

SUMMARY
Synaptic activity-induced calcium (Ca2+) influx and subsequent propagation into the nucleus is a major way in which synapses communicate with the nucleus to regulate transcriptional programs important for activity-dependent survival and memory formation. Nuclear Ca2+ shapes the transcriptome by regulating cyclic AMP (cAMP) response element-binding protein (CREB). Here, we utilize a Drosophila model of tauopathy and induced pluripotent stem cell (iPSC)-derived neurons from humans with Alzheimer’s disease to study the effects of pathogenic tau, a pathological hallmark of Alzheimer’s disease and related tauopathies, on nuclear Ca2+. We find that pathogenic tau depletes nuclear Ca2+ and CREB to drive neuronal death, that CREB-regulated genes are over-represented among differentially expressed genes in tau transgenic Drosophila, and that activation of big potassium (BK) channels elevates nuclear Ca2+ and suppresses tau-induced neurotoxicity. Our studies identify nuclear Ca2+ depletion as a mechanism contributing to tau-induced neurotoxicity, adding an important dimension to the calcium hypothesis of Alzheimer’s disease.
Graphical Abstract
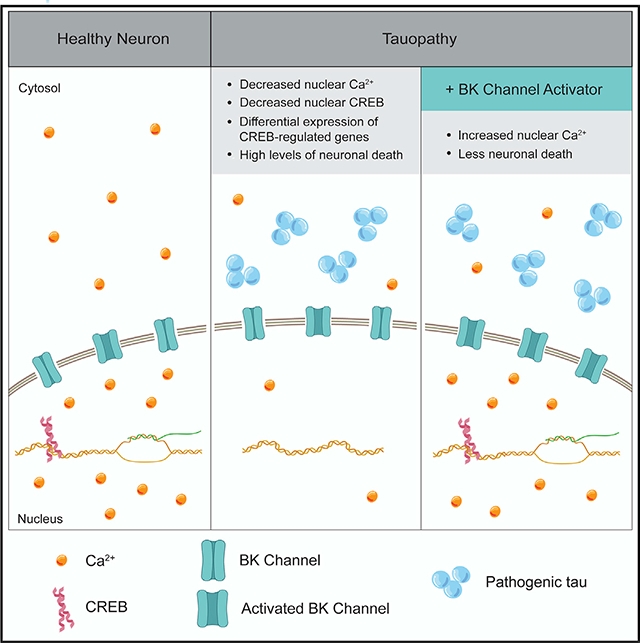
In Brief
Nuclear calcium (Ca2+) is a major mediator of communication between synapses and nuclei and is critical for CREB-dependent gene expression. Mahoney et al. identify nuclear Ca2+ depletion as a pathomechanism connecting disease-associated forms of tau to neuronal death, adding an important dimension to the long-standing Ca2+ hypothesis of Alzheimer’s disease.
INTRODUCTION
As a central signaling transducer, Ca2+ is integral to basic neuronal processes including membrane excitability and neuro-transmitter release from the synapse. In the nucleus, Ca2+ activates kinases that phosphorylate and thus activate CREB (Hardingham et al., 2001), a major transcriptional regulator of cellular programs critical for neuronal survival, plasticity, learning, and memory (Benito and Barco, 2010).
The long-standing “calcium hypothesis of Alzheimer’s disease” posits that Ca2+ dyshomeostasis is a major mediator of neuronal deterioration (Khachaturian, 1984). Neuropatho-logically, Alzheimer’s disease is defined by the presence of amyloid β plaques and neurofibrillary tau tangles in postmortem human brain samples (Braak and Braak, 1991). Although a significant decrease in CREB and pCREB levels has been reported in postmortem human Alzheimer’s disease brain tissue (Bartolotti et al., 2016; Bjorklund et al., 2012; Pugazhenthi et al., 2011), in primary hippocampal neurons from tau transgenic mice (Yin et al., 2016), and in β amyloid-based mouse models of Alzheimer’s disease (Gong et al., 2004; Pugazhenthi et al., 2011), no study to date has investigated nuclear Ca2+ in the context of Alzheimer’s disease and related tauopathies despite the well-established connection between nuclear Ca2+ and CREB activation (Hardingham et al., 2001).
To study potential links between pathogenic forms of tau and nuclear Ca2+, we utilized a Drosophila model of tauopathy and induced pluripotent stem cell (iPSC)-derived neurons from patients with sporadic Alzheimer’s disease. We selected a Drosophila model carrying a human tau transgene harboring the R406W disease-associated mutation (Wittmann et al., 2001). TauR406W is one of many mutations in the microtubule-associated protein tau (MAPT) gene that cause an autosomal dominant neurological disorder termed frontotemporal lobar degeneration (FTLD)-tau with MAPT mutation (Forrest et al., 2018; Hutton et al., 1998). In Drosophila, similar mechanisms of tau-induced toxicity are shared by transgenic expression of various disease-associated tau mutations, which model human FTLD-tau with MAPT mutation, and wild-type human tau, which models Alzheimer’s disease-associated tauopathy and other primary tauopathies not attributable to MAPT mutation (Bardai et al., 2018). The tauR406W Drosophila model has been used widely to study tau biology due to its mild toxicity at day 10 of adulthood, which is convenient for genetic analyses and precedes exponential decline in survival. To determine if our findings in tauR406W transgenic Drosophila were relevant to the wider group of human tauopathies that involve pathogenic forms of wild-type tau, we extended our studies to iPSC-derived neurons from patients with sporadic Alzheimer’s disease.
We report that panneuronal expression of human tauR406W in the adult Drosophila brain is sufficient to deplete nuclear CREB protein levels, suggesting that pathogenic forms of tau may contribute to the previously reported nuclear depletion of CREB/pCREB in neurons of post-mortem human Alzheimer’s disease brains (Bartolotti et al., 2016; Bjorklund et al., 2012; Pugazhenthi et al., 2011). We find that genes previously identified as CREB-regulated are over-represented among transcripts that are depleted in tauR406W transgenic Drosophila, suggesting that tau-induced CREB reduction significantly affects the transcriptome. We look upstream of CREB to find that both resting levels of nuclear Ca2+ and KCl-induced influx of nuclear Ca2+ are reduced as a result of human tauR406W expression in the adult Drosophila brain. We find that nuclear Ca2+ influx in response to membrane depolarization is also blunted in iPSC-derived neurons from patients with sporadic Alzheimer’s disease, suggesting that our studies in Drosophila are relevant to sporadic human tauopathies that involve pathogenic forms of wild-type tau. Finally, our studies in Drosophila identify the BK channel as a pharmacologically targetable modifier of nuclear Ca2+ signaling and neuronal death in tauopathy. Taken together, our findings highlight a key role for nuclear Ca2+ and CREB depletion in the pathogenesis of Alzheimer’s disease and related tauopathies.
RESULTS
Pathogenic TauR406W Induces Nuclear CREB Depletion in Neurons of the Adult Drosophila Brain
Previous studies report that levels of total and nuclear CREB and pCREB are reduced in postmortem human Alzheimer’s disease brains (Bartolotti et al., 2016; Bjorklund et al., 2012; Pugazhenthi et al., 2011). To determine if pathogenic forms of tau can contribute to nuclear CREB depletion, we utilized a well-described Drosophila model of tauopathy (Wittmann et al., 2001). Transgenic expression of human tauR406W in Drosophila neurons recapitulates many aspects of human Alzheimer’s disease and related tauopathies including the degeneration of synapses (Merlo et al., 2014), ectopic cell cycle activation (Khurana et al., 2006), DNA damage (Frost et al., 2014; Khurana et al., 2012), and progressive neuronal death (Khurana et al., 2006; Wittmann et al., 2001).
To directly quantify the effects of pathological tau on the Drosophila homolog of human CREB, CrebB (Usui et al., 1993; Yin et al., 1995) (referred to throughout as “CREB” for simplicity), we performed western blotting on lysates from tauR406W transgenic Drosophila heads at day 10 of adulthood, an age at which neurodegeneration is detectable, but prior to exponential decline in lifespan (Frost et al., 2016). Using an antibody that detects all CREB isoforms, we find that total CREB levels are depleted in heads of tauR406W transgenic Drosophila versus controls (Figure 1A). We next directly visualized CREB localization by co-staining control and tauR406W Drosophila brains with antibodies detecting CREB and elav, a protein restricted to neuronal nuclei. Similar to previous reports in postmortem human brains with Alzheimer’s disease (Bartolotti et al., 2016; Bjorklund et al., 2012; Pugazhenthi et al., 2011), we find that total CREB (Figure 1B) and nuclear CREB (Figure 1C) are significantly depleted in brains of adult tauR406W transgenic Drosophila.
Figure 1. TauR406W Causes Reduction of Nuclear CREB in Neurons of the Adult Drosophila Brain.
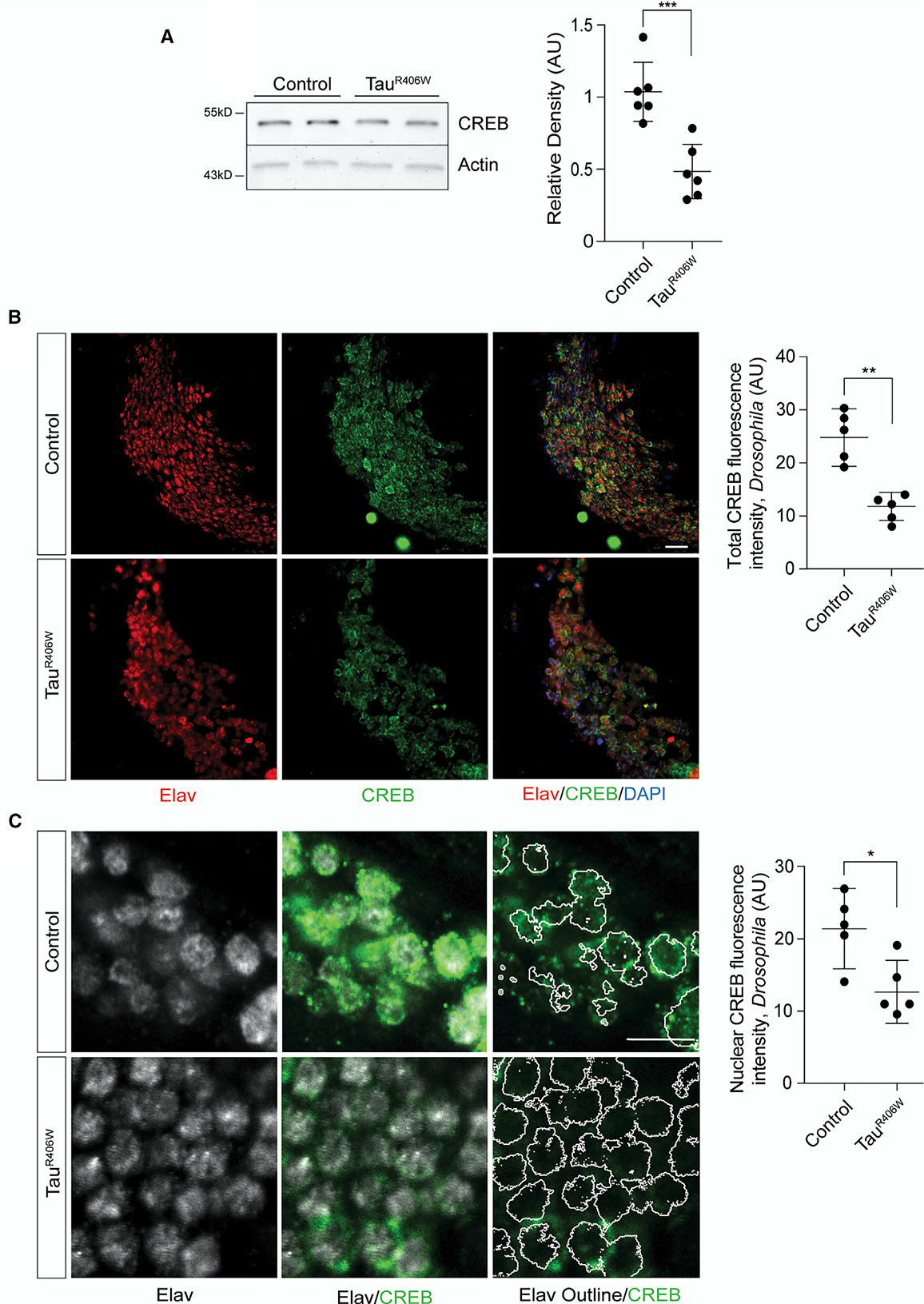
(A) CREB protein levels in control and tauR406W transgenic Drosophila head lysates based on western blotting, n = 6 biological replicates.
(B) CREB and elav immunostaining in the mushroom body of control and tauR406W transgenic Drosophila visualized by confocal microscopy. Images are from a single focal plane. n = 5 biological replicates. Scale bar, 5 μm.
(C) CREB and elav immunostaining in the mushroom body of control and tauR406W transgenic Drosophila visualized by confocal microscopy. Images are from a single focal plane. Elav-based masks (represented by white outlines) were used to measure nuclear CREB levels, n = 5 biological replicates. Scale bar, 5 mm.
All assays were performed at day 10 of adulthood. Data are presented as mean ± SEM; unpaired t test; *p < 0.05, **p < 0.01, ***p < 0.001.
CREB-Regulated Genes Are Over-Represented among Differentially Expressed Genes in TauR406W Transgenic Drosophila
As CREB is a transcription factor that is depleted in tauR406W transgenic Drosophila, we next determined if CREB-regulated genes are over-represented among transcripts that are differentially expressed in brains of tauR406W transgenic Drosophila. Genes that harbor a CREB-response element (CRE) between 3,000 bp upstream and 500 bp downstream of their transcription start site that have previously been identified as CREB targets based on chromatin immunoprecipitation sequencing (ChIP-seq) (Hirano et al., 2016; Data S1) were considered “CREB-regulated.” The antibody used for ChIP-seq recognizes both activating and repressive isoforms of Drosophila CREB (Hirano et al., 2016). Genes that are differentially expressed between control and tauR406W transgenic Drosophila at day 10 of adulthood were identified by RNA sequencing (Data S2). A hypergeometric test indicated that CREB-regulated genes are significantly over-represented among genes that are upregulated (1.84-fold enrichment, p = 3.77E–06) and downregulated (1.55-fold enrichment, p = 0.00046) in tauR406W transgenic Drosophila compared to control (Data S3, gene list and Gene Ontology [GO] analysis). Although we cannot conclude that differential expression of these genes is a direct consequence of CREB depletion, this finding is consistent with the hypothesis that tauR406W-induced CREB depletion significantly affects the transcriptome.
Physiological Aging and TauR406W Cause a Toxic Depletion of Nuclear Ca2+ in the Drosophila Brain
Given the dependence of CREB-mediated transcription on the presence of nuclear Ca2+ (Hardingham et al., 2001), we next determined the effect of pathological tauR406W on nuclear Ca2+ using a GFP-based genetically encoded Ca2+ indicator fused to a nuclear localization signal, GCaMP3.NLS (Weislogel et al., 2013). Upon binding to Ca2+, genetically encoded Ca2+ indicators undergo a conformational change that induces fluorescence (Nakai et al., 2001). We focused specifically on the cells of the mushroom body of the adult fly brain, as activation of the nuclear Ca2+ reporter can be visualized in this brain region in living flies (Weislogel et al., 2013; Figure 2A), and the mushroom body is central to Drosophila learning and memory (Heisenberg, 2003). In vivo confocal imaging reveals that tauR406W transgenic Drosophila have significantly lower resting levels of nuclear Ca2+ in the cells of the mushroom body compared to controls at day 10 of adulthood (Figure 2B). Importantly, we found that decreased nuclear Ca2+ levels are not simply a result of extensive neuronal loss (Figure S1A). As an important control, we confirmed that transgenic human tauR406W does not affect expression levels of the genetically encoded Ca2+ indicator itself (Figure S1B).
Figure 2. TauR406W Transgenic Drosophila Have a Toxic Reduction of Nuclear Ca2+.
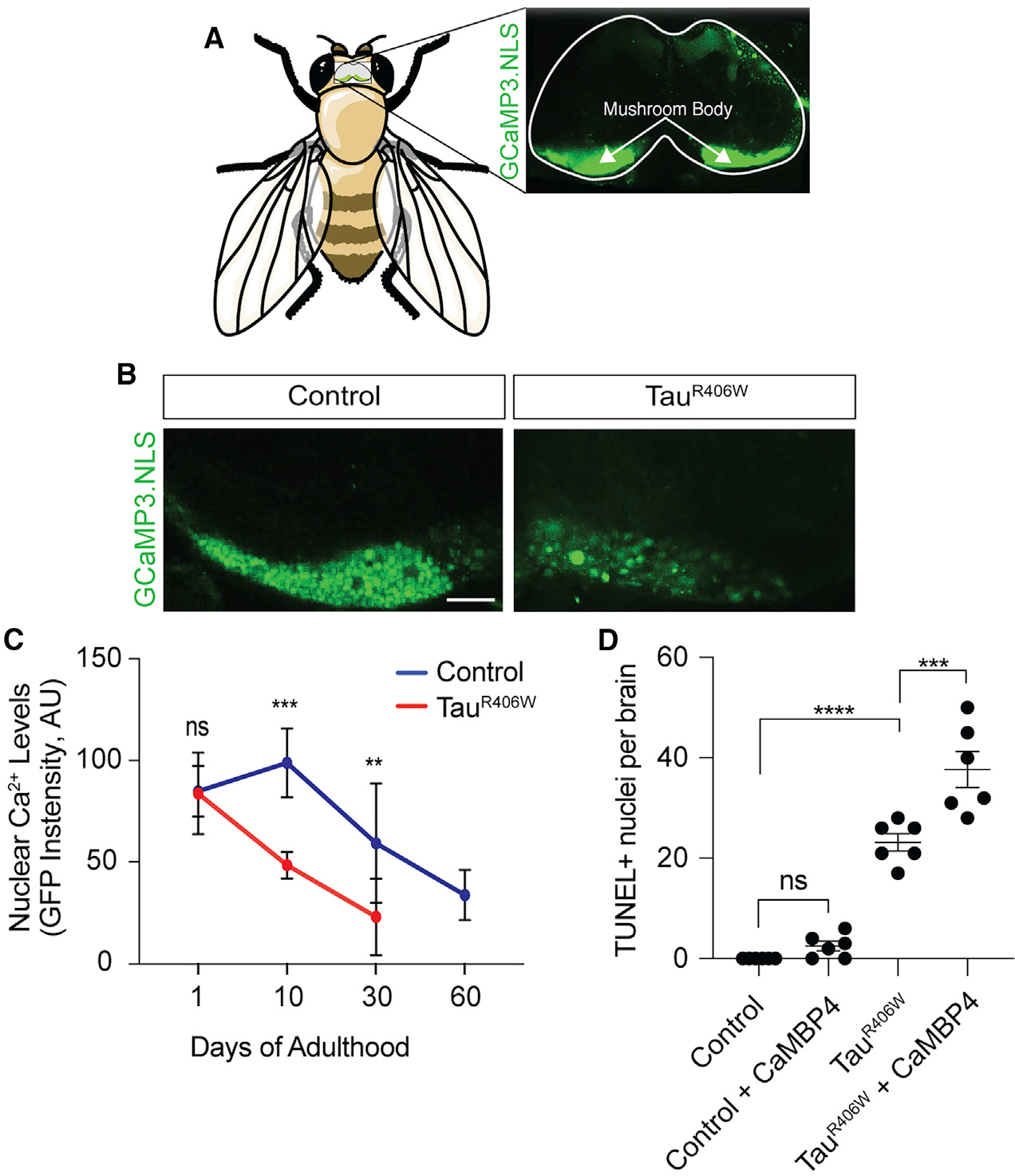
(A) Activation of the nuclear Ca2+ reporter in the mushroom body of the adult Drosophila brain based on GCaMP3.NLS in vivo imaging.
(B) Decreased levels of nuclear Ca2+ in the mushroom body of the tauR406W transgenic Drosophila brain versus control based on GCaMP3.NLS in vivo imaging. Images are of a single focal plane. Scale bar, 60 μm
(C) Quantification of nuclear Ca2+ based on GCaMP3.NLS in control and tauR406W transgenic Drosophila of the indicated age. n = 6–8 biological replicates per genotype, per age. Data are presented as mean ± SD. For visual simplicity, significance is only noted for differences between genotypes at each age. Statistical analyses of the age-dependent decline in nuclear Ca2+ within each genotype are presented in Figures S1C and S1D.
(D) Neurodegeneration assayed by TUNEL staining in brains of control and tauR406W transgenic Drosophila with and without nuclear Ca2+ blockage via panneuronal overexpression of CaMBP4.
All assays were performed at day 10 of adulthood with the exception of (C). Data are presented as mean ± SEM unless otherwise noted. One-way ANOVA with Tukey’s multiple comparison test; ***p < 0.001, ****p < 0.0001.
To determine if tauR406W-induced nuclear Ca2+ depletion is age-dependent, we quantified resting levels of nuclear Ca2+ at day 1, 10, and 30 of adulthood in control and tauR406W transgenic Drosophila. We extended our analysis to 60 days in control flies, which is close to their maximum lifespan of ~70 days, and exceeds the maximum lifespan of tauR406W transgenic Drosophila of ~35 days (Frost et al., 2016). In both genotypes, we find a significant age-dependent decrease in levels of resting nuclear Ca2+ (Figures S1C and S1D). Although nuclear Ca2+ levels do not significantly differ between control and tauR406W transgenic Drosophila at day 1 of adulthood, we find that tauR406W transgenic Drosophila have reduced levels of nuclear Ca2+ compared to controls at days 10 and 30 (Figure 2C), indicating that tauR406W exacerbates the depletion of resting nuclear Ca2+ levels that occurs with normal aging.
To determine if depletion of nuclear Ca2+ signaling is causally associated with neurodegeneration, we overexpressed a recombinant blocker of nuclear Ca2+, CaMBP4 (Weislogel et al., 2013), in neurons of the adult Drosophila brain. CaMBP4 is a nuclear protein that contains four copies of a calmodulin-binding peptide (M13). CaMBP4 binds to and inactivates Ca2+-bound calmodulin complexes, thus blocking activation of Ca2+-dependent nuclear signaling cascades. Although blocking nuclear Ca2+-dependent processes via CaMBP4 overexpression is not sufficient to induce neuronal death at day 10 of adulthood based on terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), we find that genetically blocking nuclear Ca2+ signaling in tauR406W transgenic Drosophila significantly enhances tauR406W-induced neuronal death (Figure 2D). Taken together, these data suggest that tauR406W-induced decrease in nuclear Ca2+ signaling is causally associated with neurodegeneration.
Decreased Influx of Nuclear Ca2+ in TauR406W Transgenic Drosophila and iPSC-Derived Neurons from Sporadic Human Alzheimer’s Disease Patients in Response to Membrane Depolarization
Ca2+ is a central regulator of communication between the synapse and nucleus, and Ca2+ that enters the nucleus in response to synaptic activity mediates memory formation (Bading, 2013). Having established that resting levels of nuclear Ca2+ are depleted as a consequence of pathogenic tauR406W, we next determined if influx of Ca2+ into the nucleus is affected in tauopathy when the membrane is induced to depolarize. We quantified nuclear Ca2+ upon in vivo brain exposure to 70 mM KCl, which induces membrane depolarization and opening of voltage-gated Ca2+ channels (Fiala and Spall, 2003). After normalizing to resting nuclear Ca2+ levels, we find that KCl-induced nuclear Ca2+ influx is significantly depleted in brains of tauR406W transgenic Drosophila at day 10 of adulthood compared to control (Figures 3A and 3B). Together with our previous findings, these data indicate that both resting levels and membrane depolarization-induced nuclear Ca2+ influx are depleted as a consequence of pathogenic tauR406W.
Figure 3. KCl-Induced Nuclear Ca2+ Influx Is Reduced in Brains of TauR406W Transgenic Drosophila and in iPSC-Derived Neurons from Sporadic Cases of Human Alzheimer’s Disease.
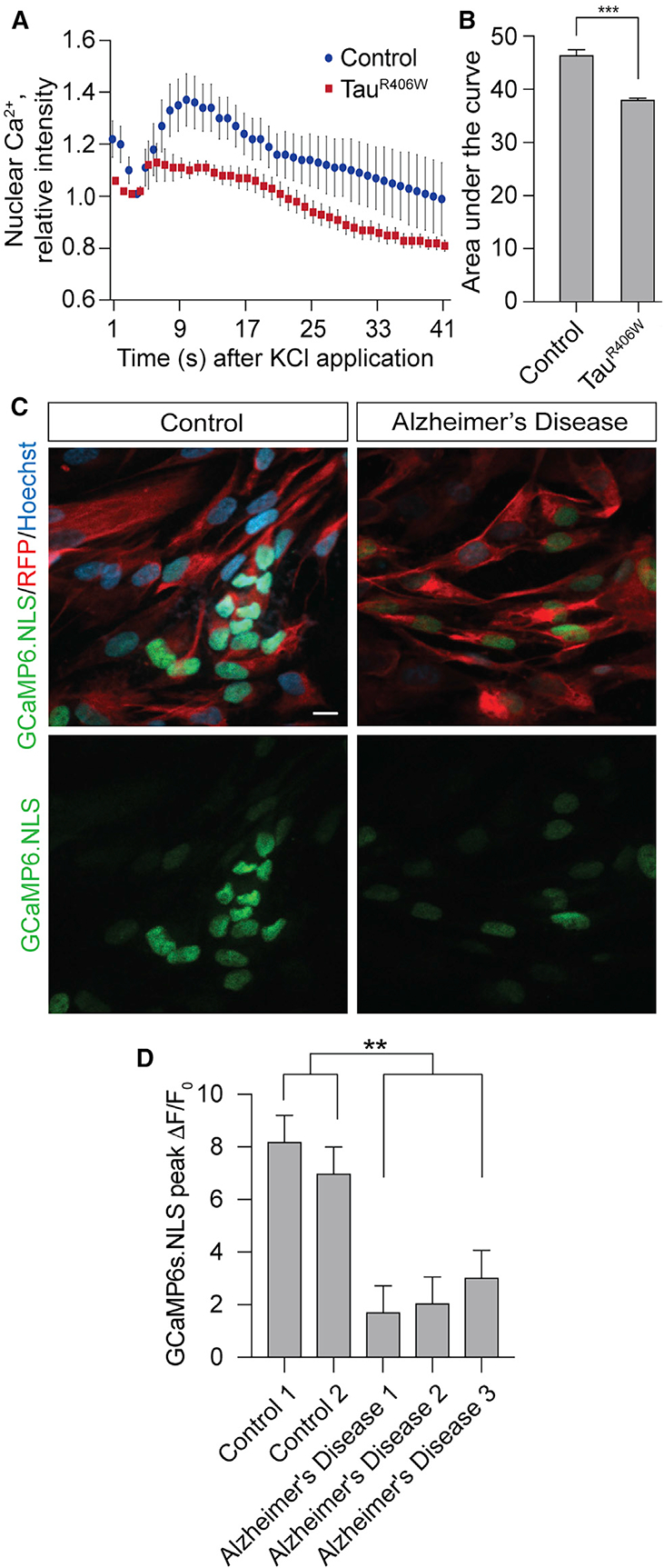
(A) Decreased depolarization-dependent influx of nuclear Ca2+ in tauR406W transgenic Drosophila compared to control in response to administration of 70 mM KCl through a cuticular window in heads of living Drosophila.
(B) Quantification of the area under the curve from (A), n = 6 biological replicates.
(C) Decreased KCl-induced release of nuclear Ca2+ in iPSC-derived neurons from patients with Alzheimer’s disease. Cells were transfected with membrane-bound RFP and the GCaMP6s.NLS nuclear Ca2+ reporter. Images show peak nuclear Ca2+ levels induced by 25 mM KCl. Images are from a single focal plane. Scale bar, 10 μm.
(D) Quantification of (C). Data are presented as peak ΔF/F0, in which ΔF is the change in GCaMP6s.NLS GFP fluorescence, and F0 is baseline GFP fluorescence. Nuclear Ca2+ was quantified in at least 50 single cells for each of six technical replicates per human sample. iPSC-derived neurons are from two control and three sporadic Alzheimer’s disease patients.
All assays in Drosophila were performed at day 10 of adulthood. Data are presented as the mean ± SEM; unpaired t test or ANOVA; **p < 0.01, ***p < 0.001.
We next utilized iPSC-derived neurons from patients with sporadic Alzheimer’s disease to determine if our findings were relevant to a human tauopathy that involves pathogenic forms of wild-type tau. As in brains of patients with Alzheimer’s disease, iPSC-derived neurons from patients with Alzheimer’s disease are reported to feature disease-associated tau phosphorylation (Israel et al., 2012; Ochalek et al., 2017). After differentiating iPSCs into excitatory forebrain neurons (Chambers et al., 2009; Reddy et al., 2016; Sproul et al., 2014), we quantified membrane depolarization-induced changes in nuclear Ca2+ levels using a GCaMP6s.NLS genetically encoded nuclear Ca2+ sensor (Hagenston and Bading, 2011). We do not observe differences in differentiation status between iPSC-derived neurons from control and Alzheimer’s disease patients (Figure S2). We find that KCl-induced increase of nucleoplasmic Ca2+ is reduced in iPSC-derived neurons from three different sporadic cases of human Alzheimer’s disease versus controls (Figures 3C and 3D), suggesting that the blunting of KCl-induced nuclear Ca2+ influx detected in tauR406W transgenic Drosophila is relevant to human Alzheimer’s disease and is not restricted to the R406W tau mutation.
Manipulation of BK Channels Modifies TauR406W-Induced Nuclear Ca2+ Reduction and Neurotoxicity
We became interested in BK channels as a potential mechanistic link between pathogenic tau and nuclear Ca2+ depletion based on a previous study reporting that BK channels regulate induced release of Ca2+ from nuclear envelope stores (Li et al., 2014). We find that oral administration of a potent activator of BK channels, BMS-191011, significantly increases resting levels of nuclear Ca2+ in cells of the mushroom body of the adult tauR406W transgenic Drosophila brain (Figures 4A and 4B). In addition, we find that BMS-191011 significantly reduces neurodegeneration in tauR406W transgenic Drosophila at day 10 of adulthood (Figure 4C).
Figure 4. BK Channels Modify Nuclear Ca2+ Release and Neurotoxicity in TauR406W Transgenic Drosophila.
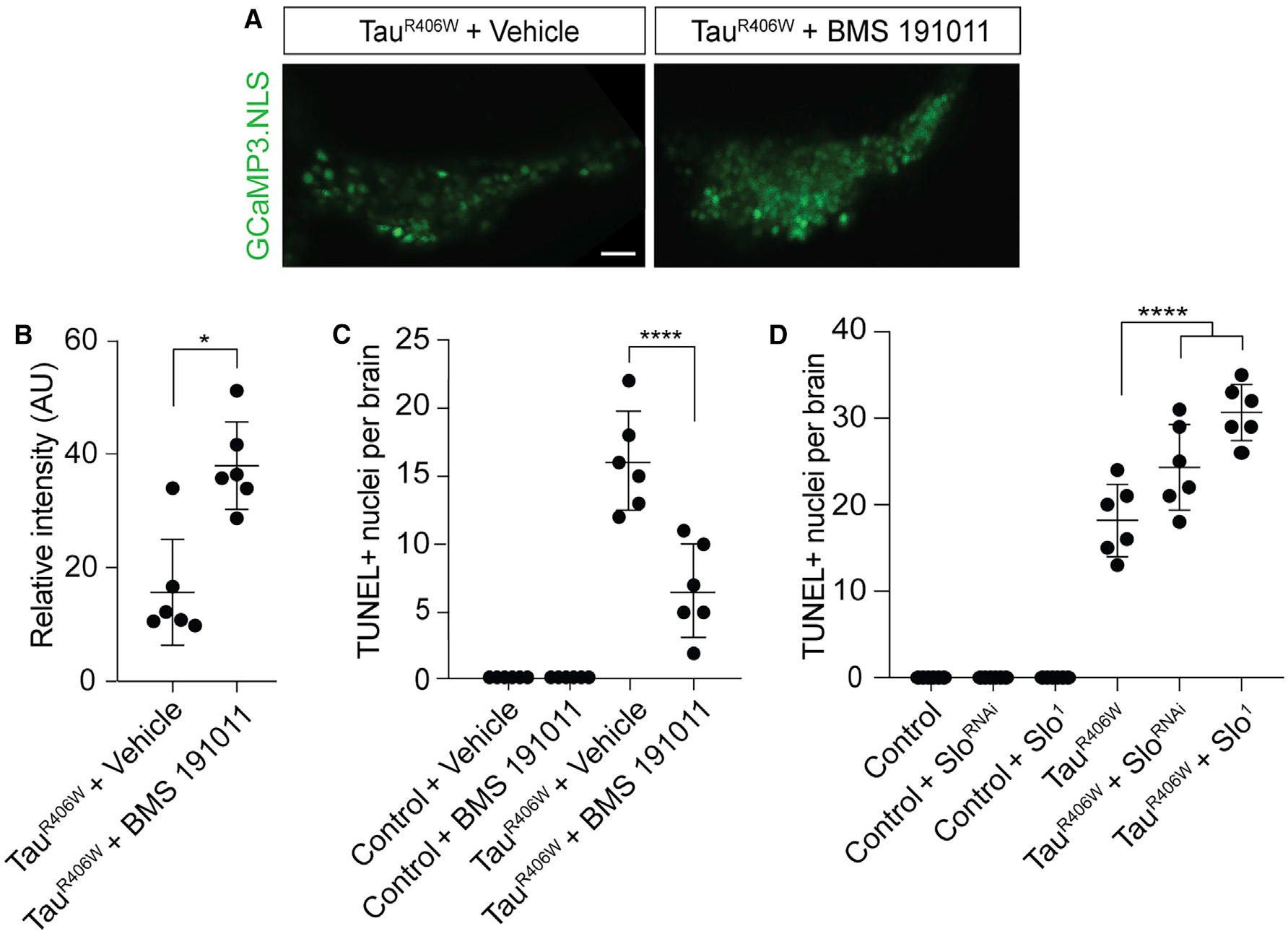
(A) Visualization of nuclear Ca2+ based on in vivo imaging of GCaMP3.NLS in tauR406W transgenic Drosophila fed either vehicle or BMS-191011 from days 2–10 of adulthood. Images are from a single focal plane. Scale bar, 10 μm.
(B) Quantification of (A), n = 6 biological replicates.
(C) Neurodegeneration assayed by TUNEL staining in the brains of control and tauR406W transgenic Drosophila with and without exposure to BMS-191011 from days 2–10 of adulthood, n = 6 biological replicates.
(D) Neurodegeneration assayed by TUNEL staining in the brains of control and tauR406W transgenic Drosophila with and without RNAi-mediated depletion or loss-of-function of slowpoke; n = 6 biological replicates.
All assays were performed at day 10 of adulthood. Data are presented as mean ± SEM; unpaired t test or ANOVA; *p < 0.05, ****p < 0.0001.
Having established that manipulation of BK channels is sufficient to increase nuclear Ca2+ and suppress neurodegeneration in tauR406W transgenic Drosophila, we next determined if genetic depletion of the Drosophila BK channel homolog, slowpoke, enhances tauR406W-induced neurodegeneration. We decreased slowpoke activity by RNAi-mediated reduction (sloRNAi) or introduction of a heterozygous loss-of-function mutation (slo1). Although neither genetic manipulation is sufficient to induce neuronal death based on TUNEL staining at day 10 of adulthood, we find that both sloRNAi and slo1 significantly enhance tauR406W-induced neuronal death (Figure 4D). Taken together, these data suggest that manipulation of BK channels can modify tauR406W-induced nuclear Ca2+ reduction and consequent neuronal death.
DISCUSSION
In the current study, we investigate the effects of pathogenic tau on nuclear Ca2+ and CREB. Our studies suggest that pathogenic tau directly contributes to CREB depletion, as we find that panneuronal expression of human transgenic tauR406W in the adult Drosophila brain is sufficient to reduce total and nuclear levels of CREB protein. Based on RNA sequencing, we detect a significant over-representation of CREB-regulated genes among transcripts that are differentially expressed in tauR406W transgenic Drosophila compared to control. As differential splicing produces both activating and repressive CREB isoforms in Drosophila (Yin et al., 1995), the overrepresentation of CREB-regulated genes that are both up- and downregulated in tauR406W transgenic Drosophila is not unexpected. Although these data are consistent with the role of CREB as a key cellular transcription factor, additional studies are required to determine if the transcriptional changes in tauR406W transgenic Drosophila are a direct result of CREB depletion.
Based on the dependence of CREB activation on nuclear Ca2+, we then visualized nuclear Ca2+ levels in neurons of live Drosophila brains using a genetically encoded, nuclear-localized Ca2+ indicator. The ability to quantify nuclear Ca2+ levels as a function of biological aging is an advantage of the Drosophila system. We found that resting-state nuclear Ca2+ levels are depleted with physiological aging, and that pathogenic tauR406W significantly exacerbates age-associated nuclear Ca2+ depletion. We found that genetic blockage of nuclear Ca2+ signaling further enhances tauR406W-induced neuronal death, suggesting that nuclear Ca2+ depletion is a causal mediator of neurodegeneration in tauopathy.
As generation of nuclear Ca2+ transients are a key route of communication between synapses and nuclei, we next asked if KCl-induced nuclear Ca2+ influx is depleted in the context of tauopathy. Using tauR406W transgenic Drosophila as well as iPSC-derived neurons from patients with sporadic Alzheimer’s disease, we find that the nuclear Ca2+ response to KCl-induced depolarization is blunted in both model systems. Our studies in tauR406W transgenic Drosophila and in human Alzheimer’s disease iPSC-derived neurons suggest that depletion of nuclear Ca2+ is neither specific to the Drosophila system nor R406W mutant tau.
We identify BK channels as a potential pharmacologically targetable link between pathogenic tau and nuclear Ca2+ depletion. Treatment of tauR406W transgenic Drosophila with a BK channel activator increases nuclear Ca2+ levels and suppresses tauR406W neurotoxicity, while genetically depleting BK channels significantly enhances tauR406W neurotoxicity. A previous study has reported that BK channels are present in the nuclear envelope and regulate nuclear Ca2+ levels, nuclear Ca2+ signaling, and activity-evoked gene expression (Li et al., 2014). While we cannot rule out the possibility that BK channels on the plasma membrane contribute to nuclear Ca2+ regulation in neurons of tauR406W transgenic Drosophila, we would expect that activation of BK channels on the plasma membrane would hyperpolarize the membrane, preventing further Ca2+ influx into the cytoplasm. We thus speculate that nuclear envelope-localized BK channels, rather than plasma membrane-localized BK channels, are the primary contributor to nuclear Ca2+ depletion in tauopathy by influencing Ca2+ stores in the nuclear envelope.
Pharmacological blockade of nuclear BK channels was previously reported to elevate nuclear Ca2+ in isolated neuronal nuclei and in cultured mouse hippocampal neurons (Li et al., 2014), which conflicts with our finding that that activation of BK channels elevates nuclear Ca2+ in brains of tauR406W transgenic Drosophila. Several differences between our respective experimental designs may underlie the discrepancy between studies. First, Li et al. (2014) analyzed nuclear Ca2+ in isolated nuclei and cultured neurons, whereas our live-imaging measurements of nuclear Ca2+ utilize intact Drosophila brains. Second, we analyzed levels of resting nuclear Ca2+ in tauR406W transgenic Drosophila in response to chronic exposure to the BK channel activator throughout adulthood, whereas Li et al. (2014) measured nuclear Ca2+ influx in response to transient BK channel blockage. Despite divergent findings between the two studies, both point toward a critical role of BK channels as a regulator of nuclear Ca2+. Our study is consistent with that of Wang et al. (2015a, 2015b), who find that drug-induced BK channel activation suppresses cognitive deficits in the 3xTg mouse model of Alzheimer’s disease, which harbors APP, PS1, and MAPT disease-associated mutant human transgenes (Oddo et al., 2003).
Why might depletion of nuclear Ca2+ be toxic to neurons? In addition to regulating a neuroprotective genetic program consisting of synaptic activity-induced “activity-regulated inhibitor of death” genes (Zhang et al., 2009), nuclear Ca2+ has been identified as a key regulator of the autolysosomal system (Reddy et al., 2016). As the autophagy-lysosome system is clearly dysfunctional in tauopathy (Uddin et al., 2018), determining the effects of tau-induced nuclear Ca2+ depletion on protein clearance pathways is an important avenue of investigation for future studies.
In summary, our study provides insight into the effects of pathogenic tau on nuclear Ca2+, which is a major mediator of communication between synapses and nuclei (Bading, 2013) and regulator of protein clearance pathways (Reddy et al., 2016). We identify nuclear Ca2+ depletion as a pathomechanism connecting disease-associated forms of tau to neuronal death, adding an important dimension to the long-standing Ca2+ hypothesis of Alzheimer’s disease.
STAR⋆ METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Bess Frost (bfrost@uthscsa.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The accession number of the raw RNA-seq files for Drosophila control versus tauR406W transgenic Drosophila reported in this paper is GEO: GSE152278.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila genetics and models
All Drosophila melanogaster crosses and aging were performed at 25°C on a 12-hour light/dark cycle. Males and females of indicated genotypes were housed in the same vial, and each experiment utilized an equal number of male and female flies. Food was made fresh weekly and flies were transferred to fresh food every two days. Transgenic flies harboring human tauR406W have been described previously (Wittmann et al., 2001). Panneuronal expression of transgenes or RNAi small hairpins were achieved using the Gal4/UAS system with the elav promoter driving expression of the Gal4 transcription factor. UAS-sloRNAi and slo1 were obtained from the Bloomington Drosophila Stock Center. UAS-GCaMP3.NLS and CaMBP4 transgenic flies were generously provided by Dr. Hilmar Bading (Weislogel et al., 2013).
iPSC-derived neurons
iPSCs from sporadic Alzheimer’s disease patients (no ApoE4 carriers) and control lines were obtained from the Coriell Institute (Camden, NJ). Neural progenitor cells were derived following established protocols (Chambers et al., 2009) and differentiated into forebrain neurons by stepwise addition (daily half-feeds for one week) of neurodifferentiation media composed of Neurobasal Medium supplemented with B-27 minus retinoic acid, Glutamax and Pen/Strep as described (Reddy et al., 2016; Sproul et al., 2014). The resulting excitatory forebrain neurons are cultured for another three weeks to allow further differentiation, which is monitored by expression of neuronal markers including MAP2 and vGluT1 (Figure S2).
METHOD DETAILS
RNA sequencing and analysis
6 biologically independent replicates were sequenced per genotype, each consisting of 15–30 ng of total RNA from 18 pooled Drosophila heads (108 heads per genotype in total). Trizol-extracted RNA was used for library preparation using the Ovation RNA-Seq System for Drosophila according to the User Guide. After quantification by Qubit and bioanalysis, libraries were pooled, purified by magnetic bead extraction and sequenced on the Illumina HiSeq 3000 platform with 100 base pair paired-end sequencing. Library quality control and RNA-sequencing was performed by the Genome Sequencing Facility at Greehey Children’s Cancer Research Institute at the University of Texas Health San Antonio.
Raw FASTQ files underwent quality control and were trimmed with Trimmomatic v.0.36 (Bolger et al., 2014) to remove adapters and low-quality reads. FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to evaluate the quality of the reads before and after trimming. Trimmed FASTQ files were mapped and aligned to the Drosophila melanogaster transcriptome (FlyBase (Thurmond et al., 2019) FB2018_6.27) using Salmon v.0.13.1 (Patro et al., 2017). Differential expression analysis was performed using DESeq2 v1.24 (Love et al., 2014). Genes with an adjusted p value of less than 0.05 were considered significant.
CRE and CREB binding-site analyses
Genes harboring a CRE were identified through the FindM program (Ambrosini et al., 2003; Bucher and Trifonov, 1986). The canonical full CRE site and the CRE half site were included. One mismatch was allowed for full sites, and no mismatches were allowed for half sites. CRE prediction sites that were between 3,000 bp upstream and 500 bp downstream of annotated genomic transcription start sites were utilized in subsequent analyses.
To validate predicted CREB target sites, CREB ChIP-seq data were downloaded from the Gene Expression Omnibus (GEO: GSE73386, samples GSM1892406 and GSM1892408) (Edgar et al., 2002; Hirano et al., 2016) and compared to predicted CRE sites. Files were converted into GRanges format and annotated with ChIPpeakAnno (Zhu et al., 2010). CREB ChIP-seq peaks that did not fall between 3,000 bp upstream and 500 bp downstream of an annotated TSS were discarded. The intersection between FindM-based CRE-containing genes, ChIP-seq-based CREB-bound genes, and genes that were up and downregulated in tauR406W transgenic Drosophila compared to control (adjusted p < 0.05) were extracted. A hypergeometric test was used to determine enrichment of CRE-containing, CREB-regulated genes in the lists of up and downregulated genes. GO analysis was performed on the CRE-containing, CREB-regulated genes that were up and downregulated in tauR406W transgenic Drosophila using the GO Enrichment Analysis tool (Ashburner et al., 2000), which utilizes the Drosophila melanogaster genome as a background gene set (Data S3). GO annotations with a false discovery rate (FDR) of less than 0.05 were considered significant.
Ca2+ imaging
To quantify resting nuclear Ca2+ levels in brains of living flies (Figures 2A–2C, 4A, and 4B), a single fly was placed on a CO2 gas pad until the fly lost postural control, then transferred into a 100% ethanol bath for 10 s. Using a small Sylgard dissection surface, the fly was then placed in cold HL3 solution (70 mM NaCl, 5 mM KCl, 10 mM NaHCO3, 5 mM trehalose, 115 mM sucrose, 5 mM HEPES, 0.5 mM CaCl2, 3 mM MgCl2) and pinned using modified minutien pins on its ventral surface (Mahoney et al., 2014). Once positioned, a small piece of cuticle was removed from the posterior side of the head (cuticular window) to reveal the underlying mushroom body (Weislogel et al., 2013). GFP fluorescence resulting from GCaMP3.NLS activation was imaged with a Zeiss LSM 780 NLO with Examiner. ImageJ was used for analysis. Six biological replicates were analyzed per group.
To quantify the nuclear Ca2+ response to KCl-induced membrane depolarization (Figures 3A and 3B), flies were first prepared for imaging in a cold HL3 solution as described above, and resting GCaMP3.NLS fluorescence levels were recorded. HL3 was removed from the exposed fly brain by pipetting and was immediately replaced with a modified HL3 solution containing 70 mM KCl. GCaMP3.NLS reporter intensity stabilizes a few seconds after KCl exposure, as flies experience some movement within the imaging system as a result of the physical administration of the buffer. After a three second recovery, baseline intensity was set to one for both control and tauR406W transgenic Drosophila. The KCl-induced nuclear Ca2+ response is presented as change from baseline (Weislogel et al., 2013). Six biological replicates were analyzed per group.
In iPSC-derived neurons, nuclear Ca2+ levels were measured using the human Synapsin 1 promoter-driven GCaMP6s.NLS (Hagenston and Bading, 2011), ensuring expression exclusively in neurons. This genetically encoded nuclear Ca2+ reporter was transfected into iPSC-derived human neurons using BioT. To assess transfection efficiency and simplify visualization of transfected cells during the experiment, cells were co-transfected with membrane-RFP (mRFP) in addition to GCaMP6s.NLS. While mRFP labels all transfected cells, GCaMP6s.NLS fluorescence is restricted to neurons and is induced following KCl-mediated depolarization. mTagRFP-Membrane-1 was a gift from Michael Davidson (Addgene plasmid #57992).
Baseline fluorescence and KCl (25 mM)-induced GCaMP6s.NLS fluorescence were measured by spinning disc confocal microscopy over time, and peak fluorescence intensities were recorded. Minima and maxima intensities were normalized to 0 or 1, respectively. Data are presented as peak ΔF/F0, in which ΔF is the change in fluorescence and F0 is baseline fluorescence. Cells with saturating F0 fluorescence are excluded from experimental measurements. To avoid measuring nuclear Ca2+ in cells that have no or very low expression of the GCaMP6s.NLS nuclear Ca2+ indicator, the microscopy field of view is set such that all cells within the field of view exhibit a baseline GFP signal. ΔF/F0 peak values presented in Figure 3D represent the maximum values of longitudinal measurements (ΔF/F0 over time). Longitudinal measurements did not plateau at their maxima at any point in time, indicating that the signal was not saturated. A technical replicate consists of the average signal from at least 50 single cells derived from one patient sample. There were six technical replicates assayed per patient-derived sample, with two biologically distinct control samples and three biologically distinct sporadic Alzheimer’s disease samples.
Drug Preparation
BMS 191011 was prepared as a stock solution in ethanol and diluted in fly food to a final concentration of 20 μM. Vehicle-treated flies were reared on food containing an equivalent volume of ethanol. Flies were treated from day 2–10 of adulthood.
Western blotting
Frozen Drosophila heads were homogenized in 20 μL 2X Laemmli sample buffer, boiled for 10 min, and run on a 4%–20% SDS-PAGE gel. Equal loading of protein was assessed by Ponceau S staining prior to blotting. After blocking membranes in PBS plus 0.05% Tween (PBSTW) and 2% milk, membranes were incubated with primary antibodies overnight at 4°C. After washing in PBSTW, membranes were incubated with their respective HRP-conjugated secondary antibodies for 2 hr at room temperature. Blots were developed with Clarity Max ECL Western Blotting Substrate. Band intensity was quantified with ImageJ. Antibodies against actin and GFP were used at 1:10,000 for western blotting, and CREB was used at 1:500.
Immunofluorescence and histology
For Drosophila studies, Drosophila brains were dissected in PBS and fixed in methanol for 20 min. After blocking with 2% milk PBSTr for 30 minutes, brains were incubated with primary antibody diluted in blocking solution overnight at 4°C. After washing with PBSTr, brains were incubated with Alexa488- or Alexa555-conjugated secondary antibodies for 2 hr at room temperature in 2% milk dissolved in PBST. Slides were washed with PBSTr and then incubated with DAPI for 2 minutes to visualize nuclei. Brains were imaged by confocal microscopy (Zeiss LSM 780 NLO with Examiner). ImageJ was used for analysis. For quantification of neuronal nuclear CREB in Drosophila, dissected brains were fixed in 100% methanol and stained with antibodies detecting Drosophila CREB and elav (1:100 and 1:5, respectively). Elav-based masks were created in ImageJ and CREB fluorescence within the elav-positive area was quantified in six control and six tauR406W transgenic dissected Drosophila brains.
TUNEL staining was performed on 4 μm sections of formalin-fixed, paraffin embedded Drosophila heads. Secondary identification of TUNEL-positive nuclei was performed using DAB. TUNEL-positive nuclei were counted throughout the entire brain by bright field microscopy.
QUANTIFICATION AND STATISTICAL ANALYSIS
A Student’s t test was used for all pairwise comparisons. A one-way ANOVA using a Tukey multiple comparisons test (alpha = 0.05) was used to compare all multiple values. For all statistical analyses, a confidence interval of 95% and normal distribution were assumed. For in vivo Drosophila experiments (Figures 1, 2, 3A, 3B, and 4), each biological replicate is one Drosophila brain. For in vitro iPSC-derived neuron experiments (Figures 4C and D), each biological replicate is one biologically distinct patient-derived cell population. Statistical analysis was performed using Prism8. Statistical details can be found in the figure legends and text, where appropriate.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE Antibodies | SOURCE | IDENTIFIER |
|---|---|---|
| Antobodies | ||
| CREB | Cell Signaling | 9197; RRID: AB_331277 |
| Actin | Developmental Studies Hybridoma Bank | JLA20; RRID: AB_528068 |
| Elav | Developmental Studies Hybridoma Bank | 9F8A9; RRID: AB_528217 |
| GFP | Thermo Fisher Scientific | CAB4211; RRID: AB_10709851 |
| MAP2 | Abcam | 5392; RRID: AB_2138153 |
| vGluT1 | Abcam | Ab77822; RRID: AB_2187677 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BMS 191011 | N/A | SML0866 |
| Critical Commercial Assays | ||
| TUNEL - FragEL DNA Fragmentation Detection Kit | EMD Millipore | QIA33 |
| Ovation RNA-seq System for Drosophila | NuGen | 0350 |
| Clarity Max ECL Western Blotting Substrate | Bio-Rad | 1705062 |
| Deposited Data | ||
| Control and tauR406W RNA-seq data, day 10 of adulthood | GEO | GEO: GSE152278 |
| Experimental Models: Cell Lines | ||
| Control 1 | Coriell Institute | GM25430; RRID: CVCL_1 N86 |
| Control 2 | Coriell Institute | GM27717 |
| Alzheimer’s Disease 1 | Coriell Institute | CW50130; RRID: CVCL_JI63 |
| Alzheimer’s Disease 2 | Coriell Institute | CW50136; RRID: CVCL_ER61 |
| Alzheimer’s Disease 3 | Coriell Institute | CW50131; RRID: CVCL_HI88 |
| Experimental Models: Organisms/Strains | ||
| Elav-GAL4 | Bloomington Drosophila Stock Center | 458; RRID: BDSC_458 |
| w1118 | Bloomington Drosophila Stock Center | 3605; RRID: BDSC_3605 |
| UAS-tauR406W | Dr. Mel Feany | Wittmann et al., 2001 |
| UAS-GCaMP3.NLS | Dr. Hilmar Bading | Weislogel et al., 2013 |
| UAS-CaMBP4 | Dr. Hilmar Bading | Weislogel et al., 2013 |
| SloRNAi | Bloomington Drosophila Stock Center | 55405; RRID: BDSC_55405 |
| Slo1 | Bloomington Drosophila Stock Center | 4587; RRID: BDSC_4587 |
| Software and Algorithms | ||
| Trimmomatic | Bolger et al., 2014 | v.0.36; RRID: SCR_011848 |
| FastQC | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | RRID: SCR_014583 |
| Salmon | Patro et al., 2017 | v.0.13.1 |
| DESeq2 | Love et al., 2014 | v1.24; RRID: SCR_015687 |
| FlyBase - Drosophila transcriptome | Thurmond et al., 2019 | FB2018_6.27; RRID: SCR_006549 |
| FindM | Ambrosini et al., 2003 | N/A |
| Gene Expression Omnibus | Edgar et al., 2002 | N/A; RRID: SCR_005012 |
| ChlPpeakAnno | Zhuetal., 2010 | N/A; RRID: SCR_012828 |
| GraphPad Prism | GraphPad Software | Prism8; RRID: SCR_002798 |
| Other | ||
| CREB ChlP-seq dataset | Hirano et al., 2016 | GEO: GSE73386, samples GSM1892406 and GSM1892408 |
| Membrane-RFP plasmid | Addgene | 57992; RRID: Addgene_57992 |
| GCaMP6s.NLS plasmid | Dr. Hilmar Bading | Hagenston and Bading, 2011 |
| BioT transfection reagent | Bioland Scientific | B01–01 |
Highlights.
TauR406W induces nuclear CREB depletion in neurons of the adult Drosophila brain
Nuclear Ca2+ decreases with aging and tauopathy in the adult Drosophila brain
Nuclear Ca2+ is depleted in iPSC-derived neurons from sporadic Alzheimer’s disease
Pharmacologic/genetic manipulation of BK channels modify tauR406W neurotoxicity
ACKNOWLEDGMENTS
We thank Dr. Hilmar Bading for providing GCaMP3.NLS and UAS-CaMBP4 Drosophila lines and GCaMP6s.NLS constructs and Peter Ngam (NuGen/Tecan) for training in use of the Ovation RNA-Seq system. The UT Health San Antonio Genome Sequencing Facility is supported by UT Health San Antonio, NIH-NCI P30 CA054174 (Cancer Center at UT Health San Antonio), NIH Shared Instrument grant 1S10OD021805 (S10 grant), and CPRIT Core Facility Award (RP160732). The Actin and GFP antibodies were obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242. The graphical abstract was created using Adobe Illustrator and Bio-render (https://www.biorender.com). This study was supported by the American Federation for Aging Research New Investigator in Alzheimer’s Disease (B.F.) and the National Institute on Aging (R01 AG062475 to R.D. and B.F.). R.M. was supported by an NIGMS-funded K12 GM111726.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107900.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ambrosini G, Praz V, Jagannathan V, and Bucher P (2003). Signal search analysis server. Nucleic Acids Res. 31, 3618–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. ; The Gene Ontology Consortium (2000). Gene ontology: tool for the unification of biology. Nat. Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bading H (2013). Nuclear calcium signalling in the regulation of brain function. Nat. Rev. Neurosci 14, 593–608. [DOI] [PubMed] [Google Scholar]
- Bardai FH, Wang L, Mutreja Y, Yenjerla M, Gamblin TC, and Feany MB (2018). A Conserved Cytoskeletal Signaling Cascade Mediates Neurotoxicity of FTDP-17 Tau Mutations In Vivo. J. Neurosci 38, 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolotti N, Bennett DA, and Lazarov O (2016). Reduced pCREB in Alzheimer’s disease prefrontal cortex is reflected in peripheral blood mononuclear cells. Mol. Psychiatry 21, 1158–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito E, and Barco A (2010). CREB’s control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci 33, 230–240. [DOI] [PubMed] [Google Scholar]
- Bjorklund NL, Reese LC, Sadagoparamanujam VM, Ghirardi V, Woltjer RL, and Taglialatela G (2012). Absence of amyloid b oligomers at the post-synapse and regulated synaptic Zn2+ in cognitively intact aged individuals with Alzheimer’s disease neuropathology. Mol. Neurodegener 7, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, and Braak E (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Bucher P, and Trifonov EN (1986). Compilation and analysis of eukaryotic POL II promoter sequences. Nucleic Acids Res. 14, 10009–10026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, and Studer L (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol 27, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala A, and Spall T (2003). In vivo calcium imaging of brain activity in Drosophila by transgenic cameleon expression. Sci. STKE 2003, PL6. [DOI] [PubMed] [Google Scholar]
- Forrest SL, Kril JJ, Stevens CH, Kwok JB, Hallupp M, Kim WS, Huang Y, McGinley CV, Werka H, Kiernan MC, et al. (2018). Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain 141, 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, Hemberg M, Lewis J, and Feany MB (2014). Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci 17, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, Bardai FH, and Feany MB (2016). Lamin Dysfunction Mediates Neurodegeneration in Tauopathies. Curr. Biol 26, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, and Arancio O (2004). Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J. Clin. Invest 114, 1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenston AM, and Bading H (2011). Calcium signaling in synapse-to-nucleus communication. Cold Spring Harb. Perspect. Biol 3, a004564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJL, and Bading H (2001). Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat. Neurosci 4, 261–267. [DOI] [PubMed] [Google Scholar]
- Heisenberg M (2003). Mushroom body memoir: from maps to models. Nat. Rev. Neurosci 4, 266–275. [DOI] [PubMed] [Google Scholar]
- Hirano Y, Ihara K, Masuda T, Yamamoto T, Iwata I, Takahashi A, Awata H, Nakamura N, Takakura M, Suzuki Y, et al. (2016). Shifting transcriptional machinery is required for long-term memory maintenance and modification in Drosophila mushroom bodies. Nat. Commun 7, 13471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, et al. (1998). Association of missense and 50-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705. [DOI] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, et al. (2012). Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachaturian Z (1984). Towards theories of brain aging In Handbook of Studies on Psychiatry and Old Age (Elsevier Science Publishers; ), pp. 7–30. [Google Scholar]
- Khurana V, Lu Y, Steinhilb ML, Oldham S, Shulman JM, and Feany MB (2006). TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr. Biol 16, 230–241. [DOI] [PubMed] [Google Scholar]
- Khurana V, Merlo P, DuBoff B, Fulga TA, Sharp KA, Campbell SD, Götz J, and Feany MB (2012). A neuroprotective role for the DNA damage checkpoint in tauopathy. Aging Cell 11, 360–362. [DOI] [PubMed] [Google Scholar]
- Li B, Jie W, Huang L, Wei P, Li S, Luo Z, Friedman AK, Meredith AL, Han M-H, Zhu X-H, and Gao TM (2014). Nuclear BK channels regulate gene expression via the control of nuclear calcium signaling. Nat. Neurosci 17, 1055–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney RE, Rawson JM, and Eaton BA (2014). An age-dependent change in the set point of synaptic homeostasis. J. Neurosci 34, 2111–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo P, Frost B, Peng S, Yang YJ, Park PJ, and Feany M (2014). p53 prevents neurodegeneration by regulating synaptic genes. Proc. Natl. Acad. Sci. USA 111, 18055–18060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai J, Ohkura M, and Imoto K (2001). A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat. Biotechnol 19, 137–141. [DOI] [PubMed] [Google Scholar]
- Ochalek A, Mihalik B, Avci HX, Chandrasekaran A, Téglási A, Bock I, Giudice ML, Táncos Z, Molnár K, László L, et al. (2017). Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res. Ther. 9, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, and LaFerla FM (2003). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421. [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugazhenthi S, Wang M, Pham S, Sze C-I, and Eckman CB (2011). Downregulation of CREB expression in Alzheimer’s brain and in Ab-treated rat hippocampal neurons. Mol. Neurodegener 6, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy K, Cusack CL, Nnah IC, Khayati K, Saqcena C, Huynh TB, Noggle SA, Ballabio A, and Dobrowolski R (2016). Dysregulation of Nutrient Sensing and CLEARance in Presenilin Deficiency. Cell Rep. 14, 2166–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sproul AA, Jacob S, Pre D, Kim SH, Nestor MW, Navarro-Sobrino M, Santa-Maria I, Zimmer M, Aubry S, Steele JW, et al. (2014). Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS ONE 9, e84547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurmond J, Goodman JL, Strelets VB, Attrill H, Gramates LS, Mary-gold SJ, Matthews BB, Millburn G, Antonazzo G, Trovisco V, et al. ; FlyBase Consortium (2019). FlyBase 2.0: the next generation. Nucleic Acids Res. 47 (D1), D759–D765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin MS, Stachowiak A, Mamun AA, Tzvetkov NT, Takeda S, Atanasov AG, Bergantin LB, Abdel-Daim MM, and Stankiewicz AM (2018). Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci 10, 04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui T, Smolik SM, and Goodman RH (1993). Isolation of Drosophila CREB-B: a novel CRE-binding protein. DNA Cell Biol. 12, 589–595. [DOI] [PubMed] [Google Scholar]
- Wang F, Zhang Y, Wang L, Sun P, Luo X, Ishigaki Y, Sugai T, Yamamoto R, and Kato N (2015a). Improvement of spatial learning by facilitating large-conductance calcium-activated potassium channel with transcranial magnetic stimulation in Alzheimer’s disease model mice. Neuropharmacology 97, 210–219. [DOI] [PubMed] [Google Scholar]
- Wang L, Kang H, Li Y, Shui Y, Yamamoto R, Sugai T, and Kato N (2015b). Cognitive recovery by chronic activation of the large-conductance calcium-activated potassium channel in a mouse model of Alzheimer’s disease. Neuropharmacology 92, 8–15. [DOI] [PubMed] [Google Scholar]
- Weislogel J-M, Bengtson CP, Müller MK, Hörtzsch JN, Bujard M, Schuster CM, and Bading H (2013). Requirement for nuclear calcium signaling in Drosophila long-term memory. Sci. Signal 6, ra33. [DOI] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, and Feany MB (2001). Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293, 711–714. [DOI] [PubMed] [Google Scholar]
- Yin JC, Wallach JS, Wilder EL, Klingensmith J, Dang D, Perrimon N, Zhou H, Tully T, and Quinn WG (1995). A Drosophila CREB/CREM homolog encodes multiple isoforms, including a cyclic AMP-dependent protein kinase-responsive transcriptional activator and antagonist. Mol. Cell. Biol 15, 5123–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Gao D, Wang Y, Wang Z-H, Wang X, Ye J, Wu D, Fang L, Pi G, Yang Y, et al. (2016). Tau accumulation induces synaptic impairment and memory deficit by calcineurin-mediated inactivation of nuclear CaMKIV/CREB signaling. Proc. Natl. Acad. Sci. USA 113, E3773–E3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S-J, Zou M, Lu L, Lau D, Ditzel DAW, Delucinge-Vivier C, Aso Y, Descombes P, and Bading H (2009). Nuclear calcium signaling controls expression of a large gene pool: identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet. 5, e1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LJ, Gazin C, Lawson ND, Pagès H, Lin SM, Lapointe DS, and Green MR (2010). ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics 11, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number of the raw RNA-seq files for Drosophila control versus tauR406W transgenic Drosophila reported in this paper is GEO: GSE152278.