

Abstract
Adaptive immunity provides the unique ability to respond to a nearly infinite range of antigenic determinants. Given the inherent plasticity of the adaptive immune system, a series of tolerance mechanisms exist to reduce reactivity toward self. While this reduces the probability of autoimmunity, it also creates an important gap in adaptive immunity: the ability to recognize microbes that look like self. As a variety of microbes decorate themselves in self-like carbohydrate antigens and tolerance reduces the ability of adaptive immunity to react with self-like structures, protection against molecular mimicry likely resides within the innate arm of immunity. In this essay, we will explore the potential consequences of microbial molecular mimicry, including factors within innate immunity that appear to specifically target microbes expressing self-like antigens and therefore provide protection against molecular mimicry.
Keywords: adaptive immunity, carbohydrates, galectins, innate immunity, lectins, microbes
INTRODUCTION:
The capacity of adaptive immunity to respond to a nearly infinite range of antigenic determinants reflects genetic diversity generated following recombination events that result in an entire array of lymphocyte receptors [1]. Stimulation of the adaptive immune system induces proliferation of antigen-specific clones [2], as well as additional genetic alterations that further refine the specificity and affinity of lymphocyte receptors [3]. This process is governed through a fascinating array of factors that provide immunological memory shaped entirely by an individual’s exposure to environmental antigens [4]. The ability of adaptive immunity to not only be molded in response to microbial exposure, but also generate immunological memory, serves as the key premise behind vaccination. The inherent plasticity of adaptive immunity not only enables an individual’s resources to be tailored to their own microbial exposure, but also provides a compelling mechanism to compete in the ongoing arms race with evolving antigenic determinants [5]. Thus, adaptive immunity provides a highly effective mechanism of immunological protection against a variety of distinct microbes.
While the unique flexibility of adaptive immunity enables individuals to effectively respond to a wide variety of potential pathogens, the same plasticity that affords adaptive immunity unique and evolving responses also increases the probability of autoimmunity. Indeed, a complex series of tolerance mechanisms exist to reduce self-reactivity, which ultimately serves to eliminate self-reactive clones or render them ineffective at mounting an immune response. Although these regulatory networks reduce the probability of autoimmunity, they also create an important gap in adaptive immunity: the ability to recognize microbes that look like self. As a result, while adaptive immunity can respond to a nearly infinite range of antigenic determinants, the Achilles’ heel of adaptive immunity is its inability to effectively recognize and respond to antigens that look like self. Not surprisingly, many microbes appear to utilize this gap in adaptive immunity by decorating themselves in host-like antigens as a form of molecular mimicry. In this essay, we will explore the intriguing paradox that confronts immunity when challenged by microbes that utilize molecular mimicry and examine data suggesting that unique innate immune factors may exist to fill this gap in adaptive immunity and thus protect individuals against microbes that utilize molecular mimicry.
Distinguishing self versus non-self
While adaptive immunity offers one of the most intriguing examples of evolutionary adaptation to microbial exposure, the plasticity afforded by acquired immunity is not without risk. Genetically-driven changes in receptor specificity may provide a nearly infinite range of potential receptors, but these alterations can also increase the risk of neoplastic transformation [6]. Many neoplastic diseases of lymphoid cells harbor mutations that likely represent misguided recombination events [6]. Equally important, the very requirement for the relatively random DNA recombination that generates this nearly limitless repertoire also increases the probability of reactivity with self. As this represents an ongoing process within an individual, effectors within adaptive immunity must learn to distinguish self from non-self within an individual’s lifetime, unlike innate immune factors that benefited from repertoire selection throughout evolutionary history [7]. This requirement must be maintained following new and unpredictable exposures that may challenge checkpoints that evolved to prevent autoimmunity [8]. Thus, while receptor diversification within adaptive immunity provides the theoretical ability to respond to an infinite range of continuously evolving antigenic determinants, it also comes with a significant risk of developing autoimmunity [8].
Given the challenge of distinguishing host from microbe, an equally impressive system evolved to reduce potential reactivity toward self and thus combat the risk of autoimmunity [9]. Fundamental in the process of preventing self-reactivity is the ability to distinguish self from non-self [10]. While evolution selected a variety of mechanisms to achieve pathogen-targeted specificity, each of these fundamentally relies on the recognition of key features that differentiate pathogenic targets from the host [10]. This resulted in a complex array of programs that reduce or eliminate cells that acquire self-reactivity, or convert these cells into vehicles that suppress immunity [8]. In contrast, innate immunity benefited from selective pressures over a long period of evolutionary history to avoid self-injury [7]. In each of these settings, immune factors either specifically recognize molecular motifs exclusively found on microbes or fail to respond if self antigens are present. While the former mechanism reflects the type of target specificity used by adaptive immunity and most innate immune factors, exclusive reactivity toward organisms that may be “missing self” serves as an ancient mechanism whereby complement and later natural killer cells distinguish self from non-self [11,12]. Regardless of the mechanism, each of these systems evolved to target cytotoxic effector molecules specifically toward potentially pathogenic organisms by discriminating key differences in the molecular signature of microbes versus self.
Microbial mechanisms of avoiding immunity
While a variety of tolerance mechanisms confer the ability to specifically direct immunity toward microbes, many microbes appear to take advantage of the inherent difficulty in differentiating self from non-self [13]. Bacteria, viruses, and parasites each utilize a variety of factors to divert or otherwise avoid host immunity by inducing self-like programs [13,14] (Table 1). Expression of immune-related functional or decoy receptors that recognize cytokines, chemokines, or other host immune factors can modulate immune function in favor of microbial survival by coopting programs designed to induce peripheral tolerance toward self [14]. Each of these mechanisms targets host immunity in a variety of ways that often cripple the ability of host immune cells to adequately recognize or respond to an offending pathogen [13,14]. Regardless of the mechanism, each of these microbe-induced pathways can significantly impair immune function and may similarly be utilized by neoplastic cells to avoid immune detection and elimination [15].
Table 1.
General microbial avoidance strategies
| Molecular mimicry | Immune deviation to a nonproductive immune response |
| Growth in immune privileged environments | Down regulation of immune activators (major histocompatibility, etc.) |
| Induction of immunological tolerance |
Inhibition of immune effectors (complement, etc.) |
General overview of some of the strategies microbes use to avoid host immunity.
In addition to actively inhibiting immune function, microbes also evolved mechanisms to simply avoid immunological detection. Several studies demonstrate that molecular mimicry of T cell epitopes might bypass T cell-mediated immune activation as a result of deletion of T cells with reactivity toward self-like peptides [16]. Presentation of host-like microbial peptides may also induce the activation of tolerogenic immune responses that could facilitate infection [16,17]. However, while most of these antigenic determinants reflect short stretches of amino acid sequences that may be very similar to host proteins, these epitopes often reside within a larger protein sequence quite foreign to the host. As a result, these forms of molecular mimicry may indeed reduce or suppress T cell help toward microbes bearing self-like peptides, but additional peptides that are unrelated to self may still facilitate immune recognition. These types of molecular mimicry may reduce an overall response to a given microbe, but do not typically render all microbes undetectable by host immunity [18]. This is especially important when considering that other antigenic determinants, in particular those on the microbial surface, often serve as the key point of initial immunological recognition [19].
Complex carbohydrates: a unique substrate for molecular mimicry
Prior to host cell-mediated microbial removal and peptide presentation, each microbe must first be recognized through cell surface interactions. Consistent with this, the most common variations observed between different microbes reflect alterations in the surface coating of each microbe; this variability likely occurred secondary to selective pressures induced by the arms race between host detection and microbial evasion [5]. Although many macromolecules could potentially decorate a microbial surface, most microbes, similar to host cells, cover themselves in complex carbohydrates, sometimes referred to as the glycocalyx [20]. As carbohydrate synthesis results from the coordinated effort of many different enzymes and a diverse array of substrates, the combinatorial diversity provided by complex carbohydrates enables the generation a variety of signature structures that decorate the microbial surface [21]. Given this plasticity and the rapid rate of microbial evolution, microbes appear to use these complex carbohydrates as the macromolecular substrates to generate antigenic diversification. Indeed, unique strains within various microbial species can be completely defined by serological discrimination of different O-carbohydrate surface antigens [22], demonstrating the significant structural diversity that these macromolecules can afford.
Unlike mammalian cell surface glycans, which often consist of a variety of different carbohydrate structures, microbes typically decorate themselves in a single type of repeating carbohydrate antigenic unit. This carbohydrate motif is often presented as the outermost sequence of lipopolysaccharide (LPS) in gram-negative microbes or as a capsular structure in both gram-negative and gram-positive organisms [22]. This provides a uniform antigenic motif enveloping the microbe. Given this unique microbial carbohydrate cloak, microbes can readily mask other antigenic determinants in this carbohydrate covering [21,23] (Figure 1). As alterations in carbohydrates can occur in the absence of significant functional changes to these macromolecules, these substrates provide a tool to allow modifications at the microbial surface without significantly altering function. In contrast, while proteins can acquire diversification, this commonly impacts protein activity and therefore would not be predicted to possess the inherent flexibility needed to change antigenicity without compromising function [24]. Furthermore, while different lipid species exist, their thermodynamic properties often preclude them from playing a significant role in direct host interactions at the microbial surface. Thus the ability of complex carbohydrates to form a shield at the cell surface not only provides a unique and diverse structural coating, but also prevents or reduces the ability of immune factors to recognize other carbohydrates, proteins, or lipids under this layer that do possess unique structural motifs required for microbial function. Thus, this carbohydrate coating enables microbes to perform functions that require specific structural components that would otherwise be potential targets of host factors if not masked by the glycocalyx [23].
Figure 1:
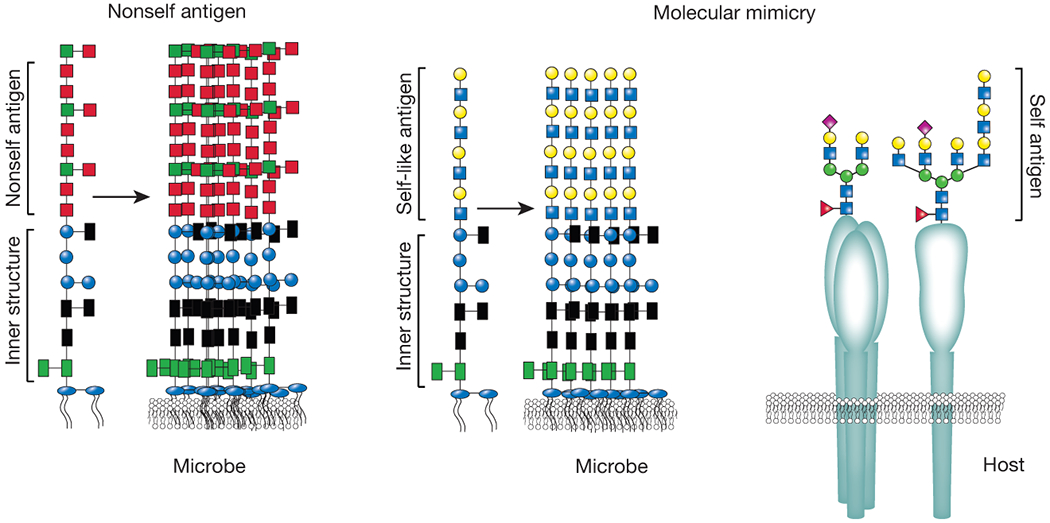
Microbial carbohydrate molecular mimicry. While many constituents at or near the membrane are very unique to microbes, outer carbohydrate structures can closely mimic those found on the surface of mammalian cells. As these structures can completely envelope a microbe and host adaptive immunity is tolerized to these structures, microbes that decorate themselves in host-like glycans can become undetectable to most host immune factors (molecular mimicry, on the right). In contrast, many microbes decorate themselves in antigen motifs that are quite distinct from the host (nonself antigens). As a result, these structures can readily become the target of host immunity (left side).
The carbohydrates covering microbes appear to reduce sensitivity to immune recognition and removal, and may also represent a strategy employed by microbes to avoid immunological detection altogether. Indeed, some microbes possess the ability to synthesize carbohydrates identical to host glycans [23] (Figure 1). As innate immune factors often recognize microbial motifs unique to pathogens, these defense mechanisms no longer possess the capacity to recognize and respond to microbes that look like self [7]. Equally important, as tolerance programs educate adaptive immunity to avoid self-reactivity, adaptive immunity likewise does not recognize self-like microbes [8,9]. Thus, despite the fascinating ability of adaptive immunity to respond to nearly any antigenic determinant [3], the necessity of immunological tolerance generates the Achilles’ heel of adaptive immunity: its inability to respond to microbes that cloak themselves in self-like antigens. As microbial recognition represents the first and foremost strategy in host protection, molecular mimicry poses a real challenge to host defense [23]. Although microbes contain thousands of unique antigenic determinants, most of these structures do not serve as the initial point of immunological detection. Indeed, as engulfment, processing, and presentation of unique underlying antigenic determinants often first requires recognition of the microbial surface, microbial decoration in carbohydrate self-like antigens would be predicted to inhibit this key initial step of host recognition. Furthermore, while some immune regulators recognize self in the context of tissue injury through damage associated molecular patterns, these interactions often reflect highly conserved carbohydrate-independent structural motifs that serve to activate the immune system following injury and therefore do not provide direct protection against molecular mimicry[25]. As a result, the mechanism for protection against microbes that decorate themselves in host-like antigens remains a fundamental question that confronts current paradigms regarding host immunity.
Microbial decoration in molecular mimics
One of the most classic examples of carbohydrate molecular mimicry occurs on Trypanosoma cruzi [26]. T. cruzi directly acquires the terminal carbohydrate structure sialic acid from host glycans and places this monosaccharide on the terminus of its own cell surface carbohydrates [26]. This trans-sialidase reaction is critical to T. cruzi survival as loss of this activity and/or loss of T. cruzi sialic acid in general results in rapid clearance of the organism [27]. Some strains of Haemophilus influenza also acquire sialic acid from the host [28]. However, instead of directly facilitating transfer from existing host glycans directly to the microbial surface, H. influenzae simply acquires host sialic acid and then funnels this substrate into existing machinery used to generate carbohydrates that decorate the cell [28].
In contrast to H. influenza and T. cruzi, which cannot synthesize their own sialic acid, other organisms appear to have evolved the ability to generate sialic acid mimics. For example, various strains of Streptococcus agalactiae generate sialic acid capping structures that also look like self [29].
Acquisition of sialic acid, the most common terminal carbohydrate structure expressed on our own cells, may not only provide a mechanism of avoiding adaptive immune recognition, but may also directly and favorably influence immune regulation for microbial survival. Engagement of sialic acid on host cells appears to reflect a key mechanism whereby hosts distinguish self from non-self [30,31]. In contrast to host cell glycans, most microbes are not decorated in sialic acid [31]. A series of unique sialic acid binding receptors, siglecs, expressed on host immune cells recognize host sialic acid. This engagement subsequently inhibits potential immune responses as a consequence of self-recognition [32]. Lack of sialic acid-siglec interactions following microbial recognition can therefore facilitate immunological removal since this siglec-mediated inhibitory process is not triggered. This program appears to be operationally similar to other systems that utilize missing self to differentiate self from non-self, such as complement and possibly Fc receptors [33,34]. In contrast, microbial decoration with sialic acid structures may not only reduce immune effector detection, but also enable microbial engagement of inhibitory siglec receptors [35]. Similar to recognition of host sialic acid structures, siglec recognition of microbial sialic acid containing glycans induces inhibitory programs that may compromise effective immune responses towards the microbes [35]. It should be noted that several sialic acid binding receptors also possess scavenging receptor activity and may have evolved, in part, to counterbalance the impact of this form of microbial mimicry [36].
ABO blood group molecular mimicry
In 1900 Karl Landsteiner described the existence of carbohydrate ABO(H) blood group polymorphisms and corresponding anti-blood group antibodies, which ultimately resolved a long-standing mystery regarding the varied outcomes of red blood cell (RBC) transfusion [37]. Following the discovery of ABO(H) blood group antigens, Landsteiner and others demonstrated that individuals given ABO(H) compatible blood displayed a therapeutic response to RBC transfusion, whereas transfusion of ABO(H) incompatible RBCs could cause adverse events that often resulted in death [38]. This discovery not only provided the foundation for modern transfusion medicine and eventually transplantation, but also represented the first and what continues to be the most common example of personalized medicine[38]. However, despite the discovery of ABO(H) blood group polymorphisms and corresponding anti-blood group antibodies over a century ago, the factors responsible for the development of these antibodies and potential consequences of ABO(H) antigen expression on an individual remained unknown for many years[38].
Unlike acquired RBC alloantibodies, which typically form following RBC transfusion or pregnancy [39,40], so-called naturally occurring anti-ABO(H) antibodies develop spontaneously within the first few months of life. Over 60 years ago, Georg Springer hypothesized that microbes may exist that decorate themselves in blood group carbohydrate antigens similar to those that decorate our own cells and that exposure to these microbes may induce naturally occurring anti-blood group antibodies [41]. To examine this, Springer and colleagues examined potential anti-A and anti-B seroreactivity toward various microbes obtained from clinical isolates. These studies demonstrated that nearly half of microbes cultured from the gut expressed some blood group antigen reactivity, with nearly 10% displaying fairly specific seroreactivity [42]. As seroreactivity was used to identify blood group positive microbes, Wang and colleagues later demonstrated that one of the strains isolated, Escherichia coli O86, indeed synthesizes the blood group B antigen [43]. To determine whether blood group positive microbes possess the capacity to induce naturally occurring anti-ABO(H) blood group antibodies, blood group negative individuals were exposed to the blood group B positive (BG B+) E. coli O86. Exposure of BG B+ E. coli resulted in a significant increase in anti-blood group antibody formation [44], strongly suggesting that microbial exposure within the first few months of life may contribute to or even be entirely responsible for the development of naturally occurring antibodies.
While the ability of BG B+ E. coli to induce anti-B antibodies in blood group O or A individuals provided a potential mechanism whereby blood group negative individuals may develop anti-blood group antibodies, these results also suggest that molecular mimicry may not be limited to microbial incorporation of sialic acid. Instead, these early studies suggested that a variety of microbes decorate themselves in a diverse range of self-like carbohydrate antigens. In doing so, these studies raised a fundamental question in immunology [42]. If BG B+ microbes exist, how do BG B+ individuals, who cannot make anti-BG B antibodies, protect themselves from these microbes? As BG B+ microbes reflect some of the earliest examples of molecular mimicry and adaptive immunity is clearly limited toward BG B in BG B+ individuals, recent studies sought to determine whether innate immune factors may exist that can recognize BG B+ microbes and in so doing provide some form of immunological protection against blood group molecular mimicry.
Unique tools needed to examine immunity against molecular mimicry
Although investigators recognized the challenge that microbial carbohydrate mimics of host glycans pose to immunity many years ago, the tools necessary to discover unique factors that may distinguish molecular mimics of host glycans only recently became available. Unlike proteins, which can be readily cloned and altered by genetic approaches, complex carbohydrates reflect the coordinated effort of a variety of glycosyltransferases and unique substrates that ultimately results in the development of a complex post-translational modification [45,46]. As a result, carbohydrates cannot be individually cloned and sequenced in a similar manner as nucleic acids and proteins. Thus, generating large libraries of carbohydrates often requires chemical and/or chemoenzymatic synthesis [47]. This process can take years and significant resources to develop a sufficient library of defined carbohydrate test ligands needed to adequately discern the carbohydrate binding specificity of a given innate immune factor [48]. Consequently, for many years, the tools needed to elucidate the carbohydrate binding specificity of innate immune factors were simply not available, despite elegant studies that provided some insight into carbohydrate binding specificity of several immune factors [49,50]. As a result, while many studies recognized and catalogued a variety of innate immune factors [7], whether any of these possessed the capacity to protect against carbohydrate molecular mimicry remained unknown.
In an effort to overcome limitations in the study of protein-carbohydrate interactions, a variety of labs synthesized large libraries of defined glycans and presented these glycans in different formats to study the specificity of carbohydrate binding proteins [51,52]. Although each of these methods clearly possesses unique advantages and limitations, printing of characterized glycans in a microarray format quickly became a practical approach to simultaneously analyze a variety of determinants while only requiring a small amount of source material [48,53–56]. These platforms completely revolutionized the study of protein-carbohydrate interactions and in so doing directly facilitated many novel discoveries, most of which directly impact our understanding of immunity [55,57–59]. In an effort to use this platform to determine whether innate immune factors exist that may specifically target blood group positive microbes, recent studies screened over 100 different immune factors with putative carbohydrate binding activity to determine whether any possess blood group binding activity. Using this approach, several members of the galectin family (Figure 2) displayed high binding to blood group antigens [52,60]. As previous studies demonstrated that several of these galectins, in particular galectin-4 (Gal-4) and galectin-8 (Gal-8), exhibited strong expression in the gut [61], the binding specificity and location of these innate immune factors suggested that they may be uniquely poised to provide immune protection against blood group molecular mimicry.
Figure 2:
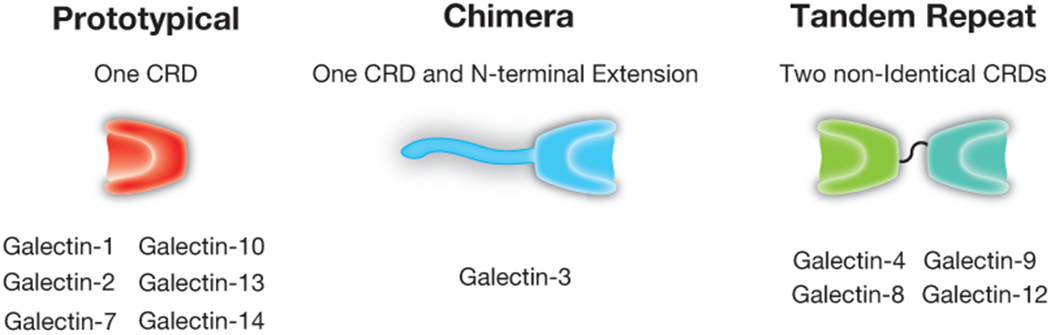
The galectin family. Human galectins have been classified into three primary classes: prototypical, chimeric, and tandem repeat. Each of these galectins possesses at least one unique carbohydrate recognition domain (CRD) that displays unique and overlapping binding specificity with other family members. Tandem repeat galectins possess two distinct carbohydrate recognition domains, each of which can exhibit very distinct binding preferences.
Galectins provide innate immunity against molecular mimicry
Gal-4 and Gal-8 displayed significant binding to blood group antigens on a glycan microarray [52,60], but their ability to recognize blood group antigens when presented on microbes remained unknown. Consistent with the binding profile of Gal-4 and Gal-8 toward characterized glycan determinants presented in an array format [52,60], these innate immune factors also recognized BG B+ E. coli. Although previous studies suggested that related galectin family members might provide antifungal activity [62], no previous studies demonstrated that these innate immune factors possessed any direct antimicrobial effect toward bacteria. Consistent with this, incubation of Gal-4 and Gal-8 with most strains of E. coli failed to result in any change in microbial viability [63,64]. However, when incubated with BG B+ E. coli, Gal-4 and Gal-8 not only bound, but this recognition resulted in rapid microbial death [63]. This study strongly suggested that these innate immune factors might fill an important gap in adaptive immunity against BG B+ microbes in BG B+ individuals.
As most innate immune factors recognize commonly occurring microbial motifs, often referred to as microbe-associated molecular patterns (MAMPs), the proclivity of Gal-4 and Gal-8 for BG B+ E. coli stood in stark contrast to previously described innate immune factors [7,10]. While the ability of Gal-4 and Gal-8 to kill BG B+ E. coli was consistent with the possibility that these innate immune factors may provide a specific form of immunity against blood group molecular mimicry, it certainly remained possible that Gal-4 and Gal-8 may recognize an unknown molecular motif on BG B+ E. coli that occurs independent of BG B antigen expression. However, genetic deletion of the ligase required for BG B antigen addition to LPS completely prevented Gal-4 and Gal-8 binding and rendered the microbes insensitive to galectin-mediated killing [63]. Ligase-deficient microbes also did not appear to be impacted by galectins in vivo. Inhibitors of galectin-microbial interactions significantly enhanced growth of BG B+ E. coli following introduction into mice, while similar inhibitor inclusion failed to alter ligase-deficient microbial viability [63]. Taken together, these findings strongly suggest that Gal-4 and Gal-8 provide a very unique form of innate immunity by targeting microbes that express distinct antigenic determinants. In so doing, these innate immune factors appear to fill an important gap in adaptive immunity against blood group molecular mimicry (Figure 3).
Figure 3:
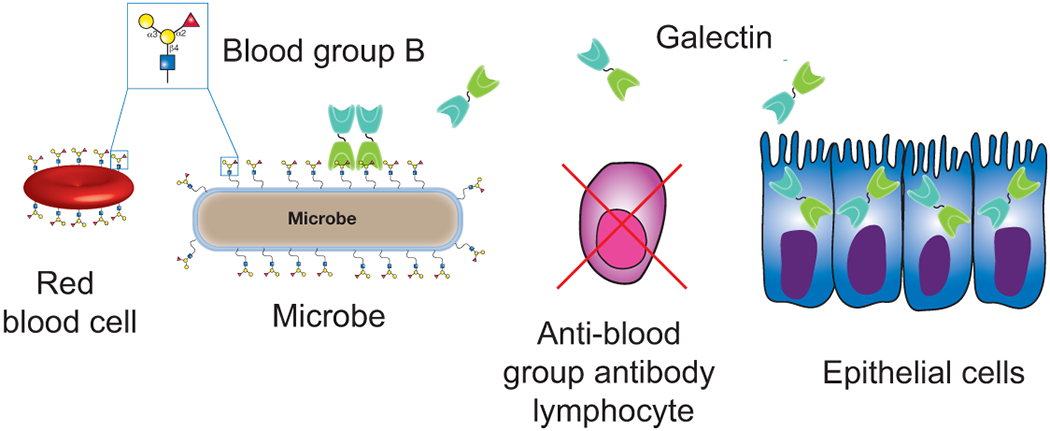
Galectins provide innate immunity against blood group molecular mimicry. Blood group expression in blood group positive individuals results in the deletion of potentially blood group reactive B cells. While this reduces the probability of autoimmunity, it also creates an important gap in adaptive immunity against blood group positive microbes. Innate immune galectins secreted by host cells, such as epithelial cells, engage and kill blood group positive microbes. In this way, galectins possess the unique ability to fill an important gap in adaptive immunity by providing innate immunity against blood group molecular mimicry.
While the ability of Gal-4 and Gal-8 to kill BG B+ E. coli provides insight into novel innate immune effector functions, as microbial molecular mimicry is not limited to blood group antigen expression, galectin-mediated immunity may extend beyond recognition of microbes that express blood group antigens. Consistent with this possibility, previous studies demonstrated that various galectin family members actually possess broad binding capacity toward many of the capping carbohydrate structures that decorate mammalian cells [51]. While this broad and rather promiscuous binding profile historically created a significant challenge when seeking to identify discrete host ligands for galectin-mediated processes [51], this binding capacity strongly suggested that these innate immune factors might actually provide a more broad form of innate immunity against molecular mimicry. To examine this, a microbial glycan microarray was generated using a similar strategy pioneered by other investigators [55,64,65]. Using this format, galectins demonstrated the unique ability to specifically recognize a variety of microbial glycans, each of which possesses mammalian-like features. In contrast, galectins failed to display significant binding to non-self microbial glycans on this array. Galectins not only recognized the isolated glycan when presented on a microarray, but also readily bound and killed microbes represented on the array that express self-like antigens [64]. In contrast, galectins failed to bind or kill related strains of microbes that do not express self-like antigenic determinants [64]. These studies suggest that the microbial glycan microarray accurately predicts previously unrecognized host microbial interactions and demonstrates that galectins possess a unique ability to specifically recognize and kill a variety of microbes that express self-like antigens. Although the potential antimicrobial activity of many galectin family members remains to be elucidated, these previous studies suggest that the galectin family provides a unique and unprecedented form of immunity against a variety of microbes that decorate themselves in mammalian-like carbohydrate antigens. In doing so, these innate immune factors appear to fill an important gap in adaptive immunity by specifically protecting individuals against molecular mimicry.
Consequences of innate immunity against molecular mimicry
Most immune factors maintain receptor specificity toward microbes through recognition of motifs that are unique to microbes [10]. In contrast, galectins appear to recognize microbes through engagement of the same exact antigenic determinant that occurs on our own cells. To examine the impact of galectin interactions with mammalian cells, previous studies determined whether galectins likewise induce changes in mammalian cell membranes following self-antigen engagement. While galectins readily engage mammalian cells, such as RBCs, this binding does not induce detectable changes in membrane integrity, while galectins induce rapid loss in the membrane integrity of BG B+ microbes [52,63,64]. The ability of galectins to recognize microbes through engagement of the same antigenic determinant that occurs on a host cell, but only kill the microbe, itself represents a new paradigm in immunological recognition (Figure 4).
Figure 4:
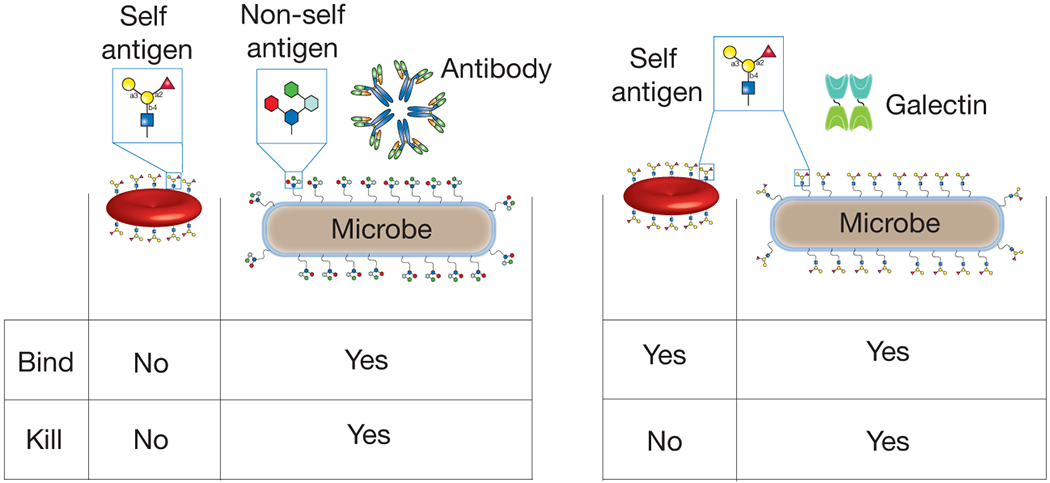
Galectins recognize the same antigenic determinant expressed on host cells and microbes but only kill microbes. The very nature of immunological protection against molecular mimicry requires immune factor recognition of very similar features on host cells and microbes. However, as most immune factors direct their activity specifically toward a microbe through engagement of molecular motifs that are unique to microbes, the ability of galectins to recognize the same antigenic determinant on a microbe and host cell, but only kill the microbe, represents a new paradigm in immunological recognition and killing.
Galectin recognition of similar antigens on microbes and host departs from fundamental rules thought to govern the specificity of host pathogen interactions. However, this very specificity is required when considering the molecular interactions needed to adequately protect against molecular mimicry. As mimics of host structures on microbes bear features that can be identical to self, immune factor protection against these microbes by definition will possess the ability to recognize both host and microbe through self-like antigen detection. As most immune factors use ligand discrimination to specifically target their activity toward potential pathogens, how galectins are able to recognize the same antigenic determinant on a microbe and host cell, but only kill the microbe, remains unknown. Other structural features unique to microbes may convey additional binding events that actually facilitate galectin-mediated killing. It should be noted that while galectins appear to possess the unique ability to provide innate immunity against molecular mimicry, other innate immune lectins might also provide a similar form of immunological protection [36]. Ongoing studies will seek to utilize unique tools, such as microbial glycan microarrays, to define the specificity and role of additional immunological factors in the process of immunological protection against molecular mimicry [66].
Galectins are pleiotropic factors involved in many biological processes
While galectins do not appear to induce direct alterations in the membrane integrity of mammalian cells, recognition of host ligands is not without consequence. As some of the only soluble lectins expressed by mammals with the ability to recognize host glycans, galectins likely served as a useful evolutionary substrate that not only provided innate immunity against molecular mimicry, but also possessed the capacity to decode host glycans to regulate a wide variety of biological processes. Galectins display expression in a variety of tissues and recognize a diverse range of cell surface glycans [51,52,60,63,67–71]. This allows galectins to induce a variety of responses following cellular engagement. Indeed, the first studies describing galectin function demonstrated one of the most well-studied properties of this family: their ability to regulate immune function. Galectin-1 (Gal-1), galectin-2 (Gal-2), galectin-3 (Gal-3), Gal-4, Gal-8, and galectin-9 (Gal-9) specifically impact T cell function, either by regulation of T cell activation and cytokine secretion, or by directly inducing apoptotic cell death [72–79]. Galectins also regulate other lymphocytes, neutrophils, mast cells, macrophages, dendritic cells and other cells in ways that can significantly impact the immunological outcome of infection, inflammation, and even neoplastic disease [77,80–88]. Each of these outcomes reflects very specific signaling events following engagement of a variety of receptors that are often differentially expressed on different immune effector cells [89–94]. Alterations in glycosylation appear to be master regulators of galectin-host glycan interactions and in so doing modulate a wide variety of immune related processes [93,95]. Galectins also mediate a variety of fundamental processes within the cell through carbohydrate independent processes [96–99], providing additional evidence regarding their highly pleiotropic activity.
In addition to the ability to regulate a broad range of biological processes within individuals, previous studies suggest that galectin-mediated immunity may not be limited to molecular mimicry, which is the subject of several excellent reviews [100–103] (Figure 5). This additional binding activity may actually represent engagement of similar motifs as occur on host cells, as the actual binding requirements for many of these interactions remain unknown. However, several examples of galectin-microbial interactions clearly suggest that in addition to significant preference for microbes that express self-like antigens, galectins also recognize non-self related determinants [65,104–106]. This additional binding activity may reflect an early ability of galectins to provide immunity in hosts where adaptive immunity is not present [100,107]. Consistent with this, previous studies suggest that galectins can recognize a variety of microbes. Early reports demonstrated that Gal-3 can bind Neisseria gonorrhoeae, Candida albicans, Toxoplasma gondii, Leishmania major, Schistosoma mansoni and T. cruzi [62,104,108–112]. Gal-1 and Gal-9 can similarly recognize L. major, while Gal-1 can also engage Trichomonas vaginalis [106,113–115]. Gal-1 also binds viruses, including Nipah, HTLV, Influenza, and HIV [114,116–118], likely through carbohydrate epitopes very similar to self. While previous studies described the specific glycan determinants and overall binding specificity of some of these interactions [104–106], most of these galectin-microbial interactions remain to be elucidated.
Figure 5:
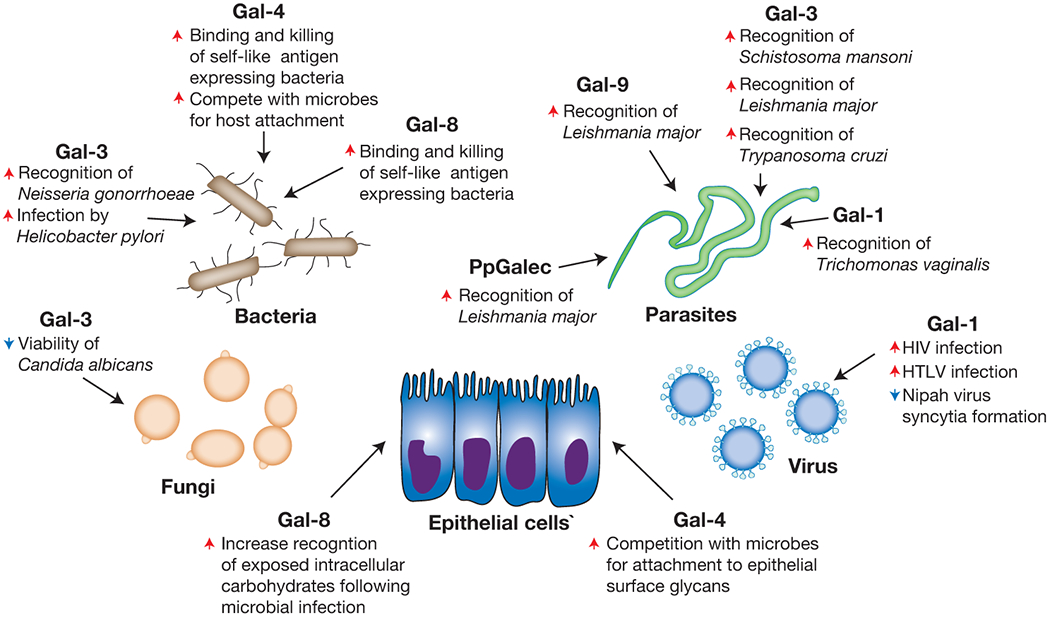
Galectin family members possess the ability to recognize many microbes with various outcomes. Galectins represent one of the most ancient mammalian lectin families described. As these proteins are present in all metazoans, it is likely that their recognition and response to invading pathogens represents one of their earliest evolutionary functions. However, microbial diversification of carbohydrate cloaks likely outstripped galectin and other innate immune factor recognition, setting the stage for the evolution of adaptive immunity. While adaptive immunity provides the ability to respond to a nearly infinite range of microbial glycans, tolerance reduces reactivity toward self. As a result, it appears that galectins maintained the ability to recognize microbes that utilize molecular mimicry, in addition to other microbes, to fill this gap in adaptive immunity and thereby protect individuals against molecular mimicry. Red arrows indicate an activity that the respective galectin increases, while blue arrows signify galectin-induced decreases in the accompanying activity.
Unlike the impact of Gal-4 and Gal-8 recognition of bacteria expressing self-like antigens, the consequence of galectin recognition of other microbes can be quite varied. While Gal-3 can induce direct antimicrobial death in C. albicans [62], similar engagement of T. cruzi facilitates macrophage recognition, leading to macrophage activation and eventual T. cruzi removal [109]. Gal-1 engagement of Influenza or Nipah viruses inhibits viral entry into host cells, providing another consequence of galectin-pathogen interactions that results in host protection without directly mediating pathogen death [114,116]. In contrast, while some intestinally expressed galectins may not actually recognize particular pathogens, galectin engagement of epithelial surface glycans may directly compete with microbial attachment and therefore inhibit colonization and subsequent invasion [119,120]. Similarly, Salmonella-induced disruption of phagolysosomes results in Gal-8-mediated interactions with exposed glycans that ultimately triggers intracellular autophagy and microbial death [121]. Antimicrobial-independent activities of galectin-mediated immune defense are not limited to mammals [122,123]. The tetravalent galectin of the oyster Crassostea virinica directly mediates pathogen tethering to hemocytes through simultaneous self-recognition on hemocytes and engagement of a variety of microbes, such as the protozoan parasite, Perkinsus marinus [107,123]. Similar studies demonstrate that additional galectin interactions may occur in other marine life [122,124]. Thus, in addition to directly inducing microbial death, galectins appear to utilize a variety of mechanisms to prevent or otherwise reduce infection.
Similar to the ability of microbes to utilize siglecs to enable survival in a mammalian host, several pathogens appear to coopt galectins to actually facilitate infection. Gal-3 appears to enhance viral infection of HSV-1 [125], although Gal-3-mediated alterations in cell surface glycoproteins may also inhibit viral entry along corneal epithelial cells [126]. Gal-1 engagement of HIV enhances viral entry, likely through interactions between GP120 and CD4 [117,118,127]. Gal-9 may also enhance HIV infection, although this requires Gal-9-mediated changes in the cell surface redox potential that favorably influence HIV engagement [128]. While Gal-1 and Gal-3 may facilitate host defense against Nipah virus and T. cruzi, these pathogens may also utilize these galectins to facilitate host cell entry or host attachment [129,130]. The timing of galectin exposure in the infectious process may in part dictate the outcome of these galectin-mediated events [129]. Gal-1 and Gal-3 engagement of HTLV also appears to enhance infection of T cells, although the mechanisms whereby each galectin facilitates viral entry may also differ [131,132]. Gal-3 similarly facilitates infection by Neisseria meningitidis [112] and Gal-3-mediated engagement of Pseudomonas aeruginosa may likewise enhance binding to corneal epithelium [133], once again potentially enabling infection. Trichomonas vaginalis may similarly use Gal-1 to adhere to vaginal epithelium and therefore increase infection [113]. Similar to the utilization of galectins in invertebrates for host defense, several organisms use galectins to enhance microbial colonization. The intestinal galectin (PgGalec) of the sand fly Phlebotomus papatasi facilitates L. Major binding in this intermediate host, thereby facilitating development and additional infection [134], while galactose-binding proteins derived from other species may modulate galectin-induced signaling pathways to drive pathogenesis in the host [135]. As a result, while galectin pathogen interactions likely evolved to provide host protection, some microbes appear to take advantage of these galectin interactions to establish host interactions and eventual infection.
Conclusions:
Although most studies examining galectin function historically focused on their apparent roles in regulating immunity [94], the ability of galectins to directly provide innate immunity likely represents one of their first and foremost evolutionary functions [100–103]. As the most ancient mammalian lectin family described, galectins likely evolved to provide innate immunity in early metazoans. Importantly, despite the apparent specificity of galectins for bacterial glycans with mammalian-like features [63,64], galectin-mediated interactions with many microbes do not appear to require the presence of mammalian-like structures [100–103], suggesting that early galectin-mediated immunity may have possessed an even broader form of microbial recognition. This may reflect a lack of adaptive immunity in these organisms and therefore a greater reliance on relatively broad binding characteristics of innate immune factors, as occurs for other innate immune lectins involved in host immunity [10]. However, as microbes evolved the ability to diversify their glycan repertoire, these unique structures likely outstripped the ability of galectins and possibly other innate immune factors to adequately recognize them. These changes likely set the stage for the selective pressures necessary for the evolution of adaptive immunity [5]. However, inherent limitations of adaptive immune reactivity against microbes coated in host-like glycans maintained the need for innate immunity against molecular mimicry. Thus, galectins appear to fill a unique and important gap in adaptive immunity, by binding and killing microbes that utilize molecular mimicry.
Acknowledgements:
The authors would like to thank Richard Cummings, PhD for introducing them to the fascinating field of glycobiology and galectin biology in particular and for providing the opportunity and encouragement to continue working in this field. We apologize for any references we may have missed owing to the limitations of the scope of an essay focused on one particular theme in galectin biology in addition to space restrictions. This work was supported in part by the National Blood Foundation, Hemophilia of Georgia, the Burroughs Wellcome Trust Career Award for Medical Scientists and the National Institutes of Health grant DP5OD019892 to SRS.
Abbreviations
- BG B+
blood group B positive
- RBC
red blood cell
REFERENCES:
- 1.Hozumi N, Tonegawa S. 1976. Evidence for somatic rearrangement of immunoglobulin genes coding for variable and constant regions. Proceedings of the National Academy of Sciences of the United States of America 73: 3628–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burnet FM. 1976. A modification of Jerne’s theory of antibody production using the concept of clonal selection. CA: a cancer journal for clinicians 26: 119–21. [DOI] [PubMed] [Google Scholar]
- 3.Kocks C, Rajewsky K. 1988. Stepwise intraclonal maturation of antibody affinity through somatic hypermutation. Proceedings of the National Academy of Sciences of the United States of America 85: 8206–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmed R, Gray D. 1996. Immunological memory and protective immunity: understanding their relation. Science 272: 54–60. [DOI] [PubMed] [Google Scholar]
- 5.Lee YK, Mazmanian SK. 2010. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 330: 1768–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsujimoto Y, Gorham J, Cossman J, Jaffe E, et al. 1985. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science 229: 1390–3. [DOI] [PubMed] [Google Scholar]
- 7.Medzhitov R, Janeway CA Jr. 1997. Innate immunity: the virtues of a nonclonal system of recognition. Cell 91: 295–8. [DOI] [PubMed] [Google Scholar]
- 8.Verkoczy LK, Martensson AS, Nemazee D. 2004. The scope of receptor editing and its association with autoimmunity. Current opinion in immunology 16: 808–14. [DOI] [PubMed] [Google Scholar]
- 9.Meffre E, Wardemann H. 2008. B-cell tolerance checkpoints in health and autoimmunity. Current opinion in immunology 20: 632–8. [DOI] [PubMed] [Google Scholar]
- 10.Medzhitov R, Janeway CA Jr. 2000. How does the immune system distinguish self from nonself? Seminars in immunology 12: 185–8; discussion 257-344. [DOI] [PubMed] [Google Scholar]
- 11.Stowell SR, Winkler AM, Maier CL, Arthur CM, et al. 2012. Initiation and regulation of complement during hemolytic transfusion reactions. Clinical & developmental immunology 2012: 307093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yokoyama WM. 1993. Recognition structures on natural killer cells. Current opinion in immunology 5: 67–73. [DOI] [PubMed] [Google Scholar]
- 13.Gershon RK, Kondo K. 1971. Infectious immunological tolerance. Immunology 21: 903–14. [PMC free article] [PubMed] [Google Scholar]
- 14.Alcami A, Koszinowski UH. 2000. Viral mechanisms of immune evasion. Trends in microbiology 8: 410–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shekarian T, Valsesia-Wittmann S, Caux C, Marabelle A. 2015. Paradigm shift in oncology: targeting the immune system rather than cancer cells. Mutagenesis 30: 205–11. [DOI] [PubMed] [Google Scholar]
- 16.Natale C, Giannini T, Lucchese A, Kanduc D. 2000. Computer-assisted analysis of molecular mimicry between human papillomavirus 16 E7 oncoprotein and human protein sequences. Immunology and cell biology 78: 580–5. [DOI] [PubMed] [Google Scholar]
- 17.Kanodia S, Fahey LM, Kast WM. 2007. Mechanisms used by human papillomaviruses to escape the host immune response. Current cancer drug targets 7: 79–89. [DOI] [PubMed] [Google Scholar]
- 18.Kanduc D 2009. “Self-nonself” peptides in the design of vaccines. Current pharmaceutical design 15: 3283–9. [DOI] [PubMed] [Google Scholar]
- 19.Lloyd DH, Viac J, Werling D, Reme CA, et al. 2007. Role of sugars in surface microbe-host interactions and immune reaction modulation. Veterinary dermatology 18: 197–204. [DOI] [PubMed] [Google Scholar]
- 20.Costerton JW, Irvin RT, Cheng KJ. 1981. The bacterial glycocalyx in nature and disease. Annual review of microbiology 35: 299–324. [DOI] [PubMed] [Google Scholar]
- 21.Comstock LE, Kasper DL. 2006. Bacterial glycans: key mediators of diverse host immune responses. Cell 126: 847–50. [DOI] [PubMed] [Google Scholar]
- 22.Lerouge I, Vanderleyden J. 2002. O-antigen structural variation: mechanisms and possible roles in animal/plant-microbe interactions. FEMS microbiology reviews 26: 17–47. [DOI] [PubMed] [Google Scholar]
- 23.Cress BF, Englaender JA, He W, Kasper D, et al. 2014. Masquerading microbial pathogens: capsular polysaccharides mimic host-tissue molecules. FEMS microbiology reviews 38: 660–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hegyi H, Gerstein M. 1999. The relationship between protein structure and function: a comprehensive survey with application to the yeast genome. Journal of molecular biology 288: 147–64. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Q, Raoof M, Chen Y, Sumi Y, et al. 2010. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Previato JO, Andrade AF, Pessolani MC, Mendonca-Previato L. 1985. Incorporation of sialic acid into Trypanosoma cruzi macromolecules. A proposal for a new metabolic route. Molecular and biochemical parasitology 16: 85–96. [DOI] [PubMed] [Google Scholar]
- 27.Tomlinson S, Pontes de Carvalho LC, Vandekerckhove F, Nussenzweig V. 1994. Role of sialic acid in the resistance of Trypanosoma cruzi trypomastigotes to complement. Journal of immunology 153: 3141–7. [PubMed] [Google Scholar]
- 28.Bouchet V, Hood DW, Li J, Brisson JR, et al. 2003. Host-derived sialic acid is incorporated into Haemophilus influenzae lipopolysaccharide and is a major virulence factor in experimental otitis media. Proceedings of the National Academy of Sciences of the United States of America 100: 8898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lewis AL, Hensler ME, Varki A, Nizet V. 2006. The group B streptococcal sialic acid O-acetyltransferase is encoded by neuD, a conserved component of bacterial sialic acid biosynthetic gene clusters. The Journal of biological chemistry 281: 11186–92. [DOI] [PubMed] [Google Scholar]
- 30.Varki A 2007. Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature 446: 1023–9. [DOI] [PubMed] [Google Scholar]
- 31.Macauley MS, Crocker PR, Paulson JC. 2014. Siglec-mediated regulation of immune cell function in disease. Nature reviews Immunology 14: 653–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crocker PR, Paulson JC, Varki A. 2007. Siglecs and their roles in the immune system. Nature reviews Immunology 7: 255–66. [DOI] [PubMed] [Google Scholar]
- 33.Girard-Pierce KR, Stowell SR, Smith NH, Arthur CM, et al. 2013. A novel role for C3 in antibody-induced red blood cell clearance and antigen modulation. Blood 122: 1793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stowell SR, Liepkalns JS, Hendrickson JE, Girard-Pierce KR, et al. 2013. Antigen modulation confers protection to red blood cells from antibody through Fcgamma receptor ligation. Journal of immunology 191: 5013–25. [DOI] [PubMed] [Google Scholar]
- 35.Carlin AF, Chang YC, Areschoug T, Lindahl G, et al. 2009. Group B Streptococcus suppression of phagocyte functions by protein-mediated engagement of human Siglec-5. The Journal of experimental medicine 206: 1691–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang YC, Olson J, Louie A, Crocker PR, et al. 2014. Role of macrophage sialoadhesin in host defense against the sialylated pathogen group B Streptococcus. Journal of molecular medicine 92: 951–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landsteiner K 1900. Zentbl Bakt Parasitkde (Abt) 27: 357–63. [Google Scholar]
- 38.Storry JR, Olsson ML. 2009. The ABO blood group system revisited: a review and update. Immunohematology / American Red Cross 25: 48–59. [PubMed] [Google Scholar]
- 39.Stowell SR, Girard-Pierce KR, Smith NH, Henry KL, et al. 2014. Transfusion of murine red blood cells expressing the human KEL glycoprotein induces clinically significant alloantibodies. Transfusion 54: 179–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stowell SR, Henry KL, Smith NH, Hudson KE, et al. 2013. Alloantibodies to a paternally derived RBC KEL antigen lead to hemolytic disease of the fetus/newborn in a murine model. Blood 122: 1494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Springer GF, Nichols JH, Callahan HJ. 1964. Galactosidase Action on Human Blood Group B Active Escherichia Coli and on Red Cell Substances. Science 146: 946–7. [DOI] [PubMed] [Google Scholar]
- 42.Springer GF, Williamson P, Brandes WC. 1961. Blood Group Activity of Gram-Negative Bacteria. The Journal of experimental medicine 113: 1077–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yi W, Shao J, Zhu L, Li M, et al. 2005. Escherichia coli O86 O-antigen biosynthetic gene cluster and stepwise enzymatic synthesis of human blood group B antigen tetrasaccharide. Journal of the American Chemical Society 127: 2040–1. [DOI] [PubMed] [Google Scholar]
- 44.Springer GF, Horton RE. 1969. Blood group isoantibody stimulation in man by feeding blood group-active bacteria. The Journal of clinical investigation 48: 1280–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stowell SR, Ju T, Cummings RD. 2015. Protein glycosylation in cancer. Annual review of pathology 10: 473–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cummings RD. 2009. The repertoire of glycan determinants in the human glycome. Molecular bioSystems 5: 1087–104. [DOI] [PubMed] [Google Scholar]
- 47.Wong CH. 2005. Protein glycosylation: new challenges and opportunities. The Journal of organic chemistry 70: 4219–25. [DOI] [PubMed] [Google Scholar]
- 48.Rillahan CD, Paulson JC. 2011. Glycan microarrays for decoding the glycome. Annual review of biochemistry 80: 797–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Merkle RK, Cummings RD. 1988. Asparagine-linked oligosaccharides containing poly-N-acetyllactosamine chains are preferentially bound by immobilized calf heart agglutinin. The Journal of biological chemistry 263: 16143–9. [PubMed] [Google Scholar]
- 50.Brewer CF. 2004. Thermodynamic binding studies of galectin-1, -3 and -7. Glycoconjugate journal 19: 459–65. [DOI] [PubMed] [Google Scholar]
- 51.Hirabayashi J, Hashidate T, Arata Y, Nishi N, et al. 2002. Oligosaccharide specificity of galectins: a search by frontal affinity chromatography. Biochimica et biophysica acta 1572: 232–54. [DOI] [PubMed] [Google Scholar]
- 52.Stowell SR, Arthur CM, Mehta P, Slanina KA, et al. 2008. Galectin-1, -2, and -3 exhibit differential recognition of sialylated glycans and blood group antigens. The Journal of biological chemistry 283: 10109–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blixt O, Head S, Mondala T, Scanlan C, et al. 2004. Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proceedings of the National Academy of Sciences of the United States of America 101: 17033–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ratner DM, Seeberger PH. 2007. Carbohydrate microarrays as tools in HIV glycobiology. Current pharmaceutical design 13: 173–83. [DOI] [PubMed] [Google Scholar]
- 55.Wang D, Liu S, Trummer BJ, Deng C, et al. 2002. Carbohydrate microarrays for the recognition of cross-reactive molecular markers of microbes and host cells. Nature biotechnology 20: 275–81. [DOI] [PubMed] [Google Scholar]
- 56.Fukui S, Feizi T, Galustian C, Lawson AM, et al. 2002. Oligosaccharide microarrays for high-throughput detection and specificity assignments of carbohydrate-protein interactions. Nature biotechnology 20: 1011–7. [DOI] [PubMed] [Google Scholar]
- 57.Stowell SR, Arthur CM, McBride R, Berger O, et al. 2014. Microbial glycan microarrays define key features of host-microbial interactions. Nature chemical biology 10: 470–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nilsson EC, Storm RJ, Bauer J, Johansson SM, et al. 2011. The GD1a glycan is a cellular receptor for adenoviruses causing epidemic keratoconjunctivitis. Nature medicine 17: 105–9. [DOI] [PubMed] [Google Scholar]
- 59.Walker LM, Huber M, Doores KJ, Falkowska E, et al. 2011. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477: 466–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carlsson S, Oberg CT, Carlsson MC, Sundin A, et al. 2007. Affinity of galectin-8 and its carbohydrate recognition domains for ligands in solution and at the cell surface. Glycobiology 17: 663–76. [DOI] [PubMed] [Google Scholar]
- 61.Gitt MA, Colnot C, Poirier F, Nani KJ, et al. 1998. Galectin-4 and galectin-6 are two closely related lectins expressed in mouse gastrointestinal tract. The Journal of biological chemistry 273: 2954–60. [DOI] [PubMed] [Google Scholar]
- 62.Kohatsu L, Hsu DK, Jegalian AG, Liu FT, et al. 2006. Galectin-3 induces death of Candida species expressing specific beta-1,2-linked mannans. Journal of immunology 177: 4718–26. [DOI] [PubMed] [Google Scholar]
- 63.Stowell SR, Arthur CM, Dias-Baruffi M, Rodrigues LC, et al. 2010. Innate immune lectins kill bacteria expressing blood group antigen. Nature medicine 16: 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stowell SR, Arthur CM, McBride R, Berger O, Razi N, Heimburg-Molinaro J, Rodrigues JP, Noll AJ, von Gunten S, Smith DF, Knirel YA, Paulson JC, Cummings RD 2014. Microbial Glycan Microarrays Define Key Features of Host-Microbial Interactions. Nature Chemical Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Knirel YA, Gabius HJ, Blixt O, Rapoport EM, et al. 2014. Human tandem-repeat-type galectins bind bacterial non-betaGal polysaccharides. Glycoconjugate journal 31: 7–12. [DOI] [PubMed] [Google Scholar]
- 66.Arthur CM, Cummings RD, Stowell SR. 2014. Using glycan microarrays to understand immunity. Curr Opin Chem Biol 18: 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lepur A, Salomonsson E, Nilsson UJ, Leffler H. 2012. Ligand induced galectin-3 protein self-association. The Journal of biological chemistry 287: 21751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stowell SR, Arthur CM, Slanina KA, Horton JR, et al. 2008. Dimeric Galectin-8 induces phosphatidylserine exposure in leukocytes through polylactosamine recognition by the C-terminal domain. The Journal of biological chemistry 283: 20547–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karmakar S, Stowell SR, Cummings RD, McEver RP. 2008. Galectin-1 signaling in leukocytes requires expression of complex-type N-glycans. Glycobiology 18: 770–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dam TK, Gabius HJ, Andre S, Kaltner H, et al. 2005. Galectins bind to the multivalent glycoprotein asialofetuin with enhanced affinities and a gradient of decreasing binding constants. Biochemistry 44: 12564–71. [DOI] [PubMed] [Google Scholar]
- 71.Kopitz J, von Reitzenstein C, Burchert M, Cantz M, et al. 1998. Galectin-1 is a major receptor for ganglioside GM1, a product of the growth-controlling activity of a cell surface ganglioside sialidase, on human neuroblastoma cells in culture. The Journal of biological chemistry 273: 11205–11. [DOI] [PubMed] [Google Scholar]
- 72.Perillo NL, Pace KE, Seilhamer JJ, Baum LG. 1995. Apoptosis of T cells mediated by galectin-1. Nature 378: 736–9. [DOI] [PubMed] [Google Scholar]
- 73.Demetriou M, Granovsky M, Quaggin S, Dennis JW. 2001. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature 409: 733–9. [DOI] [PubMed] [Google Scholar]
- 74.Toscano MA, Bianco GA, Ilarregui JM, Croci DO, et al. 2007. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nature immunology 8: 825–34. [DOI] [PubMed] [Google Scholar]
- 75.Stowell SR, Qian Y, Karmakar S, Koyama NS, et al. 2008. Differential roles of galectin-1 and galectin-3 in regulating leukocyte viability and cytokine secretion. Journal of immunology 180: 3091–102. [DOI] [PubMed] [Google Scholar]
- 76.Zhu C, Anderson AC, Schubart A, Xiong H, et al. 2005. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol 6: 1245–52. [DOI] [PubMed] [Google Scholar]
- 77.Stowell SR, Karmakar S, Stowell CJ, Dias-Baruffi M, et al. 2007. Human galectin-1, -2, and -4 induce surface exposure of phosphatidylserine in activated human neutrophils but not in activated T cells. Blood 109: 219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tribulatti MV, Mucci J, Cattaneo V, Aguero F, et al. 2007. Galectin-8 induces apoptosis in the CD4(high)CD8(high) thymocyte subpopulation. Glycobiology 17: 1404–12. [DOI] [PubMed] [Google Scholar]
- 79.Nishida A, Nagahama K, Imaeda H, Ogawa A, et al. 2012. Inducible colitis-associated glycome capable of stimulating the proliferation of memory CD4+ T cells. The Journal of experimental medicine 209: 2383–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dias-Baruffi M, Zhu H, Cho M, Karmakar S, et al. 2003. Dimeric galectin-1 induces surface exposure of phosphatidylserine and phagocytic recognition of leukocytes without inducing apoptosis. The Journal of biological chemistry 278: 41282–93. [DOI] [PubMed] [Google Scholar]
- 81.Frigeri LG, Zuberi RI, Liu FT. 1993. Epsilon BP, a beta-galactoside-binding animal lectin, recognizes IgE receptor (Fc epsilon RI) and activates mast cells. Biochemistry 32: 7644–9. [DOI] [PubMed] [Google Scholar]
- 82.Chen HY, Sharma BB, Yu L, Zuberi R, et al. 2006. Role of galectin-3 in mast cell functions: galectin-3-deficient mast cells exhibit impaired mediator release and defective JNK expression. J Immunol 177: 4991–7. [DOI] [PubMed] [Google Scholar]
- 83.Blois SM, Ilarregui JM, Tometten M, Garcia M, et al. 2007. A pivotal role for galectin-1 in fetomaternal tolerance. Nat Med. [DOI] [PubMed] [Google Scholar]
- 84.Espeli M, Mancini SJ, Breton C, Poirier F, et al. 2009. Impaired B-cell development at the pre-BII-cell stage in galectin-1-deficient mice due to inefficient pre-BII/stromal cell interactions. Blood 113: 5878–86. [DOI] [PubMed] [Google Scholar]
- 85.Tsai CM, Guan CH, Hsieh HW, Hsu TL, et al. 2011. Galectin-1 and galectin-8 have redundant roles in promoting plasma cell formation. J Immunol 187: 1643–52. [DOI] [PubMed] [Google Scholar]
- 86.Anginot A, Espeli M, Chasson L, Mancini SJ, et al. 2013. Galectin 1 modulates plasma cell homeostasis and regulates the humoral immune response. J Immunol 190: 5526–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Croci DO, Cerliani JP, Dalotto-Moreno T, Mendez-Huergo SP, et al. 2014. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell 156: 744–58. [DOI] [PubMed] [Google Scholar]
- 88.Markowska AI, Liu FT, Panjwani N. 2010. Galectin-3 is an important mediator of VEGF- and bFGF-mediated angiogenic response. The Journal of experimental medicine 207: 1981–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vespa GN, Lewis LA, Kozak KR, Moran M, et al. 1999. Galectin-1 specifically modulates TCR signals to enhance TCR apoptosis but inhibit IL-2 production and proliferation. Journal of immunology 162: 799–806. [PubMed] [Google Scholar]
- 90.Pace KE, Hahn HP, Pang M, Nguyen JT, et al. 2000. CD7 delivers a pro-apoptotic signal during galectin-1-induced T cell death. Journal of immunology 165: 2331–4. [DOI] [PubMed] [Google Scholar]
- 91.Hahn HP, Pang M, He J, Hernandez JD, et al. 2004. Galectin-1 induces nuclear translocation of endonuclease G in caspase- and cytochrome c-independent T cell death. Cell death and differentiation 11: 1277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Saravanan C, Liu FT, Gipson IK, Panjwani N. 2009. Galectin-3 promotes lamellipodia formation in epithelial cells by interacting with complex N-glycans on alpha3beta1 integrin. Journal of cell science 122: 3684–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rabinovich GA, Baum LG, Tinari N, Paganelli R, et al. 2002. Galectins and their ligands: amplifiers, silencers or tuners of the inflammatory response? Trends in immunology 23: 313–20. [DOI] [PubMed] [Google Scholar]
- 94.Cerliani JP, Stowell SR, Mascanfroni ID, Arthur CM, et al. 2011. Expanding the universe of cytokines and pattern recognition receptors: galectins and glycans in innate immunity. Journal of clinical immunology 31: 10–21. [DOI] [PubMed] [Google Scholar]
- 95.Arthur CM, Baruffi MD, Cummings RD, Stowell SR. 2015. Evolving mechanistic insights into galectin functions. Methods in molecular biology 1207: 1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yang RY, Yu L, Graham JL, Hsu DK, et al. 2011. Ablation of a galectin preferentially expressed in adipocytes increases lipolysis, reduces adiposity, and improves insulin sensitivity in mice. Proceedings of the National Academy of Sciences of the United States of America 108: 18696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vyakarnam A, Dagher SF, Wang JL, Patterson RJ. 1997. Evidence for a role for galectin-1 in pre-mRNA splicing. Molecular and cellular biology 17: 4730–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mazurek N, Conklin J, Byrd JC, Raz A, et al. 2000. Phosphorylation of the beta-galactoside-binding protein galectin-3 modulates binding to its ligands. The Journal of biological chemistry 275: 36311–5. [DOI] [PubMed] [Google Scholar]
- 99.Oka N, Nakahara S, Takenaka Y, Fukumori T, et al. 2005. Galectin-3 inhibits tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by activating Akt in human bladder carcinoma cells. Cancer research 65: 7546–53. [DOI] [PubMed] [Google Scholar]
- 100.Vasta GR. 2012. Galectins as pattern recognition receptors: structure, function, and evolution. Advances in experimental medicine and biology 946: 21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vasta GR, Ahmed H, Nita-Lazar M, Banerjee A, et al. 2012. Galectins as self/non-self recognition receptors in innate and adaptive immunity: an unresolved paradox. Frontiers in immunology 3: 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sato S, Nieminen J. 2004. Seeing strangers or announcing “danger”: galectin-3 in two models of innate immunity. Glycoconjugate journal 19: 583–91. [DOI] [PubMed] [Google Scholar]
- 103.Baum LG, Garner OB, Schaefer K, Lee B. 2014. Microbe-Host Interactions are Positively and Negatively Regulated by Galectin-Glycan Interactions. Frontiers in immunology 5: 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.van den Berg TK, Honing H, Franke N, van Remoortere A, et al. 2004. LacdiNAc-glycans constitute a parasite pattern for galectin-3-mediated immune recognition. Journal of immunology 173: 1902–7. [DOI] [PubMed] [Google Scholar]
- 105.Jouault T, El Abed-El Behi M, Martinez-Esparza M, Breuilh L, et al. 2006. Specific recognition of Candida albicans by macrophages requires galectin-3 to discriminate Saccharomyces cerevisiae and needs association with TLR2 for signaling. Journal of immunology 177: 4679–87. [DOI] [PubMed] [Google Scholar]
- 106.Pelletier I, Sato S. 2002. Specific recognition and cleavage of galectin-3 by Leishmania major through species-specific polygalactose epitope. The Journal of biological chemistry 277: 17663–70. [DOI] [PubMed] [Google Scholar]
- 107.Tasumi S, Vasta GR. 2007. A galectin of unique domain organization from hemocytes of the Eastern oyster (Crassostrea virginica) is a receptor for the protistan parasite Perkinsus marinus. Journal of immunology 179: 3086–98. [DOI] [PubMed] [Google Scholar]
- 108.Sato S, St-Pierre C, Bhaumik P, Nieminen J. 2009. Galectins in innate immunity: dual functions of host soluble beta-galactoside-binding lectins as damage-associated molecular patterns (DAMPs) and as receptors for pathogen-associated molecular patterns (PAMPs). Immunological reviews 230: 172–87. [DOI] [PubMed] [Google Scholar]
- 109.Debierre-Grockiego F, Niehus S, Coddeville B, Elass E, et al. 2010. Binding of Toxoplasma gondii glycosylphosphatidylinositols to galectin-3 is required for their recognition by macrophages. The Journal of biological chemistry 285: 32744–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Moody TN, Ochieng J, Villalta F. 2000. Novel mechanism that Trypanosoma cruzi uses to adhere to the extracellular matrix mediated by human galectin-3. FEBS letters 470: 305–8. [DOI] [PubMed] [Google Scholar]
- 111.Kleshchenko YY, Moody TN, Furtak VA, Ochieng J, et al. 2004. Human galectin-3 promotes Trypanosoma cruzi adhesion to human coronary artery smooth muscle cells. Infection and immunity 72: 6717–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Quattroni P, Li Y, Lucchesi D, Lucas S, et al. 2012. Galectin-3 binds Neisseria meningitidis and increases interaction with phagocytic cells. Cellular microbiology 14: 1657–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Okumura CY, Baum LG, Johnson PJ. 2008. Galectin-1 on cervical epithelial cells is a receptor for the sexually transmitted human parasite Trichomonas vaginalis. Cellular microbiology 10: 2078–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Levroney EL, Aguilar HC, Fulcher JA, Kohatsu L, et al. 2005. Novel innate immune functions for galectin-1: galectin-1 inhibits cell fusion by Nipah virus envelope glycoproteins and augments dendritic cell secretion of proinflammatory cytokines. Journal of immunology 175: 413–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pelletier I, Hashidate T, Urashima T, Nishi N, et al. 2003. Specific recognition of Leishmania major poly-beta-galactosyl epitopes by galectin-9: possible implication of galectin-9 in interaction between L. major and host cells. The Journal of biological chemistry 278: 22223–30. [DOI] [PubMed] [Google Scholar]
- 116.Yang ML, Chen YH, Wang SW, Huang YJ, et al. 2011. Galectin-1 binds to influenza virus and ameliorates influenza virus pathogenesis. Journal of virology 85: 10010–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mercier S, St-Pierre C, Pelletier I, Ouellet M, et al. 2008. Galectin-1 promotes HIV-1 infectivity in macrophages through stabilization of viral adsorption. Virology 371: 121–9. [DOI] [PubMed] [Google Scholar]
- 118.Ouellet M, Mercier S, Pelletier I, Bounou S, et al. 2005. Galectin-1 acts as a soluble host factor that promotes HIV-1 infectivity through stabilization of virus attachment to host cells. J Immunol 174: 4120–6. [DOI] [PubMed] [Google Scholar]
- 119.Ideo H, Seko A, Yamashita K. 2005. Galectin-4 binds to sulfated glycosphingolipids and carcinoembryonic antigen in patches on the cell surface of human colon adenocarcinoma cells. The Journal of biological chemistry 280: 4730–7. [DOI] [PubMed] [Google Scholar]
- 120.Ideo H, Fukushima K, Gengyo-Ando K, Mitani S, et al. 2009. A Caenorhabditis elegans glycolipid-binding galectin functions in host defense against bacterial infection. The Journal of biological chemistry 284: 26493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, et al. 2012. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482: 414–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shi XZ, Wang L, Xu S, Zhang XW, et al. 2014. A galectin from the kuruma shrimp (Marsupenaeus japonicus) functions as an opsonin and promotes bacterial clearance from hemolymph. PloS one 9: e91794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Feng C, Ghosh A, Amin MN, Giomarelli B, et al. 2013. The galectin CvGal1 from the eastern oyster (Crassostrea virginica) binds to blood group A oligosaccharides on the hemocyte surface. The Journal of biological chemistry 288: 24394–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rabinovich GA, Toscano MA, Jackson SS, Vasta GR. 2007. Functions of cell surface galectin-glycoprotein lattices. Current opinion in structural biology 17: 513–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Woodward AM, Mauris J, Argueso P. 2013. Binding of transmembrane mucins to galectin-3 limits herpesvirus 1 infection of human corneal keratinocytes. Journal of virology 87: 5841–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Argueso P, Guzman-Aranguez A, Mantelli F, Cao Z, et al. 2009. Association of cell surface mucins with galectin-3 contributes to the ocular surface epithelial barrier. The Journal of biological chemistry 284: 23037–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.St-Pierre C, Manya H, Ouellet M, Clark GF, et al. 2011. Host-soluble galectin-1 promotes HIV-1 replication through a direct interaction with glycans of viral gp120 and host CD4. Journal of virology 85: 11742–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bi S, Hong PW, Lee B, Baum LG. 2011. Galectin-9 binding to cell surface protein disulfide isomerase regulates the redox environment to enhance T-cell migration and HIV entry. Proceedings of the National Academy of Sciences of the United States of America 108: 10650–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Garner OB, Yun T, Pernet O, Aguilar HC, et al. 2015. Timing of galectin-1 exposure differentially modulates nipah virus entry and syncytium formation in endothelial cells. Journal of virology 89: 2520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Villalta F, Scharfstein J, Ashton AW, Tyler KM, et al. 2009. Perspectives on the Trypanosoma cruzi-host cell receptor interactions. Parasitology research 104: 1251–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gauthier S, Pelletier I, Ouellet M, Vargas A, et al. 2008. Induction of galectin-1 expression by HTLV-I Tax and its impact on HTLV-I infectivity. Retrovirology 5: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pais-Correia AM, Sachse M, Guadagnini S, Robbiati V, et al. 2010. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nature medicine 16: 83–9. [DOI] [PubMed] [Google Scholar]
- 133.Gupta SK, Masinick S, Garrett M, Hazlett LD. 1997. Pseudomonas aeruginosa lipopolysaccharide binds galectin-3 and other human corneal epithelial proteins. Infection and immunity 65: 2747–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kamhawi S, Ramalho-Ortigao M, Pham VM, Kumar S, et al. 2004. A role for insect galectins in parasite survival. Cell 119: 329–41. [DOI] [PubMed] [Google Scholar]
- 135.Rogerio AP, Cardoso CR, Fontanari C, Souza MA, et al. 2007. Anti-asthmatic potential of a D-galactose-binding lectin from Synadenium carinatum latex. Glycobiology 17: 795–804. [DOI] [PubMed] [Google Scholar]