

Abstract
Background
PCSK9 (Proprotein convertase subtilisin/kexin type 9) binds low‐density lipoprotein receptor, preventing its recycling. PCSK9 is a risk predictor and a biotarget in atherosclerosis. The PCSK9‐rs562556 variant has been reported as a gain‐of‐function mutation. The aim of this study was to determine whether the PCSK9–low‐density lipoprotein receptor–rs562556 axis is associated with carotid artery plaques between 2 visits separated by almost 20 years in a longitudinal population cohort.
Methods and Results
The STANISLAS (Suivi Temporaire Annuel Non‐Invasif de la Santé des Lorrains Assurés Sociaux) cohort is a longitudinal familial cohort from the Lorraine region of France. Participants attending 2 visits (visit 1 and visit 4) separated by 18.5 years (mean) were included (n=997). Carotid artery plaques were determined with standardized vascular echography. The mean age of the adult population at visit 1 was 42±5 years. At visit 4, 203 (20.4%) participants had arterial plaques. Participants who developed arterial plaques were older (42.7±5.4 versus 41.7±4.7 years), more often male (60% versus 49%), smokers (29% versus 18%), with diabetes mellitus (6% versus 3%), and higher cholesterol levels (low‐density lipoprotein cholesterol, 1.6±0.4 versus 1.5±0.3 g/L) (all P<0.05). The independent factors associated with arterial plaques were age, smoking, and low‐density lipoprotein cholesterol. Higher PCSK9 levels were associated with arterial plaques on top of the clinical model (odds ratio, 2.14; 95% CI,= 1.28–3.58); the missense mutation coding the single‐nucleotide polymorphism rs562556 was associated with both higher PCSK9 concentration and incident carotid arterial plaques.
Conclusions
Higher PCSK9 concentration was associated with the development of arterial plaques almost 20 years in advance in a healthy middle‐aged population. Mutations of the single‐nucleotide polymorphism rs562556 associated with both PCSK9 levels and arterial plaques reinforce the potential causality of our findings. PCSK9 inhibitors could be useful for primary cardiovascular prevention.
Keywords: STANISLAS cohort, arterial plaques, PCSK9, rs562556 mutations, LDL receptor, cholesterol
Subject Categories: Biomarkers, Ischemia, Mechanisms, Proteomics, Vascular Biology
Clinical Perspective
What Is New?
PCSK9 (Proprotein convertase subtilisin/kexin type 9) binds low‐density lipoprotein receptor, preventing its recycling; thus, PCSK9 is a risk predictor and a biotarget in atherosclerosis.
The PCSK9‐rs562556 variant has been reported as a gain‐of‐function mutation.
In this longitudinal general‐population study, higher PCSK9 levels were independently associated with incident arterial plaques on top of a well‐calibrated clinical model; the missense mutation coding the single‐nucleotide polymorphism rs562556 was associated with both higher PCSK9 concentration and incident carotid arterial plaques, reinforcing the potential causality of our findings.
What Are the Clinical Implications?
PCSK9 inhibitors could be useful for primary cardiovascular prevention.
Introduction
Atherosclerotic cardiovascular disease is the leading cause of death worldwide.1 For nearly 2 decades, several guidelines or consensus statements have highlighted that the detection of carotid plaque is an important clinical predictor of future adverse cardiovascular events2, 3 Among 13 145 participants in the ARIC (Atherosclerosis Risk in Communities) study, free of cardiovascular disease at the beginning of the follow‐up and followed for a mean of 15.1 years (accumulating a total of 1812 major cardiovascular events), the presence of carotid plaques was independently associated with incident major cardiovascular events and improved the predictive capacity of the clinical model using “classic” risk factors, such as age, smoking, and cholesterol4 Whether a strategy aimed at modifying patients’ risk factors based on the presence of carotid plaques would be effective in reducing cardiovascular risk is yet to be proven.5
Low‐density lipoprotein cholesterol (LDL‐C) is a strong, independent, and modifiable risk factor for developing cardiovascular disease6 Lowering LDL‐C, mainly with statins, has decreased the risk of cardiovascular events over the past decades7, 8 However, many patients do not achieve desired LDL‐C levels, and others experience side effects (eg, myalgia) that, despite being mild in the majority of the cases, may lead patients to abandon statin therapy or to take low and insufficient doses9, 10 Medical misinformation with rapid spread over the Internet has also contributed to the abandonment of lifesaving therapies, such as statins11 Alternative therapies may be needed for those who cannot achieve the desired LDL‐C levels or experience side effects leading to therapy low adherence and/or withdrawal.
The PCSK9 (proprotein convertase subtilisin/kexin type 9) is produced by the liver and secreted into the plasma, acting as a low‐density lipoprotein receptor (LDLR) binder at the surface of the hepatocytes that prevents the recycling of the LDLR.12 In consequence, the LDLR becomes more susceptible to degradation and less efficient in performing the clearance of the LDL‐C, thereby increasing the circulating levels of LDL‐C and the atherosclerosis risk13 Recent cardiovascular outcome trials have shown that PCSK9 inhibitors effectively reduce LDL‐C, decrease the atheroma plaque burden, and reduce the rate of major cardiovascular events in high‐risk patients with atherosclerotic cardiovascular disease and LDL‐C levels of 70 mg/dL (1.8 mmol/L) or higher who were receiving statin therapy.14, 15 The development of PCSK9 inhibitor vaccines that are administered monthly or yearly have the potential to increase the treatment adherence and to substantially decrease the negative impact of atherosclerotic cardiovascular disease16 To date, no studies have accessed the potential role of PCSK9 inhibitors for primary cardiovascular prevention. PCSK9 is a highly polymorphic gene, and some variants of the PCSK9 gene are associated with variability in LDL‐C.17 Gain‐of‐function mutations interfere with the recycling of the LDLR, reducing the LDL‐C uptake (and increasing LDL‐C levels).18 In particular, many studies have identified an association between the minor allele (A) of the rs562556 single‐nucleotide polymorphism (SNP) (located on PCSK9 gene, and responsible of missense mutation I474V) and LDL‐C levels.19
The STANISLAS (Suivi Temporaire Annuel Non‐Invasif de la Santé des Lorrains Assurés Sociaux) cohort is a single‐center familial longitudinal cohort comprising 1006 families (4295 participants) from the Nancy region of France, who were recruited from 1993 to 1995 to visit 1. Participants were then followed each 5 to 10 years, and from the initial visit, 1705 participants returned for the fourth visit (visit 4), held from 2011 to 2016, which comprised a detailed cardiovascular assessment20 The circulating levels of cholesterol, PCSK9, and LDLR were assessed both at visit1 and visit 4; and the burden of atherosclerotic plaques was assessed at visit 4. A genome‐wide association study was also performed on participants who participated to the visit 4. This design allows the determination on whether the PCSK9 and/or LDLR levels are associated with atherosclerotic plaques almost 20 years in advance, providing the background for a potential role of PCSK9 inhibitors in cardiovascular prevention. A potential causal link may be established if genetic variants associated with both PCSK9 and atherosclerotic plaques can be found.
The main aim of the present study in an initially healthy population is to study the association of PCSK9 and LDLR at visit 1 with carotid atherosclerotic plaques at visit 4, and the underlying genetic variants of PCSK9 gene; therefore, hypothesizing a role for PCSK9 inhibitors in primary cardiovascular prevention.
Methods
Study Population
The data that support the findings of this study are available from the corresponding author upon reasonable request.
A detailed description of the STANISLAS cohort has been previously published.20 The STANISLAS cohort was established with the primary objective of investigating gene–gene and gene–environment interactions in the field of cardiovascular diseases. To assess the effect of genetics on the variability of intermediate phenotypes on the transition toward pathology, the families were deemed healthy and free of declared acute and/or chronic illness at visit 1. The implementation of the fourth visit enabled a follow‐up of 18 to 23 years. The collected information was enriched with new biomarkers and detailed clinical phenotyping (including vascular echography). The STANISLAS visit 4 also allows the long‐term evaluation of clinical, biological, and morphological data, such as the advent of atherosclerotic plaques in an initially healthy population.
Institutional Review Board approval was obtained, and all patients provided informed consent to participate in the study.
In the present analysis, we included the 997 adults who attended both visit 1 and visit 4, and had also performed vascular echography and biomarker, and 949 of them were successfully genotyped.
Study Design
All participants were observed at the Centre d'Investigation Clinique Plurithématique Pierre Drouin at Nancy Hospital Center (CIC‐P de Nancy) in the morning after a 12‐ to 14‐hour fast. Blood samples were taken. Medical history, medications, anthropometric parameters, blood pressure, carotid plaques, carotid‐femoral pulse‐wave velocity (PWV), and carotid intima‐media thickness (cIMT) were recorded.
Arterial Plaques, Carotid Intima‐Media Thickness, and PWV
High‐resolution echotracking in STANISLAS visit 4 was performed to assess both carotid plaques, diameter, distention, and cIMT on the right common carotid artery.21 Intima‐media thickness was also measured in a subset of STANISLAS visit 1participants, but information regarding carotid plaques was not recorded.22 The noninvasive investigations were performed in a controlled environment at 22±1°C after 10 minutes of rest in the supine position. Four measurements were obtained per each participant. Examinations were performed with the wall track system (ESOATE, Maastricht, The Netherlands) and the ART.LAB (ESAOTE, Maastricht, The Netherlands) in immediate succession. The reproducibility and agreement (intraoperator/interoperator/devices) of the measurements were excellent.
PWV was determined with the Complior (Complior SP, Alam Medical, France) and Sphygmocor CVMS (AtCor, Australia) devices. Peripheral blood pressure was measured after at least 10 minutes of rest in the supine position, in a quiet room. Carotid‐to‐femoral PWV was assessed with Complior using the recommendations of the European Network for Noninvasive Investigation of Large Arteries23
Biomarkers and Gene‐Candidate Analysis
All samples were collected at the Centre d'Investigation Clinique Plurithématique Pierre Drouin at Nancy Hospital Center with minimally traumatic venipuncture. Standardized sample‐handling procedures enabled the collection of serum and plasma (EDTA, heparin) as well as buffy coat fraction. Blood DNA of all the participants to the STANISLAS visit 4 was extracted using Gentra Puregene Blood Kit (Qiagen, Hilden, Germany) and stored at −20°C. Genotyping was conducted at the Centre National de Recherche en Génomique Humaine (Evry, France) using 2 chips: the Illumina Global Screening Array, which is composed of 687 572 intronic and exonic markers and the Illumina Exome Array, which is composed of 244 330 SNPs, mostly exonic. All blood‐derived biosamples are stored in a central biobank facility with temperatures between −80°C and −196°C (as required).
Baseline plasma samples were analyzed for protein biomarkers by the TATAA‐biocenter using the Olink Proseek Multiplex cardiovascular II panel that includes both PCSK9 and LDLR, using a proximity extension assay technology24 where 92 oligonucleotide‐labeled antibody probe pairs per panel are allowed to bind to their respective targets in the sample in 96‐well plate format. When binding to their correct targets, they give rise to new DNA amplicons, with each ID‐barcoding their respective antigens. The amplicons are subsequently quantified using a BioMark HD real‐time polymerase chain reaction platform (Fluidigm, San Francisco, CA). The platform provides log2‐normalized protein expression data.
For the genetic analyses of the present study, we focused on the PCSK9 gene, which encodes the PCSK9 protein. First, we defined an interval that encompassed the PCSK9 gene boundaries (±20 kb) on chromosome 1 based on the reference genome built 37 from the Ensembl database (Chr1; position 55485221‐55550525; http://grch37.ensembl.org). Then, we selected all the SNPs comprised between these boundaries in the 2 chips. Twenty‐four SNPs were selected from the Global Screening Array chip and 12 from the Exome chip. However, 3 markers were duplicated between the 2 chips, and 1 of each was excluded. Hence, 33 SNPs were selected. After the quality control steps, 1 SNP was excluded for monomorphism (ie, minor allele frequency=0); no SNP had >1% of missing data (ie, all SNPs had a call rate >0.99), and no SNP deviated from the Hardy–Weinberg equilibrium at a threshold of P<1.10−8. We also excluded 7 SNPs that are rare (minor allele frequency <0.01). Tests for linkage disequilibrium in the subset of the 26 SNPs were conducted, and 4 SNPs were highly linked with r²>0.90.
Statistical Analysis
For the baseline clinical characteristics, continuous variables are expressed as means and respective SD. Categorical variables are presented as frequencies and percentages. Participant baseline characteristics were compared between those without plaques versus those with plaques at visit 4 using chi‐squared tests for categorical variables and t tests for continuous variables.
The main aim of this study was to test the association of PCSK9 and LDLR with incident carotid plaques. Logistic regression models were performed. First, a stepwise backward model including all the clinical variables with a P<0.1 from Table 1 was performed, to select the clinical features with stronger association with carotid plaques. Second, the potential association of PCSK9 and LDLR with carotid plaques was tested on top of the clinical model built in the previous step. We also tested the association of the genetic alleles on top of the clinical model plus PCSK9 and LDLR. The stability of this model was also confirmed using “partialing‐out cross‐fit” estimators controlling for all the variables with a P<0.01 from Table 1 with overlapping results (data not shown).
Table 1.
Baseline (Visit 1) Characteristics of the Study Population by the Presence of Carotid Plaques at Visit 4
| Patients’ Characteristics at Visit 1 | No Plaque | Plaque at Visit 4 | P Value |
|---|---|---|---|
| N. total=997 | 794 | 203 | |
| Age, y | 41.7±4.7 | 42.7±5.4 | 0.009 |
| Male sex | 387 (48.7%) | 122 (60.1%) | 0.004 |
| BMI, kg/m2 | 24.3±3.7 | 24.7±3.8 | 0.22 |
| Waist circumference, cm | 80.6±11.1 | 83.6±11.7 | <0.001 |
| Smoking | 140 (18.4%) | 58 (28.6%) | 0.002 |
| SBP, mm Hg | 122.1±11.8 | 124.4±13.6 | 0.015 |
| DBP, mm Hg | 74.4±9.8 | 75.6±10.3 | 0.11 |
| Heart rate, bpm | 66.3±10.3) | 64.7±10.1 | 0.056 |
| Diabetes mellitus | 21 (2.8%) | 12 (5.9%) | 0.028 |
| Glucose, g/L | 0.9±0.1 | 0.9±0.1 | 0.13 |
| Hypertension history | 69 (9.1%) | 25 (12.4%) | 0.16 |
| Total cholesterol, g/L | 2.2±0.4 | 2.3±0.4 | 0.010 |
| HDL cholesterol, g/L | 0.6±0.2 | 0.5±0.2 | 0.045 |
| LDL cholesterol, g/L | 1.5±0.3 | 1.6±0.4 | 0.001 |
| Triglycerides, g/L | 0.9±0.6 | 1.1±1.5 | 0.011 |
| Lipid‐lowering therapy | 13 (5.3%) | 3 (4.5%) | 0.79 |
| eGFR, mL/min per 1.73 m2 | 90.2±12.3 | 90.4±14.1 | 0.84 |
| PCSK9 (NPX) | 2.7±0.4 | 2.8±0.4 | <0.001 |
| LDL receptor (NPX) | 5.2±0.6 | 5.3±0.6 | 0.012 |
Median time between visit 1 and visit 4 , 18.5 years. BMI indicates body mass index; DBP, diastolic blood pressure; eGFR, estimated glomerular filtration rate calculated by the Chronic Kidney Disease Epidemiology Collaboration formula; LDL, low‐density lipoprotein; NPX, Olink log2 normalized protein expression; PCSK9, proprotein convertase subtilisin/kexin type 9; SBP, systolic blood pressure.
Since PCSK9 and LDLR proteins were measured using normalized protein expression) values on a log2 scale, the odds ratio (OR) for each protein estimates the increase in the odds of carotid plaques associated with a doubling in the protein concentration. A P<0.05 was considered statistically significant. The analyses were performed using STATA version 15 software (Stata Statistical Software, Release 15, StataCorp LP, College Station, TX).
The genetic analyses were performed using R (version 3.4.1). Association tests were performed using the R package “gaston.”25 The association of the genetic variants with the study outcomes was tested using a linear model, with age and sex used as covariates. The statistical significance level was fixed at 0.05, after applying a Benjamini–Hochberg correction for multiple testing.
Results
Characteristics of the Population
In the present analyses 997 adult participants were included. The mean age of the adult population at visit 1 was 42±5 years. Of these, 203 (20.4%) had carotid plaque(s) at visit 4. Participants who developed arterial plaques (from visit 1 to visit 4) were older than the other participants (42.7±5.4 versus 41.7±4.7 years), more often male (60% versus 49%), smokers (29% versus 18%), with diabetes mellitus (6% versus 3%), and with higher cholesterol levels (LDL‐C 1.6±0.4 versus 1.5±0.3 g/L) (P<0.05 for all) at visit 1 (Table 1. Participants who developed arterial plaques also had higher circulating PCSK9 and LDLR levels (Table 1.
The median (pct25‐75) follow‐up time from visit 1 to visit 4 was 18.5 (17.7–19.7) years. The mean age at visit 4 was 59.2±5.7 years, and the characteristics of the population with versus without plaques at visit 4 is described in the Table S1. No patient had carotid artery stenosis >50% of the lumen diameter.
Factors Associated With Carotid Plaques
The independent clinical factors at visit 1 associated with the advent (~20 years after) of arterial plaques at visit 4, were age (OR ] per 5‐year increase, 1.26; 95% CI, 1.06–1.49; P=0.007), smoking (OR, 1.79; [95% CI, 1.23–2.60; P=0.002), and LDL‐C (OR per 10 mg/dL increase, 1.72; 95% CI, 1.10–2.69; P=0.018) (Table 2. PCSK9 at visit 1 was associated with visit 4 arterial plaques on top of the clinical model (OR, 2.14, 95% CI, 1.28–3.58; P=0.004) and LDLR was not (OR, 0.91; 95% CI, 0.66–1.25; P=0.57). The PCSK9/LDLR ratio was also associated with arterial plaques driven by the PCSK9 levels (OR, 1.32; 95% CI, 1.04–1.67; P=0.023) (Table 2 and Figure. PCSK9 levels at visit 4 was also “transversally” associated with carotid plaques at visit 4 (OR, 1.66; 95% CI, 1.01–2.72; P=0.045) (Table S2).
Table 2.
Predictors (Visit 1) of Carotid Artery Plaque (at Visit 4)
| Variable | OR (95% CI) | P Value |
|---|---|---|
| “Best” clinical model | ||
| Age (per 5 y) | 1.26 (1.06–1.49) | 0.007 |
| Smoking (active) | 1.79 (1.23–2.60) | 0.002 |
| LDL‐C, g/L | 1.72 (1.10–2.69) | 0.018 |
| PCSK9 plus LDLR on top of the “best” clinical model | ||
| PCSK9 (NPX) | 2.14 (1.28–3.58) | 0.004 |
| LDLR (NPX) | 0.91 (0.66–1.25) | 0.57 |
| rs562556 polymorphism on top of the “best” clinical model plus PCSK9 and LDLR proteins | ||
| rs562556 (risk per A allele) | 1.60 (1.10–2.32) | 0.014 |
| PCSK9/LDLR ratio on top of the “best” clinical model | ||
| PCSK9/LDLR (ratio) | 1.32 (1.04–1.67) | 0.023 |
N=997; N. Plaque=203. Median time between visit 1 and visit 4 , 18.5 years. Our models presented good fit: Hosmer –Lemeshow goodness‐of‐fit test P>0.5 for all models (ie, clinical alone and clinical plus biomarkers). LDL‐C indicates low‐density lipoprotein cholesterol; LDLR, low‐density lipoprotein receptor; NPX, Olink log2 normalized protein expression; OR, odds ratio; PCSK9, proprotein convertase subtilisin/kexin type 9.
Figure 1.
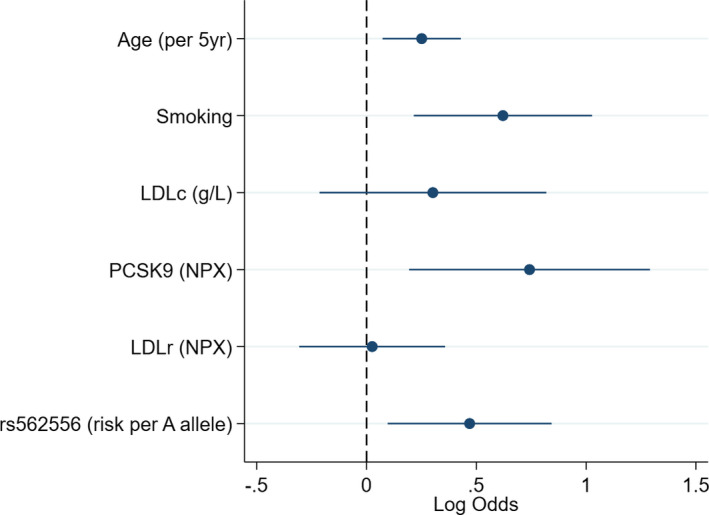
Multivariable predictors (visit 1) of carotid artery plaque (at visit 4). N =997; N. Plaque =203. Median time between visit 1 and visit 4, 18.5 years. LDL‐C indicates low‐density lipoprotein cholesterol; LDLR, LDL receptor; NPX, Olink log2 normalized protein expression; PCSK9, proprotein convertase subtilisin/kexin type 9.
Sensitivity Analysis in Children at Baseline
The proportion of children at visit 1 (n=647; mean age, 15±4 years) who developed plaques at visit 4 (mean age, 33±5 years) was 0.9% (n=6) (Table S3 and Figure S1). Despite an almost 10‐year difference, the age of these participants at visit 4 was closer to their parents at visit 1, possibly indicating that our population might have low prevalence of carotid plaques at visit 1.
Association of PCSK9 and LDLR With cIMT and PWV
At visit 1 LDLR was independently associated with visit 4 PWV as continuous variable (β‐coefficient, 0.4; 95% CI, 0.1–0.6; P=0.006). At visit 4, LDLR was independently associated with visit 4 cIMT as a continuous and categorical variable (β‐coefficient, 20.9; 95% CI, 4.5–37.2; P=0.012; and OR, 1.32; 95% CI, 1.00–1.73; P=0.042 for cIMT above the 90th percentile, respectively) (Table S4).
Genetic Considerations
We tested the association between polymorphisms of PCSK9 gene at visit 1 and presence of atherosclerotic plaques at visit 4 (Figure S2). We found 3 SNPs (rs540796, rs562556, and rs631220) associated with the presence of plaques (corrected P=0.02; Table S5). Minor alleles of these 3 SNPs are also associated with LDL‐C and PCSK9 levels both at visit 1 and visit 4. These 3 SNPs are highly correlated among them (r²>0.9) and 1 of them (rs562556) carried a missense gain‐of‐function mutation (I474V). Subjects who carried the minor allele (genotypes AA or GA) had higher cholesterol level and more often carotid plaques (Tables S6, S7, S8, S9, and S10). However, no significant association was found between PCSK9 polymorphisms and PWV or cIMT.
The partial correlation analyses suggest that rs562556 contributes with 16% to 17% of the variance of PCSK9 levels both at visit 1 and visit 4 (P<0.001 for both; (Table S11).
Discussion
As the main finding, the present study shows that increased circulating levels of PCSK9 in middle‐aged healthy people were independently associated with the presence of carotid plaques 20 years later, adjusting for important contributors of carotid plaque formation (eg, age, smoking, LDL‐C). Moreover, PCSK9 levels were similarly associated with carotid plaques “transversally” at visit 4. The identification of the SNP rs562556 (responsible for the missense mutation I474V) provides a potential causal link between the PCSK9 levels and the atherosclerotic plaques.
Higher LDL‐C concentrations have causal association with increased cardiovascular risk. Evidence derived from multiple randomized controlled trials and meta‐analyses shows a consistent and graded reduction in cardiovascular risk in response to reductions in the LDL‐C levels (grade of evidence IA).8, 26, 27, 28, 29 There is no apparent threshold at which LDL‐C lowering is not associated with reduced cardiovascular risk8 Moreover, the higher the initial LDL‐C level, the greater the absolute reduction in risk, while the relative risk reduction remains constant at any given baseline LDL‐C level30, 31 Statins effectively lower LDL‐C and are generally well tolerated. The only (rare) adverse events that have been reliably shown to be caused by statins are myalgias32 However, many patients are reluctant in taking statins, many because of medical misinformation, while many others cannot achieve the desired levels of LDL‐C despite moderate‐ to high‐intensity statin therapy11
The PCSK9, LDLR, and apolipoprotein B genes have been associated with autosomal dominant forms of familial hypercholesterolemia, caused by a “gain‐of‐function” mutation in the PCSK9 gene, resulting in excessive LDL‐C and, consequently, atherosclerotic plaques and cardiovascular events33 Mutations in the PCSK9 gene are reported to be responsible for 10% to 25% of the autosomal dominant form of familial hypercholesterolemia cases without mutations in LDLR or apolipoprotein B34 On the other hand, individuals who have a “loss‐of‐function” mutation in the PCSK9 gene express lower levels of LDL‐C and have low cardiovascular risk35 The physiological explanation is that the circulating PCSK9 binds to the LDLR, increasing its degradation in the lysosomal compartments. Inhibiting the PCSK9 activity (eg, with a monoclonal antibody) results in lower concentrations of free PCSK9, and in consequence, fewer LDLRs are degraded, thus being available for the uptake of LDL‐C, decreasing its blood concentrations36 The advent of PCSK9 inhibitors has enabled their testing in phase 3 trials. Overall, PCSK9 inhibitors significantly reduced LDL‐C regardless of patient population and/or background statin treatment37, 38
Previous to the present study, evidence regarding an independent association of PCSK9 in atherosclerosis progression was scarce. Prior cross‐sectional studies reported conflicting results regarding the association between PCSK9 levels and cIMT39, 40 In concordance with our results, in a Chinese cohort of 643 participants free of cardiovascular disease at baseline, plasma PCSK9 levels were associated with 10‐year progression of atherosclerosis (measured by the total plaque area), independently from LDL.41 However, in this study, the association of polymorphisms of the PCSK9 gene and the progression of atherosclerotic plaques were not evaluated.
In our study, we found that the rs562556 missense mutation was independently associated with incident carotid atherosclerotic plaques, LDL‐C, and PCSK9 levels. The SNP rs562556 has already been found to be associated with LDL‐C and total cholesterol, whereby patients with the mutation have higher levels of cholesterol19 Another recent study showed that genetically low LDL‐C attributable to PCSK9 variation (including the rs562556 SNP) was causally associated with low risk of cardiovascular mortality but not with low all‐cause mortality in a general population.42 These findings are in agreement with our results.
Our models retained age, smoking, and LDL‐C as the factors with stronger association with the occurrence of plaques, in concordance with previous studies43 Hence, the independent association of PCSK9 on top of these “classic” risk markers reinforces our hypothesis of targeting PCSK9 for preventing the occurrence of atherosclerotic disease. As described in the introductory section, the presence of atherosclerotic plaques has been associated with the occurrence of major cardiovascular events beyond cIMT or PWV2, 4, 44
Limitations
Several limitations should be acknowledged in the present study. First, this is an observational study; therefore, no causality can be established. Second, the presence of plaques at visit 1 was not recorded; therefore, we cannot exclude that some of these patients already had carotid plaques in their 40s; however, the near absence of plaques at visit 4 in those who started the study visit 1 as teenagers provides a robust internal control. Third, PCSK9 and LDLR were measured with the Olink technology standardized log2 normalized protein expression values, and no direct conversion to the standard “mass” values is possible.
Conclusions
PCSK9 was associated with arterial plaques almost 20 years in advance in a healthy middle‐aged population. The association of plaque and the SNP rs562556, responsible for I474V mutation, may reinforce the potential causality of our findings. Whether PCSK9 inhibitors could be useful for primary cardiovascular prevention could be worth investigating.
Sources of Funding
This biomarker study was funded by the French National Research Agency Fighting Heart Failure (ANR‐15‐RHU‐0004) and FEDER Lorraine, and all coauthors are supported by the French PIA project “Lorraine Université d'Excellence” GEENAGE (ANR‐15‐IDEX‐04‐LUE) programs, and the Contrat de Plan Etat Région Lorraine and FEDER IT2MP.
Disclosures
The authors declare not having conflicts of interest with regards to the content of this manuscript.
Supporting information
Table S1. Characteristics of the Study Population (at Visit 4) by the Presence of Carotid Plaques at Visit 4
Table S2. Factors Associated With Carotid Artery Plaque at Visit 4
Table S3. Plaque at Visit 4 in the “Children”
Table S4. Association of PCSK9 and LDLr (Visit 1 and Visit 4) With IMT and PWV (at Visit 4)
Table S5. Association Between Presence of Plaques and PCSK9 Polymorphisms (n=949)
Table S6. Crude Odd Ratios for Association Between Plaques at Visit 4 and Genotype at rs562556
Table S7. Association Between PCSK9 Polymorphisms and Circulating LDL at Visit 4 (n=895)
Table S8. Association Between PCSK9 Polymorphisms and Circulating LDL at Visit 1 (n=861)
Table S9. Association Between PCSK9 Polymorphisms and Circulating PCSK9 at Visit 1 (n=894)
Table S10. Association Between PCSK9 Polymorphisms and Circulating PCSK9 at Visit 4 (n=897)
Table S11. Partial Correlation Coefficients
Figure S1. Distribution of the STANISLAS cohort population according to age.
Figure S2. Matrix of linkage disequilibrium of the 26 SNPs for the gene PCSK9.
Acknowledgments
We acknowledge Robert Olaso and his lab “Production Platforms in Human Genomics” at the Centre National de Recherche en Génomique Humaine for genotyping data production, and Anne Boland at the Centre National de Recherche en Génomique Humaine for management of the genotyping study. We are highly grateful to the Vandoeuvre‐Lès Nancy Centre de Médecine Préventive staff, and to Dr Sophie Visvikis‐Siest (Inserm U1122) who managed the STANISLAS Cohort for the first 3 visits. The authors deeply thank the staff of the Clinical Investigation Center and other personnel involved in the STANISLAS Cohort management: Biostatisticians: Fay R, Lamiral Z, Machu JL. Computer scientists: Boucenna N, Gallina‐Muller C, Maclot PL, Sas T. Co‐investigators: Chau K, Di Patrizio P, Dobre D, Gonthier D, Huttin O, Malingrey L, Mauffrey V, Olivier A, Poyeton T, Steyer E, Watfa G. Data managers: Cimon P, Eby E, Merckle L. Data entry operators: Batsh M, Blanger O, Bottelin C, Haskour N, Jacquet V, Przybylski MC, Saribekyan Y, Thomas H, Vallee M. Echocardiographists, echographists: Ben Sassi M, Cario S, Camara Y, Coiro S, Frikha Z, Kearney‐Schwartz A, Selton‐Suty C, Watfa G. Imaging engineer: Bozec E. Laboratory engineer: Nuee‐Capiaumont J; and Technicians: Fruminet J, Kuntz M, Ravey J, Rousseau E, Tachet C. Project manager: Bouali S, Hertz C. Quality engineer: Lepage X. Registered nurses: Giansily M, Poinsignon L, Robin N, Schmartz M, Senn M, Micor‐Patrignani E, Toutlemonde M. Hospital technician: Fleurot MT. Resident doctors: Alvarez‐Vasquez R, Amiot M, Angotti M, Babel E, Balland M, Bannay A, Basselin P, Benoit P, Bercand J, Bouazzi M, Boubel E, Boucherab‐Brik N, Boyer F, Champagne C, Chenna SA, Clochey J, Czolnowski D, Dal‐Pozzolo J, Desse L, Donetti B, Dugelay G, Friang C, Galante M, Garel M, Gellenoncourt A, Guillin A, Hariton ML, Hinsiger M, Haudiquet E, Hubert JM, Hurtaud A, Jabbour J, Jeckel S, Kecha A, Kelche G, Kieffert C, Laurie`re E, Legay M, Mansuy A, Millet‐Muresan O, Meyer N, Mourton E, Naude´ AL, Pikus AC, Poucher M, Prot M, Quartino A, Saintot M, Schiavi A, Schumman R, Serot M, Sert C, Siboescu R, Terrier‐de‐la‐Chaise S, Thiesse A, Thietry L, Vanesson M, Viellard M. Secretaries: De Amorin E, Villemain C, Ziegler N. Study coordinators: Dauchy E, Laurent S; and all persons not listed above who helped to the funding, initiation, accrual, management and analysis of the fourth visit of the STANISLAS cohort. They also thank the CRB Lorrain of the Nancy CHRU for management of the biobank. Steering committee: Pierre Mutzenhardt, Mehdy Siaghy, Patrick Lacolley, Marie‐Ange Luc, Pierre Yves Marie, Jean Michel Vignaud. Advisory members: Sophie Visvikis Siest, F Zannad. Technical committee: Christiane Branlant, Isabelle Behm‐Ansmant, Jean‐Michel Vignaud, Christophe Philippe, Jacques Magdalou, Faiez Zannad, Patrick Rossignol. Scientific committee: Laurence Tiret, Denis Wahl, Athanase Benetos, Javier Diez, Maurizio Ferrari, Jean Louis Gueant, Georges Dedoussis, François Alla, François Gueyffier, Pierre‐Yves Scarabin, Claire Bonithon Kopp, Xavier Jouven, Jean‐Claude Voegel, Jan Staessen.
(J Am Heart Assoc. 2020;9:e014758 DOI: 10.1161/JAHA.119.014758.)
References
- 1. GBD 2017 Causes of Death Collaborators . Global, regional, and national age‐sex‐specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392:1736–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stein JH, Korcarz CE, Hurst RT, Lonn E, Kendall CB, Mohler ER, Najjar SS, Rembold CM, Post WS. Use of carotid ultrasound to identify subclinical vascular disease and evaluate cardiovascular disease risk: a consensus statement from the American Society of Echocardiography Carotid Intima‐Media Thickness Task Force. Endorsed by the Society for Vascular Medicine. J Am Soc Echocardiogr. 2008;21:93–111. [DOI] [PubMed] [Google Scholar]
- 3. Calonge N, Petitti DB, DeWitt TG, Gregory KD, Harris R, Isham G, LeFevre ML, Loveland‐Cherry C, Marion LN, Moyer VA, Ockene JK. Using nontraditional risk factors in coronary heart disease risk assessment: US Preventive Services Task Force recommendation statement. Ann Intern Med. 2009;151:474–482. [DOI] [PubMed] [Google Scholar]
- 4. Nambi V, Chambless L, Folsom AR, He M, Hu Y, Mosley T, Volcik K, Boerwinkle E, Ballantyne CM. Carotid intima‐media thickness and presence or absence of plaque improves prediction of coronary heart disease risk: the ARIC (Atherosclerosis Risk in Communities) study. J Am Coll Cardiol. 2010;55:1600–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stein JH, Johnson HM. Carotid intima‐media thickness, plaques, and cardiovascular disease risk: implications for preventive cardiology guidelines. J Am Coll Cardiol. 2010;55:1608–1610. [DOI] [PubMed] [Google Scholar]
- 6. Boekholdt SM, Arsenault BJ, Mora S, Pedersen TR, LaRosa JC, Nestel PJ, Simes RJ, Durrington P, Hitman GA, Welch KM, DeMicco DA, Zwinderman AH, Clearfield MB, Downs JR, Tonkin AM, Colhoun HM, Gotto AM Jr, Ridker PM, Kastelein JJ. Association of LDL cholesterol, non‐HDL cholesterol, and apolipoprotein B levels with risk of cardiovascular events among patients treated with statins: a meta‐analysis. JAMA. 2012;307:1302–1309. [DOI] [PubMed] [Google Scholar]
- 7. Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, Simes R. Efficacy and safety of cholesterol‐lowering treatment: prospective meta‐analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. [DOI] [PubMed] [Google Scholar]
- 8. Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta‐analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Avis HJ, Hutten BA, Gagne C, Langslet G, McCrindle BW, Wiegman A, Hsia J, Kastelein JJ, Stein EA. Efficacy and safety of rosuvastatin therapy for children with familial hypercholesterolemia. J Am Coll Cardiol. 2010;55:1121–1126. [DOI] [PubMed] [Google Scholar]
- 10. Thompson PD, Panza G, Zaleski A, Taylor B. Statin‐associated side effects. J Am Coll Cardiol. 2016;67:2395–2410. [DOI] [PubMed] [Google Scholar]
- 11. Hill JA, Agewall S, Baranchuk A, Booz GW, Borer JS, Camici PG, Chen PS, Dominiczak AF, Erol C, Grines CL, Gropler R, Guzik TJ, Heinemann MK, Iskandrian AE, Knight BP, London B, Luscher TF, Metra M, Musunuru K, Nallamothu BK, Natale A, Saksena S, Picard MH, Rao SV, Remme WJ, Rosenson RS, Sweitzer NK, Timmis A, Vrints C. Medical misinformation: vet the message!. Eur Heart J. 2019;40:404–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Navarese EP, Kolodziejczak M, Kereiakes DJ, Tantry US, O'Connor C, Gurbel PA. Proprotein convertase subtilisin/kexin type 9 monoclonal antibodies for acute coronary syndrome: a narrative review. Ann Intern Med. 2016;164:600–607. [DOI] [PubMed] [Google Scholar]
- 13. Shimada YJ, Cannon CP. PCSK9 (Proprotein convertase subtilisin/kexin type 9) inhibitors: past, present, and the future. Eur Heart J. 2015;36:2415–2424. [DOI] [PubMed] [Google Scholar]
- 14. Nicholls SJ, Puri R, Anderson T, Ballantyne CM, Cho L, Kastelein JJ, Koenig W, Somaratne R, Kassahun H, Yang J, Wasserman SM, Scott R, Ungi I, Podolec J, Ophuis AO, Cornel JH, Borgman M, Brennan DM, Nissen SE. Effect of evolocumab on progression of coronary disease in statin‐treated patients: the GLAGOV randomized clinical trial. JAMA. 2016;316:2373–2384. [DOI] [PubMed] [Google Scholar]
- 15. Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722. [DOI] [PubMed] [Google Scholar]
- 16. Weisshaar S, Zeitlinger M. Vaccines targeting PCSK9: a promising alternative to passive immunization with monoclonal antibodies in the management of hyperlipidaemia? Drugs. 2018;78:799–808. [DOI] [PubMed] [Google Scholar]
- 17. Schulz R, Schluter KD, Laufs U. Molecular and cellular function of the proprotein convertase subtilisin/kexin type 9 (PCSK9). Basic Res Cardiol. 2015;110:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qiu C, Zeng P, Li X, Zhang Z, Pan B, Peng ZYF, Li Y, Ma Y, Leng Y, Chen R. What is the impact of PCSK9 rs505151 and rs11591147 polymorphisms on serum lipids level and cardiovascular risk: a meta‐analysis. Lipids Health Dis. 2017;16:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chuan J, Qian Z, Zhang Y, Tong R, Peng M. The association of the PCSK9 rs562556 polymorphism with serum lipids level: a meta‐analysis. Lipids Health Dis. 2019;18:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ferreira JP, Girerd N, Bozec E, Merckle L, Pizard A, Bouali S, Eby E, Leroy C, Machu JL, Boivin JM, Lamiral Z, Rossignol P, Zannad F. Cohort Profile: rationale and design of the fourth visit of the STANISLAS cohort: a familial longitudinal population‐based cohort from the Nancy region of France. Int J Epidemiol. 2018;47:395–395j. [DOI] [PubMed] [Google Scholar]
- 21. Ferreira JP, Girerd N, Bozec E, Machu JL, Boivin JM, London GM, Zannad F, Rossignol P. Intima‐media thickness is linearly and continuously associated with systolic blood pressure in a population‐based cohort (STANISLAS Cohort Study). J Am Heart Assoc. 2016;5:e003529 DOI: 10.1161/JAHA.116.003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sass C, Herbeth B, Chapet O, Siest G, Visvikis S, Zannad F. Intima‐media thickness and diameter of carotid and femoral arteries in children, adolescents and adults from the Stanislas cohort: effect of age, sex, anthropometry and blood pressure. J Hypertens. 1998;16:1593–1602. [DOI] [PubMed] [Google Scholar]
- 23. Van Bortel LM, Laurent S, Boutouyrie P, Chowienczyk P, Cruickshank JK, De Backer T, Filipovsky J, Huybrechts S, Mattace‐Raso FU, Protogerou AD, Schillaci G, Segers P, Vermeersch S, Weber T. Expert consensus document on the measurement of aortic stiffness in daily practice using carotid‐femoral pulse wave velocity. J Hypertens. 2012;30:445–448. [DOI] [PubMed] [Google Scholar]
- 24. Lundberg M, Eriksson A, Tran B, Assarsson E, Fredriksson S. Homogeneous antibody‐based proximity extension assays provide sensitive and specific detection of low‐abundant proteins in human blood. Nucleic Acids Res. 2011;39:e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dandine‐Roulland C. Manipulation of genetic data (SNPs). Computation of GRM and dominance matrix, LD, heritability with efficient algorithms for linear mixed model (AIREML). Hum Hered. 2017‐18;83:1–29. [Google Scholar]
- 26. Boekholdt SM, Hovingh GK, Mora S, Arsenault BJ, Amarenco P, Pedersen TR, LaRosa JC, Waters DD, DeMicco DA, Simes RJ, Keech AC, Colquhoun D, Hitman GA, Betteridge DJ, Clearfield MB, Downs JR, Colhoun HM, Gotto AM Jr, Ridker PM, Grundy SM, Kastelein JJ. Very low levels of atherogenic lipoproteins and the risk for cardiovascular events: a meta‐analysis of statin trials. J Am Coll Cardiol. 2014;64:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brugts JJ, Yetgin T, Hoeks SE, Gotto AM, Shepherd J, Westendorp RG, de Craen AJ, Knopp RH, Nakamura H, Ridker P, van Domburg R, Deckers JW. The benefits of statins in people without established cardiovascular disease but with cardiovascular risk factors: meta‐analysis of randomised controlled trials. BMJ. 2009;33:b2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fulcher J, O'Connell R, Voysey M, Emberson J, Blackwell L, Mihaylova B, Simes J, Collins R, Kirby A, Colhoun H, Braunwald E, La Rosa J, Pedersen TR, Tonkin A, Davis B, Sleight P, Franzosi MG, Baigent C, Keech A. Efficacy and safety of LDL‐lowering therapy among men and women: meta‐analysis of individual data from 174,000 participants in 27 randomised trials. Lancet. 2015;385:1397–1405. [DOI] [PubMed] [Google Scholar]
- 29. Mills EJ, Rachlis B, Wu P, Devereaux PJ, Arora P, Perri D. Primary prevention of cardiovascular mortality and events with statin treatments: a network meta‐analysis involving more than 65,000 patients. J Am Coll Cardiol. 2008;52:1769–1781. [DOI] [PubMed] [Google Scholar]
- 30. Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, Goldberg AC, Gordon D, Levy D, Lloyd‐Jones DM, McBride P, Schwartz JS, Shero ST, Smith SC Jr, Watson K, Wilson PW, Eddleman KM, Jarrett NM, LaBresh K, Nevo L, Wnek J, Anderson JL, Halperin JL, Albert NM, Bozkurt B, Brindis RG, Curtis LH, DeMets D, Hochman JS, Kovacs RJ, Ohman EM, Pressler SJ, Sellke FW, Shen WK, Tomaselli GF. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129:S1–S45. [DOI] [PubMed] [Google Scholar]
- 31. Catapano AL, Graham I, De Backer G, Wiklund O, Chapman MJ, Drexel H, Hoes AW, Jennings CS, Landmesser U, Pedersen TR, Reiner Z, Riccardi G, Taskinen MR, Tokgozoglu L, Verschuren WM, Vlachopoulos C, Wood DA, Zamorano JL. 2016 ESC/EAS guidelines for the management of dyslipidaemias: the task force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS) developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Atherosclerosis. 2016;253:281–344. [DOI] [PubMed] [Google Scholar]
- 32. Collins R, Reith C, Emberson J, Armitage J, Baigent C, Blackwell L, Blumenthal R, Danesh J, Smith GD, DeMets D, Evans S, Law M, MacMahon S, Martin S, Neal B, Poulter N, Preiss D, Ridker P, Roberts I, Rodgers A, Sandercock P, Schulz K, Sever P, Simes J, Smeeth L, Wald N, Yusuf S, Peto R. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet. 2016;388:2532–2561. [DOI] [PubMed] [Google Scholar]
- 33. Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, Derre A, Villeger L, Farnier M, Beucler I, Bruckert E, Chambaz J, Chanu B, Lecerf JM, Luc G, Moulin P, Weissenbach J, Prat A, Krempf M, Junien C, Seidah NG, Boileau C. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–156. [DOI] [PubMed] [Google Scholar]
- 34. Humphries SE, Whittall RA, Hubbart CS, Maplebeck S, Cooper JA, Soutar AK, Naoumova R, Thompson GR, Seed M, Durrington PN, Miller JP, Betteridge DJ, Neil HA; Simon Broome Familial Hyperlipidaemia Register Group and Scientific Steering, C . Genetic causes of familial hypercholesterolaemia in patients in the UK: relation to plasma lipid levels and coronary heart disease risk. J Med Genet. 2006;43:943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Benn M, Nordestgaard BG, Grande P, Schnohr P, Tybjaerg‐Hansen A. PCSK9 R46L, low‐density lipoprotein cholesterol levels, and risk of ischemic heart disease: 3 independent studies and meta‐analyses. J Am Coll Cardiol. 2010;55:2833–2842. [DOI] [PubMed] [Google Scholar]
- 36. Stein EA, Mellis S, Yancopoulos GD, Stahl N, Logan D, Smith WB, Lisbon E, Gutierrez M, Webb C, Wu R, Du Y, Kranz T, Gasparino E, Swergold GD. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366:1108–1118. [DOI] [PubMed] [Google Scholar]
- 37. McKenney JM, Koren MJ, Kereiakes DJ, Hanotin C, Ferrand AC, Stein EA. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol. 2012;59:2344–2353. [DOI] [PubMed] [Google Scholar]
- 38. Giugliano RP, Desai NR, Kohli P, Rogers WJ, Somaratne R, Huang F, Liu T, Mohanavelu S, Hoffman EB, McDonald ST, Abrahamsen TE, Wasserman SM, Scott R, Sabatine MS. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE‐TIMI 57): a randomised, placebo‐controlled, dose‐ranging, phase 2 study. Lancet. 2012;380:2007–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chan DC, Pang J, McQuillan BM, Hung J, Beilby JP, Barrett PH, Watts GF. Plasma proprotein convertase subtilisin kexin type 9 as a predictor of carotid atherosclerosis in asymptomatic adults. Heart Lung Circ. 2016;25:520–525. [DOI] [PubMed] [Google Scholar]
- 40. Zhu YM, Anderson TJ, Sikdar K, Fung M, McQueen MJ, Lonn EM, Verma S. Association of proprotein convertase subtilisin/kexin type 9 (PCSK9) with cardiovascular risk in primary prevention. Arterioscler Thromb Vasc Biol. 2015;35:2254–2259. [DOI] [PubMed] [Google Scholar]
- 41. Xie W, Liu J, Wang W, Wang M, Qi Y, Zhao F, Sun J, Li Y, Zhao D. Association between plasma PCSK9 levels and 10‐year progression of carotid atherosclerosis beyond LDL‐C: a cohort study. Int J Cardiol. 2016;215:293–298. [DOI] [PubMed] [Google Scholar]
- 42. Benn M, Tybjaerg‐Hansen A, Nordestgaard BG. Low LDL cholesterol by PCSK9 variation reduces cardiovascular mortality. J Am Coll Cardiol. 2019;73:3102–3114. [DOI] [PubMed] [Google Scholar]
- 43. Cook NR, Ridker PM. Advances in measuring the effect of individual predictors of cardiovascular risk: the role of reclassification measures. Ann Intern Med. 2009;150:795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Helfand M, Buckley DI, Freeman M, Fu R, Rogers K, Fleming C, Humphrey LL. Emerging risk factors for coronary heart disease: a summary of systematic reviews conducted for the U.S. Preventive Services Task Force. Ann Intern Med. 2009;151:496–507. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of the Study Population (at Visit 4) by the Presence of Carotid Plaques at Visit 4
Table S2. Factors Associated With Carotid Artery Plaque at Visit 4
Table S3. Plaque at Visit 4 in the “Children”
Table S4. Association of PCSK9 and LDLr (Visit 1 and Visit 4) With IMT and PWV (at Visit 4)
Table S5. Association Between Presence of Plaques and PCSK9 Polymorphisms (n=949)
Table S6. Crude Odd Ratios for Association Between Plaques at Visit 4 and Genotype at rs562556
Table S7. Association Between PCSK9 Polymorphisms and Circulating LDL at Visit 4 (n=895)
Table S8. Association Between PCSK9 Polymorphisms and Circulating LDL at Visit 1 (n=861)
Table S9. Association Between PCSK9 Polymorphisms and Circulating PCSK9 at Visit 1 (n=894)
Table S10. Association Between PCSK9 Polymorphisms and Circulating PCSK9 at Visit 4 (n=897)
Table S11. Partial Correlation Coefficients
Figure S1. Distribution of the STANISLAS cohort population according to age.
Figure S2. Matrix of linkage disequilibrium of the 26 SNPs for the gene PCSK9.