

Abstract
Background
Mitochondrial transplantation has been recently explored for treatment of very ill cardiac patients. However, little is known about the intracellular consequences of mitochondrial transplantation. This study aims to assess the bioenergetics consequences of mitochondrial transplantation into normal cardiomyocytes in the short and long term.
Methods and Results
We first established the feasibility of autologous, non‐autologous, and interspecies mitochondrial transplantation. Then we quantitated the bioenergetics consequences of non‐autologous mitochondrial transplantation into cardiomyocytes up to 28 days using a Seahorse Extracellular Flux Analyzer. Compared with the control, we observed a statistically significant improvement in basal respiration and ATP production 2‐day post‐transplantation, accompanied by an increase in maximal respiration and spare respiratory capacity, although not statistically significantly. However, these initial improvements were short‐lived and the bioenergetics advantages return to the baseline level in subsequent time points.
Conclusions
This study, for the first time, shows that transplantation of non‐autologous mitochondria from healthy skeletal muscle cells into normal cardiomyocytes leads to short‐term improvement of bioenergetics indicating “supercharged” state. However, over time these improved effects disappear, which suggests transplantation of mitochondria may have a potential application in settings where there is an acute stress.
Keywords: mitochondria, mitochondrial cardiomyopathy, mitochondrial transplantation, mitochontrial respiratory chain disease, mitochondrial transfer
Subject Categories: Basic Science Research, Cardiomyopathy, Metabolism
Clinical Perspective
What Is New?
Our studies indicate that normal cellular bioenergetics is acutely enhanced after mitochondrial transplantation.
The current study delineates the potential of mitochondrial transplantation for clinical application in settings where there is an acute stress that would benefit from a boost in cellular bioenergetics.
What Are the Clinical Implications?
Given the observed bioenergetics profile of mitochondrial transplantation in normal cells, a remaining question would be if the post‐transplantation bioenergetics profile will be any different in cells with mitochondrial dysfunction.
These studies are crucial in determining the possible advantages of mitochondrial transplantation in mitigating mitochondrial diseases, if any, as a cellular biotherapy.
The mitochondrion found in most eukaryotic cells provides nearly all of the cell's energy by the oxidative phosphorylation process through the mitochondrial respiratory chain.1, 2 The eukaryotes’ mitochondrion is believed to have evolved from a small, autotrophic bacterium that was engulfed by a larger primitive, heterotrophic, eukaryotic cell.2 Owing to its origin, mitochondrion has a (maternally inherited) genome that is distinct from the cell's nuclear genome. Although mitochondrion was once a free‐living organism, through time and because of its symbiotic relationship, some of its genome has been translocated to the cell's nucleus, making it an intracellular organelle dependent on the nucleus. Like the nuclear genome, mitochondrial DNA is constantly prone to damage and mutations. However, mitochondria lack effective DNA repair mechanisms, so the mitochondrial DNA defects often clonally accumulate in subsequent mitochondria. The majority of mitochondrial diseases occur as a result of mutations in either nuclear DNA or mitochondrial DNA. Regardless, the phenotypic representations of all mitochondrial disorders are deficiencies in energy metabolism and cell function with cardiac involvement as a common manifestation, ranging from cardiomyopathy to arrhythmias and heart failure.2, 3, 4
Recently, much interest has been devoted to cellular biotherapies involving mitochondria. Elliot et al5 have successfully demonstrated that introduction of normal mitochondria into human breast cancer cells restores mitochondrial function, inhibits cancer cell proliferation, and reverses chemoresistance by increasing the sensitivity of cells to breast cancer medication. Moreover, early clinical feasibility of mitochondrial replacement therapy in humans has provided hope to mitigating inherited mitochondrial disorders,6 and transplantation of mitochondria into the ischemic zone of the myocardium has been shown to improve recovery in cardiac ischemia‐reperfusion injury.7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17 The mechanism of mitochondrial uptake is a question that is actively under investigation, and some of the suggested mechanisms are actin‐dependent endocytosis17 and macropinocytosis.18 However, little is known about the intracellular fate of mitochondria during transplantation and, in particular, the bioenergetics consequences of mitochondrial transplantation. We aimed to address this question by studying how cellular bioenergetics are affected in the short and long term after mitochondrial transplantation. The requirement for institutional review board approval was waived given that no human studies were involved.
Methods
All the data that support the findings of this study are available from the first author upon reasonable request.
Cell Culture and Mitochondrial Isolation
Cells from the H9c2 cell line derived from rat embryonic hearts, rat skeletal muscle cells from the L6 cell line, and ARPE‐19 cells from human retina were grown per manufacturer's instructions (ATCC, Manassas, VA). H9c2 and L6 cells were grown in complete growth medium, which consisted of DMEM, FBS to a final concentration of 10%, and penicillin‐streptomycin to a final concentration of 1% (Thermo Fisher Scientific, Waltham, MA). ARPE‐19 cells were grown in DMEM/F‐12 (ATCC), FBS to a final concentration of 10%, and penicillin‐streptomycin to a final concentration of 1% (Thermo Fisher Scientific). To conserve the myoblastic properties of H9c2 cells, cells were grown at a seeding density of and passaged when approximately 80% confluent (≈every 3 days).
In our studies, as schematically shown in Figure 1, we isolated mitochondria from H9c2 cells for autologous transplantation (Figure 2A and 2B) and L6 cells for non‐autologous (Figure 2C) and interspecies transplantation (Figure 2D), using a commercially available isolation kit for cultured cells (Thermo Fisher Scientific). Cardiomyocytes were plated at 50K on a μ‐slide 8‐well ibiTreat slide (ibidi, Martinsried, Germany) the day before mitochondrial isolation for imaging purposes, 40K cells onto a Seahorse Cell Culture Microplate (Agilent Technologies, Santa Clara, CA) the day before running the Seahorse assay, and 40K cells onto a 96‐well plate the day before running the MitoSOX superoxide production assay. For holotomographic microscopy (Figure 2C), a 35‐mm dish (ibidi) was used.
Figure 1.
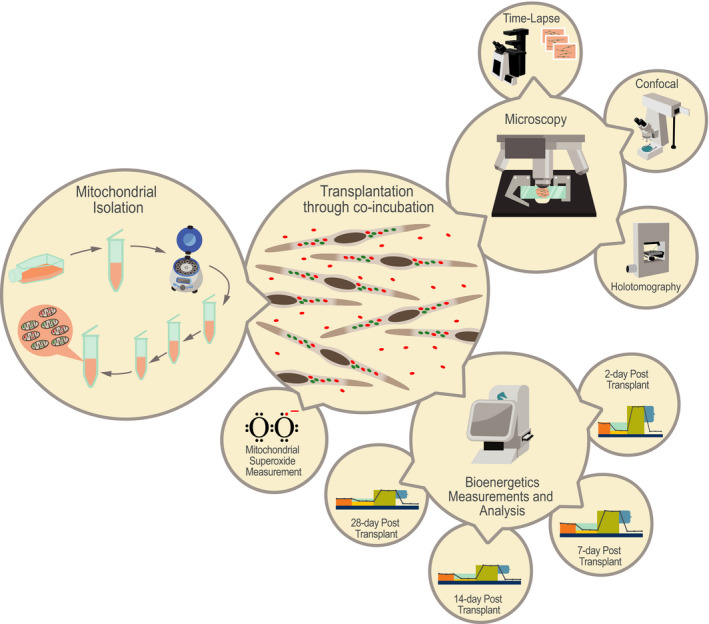
Mitochondrial transplantation studies in a glance. Feasibility of mitochondrial transplantation (autologous, non‐autologous, and interspecies) was demonstrated using (A) wide‐field time‐lapse (Olympus, Tokyo, Japan) and (B) confocal (Zeiss Microscopy, Jena, Germany) and (C) holotomography microscopy (Nanolive SA, Tolochenaz, Switzerland). Bioenergetics consequences of non‐autologous mitochondrial transplantation were studied using a Seahorse Extracellular Flux Analyzer (Agilent Technologies, Santa Clara, CA) (A) at 2, (B) 7, (C) 14, and (D) 28 days post‐transplantation. Mitochondrial superoxide was measured using MitoSOX Red Mitochondrial Superoxide Indicator (Thermo Fisher Scientific, Waltham, MA).
Figure 2.
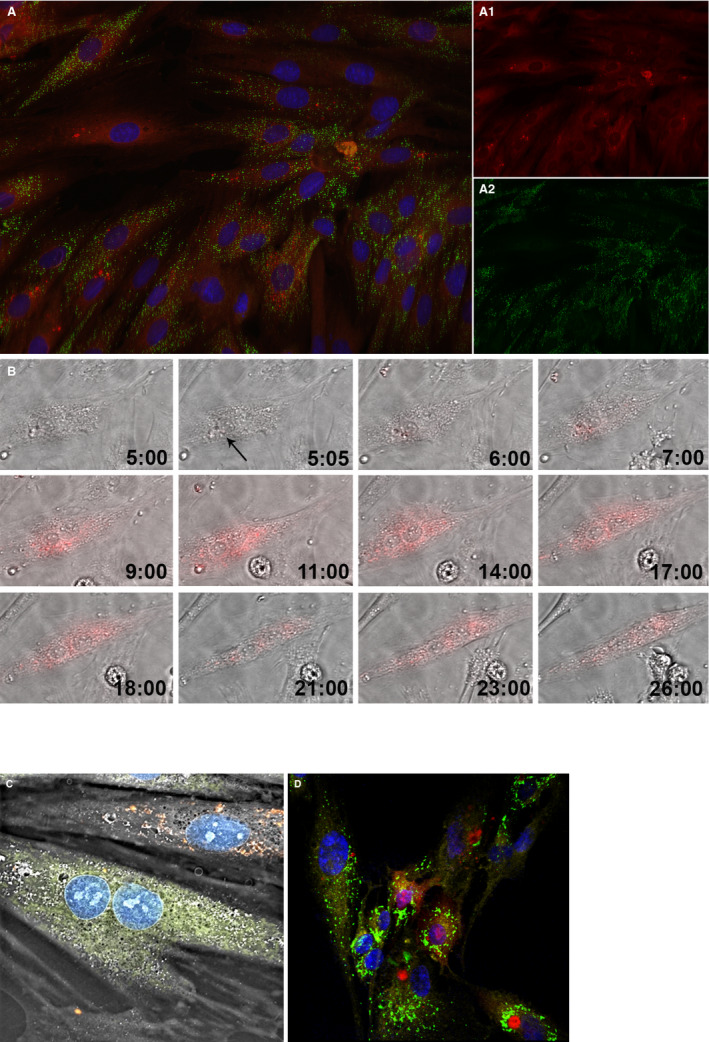
Internalization of mitochondria into H9c2 cardiomyocyte‐like cells through coincubation. (A) Feasibility of mitochondrial transplantation. The newly internalized mitochondria labeled with pHrodo Red Succinimidyl Ester (A.1.); the cell's native mitochondria labeled with MitoTracker Green FM (A.2.); the nucleus is labeled with NucBlue Live. The image was taken after 28 hours time‐lapse using a structured illumination epifluorescence microscope (Keyence, Itasca, IL). (B) Dynamics of mitochondrial internalization. Hour 5:00 represents the cardiomyocytes before mitochondrial internalization. Media contains the fluorescently labeled mitochondria. Given that pHrodo Red SE fluoresces brightly red only after it has been internalized by the cell, no florescence could yet be observed. At 5:05, the cardiomyocyte starts taking in the mitochondria, as indicated by the red fluorescence signal (black arrow). As time passes, more mitochondria are internalized into the cell. Moreover, there are interactions between the mitochondria, which are assumed to represent the dynamics of fusion and fission processes (Olympus, Tokyo, Japan). (C) Non‐autologous transplantation of mitochondria. Mitochondria from rat L6 skeletal muscle cell (shown in red) were transplanted into H9c2 cardiomyocytes (Nanolive SA, Tolochenaz, Switzerland). (D) Interspecies transplantation of mitochondria. Mitochondria from rat L6 skeletal muscle cells (shown in red) were transplanted into human ARPE‐19 retinal epithelial cells. The cell's native mitochondria are shown in green, the transpalnted mitochondria in red, the nucleus in blue, and the plasma membrane, labeled with CellMask Deep Red Plasma Membrane Stain, in yellow (Zeiss Microscopy, Jena, Germany).
Mitochondrial Labeling and Imaging
Native mitochondria were labeled with MitoTracker Green FM with excitation/emission 490/516 nm at 37°C for 45 minutes and washed twice with sterile PBS (Thermo Fisher Scientific). Isolated mitochondria were labeled with pHrodo Red Succinimidyl Ester with excitation/emission 560/585 nm for 30 minutes at 4°C, washed twice with sterile PBS (Thermo Fisher Scientific), and then coincubated with H9c2 cells. The nucleus was labeled with NucBlue Live with excitation/emission 360/460 nm (Thermo Fisher Scientific). A time‐lapse microscopy was subsequently conducted with both fluorescence and phase contrast/DIC channels to capture the dynamic behavior of mitochondrial internalization (Figure 2B; Video S1). Images were acquired either using a wide‐field (Olympus IX83; Olympus Corporation, Tokyo, Japan) or confocal microscope (Zeiss LSM 780; Zeiss Microscopy, Jena, Germany). The respective data were analyzed using Fiji (NIH, Bethesda, MD) and IMARIS software (Bitplane AG, Zurich, Switzerland).
Bioenergetics Assessment After Mitochondrial Transplantation
To investigate the bioenergetics consequences of mitochondrial transplantation, oxygen consumption rate (OCR) and extracellular acidification (ECAR) rate were measured post‐transplantation using a Seahorse XF24 Analyzer. First, H9c2 cells were plated at seeding densities of 10K, 20K, 40K, and 80K to find the optimized cell‐seeding density, which was experimentally determined to be 40K. This was followed by drug optimization tests, in which the optimal final well concentrations were experimentally determined to be 1 μmol/L for oligomycin, 1 μmol/L for carbonyl cyanide‐p‐trifluoromethoxyphenylhydrazone (FCCP), and 0.5 μmol/L of rotenone+0.5 μmol/L of antimycin A (R+A). Mitochondria from L6 skeletal cells (p.9) were isolated (from 100 cells per 1 recipient cell), using a commercially available isolation kit (Thermo Fisher Scientific), and transplanted into H9c2 cardiomyocyte cells (p.10). Isolated mitochondria were coincubated with H9c2 cells for 24 hours. After that, any mitochondria not internalized into cardiomyocytes were removed by changing the culture media. The Seahorse assays were performed in Seahorse XF DMEM supplemented with glucose to a final concentration of 10 mmol/L, sodium pyruvate to 1 mmol/L, and l‐glutamine to 2 mmol/L. Bioenergetics measurements were performed, after sequential treatment with Oligomycin, FCCP, and R+A (Agilent Technologies), at 2 (Figure 3), 7 (Figure 4), 14 (Figure 4), and 28 days (Figure 4) post‐transplantation and compared with that of the control group to investigate the effect of mitochondrial transplantation in 2 sets of independent studies, with a total of 28 observations for the experimental group and 12 for the control group. Raw data were normalized to total protein using the Micro BCA Protein Assay (Thermo Fisher Scientific).
Figure 3.
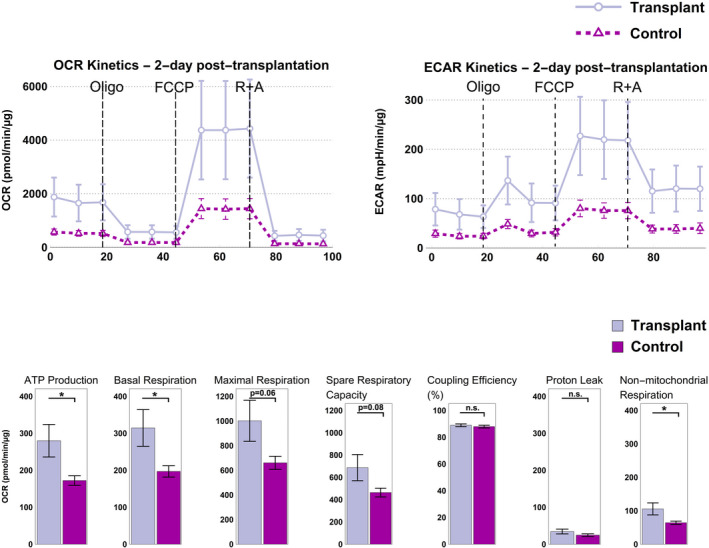
Non‐autologous mitochondrial transplantation of L6 skeletal cells into H9c2 cardiomyocytes leads to enhanced bioenergetics 2 days post‐transplantation. Top: Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) kinetics increased 2 days post‐transplantation, indicating improved metabolism after mitochondrial transplantation. Bottom: Compared with control (lavender), there was a statistically significant increase in ATP production, basal respiration, and non‐mitochondrial respiration in the transplant (purple) group. Maximal respiration and spare respiratory capacity were also enhanced, although not statistically significantly. Coupling efficiency and proton leak remained unchanged. A Welch's test was used, and results with p<0.05 were considered as statistically significant (n=28 for transplant and n=12 for control). Abbreviations: Oligo, oligomycin; FCCP, carbonyl cyanide‐p‐trifluoromethoxyphenyl hydrazone; R+A, rotenone and antimycin A; n.s., not significant.
Figure 4.
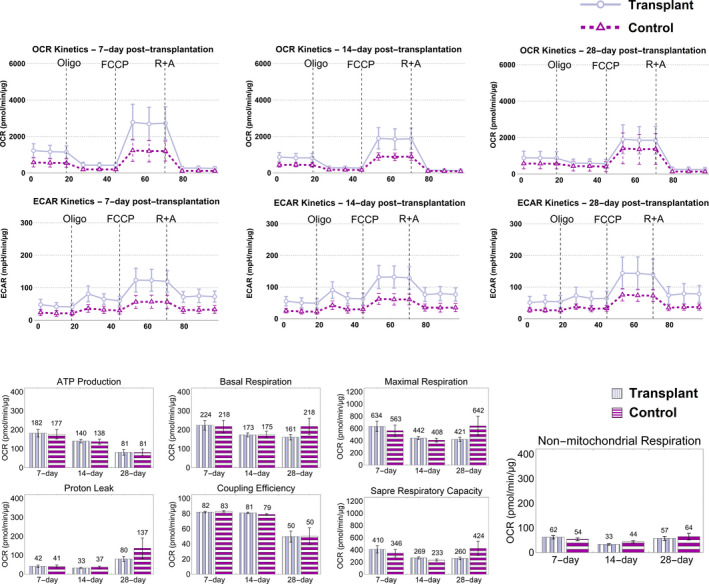
Acute enhancement in bioenergetics after mitochondrial transplantation diminishes in the long term. Top: No significant change in OCR and ECAR kinetics could be observed 7, 14, and 28 days post‐transplantation. Bottom: No significant difference was found between the transplant (lavender) and control (purple) groups in the long term. Whereas bioenergetics returned to baseline levels in the long term, no negative consequences to cells’ bioenergetics were observed because of the intervention. A Welch’s test was used, and results with p<0.05 were considered as statistically significant (n=28 for transplant and n=12 for control for t=7 days and 14 days; n=27 for transplant (because of well‐measurement failure in 1 well) and n=12 for control for t=28 days). Abbreviations: OCR, oxygen consumption rate; ECAR, extracellular acidification rate; Oligo, oligomycin; FCCP, carbonyl cyanide‐p‐trifluoromethoxyphenyl hydrazone; R+A, rotenone and antimycin A.
Quantification of Mitochondrial Superoxide Production
To assess the production of superoxide by mitochondria, MitoSOX Red Mitochondrial Superoxide Indicator (Thermo Fisher Scientific) was used. At 2 days post‐transplantation, cells were incubated with MitoSOX Red (5 μmol/L) at 37°C for 10 minutes. Cells were then washed 3 times with sterile PBS. Using a fluorescence plate reader, MitoSOX Red was excited at 485 nm and fluorescence emission was measured at 590 nm. With 10 observations in each group, fluorescence values were compared among the 2 groups of independent transplantation experiments and a control group (Figure 5).
Figure 5.
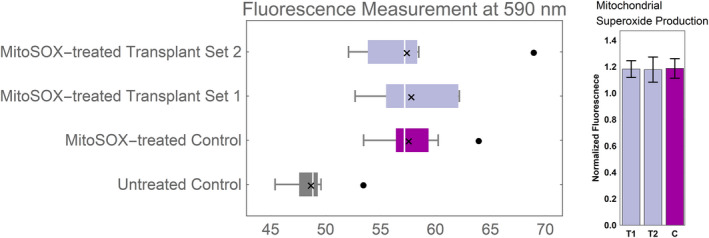
Mitochondrial superoxide production. H9c2 cells were exposed to MitoSOX Red reagent 48 hours after mitochondrial transplantation to measure superoxide production. MitoSOX Red was excited at 485 nm, and fluorescence emission was measured at 590 nm. Whisker box plot shows distribution of fluorescence readings for 10 measurements per group. No statistically significant difference was observed in the population's mean of the post‐transplant groups vs control. Fluorescence measurements of MitoSOX‐treated groups were normalized to the untreated control and are displayed as bar graphs, for the mean±SD. Symbols: White line denotes the population median, “x” indicates the population mean, and filled black dots represent outliers.
Statistical Analyses
An unpaired 2‐tailed unequal variance t test (Welch's test) was used for the mean values of Seahorse bioenergetics data in Microsoft Excel (Microsoft Corporation, Redmond, WA). Welch's test, which is a more‐stringent test than the Student t test, was used to account for: (1) the possibility of mitochondrial transplantation changing the bioenergetics both positively and negatively (2‐tailed); (2) possible different distributions of the control and transplant groups (unequal variance), and that (3) the measurements were taken independently (unpaired). To check for normality, qq plots of the data were generated in R (R Foundation for Statistical Computing, Vienna, Austria). Distribution in transplant appeared to be, in fact, different from the control group. Most observations fell within the 95% CI, and overall the data were reasonably normally distributed. Results with p<0.05 were considered as statistically significant. No statistical methods were used to predetermine the sample size. Sample size was based on experimental feasibility for proof of concept. Graphs presented in Figures 3, 4, 5 through 6 were generated in Mathematica (Wolfram Research, Champaign, IL). Data from bar graphs presented in Figures 3, 4, and 6 are means±SEM, and data from kinetic profiles in Figures 3 and 4 are mean±SD from n=2 independent experiments each with 14 biological replicates for the experimental transplant group and 6 biological replicates for the control group. For the 28‐day post‐transplantation experiment, 27 observations were recorded versus the 28 for all the other time points, because of measurement failure in one of the wells.
Figure 6.
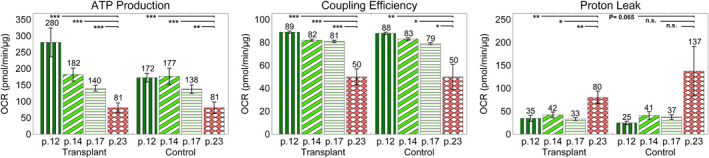
Effect of high passage number (aging) on mitochondrial function. Mitochondrial function was reduced with increasing passage number. This is indicated by the decrease in ATP production and coupling efficiency—positive indicators of mitochondrial function—and increase in proton leak— a negative indicator of mitochondrial function. An unpaired 2‐tailed unequal variance t test was used, and results with p<0.05 were considered as statistically significant (n=28 for p# 12, 14, and 17 and n=27 for p# 23 for transplant and n=12 for p# 12, 14, 17, and 23 for control). Abbreviations: OCR, oxygen consumption rate; p#, passage number, n.s., not significant.
Welch's test was used to compare post‐transplant cardiomyocytes’ mean superoxide production with that of the control at the 5% level. Fluorescence values after MitoSOX treatment were recorded from n=2 independent transplant groups and compared with n=1 control group, each with 10 biological replicates. No statistical methods were used to predetermine sample size. Fluorescence values are displayed as box and whisker plots in Figure 5. Fluorescence measurements from the mitoSOX‐treated control and transplant groups were normalized to the mean fluorescence measurement value of the untreated control. Mitochondrial superoxide production is reported as the normalized fluorescence values±SD and presented as bar graphs in Figure 5.
Results
Inspired by the endosymbiosis theory of mitochondrial origin, we hypothesized and tested whether coincubation of isolated mitochondria with cells would allow for mitochondria's uptake by the cell and enhanced cellular bioenergetics state. First, we evaluated autologous mitochondrial transplantation of rat cardiomyocyte H9c2 cells (ATCC). Based on a 28‐hour time‐lapse study (Figures 2A and 2B), we observed mitochondrial internalization at various time points and visualized the dynamics of mitochondrial internalization in time, as shown in Video S1. Figure 2B shows representative images demonstrating cellular uptake of pHrodo Red Succinimidyl Ester–labeled mitochondria (black arrow). Cells remained viable for the duration of the experiment whereas the internalized mitochondria appeared to undergo fission and fusion dynamics, based on propagation of the pHrodo Red signal to the entire cell (Figure 2B). From a mechanistic perspective, in labeling isolated mitochondria with pHrodo Red Succinimidyl Ester, the succinimidyl ester group interacts with the amine group on the mitochondria and forms a covalent amide bond. Nevertheless, to ensure specificity of the detected signal representing mitochondrial internalization, we performed a set of control experiments with 4 groups in the absence of exogenous mitochondria, as described in Table 1. Collectively, our experiments showed that the pHrodo dye by itself could not passively internalize, and consequently the pHrodo‐labeled autologous mitochondria actively internalized into the cells.
Table 1.
Validation Experiment for Mitochondrial Transplantation
| Group Descriptions | |
|---|---|
| 1. | pHrodo Red SE dye present in the media, with no cells plated |
| 2. | Cells in the absence of pHrodo Red SE dye in the media |
| 3. | Cells in the presence of pHrodo Red SE dye in the media |
| 4. | Cells in the presence of pHrodo Red SE dye in the media, and native mitochondria fluorescently labeled with MitoTracker Green FM to control for proper function of microscope and fluorescent signal detection |
pHrodo Red SE indicates pHrodo Red succinimidyl ester.
Next, we tested whether cells would similarly uptake non‐autologous mitochondria (Figure 2C). Mitochondria isolated from rat L6 skeletal muscle cells (ATCC) were internalized by rat cardiomyocyte H9c2 cells. This set of experiments shows that non‐autologous mitochondrial transplantation between the 2 rat cell lines is possible.
Lastly, we investigated interspecies mitochondrial transplantation by using isolated mitochondria from rat L6 cells and transplanted them into human ARPE‐19 retinal cells (ATCC; Figure 2D). Similar to the previous experiments, mitochondria were successfully internalized into cells, and the feasibility of mitochondrial transplantation from rat to human cells was demonstrated.
We then hypothesized and tested whether internalization of transplanted non‐autologous mitochondria would lead to enhanced bioenergetics, by conducting the Cell Mito Stress Test using a Seahorse Extracellular Flux Analyzer (Agilent). We measured the OCR, which represents the rate of oxidative phosphorylation and ECAR, which denotes the summation of acid (H+) produced from glycolysis and the tricarboxylic acid (TCA) cycle (by CO2), when obtained through the Mito Stress Test. Subsequently, we compared the results with the control groups with no transplantation event. Baseline and post‐transplant rates were measured at 4 different time points: after 2 days to measure acute effects (Figure 3); after 7 days; after 14 days to check bioenergetic indices around mitochondria turnover; and finally after 28 days to check for long‐term effect (Figure 4). We observed an upward shift in OCR kinetics 2 days post‐mitochondrial transplantation compared with baseline (Figures 3). Compared with the control, we observed a statistically significant improvement in basal respiration and ATP production 2 days post‐transplantation (p=0.031 and p=0.025, respectively; Figure 3). Maximal respiration and spare respiratory capacity—the cell's bioenergetics reserve in meeting a situation of high demand or acute/chronic stress—were also enhanced after 2 days, although not statistically significantly (p=0.060 for maximal respiration and p=0.080 for spare respiratory capacity; Figure 3). Coupling efficiency was unchanged given that both transplanted and native mitochondria were healthy (Figure 3).
Mitochondrial superoxide production was measured 2 days post‐transplantation using MitoSOX Red in control and transplant groups, and no significant difference was observed among the groups.
Discussion
H9c2 cells are phenotypically purely aerobic given that the heart tissue has a high energy demand, which is more efficiently met by aerobic respiration and oxidative phosphorylation. Increase in OCR kinetics 2 days post‐transplantation is indicative of improved bioenergetics, which is justified by more oxidative phosphorylation attributed to increased mitochondrial content (Figure 3). Based on our bioenergetics results, it appears that in normal cells, mitochondrial transplantation leads to acutely enhanced bioenergetics. However, these initial effects seem to diminish over time and return to that of the baseline control (Figure 4).
In addition to OCR, we evaluated the ECAR from the Mito Stress Test (Figures 3 and 4). In many cell types, mitochondrial activity (TCA cycle) that leads to CO2 production is a significant source of extracellular acidification.19 Given that, in our observation, the ECAR significantly decreased after R+A injection, we suppose that, in our samples, acidification was generated through the TCA cycle. However, from the Mito Stress Test, only qualitative interpretation of the changes in ECAR data can be made for kinetics after injection of R+A. If the ECAR significantly decreases (ie, low residual ECAR) in response to R+A, we infer the ECAR (before the injection of R+A) is primarily being generated by CO2 production from the TCA cycle. If the ECAR remains constant and/or increases in response to R+A, then the ECAR before R+A treatment is primarily being generated by glycolysis.20 It is also important to note that the exogenous pyruvate and glutamine supplemented in the media can be directly shunted into the TCA cycle, which leads to CO2 production independent of the CO2 produced from the pyruvate generated from glucose through glycolysis. Given that we observed a decrease in response to R+A in both control and transplant groups, we infer that CO2 is primarily being generated by the TCA cycle in this cell type. Another viewpoint indicates that in both control and transplant groups, cells were burning through the exogenous pyruvate and glutamate. However, on the contrary, we interpret our ECAR data (Figures 3 and 4) such that the increase in ECAR kinetics in transplant versus the control group (Figure 3) is attributable to CO2 production from the increased TCA cycle in the transplant group. Glycolytic rate assay can delineate the sole contribution of glycolysis or CO2 to ECAR, which we did not perform in the current experiments.
Previously, Finck and Kelly21 reported that the energetic needs of the cell finely tune the mitochondrial abundance, and too many or too few mitochondria may lead to pathology. In their review of myocardial energy metabolism, they associated myocardial diseases with perturbations of mitochondrial biogenesis. PGC‐1α (peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha) is a protein that drives mitochondrial biogenesis in cardiomyocytes by activating nuclear respiratory factor 1, which, in turn, triggers expression of activated mitochondria, leading to transcription and replication of the mitochondrial genome. Mice lacking PGC‐1α present with signs of heart failure. Alternatively, downregulation of PGC‐1α in mice leads to hypertrophic cardiomyopathy, whereas prolonged overexpression of PGC‐1α in mice leads to increased biogenesis activity and mitochondrial ultrastructural abnormalities, which ultimately leads to death from heart failure.21 Based on our observations, the enhanced bioenergetics state is temporary, which may imply that either the cell's mitochondrial content is returned to physiological levels, or the overall mitochondrial activities are reduced to reflect the cell's bioenergetics needs. Given that the spare respiratory capacity, which measures the cell's capacity in meeting a situation of high energy demand, is not significantly changed in the transplant versus the control, it is less likely that the overall mitochondrial activities have dampened and more likely that mitochondrial content has returned to physiological levels. Our observation corroborates with the findings of Finck and Kelly, and, accordingly, we hypothesize that a normal cell clears any excess mitochondria, keeping mitochondrial content at physiological levels, as the plausible mechanism for the return of the bioenergetics indices to the baseline levels in the long term. In the case of an adjustment of mitochondrial content, it is yet unclear whether the newly transplanted mitochondria are cleared from the cell or whether transplanted mitochondria have been adopted by the host and the total mitochondrial level regardless of the recipient or donor status is reduced to bring the bioenergetics levels back to the physiological state.
Moreover, the long‐term bioenergetics profile suggests that there are no negative bioenergetics consequences attributable to mitochondrial transplantation, and that the intervention can be considered safe for the cell. The question that remains to be answered is whether or not cells with damaged mitochondria would take advantage of the newly introduced mitochondria, given that those cells could uptake and adopt the mitochondria differently than a normal cell, which is already able to efficiently meet its bioenergetics demands.
It is also important to note that non‐mitochondrial respiration—an index that measures oxygen consumed by the cell by processes that are non‐mitochondrial—significantly increased after 2 days (p=0.033; Figure 3) and returned to baseline after 7, 14, and 28 days (p>0.05 for all; Figure 4). Non‐mitochondrial respiration is neither well understood nor well studied.22 Some non‐mitochondrial processes that use oxygen are superoxide production (with deleterious effects for mitochondria23, 24) and hydrogen peroxide production.25 It is known that hydrogen peroxide protects the cell from superoxide production given that it is properly degraded by catalase; otherwise, it can break apart and form hydroxyl radicals. The increase in non‐mitochondrial respiration could either suggest an improved overall metabolism or stress, which needs to be further investigated.
In a normal, tightly coupled electron transport chain, ≈1% to 3% of consumed oxygen is incompletely reduced, which can lead to superoxide production as a result of the interaction between leaky electrons and molecular oxygen.26, 27 Given that we had observed a statistically significant enhancement in bioenergetics attributable to increased oxidative phosphorylation at 2 days post‐transplantation, we used the mitochondrially targeted derivative of hydroethidine—MitoSOX Red—to detect and quantitate the level of superoxides produced by mitochondria in post‐transplant and control groups 2 days after transplantation. Compared with control, no statistically significant difference was found in superoxide production in the post‐transplant group (p=0.99 and p=0.83 for set 1 and 2 transplantations, respectively; Figure 5). The reported superoxide production was not assessed for the same Seahorse experiment, but a representative one.
Finally, we observed a reduction in mitochondrial function in the studied groups with increasing of the passage number. ATP production and coupling efficiency, positive indicators of bioenergetics, significantly decreased with increasing passage number, whereas proton leak, a negative indicator of bioenergetics, significantly increased (Figure 6). This observation corroborates with the findings of a previous study by Witek et al,28 where they observed an increase in oxidative stress in cells with passaging of cardiomyocytes. It appears that mitochondrial function is reduced in cells with a higher passage number, and thus these cells are not representative of the initial passages. The H9c2 cell line is a well‐established cell line used for assessing drug‐induced cardiotoxicity,28 and this observation indicates that only lower passages of this line should be used to consider mitochondrial adverse events.
Limitations
Given that the focus of the present study has been on mitochondrial oxidative phosphorylation, the Mito Stress Test was conducted. Thus, the ECAR kinetics presented in Figures 3 and 4 represent the summation of acid (H+) produced from glycolysis and CO2 produced from the TCA cycle. Accordingly, the ECAR data in this study are not a direct measurement of glycolysis. The XF Glycolytic Rate Assay should be performed to quantitate the changes attributable to glycolysis.
Conclusions
Our studies suggest that mitochondrial transplantation leads to transiently enhanced bioenergetics in normal cardiomyocytes, as evident by the statistically significant increase in cells’ basal respiration and ATP production. In our long‐term post‐transplantation studies, bioenergetics returned to physiological levels in normal recipient cells, and more studies are necessary to determine whether the outcome would be different in recipient cells with dysfunctional mitochondria. Although these enhancements were short‐lived in normal cells, it is evident that there were also no adverse effects attributable to the transplantation, given that the transplant cells were doing as well as the control cells in the long term, and there was no increase in mitochondrial superoxide production, which is a negative by‐product of oxidative phosphorylation.
Sources of Funding
This work is supported, in part, by an NIH Training Program in Cardiovascular Applied Research and Entrepreneurship (2T32HL116270‐06A1) to P.A., by Discovery Eye Foundation to M.C.K., and by a research award from the Council on Research Computing and Libraries (CORCL) of the University of California, Irvine to A.K. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funders.
Disclosures
None.
Supporting information
Video S1. Dynamics of mitochondrial internalization. In this video clip, dynamics of mitochondrial internalization is visualized. Isolated mitochondria labeled with pHrodo Red succinimidyl ester (SE) are being internalized into rat H9c2 cardiomyocytes through coincubation. The pHrodo Red SE label is sensitive to pH drop and fluoresces as endocytosed by the cell. The lack of fluorescence outside of the cell eliminates any doubt about the specificity of the detected signal. At around hour 5, the first instance of mitochondrial internalization is observed. Fluorescence intensity progressively decreases as the dye is segregated between the new daughter mitochondrion with each round of fission. Occasionally, fluorescence intensity increases locally, indicating new instances of mitochondrial internalization in those areas. With passing time, pHrodo Red SE signal propagates through the entire cell, perhaps attributable to mitochondrial fission and fusion events between the transplanted and host mitochondria. The cell remains viable for the duration of the time‐lapse study. Best viewed with Windows Media Player.
Acknowledgments
We thank UC Irvine's Professor Babak Shahbaba for his advice on statistical analyses, Marilyn Chwa for her help with the Seahorse flux analyzer experiments, and Agilent's Cell Analysis Team (particularly Courtney Nadeou Watts and Dr. Xiaofei Chen) for their valuable inputs related to bioenergetics studies. Furthermore, we thank Dr. Adeela Syed, UC Irvine's manager of the Optical Biology Core facility, Couryn Beleck from Keyence Corporation Life Science Microscope Team, and Matthew Bell from Nanolive Microscope Team for their assistance in imaging aspects of our studies. Additionally, some of the imaging studies were made possible through access to the Optical Biology Core Facility of the Developmental Biology Center, a shared resource supported by the Cancer Center Support Grant (CA‐62203) and Center for Complex Biological Systems Support Grant (GM‐076516) at the University of California, Irvine.
Author contributions: Ali Pour and Kheradvar designed the overall study and wrote the manuscript. Ali Pour performed the experiments and analyzed the data. Kenney provided technical expertise in the analysis and interpretation of data and reviewed the manuscript.
(J Am Heart Assoc. 2020;9:e014501 DOI: 10.1161/JAHA.119.014501.)
References
- 1. Sebastian D, Palacin M, Zorzano A. Mitochondrial dynamics: coupling mitochondrial fitness with healthy aging. Trends Mol Med. 2017;23:201–215. [DOI] [PubMed] [Google Scholar]
- 2. Svensson OL. Mitochondria: structure, functions, and dysfunctions. Nova Science Publishers, Inc., 2010. [Google Scholar]
- 3. Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. The epidemiology of mitochondrial disorders–past, present and future. Biochim Biophys Acta. 2004;1659:115–120. [DOI] [PubMed] [Google Scholar]
- 4. El‐Hattab AW, Scaglia F. Mitochondrial cardiomyopathies. Front Cardiovasc Med. 2016;3:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Elliott RL, Jiang XP, Head JF. Mitochondria organelle transplantation: introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity. Breast Cancer Res Treat. 2012;136:347–354. [DOI] [PubMed] [Google Scholar]
- 6. Zhang J, Liu H, Luo S, Lu Z, Chavez‐Badiola A, Liu Z, Yang M, Merhi Z, Silber SJ, Munne S, Konstantinidis M, Wells D, Tang JJ, Huang T. Live birth derived from oocyte spindle transfer to prevent mitochondrial disease. Reprod Biomed Online. 2017;34:361–368. [DOI] [PubMed] [Google Scholar]
- 7. Moskowitzova K, Shin B, Liu K, Ramirez‐Barbieri G, Guariento A, Blitzer D, Thedsanamoorthy JK, Yao R, Snay ER, Inkster JAH, Orfany A, Zurakowski D, Cowan DB, Packard AB, Visner GA, Del Nido PJ, McCully JD. Mitochondrial transplantation prolongs cold ischemia time in murine heart transplantation. J Heart Lung Transplant. 2019;38:92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Emani SM, McCully JD. Mitochondrial transplantation: applications for pediatric patients with congenital heart disease. Transl Pediatr. 2018;7:169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ramirez‐Barbieri G, Moskowitzova K, Shin B, Blitzer D, Orfany A, Guariento A, Iken K, Friehs I, Zurakowski D, Del Nido PJ, McCully JD. Alloreactivity and allorecognition of syngeneic and allogeneic mitochondria. Mitochondrion. 2019;46:103–115. [DOI] [PubMed] [Google Scholar]
- 10. Cowan DB, Yao R, Thedsanamoorthy JK, Zurakowski D, Del Nido PJ, McCully JD. Transit and integration of extracellular mitochondria in human heart cells. Sci Rep. 2017;7:17450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shin B, Cowan DB, Emani SM, Del Nido PJ, McCully JD. Mitochondrial transplantation in myocardial ischemia and reperfusion injury. Adv Exp Med Biol. 2017;982:595–619. [DOI] [PubMed] [Google Scholar]
- 12. McCully JD, Cowan DB, Emani SM, Del Nido PJ. Mitochondrial transplantation: from animal models to clinical use in humans. Mitochondrion. 2017;34:127–134. [DOI] [PubMed] [Google Scholar]
- 13. Emani SM, Piekarski BL, Harrild D, del Nido PJ, McCully JD. Autologous mitochondrial transplantation for dysfunction after ischemia‐reperfusion injury. J Thorac Cardiovasc Surg. 2017;154:286–289. [DOI] [PubMed] [Google Scholar]
- 14. Kaza AK, Wamala I, Friehs I, Kuebler JD, Rathod RH, Berra I, Ericsson M, Yao R, Thedsanamoorthy JK, Zurakowski D, Levitsky S, Del Nido PJ, Cowan DB, McCully JD. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J Thorac Cardiovasc Surg. 2017;153:934–943. [DOI] [PubMed] [Google Scholar]
- 15. Cowan DB, Yao R, Akurathi V, Snay ER, Thedsanamoorthy JK, Zurakowski D, Ericsson M, Friehs I, Wu Y, Levitsky S, Del Nido PJ, Packard AB, McCully JD. Intracoronary delivery of mitochondria to the ischemic heart for cardioprotection. PLoS One. 2016;11:e0160889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McCully JD, Levitsky S, del Nido PJ, Cowan DB. Mitochondrial transplantation for therapeutic use. Clin Transl Med. 2016;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pacak CA, Preble JM, Kondo H, Seibel P, Levitsky S, del Nido PJ, Cowan DB, McCully JD. Actin‐dependent mitochondrial internalization in cardiomyocytes: evidence for rescue of mitochondrial function. Biol Open. 2015;4:622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kitani T, Kami D, Matoba S, Gojo S. Internalization of isolated functional mitochondria: involvement of macropinocytosis. J Cell Mol Med. 2014;18:1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mookerjee SA, Goncalves RLS, Gerencser AA, Nicholls DG, Brand MD. The contributions of respiration and glycolysis to extracellular acid production. Biochim Biophys Acta. 2015;1847:171–181. [DOI] [PubMed] [Google Scholar]
- 20. Divakaruni AS, Paradyse A, Ferrick DA, Murphy AN, Jastroch M. Chapter sixteen—analysis and interpretation of microplate‐based oxygen consumption and pH data In: Murphy AN, Chan DC, eds. Methods in Enzymology. New York, NY: Academic; 2014:309–354. [DOI] [PubMed] [Google Scholar]
- 21. Finck BN, Kelly DP. Peroxisome proliferator‐activated receptor gamma coactivator‐1 (PGC‐1) regulatory cascade in cardiac physiology and disease. Circulation. 2007;115:2540–2548. [DOI] [PubMed] [Google Scholar]
- 22. Hill BG, Benavides GA, Lancaster JR Jr, Ballinger S, Dell'Italia L, Jianhua Z, Darley‐Usmar VM. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem. 2012;393:1485–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J. 1972;128:617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD. Mitochondrial proton and electron leaks. Essays Biochem. 2010;47:53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Batandier C, Fontaine E, Kériel C, Leverve XM. Determination of mitochondrial reactive oxygen species: methodological aspects. J Cell Mol Med. 2002;6:175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Witek P, Korga A, Burdan F, Ostrowska M, Nosowska B, Iwan M, Dudka J. The effect of a number of H9C2 rat cardiomyocytes passage on repeatability of cytotoxicity study results. Cytotechnology. 2016;68:2407–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Dynamics of mitochondrial internalization. In this video clip, dynamics of mitochondrial internalization is visualized. Isolated mitochondria labeled with pHrodo Red succinimidyl ester (SE) are being internalized into rat H9c2 cardiomyocytes through coincubation. The pHrodo Red SE label is sensitive to pH drop and fluoresces as endocytosed by the cell. The lack of fluorescence outside of the cell eliminates any doubt about the specificity of the detected signal. At around hour 5, the first instance of mitochondrial internalization is observed. Fluorescence intensity progressively decreases as the dye is segregated between the new daughter mitochondrion with each round of fission. Occasionally, fluorescence intensity increases locally, indicating new instances of mitochondrial internalization in those areas. With passing time, pHrodo Red SE signal propagates through the entire cell, perhaps attributable to mitochondrial fission and fusion events between the transplanted and host mitochondria. The cell remains viable for the duration of the time‐lapse study. Best viewed with Windows Media Player.