

A few decades ago, Professor David Barker and colleagues showed that English men and women with low birth weight had high rates of cardiac mortality in adulthood.1 That finding set off an explosion of epidemiological and basic science studies that extended Barker's findings to other populations across the globe. We now know that babies born at the extremes of the birth weight scale are at risk for ischemic heart disease, heart failure, hypertension, type 2 diabetes mellitus, and other chronic conditions. Birth weight is not the only indicator of offspring risk. The list of maternal stressors that impart risk for chronic disease in offspring has grown over 3 decades on the basis of hundreds of animal and human studies. Known maternal stressors include malnutrition, toxic chemicals, fetal hypoxia, placental shape, and a host of other detrimental social determinants.2 Even common medical conditions affecting a pregnancy are associated with elevated cardiovascular risk for both mother and child. In this issue of the Journal of the American Heart Association (JAHA), Aye and colleagues3 have documented another maternal medical condition that may portend heart disease risk in offspring: pregnancy‐induced hypertension,4 referred to herein as gestational hypertension (GH).
GH and Infant Heart Growth
Aye et al3 sought to determine the degree to which maternal hypertensive disorders are associated with indexed right and left myocardial masses, chamber volumes, and performances in the infant at birth and at 3 months. Offspring of hypertensive mothers are likely to become hypertensive themselves.5 In such offspring, blood pressure increases from childhood into adulthood and is a significant cause of high blood pressure in young adults. These hypertensive young adults have increased left ventricular (LV) wall mass and reduced chamber volume.6 A critical gap in current knowledge is the degree to which the abnormal growth trajectory of the myocardium is driven by maternal hypertension before birth versus being a postnatal response to elevated systolic load or hormonal influences.
Aye et al3 used echocardiography to make cardiac measurements in 134 infants at term birth and at 3 months of age. Fifty‐four had mothers who were normotensive over the course of gestation, and 80 had GH alone or the more severe form, preeclampsia. The echocardiographic analyses included LV and right ventricular (RV) volumes, myocardial mass, and dynamic function. Measurements were indexed to body surface area or body size. Data were analyzed by sex, body size, blood pressure, and severity of maternal disease. LV and RV masses were similar at birth, whereas end diastolic volume in the RV was some 20% smaller at birth in infants born to hypertensive mothers. At 3 months, the RV volume deficit persisted. The indexed free wall masses were 8% larger in the LV and 23% larger in the RV compared with hearts from normotensive mothers. Increases in RV mass and deficits in end diastolic volume were related to the severity of the maternal disorder. There were no functional differences between groups.
Normal and Abnormal Cardiovascular Adaptations to Pregnancy
There is no other time in life, outside of pregnancy, when the normal human body experiences such dramatic physiological changes in the cardiovascular system.7 As pregnancy proceeds, a woman's total oxygen consumption increases, and her blood volume and red cell mass expand. Cardiac output increases by some 50% as the result of increased heart rate, ventricular stroke volumes, and decreased systemic vascular input impedance. The walls of arteries, veins, and cardiac chambers are remodeled to increase compliance. Renal blood flow and glomerular filtration rates are increased by 50%.7 The uterine arteries enlarge and remodel profoundly to augment blood flow for the placenta and the growing fetus.
In some pregnancies, these cardiovascular modifications are inadequate or affected by other maternal conditions. Maladaptation may lead to more serious complications, including hypertensive syndromes.8 Hypertensive disorders of pregnancy encompass hypertension, end organ pathological conditions, and severe morbidities, including seizures and stroke.9 A subset of women with GH acquires organ‐specific pathological features that define preeclampsia, a syndrome that presents in many forms.10 Proteinuria, endothelial dysfunction, and pronounced edema are common features that can worsen to include the HELLP syndrome (hemolysis, elevated liver enzymes, and low platelet count). In the most severe cases, preeclampsia includes disease end‐organ damage or eclampsia and seizure activity; such conditions account for ≈7% of maternal deaths per year in the United States11 and many more in low‐ and middle‐income countries.
Root Causes of GH
The root causes of GH are unknown but not for lack of causative theories.12 The underlying cause of GH remains a mystery. Many cases of preeclampsia are associated with shallow implantation of the placenta, characterized by poor remodeling of spiral arteries that feed the maternal placenta and the abnormal release of exosomes and circulating factors that promote endothelial dysfunction. The placenta is also a culprit in nonpreeclampsia hypertension on the basis of its size and shape. Like most complex chronic diseases, there are genetic and environmental influences that elevate the risk of acquiring preeclampsia. Although genome‐wide association studies and other genetic studies13 have found loci associated with the disease, none of them link to a known pathophysiological process that enlightens the cause of preeclampsia. Although there is growing evidence that some women are predisposed to preeclampsia because of underlying genetic propensities or cardiovascular vulnerabilities before pregnancy,14 the strength of these findings in explaining GH is weakened by the fewer cases of preeclampsia in subsequent pregnancies. The involvement of the placenta is not a controversial concept because, in most cases, the delivery of the fetus and placenta reverses the hypertensive condition.
The GH–Fetal Heart Growth Connection
The “elephant in the room” question is: how does the gestational hypertensive syndrome associate with altered cardiac growth in the fetus? The answer is unknown. The Aye et al3 discovery of associations between maternal hypertension and cardiac remodeling in offspring is new and exciting but has no certain biological explanation.
The Figure represents a mechanistic paradigm, elements of which could be tested in a prospective study. It suggests underlying propensities in mothers, environmental, epigenetic, and genetic, that alter placentation. The role of the placenta in causing preeclampsia is unclear, but there is evidence that a poorly perfused placenta, because of either its own inadequate vascularization or poor remodeling of the uterine circulation, leads to oxidative stress within the placenta and within fetal tissues. In addition, a poorly constructed umbilical microcirculation elevates the impedance faced by the developing heart. This high resistance condition is common among babies whose intrauterine growth is restricted.15
Figure 1. A framework for understanding the origins of fetal cardiac remodeling in hypertensive pregnancies.
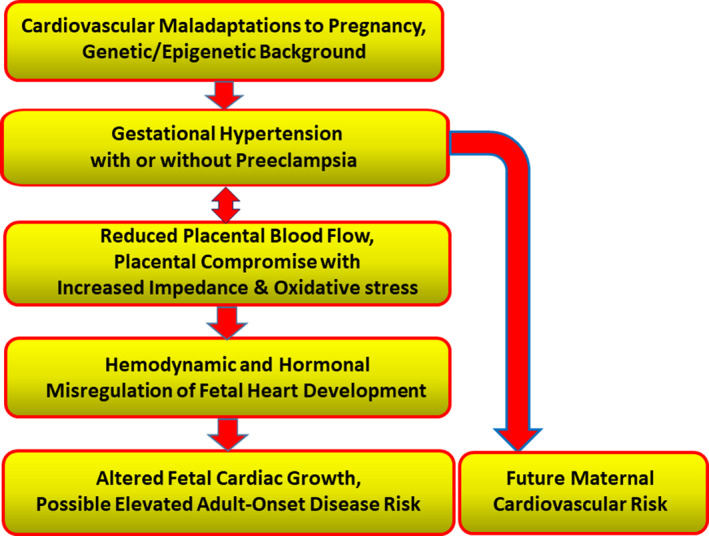
The model incorporates known or suspected physiological underpinnings of maternal hypertension likely to affect cardiac growth regulation during prenatal life. It also points to the elevation in maternal cardiovascular disease risk associated with gestational hypertension.
The fetal myocardium grows by cardiomyocyte replication with concomitant growth of the microcirculation over the first two thirds of gestation. In the last third of pregnancy, cardiomyocytes go through a maturation phase that includes terminal differentiation in which nuclear DNA goes through endoreduplication. At the time of birth, some 70% to 80% of cells have terminally differentiated and are no longer proliferating.16 Over the next few months of postnatal life, most cells in the myocardium will cease to divide. Thus, the number of cells in the myocardium around the time of birth is set and maintained over the lifespan. After birth, myocardial growth mostly depends on cardiomyocyte hypertrophy and the expansion of other constituents.
The maturation process of fetal myocardium is sensitive to 2 primary influences: hemodynamic load and hormone‐mediated regulation of proliferation and terminal differentiation. These processes are well described in large mammals, such as sheep.17 Over the last third of gestation, acutely increased systolic load in experimental animals leads to a short spurt of proliferation among fetal cardiomyocytes, followed by a steady increase in terminal differentiation and cessation of proliferation. Thus, chronically “loaded” fetal hearts have fewer cardiomyocytes at birth compared with normal hearts. Elevated systolic load augments cellular maturation and abnormal hypertrophy, resulting in altered heart chamber anatomical characteristics.
The fetal ventricles pump in parallel against a common arterial pressure. The RV is more sensitive to arterial pressure because it is the larger of the 2 chambers and has a larger radius/wall thickness ratio, higher wall stress, and a mechanical disadvantage.18 Increases in fetal arterial pressure reduce RV stroke volume compared with the smaller LV. Long‐term elevations in fetal arterial pressure lead to remodeling of the ventricles, especially the RV, which may thicken its free wall at the expense of its chamber volume, consistent with the findings of Aye et al.3
Why did the mass of the ventricles in hearts of babies whose mothers had hypertension increase abnormally over the first 3 months after birth? Under normal circumstances, systemic arterial pressure more than doubles within minutes after birth, whereas pulmonary arterial pressure decreases with dilation of the pulmonary bed and closure of the ductus arteriosus. Thus, there is a load‐driven stimulation of hypertrophy that occurs normally in the LV. In the Aye et al study,3 both the RV and the LV increased in mass over the first 3 months in infants born to hypertensive mothers. However, the RV mass outgrew the LV. One can speculate that vascular impedances were greater in infants born to hypertensive mothers even with normal increases in blood pressure, through loss of vascular compliance. The vulnerable RV would be stimulated to undergo hypertrophic growth after birth if the compromised pulmonary bed did not dilate adequately on the initiation of breathing.19 Do these changes lead to elevated risk for myocardial disease in later life? No one knows. However, on the basis of extensive animal studies, one can speculate that babies born to mothers who have GH/pregnancy‐induced hypertension will have elevated risk for ischemic heart disease and/or heart failure in later life.
What Next?
The study by the talented Leeson team3 showed a previously unknown cardiac growth response in infants born to hypertensive mothers. For this reason, the report is highly noteworthy. However, we are left wondering on 2 fronts: (1) maternal physiological profiles across gestation that were associated with the hypertension are unknown; (2) the degree to which GH alone versus hypertension with preeclampsia influence cardiac growth in offspring remains uncertain. These issues await further study.
The complexity of blood pressure regulation, as artfully explained by Arthur Guyton decades ago, is often forgotten.20 The integration of systems includes mechanisms that underlie cardiac output, peripheral resistance, and the renal regulation of blood volume, all of which are influenced by a host of circulating substances, dynamic autonomic activity, renal regulation of blood volume, and structural features of the vascular tree. Although our understanding of blood pressure regulatory mechanisms in adults has progressed over the past 4 decades, it has not led to many large systematic studies of hypertension in pregnancy. The field is in need of a comprehensive prospective study that includes women expecting to become pregnant and then followed up at frequent intervals over the entire course of their pregnancy. With the acquisition of all‐encompassing biological data, including “omic” data across the whole of pregnancy, it would it be possible to uncover many currently mysterious regulators of hypertensive pregnancies and their effects on offspring. Although such studies would be expensive, the resulting savings in morbidity, mortality, and human suffering would make such an effort cost‐effective indeed.
Disclosures
None.
(J Am Heart Assoc. 2020;9:e016538 DOI: 10.1161/JAHA.120.016538.)
The opinions expressed in this article are not necessarily those of the editors or of the American Heart Association.
For Disclosures, see page 4.
References
- 1. Barker DJ. Fetal origins of coronary heart disease. BMJ. 1995;311:171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barker DJ, Thornburg KL. The obstetric origins of health for a lifetime. Clin Obstet Gynecol. 2013;56:511–519. [DOI] [PubMed] [Google Scholar]
- 3. Aye CYL, Lewandowski AJ, Lamata P, Upton R, Davis E, Ohuma EO, Kenworthy Y, Boardman H, Frost AL, Adwani S, et al. Prenatal and postnatal cardiac development in offspring of hypertensive pregnancies. J Am Heart Assoc. 2020;9:e014586 DOI: 10.1161/JAHA.119.014586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roberts JM, Pearson G, Cutler J, Lindheimer M. Summary of the NHLBI working group on research on hypertension during pregnancy. Hypertension. 2003;41:437–445. [DOI] [PubMed] [Google Scholar]
- 5. Davis EF, Lewandowski AJ, Aye C, Williamson W, Boardman H, Huang RC, Mori TA, Newnham J, Beilin LJ, Leeson P. Clinical cardiovascular risk during young adulthood in offspring of hypertensive pregnancies: insights from a 20‐year prospective follow‐up birth cohort. BMJ Open. 2015;5:e008136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Timpka S, Macdonald‐Wallis C, Hughes AD, Chaturvedi N, Franks PW, Lawlor DA, Fraser A. Hypertensive disorders of pregnancy and offspring cardiac structure and function in adolescence. J Am Heart Assoc. 2016;5:e003906 DOI: 10.1161/JAHA.116.003906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thornburg KL, Bagby SP, Giraud GD. Maternal Adaptations to Pregnancy In: Knobil and Neill's Physiology of Reproduction. Elsevier Inc.; Vol 2, 2015:1927–1955. [Google Scholar]
- 8. Bernstein IM, Meyer MC, Osol G, Ward K. Intolerance to volume expansion: a theorized mechanism for the development of preeclampsia. Obstet Gynecol. 1998;92:306–308. [DOI] [PubMed] [Google Scholar]
- 9. ACOG Practice Bulletin No. 202: gestational hypertension and preeclampsia. Obstet Gynecol. 2019;133:e1–e25. [DOI] [PubMed] [Google Scholar]
- 10. Burton GJ, Redman CW, Roberts JM, Moffett A. Pre‐eclampsia: pathophysiology and clinical implications. BMJ. 2019;366:l2381. [DOI] [PubMed] [Google Scholar]
- 11. Creanga AA, Syverson C, Seed K, Callaghan WM. Pregnancy‐related mortality in the United States, 2011–2013. Obstet Gynecol. 2017;130:366–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Phipps EA, Thadhani R, Benzing T, Karumanchi SA. Pre‐eclampsia: pathogenesis, novel diagnostics and therapies. Nat Rev Nephrol. 2019;15:275–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gammill HS, Chettier R, Brewer A, Roberts JM, Shree R, Tsigas E, Ward K. Cardiomyopathy and preeclampsia. Circulation. 2018;138:2359–2366. [DOI] [PubMed] [Google Scholar]
- 14. Mol BWJ, Roberts CT, Thangaratinam S, Magee LA, de Groot CJM, Hofmeyr GJ. Pre‐eclampsia. Lancet. 2016;387:999–1011. [DOI] [PubMed] [Google Scholar]
- 15. Nardozza LM, Caetano AC, Zamarian AC, Mazzola JB, Silva CP, Marcal VM, Lobo TF, Peixoto AB, Araujo Junior E. Fetal growth restriction: current knowledge. Arch Gynecol Obstet. 2017;295:1061–1077. [DOI] [PubMed] [Google Scholar]
- 16. Thornburg K, Jonker S, O'Tierney P, Chattergoon N, Louey S, Faber J, Giraud G. Regulation of the cardiomyocyte population in the developing heart. Prog Biophys Mol Biol. 2011;106:289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jonker SS, Louey S. Endocrine and other physiologic modulators of perinatal cardiomyocyte endowment. J Endocrinol. 2016;228:R1–R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zheng M, Schaal M, Chen Y, Li X, Shentu W, Zhang P, Ashraf M, Ge S, Sahn DJ. Real‐time 3‐dimensional echocardiographic assessment of ventricular volume, mass, and function in human fetuses. PLoS One. 2013;8:e58494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jayet PY, Rimoldi SF, Stuber T, Salmon CS, Hutter D, Rexhaj E, Thalmann S, Schwab M, Turini P, Sartori‐Cucchia C, et al. Pulmonary and systemic vascular dysfunction in young offspring of mothers with preeclampsia. Circulation. 2010;122:488–494. [DOI] [PubMed] [Google Scholar]
- 20. Montani JP, Van Vliet BN. Understanding the contribution of Guyton's large circulatory model to long‐term control of arterial pressure. Exp Physiol. 2009;94:382–388. [DOI] [PubMed] [Google Scholar]