

Abstract
Background
Patients with peripheral artery disease (PAD) undergo frequent episodes of ischemia‐reperfusion in lower extremity muscles that may negatively affect mitochondrial health and are associated with impaired mobility. We hypothesized that skeletal muscle from PAD patients will show high mitochondrial DNA heteroplasmy, especially in regions more susceptible to oxidative damage, such as the displacement loop, and that the degree of heteroplasmy will be correlated with the severity of ischemia and mobility impairment.
Methods and Results
Mitochondrial mutations and deletions and their relative abundance were identified by targeted mitochondrial DNA sequencing in biopsy specimens of gastrocnemius muscle from 33 PAD (ankle brachial index <0.9) and 9 non‐PAD (ankle brachial index >0.9) subjects aged ≥60 years. The probability of heteroplasmy per DNA base was significantly higher for PAD subjects than non‐PAD within each region. In adjusted models, PAD was associated with higher heteroplasmy than non‐PAD (P=0.003), but the association was limited to microheteroplasmy, that is heteroplasmy found in 1% to 5% of all mitochondrial genomes (P=0.004). Heteroplasmy in the displacement loop and coding regions were significantly higher for PAD than non‐PAD subjects after adjustment for age, sex, race, and diabetes mellitus (P=0.037 and 0.004, respectively). Low mitochondrial damage, defined by both low mitochondrial DNA copy number and low microheteroplasmy, was associated with better walking performance.
Conclusions
People with PAD have higher “low frequency” heteroplasmy in gastrocnemius muscle compared with people without PAD. Among people with PAD, those who had evidence of least mitochondrial damage, had better walking performance than those with more mitochondrial damage.
Registration
URL: http://www.clinicaltrials.gov. Unique identifier: NCT02246660.
Keywords: ankle brachial index, D‐loop, heteroplasmy, mtDNA, mtDNA copy number, peripheral artery disease
Subject Categories: Clinical Studies, Ischemia, Oxidant Stress, Pathophysiology, Translational Studies
Nonstandard Abbreviations and Acronyms
- ABI
ankle brachial index
- D‐loop
displacement‐loop region
- IR
ischemia‐reperfusion
- mtDNA
mitochondrial DNA
- PAD
peripheral arterial disease
- RESTORE
Resveratrol to Improve Outcomes in Older People With Peripheral Arterial Disease (Clinical Trial)
Clinical Perspective
What Is New?
This is the first study showing that mitochondrial DNA (mtDNA) global heteroplasmy, the coexistence of different mtDNA sequences in a tissue and the abundance of mtDNA sequence variability, are higher in skeletal muscle of people with peripheral artery disease (PAD); mtDNA heteroplasmic variants are not homogeneously distributed across regions of the mitochondrial genome, with greater frequency in the displacement loop region, in PAD patients.
Our study shows that the displacement loop is a hot spot for heteroplasmy accumulation, but also identified cytochrome b coding regions as an another heteroplasmy hot spot in PAD.
Among people with PAD, those who had evidence of least mitochondrial damage, defined as both low mtDNA copy number and low heteroplasmy in calf muscle, had better walking performance than those with more mitochondrial damage.
What Are the Clinical Implications?
Our findings may suggest that both mtDNA heteroplasmy and copy number could be used as biomarkers for mitochondrial damage in the muscle of patients with PAD.
This might be useful to identify patients with a worst prognosis of the disease and to develop interventions focused to improve the mobility and walking performance in these patients.
Introduction
Peripheral artery disease (PAD) is typically an atherosclerotic disorder of the lower limb arteries that affects mobility and is associated with high morbidity and mortality.1, 2, 3, 4 PAD patients undergo ischemia‐reperfusion (IR) episodes during walking; walking induced ischemia limits oxygen supply to lower extremity muscle mitochondria and reperfusion induces a burst of radical species production that causes oxidative damage to mitochondria and disrupts ATP production. Damaged mitochondria release damage‐associated molecular pattern molecules, such as fragmented mtDNA, which trigger an inflammatory response that contributes to muscle pathology.5, 6 The specific mechanisms by which oxidative stress damages mitochondria and impairs their function have not been fully elucidated.7, 8, 9, 10, 11
In a previous study including people with and without PAD we found that lower ankle brachial index (ABI) was associated with higher mitochondrial DNA copy number (mtDNA) in skeletal muscle biopsies.12 Further, among people with PAD, those with higher muscle mtDNA copy number had worse walking performance, whereas in those without PAD, higher mtDNA copy number was associated with better walking performance. Based on these results, we hypothesize that increased mtDNA copy number in people with PAD may represent a compensatory mechanism to increase mitochondrial abundance in ischemic calf muscle. However, because of damage to mtDNA, including fragmentation and increased point mutation frequency attributable to excessive oxidative stress, this compensatory mechanism might fail to increase mitochondrial function and consequently walking performance.
MtDNA is a small circular molecule of about 16.5k bases, with multiple copies contained within each mitochondrion and thousands contained within each cell. MtDNA includes coding regions for 13 proteins, 22 tRNAs, 2 rRNAs and a main non‐coding region called the displacement loop (D‐loop), that is considered a critical region for DNA replication.13, 14 It has been estimated that mutations occur in mtDNA at a rate ≈10 to 17 times higher than in nuclear DNA probably because of the close vicinity of reactive oxygen species produced by the mitochondrial electron transport chain complexes.14 Random mtDNA mutations accumulate with aging and undergo clonal expansion, especially in tissues with high energy demand such as skeletal muscle, causing a mixture of wild‐type and mutated mtDNA sequences known as heteroplasmy.15, 16, 17, 18, 19 Repeated episodes of IR in calf muscle in people with PAD may elevate mtDNA heteroplasmy over the threshold for functional consequences.20, 21, 22, 23 Point mutations of mtDNA lead to amino acid substitutions of respiratory chain subunits, which in some cases may affect the functionality of respiratory chain complexes further increasing ROS production and curtailing energy production with deleterious effects on skeletal muscle function.14, 24, 25, 26, 27, 28, 29 It has been suggested that the D‐loop region of mtDNA is especially sensitive to oxidative damage that occurs during IR episodes, and oxidized D‐loop shows enhanced affinity for the transcription factor A mitochondrial (TFAM), the major transcription factor that regulates mtDNA replication.30, 31, 32, 33, 34 It is possible that in people with PAD, IR may cause damage in the skeletal muscle mtDNA D‐loop region and consequent increased affinity for TFAM leading to higher mtDNA copy number, but this hypothesis has not been formally tested.12
The purpose of this study was to determine whether there is higher mtDNA heteroplasmy in lower extremity muscle from people with PAD compared with people without PAD, and whether the degree of heteroplasmy is proportional to the degree of lower extremity ischemia and walking ability in people with PAD. We measured mtDNA copy number and degree of heteroplasmy in biopsy specimens of gastrocnemius muscle from PAD patients and individuals free of PAD. We hypothesized that skeletal muscle from PAD patients will show high mtDNA heteroplasmy, especially in regions more susceptible to oxidative damage, such as the D‐loop, and that the degree of heteroplasmy will be correlated with the severity of ischemia and mobility impairment in PAD patients.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Study Population
This study uses data from baseline muscle biopsies from subjects who participated in the RESTORE (Resveratrol to Improve Outcomes in Older People With PAD) trial, a pilot randomized controlled trial of resveratrol to improve mobility function in PAD patients.35 Non‐PAD controls were identified from among those evaluated for eligibility in RESTORE and other randomized trials of PAD participants or observational studies of participants with and without PAD.10, 12 The Institutional Review Board of Northwestern University approved the protocol. Participants provided written informed consent.
Inclusion and Exclusion Criteria
All participants were aged ≥60 years. PAD participants were included if they had an ABI <0.90 and controls if they had an ABI between 0.90 and 1.40. Exclusion criteria are reported elsewhere.10, 12, 35, 36, 37 Briefly, PAD patients with a below‐knee or above‐knee amputation in the next 6 months were excluded, as well as participants using a walking aid or wheelchair or who had severe hearing or visual impairment. Participants with a recent cardiovascular event (within the past 3 months) or a poor prognosis because of cancer or other significant medical illness were excluded as well as participants with a score <23 at baseline in the Mini‐Mental Status.
Ankle Brachial Index Measures
The ankle brachial index (ABI) was measured as previously described.38, 39 A hand‐held Doppler probe (Nicolet Vascular Pocket Dop II, Golden, CO) was used to obtain systolic pressures at different locations, twice in the right and left brachial, dorsalis pedis, and posterior tibial arteries using established methods. The ABI was calculated by dividing the mean of ankle systolic pressure by the mean of the arm systolic pressure, which is an indicator of the severity of ischemia in lower extremity muscle.
Medical History
Medical history, race, and demographics were obtained using a questionnaire administered at baseline by a certified and trained health practitioner.
Six‐Minute Walk Test
Participants were instructed to walk continuously for 6 minutes to cover as much distance as possible, as described elsewhere.10, 12 Briefly, participants walked back and forth over a 100‐foot hallway for 6 minutes, to complete as many laps as possible. The distance completed after 6 minutes as well as the distance at onset of leg symptoms were recorded.
Four‐Meter Walking Velocity
Participants were instructed to walk 4 m at a “normal” and “fast” pace to measure walking velocity, as previously described.10, 12
Skeletal Muscle Biopsy Procedures
Skeletal muscle biopsies were taken from the medial head of the gastrocnemius muscle of the leg with lower ABI, as described elsewhere.10 Briefly, an open muscle biopsy was performed under anesthesia with subcutaneous lidocaine. Adipose and subcutaneous tissues were dissected until muscle was identified and biopsied. One hundred milligrams of muscle tissue was collected and immediately frozen in liquid‐nitrogen and stored at −80°C.
Mitochondrial DNA Isolation
Total DNA was isolated from human muscle using the Wizard Genomic DNA Purification Kit according to the manufacturer's instructions (Promega, Madison, WI, USA). After purification, DNA was evaluated spectrophotometrically by NanoDrop 1000 (Thermo Scientific, Rockford, IL, USA)
Mitochondrial DNA Genome Sequencing
Mitochondrial mutations and deletions and their relative abundance were identified by targeted mtDNA sequencing (mtDNA‐Seq) performed at the DNA Methylation and Mitochondrial Heteroplasmy Core of the Nathan Shock Center (Reynolds Center on Aging, Oklahoma Health Sciences Center, Oklahoma City, OK, USA). Briefly, 1 ng of DNA isolated from gastrocnemius muscle was used for the analysis. Polymerase chain reaction was used to enrich mtDNA from total genomic DNA, followed by Next‐Generation Sequencing of the entire mitochondrial genome at depths of ≥1000x allowing the identification of (1) large‐scale deletions and breakpoints, (2) homozygous or inherent variants, and (3) heterozygous or low frequency somatic mutations. For the present analyses, heteroplasmy was quantified as counts of positions where heterozygous or somatic mutations with a frequency of >1% were identified. There is evidence that maternally inherited mtDNAs mutations are characterized by high frequency heteroplasmy, while somatic mutation that accumulate during the lifetime, especially in post‐mitotic tissues, are characterized by low frequency heteroplasmy.40 Thus, mtDNA heteroplasmies were further classified as microheteroplasmy (heteroplasmies that affect between 1% and <5% of the mtDNA genomes) and mtDNA heteroplasmies at high frequency (heteroplasmies that affect >10% of mtDNA genomes).41 MtDNA heteroplasmy was also classified by region of the mitochondrial genome with counts of heteroplasmic positions in the D‐loop and in gene regions including protein coding, rRNA, and tRNA regions also quantified. The variants in the mtDNA were characterized in comparison with the Revised Cambridge Reference Sequence (NC_012920): The Human Mitochondrial Resequencing Array 2.0 (Affymetrix, Santa Clara, CA)42, 43 and classified as single nucleotide variants, multiple nucleotide variants, and deletions. mtDNA copy number was quantified at the Nathan Shock Center (Oklahoma Health Sciences Center, Oklahoma City, OK, USA) with a chip‐based digital polymerase chain reaction followed by fluorogenic assays that allow counting of mtDNA copies, and expressed as the ratio between mitochondrial DNA to nuclear DNA copy number (mtDNA/nDNA).
Statistical Analyses
Baseline characteristics of participants with versus without PAD were summarized as means and SDs for continuous variables and as frequencies and percentages for categorical variables. T tests were used to compare continuous characteristics and Fisher exact tests were used to compare categorical characteristics of participants with versus without PAD, when appropriate. The heteroplasmy per base was calculated for different mtDNA coding and non‐coding regions. Differences in heteroplasmy by regions were tested by mixed effect models adjusted for age, sex, PAD status, and diabetes mellitus. Average number of heteroplasmy counts according to PAD status were plotted within different mtDNA regions and compared by Student t tests. Association of PAD status with the natural log of mtDNA heteroplasmy count was assessed in multivariable linear regression models adjusted for age, sex, race, and diabetes mellitus. The association between natural log count of low‐ and high‐frequency variants, as well as of variants in the D‐loop and in gene regions were evaluated in separate models. For participants with no high frequency variants, log values were imputed equal to 0.1. Age was centered at 70 and scaled to 10 years. Relationship of mtDNA characteristics with ABI and walking performance were examined by mixed effect models adjusted for sex, age, race, body mass index, smoking status, and diabetes mellitus. In additional adjusted models, participants with PAD were cross‐classified by the median of mtDNA copy number (mtDNA/nDNA) and heteroplasmy count, and those with low mtDNA copy number and low heteroplasmy count (considered having low mitochondrial deficit) were compared with other participants in t tests and multivariable linear regression models adjusted for sex, age, race, body mass index, smoking status, and diabetes mellitus. Age was centered and scaled as above, and body mass index was centered at 30. All analyses were performed using R, version 3.6.0 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Participant Characteristics
A total of 44 participants consented to the muscle biopsy. One participant was excluded because of poor quality mtDNA and another because of incomplete demographics and clinical data. Therefore, the final sample for this analysis was 42 participants; 33 people with PAD and 9 without PAD. The main characteristics of the study population, including demographics, clinical and mtDNA copy number and heteroplasmy levels are summarized in Table 1.
Table 1.
Characteristics of Non‐PAD and PAD Participants
| Non‐PAD (n=9) | PAD (n=33) | P Value | |
|---|---|---|---|
| Age, y | 73.3 (4.9) | 73.7 (6.5) | 0.858 |
| Men (%) | 2 (22.2) | 24 (72.7) | 0.016 |
| Black race, n (%) | 5 (55.6) | 18 (54.5) | 1 |
| ABI | 1.14 (0.07) | 0.67 (0.15) | Not applicable |
| Smoking status, n (%) | |||
| Never | 3 (33.3) | 6 (18.2) | |
| Former | 6 (66.7) | 18 (54.5) | 0.188 |
| Current | 0 (0) | 9 (27.3) | |
| BMI | 27.6 (5.1) | 29.6 (4.1) | 0.294 |
| Diabetes mellitus, n (%) | 1 (11.1) | 15 (45.5) | 0.119 |
| Other non‐diabetes mellitus comorbiditya | 2 (22.2) | 10 (30.3) | 1 |
| Rapid 4‐m walk speed, m/s | 1.16 (0.25) | 1.14 (0.20) | 0.841 |
| Normal 4‐m walk speed, m/s | 0.92 (0.25) | 0.82 (0.12) | 0.282 |
| 6‐min walk distance, m | 1387 (488) | 1207 (206) | 0.307 |
| mtDNA copy number | 1472 (929) | 4456 (5325) | 0.004 |
| Heteroplasmyb | |||
| Count | 21.7 (5.2) | 42.6 (21.9) | <0.001 |
| Microheteroplasmy (>1% & <5%) count | 18.2 (4.3) | 34.3 (15.9) | <0.001 |
| High frequency heteroplasmy (>10%) count | 2.6 (1.4) | 3.2 (3.0) | 0.407 |
| Displacement loop heteroplasmy count | 9.1 (3.3) | 13.5 (6.2) | 0.008 |
| Gene region heteroplasmy count | 12.6 (4.8) | 28.8 (17.7) | <0.001 |
Values are mean±SD. ABI indicates ankle brachial index; and BMI, body mass index.
Angina, myocardial infarction, heart failure, or pulmonary disease.
Heteroplasmy count corresponds to the count of all heteroplasmic variants (single nucleotide polymorphism [SNP], insertion, or deletion) observed with a minor frequency >1% in a participant sample. Low and high frequency counts reflect the number of variants with a frequency of the minor variant within the specified ranges. Displacement loop and gene region are counts of the subsets of all variants within the displacement loop and region of any mtDNA gene (coding, rRNA, tRNA), respectively. (N=42, exclusions: mtDNA copy number=1, covariates=1).
Participants With PAD have Higher mtDNA Copy Number and mtDNA Heteroplasmy in Gastrocnemius Muscle Compared With Non‐PAD
One hundred sixty‐two mtDNA variants were identified across the entire population (89.5% single nucleotide variants; 8.64% deletions and 1.85% multiple nucleotide variants) (Table S1). After adjusting for age, sex, PAD, and diabetes mellitus, the probability of mtDNA heteroplasmy per base was higher in the D‐loop and in the cytochrome b coding region compared with all other regions (Figure 1A, Table S2). Microheteroplasmy, namely heteroplasmy that involved between 1% and 5% of all mtDNA genomes, was the most common heteroplasmy for all participants (Figure 1B). The number of mtDNA heteroplasmic variants was higher in skeletal muscle from patients with PAD compared with non‐PAD (P<0.001; Table 1; Figure 2 [inset]) overall, as well as within each one of different mtDNA regions (Figure 2).
Figure 1. Distribution of heteroplasmic variants by region and frequency in the whole study sample (n=42).
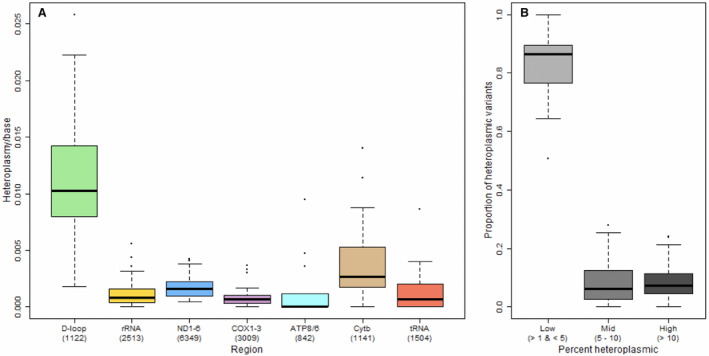
A, Rate of heteroplasmy (heteroplasmy count per base) by mitochondrial DNA region among all participants; (B) proportion of heteroplasmic variants categorized as low (microheteroplasmy), mid, or high frequency. Parentheses indicate the number of bases used to calculate rate for each region. D‐loop indicates displacement loop; and mtDNA, mitochondrial DNA.
Figure 2. Heteroplasmy count by mitochondrial DNA region and peripheral artery disease status (n=42, darker boxes=non‐peripheral artery disease; lighter boxes=peripheral artery disease; *P<0.05 and # P<0.01).
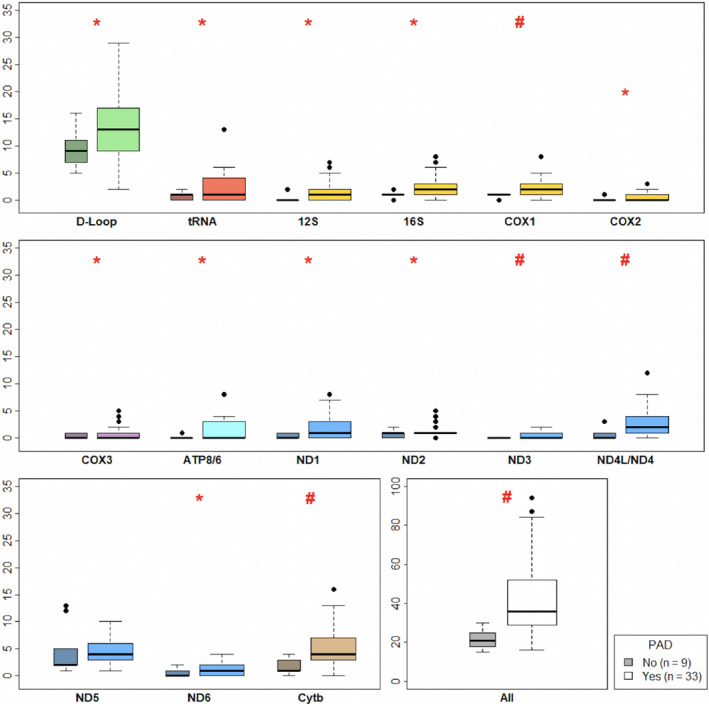
mtDNA indicates mitochondrial DNA; and PAD, peripheral artery disease.
Adjusting for age, sex, race and diabetes mellitus, the presence of PAD was associated with higher levels of heteroplasmy in skeletal muscle compared with those without PAD (P=0.003) (Table 2). The count of microheteroplasmy positions, and counts of variants located in the D‐loop or coding regions were significantly associated with PAD after adjustment for age, sex, race and diabetes mellitus (β=0.58, P=0.004; β=0.49, P=0.037 and β=0.72, P=0.004, respectively) (Table 2). No statistically significant difference in heteroplasmy at high frequency were found between PAD and non‐PAD in skeletal muscle (β=−0.20, P=0.664). After excluding participants affected by other chronic disease such as angina, myocardial infarction, heart failure, or pulmonary disease (12 of 42 participants), the association between heteroplasmy frequency and PAD remained substantially unchanged, although difference in heteroplasmy by PAD in the D‐loop region only approached statistical significance (β=0.55, P=0.067; Table S3).
Table 2.
Coefficients From Multivariable Linear Regression Models Estimating the Association Between the Natural Log of mtDNA Heteroplasmy Count and PAD Status by mtDNA Region and Frequency, Adjusting for Age, Sex, Race, and Diabetes Mellitus Status (n=42)
| Coefficient | All | All | D‐Loop | Gene Region | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Microheteroplasmy | High Frequency | |||||||||
| β (SE) | P Value | β (SE) | P Value | β (SE) | P Value | β (SE) | P Value | β (SE) | P Value | |
| (Intercept) | 2.96 (0.19) | <0.001 | 2.84 (0.19) | <0.001 | 0.39 (0.44) | 0.387 | 2.13 (0.22) | <0.001 | 2.34 (0.23) | <0.001 |
| PAD | 0.62 (0.20) | 0.003 | 0.58 (0.19) | 0.004 | −0.20 (0.45) | 0.664 | 0.49 (0.23) | 0.037 | 0.72 (0.23) | 0.004 |
| Agea | 0.03 (0.12) | 0.815 | 0.05 (0.12) | 0.649 | −0.28 (0.27) | 0.319 | 0.01 (0.14) | 0.925 | 0.08 (0.14) | 0.581 |
| Men | 0.12 (0.16) | 0.465 | 0.09 (0.16) | 0.564 | 0.30 (0.38) | 0.427 | −0.15 (0.19) | 0.445 | 0.27 (0.19) | 0.165 |
| Black | 0.15 (0.15) | 0.331 | 0.04 (0.15) | 0.785 | 0.76 (0.35) | 0.036 | 0.14 (0.17) | 0.436 | 0.16 (0.18) | 0.387 |
| Diabetes mellitus | −0.27 (0.15) | 0.082 | −0.23 (0.15) | 0.132 | −0.05 (0.35) | 0.878 | −0.25 (0.17) | 0.166 | −0.38 (0.18) | 0.041 |
D‐loop indicates displacement loop; and PAD, peripheral artery disease.
Age centered at 70 years and scaled to 10 years.
Among Participants With PAD, Those With the Least mtDNA Damage had Better Walking Performance
Participants with PAD had higher mtDNA copy number per cell (mtDNA/n/DNA) compared with non‐PAD (P=0.004, Figure S1A). However, heteroplasmy count was not associated with mtDNA copy number among participants with PAD (Figure S1B). Heteroplasmy was also not correlated to ABI (Figure S2).
In unadjusted and adjusted analyses, neither mtDNA microheteroplasmy nor mtDNA copy number were significantly associated with performance in the 4‐m walk at usual and fastest pace, nor in the 6‐minute walk test (Figure S3 and Table S4). To understand whether mtDNA microheteroplasmy and mtDNA copy number provide complementary information as biomarkers of mitochondrial impairment, PAD participants were cross‐classified by mtDNA microheteroplasmy (≤ versus >36, median count level) and copy number (≤ versus >3041, median level mtDNA/nDNA). PAD participants with low copy number and low microheteroplasmy did not, on average, differ in ABI (P t‐test=0.854), but did have higher walking speed than those in the other 3 groups (P t‐test=0.049) (Figure 3, Table S5). The difference in walking performance was consistent across measures of walking performance, rapid walking speed, and distance walked in 6 minutes, and after adjustment for sex, age, race, body mass index, smoking status, diabetes mellitus in linear regression models (Table S5).
Figure 3. Distribution of ankle brachial index (top panel) and normal 4‐m walking speed (bottom panel) by categories of heteroplasmy count and copy number among participants with peripheral artery disease (n=33).
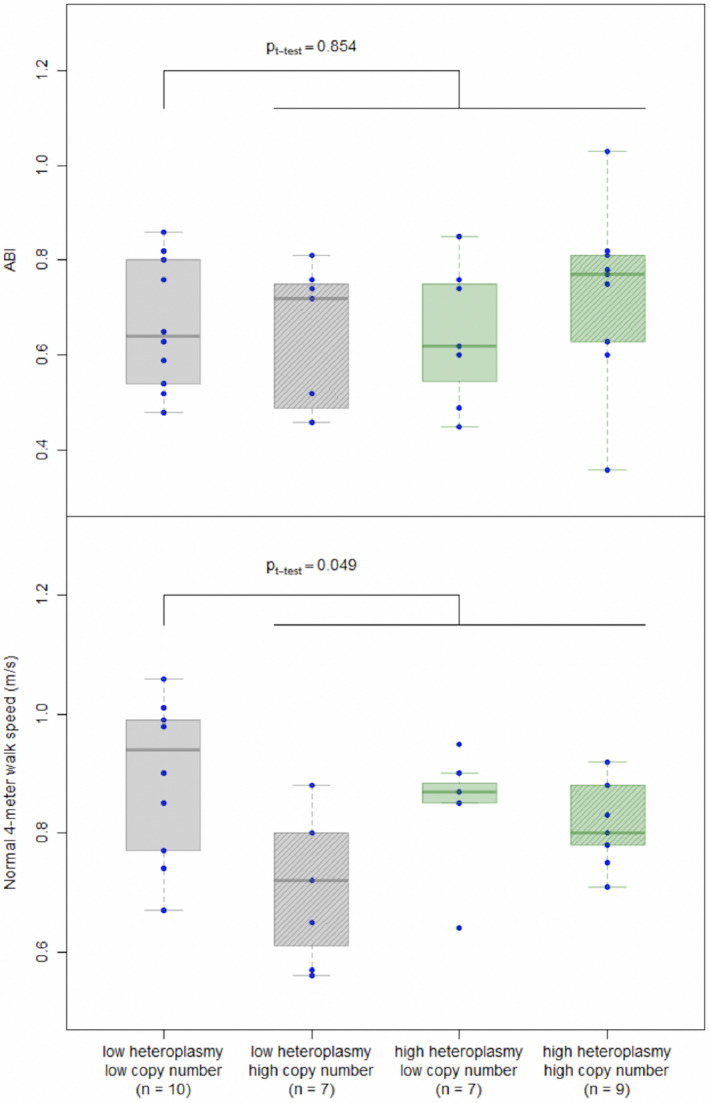
Participants were cross‐classified by heteroplasmy count ≤ vs >36 (median level) and mitochondrial DNA copy number ≤ vs >3041 (median level). ABI indicates ankle brachial index.
Discussion
In a previous study we found that lower ABI was associated with higher mitochondrial mtDNA copy number in gastrocnemius muscle biopsies of PAD patients compared with non‐PAD.12 Also, mtDNA copy number was negatively associated with mobility performance in PAD participants, whereas it was positively related to performance in non‐PAD participants. To better understand the significance of these findings, in this study, we used next generation sequencing to comprehensively characterize mtDNA variants. To our knowledge, this is the first study showing that mtDNA global heteroplasmy, the coexistence of different mtDNA sequences in a tissue and the abundance of mtDNA sequence variability, were higher in the gastrocnemius muscle of people with PAD compared with non‐PAD.
In keeping with our original hypothesis, we found that mtDNA heteroplasmic variants were not homogeneously distributed across regions of the mitochondrial genome, with greater frequency in the D‐loop region (Figure 1). We also observed higher heteroplasmy among participants with PAD compared with non‐PAD. Consistent with our findings, a previous study assessed the frequency of a specific 4977–base pair mtDNA deletion mutations in muscle of the ischemic limb of PAD patients compared with the less ischemic limb and to muscle from sedentary non‐PAD controls.20 They found that PAD patients had a higher deletion frequency than controls, although there was no significant difference between affected and unaffected limbs. Compared with the prior study, the current study included a substantially larger sample size and included a more comprehensive assessment of heteroplasmy, including both deletions and mutations, low and high frequency heteroplasmy, and heteroplasmy in different mtDNA regions. To our knowledge, no prior work has studied microheteroplasmy versus high frequency heteroplasmy or mtDNA regional heteroplasmy in patients with versus without PAD.
Several mechanisms may contribute to elevated abundance of mtDNA microheteroplasmy variants in gastrocnemius muscle in PAD. For example, PAD patients undergo repeated episodes of ischemia‐reperfusion, with production of reactive oxygen species that cause oxidative damage in surrounding macromolecules, including random mutations in mtDNA. This hypothesis is consistent with the fact that high frequency mutations, which are probably maternally inherited, did not differ between PAD and non‐PAD. This is also consistent with data showing that in patients with PAD increased mitochondria ROS production is associated with elevated intramuscular oxidative stress and is associated with disease severity.8, 44
Oxidative stress also upregulates Peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha PGC‐1α that induces mitochondrial biogenesis by activating nuclear respiratory factors 1 and 2, which promote the expression of TFAM.45, 46, 47 TFAM drives the transcription and replication of mtDNA, the early step in mitochondrial biogenesis, by binding to the D‐loop region of mtDNA. Several previous studies have shown that the D‐loop is particularly sensitive to oxidative damage and is a frequent site of heteroplasmies in other chronic diseases. We had hypothesized that the enhanced affinity for TFAM of oxidized D‐loop may overdrive mtDNA replication and cause higher mtDNA copy number.12 Our results support the notion that the D‐loop is a mutational hot spot contributing to heteroplasmy, and also identified the cytochrome b coding region as another hot spot. In these hot spots as well in other regions of mtDNA heteroplasmy was higher in PAD than non‐PAD.31, 34 In addition, we found no evidence of a relationship between heteroplasmy in the D‐loop (or anywhere else) with mtDNA copy number, nor degree of ischemia operationalized as ABI, therefore failing to confirm our original hypothesis.
In the RESTORE PAD cohort study we found that citrate synthase activity, a proxy of mitochondrial number and function, was positively correlated with mtDNA copy number and we speculated that the association of higher mtDNA copy number with poor walking performance, specific to PAD, may represent a compensatory mechanism for decreased mitochondrial activity because of oxidative stress.12 In particular, we hypothesized that the increased mtDNA mutational load in regions coding for proteins involved in mitochondrial electron transport chain in PAD may result in newly created mitochondria being of poorer quality and less functional, which may contribute to the association of higher mtDNA copy number with poorer walking performance specifically in PAD. An alternative hypothesis prompted by this study is that ROS‐mediated mitochondrial damage causes the release of mtDNA fragments from damaged cells and, therefore, mtDNA copy number in the muscle of PAD patients should be considered a biomarker of mitochondrial damage accumulation.48
In this study, total heteroplasmy, microheteroplasmy, and mtDNA copy number were not associated with differential performance in the 3 walking tests investigated. However, compared with other participants in our study sample, PAD participants with both mtDNA heteroplasmy and mtDNA copy number below the median value had significantly better walking performance in the 3 walking tests considered. These findings may suggest that both mtDNA microheteroplasmy and copy number are biomarkers of mitochondrial damage in the muscle of patients with PAD. Because of the small sample size and the cross‐sectional nature of this study, the findings presented here should be interpreted with caution and do not prove causality between mtDNA heteroplasmy and mobility impairment in PAD. However, our findings are consistent with the notion that severity of mtDNA damage could be implicated in the functional damage induced by PAD and this hypothesis should be tested in future larger, longitudinal studies to establish whether preventing DNA damage and reducing mtDNA copy number is associated with improved functional performance in those with PAD.
Interestingly, point mutations are frequent during embryogenesis in mtDNA mutator mice, but the abnormal phenotype is not observed until early adult life.15 Thus, an alternative hypothesis is that mtDNA heteroplasmy that emerges earlier in life predisposes individuals to PAD. This is consistent with the finding that lower ABI within the normal range is already associated with mitochondrial dysfunction.49 To address this possibility, future studies will assess mtDNA heteroplasmy, mtDNA copy number, and mitochondrial function both in lower extremity skeletal muscle affected by PAD as well as in upper extremity skeletal muscle that should not be affected by PAD. Comparable mtDNA heteroplasmy in upper and lower extremity muscles would suggest mutations are independent of IR‐induced oxidative stress, and may identify individuals at an early stage at risk of developing more severe PAD.
Our study has limitations. First, the small sample size and the fact that there were more people with PAD than non‐PAD may have limited our ability to uncover important differences. Second, the percentage of males with PAD was higher than the non‐PAD; although all analyses presented were adjusted for sex, we cannot exclude the persistence of residual confounding. Third, the study was cross‐sectional and no causal inferences can be made. In spite of the limitations, our study provides convincing evidence that PAD is associated with high mtDNA microheteroplasmy, and results are consistent with the hypothesis that mtDNA damage affects functional performance in PAD.
Conclusions
People with PAD have greater abundance of mtDNA mutations than non‐PAD controls across the entire mitochondrial genome. Our findings suggest that both load of mtDNA microheteroplasmy and increased mtDNA copy number are biomarkers of mitochondrial damage, and in combination are correlated with impaired mobility performance. Longitudinal studies are needed to test the hypothesis that people with PAD with low mtDNA microheteroplasmy and low copy number have a better clinical course of PAD and better preservation of mobility performance over time.
Sources of Funding
This work was supported by the National Institute on Aging (R21‐AG047510), by the National Heart, Lung, and Blood Institute (R01‐HL107510, R01HL109244, R01‐HL088589, R01HL122846), the Office of Dietary Supplements, and the Nathan Shock Center for Aging.
Disclosures
None.
Supporting information
Tables S1–S5
Figures S1–S3
Acknowledgments
We thank Willard “Bill” Freeman, from the DNA Methylation and Mitochondrial Heteroplasmy Core of the Nathan Shock Center (Reynolds Center on Aging, Oklahoma City, OK, USA), for his help with the analysis of the mitochondrial DNA genome sequencing.
J Am Heart Assoc. 2020;9:e015197 DOI: 10.1161/JAHA.119.015197
Contributor Information
Marta Gonzalez‐Freire, Email: martagonzalezfreire@gmail.com.
Luigi Ferrucci, Email: ferruccilu@mail.nih.gov.
References
- 1. Criqui MH, Aboyans V. Epidemiology of peripheral artery disease. Circ Res. 2015;116:1509–1526. [DOI] [PubMed] [Google Scholar]
- 2. McDermott MM. Lower extremity manifestations of peripheral artery disease: the pathophysiologic and functional implications of leg ischemia. Circ Res. 2015;116:1540–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McDermott MM, Greenland P, Liu K, Guralnik JM, Celic L, Criqui MH, Chan C, Martin GJ, Schneider J, Pearce WH, et al. The ankle brachial index is associated with leg function and physical activity: the Walking and Leg Circulation Study. Ann Intern Med. 2002;136:873–883. [DOI] [PubMed] [Google Scholar]
- 4. McDermott MM, Liu K, Greenland P, Guralnik JM, Criqui MH, Chan C, Pearce WH, Schneider JR, Ferrucci L, Celic L, et al. Functional decline in peripheral arterial disease: associations with the ankle brachial index and leg symptoms. JAMA. 2004;292:453–461. [DOI] [PubMed] [Google Scholar]
- 5. Picca A, Lezza AMS, Leeuwenburgh C, Pesce V, Calvani R, Bossola M, Manes‐Gravina E, Landi F, Bernabei R, Marzetti E. Circulating mitochondrial DNA at the crossroads of mitochondrial dysfunction and inflammation during aging and muscle wasting disorders. Rejuvenation Res. 2018;21:350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol. 2011;8:292–300. [DOI] [PubMed] [Google Scholar]
- 7. Brass EP, Hiatt WR, Green S. Skeletal muscle metabolic changes in peripheral arterial disease contribute to exercise intolerance: a point‐counterpoint discussion. Vasc Med. 2004;9:293–301. [DOI] [PubMed] [Google Scholar]
- 8. Hart CR, Layec G, Trinity JD, Kwon OS, Zhao J, Reese VR, Gifford JR, Richardson RS. Increased skeletal muscle mitochondrial free radical production in peripheral arterial disease despite preserved mitochondrial respiratory capacity. Exp Physiol. 2018;103:838–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McDermott MM, Hoff F, Ferrucci L, Pearce WH, Guralnik JM, Tian L, Liu K, Schneider JR, Sharma L, Tan J, et al. Lower extremity ischemia, calf skeletal muscle characteristics, and functional impairment in peripheral arterial disease. J Am Geriatr Soc. 2007;55:400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. White SH, McDermott MM, Sufit RL, Kosmac K, Bugg AW, Gonzalez‐Freire M, Ferrucci L, Tian L, Zhao L, Gao Y, et al. Walking performance is positively correlated to calf muscle fiber size in peripheral artery disease subjects, but fibers show aberrant mitophagy: an observational study. J Transl Med. 2016;14:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhou T, Prather ER, Garrison DE, Zuo L. Interplay between ROS and antioxidants during ischemia‐reperfusion injuries in cardiac and skeletal muscle. Int J Mol Sci. 2018;19:E417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McDermott MM, Peterson CA, Sufit R, Ferrucci L, Guralnik JM, Kibbe MR, Polonsky TS, Tian L, Criqui MH, Zhao L, et al. Peripheral artery disease, calf skeletal muscle mitochondrial DNA copy number, and functional performance. Vasc Med. 2018;23:340–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nicholls TJ, Minczuk M. In D‐loop: 40 years of mitochondrial 7S DNA. Exp Gerontol. 2014;56:175–181. [DOI] [PubMed] [Google Scholar]
- 14. Pastukh VM, Gorodnya OM, Gillespie MN, Ruchko MV. Regulation of mitochondrial genome replication by hypoxia: the role of DNA oxidation in D‐loop region. Free Radic Biol Med. 2016;96:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. [DOI] [PubMed] [Google Scholar]
- 16. Payne BAI, Wilson IJ, Yu‐Wai‐Man P, Coxhead J, Deehan D, Horvath R, Taylor RW, Samuels DC, Santibanez‐Koref M, Chinnery PF. Universal heteroplasmy of human mitochondrial DNA. Hum Mol Genet. 2013;22:384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly‐Y M, Gidlof S, Oldfors A, Wibom R, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. [DOI] [PubMed] [Google Scholar]
- 18. Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, Loeb LA. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet. 2008;40:392–394. [DOI] [PubMed] [Google Scholar]
- 19. Wachsmuth M, Hubner A, Li M, Madea B, Stoneking M. Age‐related and heteroplasmy‐related variation in human mtDNA copy number. PLoS Genet. 2016;12:e1005939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bhat HK, Hiatt WR, Hoppel CL, Brass EP. Skeletal muscle mitochondrial DNA injury in patients with unilateral peripheral arterial disease. Circulation. 1999;99:807–812. [DOI] [PubMed] [Google Scholar]
- 21. Fetterman JL, Holbrook M, Westbrook DG, Brown JA, Feeley KP, Breton‐Romero R, Linder EA, Berk BD, Weisbrod RM, Widlansky ME, et al. Mitochondrial DNA damage and vascular function in patients with diabetes mellitus and atherosclerotic cardiovascular disease. Cardiovasc Diabetol. 2016;15:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hefti E, Blanco JG. Mitochondrial DNA heteroplasmy in cardiac tissue from individuals with and without coronary artery disease. Mitochondrial DNA A DNA Mapp Seq Anal. 2018;29:587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sobenin IA, Zhelankin AV, Sinyov VV, Bobryshev YV, Orekhov AN. Mitochondrial aging: focus on mitochondrial DNA damage in atherosclerosis—a mini‐review. Gerontology. 2015;61:343–349. [DOI] [PubMed] [Google Scholar]
- 24. Grady JP, Pickett SJ, Ng YS, Alston CL, Blakely EL, Hardy SA, Feeney CL, Bright AA, Schaefer AM, Gorman GS, et al. mtDNA heteroplasmy level and copy number indicate disease burden in m.3243A>G mitochondrial disease. EMBO Mol Med. 2018;10:e8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015;16:530–542. [DOI] [PubMed] [Google Scholar]
- 26. Wallace DC, Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol. 2013;5:a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barbieri E, Sestili P. Reactive oxygen species in skeletal muscle signaling. J Signal Transduct. 2012;2012:982794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gonzalez‐Freire M, de Cabo R, Bernier M, Sollott SJ, Fabbri E, Navas P, Ferrucci L. Reconsidering the role of mitochondria in aging. J Gerontol A Biol Sci Med Sci. 2015;70:1334–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Powers SK, Ji LL, Kavazis AN, Jackson MJ. Reactive oxygen species: impact on skeletal muscle. Compr Physiol. 2011;1:941–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chimienti G, Picca A, Sirago G, Fracasso F, Calvani R, Bernabei R, Russo F, Carter CS, Leeuwenburgh C, Pesce V, et al. Increased TFAM binding to mtDNA damage hot spots is associated with mtDNA loss in aged rat heart. Free Radic Biol Med. 2018;124:447–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Del Bo R, Bordoni A, Martinelli Boneschi F, Crimi M, Sciacco M, Bresolin N, Scarlato G, Comi GP. Evidence and age‐related distribution of mtDNA D‐loop point mutations in skeletal muscle from healthy subjects and mitochondrial patients. J Neurol Sci. 2002;202:85–91. [DOI] [PubMed] [Google Scholar]
- 32. Milenkovic D, Matic S, Kuhl I, Ruzzenente B, Freyer C, Jemt E, Park CB, Falkenberg M, Larsson N‐G. TWINKLE is an essential mitochondrial helicase required for synthesis of nascent D‐loop strands and complete mtDNA replication. Hum Mol Genet. 2013;22:1983–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Picca A, Lezza AMS. Regulation of mitochondrial biogenesis through TFAM‐mitochondrial DNA interactions: useful insights from aging and calorie restriction studies. Mitochondrion. 2015;25:67–75. [DOI] [PubMed] [Google Scholar]
- 34. Stoccoro A, Mosca L, Carnicelli V, Cavallari U, Lunetta C, Marocchi A, Migliore L, Coppede F. Mitochondrial DNA copy number and D‐loop region methylation in carriers of amyotrophic lateral sclerosis gene mutations. Epigenomics. 2018;10:1431–1443. [DOI] [PubMed] [Google Scholar]
- 35. McDermott MM, Leeuwenburgh C, Guralnik JM, Tian L, Sufit R, Zhao L, Criqui MH, Kibbe MR, Stein JH, Lloyd‐Jones D, et al. Effect of resveratrol on walking performance in older people with peripheral artery disease: the RESTORE randomized clinical trial. JAMA Cardiol. 2017;2:902–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McDermott MM, Liu K, Carr J, Criqui MH, Tian L, Li D, Ferrucci L, Guralnik JM, Kramer CM, Yuan C, et al. Superficial femoral artery plaque, the ankle‐brachial index, and leg symptoms in peripheral arterial disease: the walking and leg circulation study (WALCS) III. Circ Cardiovasc Imaging. 2011;4:246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McDermott MM, Greenland P, Liu K, Tian L, Green D, Shah SJ, Huffman M, Wilkins J, Kibbe M, Liao Y, et al. Vulnerable blood in high risk vascular patients: study design and methods. Contemp Clin Trials. 2014;38:121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McDermott MM, Criqui MH, Liu K, Guralnik JM, Greenland P, Martin GJ, Pearce W. Lower ankle/brachial index, as calculated by averaging the dorsalis pedis and posterior tibial arterial pressures, and association with leg functioning in peripheral arterial disease. J Vasc Surg. 2000;32:1164–1171. [DOI] [PubMed] [Google Scholar]
- 39. McDermott MM, Tian L, Liu K, Guralnik JM, Ferrucci L, Tan J, Pearce WH, Schneider JR, Criqui MH. Prognostic value of functional performance for mortality in patients with peripheral artery disease. J Am Coll Cardiol. 2008;51:1482–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet. 2012;13:878–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smigrodzki RM, Khan SM. Mitochondrial microheteroplasmy and a theory of aging and age‐related disease. Rejuvenation Res. 2005;8:172–198. [DOI] [PubMed] [Google Scholar]
- 42. Maitra A, Cohen Y, Gillespie SED, Mambo E, Fukushima N, Hoque MO, Shah N, Goggins M, Califano J, Sidransky D, et al. The Human MitoChip: a high‐throughput sequencing microarray for mitochondrial mutation detection. Genome Res. 2004;14:812–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou S, Kassauei K, Cutler DJ, Kennedy GC, Sidransky D, Maitra A, Califano J. An oligonucleotide microarray for high‐throughput sequencing of the mitochondrial genome. J Mol Diagn. 2006;8:476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pipinos II, Judge AR, Zhu Z, Selsby JT, Swanson SA, Johanning JM, Baxter BT, Lynch TG, Dodd SL. Mitochondrial defects and oxidative damage in patients with peripheral arterial disease. Free Radic Biol Med. 2006;41:262–269. [DOI] [PubMed] [Google Scholar]
- 45. Austin S, St‐Pierre J. PGC1alpha and mitochondrial metabolism—emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci. 2012;125:4963–4971. [DOI] [PubMed] [Google Scholar]
- 46. Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:69–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab. 2012;23:459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. [DOI] [PubMed] [Google Scholar]
- 49. AlGhatrif M, Zane A, Oberdier M, Canepa M, Studenski S, Simonsick E, Spencer RG, Fishbein K, Reiter D, Lakatta EG, et al. Lower mitochondrial energy production of the thigh muscles in patients with low‐normal ankle‐brachial index. J Am Heart Assoc. 2017;6:e006604 DOI: 10.1161/JAHA.117.006604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S5
Figures S1–S3