

Abstract
Background
In spontaneously hypertensive rats (SHR) we observed profound myocardial metabolic changes during early hypertension before development of cardiac dysfunction and left ventricular hypertrophy. In this study, we evaluated whether metformin improved myocardial metabolic abnormalities and simultaneously prevented contractile dysfunction and left ventricular hypertrophy in SHR.
Methods and Results
SHR and control Wistar–Kyoto rats were treated with metformin from 2 to 5 months of age, when SHR hearts exhibit metabolic abnormalities and develop cardiac dysfunction and left ventricular hypertrophy. We evaluated the effect of metformin on myocardial glucose uptake rates with dynamic 2‐[18F] fluoro‐2‐deoxy‐D‐glucose positron emission tomography. We used cardiac MRI in vivo to assess the effect of metformin on ejection fraction, left ventricular mass, and end‐diastolic wall thickness, and also analyzed metabolites, AMP‐activated protein kinase and mammalian target‐of‐rapamycin activities, and mean arterial blood pressure. Metformin‐treated SHR had lower mean arterial blood pressure but remained hypertensive. Cardiac glucose uptake rates, left ventricular mass/tibia length, wall thickness, and circulating free fatty acid levels decreased to normal, and ejection fraction improved in treated SHR. Hearts of treated SHR exhibited increased AMP‐activated protein kinase phosphorylation and reduced mammalian target‐of‐rapamycin activity. Cardiac metabolite profiling demonstrated that metformin decreased fatty acyl carnitines and markers of oxidative stress in SHR.
Conclusions
Metformin reduced blood pressure, normalized myocardial glucose uptake, prevented left ventricular hypertrophy, and improved cardiac function in SHR. Metformin may exert its effects by normalizing myocardial AMPK and mammalian target‐of‐rapamycin activities, improving fatty acid oxidation, and reducing oxidative stress. Thus, metformin may be a new treatment to prevent or ameliorate chronic hypertension–induced left ventricular hypertrophy.
Keywords: cardiac hypertrophy, cardiac MRI, dynamic FDG PET, metformin, spontaneously hypertensive rats
Subject Categories: Hypertension, Metabolism, Hypertrophy, Nuclear Cardiology and PET, Magnetic Resonance Imaging (MRI)
Introduction
Left ventricular hypertrophy (LVH) develops in >35% of hypertensive individuals1 and is a key risk factor for heart failure.2, 3 Antihypertensive drugs reduce, but do not eliminate, LVH and risk for complications.4 In a recent study we observed that profound metabolic changes are present in the early stages of hypertension in hearts of spontaneously hypertensive rats (SHR), before manifestation of cardiac dysfunction and LVH.5 The sequence of events corroborates the concept that early metabolic remodeling precedes hypertension‐induced LVH. Accordingly, targeting metabolism may offer a novel approach to prevent cardiac contractile dysfunction and structural remodeling in patients with hypertension. Metformin, a drug approved by the US Food and Drug Administration, has multiple metabolic actions6 and is widely used as a first‐line treatment of type 2 diabetes. Cittadini et al7 recently demonstrated that metformin improved cardiac function and ameliorated development of structural changes in the hearts of spontaneously hypertensive and insulin‐resistant rats. However, the authors did not evaluate the metabolic effects of metformin on the heart itself. In an earlier study, we observed that metformin, most likely by improving glucose oxidation, decreased mammalian target‐of‐rapamycin (mTOR) activation, and improved contractile function in isolated working rat hearts ex vivo.8 In the present study we tested whether metformin could improve myocardial metabolic abnormalities and simultaneously prevent cardiac contractile dysfunction and LVH in SHR.
Methods
The data that support the findings of this study are available from the corresponding authors upon request.
SHR (n=6) and control Wistar–Kyoto rats (WKY) (n=6) (Charles River, Kingston, NY) were treated with metformin (300 mg/kg per day in drinking water), starting at 2 months of age (baseline), when we had observed significant metabolic changes.5 For comparison, we also evaluated untreated SHR (n=6) and WKY (n=6). Serial positron emission tomography (PET) and cardiac MRI (CMR) measurements were performed at baseline and after 3 months of treatment (at 5 months of age) for all groups of rats. The metformin dose was selected based on the following guidelines. A dose of 30 mg/kg per day used in humans converts to ≈150 mg/kg per day in rats due to a higher metabolic rate in rodents. We used a 2‐fold higher dose because metabolomics data in SHR after treatment with 100 mg/kg per day metformin indicated incomplete normalization of several metabolites (including branched‐chain amino acids [BCAAs] leucine, isoleucine, and valine, and the glucose metabolites pyruvate and lactate) (data not shown). We assessed the effect of metformin treatment on cardiac metabolism, function, and structure using dynamic 2‐[18F] fluoro‐2‐deoxy‐d‐glucose (FDG)‐PET and CMR. Dynamic FDG‐PET imaging of rat hearts was performed for 60 minutes using a Bruker Albira Si Trimodal imager (150‐mm axial field of view). Analysis of image data followed methods recently described by our laboratory.9 Cardiac gated CMR with 7T Bruker–Siemens ClinScan MRI5 was used to measure left ventricular mass (LVM), ejection fraction, and end‐diastolic wall thickness (EDWT). LVM was normalized to tibia length (TL) to yield LVM/TL ratios.
Mean arterial pressure (MAP) measurements5 were performed in separate groups of rats at 2 months (baseline) and at 5 months of age (n=6 for each group at each timepoint). Hearts harvested after terminal MAP measurements were used for molecular and metabolic analyses as described elsewhere.5 Rats were fasted for 6 hours prior to all experiments except CMR. AMP‐activated protein kinase (AMPK) and acetyl‐CoA carboxylase phosphorylation, as measures for AMPK activity, and mTOR activity were evaluated as described elsewhere.5, 8 Blood was collected from the same rats to determine circulating insulin, glucose, free fatty acids, and BCAA levels.5
To assess changes in cardiac metabolites, hearts were harvested from SHR treated with metformin (100 mg/kg per day) for 1 month from 1 to 2 months of age (SHR+metformin [SHR+M], n=10), and from untreated WKY (n=10) and SHR (n=9). All animal experiments were approved by the institutional animal care and use committee of the University of Virginia and performed according to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD).
Statistical Analysis
Due to the invasive and terminal method used for MAP measurements, MAP was measured repetitively at a single timepoint (age) for each rat, not repetitively over time. The MAP data were thus analyzed by 2‐way ANOVA with replicate measurements for each rat considered as subsampling units. In terms of the 2‐way ANOVA model, the dependent variable was MAP, and the independent variables were rat type (SHR and WKY) and age at assessment (2 and 5 months). For hypothesis testing, a set of linear contrasts of the ANOVA least‐squares means were utilized to conduct between rat‐type comparisons of MAP distribution means at 2 and 5 months. A Bonferroni‐corrected P<0.05 decision rule was utilized as the null hypothesis rejection rule. The PET, CMR serial imaging data (glucose uptake [Ki], ejection fraction, LVM/TL, and EDWT), and body‐weight (BW) measurements were analyzed by way of linear mixed models for timepoint by timepoint between and within rat‐type comparisons. Linear mixed‐model analyses for both comparisons were performed taking into account the 2 timepoints (2 and 5 months). Hypothesis testing was performed with respect to the mean response, and a comparison‐wise P<0.05 decision rule was used for the null hypothesis rejection criterion for between and within rat‐type comparisons. Indices for homeostatic model for insulin resistance, immunoblots, circulating insulin, glucose, FFA, and BCAA levels were analyzed using paired Student t tests, and metabolomics data with Welch's 2‐sample t tests. A P<0.05 decision rule was used as the null hypothesis rejection criterion. SAS version 9.4 (SAS Institute, Inc, Cary, NC) and GraphPad Prism 7 (GraphPad, Inc, La Jolla, CA) were used to conduct all statistical analyses. All data are shown as mean±standard error (SE). Results are only shown for rats at 5 months of age, comparing 3 groups (SHR, WKY, SHR+M), as metformin did not have any effects on control WKY rats.
Results and Discussion
In metformin‐treated SHR, MAP was lower than in untreated SHR, but remained in the hypertensive range (Figure 1A). Meanwhile, glucose uptake rates (Ki) in treated SHR returned to normal (P=NS from WKY) (Figure 1B and 1C). Ejection fraction improved (Figure 2A and 2B) and LVM/TL and EDWT were reduced in treated SHR when compared with untreated SHR (Figure 2C and 2D). Both LVM/TL and EDWT in treated SHR were comparable with those for WKY rats (P=NS). Body weights were not significantly different between metformin‐treated SHR and untreated SHR at 5 months of age (334.5±11.9 vs 360.5±16.7 g; Bonferroni‐corrected, P=0.129). Hearts of treated SHR exhibited increased phosphorylation of AMPK and of the AMPK target acetyl‐CoA carboxylase when compared with untreated SHR (Figure 3A–3D). Myocardial mTOR activity toward p70S6K (p‐p70S6K) in treated SHR hearts was reduced (Figure 3E and 3F). Circulating BCAA levels were increased in untreated SHR (P=0.021 versus WKY) and remained elevated with metformin treatment (data not shown). However, circulating FFA levels that were increased in untreated SHR were normalized in treated SHR (Figure 3G). Glucose and insulin levels were normal in treated and untreated SHR (data not shown). Also, there were no significant differences in homeostatic model for insulin resistance indices between metformin‐treated SHR (12.88±3.84) and untreated SHR (17.13±7.72) (P=0.26) or between metformin‐treated SHR and control WKY (17.5±4.55) (P=0.09). The overall higher homeostatic model for insulin resistance indices were due to higher insulin and glucose levels in circulation after only 6 hours of fasting. Metformin treatment had no effect on any of the measured parameters in WKY rats and hearts (data not shown).
Figure 1. Metformin treatment reduced blood pressure and normalized in vivo glucose uptake rates in SHR hearts.
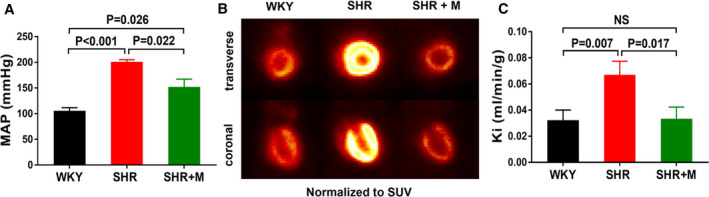
A, MAP in WKY rats (n=6), SHR (n=6), and SHR treated with metformin (300 mg/kg per day) for 3 months (SHR+M) (n=6). B, In vivo FDG PET images normalized to (SUV). C, Glucose uptake rates (Ki) computed from in vivo dynamic FDG PET images. All data are shown as mean±SE. FDG PET, fluorodeoxyglucose positron emission tomography; MAP, mean arterial pressure; SHR, spontaneously hypertensive rats; SUV, standardized uptake value; WKY, Wistar–Kyoto.
Figure 2. Metformin treatment improved cardiac function and normalized cardiac structure in SHR in vivo.
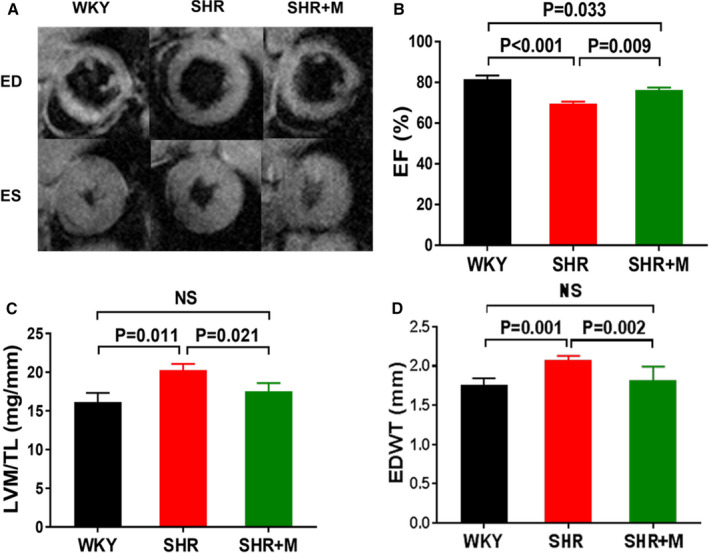
A, Short‐axis CMR images at ED and ES. B, EF, (C) LVM normalized to tibia length (TL), and (D) EDWT measured by CMR for WKY (n=6), SHR (n=6), and SHR treated with metformin (300 mg/kg per day) for 3 months (SHR+M) (n=6). All data are shown as means±SE. CMR indicates cardiac MRI; ED indicates end‐diastole; EF, ejection fraction; EDWT, end‐diastolic wall thickness; ES, end‐systolic; LVM, left ventricular mass; M, metformin; SHR, spontaneously hypertensive rats; WKY, Wistar–Kyoto.
Figure 3. Effects of metformin treatment on cardiac AMPK and ACC phosphorylation and mTOR activity, and on cardiac and circulating metabolites in SHR.
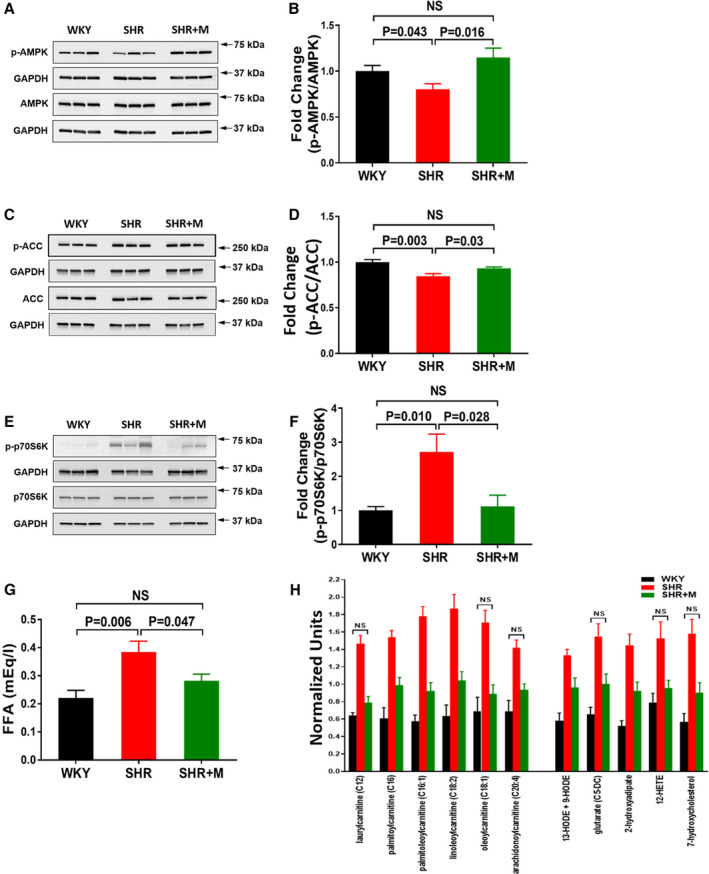
A, Immunoblots for p‐AMPK and (B) signal intensities for p‐AMPK and AMPK normalized to GAPDH and p‐AMPK/AMPK ratios determined. C, Immunoblots for p‐ACC and (D) signal intensities for p‐ACC and ACC normalized to GAPDH and p‐ACC/ACC ratios determined. E, Immunoblots for p‐p70S6K, a marker for mTOR activity, and (F) signal intensities for p‐p70S6K and p70S6K normalized to GAPDH and p‐p70S6K/p70S6K ratios determined. G, FFA in circulation. A through G, Data for WKY (n=6), SHR (n=6), and SHR treated with metformin (300 mg/kg per day) for 3 months (SHR+M [n=6]). H, Changes in cardiac fatty acyl carnitines (left) and lipid oxidation and peroxidation products (right) in SHR treated with metformin (100 mg/kg per day) (n=10) for 1 month from 1 to 2 months of age when compared with untreated WKY (n=10) and SHR (n=9). Data for biochemicals in raw area counts were rescaled to set medians equal to 1 and plotted. Data for untreated SHR and WKY were reported in our earlier study5 and are shown here for comparison with treated SHR. Data for WKY vs SHR, SHR vs SHR+M, and WKY vs SHR+M were statistically significantly different at P<0.05 except for the ones marked NS (WKY vs SHR+M). All data are shown as mean±SE. ACC indicates acetyl‐CoA carboxylase; FFA, free fatty acids; M, metformin; mTOR, mammalian target of rapamycin; SHR, spontaneously hypertensive rats; WKY, Wistar–Kyoto.
To discern whether the insignificant findings between the SHR+M versus WKY comparisons of Ki, LVM/TL, and EDWT were simply a consequence of the small number of replicates used in our study (n=6), we calculated the effect size and the sample sizes that would be needed to have ≥0.80 statistical power to test the same set of hypotheses if the study was replicated prospectively. For each imaging metric (Ki, LVM/TL, and EDWT), column 3 in Table shows the minimum detectable underlying absolute differences between the means of the SHR+M and WKY imaging metric measurement distributions at 5 months that would lead to a ≥80% incidence of rejecting the null hypothesis that the imaging metric mean of the SHR+M group is equal to the imaging metric mean of WKY group, with no more than a 5% chance of committing a type I error. For each imaging metric (Ki, LVM/TL, and EDWT), column 5 in Table lists the number of animals that would be required per group (SHR+M and WKY) to have a ≥80% chance—with type I error ≤0.05—of rejecting the null hypothesis that the mean of the imaging metric measurement distribution of the SHR+M group is equal to the mean of the imaging metric measurement distribution of the WKY group, if the true underlying difference between the means of the 2 imaging metric measurement distributions is equal to the difference observed in the current study (column 4).
Table 1.
Statistical Power Estimates
| Imaging Metric | Observed Standard Deviation | Minimum Detectable Absolute Difference With 6 Animals per Group | Observed Absolute Difference in Means | Sample Size Requirement per Group to Detect Observed Difference |
|---|---|---|---|---|
| Ki | 0.033 | 0.053 | 0.001 | 17 095 |
| LVM/tibia length | 2.60 | 4.21 | 1.06 | 95 |
| ED wall thickness | 0.184 | 0.298 | 0.063 | 134 |
ED indicates end‐diastolic; Ki, rate of myocardial 2‐[18F] fluoro‐2‐deoxy‐D‐glucose uptake; and LVM, left ventricular mass.
Early intervention with metformin normalized myocardial glucose uptake, simultaneously improved cardiac function, and prevented the development of LVH in SHR. Although blood pressure was reduced with metformin treatment, it remained in the hypertensive range, at a level associated with increased glucose uptake and profound changes in cardiac metabolites in our earlier study.5 Thus, the beneficial effects of metformin cannot be attributed to decreased blood pressure alone. Normalization of glucose uptake by metformin suggests that metabolic improvements contribute to the beneficial effects of metformin. Metformin exerts multiple AMPK‐dependent and ‐independent effects on metabolism.6 Increased AMPK activity improves glucose and fatty acid oxidation,8, 10 whereas AMPK‐independent effects include inhibition of mitochondrial complex I and protection against oxidative stress.11 In our study, metformin restored AMPK phosphorylation in SHR to levels found in control WKY. Preliminary cardiac metabolite analysis, using methods described in our earlier work,5 showed decreased levels of fatty acyl carnitines and markers of oxidative stress (lipid oxidation and peroxidation products) in SHR treated with a lower dose of metformin (100 mg/kg per day) for 1 month, from 1 to 2 months of age (Figure 3H). These observations suggest that metformin can improve fatty acid oxidation and decrease oxidative stress in SHR hearts.
Improved AMPK activity may lead to the observed decreased mTOR activity, thereby suppressing cell growth and preventing development of LVH.8, 12 However, other AMPK targets may also play roles in inhibiting cardiac hypertrophy in response to metformin. Gelinas et al13 revealed that the anti‐hypertrophic actions of metformin on hearts of angiotensin II–treated mice were due to AMPK‐dependent inhibition of protein O‐GlcNAcylation and independent of changes in mTOR activity. The effect of metformin on protein O‐GlcNAcylation in SHR, as well as on other signaling pathways that regulate the development of cardiac hypertrophy, such as the calcineurin/nuclear factor of activated T cells and ubiquitin–proteasome pathways,13, 14 will need to be evaluated in the future. A recent study revealed that mitogen‐activated protein kinase is activated in SHR and that inhibition of mitogen‐activated protein kinase signaling improved pressure overload–induced cardiac remodeling.15 However, we observed no significant differences between metformin‐treated and untreated SHR and WKY in the phosphorylation of mitogen‐activated protein kinase (data not shown).
Metformin is known to improve cardiovascular disease outcomes (as reviewed by Nesti and Natali11). A recent clinical study tested metformin in insulin‐resistant prediabetic patients with coronary artery disease and found that metformin treatment lowered blood pressure, decreased LVH, and reduced oxidative stress.16 However, the use of metformin in chronic hypertension and during the early development of LVH has not been reported. Our study suggests that metformin may serve as a novel treatment approach to ameliorate cardiac metabolic abnormalities that develop in response to chronic pressure overload and thereby lessen hypertension‐induced LVH, even in patients without diabetes.
Sources of Funding
This work was supported by grants from the National Institutes of Health (R01 HL123627 to Kundu, R01 EB 001763 to Epstein, R01 HL128189 to Carey, and R01 HL61483 to Taegtmeyer) as well as start‐up funds from the Department of Radiology and Medical Imaging, University of Virginia (to Kundu).
Disclosures
None.
Acknowledgments
The authors thank Jeremy Gatesman from the Center for Comparative Medicine, University of Virginia, for tail‐vein catheterizations during the FDG‐PET imaging studies.
(J Am Heart Assoc. 2020;9:e015154 DOI: 10.1161/JAHA.119.015154.)
For Sources of Funding and Disclosures, see page 6.
Contributor Information
Susanna R. Keller, Email: srk4b@virginia.edu.
Bijoy K. Kundu, Email: bkk5a@virginia.edu.
References
- 1. Cuspidi C, Facchetti R, Bombelli M, Tadic M, Sala C, Grassi G, Mancia G. High normal blood pressure and left ventricular hypertrophy echocardiographic findings from the PAMELA population. Hypertension. 2019;73:612–619. [DOI] [PubMed] [Google Scholar]
- 2. Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. [DOI] [PubMed] [Google Scholar]
- 3. Verdecchia P, Carini G, Circo A, Dovellini E, Giovannini E, Lombardo M, Solinas P, Gorini M, Maggioni AP. Left ventricular mass and cardiovascular morbidity in essential hypertension: The MAVI study. J Am Coll Cardiol. 2001;38:1829–1835. [DOI] [PubMed] [Google Scholar]
- 4. Devereux RB, Wachtell K, Gerdts E, Boman K, Nieminen MS, Papademetriou V, Rokkedal J, Harris K, Aurup P, Dahlof B. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA. 2004;292:2350–2356. [DOI] [PubMed] [Google Scholar]
- 5. Li J, Kemp BA, Howell NL, Massey J, Minczuk K, Huang Q, Chordia MD, Roy RJ, Patrie JT, Davogustto GE, et al. Metabolic changes in spontaneously hypertensive rat hearts precede cardiac dysfunction and left ventricular hypertrophy. J Am Heart Assoc. 2019;8:e010926 DOI: 10.1161/JAHA.118.010926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60:1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cittadini A, Napoli R, Monti MG, Rea D, Longobardi S, Netti PA, Walser M, Sama M, Aimaretti G, Isgaard J, et al. Metformin prevents the development of chronic heart failure in the SHHF rat model. Diabetes. 2012;61:944–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sen S, Kundu BK, Wu HC, Hashmi SS, Guthrie P, Locke LW, Roy RJ, Matherne GP, Berr SS, Terwelp M, et al. Glucose regulation of load‐induced mTOR signaling and ER stress in mammalian heart. J Am Heart Assoc. 2013;2:e004796 DOI: 10.1161/JAHA.113.004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang Q, Massey JC, Minczuk K, Li J, Kundu BK. Non‐invasive determination of blood input function to compute rate of myocardial glucose uptake from dynamic FDG PET images of rat heart in vivo: comparative study between the inferior vena cava and the left ventricular blood pool with spill over and partial volume corrections. Phys Med Biol. 2019;64:165010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Canto C, Auwerx J. AMP‐activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci. 2010;67:3407–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nesti L, Natali A. Metformin effects on the heart and the cardiovascular system: a review of experimental and clinical data. Nutr Metab Cardiovasc Dis. 2017;27:657–669. [DOI] [PubMed] [Google Scholar]
- 12. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gelinas R, Mailleux F, Dontaine J, Bultot L, Demeulder B, Ginion A, Daskalopoulos EP, Esfahani H, Dubois‐Deruy E, Lauzier B, et al. AMPK activation counteracts cardiac hypertrophy by reducing O‐GlcNAcylation. Nat Commun. 2018;9:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baskin KK, Taegtmeyer H. AMP‐activated protein kinase regulates E3 ligases in rodent heart. Circ Res. 2011;109:1153–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu X, Chen K, Zhuang Y, Huang Y, Sui Y, Zhang Y, Lv L, Zhang G. Paeoniflorin improves pressure overload‐induced cardiac remodeling by modulating the MAPK signaling pathway in spontaneously hypertensive rats. Biomed Pharmacother. 2019;111:695–704. [DOI] [PubMed] [Google Scholar]
- 16. Mohan M, Al‐Talabany S, McKinnie A, Mordi IR, Singh JSS, Gandy SJ, Baig F, Hussain MS, Bhalraam U, Khan F, et al. A randomized controlled trial of metformin on left ventricular hypertrophy in patients with coronary artery disease without diabetes: the MET‐REMODEL trial. Eur Heart J. 2019;40:3409–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]