

Abstract
Background
Alteration in hemodynamic shear stress at atheroprone sites promotes endothelial paracellular pore formation and permeability. The molecular mechanism remains unknown.
Methods and Results
We show that Nck (noncatalytic region of tyrosine kinase) deletion significantly ameliorates disturbed flow‐induced permeability, and selective isoform depletion suggests distinct signaling mechanisms. Only Nck1 deletion significantly reduces disturbed flow‐induced paracellular pore formation and permeability, whereas Nck2 depletion has no significant effects. Additionally, Nck1 re‐expression, but not Nck2, restores disturbed flow‐induced permeability in Nck1/2 knockout cells, confirming the noncompensating roles. In vivo, using the partial carotid ligation model of disturbed flow, Nck1 knockout prevented the increase in vascular permeability, as assessed by Evans blue and fluorescein isothiocyanate dextran extravasations and leakage of plasma fibrinogen into the vessel wall. Domain swap experiments mixing SH2 (phosphotyrosine binding) and SH3 (proline‐rich binding) domains between Nck1 and Nck2 showed a dispensable role for SH2 domains but a critical role for the Nck1 SH3 domains in rescuing disturbed flow‐induced endothelial permeability. Consistent with this, both Nck1 and Nck2 bind to platelet endothelial adhesion molecule‐1 (SH2 dependent) in response to shear stress, but only Nck1 ablation interferes with shear stress–induced PAK2 (p21‐activated kinase) membrane translocation and activation. A single point mutation into individual Nck1 SH3 domains suggests a role for the first domain of Nck1 in PAK recruitment to platelet endothelial cell adhesion molecule‐1 and activation in response to shear stress.
Conclusions
This work provides the first evidence that Nck1 but not the highly similar Nck2 plays a distinct role in disturbed flow‐induced vascular permeability by selective p21‐activated kinase activation.
Keywords: adaptor proteins Nck1 and Nck2, p21‐activated kinase, shear stress, vascular permeability
Subject Categories: Atherosclerosis, Vascular Disease
Nonstandard Abbreviations and Acronyms
- DKO
double knockout
- EBA
Evans blue albumin
- FITC
fluorescein isothiocyanate
- HAECs
human aortic endothelial cells
- Nck
noncatalytic region of tyrosine kinase
- KO
knockout
- OSS
oscillatory shear stress
- PAK
p21‐activated kinase
- PCL
partial carotid ligation
- PCR
polymerase chain reaction
- PECAM‐1
platelet endothelial cell adhesion molecule‐1
- shRNA
short hairpin RNA
- VEGF
vascular endothelial growth factor
Clinical Perspective
What Is New?
Disturbed flow at arterial curvatures and bifurcations promotes endothelial permeability and atherosclerotic plaque formation.
Understanding this process at a molecular level is critical to investigate plaque formation, regression, and treatment.
What Are the Clinical Implications?
Our data suggest an important role for noncatalytic region of tyrosine kinase 1 signaling in disturbed flow‐induced permeability.
By understanding how noncatalytic region of tyrosine kinase 1 mediates vascular permeability, there is potential for new drug development to limit vascular leak in a variety of cardiovascular pathologies.
The earliest stages of atherosclerosis involve local endothelial cell activation, characterized by enhanced endothelial permeability to plasma proteins and elevated expression of proinflammatory genes that facilitate leukocyte recruitment. Leak of plasma proteins into atherosclerosis‐prone regions precedes plaque formation,1, 2 and plasma protein exudate into coronary arteries is a sensitive index of human coronary atherosclerosis.3 In addition, multiple lines of evidence suggest that plasma protein leak into the subendothelial space perpetuates endothelial cell activation. Accumulation of albumin in the plaque may affect endothelial function by serving as a local source of advanced glycation end products or as a carrier for a variety of bioactive substances in the plasma.4 Plasma contains high levels of the provisional matrix proteins fibronectin and fibrinogen that leak into peripheral tissues following vessel injury, where they serve as a permissive matrix for local tissue remodeling.5 However, both fibronectin and fibrinogen promote endothelial activation associated with elevated permeability, proinflammatory signaling, and proinflammatory gene expression,1, 6, 7, 8 and mice deficient for plasma fibronectin show reduced atherogenic inflammation and atherosclerotic plaque formation.9 Therefore, methods to limit endothelial permeability may affect multiple aspects of endothelial activation and early plaque formation in vivo.
While most known risk factors for atherosclerosis should systemically affect vessel remodeling, atherosclerotic plaque formation occurs at distinct sites of blood vessels exposed to specific hemodynamics. Flow patterns regulate multiple aspects of atherosclerotic plaque formation through dynamic changes in endothelial permeability and leukocyte recruitment.10 Endothelial cells sense changes in wall shear stress and remodel their cytoskeleton to adapt to the applied forces.11 In atheroprotected regions, endothelial cells are aligned in the direction of flow, and the relatively uniform unidirectional laminar flow drives endothelial cells to adopt an atheroprotective phenotype with low endothelial permeability.11, 12, 13 In contrast, disturbed flow observed at atheroprone regions14 supports a sustained cytoskeletal remodeling response that compromises endothelial layer integrity, resulting in enhanced permeability to macromolecules such as albumin, fibronectin, and fibrinogen.2, 6, 11 Endothelial permeability to macromolecules can occur through multiple responses, such as transcellular vesicular transport (eg, low‐density lipoprotein) or paracellular pore formation (eg, plasma proteins).15 Disturbed flow induces paracellular pore formation through cytoskeletal remodeling and turnover of junctional proteins,16, 17 and regions of disturbed flow in vivo show discontinuous tight junctions.18, 19 Multiple molecular mechanisms have been linked to disturbed flow‐enhanced permeability, including activation of the serine/threonine kinase p21‐activated kinase (PAK) in both in vitro6 and in vivo16 atherogenic models. Activated PAK localizes to cell‐cell junctions and promotes paracellular pore formation through myosin‐dependent cytoskeletal contraction and adherens junction destabilization.16, 17 However, the mechanisms by which PAK is recruited to cell‐cell junctions in response to disturbed flow remains unknown.
The Nck (noncatalytic region of tyrosine kinase) family of adaptor proteins includes 2 proteins (Nck1 and Nck2) composed exclusively of SH2/SH3 domains that lack enzymatic functions but mediate protein‐protein interactions.20 Interactions between Nck's SH2 domain and tyrosine phosphorylated proteins recruits Nck to sites of active cell signaling, where it brings in other signaling mediators through interactions between its SH3 domains and proline‐rich sequences in target proteins.21 Nck signaling classically couples tyrosine phosphorylation to induction of actin cytoskeletal reorganization and cell movement.22 Consistent with this, genetic deletion of both Nck1 and Nck2, but not individual isoforms, impairs vascular development and endothelial angiogenesis.23 While the 2 Nck isoforms were believed to have overlapping functions, noncompensating roles have emerged in multiple pathological settings. For example, Nck1 and Nck2 can act independently in dermal fibroblasts through distinct Rho GTPase family members; Nck1 promoted Cdc42 signaling for filopodium formation and Nck2 promoted Rho signaling to induce stress fibers.24
Previous studies have shown that pretreating endothelial cells with a membrane‐permeable peptide corresponding to the Nck‐binding, proline‐rich sequence of PAK (Pak‐Nck peptide) reduces PAK targeting to cell‐cell junctions and blunts vascular permeability in atherosclerosis, ischemia‐reperfusion injury, and acute lung injury.6, 17, 25 However, the specificity of this peptide to Nck and the individual roles of Nck1 and Nck2 in disturbed flow‐induced permeability remain unknown. Therefore, we utilized both cell culture and animal models of selective Nck1 and Nck2 depletion to characterize Nck signaling in disturbed flow‐induced endothelial permeability.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
All reagents were provided from Gibco, USA, unless otherwise stated. All lentiviral vectors were designed and obtained using VectorBuilder website.
Cell Culture, Plasmids, and RNA Interference
Human aortic endothelial cells (HAECs; Cell Applications, Inc) were maintained in MCDB131 containing 10% (v/v) fetal bovine serum and cultured as we previously described.25 Nck1 and Nck2 were depleted from endothelial cells using lentiviral short hairpin RNA (shRNA) or CRISPR/Cas9 gene editing. The lentiviral vectors included pLV‐(shRNA)‐mCherry:T2A:Puro‐U6 for Nck1 (target seq: GGGTTCTCTGTCAGAGAAA) and Nck2 (target seq: CTTAAAGCGTCAGGGAAGA) with third‐generation lentivirus components provided from Didier Trono; pMD2.G (Addgene #12259), pRSV‐Rev (Addgene #12253), and pMDLg/pRRE (Addgene #12251).26 The lentiviral CRISPR/Cas9 plasmids utilized the pLV[CRISPR]‐hCas9:T2A:Puro‐U6>gRNA with the single‐guide RNA sequences targeting Nck1 (target seq: GTCGTCAATAACCTAAATAC), Nck2 (target seq: TGACGCGCGACCCCTTCACC) or scrambled single‐guide RNA (target seq: GCACTACCAGAGCTAACTCA). Transfection with SMARTpool small interfering RNA targeting Nck1 (L‐006354) and Nck2 (L‐019547; IDT) was performed using Lipofectamin 3000 transfection reagent (Invitrogen) according to the manufacturer's instructions. The lentiviral vectors encoding full‐length Nck1 and Nck2 were generated in the pLV‐EXP‐mCherry:T2A:Puro‐CMV>Nck vector. For the domain swap experiments, pLV‐EXP‐mCherry:T2A:Puro‐CMV>Nck constructs were generated containing the Nck1 SH2 domain and Nck2 SH3 domains or the Nck2 SH2 domain and Nck1 SH3 domains. Single point mutations in Nck1 SH3 domains were performed by the Redox Molecular Signaling Core (COBRE Center for Cardiovascular Diseases and Sciences, Louisiana State University Health Sciences Center Shreveport). Nck1/2 domains’ point mutations have been verified by cloning polymerase chain reaction (PCR) and sequencing (Eurofins Genomics) before being used for experiments. All cell lines were tested for the respective gene knocked down/knocked out before used for the experiments. For Nck1 SH3.1 inhibitor, HAECs were pretreated with AX‐024 for 30 minutes before shear stress exposure (50 nmol/L,27 Axon Medchem). Shear stress experiments were performed using the parallel plate flow chambers as we previously published.1, 16
In Vitro Permeability Assays and Immunocytochemistry
Endothelial cell barrier function was determined by assessing paracellular pore formation following immunostaining for PECAM (platelet endothelial cell adhesion molecule) (ab9498, 5 μg/mL; Abcam) and ß‐catenin (ab32572, 10 μg/mL; Abcam) as previously described.25 In vitro permeability was assessed using the streptavidin/biotinylated‐gelatin trapping assay as previously described.28 Briefly, silanized slides (Corning) were coated with biotinylated gelatin at 0.25 mg/mL overnight at 4°C. On the second day, endothelial cells were plated to confluence at equal seeding density of 2×106. The cells were subjected to oscillatory shear stress (OSS) 24 hours later using a syringe pump (±5 dynes/cm2 with a superimposed 1 dynes/cm2 for waste exchange for 18 hours). Immediately after cessation of flow, Streptavidin‐Alexa Fluor 647 (1:1000; Invitrogen) was added to the cells for 1 minute before the fixation by the addition of 3.7% (v/v) paraformaldehyde. Following subsequent washing and blocking, Alexa Fluor 488 Phalloidin (1:200; Invitrogen) was added to stain F‐actin filaments. Proximity ligation assay was performed as previously described25 and according to the manufacturer's recommendation. Images were captured from 5 random fields and analyzed using NIS‐Elements software (Nikon Instruments Inc).
Immunoprecipitation and Immunoblotting
For immunoprecipitation, cells were lyzed as previously described.25 After a preclearing incubation with Gammabind G beads (GE Healthcare Life Sciences) for 30 minutes and centrifugation, lysates were incubated for 2 hours with anti‐Nck1/2 (#06‐288, 0.5 μg/mL; Millipore), anti‐Nck1 (sc‐20026, 1:100; Santa Cruz), or anti–PECAM‐1 (ab9498, 10 μg/mL; Abcam). Cell lysates were then incubated with the Gammabind G beads for an additional 2 hours, and subsequently the beads were washed 4 times with the lysis buffer solution. The immunoprecipitated proteins were dissociated from the beads by addition of 2X laemmli buffer and boiling at 94°C for 5 minutes. Whole cell lysates or immunoprecipitated proteins were separated at equal protein concentration using SDS‐PAGE gel and transferred to polyvinylidene fluoride membranes (BioRad). Nonspecific antibody binding was blocked with 5% (w/v) nonfat milk/TBST and membranes were incubated with the following antibodies overnight at 4°C: anti‐Nck1 (#2319, 1:1000, Cell Signaling Technology, Inc), anti‐Nckß (ab109239, 0.45 μg/mL, Abcam), anti‐Nck1/2 (#06‐288, 0.1 μg/mL, Millipore), anti‐GAPDH (#2118, 1:10 000, Cell Signaling), anti–phospho‐PAK1/2/3 (Ser141, #44‐940G, 1:10 000, Invitrogen), or anti‐PAK2 (#2608, 1:1000, Cell Signaling Technology, Inc). After secondary incubation and chemiluminescence detection, densitometry was analyzed using ImageJ software (National Institutes of Health).
Animal Model
All animal work was performed according to the National Research Council's Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at LSU Health Sciences Center—Shreveport.
In Vivo Endothelial Nck1/2 Knockout
Male ApoE−/− mice on the C57BI/6J backgrounds were purchased from Jackson Laboratory. Mice that contained alleles Nck1−/− and Nck2fl/fl were a gift from Tony Pawson (Lunenfeld‐Tanenbaum Research Institute, University of Toronto). Nck1fl/fl were purchased from Cyagen Biosciences. To generate Nck1fl/fl, loxP sites flanking exon 2 were inserted in C57BI/6 embryonic stem cells. The targeted embryonic stem cell clones were then injected into C57BI/6 albino embryos, which were then reimplanted into CD‐1 pseudopregnant females. Their germline transmission was confirmed by breeding with C57BI/6J females and subsequent genotyping of their offspring. PCR screenings were then performed for loxP and neomycin deletion. Positive targeted mice were generated (homozygous) and then bred with tissue‐specific Cre delete mice to generate mice that are heterozygous for a targeted allele and a homozygous/heterozygous for the Cre transgene, for which they were inbred together to generate Nck1fl/fl animals. The tissue‐specific gene deletion was confirmed by a PCR assay using the following primers (loxP‐F (F1): 5′‐ATGTTGTCTAGGCCTCAGAGTTG‐3′, Neo‐del‐F (F2): 5′‐ACACAGGCATTTGAAGTAAAGCAAG‐3′, Neo‐del‐R (R2): 5′‐GATCACTGTTTCCTTAGGCTTTCTG‐3′. Mice that contained vascular endothelial cadherin (VE‐Cad CreERT2) were kindly provided from Dr Luisa Iruela‐Arispe (UCLA). Mice were crossed with ApoE−/− and VE‐Cad CreERT2 to generate endothelial‐specific (iEC) control mice (iEC‐control; ApoE−/−, VE‐Cad CreERT2tg/?), (iEC) Nck1 knockout (KO) mice (ApoE−/−, VE‐Cad CreERT2tg/?, Nck1fl/fl), (iEC) Nck2 KO mice (iEC‐Nck2 KO; ApoE−/−, VE‐Cad CreERT2tg/?, Nck2fl/fl), and (iEC) Nck1/2 double KO (DKO) mice (iEC‐Nck1/2 DKO; ApoE−/−, VE‐Cad CreERT2tg/?, Nck2fl/fl, Nck1−/−). At 8 to 9 weeks of age, all experimental mice were intraperitoneally injected with tamoxifen (1 mg/kg, Sigma) for 5 subsequent days to induce nuclear translocation of the CreERT2 transgene in the endothelium, resulting in excision and deletion of the floxed (loxP flanked) genes.
Method of Anesthesia
The surgery was performed under 4% (v/v) isoflurane/O2 to induce the anesthesia and then 2% (v/v) isoflurane/O2 to maintain the anesthesia. The level of anesthesia during surgery was assessed by absence of carpopedal reflexes.
Partial Carotid Ligation Model of Disturbed Flow
After 2 weeks of recovery from tamoxifen injection, the animals were subjected to partial carotid ligation (PCL) surgery as previously described.29 Briefly, after the induction of anesthesia, mouse neck was exposed and disinfected with betadine and a ventral midline incision was made. Left common carotid artery was exposed by blunt dissection and 3 of its 4 caudal branches were ligated, including left external carotid, internal carotid and occipital branch. The ligation was performed with 6‐0 sutures. The superior thyroid artery was left intact and that was to create an area of low OSS just below the ligation. The right carotid artery was left unligated and served as an internal control. The incision was then closed and mice were monitored in a heating pad chamber until recovery. A single subcutaneous injection of carprofen (0.5 mg/mL) was given as an analgesic immediately after surgery.
Disturbed flow patterns 2 days after ligation were assessed using high‐resolution Doppler ultrasound (VisualSonics VEVO3100 System) as we previously showed.29
In Vivo Permeability Assays
One week after ligation, in the ligated carotids just below the area of ligation, in vivo permeability was assessed with either Evans blue albumin (EBA) dye or fluorescein isothiocyanate (FITC)‐dextran (70 kDa) as previously reported30, 31 with slight modifications. Briefly, to assess permeability with EBA, mice were injected with 2% (w/v) EBA (200 μL, Sigma, E2129) retro‐orbitally. After 30 minutes, the animals were euthanized by isoflurane overdose and pneumothorax, and the vessels were flushed by intracardiac injection of PBS to remove residual intraluminal dye or tracer. The ligated left and unligated right carotid arteries were collected. The amount of EBA was assessed as we previously described.32 Collected tissues were air‐dried and incubated for 18 hours at 60°C with formamide (Fisher, 2 mL per 1 g of dry tissue). After centrifugation at 12 000 g for 30 minutes, supernatants were collected and concentrations of extracted EBA were calculated against a standard curve (ng/μL) using a spectrophotometer at a wavelength of 620 nm. To assess endothelial permeability with FITC‐dextran (70 kDa, Sigma, 46945), mice were retro‐orbitally injected with 200 μL of FITC‐dextran at a dose of 100 mg/mL. Mice were euthanized 30 minutes later, intracardically perfused with 10 mL of PBS. The right and left carotids were isolated, dry weighted, homogenized in PBS 1 mL per 1 g dry tissue, and centrifuged at 5000 g for 30 minutes. FITC‐dextran fluorescence (excitation 490 nm and emission 520 nm) was quantified using fluorescence microplate reader. Amount of FITC‐dextran (ng/μL) was calculated from a standard curve. All of the studies were performed on ApoE−/− background; mice were fed a normal chow diet for the PCL model since we aimed to investigate the effect of induced disturbed flow in the ligated left carotid arteries on the endothelial barrier.
Immunohistochemistry
Paraffin‐embedded sections of carotid arteries were stained as previously described.33 Heat‐mediated antigen retrieval pretreatment using 10 mmol/L Sodium Citrate buffer (Vector Biolabs) were used. Primary antibodies (CD31, sc‐1506, 1 μg/mL, Fibrinogen, Abcam, ab34269, 5 μg/mL) were incubated at 4°C overnight. 4′,6‐Diamidino‐2‐phenylindole was used as a nuclear counterstain. Images were captured with a Nikon microscope and analyzed using NIS‐Elements software.
Isolation of mRNA and Quantitative Real‐Time PCR
Total RNAs were extracted from mouse right and left carotid arteries using TRIzol flush as we previously reported.34 Intimal and medial/adventitial RNAs were snapped frozen in liquid nitrogen until analysis. iScript kit (Bio‐Rad Laboratories, Inc) was used for the reverse transcription reaction, and quantitative real‐time PCR was performed using SYBR Green Master Mix (Bio‐Rad Laboratories, Inc) according to the manufacturer's recommendations. Relative expressions of Nck1/2 in intimal and medial/adventitial tissues were normalized to ribosomal protein L13a (RPL13a) and analyzed by the delta‐delta‐CT method. The primers used were the following: mNCK1 (fwd): TCCTGCTGATGATAGCTTTGTTG, mNCK1 (rev): ACGATCACCTTGGTCCCTTTTAT, mNCK2 (fwd): GTCATAGCCAAGTGGGACTACA, mNCK2 (Rev): GCACGTAGCCTGTCCTGTT, RPL13a (fwd): GGGCAGGTTCTGGTATTGGAT, and RPL13a (rev): GGCTCGGAAATGGTAGGGG.
Plasma Cholesterol Measurement
Plasma total cholesterol was analyzed using a commercially provided kit as previously described.34
The investigators were blinded to the animal groups during the process of data collection and analysis.
Statistical Analysis
Data were analyzed using GraphPad Prism 8.0 (GraphPad Software) and are presented as median and interquartile range. Data were first tested for normality (Shapiro–Wilk test). For data passed the normality check, multiple comparisons, 1‐way ANOVA followed by Tukey post‐test or 2‐way ANOVA followed by Bonferroni post‐test were used. Data that did not show normal distribution were assessed by Kruskal–Wallis test. Statistical significance was achieved only when P<0.05.
Results
Deletion of Nck1/2 Reduces OSS‐Induced Endothelial Permeability
To test whether Nck1 and Nck2 are involved in shear stress–induced permeability, we first deleted Nck1 and Nck2 in HAECs using the lenti‐silencing system. Complete endothelial Nck1 and Nck2 deletion was confirmed using Western blot (Figure 1A). We previously demonstrated that shear stress increased endothelial permeability by paracellular pore formation.6 Compared with static controls, scrambled shRNA‐treated endothelial cells subjected to OSS showed a remarkable increase in paracellular pore formation that was significantly ablated in the Nck1/2 shRNA‐treated cells (Figure 1B and 1C, gaps between cells indicated by white arrows). Endothelial permeability was also assessed by 647 Alexa/streptavidin/biotinylated‐gelatin trapping assay. While there were no significant differences between the 2 cell types (scrambled shRNA versus Nck1/2 shRNA) at basal levels, there was significantly less permeability following shear stress in the Nck1/2 shRNA cells (Figure 1D and 1E). A similar observation was seen in Nck1/2 deleted genes in endothelial cells using the CRISPR/Cas9 system (Figure S1), suggesting that Nck1/2 adaptor proteins play a role in OSS‐induced endothelial permeability.
Figure 1. Ablation of Nck (noncatalytic region of tyrosine kinase) 1 and Nck2 adaptor proteins reduces shear stress–induced endothelial permeability.
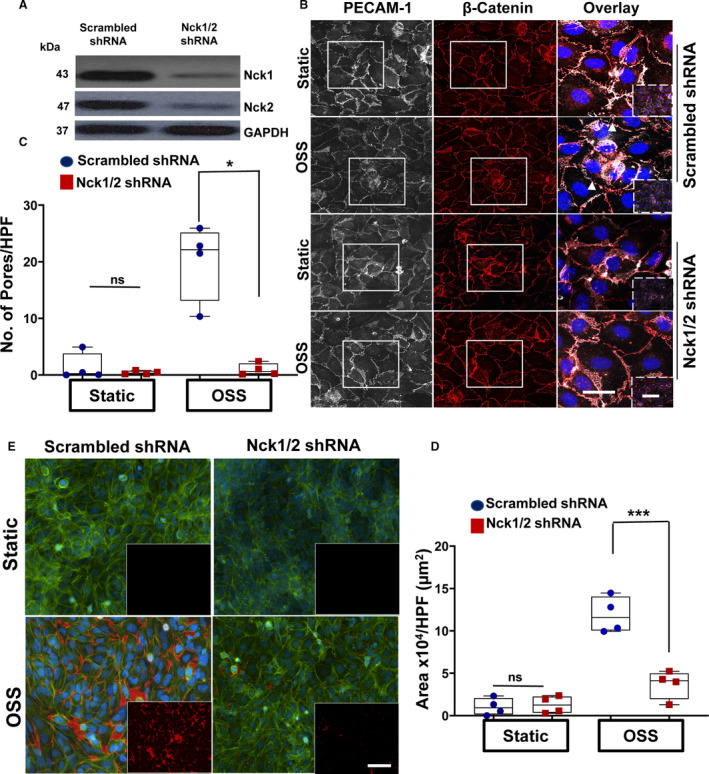
A, Representative blots showing the efficiency of the knocking down of Nck1/2 in human aortic endothelial cells (HAECs) after Nck1/2 short hairpin RNA (shRNA) or scrambled shRNA control transduction. B and C, PECAM‐1 (platelet endothelial cell adhesion molecule‐1; white)/ß‐catenin (red)–stained cells showing paracellular formation after oscillatory shear stress (OSS; ±5 dynes/cm2 with 1 dyne/cm2 forward flow) exposure. Images in the boxes indicate areas of higher magnifications, whereas areas in the dashed boxes indicate lower magnifications. Data analyzed by Kruskal‐Wallis test (*P=0.03). White arrowheads indicate paracellular pores. D/E, Nck1/2 knocking down prevents flow‐induced permeability using biotinylated gelatin/Alexa 647‐streptavidin assay. 4′,6‐Diamidino‐2‐phenylindole (blue; nuclei), phalloidin (green; F‐actin fibers), and streptavidin (red; exposed biotinylated gelatin). Images analyzed using NIS‐Elements software. Scale bars=50‐100 μm. Data are from n=4 independent experiments. Data are reported using box and whisker plots and analyzed by 2‐way ANOVA and Bonferroni posttest (***P<0.001).
Nck1 But Not Nck2 Ablation Decreases OSS‐Induced Permeability
Nck1 and Nck2 are known to have both redundant23 and nonredundant35 functions; therefore, we sought to determine whether the individual Nck proteins may differentially mediate this response. Using lentiviral shRNA delivery, we specifically knocked down Nck1 and Nck2 in endothelial cells and confirmed the depletion was isoform‐specific (Figure 2A). Only Nck1‐deleted cells showed significant amelioration in shear stress–induced paracellular pore formation, whereas Nck2‐ablated cells showed enhanced pore formation (Figure 2B and 2C). Similarly, the shear‐induced increase in streptavidin leak was significantly decreased in Nck1‐, but not Nck2‐, depleted cells (Figure 2D and 2E). Acute small interfering RNA–mediated Nck1 and Nck2 knockdown produced similar results (Figure S2), confirming that the Nck1 rather than Nck2 regulates endothelial permeability by oscillatory flow.
Figure 2. Deletion of Nck 1 but not Nck2 adaptor protein blunts shear stress–induced endothelial permeability.
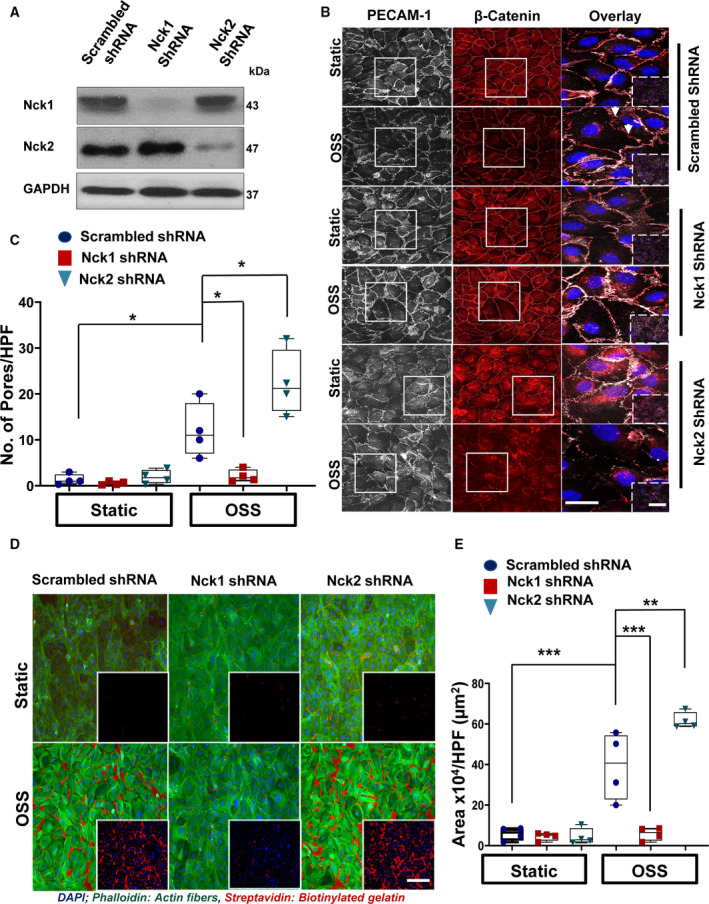
A, Representative blots showing the efficiency of the knocking down of Nck1/2 in human aortic endothelial cells (HAECs) after Nck1 short hairpin RNA (shRNA), Nck2 shRNA, or scrambled shRNA control transduction. B and C, PECAM‐1 (platelet endothelial cell adhesion molecule‐1; white)/ß‐catenin (red)–stained cells showing paracellular formation (white arrowheads) after oscillatory shear stress (OSS) exposure. D and E, Nck1 but not Nck2 knocking down prevents flow‐induced permeability using biotinylated gelatin/Alexa 647‐streptavidin assay. 4′,6‐Diamidino‐2‐phenylindole (blue; nuclei), phalloidin (green; F‐actin fibers), and streptavidin (red; exposed biotinylated gelatin). Images were analyzed using NIS‐Elements software. Scale bars=50 to 100 μm. Data are from n=4 and analyzed as mean±SEM by 2‐way ANOVA and Bonferroni posttest (*P<0.05, **P<0.01, ***P<0.001). Images in the boxes indicate areas of higher magnifications, whereas areas in the dashed boxes indicate lower magnifications.
To test the effect of endothelial Nck1 and Nck2 deletion on permeability in vivo, we generated conditional Nck1 KO mice by inserting loxP sites flanking exon 2 in C57BI/6 embryonic stem cells (Cyagen Biosciences) (Figure 3A). In vivo, areas of disturbed flow show elevated endothelial permeability.13 Therefore, to study the effects of Nck1 and Nck2 on OSS‐induced permeability in vivo, we elected to perform PCL to induce disturbed flow in the left common carotid as previously described.29 Following tamoxifen injection, endothelial‐specific deletion of Nck1 and Nck2 were verified in pulmonary endothelial cells isolated from iEC‐control, iEC‐Nck1, iEC‐Nck2 KO, and iEC‐Nck1/2 DKO using immunoblotting (Figure 3B). The inducible deletions of Nck1/2 were also verified in mRNA isolated from the carotid intima and medial/adventitia, showing a specific deletion of intimal Nck1/2 mRNA after tamoxifen injection (Figure S3A/B). There were no significant effects on medial/adventitial Nck1/2 mRNAs (Figure S3C/D). The 4 groups of mice then underwent PCL and the left carotid was subjected to disturbed flow for 7 days. The unligated right carotid serves as an internal control for nondisturbed flow (Figure 3C). Disturbed blood flow after ligation was confirmed using doppler ultrasound (Figure S4A/B). Among experimental groups, there were no significant changes in plasma cholesterol levels (Figure S4C).
Figure 3. Conditional endothelial deletion of Nck (noncatalytic region of tyrosine kinase) 1 but not Nck2 blunts partial carotid ligation–induced permeability.
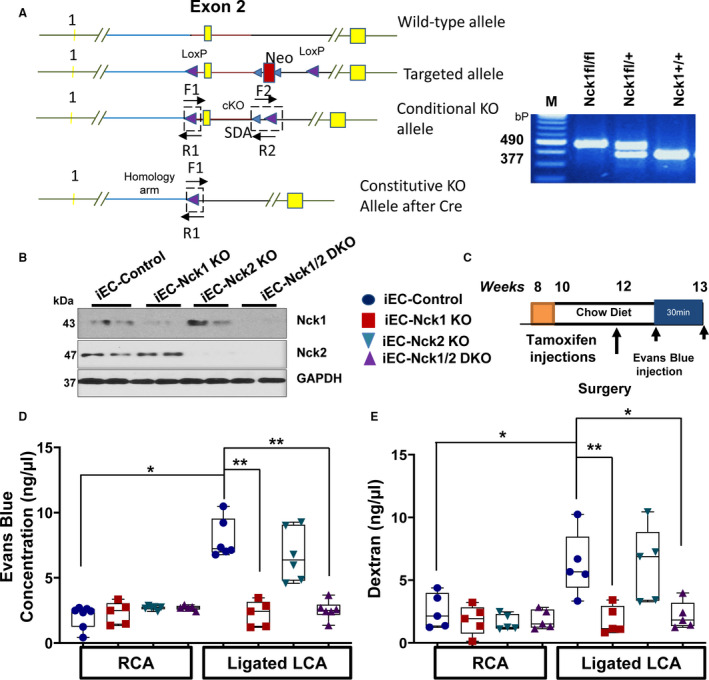
A, Schematic of targeting strategy to construct Nck1 fl/fl mice. Nucleotide substitutions were made in exon 2. Embryonic stem cells transfected with the targeting constructs were screened for homologous recombination by polymerase chain reaction (PCR). PCR showing heterozygous Nck1 fl/+ and homozygous Nck1 fl/fl mice with the expected products at 490 bp (targeted alleles) and wild‐type (Nck1+/+) at 377 bp. B, Pulmonary endothelial cells isolated from different experimental groups after tamoxifen injection and 2 weeks’ recovery, Nck1 and Nck2 expression were assessed by immunoblotting n=2 per group. C, Schematic of the study in which 4 groups of mice were subjected to the ligation surgery as indicated animal genotypes and time of surgery. D, Nck1 but not Nck2 knockout mice showed less Evans blue extravasation and (E) fluorescein isothiocyanate (FITC)‐dextran in ng/μL after ligation, n=5 or 6 per group. Data are expressed as mean±SEM and analyzed by 2‐way ANOVA and Bonferroni posttest (*P<0.05, **P<0.01) cKO indicates conditional knockouts; M, marker; Neo, neomycin; SDA, self‐deletion anchor site.
After 1 week, permeability was assessed by either EBA dye or FITC‐Dextran injections (Figure 3D and 3E). While there were no significant changes in EBA extravasation in the right (unligated) carotids among all experimental groups, only iEC‐Nck1 KO and iEC‐Nck1/2 DKO mice showed a 2‐fold reduction in EBA accumulation in the ligated left carotid (Figure 3D). Consistent with this, we also observed a significant accumulation of FITC‐dextran within the ligated left carotid in iEC‐control and iEC‐Nck2 KO mice, that was significantly less in iEC‐Nck1 KO and iEC‐Nck1/2 DKO mice (Figure 3E). To verify this isoform selective permeability effect, permeability was also assessed by measuring the leak of the plasma protein fibrinogen into the ligated carotid arteries. As expected, the leakage of plasma fibrinogen was significantly ameliorated in Nck1 KO and iEC‐Nck1/2 DKO mice compared with controls. However, Nck2 KO mice did not significantly affect fibrinogen staining (Figure S5). Taken together, these results indicate a crucial role for Nck1 but not Nck2 in regulating vascular permeability in vivo under disturbed flow.
Nck1 Regulates OSS‐Induced Permeability via its SH3 Domains’ Interactions
To characterize why Nck1 but not the highly homologous Nck2 is capable of regulating endothelial barrier function in response to OSS, we conducted domain swap experiments with chimeric proteins in which the Nck1 SH2 domain was paired with Nck2 SH3 domains or the Nck2 SH2 domain was paired with the Nck1 SH3 domains (Figure 4A). Re‐expression of full‐length Nck1, Nck2, or the chimeric proteins Nck1 SH2/Nck2 SH3 or Nck2 SH2/Nck1 SH3 in Nck1/2 DKO HAECs were confirmed by mCherry fluorescence (data not shown) and Western blotting and was comparable to the Nck1/2 endogenous levels in wild‐type cells (Figure 4B). The re‐expression of Nck1 but not Nck2 in Nck1/2 DKO cells rescued the permeability response to OSS (Figure 4C and 4D), confirming the noncompensating roles. The chimera containing the Nck1 SH2 domain and Nck2 SH3 domains failed to rescue the permeability response, whereas the chimera containing the Nck2 SH2 and Nck1 SH3 domains significantly enhanced disturbed flow‐induced endothelial permeability (Figure 4C and 4D). The data suggest that Nck1 SH3 domains are critical to the permeability response of OSS, whereas the Nck1 and Nck2 SH2 domains are both capable of targeting Nck to plasma membrane (Figure 4E). Together, these data reveal that Nck1 SH3 domains (1–3) mediate the isoform‐selective effects of Nck1 on OSS‐induced permeability, whereas the SH2 domains of Nck1 and Nck2 are redundant in this regard.
Figure 4. SH3 domains of Nck (noncatalytic region of tyrosine kinase) 1 are essential for flow‐induced endothelial permeability.
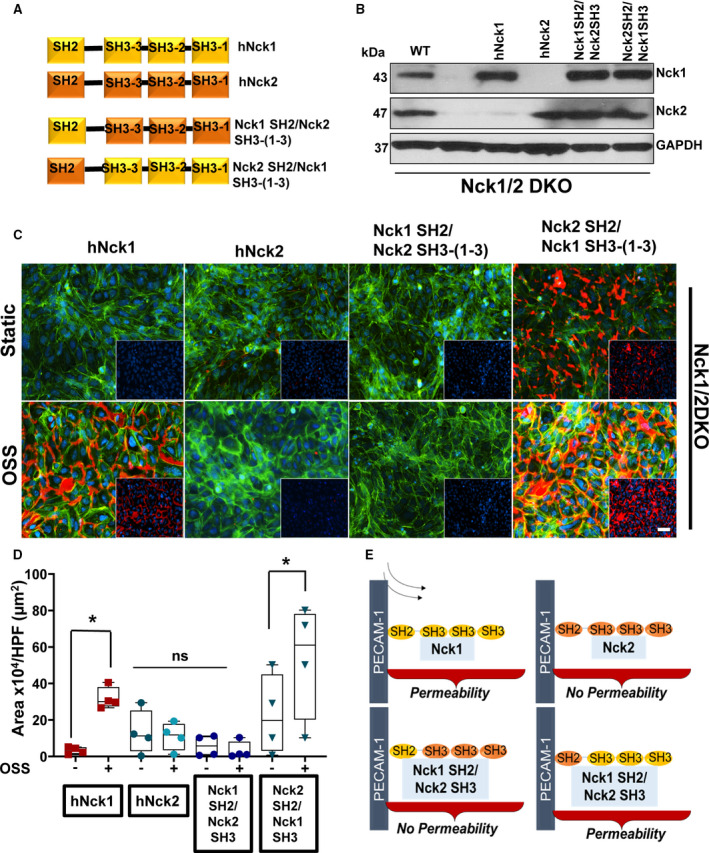
A, Schematic showing the domain structure of Nck1 and Nck2 and 2 chimera of Nck1 SH2/SH3 and Nck1 SH3/Nck2 SH2. B, Western blot analysis showing comparable Nck1/2 re‐expression following transduction of constructs in (A) in Nck1/2 double knockout (DKO) cells. C and D, Permeability of endothelial cells after oscillatory shear stress (OSS) as assessed by streptavidin‐biotinylated gelatin trapping assay. Data are analyzed by 2‐way ANOVA and Bonferroni posttest (*P<0.05; ns, nonsignificant, from n=4). E, Schematic diagram suggesting that Nck1/2 bind to PECAM‐1 (platelet endothelial cell adhesion molecule‐1) in response to shear stress, but only Nck1 through its SH3 domains regulates the permeability.
Nck1 Mediates Permeability by Promoting PAK Recruitment to PECAM‐1
We previously showed that the tyrosine phosphorylated PECAM‐1 in the endothelial adherens junction recruits Nck1/2 in response to oxidative stress36; however, its role in Nck1/2 recruitment in response to OSS is unclear. Following shear stress, there was a significant increase of PECAM‐1 (130 kDa) interactions with Nck1 (43 kDa) and Nck2 (47 kDa) in HAECs as shown by coimmunopreciptation (Figure 5A and 5B). The interactions between Nck1/2 and PECAM‐1 were also confirmed using immunostaining (Figure 5C). Our data presented so far suggested that even though in response to shear stress both Nck1 and Nck2 bind to PECAM‐1 through their SH2 domains, the permeability induced by shear stress is indeed Nck1‐mediated. Previous reports suggested a critical role for endothelial PAK2 in regulating endothelial permeability,6, 16, 37 and Nck1 and Nck2 signaling can promote PAK's membrane's translocation enhancing its activation.36, 38 To test whether shear stress induces PECAM‐1/PAK interactions, we performed coimmunoprecipitation experiments and detected a time‐dependent increase in phospho‐PAK S141 (active PAK) coimmunoprecipitation with PECAM‐1 (Figure 5D). To investigate whether shear stress enhances PAK/PECAM‐1 interactions in a Nck1‐dependent fashion, we performed a proximity ligation assay between PAK and PECAM‐1 in the presence or absence of Nck1. Consistent with coimmunoprecipitation experiments, shear stress enhanced PAK interaction with PECAM‐1 as assessed by proximity ligation assay (Figure 5E). However, this interaction was ablated by Nck1 depletion (Figure 5E). Furthermore, Nck1 knockdown significantly decreased PAK activation following both acute‐onset shear stress (Figure 5F and 5G) and chronic OSS (Figure S6). Importantly, Nck2 has no role in PAK activation by shear stress as Nck2‐depleted cells did not show any significant effects (Figure 5F and 5G). Taken together, the data suggest that Nck1 is required to promote OSS‐induced PAK activation and interaction with PECAM‐1.
Figure 5. Nck (noncatalytic region of tyrosine kinase) 1 and Nck2 bind to PECAM‐1 (platelet endothelial cell adhesion molecule‐1) in response to shear stress but Nck1 is required for bridging PAK (p21‐activated kinase) 2/PECAM‐1 and activation.
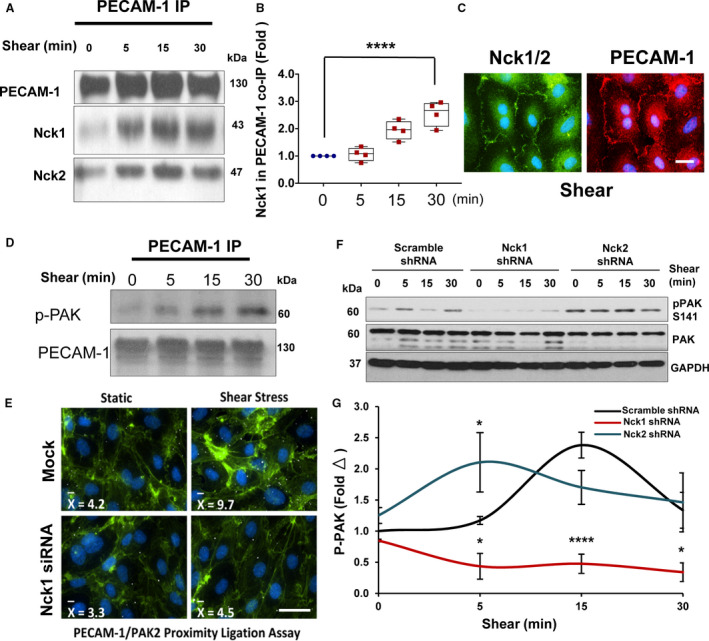
A, Immunoblotting showing PECAM‐1 coimmunoprecipitations and confirms the interactions with Nck1 and Nck2 are shear stress dependent. B, Graphical representation of the levels of Nck1 in PECAM‐1 coimmunopreciptation showing statistical significance. C, Immunofluorescence staining to PECAM‐1 (red) and Nck1/2 (green) after shears stress. Scale bar=50 μm. D, phospho‐PAK2 is detected in PECAM‐1 coimmunopreciptation and (E) PAK membrane translocation and PECAM‐1 binding is enhanced by shear stress and impaired after knocking down of Nck1. F and G, PAK2 activation as assessed by phosphorylation is downregulated by Nck1 ablation but not by Nck2 deletion. Western blots are from n=4 independent experiments. Data are mean±SEM analyzed by either 1‐way ANOVA and Tukey posttest or 2‐way ANOVA and Bonferroni posttest (*P<0.05, **** P<0.0001).
SH3 Domain 1 of Nck1 is Required to Regulate Permeability and PAK/PECAM‐1 Interaction
Since Nck1 is required for OSS‐induced PAK activation and Nck1 SH3 domains are capable of binding to PAK,36 we next sought to isolate which of the 3 domains are most critical to the PAK‐Nck interaction in response to shear stress. To test this, we re‐expressed either a full‐length Nck1 or Nck1 with individual point mutations that inactivate the proline‐binding functions of the 3 SH3 domains: (Nck1 SH3.3*; W229K), (Nck1 SH3.2*; W143K), and (Nck1 SH3.1*; W38K) (Figure 6A). After confirming the individual re‐expression efficiency of the Nck1 constructs in HAECs that lack Nck1/2 (Nck1/2 DKO) (Figure 6B), the cells were subjected to OSS and tested for permeability. While cells expressing Nck1 and the variants Nck1 SH3.3* and Nck1 SH3.2* still showed normal disturbed flow‐induced permeability, cells expressing the Nck1 SH3.1* mutant did not restore disturbed flow‐induced endothelial permeability (Figure 6C and 6D), suggesting that Nck1 regulates permeability through its first SH3 domain. Consistent with an important role for this domain in disturbed flow‐induced permeability, shear stress–induced PECAM‐1/PAK coimmunoprecipitation (Figure 6E) and PAK activation (Figure 6F) in endothelial cells expressing wild‐type Nck1 but not the Nck1 SH3.1* mutant (Figure 6E and 6F). Interestingly, pretreatment of HAECs with AX‐024 (an inhibitor to Nck1 SH3.1) reduced PAK‐Nck1 interactions in response to shear stress (Figure S7), revealing that Nck1 interacts with PAK through its SH3.1 to mediate its activation in response to OSS.
Figure 6. Nck (noncatalytic region of tyrosine kinase) 1 first SH3 domain regulates PAK2 (p21‐activated kinase 2)‐induced endothelial permeability.
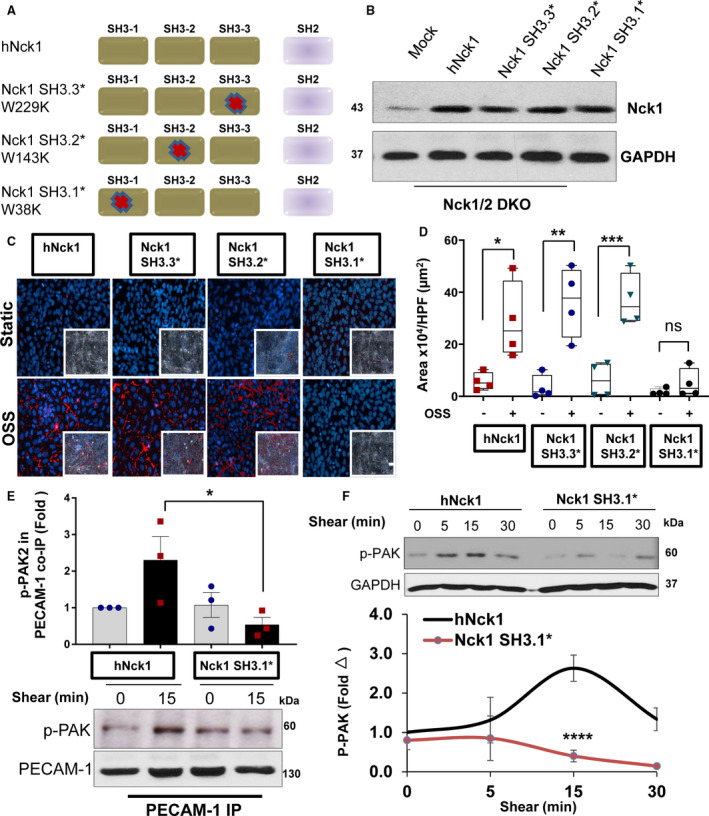
A, Schematic of Nck1 point mutations in each of the SH3 domains and the nomenclature used to inactivation of SH3 domains. B, Lysates from human aortic endothelial cells (HAECs) transiently transfected with Nck1 variants with inactivated SH3 domains. C and D, Flow‐induced permeability using biotinylated gelatin/Alexa 647‐streptavidin assay. 4′,6‐Diamidino‐2‐phenylindole (blue; nuclei), phalloidin (green; F‐actin fibers), and streptavidin (red; exposed biotinylated gelatin) Images analyzed using NIS‐Elements software from n=4. Scale bar=200 μm. Data were reported as box and whiskers with scatter plots and analyzed by Kruskal–Wallis test (*P<0.05,**P<0.01, ***P<0.001). E, Lysates and graphical quantifications from HAECs Nck1/2 double knockout (DKO) transiently transfected with either hNck1 or Nck1 SH3.1* and were immunoprecipitated for PECAM‐1 (platelet endothelial cell adhesion molecule‐1). Blotted for phospho‐PAK2 S141. F, HAECs Nck1/2 DKO either transfected with hNck1 or Nck1 SH3.1* were lyzed after shear stress exposure (0–30 minutes) and immunoblotted for phospho‐PAK2 S141 and GAPDH served as a loading control. Data are from n=3‐4, expressed as mean±SEM, and analyzed by 2‐way ANOVA and Bonferroni posttest (*P<0.05,**** P<0.0001).
Discussion
In summary, we have identified a novel role for the Nck1 adaptor protein in regulating disturbed flow‐induced endothelial permeability, and we showed through its interaction with PECAM‐1 that it recruits PAK to PECAM‐1, facilitates PAK's activation. To date, the association of Nck and PAK has been ascribed functional significance primarily as a signaling interaction upstream of actin cytoskeletal reorganization.25, 36 Here, we extend this interaction by showing that Nck1 but not Nck2 is capable of using its SH3.1 domain to bridge PAK with PECAM‐1 to promote OSS‐induced endothelial permeability.
The vascular endothelium regulates the selective exchange of water, solutes, and plasma proteins between flowing blood and surrounding tissues.39 A significant limitation to our model system is the limited analysis of only permeability to plasma proteins and not the permeability to water and solutes. However, the role of permeability in atherosclerotic inflammation primarily involves the leak of plasma proteins into the growing neointima, whereas solute exchange and water flow out of the vessel play a major role in tissue edema during inflammatory responses.15 Hemodynamic shear stress acts locally to regulate vascular permeability by controlling cytoskeletal remodeling and endothelial cell‐cell junctional stability.40 While laminar flow improves the barrier integrity, dysregulation of this barrier has been observed at atheroprone areas secondary to disturbed oscillatory blood flow.41 A second limitation to this model is that the changes in endothelial layer were compared with that of static conditions in vitro and to areas of stable laminar flow in vivo. However, both models involve endothelial cells responding to a change in their flow environment, and the current models of endothelial mechanotransduction suggest that these changing hemodynamic forces dominantly regulate the endothelial activation response to disturbed flow at atheroprone sites.11 A final limitation to the study is the sole focus on oscillatory flow in the PCL model, instead of other potential mechanical stimuli such as an increase in local pressure. Consistent with enhanced pressure in this model, extending the model to time points beyond our current study (≈3 weeks) results in vessel stiffening.42 However, local pressure was shown to not affect the uptake of albumin and low‐density lipoprotein in the porcine aorta,43 suggesting that any change in pressure is not likely to play a dominant role in the local permeability response.
In the present study we reveal that formation of paracellular pores and subsequently enhanced permeability in response to disturbed flow is prevented by selective depletion of Nck1 but not Nck2. The 2 highly similar Nck proteins (Nck1 and Nck2) are expressed by different genes44 that play redundant roles during development, as deletion of both Nck isoforms results in embryonic lethalities caused by impaired vasculogenesis, while deletion of only one isoform does not.23 Nck1 and Nck2 also play redundant roles in regulating angiogenesis in mouse models of retinopathy45; however, individual nonredundant roles have recently emerged. For example, Nck2 plays a dominant role in platelet‐derived growth factor–induced actin polymerization in NTH3T346 and nerve growth factor–induced axon and dendrite tree in primary rat cortical neurons.24 By contrast, Nck1 plays a more prominent role in T‐cell receptor–induced ERK activation and inflammation,47 suggesting the noncompensating roles are during phenotypic regulation postdevelopment and points to differences in specific binding affinities between Nck1 and Nck2, despite their overall sequence homology.22 The physiological significance of the noncompensating roles of Nck1/2 in cardiovascular disease has yet to be determined. Our study shows for the first time that in vivo endothelial deletion of Nck1 remarkably reduced the permeability induced by disturbed flow underscoring the physiological relevance of Nck1 within the vascular endothelium. Even though single Nck2 KO showed a slight increase in endothelial permeability, Nck1/2 DKO showed less permeability. While we did not observe any remarkable changes in their endogenous levels following either Nck1 or Nck2 deletion, Nck2 depletion might alter Nck1 signaling interactions and thus promots the slight increase in permeability. In support of this, a previous report by Kebache and colleagues48 showed that Nck2 depletion in HEK293 cells enhanced Nck1 signaling activation.
Nck1 and Nck2 share ≈68% amino acid identity22 and it was suggested that they differ with respect to their SH2 (phosphotyrosine) or SH3 (proline‐rich) binding properties.49 For example, Nck2 through its SH2 domain preferentially binds to platelet‐derived growth factor to regulate actin polymerization.46 Nck1, on the other hand, is shown to interact with RhoA through its SH3 domains to promote actin stress fibers.50 This points to the differences in the specific binding affinities of Nck1 and Nck2 SH2/SH3 domains. With the domain‐swapping experiments, now we provide evidence for the redundant binding of Nck1/2's SH2 domains and a nonredundant function for Nck1 SH3 domains. Recruitment of Nck to the plasma membrane typically involves enhanced interactions between Nck's SH2 domain and phosphotyrosine residues.51 Although multiple proteins in the endothelial cell adherens junctions can be tyrosine phosphorylated, shear stress stimulates phosphorylation of the vascular endothelial growth factor (VEGF) receptor VEGFR2 and the cell‐cell adhesion molecule PECAM‐1. Nck recruitment to VEGFR2 mediates VEGF‐induced focal adhesion turnover, actin remodeling, and cell migration.52 However, shear stress does not stimulate VEGFR2 interactions with Nck.53 Alternatively, we found that OSS promotes the coimmunopreciptation of Nck1/2 and PECAM‐1. Moreover, we and others demonstrated that Nck binds to tyrosine phosphorylated PECAM‐1 via its SH2 domain.25, 54 We now show a nonredundant role for Nck1 in endothelial permeability by illustrating that despite the fact that both Nck1 and Nck2 bind to PECAM‐1, only Nck1 SH3 (1–3) domains are involved in regulating endothelial permeability, demonstrating that a Nck1‐specific downstream mediator is involved in OSS‐induced endothelial permeability.
Previous work from our group and others demonstrates an important role for the Ser/Thr kinase PAK in endothelial permeability.6, 16 PAK signaling can promote endothelial barrier function55, 56; however, it has been demonstrated that endothelial deletion of PAK2 increased vascular permeability.57 These conflicting data suggest that endothelial PAK signaling can both promote and limit endothelial permeability, with the signaling context likely tuning PAK's functional effects. Nck binding is likely to contribute to context‐dependent PAK signaling, as a PAK‐Nck–blocking peptide reduces permeability and inflammation in a variety of pathological conditions.6, 25 Both Nck1 and Nck2 can promote PAK membrane translocation and activation,36 yet, depending on the cellular context, PAK could be preferentially activated by either one of them. For instance, Thevenot et al58 demonstrated that Nck2 rather than Nck1 preferably recruits PAK to mediate synaptic transmission. It is intriguing to speculate that OSS enables Nck1 (and not Nck2) to mediate the changes in PAK‐induced endothelial permeability. Therefore, Nck1 deletion may not limit all PAK activity but rather the specific PAK signaling at the cell‐cell junctions that is required to promote permeability.
Moreover, here we propose that the nonredundant effect of Nck1 on PAK activation is caused by the first SH3 domain of Nck1. Previous studies suggest that Nck binds to PAK through its second SH3 domain.38, 59 Here we make the unanticipated observation that specific point mutations to inactivate Nck1 SH3.2 did not show any effect on endothelial permeability, whereas the first Nck1 SH3 domain is required for the permeability effect. A limitation to previous studies on PAK binding to Nck SH3.2 were that Nck1 domains and PAK interactions have been tested outside a cellular environment, in vitro. In support of Nck1 SH3.1 and PAK, Bokoch et al60 showed that each domain of Nck SH3 could bind with PAK depending on the availability of domains to bind. Future studies are clearly warranted, however, to identify the impact of PAK binding on the second and third domains of Nck and whether that might mediate some other cellular responses. However, at least in OSS‐induced permeability, Nck1 SH3.1 domain interactions are required for PAK activation.
Nck1 SH3.1 differs from Nck2 SH3.1 in that it exhibits a significantly weaker negative charge22 because of the presence of a DY pocket,61 a shallow pocket in SH3.1, but not in other domains of Nck SH3(2‐3). The DY pocket is uncommon and has only been described in a handful of proteins, including the Eps8 family of proteins.62 The physiological relevance of this pocket in Nck is unclear. Interestingly, it has been reported that a small molecule inhibitor of the Nck1 SH3.1 DY pocket (AX‐024) limits T‐cell function and reduces inflammation.27 In our system, pretreatment of endothelial cells with AX‐024 reduces PAK/Nck1 interactions, suggesting that AX‐024 may serve as a therapy to reduce endothelial permeability in vivo. A limitation to inhibiting Nck signaling in atherosclerosis is that Nck adaptor proteins critically regulate angiogenesis, suggesting that Nck inhibition would be detrimental in conditions where angiogenesis promotes proper healing, such as ischemic injury. Since multiple groups have shown that both Nck1 and Nck2 must be inhibited to reduce angiogenesis,45 our data suggest that by selective inhibition of Nck1 may allow for improved barrier function without compromising the angiogenic response.
Conclusions
It has been widely hypothesized that PECAM‐1 must be involved in sensing and responding to mechanical signals, including hemodynamic shear stress.63 We now present Nck1 as a molecular bridge, integrating PECAM‐1 tyrosine phosphorylation at the adherens junction and PAK Ser/Thr kinase activation. These findings support the burgeoning role of Nck1 in mechanosensing, and they have implications for our understanding of the endothelial response to shear stress and endothelial barrier regulation.
Sources of Funding
This work was supported by a Malcolm Feist Postdoctoral Fellowship and an American Heart Association Postdoctoral Fellowship (grant number 20POST35120288) to Alfaidi and the National Institutes of Health (grants HL098435, HL133497, HL141155, and GM121307) to Orr.
Disclosures
None.
Supporting information
Figures S1–S7
Acknowledgments
The authors thank the late Dr Tony Pawson (Lunenfeld‐Tanenbaum Research Institute, University of Toronto) (Nck1ko and Nck2flox/flox mice), and Dr Luisa Iruela‐Arispe (UCLA) (VeCad Cre mice). Special thanks to Redox Molecular Signaling Core (P20GM121307) (COBRE Center for Cardiovascular Diseases and Sciences, LSU Health Sciences Center—Shreveport) for the site‐directed mutagenesis. Author contributions: M.A. performed experiments, data collection, interpretation of data and analysis, and article writing; U.B. performed PECAM‐1 coimmunopreciptation experiments and collect data; and A.W.O., performed experimental design and article writing.
(J Am Heart Assoc. 2020;9:e016099 DOI: 10.1161/JAHA.120.016099.)
For Sources of Funding and Disclosures, see page 15.
References
- 1. Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA. The subendothelial extracellular matrix modulates NF‐kappaB activation by flow: a potential role in atherosclerosis. J Cell Biol. 2005;169:191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. LaMack JA, Himburg HA, Li XM, Friedman MH. Interaction of wall shear stress magnitude and gradient in the prediction of arterial macromolecular permeability. Ann Biomed Eng. 2005;33:457–464. [DOI] [PubMed] [Google Scholar]
- 3. Zhang YX, Cliff WJ, Schoefl GI, Higgins G. Plasma‐protein insudation as an index of early coronary atherogenesis. Am J Pathol. 1993;143:496–506. [PMC free article] [PubMed] [Google Scholar]
- 4. Popov D, Simionescu M. Cellular mechanisms and signalling pathways activated by high glucose and age‐albumin in the aortic endothelium. Arch Physiol Biochem. 2006;112:265–273. [DOI] [PubMed] [Google Scholar]
- 5. Barker TH, Engler AJ. The provisional matrix: setting the stage for tissue repair outcomes. Matrix Biol. 2017;60–61:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Orr AW, Stockton R, Simmers MB, Sanders JM, Sarembock IJ, Blackman BR, Schwartz MA. Matrix‐specific p21‐activated kinase activation regulates vascular permeability in atherogenesis. J Cell Biol. 2007;176:719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cao Y, Zhou X, Liu H, Zhang Y, Yu X, Liu C. The Nf‐kappa B pathway: regulation of the instability of atherosclerotic plaques activated by Fg, Fb, and FDPS. Mol Cell Biochem. 2013;383:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tyagi N, Roberts AM, Dean WL, Tyagi SC, Lominadze D. Fibrinogen induces endothelial cell permeability. Mol Cell Biochem. 2008;307:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rohwedder I, Montanez E, Beckmann K, Bengtsson E, Duner P, Nilsson J, Soehnlein O, Faessler R. Plasma fibronectin deficiency impedes atherosclerosis progression and fibrous cap formation. EMBO Mol Med. 2012;4:564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tabas I, Garcia‐Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol. 2009;10:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Berk BC, Min W, Yan C, Surapisitchat J, Liu YM, Hoefen R. Atheroprotective mechanisms activated by fluid shear stress in endothelial cells. Drug News Perspect. 2002;15:133–139. [DOI] [PubMed] [Google Scholar]
- 13. Himburg HA, Grzybowski DM, Hazel AL, LaMack JA, Li XM, Friedman MH. Spatial comparison between wall shear stress measures and porcine arterial endothelial permeability. Am Physiol Heart Circ Physiol. 2004;286:H1916–H1922. [DOI] [PubMed] [Google Scholar]
- 14. Alfaidi MA, Chamberlain J, Rothman A, Crossman D, Villa‐Uriol MC, Hadoke P, Wu J, Schenkel T, Evans PC, Francis SE. Dietary docosahexaenoic acid reduces oscillatory wall shear stress, atherosclerosis, and hypertension, most likely mediated via an IL‐1‐mediated mechanism. J Am Heart Assoc. 2018;7:e008757 DOI: 10.1161/JAHA.118.008757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mundi S, Massaro M, Scoditti E, Carluccio MA, van Hinsbergh VM, Iruela‐Arispe ML, De Caterina R. Endothelial permeability, LDL deposition, and cardiovascular risk factors‐a review. Cardiovasc Res. 2018;114:35–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Orr AW, Hahn C, Blackman BR, Schwartz MA. P21‐activated kinase signaling regulates oxidant‐dependent NF‐kappa B activation by flow. Circ Res. 2008;103:671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stockton RA, Schaefer E, Schwartz MA. P21‐activated kinase regulates endothelial permeability through modulation of contractility. J Biol Chem. 2004;279:46621–46630. [DOI] [PubMed] [Google Scholar]
- 18. Ogunrinade O, Kameya GT, Truskey GA. Effect of fluid shear stress on the permeability of the arterial endothelium. Ann Biomed Eng. 2002;30:430–446. [DOI] [PubMed] [Google Scholar]
- 19. Okano M, Yoshida Y. Junction complexes of endothelial‐cells in atherosclerosis‐prone and atherosclerosis‐resistant regions on flow dividers of brachiocephalic bifurcations in the rabbit aorta. Biorheology. 1994;31:155–161. [DOI] [PubMed] [Google Scholar]
- 20. Birge RB, Knudsen BS, Besser D, Hanafusa H. Sh2 and SH3‐containing adaptor proteins: redundant or independent mediators of intracellular signal transduction. Genes Cells. 1996;1:595–613. [DOI] [PubMed] [Google Scholar]
- 21. McCarty JH. The Nck SH2/SH3 adaptor protein: a regulator of multiple intracellular signal transduction events. BioEssays. 1998;20:913–921. [DOI] [PubMed] [Google Scholar]
- 22. Lettau M, Pieper J. Janssen O. Nck adapter proteins: Functional versatility in t cells. Cell Communication and Signaling; 2009:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bladt F, Aippersbach E, Gelkop S, Strasser GA, Nash P, Tafuri A, Gertler FB, Pawson T. The murine Nck SH2/SH3 adaptors are important for the development of mesoderm‐derived embryonic structures and for regulating the cellular actin network. Mol Cell Biol. 2003;23:4586–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guan S, Chen M, Woodley D, Li W. Nck beta adapter controls neuritogenesis by maintaining the cellular paxillin level. Mol Cell Biol. 2007;27:6001–6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen J, Leskov IL, Yurdagul A, Thiel B, Kevil CG, Stokes KY, Orr AW. Recruitment of the adaptor protein Nck to PECAM‐1 couples oxidative stress to canonical NF‐kappa B signaling and inflammation. Sci Signal. 2015;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third‐generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Borroto A, Reyes‐Garau D, Jimenez MA, Carrasco E, Moreno B, Martinez‐Pasamar S, Cortes JR, Perona A, Abia D, Blanco S, et al. First‐in‐class inhibitor of the T cell receptor for the treatment of autoimmune diseases. Sci Transl Med. 2016;8. [DOI] [PubMed] [Google Scholar]
- 28. Dubrovskyi O, Birukova AA, Birukov KG. Measurement of local permeability at subcellular level in cell models of agonist‐ and ventilator‐induced lung injury. Lab Invest. 2013;93:254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yuan S, Yurdagul A, Peretik JM, Alfaidi M, Al Yafeai Z, Pardue S, Kevil CG, Orr AW. Cystathionine γ‐lyase modulates flow‐dependent vascular remodeling. Arterioscler Thromb Vasc Biol. 2018;38:2126–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peng XQ, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, Pearse D, Tuder RM, Garcia JG. Protective effects of sphingosine 1‐phosphate in murine endotoxin‐induced inflammatory lung injury. Am J Respir Crit Care Med. 2004;169:1245–1251. [DOI] [PubMed] [Google Scholar]
- 31. Fabis MJ, Phares TW, Kean RB, Koprowski H, Hooper DC. Blood‐brain barrier changes and cell invasion differ between therapeutic immune clearance of neurotrophic virus and CNS autoimmunity. Proc Natl Acad Sci USA. 2008;105:15511–15516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Radu M, Chernoff J. An in vivo assay to test blood vessel permeability. J Vis Exp. 2013;73:e50062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yurdagul A, Green J, Albert P, McInnis MC, Mazar AP, Orr AW. α5β1 integrin signaling mediates oxidized low‐density lipoprotein‐induced inflammation and early atherosclerosis. Arterioscler Thromb Vasc Biol. 2014;34:1362–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Al‐Yafeai Z, Yurdagul A, Peretik JM, Alfaidi M, Murphy PA, Orr AW. Endothelial FN (fibronectin) deposition by 51 integrins drives atherogenic inflammation. Arterioscler Thromb Vasc Biol. 2018;38:2601–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aryal AC, Miyai K, Hayata T, Notomi T, Nakamoto T, Pawson T, Ezura Y, Noda M. Nck1 deficiency accelerates unloading‐induced bone loss. J Cell Physiol. 2013;228:1397–1403. [DOI] [PubMed] [Google Scholar]
- 36. Bokoch GM. Biology of the p21‐activated kinases. Annu Rev Biochem. 2003;72:743–781. [DOI] [PubMed] [Google Scholar]
- 37. Zhou GL, Zhuo Y, King CC, Fryer BH, Bokoch GM, Field J. Akt phosphorylation of serine 21 on Pak1 modulates Nck binding and cell migration. Mol Cell Biol. 2003;23:8058–8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Galisteo ML, Chernoff J, Su YC, Skolnik EY, Schlessinger J. The adaptor protein Nck links receptor tyrosine kinases with the serine‐threonine kinase Pak1. J Biol Chem. 1996;271:20997–21000. [DOI] [PubMed] [Google Scholar]
- 39. Zihni C, Mills C, Matter K, Balda MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol. 2016;17:564–580. [DOI] [PubMed] [Google Scholar]
- 40. Osborn EA, Rabodzey A, Dewey CF, Hartwig JH. Endothelial actin cytoskeleton remodeling during mechanostimulation with fluid shear stress. Am J Physiol Cell Physiol. 2006;290:C444–C452. [DOI] [PubMed] [Google Scholar]
- 41. Davies PF, Civelek M, Fang Y, Fleming I. The atherosusceptible endothelium: endothelial phenotypes in complex haemodynamic shear stress regions in vivo. Cardiovasc Res. 2013;99:315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim CW, Pokutta‐Paskaleva A, Kumar S, Timmins LH, Morris AD, Kang DW, Dalal S, Chadid T, Kuo KM, Raykin J, et al. Disturbed flow promotes arterial stiffening through thrombospondin‐1. Circulation. 2017;136:1217–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fry DL, Herderick EE, Johnson DK. Local intimal‐medial uptakes of I‐125 albumin, I‐125 LDL, and parenteral evans blue‐dye protein complex along the aortas of normocholesterolemic minipigs as predictors of subsequent hypercholesterolemic atherogenesis. Arterioscler Thromb. 1993;13:1193–1204. [DOI] [PubMed] [Google Scholar]
- 44. Pawson T. Protein modules and signaling networks. Nature. 1995;373:573–580. [DOI] [PubMed] [Google Scholar]
- 45. Dubrac A, Kunzel SE, Kunzel SH, Li JY, Chandran RR, Martin K, Greif DM, Adams RH, Eichmann A. Nck‐dependent pericyte migration promotes pathological neovascularization in ischemic retinopathy. Nat Commun. 2018;9:3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen M, She HY, Kim A, Woodley DT, Li W. Nck beta adapter regulates actin polymerization in NIH 3T3 fibroblasts in response to platelet‐derived growth factor bb. Mol Cell Biol. 2000;20:7867–7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ngoenkam J, Paensuwan P, Preechanukul K, Khamsri B, Yiemwattana I, Beck‐Garca E, Minguet S, Schamel WW, Pongcharoen S. Non‐overlapping functions of Nck1 and Nck2 adaptor proteins in T cell activation. Cell Commun Signal. 2014;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kebache S, Cardin E, Nguyen DT, Chevet E, Larose L. Nck‐1 antagonizes the endoplasmic reticulum stress‐induced inhibition of translation. J Biol Chem. 2004;279:9662–9671. [DOI] [PubMed] [Google Scholar]
- 49. Lettau M, Pieper J, Gerneth A, Lengl‐Janssen B, Voss M, Linkermann A, Schmidt H, Gelhaus C, Leippe M, Kabelitz D, et al. The adapter protein Nck: role of individual SH3 and SH2 binding modules for protein interactions in T lymphocytes. Protein Sci. 2010;19:658–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Buvall L, Rashmi P, Lopez‐Rivera E, Andreeva S, Weins A, Wallentin H, Greka A, Mundel P. Proteasomal degradation of Nck1 but not Nck2 regulates rhoa activation and actin dynamics. Nat Commun. 2013;4:2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chaki SP, Barhoumi R, Berginski ME, Sreenivasappa H, Trache A, Gomez SM, Rivera GM. Nck enables directional cell migration through the coordination of polarized membrane protrusion with adhesion dynamics. J Cell Sci. 2013;126:1637–1649. [DOI] [PubMed] [Google Scholar]
- 52. Stoletov KV, Ratcliffe KE, Spring SC, Terman BI. Nck and Pak participate in the signaling pathway by which vascular endothelial growth factor stimulates the assembly of focal adhesions. J Biol Chem. 2001;276:22748–22755. [DOI] [PubMed] [Google Scholar]
- 53. Wang YX, Chang J, Chen KD, Li S, Li JYS, Wu CY, Chien S. Selective adapter recruitment and differential signaling networks by VEGF vs. shear stress. Proc Natl Acad Sci USA. 2007;104:8875–8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Machida K, Thompson CM, Dierck K, Jablonowski K, Karkkainen S, Liu B, Zhang HM, Nash PD, Newman DK, Nollau P, et al. High‐throughput phosphotyrosine profiling using SH2 domains. Mol Cell. 2007;26:899–915. [DOI] [PubMed] [Google Scholar]
- 55. Birukov KG, Birukova AA, Dudek SM, Verin AD, Crow MT, Zhan X, DePaola N, Garcia JG. Shear stress‐mediated cytoskeletal remodeling and cortactin translocation in pulmonary endothelial cells. Am J Respir Cell Mol Biol. 2002;26:453–464. [DOI] [PubMed] [Google Scholar]
- 56. Birukova AA, Alekseeva E, Cokic I, Turner CE, Birukov KG. Cross talk between paxillin and Rac is critical for mediation of barrier‐protective effects by oxidized phospholipids. Am J Physiol Lung Cell Mol Physiol. 2008;295:L593–L602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Radu M, Lyle K, Hoeflich KP, Villamar‐Cruz O, Koeppen H, Chernoff J. P21‐activated kinase 2 regulates endothelial development and function through the Bmk1/Erk5 pathway. Mol Cell Biol. 2015;35:3990–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Thevenot E, Moreau AW, Rousseau V, Combeau G, Domenichini F, Jacquet C, Goupille O, Amar M, Kreis P, Fossier P, et al. P21‐activated kinase 3 (Pak3) protein regulates synaptic transmission through its interaction with the Nck2/Grb4 protein adaptor. J Biol Chem. 2011;286:40044–40059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lu WG, Katz S, Gupta R, Mayer BJ. Activation of Pak by membrane localization mediated by an SH3 domain from the adaptor protein Nck. Curr Biol. 1997;7:85–94. [DOI] [PubMed] [Google Scholar]
- 60. Bokoch GM, Wang Y, Bohl BP, Sells MA, Quilliam LA, Knaus UG. Interaction of the Nck adapter protein with P21‐activated kinase (Pak1). J Biol Chem. 1996;271:25746–25749. [DOI] [PubMed] [Google Scholar]
- 61. Teyra J, Huang HM, Jain S, Guan XY, Dong AP, Liu YL, Tempel W, Min JR, Tong YF, Kim PM, et al. Comprehensive analysis of the human SH3 domain family reveals a wide variety of non‐canonical specificities. Structure. 2017;25:1598–1610.e3. [DOI] [PubMed] [Google Scholar]
- 62. Aitio O, Hellman M, Kesti T, Klein I, Samuilova O, Paakkonen K, Tossavainen H, Saksela K, Permi P. Structural basis of PxxDY motif recognition in SH3 binding. J Mol Biol. 2008;382:167–178. [DOI] [PubMed] [Google Scholar]
- 63. Tzima E, Irani‐Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao GY, DeLisser H, Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437:426–431. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S7