammatory cytokine release during the ischemic phase of I‐R injury but also identified that the promoting effect of CARD3 on the activation NF‐κB signaling depends on the activation of TAK1. These results pose a therapeutic niche into the CARD3‐TAK1‐NF‐κB regulatory axis during the development of I‐R induced brain injury.
Previous reports have shown that CARD3 can mediate different MAPK signaling pathways in a variety of tissues in response to different stimuli. For example, CARD3 activates the p38 MAPK signaling axis to increase cardiac infarct area44 and knocking‐out CARD3 significantly shrinks infarct area and improves left ventricular function.16 Activation of the JNK signaling protects the heart against reperfusion injury, but this protective effect disappeared in CARD3−/− mice,30 implying that CARD3 regulates the JNK activation. Additionally, transfection of 293 cells with flag‐tagged CARD3 plasmid dramatically enhanced the TNF‐inducing Raf/ERK axis but had no effect on JNK.45 ERK1/2, JNK, and p38 MAPK has also been reported to participate in stroke‐related injury.27, 28, 46, 47 Thus, it is not clear whether CARD3 contributes to the pathophysiology of stroke via MAPKs activation. We have nevertheless demonstrated that CARD3 can upregulate activation of both p38 and JNK, but not upregulate ERK activation, following I‐R insult. Moreover, this upregulation is mediated by TAK1, so that inhibition of TAK1 blocks the positive effect of CARD3 on MAPKs.
Multiple pathophysiological events, such as excitotoxicity, inflammation, apoptosis, autophagy immune response, oxidative and nitrosative stress, and endoplasmic reticulum stress, were involved in I‐R injury.5, 8 Various unsuccessful clinical trials simply focus on the downstream signal pathway, such as inflammation and oxidative stress. In our study, we demonstrated that CARD3 not only regulates the inflammation but also promotes the neuron apoptosis to induce I‐R injury. Therefore, CARD3 may serve as the key upstream regulator to mediate the two aspects. Targeting CARD3 may have great potential to alleviate the I‐R injury, although developing a successful blood–brain barrier penetrating CARD3 antagonist could be a potential limitation.
In conclusion, our research has revealed a novel function of CARD3 in mediating I‐R damage via activating TAK1 signaling cascade. Therefore, targeting CARD3 might be insightful for exploring new drugs and therapeutic strategies for ameliorating the effects of stroke in human beings.
Sources of Funding
This study was supported by awards from the National Key Research and Development Program of China (No. 2016YFF0101504) for data collection, the National Natural Science Foundation of China (No. 81571146 and No. 81771280) and the Hubei Science and Technology Support Project (No. 2017BEC015) for trial design, and the Fundamental Research Funds for the Central Universities (No. 2042017kf0198) for data analysis and interpretation. The authors have not been paid to write this article by a pharmaceutical company or other agency. The corresponding author had full access to all data in this study and final responsibility for the decision to submit for publication.
Disclosures
None.
Supporting information
Tables S1–S3
Figure S1
(J Am Heart Assoc. 2020;9:e014920 DOI: 10.1161/JAHA.119.014920.)
For Sources of Funding and Disclosures, see page 13.
Contributor Information
Jincao Chen, Email: chenjincao2012@hotmail.com.
Zhi‐Gang She, Email: zgshe@whu.edu.cn.
References
- 1. Hankey GJ. Stroke. Lancet. 2017;389:641–654. [DOI] [PubMed] [Google Scholar]
- 2. Writing Group M , Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, et al. Heart disease and stroke statistics—2016 update: a report from the American Heart Association. Circulation. 2016;133:e38–e360. [DOI] [PubMed] [Google Scholar]
- 3. Kondziella D, Cortsen M, Eskesen V, Hansen K, Holtmannspotter M, Hojgaard J, Stavngaard T, Sondergaard H, Wagner A, Welling KL. Update on acute endovascular and surgical stroke treatment. Acta Neurol Scand. 2013;127:1–9. [DOI] [PubMed] [Google Scholar]
- 4. Powers WJ, Derdeyn CP, Biller J, Coffey CS, Hoh BL, Jauch EC, Johnston KC, Johnston SC, Khalessi AA, Kidwell CS, et al. 2015 American Heart Association/American Stroke Association focused update of the 2013 guidelines for the early management of patients with acute ischemic stroke regarding endovascular treatment: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2015;46:3020–3035. [DOI] [PubMed] [Google Scholar]
- 5. Chamorro A, Dirnagl U, Urra X, Planas AM. Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016;15:869–881. [DOI] [PubMed] [Google Scholar]
- 6. Gottlieb M, Hunter B. Update: is endovascular therapy effective in the treatment of acute ischemic stroke? Ann Emerg Med. 2015;66:613–615. [DOI] [PubMed] [Google Scholar]
- 7. Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. 2011;17:1391–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Prentice H, Modi JP, Wu J‐Y. Mechanisms of neuronal protection against excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction in stroke and neurodegenerative diseases. Oxid Med Cell Longev. 2015;2015:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nagy Z, Nardai S. Cerebral ischemia/repefusion injury: from bench space to bedside. Brain Res Bull. 2017;134:30–37. [DOI] [PubMed] [Google Scholar]
- 10. Thome M, Hofmann K, Burns K, Martinon F, Bodmer JL, Mattmann C, Tschopp J. Identification of CARDIAK, a RIP‐like kinase that associates with caspase‐1. Curr Biol. 1998;8:885–888. [DOI] [PubMed] [Google Scholar]
- 11. Dorsch M, Wang A, Cheng H, Lu C, Bielecki A, Charron K, Clauser K, Ren H, Polakiewicz RD, Parsons T, et al. Identification of a regulatory autophosphorylation site in the serine‐threonine kinase RIP2. Cell Signal. 2006;18:2223–2229. [DOI] [PubMed] [Google Scholar]
- 12. Hollenbach E, Neumann M, Vieth M, Roessner A, Malfertheiner P, Naumann M. Inhibition of p38 MAP kinase‐ and RICK/NF‐kappaB‐signaling suppresses inflammatory bowel disease. FASEB J. 2004;18:1550–1552. [DOI] [PubMed] [Google Scholar]
- 13. Inohara N, del Peso L, Koseki T, Chen S, Nunez G. RICK, a novel protein kinase containing a caspase recruitment domain, interacts with CLARP and regulates CD95‐mediated apoptosis. J Biol Chem. 1998;273:12296–12300. [DOI] [PubMed] [Google Scholar]
- 14. Rahman MA, Sundaram K, Mitra S, Gavrilin MA, Wewers MD. Receptor interacting protein‐2 plays a critical role in human lung epithelial cells survival in response to Fas‐induced cell‐death. PLoS One. 2014;9:e92731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andersson L, Scharin Tang M, Lundqvist A, Lindbom M, Mardani I, Fogelstrand P, Shahrouki P, Redfors B, Omerovic E, Levin M, et al. Rip2 modifies VEGF‐induced signalling and vascular permeability in myocardial ischaemia. Cardiovasc Res. 2015;107:478–486. [DOI] [PubMed] [Google Scholar]
- 16. Li L, Wang X, Chen W, Qi H, Jiang DS, Huang L, Huang F, Wang L, Li H, Chen X. Regulatory role of CARD3 in left ventricular remodelling and dysfunction after myocardial infarction. Basic Res Cardiol. 2015;110:56. [DOI] [PubMed] [Google Scholar]
- 17. Shaw PJ, Barr MJ, Lukens JR, McGargill MA, Chi H, Mak TW, Kanneganti TD. Signaling via the RIP2 adaptor protein in central nervous system‐infiltrating dendritic cells promotes inflammation and autoimmunity. Immunity. 2011;34:75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khursigara G, Bertin J, Yano H, Moffett H, DiStefano PS, Chao MV. A prosurvival function for the p75 receptor death domain mediated via the caspase recruitment domain receptor‐interacting protein 2. J Neurosci. 2001;21:5854–5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kisiswa L, Fernandez‐Suarez D, Sergaki MC, Ibanez CF. RIP2 gates TRAF6 interaction with death receptor p75(NTR) to regulate cerebellar granule neuron survival. Cell Rep. 2018;24:1013–1024. [DOI] [PubMed] [Google Scholar]
- 20. Peel AL, Zolotukhin S, Schrimsher GW, Muzyczka N, Reier PJ. Efficient transduction of green fluorescent protein in spinal cord neurons using adeno‐associated virus vectors containing cell type‐specific promoters. Gene Ther. 1997;4:16–24. [DOI] [PubMed] [Google Scholar]
- 21. Li T, Qin JJ, Yang X, Ji YX, Guo F, Cheng WL, Wu X, Gong FH, Hong Y, Zhu XY, et al. The ubiquitin E3 ligase TRAF6 exacerbates ischemic stroke by ubiquitinating and activating Rac1. J Neurosci. 2017;37:12123–12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xia CF, Smith RS, Shen B, Yang ZR, Borlongan CV, Chao L, Chao J. Postischemic brain injury is exacerbated in mice lacking the kinin B2 receptor. Hypertension. 2006;47:752–761. [DOI] [PubMed] [Google Scholar]
- 23. Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral‐ischemia model in rats. Stroke. 1993;24:117–121. [DOI] [PubMed] [Google Scholar]
- 24. Chen HZ, Guo S, Li ZZ, Lu Y, Jiang DS, Zhang R, Lei H, Gao L, Zhang X, Zhang Y, et al. A critical role for interferon regulatory factor 9 in cerebral ischemic stroke. J Neurosci. 2014;34:11897–11912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gong J, Li ZZ, Guo S, Zhang XJ, Zhang P, Zhao GN, Gao L, Zhang Y, Zheng A, Zhang XF, et al. Neuron‐specific tumor necrosis factor receptor‐associated factor 3 is a central regulator of neuronal death in acute ischemic stroke. Hypertension. 2015;66:604–616. [DOI] [PubMed] [Google Scholar]
- 26. Zhang YM, Qu XY. Xingnaojing injection ameliorates cerebral ischaemia/reperfusion injury via SIRT1‐mediated inflammatory response inhibition. Pharm Biol. 2020;58:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun J, Nan G. The mitogen‐activated protein kinase (MAPK) signaling pathway as a discovery target in stroke. J Mol Neurosci. 2016;59:90–98. [DOI] [PubMed] [Google Scholar]
- 28. Liu R, Tang JC, Pan MX, Zhuang Y, Zhang Y, Liao HB, Zhao D, Lei Y, Lei RX, Wang S, et al. ERK 1/2 activation mediates the neuroprotective effect of BpV(pic) in focal cerebral ischemia‐reperfusion injury. Neurochem Res. 2018;43:1424–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang D, Yan H, Li H, Hao S, Zhuang Z, Liu M, Sun Q, Yang Y, Zhou M, Li K, et al. TGFbeta‐activated kinase 1 (TAK1) inhibition by 5Z‐7‐oxozeaenol attenuates early brain injury after experimental subarachnoid hemorrhage. J Biol Chem. 2015;290:19900–19909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sicard P, Jacquet S, Kobayashi KS, Flavell RA, Marber MS. Pharmacological postconditioning effect of muramyl dipeptide is mediated through RIP2 and TAK1. Cardiovasc Res. 2009;83:277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Neubert M, Ridder DA, Bargiotas P, Akira S, Schwaninger M. Acute inhibition of TAK1 protects against neuronal death in cerebral ischemia. Cell Death Differ. 2011;18:1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qi Z, Shen L, Zhou H, Jiang Y, Lan L, Luo L, Yin Z. Phosphorylation of heat shock protein 27 antagonizes TNF‐alpha induced HeLa cell apoptosis via regulating TAK1 ubiquitination and activation of p38 and ERK signaling. Cell Signal. 2014;26:1616–1625. [DOI] [PubMed] [Google Scholar]
- 33. Zhou J, Zhong J, Huang Z, Liao M, Lin S, Chen J, Chen H. TAK1 mediates apoptosis via p38 involve in ischemia‐induced renal fibrosis. Artificial cells, nanomedicine, and biotechnology. 2018;46(sup 1):1016–1025. [DOI] [PubMed] [Google Scholar]
- 34. Ridder DA, Schwaninger M. TAK1 inhibition for treatment of cerebral ischemia. Exp Neurol. 2013;239:68–72. [DOI] [PubMed] [Google Scholar]
- 35. Yang Y, Xia F, Hermance N, Mabb A, Simonson S, Morrissey S, Gandhi P, Munson M, Miyamoto S, Kelliher MA. A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF‐kappaB and p38 mitogen‐activated protein kinase (MAPK)/MAPK‐activated protein 2 responses to DNA damage. Mol Cell Biol. 2011;31:2774–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vanlangenakker N, Vanden Berghe T, Bogaert P, Laukens B, Zobel K, Deshayes K, Vucic D, Fulda S, Vandenabeele P, Bertrand MJ. cIAP1 and TAK1 protect cells from TNF‐induced necrosis by preventing RIP1/RIP3‐dependent reactive oxygen species production. Cell Death Differ. 2011;18:656–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Windheim M, Lang C, Peggie M, Plater LA, Cohen P. Molecular mechanisms involved in the regulation of cytokine production by muramyl dipeptide. Biochem J. 2007;404:179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. White BJ, Tarabishy S, Venna VR, Manwani B, Benashski S, McCullough LD, Li J. Protection from cerebral ischemia by inhibition of TGFbeta‐activated kinase. Exp Neurol. 2012;237:238–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Anrather J, Iadecola C. Inflammation and stroke: an overview. Neurotherapeutics. 2016;13:661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schneider A, Martin‐Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF‐kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. [DOI] [PubMed] [Google Scholar]
- 41. Nurmi A, Lindsberg PJ, Koistinaho M, Zhang W, Juettler E, Karjalainen‐Lindsberg ML, Weih F, Frank N, Schwaninger M, Koistinaho J. Nuclear factor‐kappaB contributes to infarction after permanent focal ischemia. Stroke. 2004;35:987–991. [DOI] [PubMed] [Google Scholar]
- 42. Kobayashi K, Inohara N, Hernandez LD, Galan JE, Nunez G, Janeway CA, Medzhitov R, Flavell RA. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature. 2002;416:194–199. [DOI] [PubMed] [Google Scholar]
- 43. Hasegawa M, Fujimoto Y, Lucas PC, Nakano H, Fukase K, Nunez G, Inohara N. A critical role of RICK/RIP2 polyubiquitination in Nod‐induced NF‐kappaB activation. EMBO J. 2008;27:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jacquet S, Nishino Y, Kumphune S, Sicard P, Clark JE, Kobayashi KS, Flavell RA, Eickhoff J, Cotten M, Marber MS. The role of RIP2 in p38 MAPK activation in the stressed heart. J Biol Chem. 2008;283:11964–11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Navas TA, Baldwin DT, Stewart TA. RIP2 is a Raf1‐activated mitogen‐activated protein kinase kinase. J Biol Chem. 1999;274:33684–33690. [DOI] [PubMed] [Google Scholar]
- 46. Nito C, Kamada H, Endo H, Niizuma K, Myer DJ, Chan PH. Role of the p38 mitogen‐activated protein kinase/cytosolic phospholipase A2 signaling pathway in blood‐brain barrier disruption after focal cerebral ischemia and reperfusion. J Cereb Blood Flow Metab. 2008;28:1686–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Maddahi A, Edvinsson L. Cerebral ischemia induces microvascular pro‐inflammatory cytokine expression via the MEK/ERK pathway. J Neuroinflammation. 2010;7:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S3
Figure S1