

Abstract
Background
Hyperoxia during cardiopulmonary resuscitation (CPR) may lead to oxidative injury from mitochondrial‐derived reactive oxygen species, despite guidelines recommending 1.0 inspired oxygen during CPR. We hypothesized exposure to 1.0 inspired oxygen during CPR would result in cerebral hyperoxia, higher mitochondrial‐derived reactive oxygen species, increased oxidative injury, and similar survival compared with those exposed to 21% oxygen.
Methods and Results
Four‐week‐old piglets (n=25) underwent asphyxial cardiac arrest followed by randomization and blinding to CPR with 0.21 (n=10) or 1.0 inspired oxygen (n=10) through 10 minutes post return of spontaneous circulation. Sham was n=5. Survivors received 4 hours of protocolized postarrest care, whereupon brain was obtained for mitochondrial analysis and neuropathology. Groups were compared using Kruskal‐Wallis test, Wilcoxon rank‐sum test, and generalized estimating equations regression models. Both 1.0 and 0.21 groups were similar in systemic hemodynamics and cerebral blood flow, as well as survival (8/10). The 1.0 animals had relative cerebral hyperoxia during CPR and immediately following return of spontaneous circulation (brain tissue oxygen tension, 85% [interquartile range, 72%–120%] baseline in 0.21 animals versus 697% [interquartile range, 515%–721%] baseline in 1.0 animals; P=0.001 at 10 minutes postarrest). Cerebral mitochondrial reactive oxygen species production was higher in animals treated with 1.0 compared with 0.21 (P<0.03). Exposure to 1.0 oxygen led to increased cerebral oxidative injury to proteins and lipids, as evidenced by significantly higher protein carbonyls and 4‐hydroxynoneals compared with 0.21 (P<0.05) and sham (P<0.001).
Conclusions
Exposure to 1.0 inspired oxygen during CPR caused cerebral hyperoxia during resuscitation, and resultant increased mitochondrial‐derived reactive oxygen species and oxidative injury following cardiac arrest.
Keywords: brain, cardiac arrest, cardiopulmonary resuscitation, mitochondria, neuroprotection, oxygen
Subject Categories: Cardiopulmonary Arrest, Cardiopulmonary Resuscitation and Emergency Cardiac Care, Animal Models of Human Disease, Oxidant Stress, Metabolism
Nonstandard Abbreviations and Acronyms
- CA
cardiac arrest
- CA100
cardiac arrest with cardiopulmonary resuscitation in 1.0 fraction of inspired oxygen
- CA21
cardiac arrest with cardiopulmonary resuscitation in 0.21 fraction of inspired oxygen
- CI
complex I
- CII
complex II
- CIV
complex IV
- CoPP
coronary perfusion pressure
- LPR
lactate/pyruvate ratio
- MtROS
mitochondrial‐derived reactive oxygen species
- PbtO2
brain tissue oxygen tension
Clinical Perspective
What Is New?
Despite increased survival after pediatric cardiac arrest, neurologic injury remains a major challenge, which can be attributed to global ischemia‐reperfusion injury, often referred to as the post–cardiac arrest syndrome; these impairments have significant impact on long‐term quality of life and resources.
Mechanisms of postarrest neurologic and myocardial injury include mitochondrial reactive oxygen species generation, cytokine elevation, and activation of apoptotic pathways.
Molecular oxygen may serve as a substrate for amplifying reperfusion injury and post–cardiac arrest syndrome, as observational clinical studies have suggested that limiting oxygen delivery in the postarrest period (and therefore reducing iatrogenic arterial hyperoxia) can attenuate reperfusion injury and improve arrest outcomes.
What Are the Clinical Implications?
This study establishes that limiting oxygen delivery during cardiopulmonary resuscitation in immature swine can decrease cerebral mitochondrial reactive oxygen species and oxidative injury.
With further study, this may lay the scientific foundations for future clinical trials because of the fact that modification of oxygen delivery represents a relatively simple and inexpensive care strategy, which could be readily implemented in resuscitation guidelines.
Thousands of children each year experience a pediatric cardiac arrest (CA).1, 2 Despite improvements in survival,3 neurologic disability is common.4 Exposure to hyperoxia during and after cardiopulmonary resuscitation (CPR) has been implicated as a potential mediator of postarrest neurologic dysfunction. The developing brain may be particularly susceptible to hyperoxia,5 and resuscitation with room air is now the standard of care for neonatal asphyxia, on the basis of translational and clinical studies.6 In contrast, current neonatal CA guidelines add supplemental inspired oxygen (FiO2) during CPR, and pediatric and adult CPR guidelines recommend 1.0 FiO2,7, 8 despite a paucity of data to support this practice.
Observational clinical studies in adults establish that arterial hyperoxia after arrest is associated with worse survival and neurologic outcomes.9, 10 Several animal studies of CA demonstrate worse brain injury and neurologic outcomes with 1.0 FiO2 compared with room air, but these prior models were unable to differentiate whether the injury occurs from hyperoxic exposure during CPR and the first several minutes after return of spontaneous circulation (ROSC) versus prearrest and postarrest hyperoxic exposures.11, 12 This important gap in knowledge is especially pertinent for the timing of interventions to mitigate brain injury.
Although the evidence overall suggests an association between hyperoxia and worse outcomes from CA, the underlying mechanisms and time course remain poorly defined. Alterations in mitochondrial bioenergetics and mitochondrial‐derived reactive oxygen species (mtROS) may play a critical role.13, 14, 15, 16 To this end, we conducted a randomized blinded trial of 0.21 FiO2 compared with 1.0 FiO2 focused on the CPR period in a high‐fidelity, large animal model of pediatric asphyxia‐associated ventricular fibrillation (VF) CA. In an effort to understand the relationship between hyperoxia, alterations in mitochondrial function, oxidative injury, and neurologic injury, we continuously monitored cerebral oxygen tension and cerebral microdialysis biomarkers and performed ex vivo analyses of cerebral mtROS, mitochondrial bioenergetics, oxidative injury, and mitochondrial RNA expression. We hypothesized that relative to treatment with 0.21 FiO2, exposure to 1.0 FiO2 during CPR would result in cerebral hyperoxia, higher cerebral mtROS production, and worse cerebral oxidative injury.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request. The Children's Hospital of Philadelphia Institutional Animal Care and Use Committee approved all procedures in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Twenty‐five 4‐week‐old female Yorkshire piglets (Sus scrofus domestica) were block randomized into 3 groups: (1) CA with CPR in 0.21 FiO2 (CA21; n=10), (2) CA with CPR in 1.0 FiO2 (CA100; n=10), and (3) anesthetized sham with no CA (n=5). Animals were randomized in blocks of 5. CA animals were randomized in a 1:1 manner, with 2 animals per CA group in each block, and 1 sham animal (eg, AABBC, whereby A=CA100, B=CA21, and C=sham). To account for attrition and exclusion, additional animals (n=7) were randomized in a final block without unblinding the original treatment allocation at any point. All study personnel were blinded to the randomization, except for one team member who determined the assignments and personally set the blinded, randomized FiO2 during each experiment. The FiO2 and pulse oximetry were concealed during CPR and through 10 minutes post‐ROSC and could not be determined by other laboratory personnel. A separate study team member not present during the resuscitation performed the mitochondrial analyses.
Model
We selected a pediatric porcine model to study CA‐related ischemia‐reperfusion injury in the immature brain because piglet central nervous system development, gross anatomical features, and cerebral physiological features closely mirror those of human children.17, 18 From a CPR mechanics standpoint, swine are commonly used for CPR research because porcine and human chests have comparable anterior‐posterior diameter and stiffness.19 Our protocol was devised to simulate a CA following a respiratory deterioration in a young child, which is the most common cause and age group for pediatric in‐hospital CA.4, 20 We studied 4‐week‐old, 10‐kg piglets to model the human toddler in weight, body mass index, and brain structure and development.21 We chose a 7‐minute period of asphyxia followed by 10 minutes of hemodynamic‐directed CPR to model both a clinically relevant insult22 and administration of high‐quality CPR.23, 24 In keeping with our prior published experiments,23, 25 VF was induced to maintain a consistent, 10‐minute minimum duration of CPR to evaluate alternative CPR techniques, because most in‐hospital CPR is ≥10 minutes in duration and because effective CPR for asphyxia‐induced CA often results in ROSC within 2 to 4 minutes. Therefore, this method ensured a controlled CA in which to study the treatment interventions. We maintained the blinded, randomized oxygen therapy for only 10 minutes post‐ROSC to isolate the exposure to the CPR period yet simulate a clinically plausible scenario. After the treatment period, oxygen therapy was titrated to maintain normoxemia, as described below. Comprehensive descriptions of the CA model23 and hemodynamic‐directed CPR26 are available in our previous publications.
Animal Preparation
Anesthesia
All animals received anesthesia with intramuscular ketamine (20–40 mg/kg) and xylazine (1.5–3 mg/kg) followed by inhaled isoflurane in 1.0 FiO2 via a snout mask. Following tracheal intubation, mechanical ventilation was provided in a mixture of 0.21 FiO2 and isoflurane (1%–2%) to maintain anesthesia. Animals were ventilated with tidal volumes of 10 mL/kg, positive end‐expiratory pressure of 6 cm H2O, and respiratory rate adjusted to achieve end‐tidal carbon dioxide values between 38 and 42 mm Hg. Rectal temperature, 3‐lead ECG, peripheral capillary oxygen saturation, and end‐tidal carbon dioxide were continuously monitored and recorded (NICO; Novametrix Medical Systems Inc, Wallingford, CT).
Neuromonitoring
A brain tissue oxygen catheter (Licox; Integra LifeSciences, Plainsboro, NJ) was inserted via a frontal burr hole ≈1 cm into the brain to terminate at the gray‐white matter junction. A second burr hole for intracranial pressure monitoring (NEUROVENT PTO; Raumedic, Germany) was created in the right parietal bone. A laser Doppler probe (Periflux; Perimed Inc, Ardmore, PA) to measure cerebral blood flow was inserted into a third burr hole and affixed atop intact cortical dura. Brain tissue oxygen tension (PbtO2), intracranial pressure, and cerebral blood flow were continuously monitored and recorded (Moberg CNS, Moberg ICU Solutions, Ambler, PA). A cerebral microdialysis catheter (CMA 71 Elite; mDialysis, Sweden) was placed 1 cm deep in the brain parenchyma through a fourth burr hole in the right parietal bone. Sterile saline was perfused at 1 μL/min, and after a calibration period, samples were collected in 30‐minute intervals throughout the experiment and immediately frozen at −80°C. Pyruvate, lactate, glycerol, and glucose levels were analyzed using the automated ISCUS Flex Microdialysis Analyzer (mDialysis) and processed using LABpilot software (mDialysis).
Hemodynamic Monitoring
Bilateral femoral veins and one femoral artery were percutaneously cannulated (Cordis Corporation, Fremont, CA) under ultrasound guidance. High‐fidelity, solid‐state, micromanometer‐tipped catheters (Millar Instruments, Houston, TX) were advanced to thoracic positions to obtain continuous aortic and right atrial pressures (PowerLab; ADInstruments, New Zealand). A normal saline (0.9% sodium chloride) bolus of 20 mL/kg was administered intravenously to all animals to account for fluid losses during the preexperimental fast.
Experimental Protocol: CA Animals
Summarized in Figure 1A, following initiation of fentanyl for analgesia and baseline measurements, asphyxia was induced in CA animals by clamping their endotracheal tube and disconnecting the ventilator, and confirmed by the absence of exhaled CO2. During this time, the ventilator was immediately set to either 1.0 or 0.21 FiO2 by a single study team member not involved in the resuscitation. The remainder of the investigative team was blinded to both the ventilator settings and peripheral capillary oxygen saturation. After 7 minutes of asphyxia, VF was induced via transthoracic intracardiac needles. On confirmation of VF, the endotracheal tube was unclamped and reconnected to the ventilator delivering the randomized FiO2, and CPR was conducted in accordance with the hemodynamic‐directed CPR algorithm.26 During CPR, animals were mechanically ventilated with a respiratory rate of 10 breaths per minute, positive end‐expiratory pressure of 6 cm H2O, tidal volume of 10 mL/kg, and, depending on their blinded randomization group, either 1.0 or 0.21 FiO2. After 10 minutes of CPR, an initial biphasic defibrillation attempt was performed by delivering 50 J (5 J/kg). CPR was continued until sustained ROSC occurred or no ROSC occurred after 20 minutes of CPR. Animals who obtained ROSC received protocolized post‐CA intensive care for 4 hours before euthanasia.
Figure 1. Experimental protocol and study flow diagram.
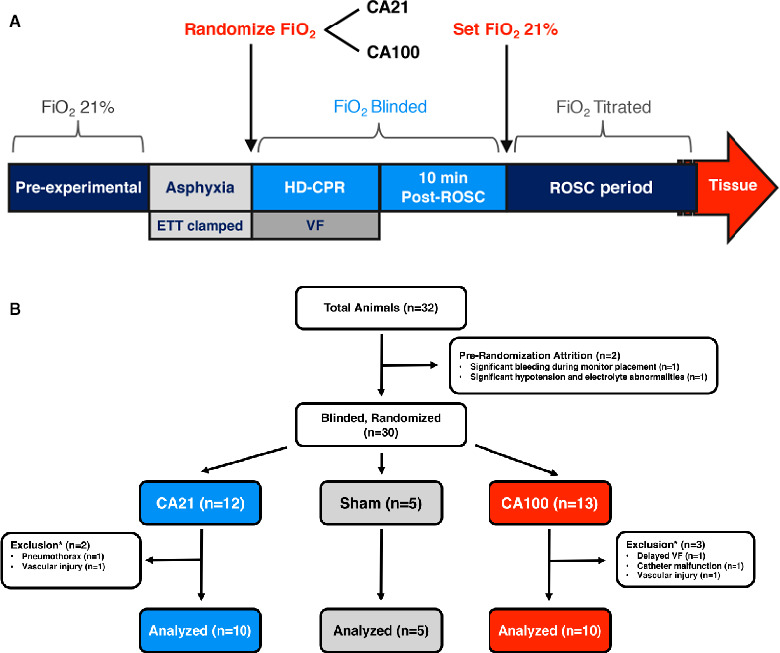
A, During hemodynamic‐directed cardiopulmonary resuscitation (HD‐CPR), depth was titrated to achieve systolic blood pressure >90 mm Hg, and vasopressors were given if coronary perfusion pressure <20 mm Hg. Dosing order was epinephrine (0.02 mg/kg), epinephrine, and vasopressin (0.4 µ/kg). Dosing interval was a minimum of 1 minute after epinephrine and 2 minutes after vasopressin. B, *Excluded animals were rerandomized without unblinding the original treatment allocation at any point. CA21 indicates cardiac arrest (CA) with CPR in 0.21 fraction of FiO2; CA100, CA with CPR in 1.0 fraction of FiO2; ETT, endotracheal tube; FiO2, inspired oxygen; ROSC, return of spontaneous circulation; and VF, ventricular fibrillation.
Post‐ROSC, animals remained in their blinded, randomized CA groups for 10 minutes. After 10 minutes, the FiO2 was set to 0.21 in all animals and the peripheral capillary oxygen saturation was unblinded to allow titration of FiO2 to attain a peripheral capillary oxygen saturation of 92% to 96% for the duration of the post‐ROSC period. On ROSC, the respiratory rate was returned to its prearrest setting and titrated to maintain normocapnia. Intravenous epinephrine (infusion and bolus dosing) was administered if needed to maintain mean arterial pressure (MAP) ≥45 mm Hg. Inhaled isoflurane was restarted and titrated to inhibit pinch‐blink response. Post‐ROSC care was maintained for 4 hours.
Experimental Protocol: Sham Animals
Animals block randomized to the sham group received identical procedures and monitoring but did not undergo asphyxia or CA. Sham animals were maintained with the same protocol and for the same duration of anesthesia as CA animals.
Cerebral Mitochondrial Assessment
A bilateral craniectomy was performed to expose the brain following 4 hours of post‐ROSC intensive care or equivalent duration of anesthesia in sham animals. Fresh tissue samples from cortex and hippocampus were rapidly extracted and prepared for mitochondrial assessment, according to established protocols.24, 27
Measurement of ROS
MtROS production from cortex and hippocampus was measured simultaneously in side‐by‐side chambers on a high‐resolution respirometer fitted with optical sensors (O2k‐Fluo LED2‐Module; OROBOROS Instruments, Austria). Hydrogen peroxide (H2O2) was detected using an Amplex UltraRed assay (Invitrogen, Carlsbad, CA). Amplex Red is a nonfluorescent, colorless substrate that reacts with H2O2 in the presence of horseradish peroxidase to produce red‐fluorescent resorufin on oxidation. The most proximal ROS produced by mitochondrial respiration is superoxide.28 Superoxide dismutase was therefore added to transform all mitochondrial superoxide into H2O2 for direct measurement by fluorometry. Following Amplex Red, horseradish peroxidase, and superoxide dismutase, 100 nmol/L of H2O2 was added for instrument calibration before addition of 22 µL of homogenized brain tissue. H2O2 flux was measured concurrently with mitochondrial respiration using DatLab software (OROBOROS Instruments).
Measurement of Mitochondrial Respiration
As described in our previous publications, assessment of mitochondrial respiration was performed using a high‐resolution respirometer (Oxygraph‐2k; OROBOROS Instruments), as previously described.13, 24, 27 Oxygen consumption was recorded with DatLab (OROBOROS Instruments) and expressed as pmol/(s/mg of tissue homogenate). A substrate‐uncoupler‐inhibitor titration protocol optimized for porcine brain tissue24 was used to assess the individual respiratory capacities of complex I (CI) and complex II (CII), as well as convergent electron flow (CI+CII) in the electron transfer system. The substrate‐uncoupler‐inhibitor titration protocol was identical for sham and injured tissues.
Cerebral Oxidative Injury
Measurement of Protein Carbonyls
A protein carbonyl ELISA kit (No. ALX‐850‐312‐KI01; Enzo Life Sciences, Farmingdale, NY) was used to measure protein carbonyls from isolated mitochondrial fraction from frozen tissue samples. Samples were diluted 10:1 in ELISA buffer for a protein concentration of 4 mg/mL, and procedures were performed in accordance with the manufacturer's instructions.
Measurement of Lipid Peroxidation
Western blot analysis was used to measure 4‐hydroxynoneal, an index of lipid peroxidation. Using frozen tissue whole cell lysates, equal amounts of protein were denatured in 4× Laemmli sample buffer with 1:10 dithiothreitol (D0632‐1G; Sigma Aldrich, St. Louis, MO) and resolved by SDS‐PAGE (12% Bis‐Tris; No. NP0349BOX; Thermo Fisher, Carlsbad, CA) and transferred to nitrocellulose membranes (No. 88024; Thermo Scientific, Rockford, IL). Membranes were blocked with Odyssey Blocking Buffer (No. 927‐40000; Li‐Cor, Lincoln, NE) and incubated with primary antibody overnight for 4‐hydroxynoneal (1:1000; AB5605; Millipore Sigma, Burlington, MA). After washing, membranes were stained with IRDye secondary antibody (1:15 000; No. 925‐32211; Li‐Cor) for 1 hour, then imaged using an Odyssey Scanner (Li‐Cor). Relative band densities were determined by digital densitometry using Li‐Cor Odyssey Application Software v3.
Mitochondrial RNA Expression
RNA sequencing was performed to assess mitochondrial genes related to respiratory function. As previously described,29 total RNA was isolated from flash frozen cortex using an automated RNA extraction robot (QIAsymphony; Qiagen, Hilden, Germany), reverse transcribed, and RNA depleted, followed by sequencing using a HiSeq instrument (Illumina, San Diego, CA) in 2×150 paired end configuration. Reads were aligned to the SusScrofa11.1 reference genome. Total RNA was extracted using a QIAsymphony automated RNA extraction robot and reverse transcribed with a high‐capacity cDNA reverse transcription kit with RNase inhibitor (catalog No. 4387406; Life Technologies, NY). rRNA depletion was conducted using a Ribozero rRNA Removal Kit (human/mouse/rat probe) (Illumina), followed by library preparation with NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, Ipswich, MA). Sequencing was performed in 2×150 paired end configuration with an Illumina HiSeq instrument. Reads were aligned to the SusScrofa11.1 reference genome using STAR.30, 31 Transcripts at the gene level were quantified with featureCounts from the subreads package.32 Data visualization was performed with igraph33 and ggplots.34 Downstream analyses were conducted with the R packages DESeq2.35
Statistical Analysis
Sample Size Calculation
A sample size of 10 animals in each CA group provided >80% power to detect a 20% reduction in mean mtROS between CA groups, using a 10% SD derived from pilot mitochondrial data from prior experiments (unpublished data; Kilbaugh, TJ 2018). Five sham animals were chosen a priori. Type I error, set at 0.05, was not corrected for multiple comparisons across the 3 randomized arms.
Statistical Analysis
Descriptive statistics were used to characterize baseline variables. A skewness‐kurtosis test was performed to assess normality of continuous variables. Differences between groups were evaluated with Student t test or ANOVA for normally distributed variables and Wilcoxon–rank sum or Kruskal‐Wallis with Dunn's multiple comparison tests for nonnormally distributed variables, and expressed as mean±SEM or median with interquartile range, as appropriate. The method of generalized estimating equations fitted with an exchangeable correlation structure was used to assess continuous variables while accounting for clustering of data points within individual animals. PbtO2 and cerebral blood flow were described as percentage baseline. Categorical outcomes (eg, mortality and ROSC) were reported with Fisher's exact testing. Differential expression analysis was performed for RNA sequencing data using physiologic variables and RNA quality as covariates, and adjusted P<0.05 was considered significant (Benjamini‐Hochberg procedure for controlling false discovery rate). Some cerebral microdialysis data points were coded as missing if they were deemed inaccurate by a blinded team member who processed the microdialysis and reviewed it for quality control. Missing values were not imputed/replaced, and therefore sample sizes used to compare groups at various time points were likewise updated to include only animals with accurate data. Analyses were conducted using STATA (Version 15.1; Statacorp LP, College Station, TX), GraphPad Prism (Version 7; GraphPad, San Diego, CA), and DESeq2 package for R (Version X; R Foundation for Statistical Computing, Austria). The block randomization was only revealed after statistical analysis.
Results
Of 32 initial animal experiments, there were 2 prerandomization attritions and 5 exclusions (Figure 1B), resulting in 25 successful experiments (n=10 in each treatment group; n=5 in sham group). Animals were excluded for vascular access‐associated hemorrhage (n=2), pneumothorax (n=1), delayed ability to induce VF (n=1), and aortic catheter malfunction (n=1). Rates of survival were not different, with 8 of 10 (80%) in both CA21 and CA100 treatment groups achieving sustained ROSC and 4‐hour survival.
Systemic Physiological Characteristics
Prerandomization characteristics are described in Table 1. There were no differences in weight, heart rate, arterial blood pressure (BP), coronary perfusion pressure (CoPP), Pao 2, or Paco 2 at baseline or during asphyxia. After 6 minutes of asphyxia, animals were hypotensive (mean MAP, 28±17 mm Hg) and hypoxemic (mean Pao 2, <10 mm Hg). Pulseless electrical activity occurred in 9 of 20 (45%) before VF induction and was not different between treatment groups.
Table 1.
Prerandomization Characteristics
| Variable | CA21 (n=10) | CA100 (n=10) | P Value |
|---|---|---|---|
| Baseline | |||
| Weight, kg | 10.4 | 10.3 | 0.78 |
| Body temperature, °C | 37.4 (37.1–37.7) | 37.6 (37.3–37.7) | 0.54 |
| Heart rate, bpm | 138 (9.4) | 120 (8.9) | 0.18 |
| Systolic aortic BP, mm Hg | 76.2 (3.2) | 76.9 (3.4) | 0.56 |
| Diastolic aortic BP, mm Hg | 53.9 (51.1–56.3) | 51.1 (50.4–57.9) | 0.65 |
| Pao 2, mm Hg | 114 (108–119) | 115 (108–118) | 0.76 |
| SpO2, % | 96.6 (0.44) | 96.6 (0.48) | 0.95 |
| Paco 2, mm Hg | 114 (108–119) | 115 (108–118) | 0.76 |
| ETCO2, mm Hg | 39.9 (38.5–43.5) | 39.1 (38.2–40.4) | 0.29 |
| Brain temperature, °C | 36.9 (36.4–37.0) | 37.0 (36.4–37.1) | 0.74 |
| Intracranial pressure, mm Hg | 13.1 (2.3) | 14.1 (1.3) | 0.61 |
| Asphyxia | |||
| Body temperature, °C | 37.4 (37.1–37.8) | 37.6 (37.5–37.7) | 0.40 |
| Heart rate, bpm | 108.7 (16.5) | 93.5 (16.9) | 0.53 |
| Systolic aortic BP, mm Hg | 38.2 (7.9) | 41.3 (10.7) | 0.82 |
| Diastolic aortic BP, mm Hg | 23.6 (3.6) | 21.4 (4.0) | 0.69 |
| Mean arterial BP, mm Hg | 28.5 (4.9) | 28.0 (6.2) | 0.95 |
| Achieved pulseless electrical activity, n (%) | 4 (40) | 5 (50) | 0.67 |
| Pao 2, mm Hg | 7.8 (1.3) | 6.4 (1.9) | 0.55 |
| SpO2, % | 15.5 (7.4) | 14.7 (5.9) | 0.94 |
| Paco 2, mm Hg | 62.2 (4.5) | 60.8 (4.6) | 0.83 |
| ETCO2, mm Hg | 0.94 (0.22–1.7) | 0.71 (0.21–2.88) | 0.88 |
| Brain temperature, °C | 37.0 (36.0–37.3) | 37.1 (36.7–37.2) | 0.82 |
| Intracranial pressure, mm Hg | 11.9 (1.9) | 13.8 (1.2) | 0.38 |
| Brain tissue oxygen tension, % baseline | 8.2 (3.9) | 9.6 (3.3) | 0.80 |
| Cerebral blood flow, % baseline | 27.3 (9.8) | 22.7 (7.7) | 0.72 |
Comparison between groups at baseline (mean values during 2 minutes preceding start of asphyxia) and during asphyxia, before induction of ventricular fibrillation and randomization to treatment group. Student t test is used for normally distributed data. Wilcoxon rank‐sum test is used for nonnormally distributed data. Values are mean (SEM) or median (interquartile range). BP indicates blood pressure; bpm, beats per minute; CA21, cardiac arrest (CA) with cardiopulmonary resuscitation (CPR) in 0.21 fraction of inspired oxygen (FiO2); CA100, CA with CPR in 1.0 fraction of FiO2; ETCO2, end‐tidal carbon dioxide; and SpO2, peripheral capillary oxygen saturation.
During CPR, there were no differences in hemodynamics, chest compression mechanics, or the number of vasopressor doses administered between CA21 and CA100 treatment groups (Table 2). Survivors achieved higher CoPP (20.6±2.8 versus 12.8±2.5 mm Hg; P<0.01) and higher diastolic BP (30.1±3.0 versus 22.1±2.7 mm Hg; P<0.01) compared with nonsurvivors, consistent with previous investigations with hemodynamic‐directed CPR.13, 23 Systemic oxygenation was not significantly different between groups during CPR (Pao 2, 55 [interquartile range {IQR}, 54–80] mm Hg in CA21 versus 77 [IQR, 64–191] mm Hg in CA100; P=0.10).
Table 2.
CPR Characteristics
| Variable | CA21 (n=10) | CA100 (n=10) | P Value |
|---|---|---|---|
| Mechanics | |||
| Chest compression rate, bpm | 100.4 (0.14) | 100.8 (0.16) | 0.05 |
| Chest compression depth, cm | 42.3 (1.7) | 38.5 (2.9) | 0.29 |
| Chest compression fraction, % | 96 (0.4) | 96 (0.7) | 0.28 |
| Release velocity, m/s | 261 (10.7) | 236 (17.1) | 0.25 |
| No. of vasopressors | 5 (4–5) | 6 (3–6) | 0.73 |
| Systemic hemodynamics | |||
| Systolic aortic BP, mm Hg | 82.8 (2.2) | 88.4 (3.2) | 0.08 |
| Diastolic aortic BP, mm Hg | 30.7 (2.5) | 32.7 (3.6) | 0.57 |
| Coronary perfusion pressure, mm Hg | 20.9 (2.3) | 22.4 (3.3) | 0.65 |
| Pao 2, mm Hg | 55 (54–80) | 77 (64–191) | 0.10 |
| Paco 2, mm Hg | 45.2 (4.3) | 49.7 (5.7) | 0.54 |
| ETCO2, mm Hg | 33.0 (2.7) | 29.8 (3.9) | 0.41 |
| Cerebral physiological features | |||
| Brain temperature, °C | 36.4 (0.41) | 36.2 (0.59) | 0.72 |
| Intracranial pressure, mm Hg | 17.7 (2.2) | 20.3 (3.0) | 0.38 |
| Brain tissue oxygen tension, % baseline | 37.1 (29.0) | 121.2 (40.0) | 0.04 |
| Cerebral blood flow, % baseline | 58.8 (9.93) | 48.9 (14.0) | 0.48 |
Student t test was used for normally distributed data. Wilcoxon rank‐sum test was used for nonnormally distributed data. Continuous physiologic variables were compared during the last 2 minutes of CPR using a generalized estimating equations regression model. Values are mean (SEM) or median (interquartile range). BP indicates blood pressure; bpm, beats per minute; CA21, cardiac arrest (CA) with CPR in 0.21 fraction of inspired oxygen (FiO2); CA100, CA with CPR in 1.0 fraction of FiO2; CPR, cardiopulmonary resuscitation; and ETCO2, end‐tidal carbon dioxide.
At 10 minutes post‐ROSC, CA21 animals had an increase in arterial oxygen tension following CPR; however, they remained normoxemic, with Pao 2 of 90 (IQR, 82–100) mm Hg. CA100 animals were hyperoxemic and had significantly increased arterial oxygen tension (409 [IQR, 398–448] mm Hg) compared with CA21 (P<0.001). No CA21 animals were hypoxemic (lowest Pao 2, 79 mm Hg) post‐ROSC. During the 4 hours post‐ROSC, there were some hemodynamic differences between treatment groups (Table 3). At 1 hour post‐ROSC, CA100 animals had higher BP compared with CA21 animals (MAP, 57.6±3.19 mm Hg in CA21 versus 70.6±3.75 mm Hg in CA100; P=0.02) and had lower heart rate (193.1±14.3 beats per minute in CA21 versus 156.4±10.6 beats per minute in CA100; P=0.06), but these differences did not persist at later time points post‐ROSC. All BPs remained above the a priori goal MAP ≥45 mm Hg.
Table 3.
Post‐ROSC: Systemic Hemodynamics
| Variable | CA21 (n=8) | CA100 (n=8) | P Value |
|---|---|---|---|
| 10 mina | |||
| Heart rate, bpm | 161.0 (12.3) | 128.1 (3.39) | 0.02 |
| Systolic aortic BP, mm Hg | 106.7 (11.6) | 120.6 (6.18) | 0.31 |
| Diastolic aortic BP, mm Hg | 79.6 (8.45) | 91.0 (4.28) | 0.25 |
| Mean BP, mm Hg | 88.7 (9.41) | 100.9 (4.87) | 0.27 |
| Pao 2, mm Hg | 90 (82–100) | 409 (398–448) | <0.001 |
| SpO2, % | 94.0 (92.3–96.8) | 96.6 (95.2–98.7) | 0.17 |
| Paco 2, mm Hg | 40.3 (2.57) | 40.4 (2.75) | 0.97 |
| ETCO2, mm Hg | 43.1 (2.30) | 42.7 (1.61) | 0.91 |
| 30 min | |||
| Heart rate, bpm | 181.1 (11.8) | 158.0 (9.60) | 0.15 |
| Systolic aortic BP, mm Hg | 76.4 (3.93) | 77.3 (3.49) | 0.87 |
| Diastolic aortic BP, mm Hg | 59.0 (4.06) | 58.1 (3.35) | 0.87 |
| Mean BP, mm Hg | 64.8 (3.98) | 64.5 (3.36) | 0.95 |
| Pao 2, mm Hg | 90 (82–100) | 409 (398–448) | <0.001 |
| SpO2, % | 95.6 (94.5–96.7) | 95.7 (94.6–96.4) | >0.99 |
| Paco 2, mm Hg | 40.3 (2.57) | 40.4 (2.75) | 0.97 |
| ETCO2, mm Hg | 38.9 (37.3–41.3) | 39.5 (38.0–41.5) | 0.53 |
| 1 h | |||
| Heart rate, bpm | 193.1 (14.3) | 156.4 (10.6) | 0.06 |
| Systolic aortic BP, mm Hg | 70.3 (3.48) | 86.1 (4.80) | 0.02 |
| Diastolic aortic BP, mm Hg | 51.2 (3.31) | 62.9 (3.34) | 0.03 |
| Mean BP, mm Hg | 57.6 (3.19) | 70.6 (3.75) | 0.02 |
| Pao 2, mm Hg | 103.4 (5.46) | 108.3 (2.40) | 0.43 |
| SpO2, % | 96.8 (95.3–97.6) | 95.7 (95.2–96.5) | 0.29 |
| Paco 2, mm Hg | 34.8 (0.82) | 35.1 (0.69) | 0.77 |
| ETCO2, mm Hg | 38.2 (1.0) | 39.9 (1.23) | 0.86 |
| 2 h | |||
| Heart rate, bpm | 217.1 (14.1) | 189.1 (14.2) | 0.18 |
| Systolic aortic BP, mm Hg | 80.3 (77.3–83.0) | 85.7 (81.0–91.8) | 0.07 |
| Diastolic aortic BP, mm Hg | 56.0 (1.65) | 61.3 (3.07) | 0.15 |
| Mean BP, mm Hg | 65.8 (60.9–66.5) | 67.5 (63.9–74.9) | 0.29 |
| PaO2, mm Hg | 97.4 (3.73) | 109 (3.19) | 0.03 |
| SpO2, % | 95.8 (92.9–97.9) | 96.3 (95.4–96.9) | 0.92 |
| Paco 2, mm Hg | 36.5 (1.27) | 35.6 (1.41) | 0.64 |
| ETCO2, mm Hg | 39.8 (1.14) | 39.2 (3.98) | 0.77 |
| 3 h | |||
| Heart rate, bpm | 209.4 (15.6) | 192.9 (11.9) | 0.42 |
| Systolic aortic BP, mm Hg | 80.5 (3.72) | 88.2 (3.65) | 0.16 |
| Diastolic aortic BP, mm Hg | 57.1 (2.90) | 60.8 (3.10) | 0.40 |
| Mean BP, mm Hg | 64.9 (2.96) | 69.9 (3.13) | 0.26 |
| Pao 2, mm Hg | 97.1 (3.35) | 105 (3.94) | 0.15 |
| SpO2, % | 94.9 (61.9–96.7) | 95.4 (95.1–96.5) | 0.67 |
| Paco 2, mm Hg | 36.1 (0.93) | 36.6 (1.60) | 0.78 |
| ETCO2, mm Hg | 39.3 (38.3–42.0) | 39.5 (38.3–40.7) | 0.83 |
| 4 h | |||
| Heart rate, bpm | 211.2 (15.9) | 193.2 (11.0) | 0.37 |
| Systolic aortic BP, mm Hg | 78.2 (3.08) | 80.0 (4.36) | 0.75 |
| Diastolic aortic BP, mm Hg | 54.4 (2.24) | 54.5 (4.38) | 0.98 |
| Mean BP, mm Hg | 62.3 (2.23) | 63.0 (4.29) | 0.89 |
| Pao 2, mm Hg | 96.3 (4.0) | 106.9 (5.03) | 0.12 |
| SpO2, % | 95.1 (84.4–96.4) | 95.6 (94.6–96.7) | 0.60 |
| PaCO2, mm Hg | 37.3 (1.44) | 37.0 (1.35) | 0.92 |
| ETCO2, mm Hg | 40.6 (1.32) | 40.1 (1.14) | 0.75 |
BP indicates blood pressure; bpm, beats per minute; CA21, cardiac arrest (CA) with cardiopulmonary resuscitation (CPR) in 0.21 fraction of inspired oxygen (FiO2); CA100, CA with CPR in 1.0 fraction of FiO2; ETCO2, end‐tidal carbon dioxide; ROSC, return of spontaneous circulation; and SpO2, peripheral capillary oxygen saturation.
Groups were compared at indicated time points post‐ROSC, and values were analyzed over the 10 minutes preceding each time point, except preceding the 10‐minute post‐ROSC time point when values were analyzed over 2 minutes. Student t test is used for normally distributed data. Wilcoxon rank‐sum test is used for nonnormally distributed data. Values are mean (SEM) or median (interquartile range).
Cerebral Physiological Characteristics
There were no differences in intracranial temperature, intracranial pressure, or cerebral blood flow between treatment groups at any time (Table 4). Figure 2 compares PbtO2 between CA groups during the experimental period. At the end of the asphyxial period, all animals developed cerebral hypoxia (mean PbtO2, <10% baseline). During CPR, PbtO2 was significantly higher in CA100 animals compared with CA21 animals starting at 5 minutes of CPR (34.4±19.4% baseline in CA21 versus 89.3±26.7% baseline in CA100; P=0.04). At 10 minutes post‐ROSC, CA100 demonstrated significant cerebral hyperoxia compared with CA21 (PbtO2, 85% [IQR, 72%–120%] baseline in CA21 versus 697% [IQR, 515%–721%] baseline in CA100; P=0.001).
Table 4.
Post‐ROSC: Cerebral Hemodynamics
| Variable | CA21 (n=8) | CA100 (n=8) | P Value |
|---|---|---|---|
| 10 mina | |||
| Intracranial temperature, °C | 36.4 (35.7–36.8) | 36.7 (36.5–37.1) | 0.23 |
| Intracranial pressure, mm Hg | 16.1 (3.07) | 16.09 (1.88) | 0.99 |
| Brain tissue oxygen tension, % baseline | 86.3 (68.7–122) | 711 (541–766) | 0.001 |
| Cerebral blood flow, % baseline | 118.7 (31.4) | 141.4 (27.8) | 0.60 |
| 30 min | |||
| Intracranial temperature, °C | 36.0 (35.4–37.1) | 36.7 (36.2–37.1) | 0.34 |
| Intracranial pressure, mm Hg | 11.0 (1.59) | 12.6 (1.14) | 0.43 |
| Brain tissue oxygen tension, % baseline | 85.9 (9.82) | 93.7 (7.31) | 0.53 |
| Cerebral blood flow, % baseline | 104.8 (21.6) | 106.1 (12.1) | 0.96 |
| 1 h | |||
| Intracranial temperature, °C | 36.1 (0.53) | 37.0 (0.21) | 0.16 |
| Intracranial pressure, mm Hg | 12.3 (10.8–13.6) | 11.2 (9.61–13.5) | 0.67 |
| Brain tissue oxygen tension, % baseline | 85.9 (9.82) | 93.7 (7.31) | 0.53 |
| Cerebral blood flow, % baseline | 71.2 (13.3) | 94.9 (14.5) | 0.25 |
| 2 h | |||
| Intracranial temperature, °C | 36.3 (0.50) | 37.4 (0.14) | 0.06 |
| Intracranial pressure, mm Hg | 14.0 (2.77) | 12.8 (0.87) | 0.69 |
| Brain tissue oxygen tension, % baseline | 76.8 (10.6) | 93.9 (5.22) | 0.14 |
| Cerebral blood flow, % baseline | 68.4 (20.6) | 92.8 (17.1) | 0.38 |
| 3 h | |||
| Intracranial temperature, °C | 37.1 (35.7–37.8) | 37.6 (37.0–38.1) | 0.34 |
| Intracranial pressure, mm Hg | 13.8 (2.31) | 14.1 (0.91) | 0.91 |
| Brain tissue oxygen tension, % baseline | 87.9 (12.2) | 93.1 (5.35) | 0.68 |
| Cerebral blood flow, % baseline | 70.0 (20.6) | 103.9 (21.9) | 0.28 |
| 4 h | |||
| Intracranial temperature, °C | 37.6 (35.2–38.8) | 37.1 (37.1–38.4) | 0.91 |
| Intracranial pressure, mm Hg | 14.2 (10.9–16.1) | 14.5 (11.7–16.6) | 0.78 |
| Brain tissue oxygen tension, % baseline | 92.6 (12.1) | 96.1 (10.2) | 0.83 |
| Cerebral blood flow, % baseline | 60.3 (45.6–103.7) | 80.5 (64.3–123.8) | 0.22 |
Wilcoxon rank‐sum test is used for nonnormally distributed data. Values are mean (SEM) or median (interquartile range). CA21 indicates cardiac arrest (CA) with cardiopulmonary resuscitation (CPR) in 0.21 fraction of inspired oxygen (FiO2); CA100, CA with CPR in 1.0 fraction of FiO2; and ROSC, return of spontaneous circulation.
Groups were compared at indicated time points post‐ROSC, and values were analyzed over the 10 minutes preceding each time point, except preceding the 10‐minute post‐ROSC time point when values were analyzed over 2 minutes. Student t test is used for normally distributed data.
Figure 2. Brain tissue oxygen tension (PbtO2) during the experimental period.
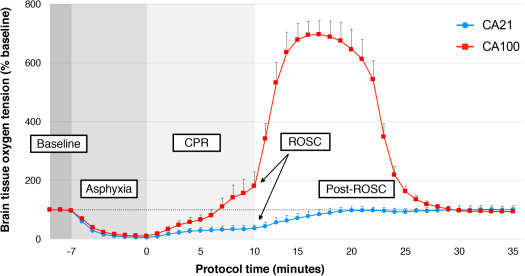
PbtO2, expressed as percentage baseline, is described between cardiac arrest (CA) with cardiopulmonary resuscitation (CPR) in 0.21 fraction of inspired oxygen (FiO2) (CA21) (blue) and CA with CPR in 1.0 fraction of FiO2 (CA100) (red) treatment groups during baseline, asphyxia, CPR, and through 25 minutes after return of spontaneous circulation (ROSC). Error bars represent SEM. Comparison between groups performed with generalized estimating equations regression model.
Cerebral Mitochondrial Respiration and Cerebral Microdialysis
Maximal, coupled oxidative phosphorylation from CI and CII–driven respiration, compared per mg of tissue and normalized to citrate synthase, was not different between treatment groups (Table 5). However, maximal, coupled oxidative phosphorylation was significantly lower following CA in both treatment groups compared with sham in cortex (sham versus CA21, P<0.01; sham versus CA100, P<0.05) as well as in hippocampus (sham versus CA21, P<0.01; sham versus CA100, P<0.05). CI‐driven respiration (oxidative phosphorylation capacity of CI) was also lower in both CA groups compared with sham in cortex (sham versus CA21, P<0.01; sham versus CA100, P<0.05) as well as in hippocampus (sham versus CA21, P<0.01; sham versus CA100, P<0.05). State 4° respiration without ATP production did not differ between CA groups or compared with sham for either region. Finally, the respiratory control ratio for maximal oxidative phosphorylation (maximal, coupled oxidative phosphorylation/state 4° respiration without ATP production), which evaluates phosphorylation coupling efficiency and is a measure of global mitochondrial function, was significantly lower in CA groups compared with sham in cortex (sham versus CA21, P<0.001; sham versus CA100, P<0.01) as well as in hippocampus (sham versus CA21, P<0.01; sham versus CA100, P<0.05), but was not different between CA groups.
Table 5.
Mitochondrial Respiration at 4 Hours Following CA
| Respiratory Parameters (Normalized to CS) | CA21 (n=8) | CA100 (n=8) | Sham (n=5) |
|---|---|---|---|
| Cortex | |||
| OXPHOSCI | 2.29 (1.9–2.73)a | 2.42 (2.24–2.80)a | 4.05 (3.3–4.67) |
| OXPHOSCI+CII | 4.48 (4.26–4.82)a | 4.61 (4.31–5.08)a | 5.87 (5.62–6.3) |
| LEAKCI+CII | 0.76 (0.65–0.93) | 0.67 (0.29–0.74) | 0.77 (0.63–1.02) |
| ETCCI+CII | 4.40 (4.38–4.44)a | 4.49 (4.37–4.51)a | 5.52 (5.47–5.79) |
| ETCCII | 3.17 (2.79–3.37)a | 3.33 (3.21–3.55)a | 3.89 (3.68–3.97) |
| CIV | 8.05 (7.65–8.87)a | 8.24 (7.89–8.44)a | 9.35 (8.78–9.99) |
| RCROXPHOSCI+CII | 5.29 (4.64–5.83)a | 5.82 (5.08–8.15)a | 9.42 (8.16–9.95) |
| Hippocampus | |||
| OXPHOSCI | 2.67 (2.13–3.57)a | 3.15 (2.62–3.77)a | 3.73 (3.06–3.99) |
| OXPHOSCI+CII | 5.6 (5.15–6.33)a | 5.9 (5.57–6.22)a | 8.49 (8.43–9.3) |
| LEAKCI+CII | 0.83 (0.69–0.94) | 0.86 (0.60–0.92) | 0.77 (0.75–1.0) |
| ETCCI+CII | 5.51 (5.45–5.37)a | 5.79 (5.74–5.87)a | 8.22 (8.12–8.37) |
| ETCCII | 3.13 (3.02–3.28)a | 3.33 (3.12–3.38)a | 3.65 (3.41–3.69) |
| CIV | 8.0 (7.2–8.4)a | 9.12 (8.4–9.7)a | 10.8 (10.1–11.3) |
| RCROXPHOSCI+CII | 6.21 (5.56–6.59)a | 6.74 (5.93–7.45)a | 7.97 (7.75–8.06) |
CA indicates cardiac arrest; CA21, CA with cardiopulmonary resuscitation (CPR) in 0.21 fraction of inspired oxygen (FiO2); CA100, CA with CPR in 1.0 fraction of FiO2; CIV, complex IV; CS, citrate synthase; ETC, electron transport chain; LEAKCI+CII, state 4° respiration without ATP production; OXPHOSCI, oxidative phosphorylation capacity of complex I; OXPHOSCI+CII, maximal coupled, phosphorylating respiration stimulated by both complex I and complex II substrates; and RCROXPHOSCI+CII, respiratory control ratio for maximal, oxidative phosphorylation.
P<0.05 compared with sham. There were no differences between CA21 and CA100 treatment groups. Respiration is expressed per mg of tissue (pmol O2/s/mg) normalized per CS activity, a marker of mitochondrial content. Wilcoxon rank‐sum test was used for nonnormally distributed data. Values are median (interquartile range).
Glucose, pyruvate, lactate, and glycerol were measured from extracellular fluid via a cerebral microdialysis catheter at predefined intervals throughout the experimental period. There were no differences in metabolites between CA21 and CA100 animals at any time points (Figure 3 and Table 6). During the first 30 minutes post‐ROSC, lactate/pyruvate ratio (LPR) was increased in CA animals compared with sham (CA21, P=0.2; CA100, P<0.05). Glycerol, a marker of neuronal injury, was also higher in CA animals compared with sham at 30 minutes post‐ROSC (CA21, P<0.05; CA100, P=0.29) in the sample drawn 30 minutes post‐ROSC.
Figure 3. Cerebral microdialysis lactate/pyruvate ratio (LPR) and glycerol during the experimental period across groups.
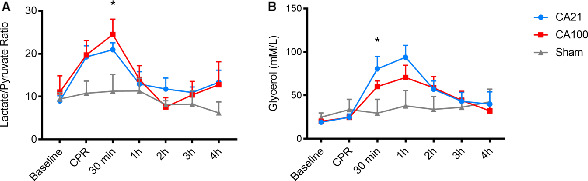
There were no differences in LPR or glycerol between cardiac arrest (CA) with cardiopulmonary resuscitation (CPR) in 0.21 fraction of inspired oxygen (FiO2) (CA21) and CA with CPR in 1.0 fraction of FiO2 (CA100) at any time points. A, LPR was significantly increased at 30 minutes after return of spontaneous circulation (ROSC) in CA100 compared with sham (P<0.05). B, Glycerol was significantly increased at 30 minutes post‐ROSC in CA21 compared with sham (P<0.05). Comparisons performed using ANOVA with Bonferroni correction.
Table 6.
Cerebral Microdialysis
| Parameter | CA21 | CA100 | Sham |
|---|---|---|---|
| Lactate/pyruvate ratio | |||
| Baseline | 8.99 (1.23) (n=9) | 11.1 (3.81) (n=9) | 9.43 (1.52) (n=3) |
| CPR | 19.3 (2.59) (n=9) | 19.1 (3.35) (n=9) | 10.8 (2.83) (n=4) |
| Post‐ROSC 30 min | 21.0 (1.60) (n=7) | 24.5 (3.59)a (n=8) | 11.3 (3.93) (n=4) |
| Post‐ROSC 1 h | 12.9 (2.88) (n=6) | 14.0 (3.21) (n=8) | 11.3 (2.94) (n=5) |
| Post‐ROSC 2 h | 11.8 (2.56) (n=5) | 7.59 (2.15) (n=6) | 8.12 (0.92) (n=4) |
| Post‐ROSC 3 h | 11.0 (1.28) (n=3) | 10.4 (3.15) (n=5) | 8.23 (3.18) (n=3) |
| Post‐ROSC 4 h | 13.3 (2.83) (n=3) | 12.8 (5.38) (n=2) | 6.22 (2.55) n=3 |
| Glycerol | |||
| Baseline | 19.2 (2.63) (n=9) | 20.2 (2.55) (n=10) | 24.9 (4.90) (n=5) |
| CPR | 24.8 (3.76) (n=8) | 24.6 (2.64) (n=9) | 33.6 (11.8) (n=5) |
| Post‐ROSC 30 min | 80.6 (14.3)a (n=6) | 60.1 (6.61) (n=7) | 29.5 (15.9) (n=5) |
| Post‐ROSC 1 h | 94.0 (13.6) (n=7) | 70.6 (14.1) (n=7) | 38.0 (17.7) (n=5) |
| Post‐ROSC 2 h | 57.1 (9.64) (n=7) | 58.8 (12.8) (n=8) | 33.9 (14.8) (n=5) |
| Post‐ROSC 3 h | 43.3 (10.3) (n=7) | 44.2 (10.5) (n=8) | 36.3 (11.5) (n=5) |
| Post‐ROSC 4 h | 39.9 (14.2) (n=7) | 32.2 (7.83) (n=7) | 42.6 (14.7) (n=4) |
There were no differences in lactate/pyruvate ratio or glycerol between CA21 and CA100 at any time points. Comparisons between groups performed using ANOVA with Bonferroni correction. Values are mean (SEM). CA21 indicates cardiac arrest (CA) with CPR in 0.21 fraction of inspired oxygen (FiO2); CA100, CA with CPR in 1.0 fraction of FiO2; CPR, cardiopulmonary resuscitation; and ROSC, return of spontaneous circulation.
P<0.05 compared with sham.
Cerebral mtROS and Cerebral Oxidative Injury
MtROS production was significantly increased in CA100 animals compared with CA21 animals in cortical tissue (0.283 [IQR, 0.276–0.290] pmol H2O2/[s/mg] versus 0.224 [IQR, 0.205–0.239] pmol H2O2/[s/mg]; P<0.05). Hippocampal mtROS was also increased (0.197 [IQR, 0.152–0.236] pmol H2O2/[s/mg] in CA100 versus 0.176 [IQR, 0.158–0.186] pmol H2O2/[s/mg] in CA21; P=0.08), but did not reach significance. MtROS was higher in CA100 animals compared with sham in both regions (cortex, P<0.001; hippocampus, P<0.05). MtROS was not significantly different between CA21 animals and sham (cortex, P=0.20; hippocampus, P=0.13).
CA100 animals demonstrated significantly increased protein carboxylation (0.84 [IQR, 0.72–0.89] nmol/mg protein) compared with CA21 animals (0.52 [IQR, 0.50–0.56] nmol/mg protein; P<0.05) and compared with sham (0.35 [IQR, 0.31–0.38] nmol/mg protein; P<0.001). CA100 animals demonstrated a significant increase in 4‐hydroxynoneal (1.30 [IQR, 1.12–1.55] absorbance at 450 nm) compared with CA21 animals (0.74 [IQR, 0.64–0.99] absorbance at 450 nm; P<0.05) and compared with sham (0.39 [IQR, 0.30–0.43] absorbance at 450 nm; P<0.001). There were no significant differences in protein carbonyls or 4‐hydroxynoneal between CA21 and sham (Figure 4).
Figure 4. Mitochondrial‐derived reactive oxygen species (mtROS) and oxidative brain injury across groups.
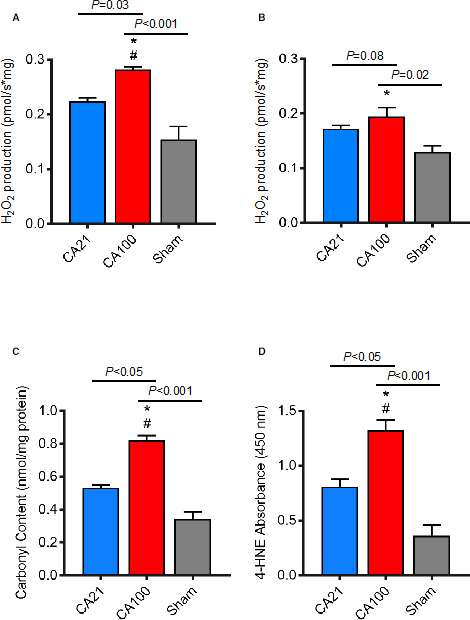
MtROS (hydrogen peroxide [H2O2] production) during maximal oxidative phosphorylation was significantly increased in cardiac arrest (CA) with cardiopulmonary resuscitation (CPR) in 1.0 fraction of inspired oxygen (FiO2) (CA100) compared with CA with CPR in 0.21 fraction of FiO2 (CA21) (*P=0.03) and sham (# P<0.001) in cerebral cortex (A) and significantly increased in CA100 compared with sham (*P=0.02) in hippocampus (B). MtROS was not significantly different in CA21 compared with sham in either region. C, Protein carboxylation (4‐hydroxynoneal [4‐HNE]) measured in cortex. CA100 animals demonstrated a significant increase in protein carboxylation compared with animals treated with CA21 (# P<0.05) and sham (P<0.001). D, Lipid peroxidation measured by 4‐HNE absorbance. CA100 animals demonstrated a significant increase in 4‐HNE compared with animals treated with CA21 (# P<0.05) and sham (*P<0.001).
Mitochondrial RNA Expression
Thirteen mitochondrial‐encoded genes were examined. There were no differences in gene expression between CA21 and sham animals. Compared with CA21 and sham, CA100 animals exhibited significantly reduced expression of >90% of the mitochondrial genes involved in oxidative phosphorylation, with CI predominately affected (Figure 5).
Figure 5. Mitochondrial RNA expression is downregulated in cardiac arrest (CA) with cardiopulmonary resuscitation (CPR) in 1.0 fraction of inspired oxygen (FiO2) (CA100).
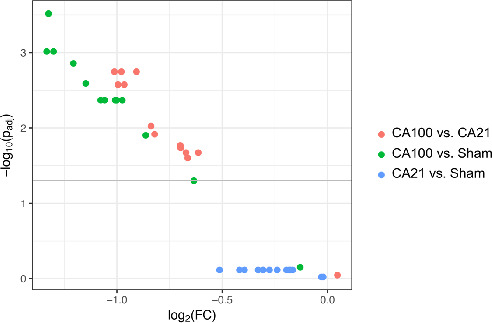
This volcano plot is a type of scatterplot used to quickly visualize significant genes in RNA‐sequencing results. It displays statistical significance against magnitude of change (ie, fold change [FC]). The gray line indicates threshold of significance (adjusted P<0.05, using Benjamini‐Hochberg correction for multiple comparisons). The negative log of the P values is used for the y axis so that the smallest P values (most significant) are at the top of the plot. Therefore, the clustering in the upper left corner of the graph displays significant downregulation of multiple mitochondrial respiratory chain genes in CA100 animals compared with CA with CPR in 0.21 fraction of FiO2 (CA21) and sham.
Discussion
In this randomized, blinded, preclinical trial comparing oxygenation strategies during pediatric CPR, animals treated with 1.0 FiO2 had relative cerebral hyperoxia during and immediately following CPR, higher mtROS production, and downstream oxidative injury in the brain 4 hours following ROSC. Notably, animals treated with 0.21 FiO2 during CPR were equally likely to survive and had similar intra‐arrest systemic and cerebral hemodynamics compared with animals treated with 1.0 FiO2. Both groups had significantly decreased cerebral mitochondrial bioenergetics 4 hours after ROSC compared with sham, but there were no detectable differences between 21 and 1.0 FiO2. Cumulatively, these findings suggest that minimizing exposure to potentially injurious high concentrations of FiO2 or adding a therapeutic intervention to mitigate hyperoxic cerebral injury during CPR may improve outcomes from pediatric CA. Perhaps, attempting to mitigate oxygen toxicity after ROSC may be too late.
Animal models of adult VF CA have compared lower FiO2 fractions against 1.0, reporting equivalent survival rates with room air.12, 36, 37 These findings may not be generalizable to a pediatric CA scenario, in which respiratory deterioration is typically the inciting event.38 This study of pediatric asphyxia‐associated CA adds relevance for the pediatric population by mimicking severe systemic hypoxemia and inadequate tissue perfusion immediately before CPR and by showing equal survival with 0.21 FiO2. Although systemic oxygenation was not significantly different between groups during CPR, there was a trend toward higher Pao 2 in the CA100 group (P=0.10). This may have reached significance with a larger sample size. Although the precise oxygen fraction required to achieve ROSC is unknown,39 animal and human data have established that CoPP and aortic diastolic pressure (the main determinant of CoPP) are the critical drivers for myocardial blood flow, ROSC, and longer‐term survival.40, 41 As described above, our study provided protocolized, hemodynamically targeted CPR. We found that CoPP and diastolic BP, not systemic oxygenation (ie, Pao 2, Mixed Venous Oxygen Saturation (SvO2), acid‐base balance), were the only parameters that were significantly associated with survival. In the setting of high‐quality CPR, it is reasonable to conclude that 0.21 FiO2 may be safe and effective for some patients and may limit neurologic injury.
Given that most children with CA initially survive but many of them have neurologic morbidity or do not survive to hospital discharge, often because of neurologic injury,4 we sought to understand the impact of oxygen resuscitation strategies on brain tissue oxygenation during CPR, cerebral mitochondrial function, and metabolic markers of cerebral damage. Compared with sham animals, animals in both CA groups had significantly lower CI and CII–driven oxidative phosphorylation and respiratory control ratio. This is consistent with previous reports of mitochondrial bioenergetics failure in preclinical models of CA.13, 24 Animals resuscitated with 1.0 FiO2 had cerebral hyperoxia during and following CPR compared with the 0.21 FiO2 group, demonstrating a relationship between oxygen resuscitation strategy and brain tissue oxygenation. However, the 2 CA groups did not differ in either cerebral cortex or hippocampus mitochondrial respiration, or in cerebral metabolism measured via real‐time cerebral microdialysis markers. These findings demonstrate that reducing oxygen supply does not necessarily impair mitochondrial bioenergetics or precipitate a metabolic crisis.42
The correlation between mitochondrial dysfunction and microdialysis markers of metabolic crisis has been previously reported in a preclinical model of traumatic brain injury43 and in cardiac surgical models of deep hypothermic circulatory arrest,44 but to our knowledge, this is the first in CA. In this study, LPR and glycerol were increased following CA. These biochemical markers have been associated with mortality and poor neurologic outcome in adult traumatic brain injury.45 LPR reflects the extent of anaerobic glycolysis, which may occur because of disturbed oxygen delivery (ischemia) or use (mitochondrial dysfunction).43, 46 Our data suggest that the primary driver of increased LPR following CA may be mitochondrial bioenergetics failure and not ischemia, as LPR was inversely proportional to mitochondrial function, but unaffected by cerebral oxygen tension following reperfusion. Other explanations for increased lactate production could be increased cell metabolism,47 glycolysis‐stimulating effects of circulating epinephrine following ROSC, or inactivation of the pyruvate dehydrogenase complex by oxidative injury during reperfusion.48 Invoking this latter mechanism, hyperoxic resuscitation has been shown to exacerbate loss of pyruvate dehydrogenase complex activity and increase brain tissue lactate levels in hippocampus.37, 49 In contrast, we did not observe differences in LPR between CA groups. This may be attributed to differences in technique (microdialysis versus nuclear magnetic resonance spectroscopy), location (cortex versus hippocampus), or duration of reperfusion hyperoxia exposure (10 minutes versus 1 hour post‐ROSC). More important, impairments in mitochondrial bioenergetics observed in this study are not fully explained by altered metabolism, because our protocol provided mitochondria with substrates at saturating concentrations, including pyruvate dehydrogenase complex–independent substrates malate, glutamate, and succinate. Last, our data show that increasing systemic oxygenation and cerebral extracellular oxygen tension (ie, Pao 2 and PbtO2) above normal levels does not improve mitochondrial function or cerebral metabolism, at least early after CA. Thus, there does not appear to be a metabolic benefit with the administration of 1.0 compared with 0.21 FiO2 during CA measured at these time points.
During ischemia, the respiratory chain and electron carrier pools become maximally reduced, and mitochondria become progressively compromised because of ATP depletion, loss of ion homeostasis, calcium overload, and pH changes.50 On reperfusion with oxygenated blood, the inappropriately reduced and damaged mitochondrial components will spill electrons onto oxygen to form ROS. Mitochondria are both the primary producers of ROS during reperfusion and the primary target of ROS‐induced damage. Therefore, therapeutic strategies that minimize mtROS production may ultimately preserve mitochondrial integrity and neurological function following CA. Animals treated with 1.0 FiO2 had higher levels of mtROS compared with those treated with 0.21 FiO2 and the sham group. Notably, mtROS did not significantly differ between those treated with 0.21 FiO2 and the non‐CA sham group, which did not undergo CA. Exposure to excessive oxygen has been shown in vitro to accelerate mtROS production from the electron transport chain.50, 51 This may occur because oxygen is a substrate for superoxide (O2 .‐) production, and mitochondrial oxygen concentration is determined by extracellular tissue oxygen tension.52 These data establish that excessive oxygen during resuscitation can lead to harmful mtROS production, and that this may be prevented by avoiding cerebral hyperoxia during and immediately after resuscitation.
Animals treated with 1.0 FiO2 had worse oxidative injury, evidenced by increased protein carboxylation and lipid peroxidation in this group. A striking finding was that in CA100 animals, nearly all mitochondrial‐encoded genes were downregulated, potentially serving to dampen oxidative stress produced by damaged mitochondria in a high oxygen environment. These findings augment existing data12, 15, 16, 37 by correlating mtROS measurements and downstream pathologic effects, revealing oxidative injury even over the short time frame of these experiments.53 Although this may initially reduce oxidative stress, persistently decreased CI activity may ultimately limit cerebral recovery and promote neurodegeneration, thus contributing to long‐term cognitive dysfunction post‐CA.13, 16, 54, 55, 56, 57 In this study, 1.0 FiO2 was continued post‐ROSC for a relatively brief period (ie, 10 minutes). It is plausible that children who are exposed to 1.0 FiO2 for longer periods post‐ROSC may have exaggerated mtROS production and oxidative brain injury, because most mtROS production occurs during reperfusion.14, 16 Further investigation into the precise time course of mtROS production and oxidative injury under varying oxygen exposure is needed. Given the known benefits of titrating resuscitation efforts to patient‐specific pathophysiological features in real‐time, future work should focus on titrating intra‐arrest oxygen therapy to actual markers of cerebral bioenergetics and metabolism.
This study has limitations. First, although we observed no differences in survival between CA groups, our study was not powered a priori to detect survival differences. Second, our subjects were a relatively homogeneous group of healthy female swine with normal lungs. Children with significant lung disease who experience CA may require higher FiO2 because of ventilation‐perfusion mismatch, diffusion restriction, and possibly pulmonary vasoreactivity. However, respiratory arrest leading to CA can occur without significant lung pathological features, as in mucous plugging, endotracheal tube dislodgement, and other airway obstructions. Future experiments will focus on understanding which subsets of patients could achieve ROSC with lower FiO2. Third, time course may play an important role in the interplay between mtROS, oxidative stress, and mitochondrial bioenergetic changes following CA. Last, mitochondrial respiration, microdialysis markers of cerebral metabolism, and mtROS are markers of brain damage but only surrogates for brain function. Nevertheless, these metrics have been correlated with functional neurologic outcome post‐CA,13 are important for mechanistic understanding of hyperoxia‐induced brain injury, and represent potential targets for intervention. In this model, we prioritized invasive neuromonitoring and measurement of fresh brain tissue mitochondrial bioenergetics and mtROS, which required a craniotomy. Thus, for these initial proof‐of‐concept studies, we focused on short‐term time course to investigate mechanisms. With our findings, the next critical steps will be to assess longer‐term behavior, neuroimaging, and biomarkers to simulate a clinical trial.
In conclusion, immature swine treated with 1.0 FiO2 during CPR had increased cerebral mtROS and oxidative injury in the brain compared with animals treated with 0.21 FiO2. The 0.21 FiO2 group was equally likely to survive and had similar intra‐arrest hemodynamics, mitochondrial bioenergetics, and cerebral metabolism compared with animals treated with 1.0 FiO2.
Sources of Funding
Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health (NIH) under award number TL1TR001880 (Dr Marquez), NIH National Institute of Child Health and Human Development F31HD085731 (Dr Ko), NIH National Heart, Lung, and Blood Institute T32HL007915 (Dr Ko), Tim Raymond Family Fund (Dr Kilbaugh), NIH National Heart, Lung, and Blood Institute R01HL141386 (Dr Kilbaugh), and Department of Defense W81XWH‐16‐PRMRP‐TTDA (Dr Kilbaugh).
Disclosures
Dr Marquez received a research grant from the National Institutes of Health (NIH) National Center for Advancing Translational Sciences TL1TR001880 (significant). Dr Morgan received research grants from the NIH National Institute of Child Health and Human Development R21HD089132 (modest), NIH National Heart, Lung, and Blood Institute R01HL147616 (modest), and NIH National Heart, Lung, and Blood Institute R01HL141386 (modest). Dr Ko received research grants from the NIH National Institute of Child Health and Human Development F31HD085731 (significant) and NIH National Heart, Lung, and Blood Institute T32HL007915 (significant). Dr McManus received a research grant from the Department of Defense W81XWH‐16‐PRMRP‐TTDA (significant). Dr Licht received a research grant from the NIH National Institute of Neurological Disorders and Stroke R01NS060653 (significant). Dr Berg received the following research grants: NIH R21HD089132 (significant); NIH National Heart, Lung, and Blood Institute R01HL147616 (significant); NIH National Heart, Lung, and Blood Institute R01HL141386 (modest); and NIH National Heart, Lung, and Blood Institute R01HL131544 (modest). Dr Sutton holds a Zoll speaking honorarium and received the following research grants: NIH National Heart, Lung, and Blood Institute R01HL131544 (significant); NIH National Heart, Lung, and Blood Institute R01HL147616 (significant); and NIH National Heart, Lung, and Blood Institute R01HL141386 (significant). Dr Kilbaugh received the following research grants: NIH National Heart, Lung, and Blood Institute R01HL141386 (significant); Department of Defense W81XWH‐16‐PRMRP‐TTDA (significant); and Tim Raymond Family Fund (modest). The remaining authors have no disclosures to report.
Acknowledgments
We wish to thank Children's Hospital of Philadelphia Department of Veterinary Resources, including Drs Katharine Tuohy, Travis Seymour, and Laike Stewart for their hard work and dedication to our laboratory and animal welfare.
(J Am Heart Assoc. 2020;9:e015032 DOI: 10.1161/JAHA.119.015032.)
For Sources of Funding and Disclosures, see page 15.
References
- 1. Atkins DL, Everson‐Stewart S, Sears GK, Daya M, Osmond MH, Warden CR, Berg RA. Epidemiology and outcomes from out‐of‐hospital cardiac arrest in children: the Resuscitation Outcomes Consortium Epistry‐Cardiac Arrest. Circulation. 2009;119:1484–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Knudson JD, Neish SR, Cabrera AG, Lowry AW, Shamszad P, Morales DLS, Graves DE, Williams EA, Rossano JW. Prevalence and outcomes of pediatric in‐hospital cardiopulmonary resuscitation in the United States. Crit Care Med. 2012;40:2940–2944. [DOI] [PubMed] [Google Scholar]
- 3. Girotra S, Spertus JA, Li Y, Berg RA, Nadkarni VM, Chan PS. Survival trends in pediatric in‐hospital cardiac arrests: an analysis from get with the guidelines‐resuscitation. Circ Cardiovasc Qual Outcomes. 2013;6:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berg RA, Nadkarni VM, Clark AE, Moler F, Meert K, Harrison RE, Newth CJ, Sutton RM, Wessel DL, Berger JT, et al. Incidence and outcomes of cardiopulmonary resuscitation in PICUs. Crit Care Med. 2016;44:798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saugstad OD, Ramji S, Soll RF, Vento M. Resuscitation of newborn infants with 21% or 100% oxygen: an updated systematic review and meta‐analysis. Neonatology. 2008;94:176–182. [DOI] [PubMed] [Google Scholar]
- 6. Wyckoff MH, Aziz K, Escobedo MB, Kapadia VS, Kattwinkel J, Perlman JM, Simon WM, Weiner GM, Zaichkin JG. Part 13: neonatal resuscitation. Circulation. 2015;132:S543–S560. [DOI] [PubMed] [Google Scholar]
- 7. de Caen AR, Maconochie IK, Aickin R, Atkins DL, Biarent D, Guerguerian A‐M, Kleinman ME, Kloeck DA, Meaney PA, Nadkarni VM, et al. Part 6: pediatric basic life support and pediatric advanced life support. Circulation. 2015;132:S177–S203. [DOI] [PubMed] [Google Scholar]
- 8. Callaway CW, Soar J, Aibiki M, Bottiger BW, Brooks SC, Deakin CD, Donnino MW, Drajer S, Kloeck W, Morley PT, et al. Part 4: advanced life support: 2015 international consensus on cardiopulmonary resuscitation and emergency cardiovascular care science with treatment recommendations. Circulation. 2015;132:S84–S145. [DOI] [PubMed] [Google Scholar]
- 9. Kilgannon JH. Association between arterial hyperoxia following resuscitation from cardiac arrest and in‐hospital mortality. JAMA. 2010;303:2165. [DOI] [PubMed] [Google Scholar]
- 10. Wang C‐H, Chang W‐T, Huang C‐H, Tsai M‐S, Yu P‐H, Wang A‐Y, Chen N‐C, Chen W‐J. The effect of hyperoxia on survival following adult cardiac arrest: a systematic review and meta‐analysis of observational studies. Resuscitation. 2014;85:1142–1148. [DOI] [PubMed] [Google Scholar]
- 11. Pilcher J, Weatherall M, Shirtcliffe P, Bellomo R, Young P, Beasley R. The effect of hyperoxia following cardiac arrest—a systematic review and meta‐analysis of animal trials. Resuscitation. 2012;83:417–422. [DOI] [PubMed] [Google Scholar]
- 12. Liu Y, Rosenthal RE, Haywood Y, Miljkovic‐Lolic M, Vanderhoek JY, Fiskum G. Normoxic ventilation after cardiac arrest reduces oxidation of brain lipids and improves neurological outcome. Stroke. 1998;29:1679–1686. [DOI] [PubMed] [Google Scholar]
- 13. Lautz AJ, Morgan RW, Karlsson M, Mavroudis CD, Ko TS, Licht DJ, Nadkarni VM, Berg RA, Sutton RM, Kilbaugh TJ. Hemodynamic‐directed cardiopulmonary resuscitation improves neurologic outcomes and mitochondrial function in the heart and brain. Crit Care Med. 2019;47:e241–e249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chouchani ET, Pell VR, James AM, Work LM, Saeb‐Parsy K, Frezza C, Krieg T, Murphy MP. A unifying mechanism for mitochondrial superoxide production during ischemia‐reperfusion injury. Cell Metab. 2016;23:254–263. [DOI] [PubMed] [Google Scholar]
- 15. Piantadosi CA, Zhang J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke. 1996;27:327–331; discussion 332. [DOI] [PubMed] [Google Scholar]
- 16. Niatsetskaya ZV, Sosunov SA, Matsiukevich D, Utkina‐Sosunova IV, Ratner VI, Starkov AA, Ten VS. The oxygen free radicals originating from mitochondrial complex I contribute to oxidative brain injury following hypoxia‐ischemia in neonatal mice. J Neurosci. 2012;32:3235–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dickerson JW, Dobbing J. Prenatal and postnatal growth and development of the central nervous system of the pig. Proc R Soc London Ser B Biol Sci. 1967;166:384–395. [DOI] [PubMed] [Google Scholar]
- 18. Duhaime AC, Margulies SS, Durham SR, O'Rourke MM, Golden JA, Marwaha S, Raghupathi R. Maturation‐dependent response of the piglet brain to scaled cortical impact. J Neurosurg. 2000;93:455–462. [DOI] [PubMed] [Google Scholar]
- 19. Neurauter A, Nysæther J, Kramer‐Johansen J, Eilevstjønn J, Paal P, Myklebust H, Wenzel V, Lindner KH, Schmölz W, Pytte M, et al. Comparison of mechanical characteristics of the human and porcine chest during cardiopulmonary resuscitation. Resuscitation. 2009;80:463–469. [DOI] [PubMed] [Google Scholar]
- 20. Reis AG, Nadkarni V, Perondi MB, Grisi S, Berg RA. A prospective investigation into the epidemiology of in‐hospital pediatric cardiopulmonary resuscitation using the international Utstein reporting style. Pediatrics. 2002;109:200–209. [DOI] [PubMed] [Google Scholar]
- 21. WHO child growth standards: length/height‐for‐age, weight‐for‐age, weight‐for‐length, weight‐for‐height and body mass index‐for‐age: methods and development. 2014. Available at: https://www.who.int/childgrowth/standards/Technical_report.pdf. Accessed April 18, 2020.
- 22. Matos RI, Watson RS, Nadkarni VM, Huang H‐H, Berg RA, Meaney PA, Carroll CL, Berens RJ, Praestgaard A, Weissfeld L, et al. Duration of cardiopulmonary resuscitation and illness category impact survival and neurologic outcomes for in‐hospital pediatric cardiac arrests. Circulation. 2013;127:442–451. [DOI] [PubMed] [Google Scholar]
- 23. Morgan RW, Kilbaugh TJ, Shoap W, Bratinov G, Lin Y, Hsieh T‐C, Nadkarni VM, Berg RA, Sutton RM; Pediatric Cardiac Arrest Survival Outcomes PiCASO Laboratory Investigators . A hemodynamic‐directed approach to pediatric cardiopulmonary resuscitation (HD‐CPR) improves survival. Resuscitation. 2017;111:41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kilbaugh TJ, Sutton RM, Karlsson M, Hansson MJ, Naim MY, Morgan RW, Bratinov G, Lampe JW, Nadkarni VM, Becker LB, et al. Persistently altered brain mitochondrial bioenergetics after apparently successful resuscitation from cardiac arrest. J Am Heart Assoc. 2015;4:e002232 DOI: 10.1161/JAHA.115.002232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sutton RM, Friess SH, Bhalala U, Maltese MR, Naim MY, Bratinov G, Niles D, Nadkarni VM, Becker LB, Berg RA. Hemodynamic directed CPR improves short‐term survival from asphyxia‐associated cardiac arrest. Resuscitation. 2013;84:696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sutton RM, Friess SH, Naim MY, Lampe JW, Bratinov G, Weiland TR, Garuccio M, Nadkarni VM, Becker LB, Berg RA. Patient‐centric blood pressure–targeted cardiopulmonary resuscitation improves survival from cardiac arrest. Am J Respir Crit Care Med. 2014;190:1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kilbaugh TJ, Karlsson M, Duhaime A‐C, Hansson MJ, Elmer E, Margulies SS. Mitochondrial response in a toddler‐aged swine model following diffuse non‐impact traumatic brain injury. Mitochondrion. 2016;26:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murphy Michael P. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hefti MM, Farrell K, Kim S, Bowles KR, Fowkes ME, Raj T, Crary JF. High‐resolution temporal and regional mapping of MAPT expression and splicing in human brain development. PLoS One. 2018;13:e0195771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baruzzo G, Hayer KE, Kim EJ, Di Camillo B, FitzGerald GA, Grant GR. Simulation‐based comprehensive benchmarking of RNA‐seq aligners. Nat Methods. 2017;14:135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. [DOI] [PubMed] [Google Scholar]
- 33. Csárdi G, Nepusz T. The igraph software package for complex network research. InterJournal Complex Syst. 2006;1695:1–9. [Google Scholar]
- 34. Wickham H. Ggplot2: Elegant Graphics for Data Analysis. Springer; 2009. [Google Scholar]
- 35. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zwemer CF, Whitesall SE, D'Alecy LG. Cardiopulmonary‐cerebral resuscitation with 100% oxygen exacerbates neurological dysfunction following nine minutes of normothermic cardiac arrest in dogs. Resuscitation. 1994;27:159–170. [DOI] [PubMed] [Google Scholar]
- 37. Vereczki V, Martin E, Rosenthal RE, Hof PR, Gloria E. Normoxic resuscitation after cardiac arrest protects against hippocampal oxidative stress, metabolic dysfunction, and neuronal death. J Cereb Blood Flow Metab. 2006;26:821–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nadkarni VM, Larkin GL, Peberdy MA, Carey SM, Kaye W, Mancini ME, Nichol G, Lane‐Truitt T, Potts J, Ornato JP, et al. First documented rhythm and clinical outcome from in‐hospital cardiac arrest among children and adults. JAMA. 2006;295:50–57. [DOI] [PubMed] [Google Scholar]
- 39. Yeh ST, Cawley RJ, Aune SE, Angelos MG. Oxygen requirement during cardiopulmonary resuscitation (CPR) to effect return of spontaneous circulation. Resuscitation. 2009;80:951–955. [DOI] [PubMed] [Google Scholar]
- 40. Paradis NA, Martin GB, Rivers EP, Goetting MG, Appleton TJ, Feingold M, Nowak RM. Coronary perfusion pressure and the return of spontaneous circulation in human cardiopulmonary resuscitation. JAMA. 1990;263:1106–1113. [PubMed] [Google Scholar]
- 41. Berg RA, Sutton RM, Reeder RW, Berger JT, Newth CJ, Carcillo JA, McQuillen PS, Meert KL, Yates AR, Harrison RE, et al. Association between diastolic blood pressure during pediatric in‐hospital cardiopulmonary resuscitation and survival. Circulation. 2018;137:1784–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kochanek PM, Clark RSB, Ruppel RA, Adelson PD, Bell MJ, Whalen MJ, Robertson CL, Satchell MA, Seidberg NA, Marion DW, et al. Biochemical, cellular, and molecular mechanisms in the evolution of secondary damage after severe traumatic brain injury in infants and children: lessons learned from the bedside. Pediatr Crit Care Med. 2000;1:4–19. [DOI] [PubMed] [Google Scholar]
- 43. Kilbaugh TJ, Bhandare S, Lorom DH, Saraswati M, Robertson CL, Margulies SS. Cyclosporin A preserves mitochondrial function after traumatic brain injury in the immature rat and piglet. J Neurotrauma. 2011;28:763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mavroudis CD, Karlsson M, Ko T, Hefti M, Gentile JI, Morgan RW, Plyler R, Mensah‐Brown KG, Boorady TW, Melchior RW, et al. Cerebral mitochondrial dysfunction associated with deep hypothermic circulatory arrest in neonatal swine. Eur J Cardiothorac Surg. 2018;54:162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zeiler FA, Thelin EP, Helmy A, Czosnyka M, Hutchinson PJA, Menon DK. A systematic review of cerebral microdialysis and outcomes in TBI: relationships to patient functional outcome, neurophysiologic measures, and tissue outcome. Acta Neurochir. 2017;159:2245–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Menon DK, Coles JP, Gupta AK, Fryer TD, Smielewski P, Chatfield DA, Aigbirhio F, Skepper JN, Minhas PS, Hutchinson PJ, et al. Diffusion limited oxygen delivery following head injury. Crit Care Med. 2004;32:1384–1390. [DOI] [PubMed] [Google Scholar]
- 47. Pellerin L, Bouzier‐Sore A‐K, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ. Activity‐dependent regulation of energy metabolism by astrocytes: an update. Glia. 2007;55:1251–1262. [DOI] [PubMed] [Google Scholar]
- 48. Martin E, Rosenthal RE, Fiskum G. Pyruvate dehydrogenase complex: metabolic link to ischemic brain injury and target of oxidative stress. J Neurosci Res. 2005;79:240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Richards EM, Fiskum G, Rosenthal RE, Hopkins I, McKenna MC. Hyperoxic reperfusion after global ischemia decreases hippocampal energy metabolism. Stroke. 2007;38:1578–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide: general properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hoffman DL, Salter JD, Brookes PS. Response of mitochondrial reactive oxygen species generation to steady‐state oxygen tension: implications for hypoxic cell signaling. Am J Physiol Heart Circ Physiol. 2007;292:H101–H108. [DOI] [PubMed] [Google Scholar]
- 52. Erecińska M, Silver IA. Tissue oxygen tension and brain sensitivity to hypoxia. Respir Physiol. 2001;128:263–276. [DOI] [PubMed] [Google Scholar]
- 53. Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time course of post‐traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab. 2006;26:1407–1418. [DOI] [PubMed] [Google Scholar]
- 54. Navarro A, Boveris A. Brain mitochondrial dysfunction in aging, neurodegeneration and Parkinson's disease. Front Aging Neurosci. 2010;2:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Walker KR, Tesco G. Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Front Aging Neurosci. 2013;5:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. [DOI] [PubMed] [Google Scholar]
- 57. Keeney PM, Xie J, Capaldi RA, Bennett JP. Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26:5256–5264. [DOI] [PMC free article] [PubMed] [Google Scholar]